

Abstract
Lipocalin prostaglandin D synthase (L-PGDS) regulates synthesis of an important inflammatory and signaling mediator, prostaglandin D2 (PGD2). Here, we used structural, biophysical, and biochemical approaches to address the mechanistic aspects of substrate entry, catalysis, and product exit of this enzyme. Structure of human L-PGDS was solved in a complex with a substrate analog (SA) and in ligand-free form. Its catalytic Cys 65 thiol group was found in two different conformations, each making a distinct hydrogen bond network to neighboring residues. These help in elucidating the mechanism of the cysteine nucleophile activation. Electron density for ligand observed in the active site defined the substrate binding regions, but did not allow unambiguous fitting of the SA. To further understand ligand binding, we used NMR spectroscopy to map the binding sites and to show the dynamics of protein-substrate and protein-product interactions. A model for ligand binding at the catalytic site is proposed, showing a second binding site involved in ligand exit and entry. NMR chemical shift perturbations and NMR resonance line-width alterations (observed as changes of intensity in two-dimensional cross-peaks in [1H,15N]-transfer relaxation optimization spectroscopy) for residues at the Ω loop (A-B loop), E-F loop, and G-H loop besides the catalytic sites indicate involvement of these residues in ligand entry/egress.
Keywords: lipid signaling, X-ray crystallography, nuclear magnetic resonance spectroscopy, lipophilic substrate, protein-lipid interaction, β trace protein
Prostaglandins are members of the eicosanoid family alongside leukotrienes, lipoxins, epoxy fatty acids, and thromboxane (1, 2). They are involved in various biological responses such as inflammation, allergy, nociception, cell growth, cell proliferation, and tumorigenesis (3, 4). Hormonal or stress stimuli induce hydrolysis of glycerophospholipids to free arachidonic acid (AA) by cytoplasmic phospholipase A2. The substrate AA enters the cyclooxygenase (COX) pathway to yield prostaglandin H2 (PGH2) (5), which acts as a substrate for various downstream cell-specific enzymes producing prostaglandin D2 (PGD2), prostaglandin I2 (PGI2), prostaglandin E2, prostaglandin F2α, and thromboxane. There are two types of prostaglandin D synthase (PGDS), namely hematopoietic PGDS (H-PGDS) and lipocalin prostaglandin D synthase (L-PGDS). They are distributed in different tissues and distinct in structure; nonetheless both perform the same catalytic reaction to produce PGD2.
L-PGDS is a major PGD2 synthase in the central nervous system, heart, and reproductive tissue, catalyzing the isomerization of PGH2 to PGD2 (6). This protein is involved in pain induction, nephropathy, immunomodulation, adipocyte differentiation, lipid and carbohydrate metabolism, as well as sleep-wake regulation (7–11). Similar to other prostaglandin synthases such as COX1, COX2, prostaglandin E synthase (PGES), and prostaglandin F synthase, L-PGDS is also associated with cancer progression (4). However, unlike PGES's prominent role in enhancing tumorigenesis and metastasis, PGDS expression is downregulated in cerebral spinal fluid of patients suffering from a brain tumor (12). PGD2 production by L-PGDS activates peroxisome proliferator-activated receptor (PPAR)γ to inhibit tumor cell growth (13). Apart from its anti-tumorigenic function, PGD2 has also been identified as a potential diagnostic marker for hypertension-related renal injury and gentamicin-induced renal impairment (14, 15). Thus, L-PGDS knockout mice have been used as a model for diabetic studies (16). In addition, studies by various research groups have proposed the diagnostic application of L-PGDS in obstructive azoospermia (17), and neurological disorders (18). Recently L-PGDS and PGD2 were linked to hair loss and were found to be overexpressed on the bald scalp of patients suffering from androgenetic alopecia (19).
L-PGDS, in addition to its enzymatic role, also functions as a transporter for lipophilic ligands. Interestingly, chicken and fish L-PGDS lack enzymatic activity and operate primarily as lipophilic ligand vehicles (20). Human L-PGDS binds to a range of all-trans-retinoic acids (RAs), biliverdin, bilirubin, and thyroid hormones with good affinity (21). Hence, it has been proposed to be a vital RA transporter and scavenger for bile pigments (22, 23). Lipocalin family members often display promiscuous binding to other lipophilic compounds beside their endogenous ligands (24). Its broad specificity in ligand binding suggests that L-PGDS could be exploited as a drug delivery system for lipophilic molecules (25).
Previous structural studies have been achieved using mutants whereby the catalytic cysteine residue (Cys 65 in human L-PGDS) has been substituted for alanine. Structures of mouse L-PGDS determined using both NMR and X-ray crystallography established the lipocalin family fold of the protein as well as the presence of a large cavity for substrate and transport-ligand binding (26–28). NMR titration experiments also predict the binding of substrate and hydrophobic transport-ligand to the large cavity (29). Mutation analysis has identified conserved Cys 65 as the critical residue that confers enzymatic activity (27). Subsequently, the crystal structure of mutant human L-PGDS (C65A) with bound fatty acids was determined, revealing an extended binding mode of two associated lipids in the L-PGDS pocket (30). Despite extensive efforts from several groups, detailed understanding of L-PGDS substrate binding and catalytic mechanism is still limited. No informative structural complex with substrate or product analogs has been obtained thus far.
We have attempted to determine the substrate binding modes of L-PGDS with substrate and product analogs using X-ray crystallography. Our data define the substrate/product binding regions in the active site and identify potential interactions of the substrate with the protein. Comparison of structures with and without analog revealed conformational changes in the protein, mediating substrate entry, enzyme catalysis, and product egress. This is the first structure of wild-type human L-PGDS with substrate analog (SA) U44069, allowing a detailed description of the environment of the catalytic cysteine. Finally, NMR titration studies with substrate and product analogs as well as membrane mimetics further elucidated the dynamics of substrate binding and a potential mode of interaction with the membrane.
MATERIALS AND METHODS
Constructs were obtained from the Mammalian Genomic Consortium. All prostaglandin analogs and chemical assays were purchased from Cayman Chemicals.
Cultivation and expression
The sequence encoding human L-PGDS was subcloned into pNIC-CH2 vector with C-terminal 6xHis tag and expressed in Rosetta BL21-DE3 Escherichia coli in Terrific Broth media consisting of tryptone, yeast extracts, and glycerol. Cells were grown at 37°C until OD600 nm = 1.0 and induced with 0.5 mM isopropyl-beta-D-1-thiogalactopyranoside (IPTG) at 18°C for 18 h. Cells were resuspended and sonicated in buffer containing 20 mM HEPES pH 8.0, 300 mM NaCl, 2 mM tris-(2-carboxyethyl)phosphine (TCEP), EDTA-free protease inhibitor cocktail (Merck), and 1 μl of Benzonase (Merck) per 1 l culture. After centrifugation, the lysate was loaded onto a 1 ml Nickel-affinity column equilibrated with Buffer A (20 mM HEPES pH 8.0, 300 mM NaCl, 2 mM TCEP, 10 mM imidazole) and eluted with Buffer B (20 mM HEPES pH 8.0, 300 mM NaCl, 2 mM TCEP, 500 mM imidazole). Fractions containing L-PGDS were pooled and further purified by gel filtration using HiLoad 16/60 Superdex 75 equilibrated with Buffer C (20 mM HEPES pH 6.5, 150 mM NaCl, 2 mM TCEP). The color of protein fractions changed from yellow to colorless with increasing elution time. Protein elute later appeared less yellow and showed good dispersion in 15N-heteronuclear single quantum correlation (HSQC) measurement. Only colorless fractions were pooled and concentrated to 5.5 mg/ml for crystallization trials. Protein identity was confirmed by mass spectrometry and Western blot analysis.
Crystallization
L-PGDS was cocrystallized with SA U44069 9,11-epoxymethano PGH2 (Table 1) in condition A (0.1 M potassium thiocyanate and 30% PEG-MME 2000) in 1:1 protein-reservoir ratio. Crystals appeared after 5 days of incubation at 4°C by hanging drop vapor diffusion. Cocrystals were also obtained in condition B (1.4 M tri-sodium citrate pH 6.5) using a similar method except in 2:1 protein-reservoir ratio. Micro-crystals from condition A were used to seed crystallization of ligand-free L-PGDS in the same condition but in the absence of SA U44069. Crystals from condition A were cryo-protected using reservoir with 25% glycerol added while crystals from condition B were cryo-protected with 1.6 M tri-sodium citrate solution.
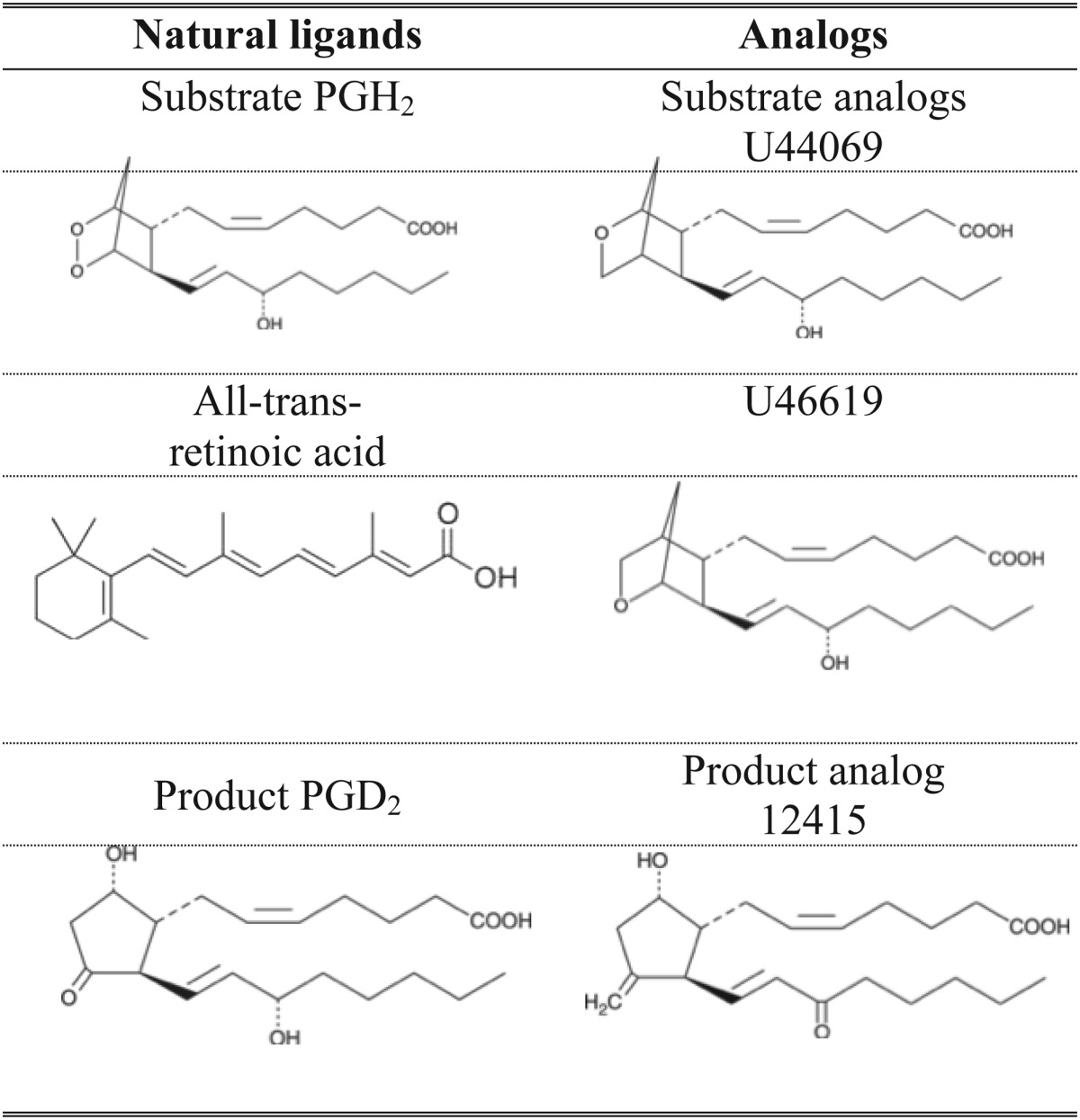
TABLE 1.
Chemical representation of natural substrates, ligands, and analogs used in this study
Data collection and processing
Native data sets were collected at beam line (BL)13C1 and BL13B1 at the National Synchrotron Radiation Research Center, Taiwan, Republic of China. Data sets were processed using HKL-2000 (31) and iMosflm (32), phases were generated by molecular replacement (MR, Phaser) (33) with mouse L-PGDS (PDB ID: 2CZT). Automatic building of the structure was carried out using ARP/wARP 7.3 (34), ligand fitting was performed in Coot 0.6.2 (35), and refinement was performed using autoBUSTER (Global Phasing Limited) and REFMAC5 (36) in the CCP4 suite (37). Table 2 lists the final statistics for L-PGDS-ligand structure.
TABLE 2.
Data collection and refinement statistic
| SA Complex 1 (4IMO) | APO (4IMN) | |
| Crystallization condition | 0.1M potassium thiocyanate, 30% PEG MME 2000 | 0.1M potassium thiocyanate, 30% PEG MME 2000 |
| Data collection | ||
| Space group | P212121 | P212121 |
| Cell dimension | ||
| a, b, c (Å) | 36.24, 56.38, 72.92 | 36.41, 56.41, 72.99 |
| α, β, γ | 90, 90, 90 | 90, 90, 90 |
| Wavelength resolution (Å) | 44.6–1.88 (1.92–1.88) | 44.6–2.09 (2.09–2.16) |
| Rsym | 0.062 (0.412) | 0.060 (0.303) |
| I/σI | 17.8 (3.6) | 18.6 (3.23) |
| Completeness (%) | 100 (99.2) | 94.6 (95.3) |
| Redundancy | 5.4 (5.5) | 5.0 (4.7) |
| Refinement | ||
| Resolution (Å) | 1.88 | 2.09 |
| No. reflections | 13,902 | 4,675 |
| Rwork/Rfree | 0.184/0.240 | 0.174/0.243 |
| No of atoms | 1,386 | 1,303 |
| Protein | 1,241 | 1,221 |
| Ligand | 56 | 17 |
| Water | 89 | 65 |
| B-factors | 22.9 | 33.9 |
| R.m.s deviations | ||
| Bond length (Å) | 0.020 | 0.018 |
| Bond angles (°) | 2.012 | 1.969 |
| Ramachandran plot | ||
| Most favored (%) | 96.8 | 97.4 |
| Allowed (%) | 3.2 | 2.6 |
| Disallowed (%) | 0.0 | 0.0 |
Reported datasets are obtained from two different crystal structures. Values in parentheses are for highest-resolution shell. APO, apoenzyme.
NMR protein sample preparation
15N single-labeled protein was prepared in M9 minimal media supplied with 15N-labeled ammonium chloride. Protein was induced at OD600 nm = 0.7 with 0.5 mM IPTG at 18°C for 18 h. 13C and 15N double-labeled sample was prepared by supplementing 13C-labeled glucose at the beginning of cultivation and inducing at OD600 nm = 0.6 with 0.5 mM IPTG at 18°C for 20 h. The final protein concentration was 0.3 mM for the 15N-labeled sample and 0.27 mM for the double-labeled sample. Both samples were in buffer consisting of 20 mM HEPES pH 6.5, 150 mM NaCl, 2 mM TCEP, 10% D2O, and 0.5 mM 4,4-dimethyl-4-silapentane-1-sulfonic acid for NMR experiments. Selective labeling of 1-13C amino acids, leucine, and alanine in the background of [1H-15N] were carried out as described in the study by Wagner et al. (38).
NMR spectroscopy
NMR spectroscopy experiments were measured using Bruker Avance 700 MHz with triple resonance z-axis gradient cryoprobe at 298 K. All experiments were recorded using a protein concentration of 0.3–0.5 mM. Data were processed by Topspin 2.2 (Bruker Corporation) and analyzed by CARA (www.nmr.ch) (39). 15N-nuclear Overhauser effect spectroscopy (NOESY)-HSQC was measured at the Swedish NMR Center, Gothenburg using Varian 800 MHz. The backbone resonances were assigned using transfer relaxation optimization spectroscopy (TROSY)-HNCA, CBCA(CO)NH, and 15N-NOESY-HSQC experiments. TROSY-HN(CO) experiments were applied to further aid in residue-specific resonance assignment.
NMR titration
Unlabeled ligands SA U44069, SA U46619, and PA 12415 (Table 1) were titrated in 15N-labeled protein sample and a series of 15N-HSQC experiments were measured for analysis of ligand binding interactions at concentrations of 2 mM, 3 mM, and 4 mM. Protein-ligand complex and detergent interactions were evaluated by adding dodecylphosphocholine (DPC) into the protein-ligand solution. DPC stock solution of 60 mM was prepared by dissolving the detergent in Millipore water. The samples contained 0.35 mM protein, 4 mM SA U44069 or PA 12415, and 3 mM DPC [critical micelle concentration (c.m.c.), 1.2 mM]. Chemical shift changes were mapped and the perturbations were calculated using the following equation,
Differential static light scattering assay
Protein thermal stability was measured using StarGazer-384 from Harbinger Biotechnology and Engineering. Under heat-induced denaturation, protein slowly unfolds and aggregates. The rate of protein aggregation can be measured by differential static light scattering and the intensity of scattering is plotted against temperature to obtain a temperature aggregation regression curve. This assay was carried out and analyzed according to the protocol stated in a previous study (40). Each well of a 384-well black assay plate (Nunc) had 45 μl of 0.3 mg/ml L-PGDS protein in buffer containing 20 mM HEPES pH 6.5, 150 mM NaCl, 2 mM TCEP, and 0.5 μl of ligand(s) dissolved in DMSO. The protein-ligand molar ratio was 1:10 for all ligands. To screen for stabilizing factors of L-PGDS, we used ligands such as RA, SA U44069, PA 12415, MgCl2, CaCl2, and in any combination of two to study if a mutually stabilizing effect could be observed.
Isothermal titration calorimetry
L-PGDS (200 μM) in Buffer C was titrated with 2.5 mM of product PGD2 in the same buffer and concentration of DMSO to measure the binding affinity of the protein with product using the MicroCal iTC200 system (Northampton, MA). The experiment was initiated with 0.5 μl of titrant injection and followed by sixteen 2.4 μl injections, each in 180 s intervals at 27°C. Stirring speed for the titration event was 1,000 rpm. Integration of heat peaks and regression analysis were performed with Origin software.
Enzymatic assay
Functional activity of L-PGDS was tested with Cayman Chemicals PGD2-methoxylamine (MOX) ELISA kit assay (catalog number 512011). L-PGDS concentration was diluted according to the protocol for optimal enzymatic reaction. Detection of the PGD2-MOX concentration was measured by the intensity of specific secondary fluorescence antibody provided.
RESULTS
Ligand binding to human L-PGDS
Because the biological substrate (PGH2) and product (PGD2) are chemically unstable, analogs were used for binding studies. To establish structural information of L-PGDS in complex with the substrate and product analog, we first wanted to characterize the binding of these ligands to L-PGDS and to confirm the catalytic activity of the recombinant protein. To identify suitable ligands/analogs and conditions for optimal binding, we used a thermal shift assay (TSA) based on a differential static light scattering assay (DSLS, Stargazer, Harbinger Biotech). This assay monitors the rate of protein unfolding by measuring aggregation formed after thermally induced unfolding (41, 42). The thermal shifts for L-PGDS with SA U44069, PA 12415, and RA and any two combinations of the above were measured (Fig. 1A). SA U44069 binding to L-PGDS induced an 8°C shift of its aggregation temperature (Tagg), indicative of strong binding. PA 12415 and RA also increased the aggregation temperature of the protein, in the range of 2–3°C. It was previously reported that H-PGDS activity is Mg2+ dependent (43). However, neither Mg2+ nor other divalent cations like Ca2+ stabilized L-PGDS, so a specific binding site on the protein is not likely (data not shown). The combination of RA with SA U44069 or PA 12415 did not further increase the aggregation temperature suggesting overlapping binding sites (Fig. 1A).
Fig. 1.

A: Thermal shift assay of L-PGDS in the presence of SA U44069, PA 12415, and RA shows various degrees of stability induced by these ligands. B: Recombinant protein is able to produce PGD2 when supplemented with substrate PGH2. Its catalytic activity is efficiently inhibited by SA U44069, PA 12415, and RA. WT, wild type. C: Human L-PGDS binds to its product PGD2 at a Kd of 154 μM, suggesting that the protein binds to its product even postcatalysis. Asterisk (*) indicates test injection; data was not included in integration.
The enzymatic activity of wild-type recombinant L-PGDS was measured based on detection of the product PGD2 after incubation with substrate PGH2 using Cayman Chemical's PGD2-MOX ELISA kit. Recombinant L-PGDS was shown to be active (Fig. 1B) and a fixed time point assay of L-PGDS measured a Vmax of 3.66 μmol/μg/s and Km of 4.15 μM. These values are in a similar range to those previously reported for recombinant mouse and human L-PGDS (27, 30). In addition, our data also show that SA U44069, PA 12415, and RA can inhibit the catalytic activity. This result agrees with Shimamoto et al. (29) who showed that RA inhibits mouse L-PGDS. In their study, they modeled two separate binding pockets for substrate and RA respectively. However the two sites were proposed to share one amino acid; it is not certain that the residue facilitates binding of both substrate and RA. Nonetheless, their Lineweaver-Burk analysis of a kinetic study claimed that the inhibition was noncompetitive (29). L-PGDS also inherently binds its product with a Kd of 154 μM (Fig. 1C).This is an interesting observation because most enzymes are designed to bind weakly to their products to facilitate release from the active site.
Cystal structure of L-PGDS in complex with substrate analog and in substrate analog free form
Human L-PGDS was cocrystallized with SA U44069 in a P212121 unit cell with one monomer in the asymmetric unit. The overall structure had the classical lipocalin fold comprising an eight-stranded β-barrel with two α-helices. One of the helices, known as helix 2 on the Ω loop, lines the “entrance” of the large substrate and ligand binding cavity (Fig. 2A). The electron density map reveals two potential ligand binding sites in the cavity; one site near to the catalytic Cys 65 (site A, Fig. 2C) and one site beneath Trp 112 (site B, Fig. 2D). Crystals of L-PGDS (in the same crystal form) without SA U44069, were generated by micro-seeding with crystals grown in the presence of SA U44069 into a crystallization solution lacking the analog. The overall structure of L-PGDS without SA U44069 is very similar to the structure with it, except for distinct conformational changes in the region of the Ω loop (Fig. 3). Density at site A is lacking in the SA U44069 free form, instead a long extended density is found stretching from site B into the cavity. We have interpreted this density as a bound polyethylene glycol (truncated PEG MME 2000, supplementary Fig. ID), a component found in the crystallization reservoir. The atomic coordinates and experimental data for the SA U44069 bound protein crystal, SAC (PDB ID: 4IMO) and ligand-free-form crystal, APO (PDB ID: 4IMN) have been deposited in the Protein Data Bank (www.wwpdb.org).
Fig. 2.

A: Crystal structure of L-PGDS in complex with SA U44069 at site A and site B. β sheet A–H form the core barrel with four prominent inter-sheet loops namely the Ω loop (which consists of helix 2), C-D loop, E-F loop, G-H loop, and one long α helix H3. B: Detailed view of the binding site. Two SA U44069 were modeled into two regions of observed densities in site A and site B respectively. C: A zoomed in view of binding pocket A with two configurations that differ in the location of the carboxylate group at position A1. The head group of the bicyclopentane ring can also be positioned in A2, the Fo-Fc map at sigma level = 3.0 while the 2Fo-Fc map = 1.0. D: Binding pocket B with corresponding Fo-Fc map and 2Fo-Fc map at sigma levels 3.0 and 1.0, respectively.
Fig. 3.

A: Overlay of our substrate analog cocrystal (SAC) crystal structure in blue with apoenzyme (APO) structure in beige, root means square deviation values: 0.358. B: Ω loop of SAC structure, which is tilted outward in the presence of SA U44069; Trp 54 and Lys 59 on the Ω loop are observed to undergo major configuration changes.
Detailed experimental data on substrate binding in L-PGDS has been elusive. Hence, we attempted to generate a molecular framework by solving the structure of human L-PGDS cocrystallized with SA U44069. The difference density map revealed a number of residual densities in the large hydrophobic cavity of L-PGDS that represent potential SA U44069 binding sites (Fig. 2C, D). Judging from the local interactions with the protein and in comparison with the analog free crystals in the same form, these density peaks are not likely to correspond to other buffer molecules but instead must originate from bound SA U44069. Even though the electron density is not sufficiently continuous for unambiguous modeling of SA U44069, it is very likely that the strongest density patches correspond to the SA head groups. Based on this hypothesis we have generated putative binding models for SA U44069 binding to L-PGDS at site A and site B.
Site A shows the major distinct prostaglandin head group density with a distance of 6 Å above the catalytic Cys 65 positioning its bicycloheptane ring in interaction with the Phe 83 side chain at site A1 (Fig. 2B). A second binding site, A2, is also possible; this model positions the SA U44069 further down in the pocket with less than 5 Å from the head group to Cys 65. The carboxylic group of SA U44069, bound in site A1, appears to extend into a polar patch formed by His 111 and Tyr 116. The hydroxyl group of the prostaglandin ω chain is in close proximity with Cys 65 and the aliphatic tail in this model is inserted into a hydrophobic pocket of Leu 79, Leu 131, and Tyr 149. The binding mode of SA U44069 in site A1 is not likely to be identical to the binding mode of the substrate during catalysis, as the bicycloheptane ring is too far from the catalytic Cys 65. The bicycloheptane in site A2, however, is more representative of a productive substrate binding mode. With the head group in this second position, the aliphatic chain could extend deeper into the hydrophobic pocket, allowing the hydroxyl group of the prostaglandin ω chain to interact with Tyr 149 and Thr 147, equivalent to what has been proposed from substrate modeling in mouse L-PGDS (27). The carboxyl group of the substrate could still reach the polar patch containing His 111 and Tyr 116. Alternatively, it could reach Arg 92 and Arg 85 lining the groove of the cavity, as proposed for the mouse protein (27). In our L-PGDS structure, Arg 92 forms a salt bridge with Glu 35 of a neighboring molecule in the crystal lattice.
In site B, the prostaglandin head group is sandwiched between Trp 112 on the E-F loop and Phe143 of the H β-strand through stacking interactions. Modeling of the SA U44069 into this density enables its hydroxyl group to make a hydrogen bond with the hydroxyl group of Tyr 116 (Fig. 2C). Both the aliphatic and the carboxylate arms of the analog are exposed to the solvent region. In the SA U44069 free form, we could see a potential PEG MME 2000 molecule binding in site B, extending further into the large cavity. Similarly, in the structure of fatty acid-coordinated human L-PGDS, the fatty acid is clamped between Trp 112 and Phe 143 and extends into the large cavity. Previously, there was no direct evidence showing that the E-F loop containing Trp 112 and G-H loop containing Phe 143 are involved in the binding of substrate or product, although it has been proposed to facilitate the exit of product from the protein (27). Binding in site B reveals ligand stabilization of the E-F and G-H loops, and it is highly possible that the substrate and/or the product use this site as an intermediate site before exit/entrance.
The noncontinuous ligand density of the SA U44069 cocrystallized structure is most likely due to a combination of multiple binding modes and flexibility of the aliphatic moieties of the ligand. After a test refinement with models in sites A1 and B, the B factor values of the modeled ligands at both sites are in the range of 45–55 Å2, while B factor values of neighboring residues are in the range of 20–25 Å2. The most notable structural change upon SA U44069 binding is the rotation of helix 2 in the Ω loop, which unfolds outward, partially losing α helical rigidity. The Trp 54 side chain, as well as its main chain, showed high temperature B factor (110–120 Å2) in comparison with neighboring residues (B factor: 20–30 Å2). However, in the SA U44069 free structure it has a B factor of 30–44 Å2. Hence, Trp 54 could serve as a gate for substrate upon entry/egress. Conformational change in the Ω loop is also observed when the SA U44069 bound structure is compared with the recently solved fatty acid bound C65A mutant structure (PDB ID: 3O22). Binding sites A and B of SA U44069 in our structure coincide with the binding sites for oleic acid and palmitic acid in the fatty acid bound structures.
Most residues such as Trp 54, Phe 83, Tyr 107, His 111, Trp 112, Tyr 116, and Phe 143 interacting with SA U44069 in our structure are also preserved in close homologs (Fig. 4A). Even though Trp 54 is highly conserved across several species, mutation of Trp 54 in mouse L-PGDS only moderately decreases the catalytic activity of L-PGDS (27). Although Trp 112 is conserved in mammals and fish, it has been replaced by tyrosine in most other species, therefore retaining the basic aromatic structure required for base stacking interaction. Phe 83 is conserved in mammals, but substituted by tyrosine in Xenopus laevis. We agree with the prediction by Zhou et al. (30) that the aromatic side chain is crucial in holding the bicyclopentane head group of the substrate as shown in our structure.
Fig. 4.

A: Multiple sequence alignment of human L-PGDS with homologs. Residues proposed to interact with substrates from crystal structure results are shown as purple stars; mutations made by previous studies are highlighted as blue stars; and catalytic cysteine is indicated as a red star. B: Catalytic site comparison of Cys 65 environment in substrate analog cocrystal (SAC) and apoenzyme (APO) structure with respect to its neighboring serines. C: Schematic of L-PGDS catalytic reaction. Production of PGD2 requires correct stereochemical positioning of SA U44069 in the catalytic site.
Structure of human L-PGDS catalytic site
The resolution of the structures allows for a detailed analysis of the environment of the catalytic cysteine. Previous crystal and NMR structures of mouse (27, 29) and human (30) L-PGDS were obtained from protein with the catalytic cysteine mutated to alanine. The structure of the wild-type human L-PGDS without substrate reveals that Cys 65 has two potential side chain conformations. One of them has higher occupancy and is hydrogen bonded to the main chain carbonyl group of residue Ser 63. The thiol sulfur is not well exposed to the active site pocket but instead extends into a hydrophobic patch formed by Leu 62 and is exposed to the solvent outside the protein.
In the structure of SA U44069 bound L-PGDS, Cys 65 adopts two conformations and the dominant conformation is exposed into the pocket of L-PGDS, making a strong hydrogen bond to Ser 45 (2.8 Å) (Fig. 4B). Ser 45 also has two conformations. Interestingly, a distinct conformational switch is induced in the region of Lys 59 and Leu 62 upon substrate binding, resulting in a complete inversion of Lys 59 (Fig. 3B). This inversion is most likely due to the conformational changes observed in the Ω loop upon substrate binding, as discussed above. Mutation of K59A has been shown to increase enzymatic effect (23) by mimicking the effect of Lys 59 twisting outward to accommodate substrate binding. Shifting of the Leu 62 side chain results in the Cys 65 thiol group becoming more exposed to the substrate binding cavity with its side chain forming strong hydrogen bonding to Ser 45, thus activating Cys 65 for the subsequent reaction steps. Mutagenesis studies revealed the involvement of Ser 67 and Ser 81 in catalysis (23), but we did not observe any direct hydrogen bonding to the Cys thiol group in the structure presented here. However, we do not exclude the possibility that such hydrogen bonds could be formed upon productive substrate binding. In our structure, we identified a highly conserved residue, Leu 62, which was shown to play an important role in unraveling Cys 65 for substrate binding.
The picture that emerges from the structural data shows that substrate binding induces a distortion of helix 2 on the Ω loop and Leu 62, which exposes the Cys 65 thiol group into the substrate binding site. This allows Cys 65 to form a strong hydrogen bond to Ser 45, which helps in activating the cysteine nucleophile. The close juxtaposition of the nucleophile with substrate peroxide promotes the nucleophilic attack on C11 of the substrate (Fig. 4C). This might act in concert with the protonation of O9, to minimize net charge buildup along the reaction trajectory.
Mapping of L-PGDS interactions with substrate and PAs using NMR titration
NMR titration was used to further understand the dynamics of substrate binding to L-PGDS. L-PGDS was uniformly labeled with 15N isotope for [1H,15N]TROSY experiments and double-labeled with 15N and 13C for residue-specific assignment of backbone resonances. Reference spectra were acquired at 2 mM of SA U46619, an analog that has been used for NMR titration against the mouse protein (29). The analog saturation point was found to be at 2 mM. The spectra showed 93.3% of the expected number of cross-peaks (supplementary Table I and supplementary Fig. II). Subsequently, two resonance assignment experiments, namely HNCA and CBCA(CO)NH, together with a 15N-NOESY HSQC spectra were collected for residue-specific assignment. Overall, 75% of protein assignment was achieved for L-PGDS (supplementary Fig. II).
A series of titration studies were performed with SA U44069 and PA 12415 whereby similarly the saturation point was observed at ligand concentrations at or above 2 mM. Chemical shift differences above a threshold of 0.1 ppm were mapped onto the crystal structure (Fig. 5A, B). It was observed that both SA U44069 and PA 12415 induced similar chemical shift perturbations in L-PGDS. Resonance intensities for most cross-peaks were significantly increased when either SA U44069 or PA 12415 was added (supplementary Fig. III and supplementary Table II). This reflects a narrower line-width with the concomitant reduction of the conformational exchange-induced line broadening. In the presence of the SA U44069, residues Ala 49, Trp 54, Arg 56, and Glu 57 on the Ω loop as well as Trp 112 and Tyr 116 on the E-F loop showed large changes in chemical shifts (>0.1 ppm) consistent with the conformational changes seen in our crystal structures (Fig. 5A). This was accompanied by the lower chemical shift differences (>0.08 ppm) observed for Gly 140, Asp 142, and Arg 144 in the G-H loop (Fig. 5A). As expected, the chemical environment of the catalytic site residue, Cys 65, was altered and chemical shift changes of neighboring residues Met 64, Thr 147, and Leu 148 were also perturbed.
Fig. 5.

A: Chemical shift perturbations induced by addition of SA U44069. In the crystal structure below, residues shifted 0.1–0.3 ppm are represented in red while residues peaks that appeared or disappeared are represented in dark red. B: Similar thresholds were set for perturbations observed in the presence of PA 12415. In the cartoon structure, chemical shift perturbations of 0.1–0.3 ppm are in dark blue while residues peaks with intensity attenuation are highlighted in blue. Secondary structure of L-PGDS was represented as follow: yellow blocks for the β sheet. orange block for the α helix; and white blocks for the inter-sheet loop.
In the presence of PA 12415, changes in chemical shift at the Ω loop were only slightly less than that of SA (Fig. 5B). Two residues on the β-strand H, Met 145 and Ala 146, showed perturbations specific to PA. Chemical shifts of residues Asp 37, Trp 43, Phe 39, and Tyr 128 were consistently perturbed when both analogs were added. These four residues are situated at the narrow opening of the barrel structure, opposite to the Ω loop. It is likely that these residues alter their side-chain conformations to form a flexible plug during ligand entry/egress and binding.
Evaluation of the resonance peaks’ intensity increments between bound and unbound L-PGDS are shown in supplementary Fig. III. We attribute this general increase of 2D cross-peak intensities caused by the addition of SA U44069 to the narrowing of the apparent resonance line-widths. This is due to the more restricted conformational space spawned by the protein's backbone and concomitant structural stabilization. Structural accretion due to ligand binding reduces conformational exchange-induced line broadening and this effect is also shown in the case of human protein disulphide isomerase (PDI) caused by peptide ligand binding (44). In L-PGDS approximately 24% of the peaks have intensity increases of more than 50%. These cross-peaks correspond to residues that undergo significant chemical shift perturbations. It is not known whether these residues are in direct or indirect contact with ligands because both intensity and chemical shifts were altered (45). Nonetheless, line-width reduction accompanied with increased intensity has been observed in the complex formation of the troponin C regulatory domain and troponin I (46). This suggests a stable and tight complex formation of the troponin protein. Likewise in the L-PGDS-analog complex, these NMR results are consistent with our TSA data and previous small angle X ray scattering data (28) showing that L-PGDS is greatly stabilized by SA U44069 and PA 12415.
By utilizing both X-ray crystallography and NMR spectroscopy data, we were able to provide a glimpse into L-PGDS substrate binding mechanics. Both methods suggested three key points of L-PGDS binding. First, substrate entry and exit was facilitated by both the E-F loop and the Ω loop. Second, residues in reaction center proximity, Met 64, Cys 65, and Ser 67, changed their rotamer configuration and backbone conformation upon ligand binding. β-sheet B (Fig. 2A), which is comprised of catalytic residues such as Met 64, Cys 65, and Ser 67, became disordered upon ligand addition. Third, N-terminal residues of the barrel cavity, usually described as the bottom of the calyx structure underwent “plugging” upon ligand binding. Phe 34 had been speculated to be involved in such an event due to poor electron density being observed (35). However our crystal data had clear density of Phe 34 in the presence and absence of SA U44069. In our NMR data, residues Asp 37, Phe 39, Trp 43, and Tyr 128 underwent chemical shift perturbations (Fig. 5A, B) while residues Thr 73, Gly 75, Gly 76, and Leu 77, which corresponded to the B-C loop, showed an increase in intensities upon ligand binding (supplementary Fig. III). We propose that the bulky side chains of Trp 43, Phe 39, and the hydrophobic side chain of Leu 77, together with the B-C loop, form a barrier at the barrel opening. Table 3 summarizes the key residues involved in substrate recognition, proposed role in catalysis, product release, and plugging observed from both crystal data and NMR results.
TABLE 3.
Summary of residues involve in the substrate binding, catalysis and exit
| Residues | Conservation/Substitution in Orthologs | Proposed Function |
| Phe 83 | Highly/tyrosine | Position bicyclopentane ring of substrate |
| Leu 62 | Highly/methionine | Expose Cys 65 sulfur group |
| Lys 59 | Conserved | Facilitate substrate entry |
| Cys 65 | Conserved | Nucleophile |
| Ser 45 | Partial/threonine | Deprotonate Cys 65 |
| Ser 81 | Highly/alaninea | Catalysis |
| Ser 67 | Partial/threonine | Catalysis |
| Trp 54 | Conserved | Gating/product exit |
| Trp 112 | Highly/tyrosine or serine | Gating/product exit |
| His 111 | Highly/arginine | Position carboxylic tail of substrate |
| Tyr 107 | Highly/phenyalanine | Position carboxylic tail of substrate |
| Leu 79 | Conserved | Positioning aliphatic tail of substrate |
| Leu 131 | Conserved | Positioning aliphatic tail of substrate |
| Tyr 149 | Conserved | Positioning aliphatic tail of substrate |
| Asp 37 | Highly/glutamate | Plugging of clayx |
| Trp 43 | Conserved | Plugging of clayx |
| Tyr 128 | Conserved | Plugging of clayx |
| Thr 73 | Highly/alanine | Plugging of clayx |
| Gly 75 | Conserved | Plugging of clayx |
| Leu 77 | Highly/methionine | Plugging of clayx |
Only in Xenopus.
L-PGDS interactions with dedecylphosphocholine to model membrane interactions
Besides protein-ligand interaction studies, protein-detergent interactions were studied in the absence and presence of ligand in order to clarify how L-PGDS interacts with a mimetic membrane and how this might regulate its activity. DPC has been commonly used as a mimetic membrane study model in solution NMR (47). Undeuterated DPC of 3 mM concentration was added to isotopically labeled protein premixed with 2 mM of SA U44069 or PA 12415. DPC micelles usually constitute a 70-80mer aggregation moiety at 25°C in 50 mM NaCl, which corresponds to a size of about 26 kDa (48). Signal disruptions of the labeled protein residues by undeuterated DPC can be detected in two-dimensional 1H-15N-HSQC experiments. Perturbations resulting from DPC are distinct in the presence of SA U44069 and PA 12415, or in the absence of any ligands.
Figure 6A, B shows the magnitude of chemical shift differences when DPC was added in solution under two conditions, SA U44069 with DPC (SA-DPC) or PA 12415 with DPC (PA-DPC). 15N-HSQC spectra were also acquired for all three samples with titrations of 3 mM and 6 mM DPC. Samples were measured for a total of 6 h with spectra being recorded hourly to observe protein state over time. There were no significant changes in chemical shifts of the protein within the period of 6 h or at higher concentrations of DPC. No degradation of protein was observed throughout the experiments. Resonance intensity attenuation after addition of DPC was measured in both conditions (Fig. 6C, D).
Fig. 6.

Interactions of L-PGDS with DPC micelles in the presence of SA U44069 (SA-DPC) and PA 12415 (PA-DPC). Panel (A) shows the chemical shift perturbations for SA-DPC and (B) for PA-DPC. The residues involved are mapped to crystal structure in the middle. C: Intensity attenuation of L-PGDS cross-peaks in the event of DPC interaction either with SA U44069 or PA 12415 in (D). The residues involved were mapped to the crystal structure: blue, fully attenuated; dark blue, ≥50% attenuation; red, ≥50% intensity enhancement.
For the SA-DPC sample, residues Trp 54, Arg 56, and Ser 119 experienced alterations (>0.1 ppm) of chemical environment when compared with only SA U44069 titrated L-PGDS. Other perturbed residues included Asp 37, Ser 50, Thr 91, Ser 114, and Thr 123, which shifted between 0.08 and 0.1 ppm. Together with Ser 119, they corresponded to residues with a side chain facing the external environment of the protein. Hence, it is likely that the signal change at these residues is due to nonspecific interactions with the DPC micelles.
Overall, the effect of DPC was more pronounced in PA-DPC samples. Residues perturbed in PA-DPC (Fig. 6B) show similarity to residues perturbed in solely ligand titrated samples (Fig. 5A, B), with alterations concentrated at the E-F loop and the Ω loop. The cross-peak intensities which increased upon addition of PA 12415 (supplementary Fig. III) were attenuated to a larger extent when DPC was added (Fig. 6D). For example, residues Asp 37, Lys 59, Lys 60, Met 64, Leu 131, Asp 142, Leu 159, Glu 161, Phe 172, Ile 177, Asp 184, and Met 187 had signal reductions of more than 50% while signals of Lys 38, Ser 50, Glu 57, and Trp 112 were fully attenuated. In contrast, residues Ala 49, Glu 90, Thr 123, Ser 133, Arg 144, and Leu 148 showed more than a 50% increase in intensities (Fig. 6D). The overall decrease in intensities, which are coupled with the broadening of peaks, are due to the protein undergoing conformational exchange to release stabilization imposed by ligand binding. These findings suggest a plausible mode of product release from the protein.
When the apoenzyme was titrated with DPC, amide cross-peaks became more resolved with a global increase in resonance intensities and chemical shift perturbations (supplementary Fig. IV). This pattern is similar to the observations made when SA U44069, SA U46619, or PA 12415 were added to the protein (Fig. 5B), which is a general stabilization of the protein. Judging from these titrations, it is likely that L-PGDS comprises intrinsic interactions with detergent micelles, and its ligand binding state could affect its interaction with membranes.
DISCUSSION
Analog bound L-PGDS cocrystals revealed putative binding sites for SA U44069 in the large cavity, where binding is accompanied by protein conformational change. Because both SA U44069-bound and analog free structures were determined in the same space group with similar crystallization buffer, this minimized the risk of artifacts, indicating that the observed structural changes of the protein are indeed due to substrate binding. The noncontinuous ligand densities are likely to be due to multiple binding modes and flexible interactions of aliphatic moieties of the SA U44069 with L-PGDS. Still, the strongest densities are most likely due to the coordinated head groups, which have allowed tentative modeling of the SA U44069 in the binding pocket. It should be noted that structures with well-defined binding of lipophilic ligands are relatively rare (49). Similar partial electron densities were observed for substrates in mouse urinary protein-pheromone complex (50), phospholipids in saposin B (53), and in 5-lipoxygenase-substrate complex (52).
Previous modeling of substrate binding in mouse L-PGDS and structural comparison with the catalytic site of H-PGDS have suggested that residues mPhe 83, mSer 81, mCys 65, mThr 67, and mSer 45 constitute the catalytic site in L-PGDS (27).3 In our crystal structure we observed the ligand density near Phe 83, Cys 65, and Ser 67. The aromatic ring of the conserved Phe 83 is positioned facing the cyclopentane ring of the modeled substrate analog in site A1 (as well as in site A2). This interaction appears crucial to position the head group of the substrate for catalysis. Mutant F83A has been shown to reduce L-PGDS enzymatic activity by more than 80% (30). Interestingly, we observed that binding of the SA U44069 induces a conformational change of Cys 65 and its neighboring residue. The side chain of Cys 65 was moved into the catalytic center and potentially assists in activating the thiol by establishing strong hydrogen bonding to Ser 67 for the attack of the cyclopentane peroxide bond of the substrate and subsequently produce PGD2 (30). When no other suitable proton source is available in the active site, it is likely that Cys 65 also provides a proton to the substrate where nucleophilic attack and proton transfer might be concerted to minimize charge build-up.
Apart from the expected binding at the catalytic site, our study reports a pseudo-binding site observed at the E-F loop of L-PGDS. This site binds polyethylene glycol in our apoenzyme structure and coincides with fatty acids binding in the structure from Zhou et al. (30). Residues of the E-F loop binding site are highly conserved across homologs of different organisms, making this a potential interaction point to facilitate the entry of ligand. The role of the E-F loop in ligand binding is highlighted in NMR titration studies in the presence of either SA U44069 or PA 12415. Chemical shift perturbations coupled with intensities augmentation upon ligand addition suggest ligand stabilization of the E-F loop. Because movement of E-F loop is coupled with conformational changes on the Ω loop and catalytic residues upon ligand titration, we speculate that the E-F loop either serves to usher the substrate into the catalytic site upon ligand binding or facilitate the release of product.
This then leads to the question of how PGD2 is released into the right environment for its functional purpose. Because PGD2 is produced in the lumen of ER by L-PGDS or H-PGDS, it has to be secreted into the extracellular compartment to reach its target, the receptor DP1. Even though de Waart et al. (53) suggested that PGD2 transport through the plasma membrane might also be mediated by multidrug resistance associated protein that transport PGE2, hitherto there had been no studies describing the transfer of PDG2 from the lumen of endoplasmic reticulum (ER) to the cytoplasm side. Members of the prostaglandin synthases like PGI2 synthase (PGIS) and thromboxane A2 synthase (TXAS) are single-pass membrane proteins that bind their substrate on the luminal side of the ER membrane and release their product on the cytoplasmic side (54). However, as prostaglandin D synthase is a soluble protein, this model clearly does not apply.
Based on our data we hypothesized that L-PGDS could potentially regulate its product release through its ability to interact with membranes. Significant chemical shift perturbations are observed between the E-F loop and the Ω loop in the presence of DPC when L-PGDS is bound with PA 12415 but not with SA U44069. This is especially interesting as both the PA 12415 and SAU44069 are structurally similar (Table 1). Nonetheless, we cannot rule out the possibility that free detergent would interact with the hydrophobic core of the L-PGDS barrel especially in the apoenzyme state. Nevertheless, chemical shift perturbations and resonance intensity alterations in titration experiments with DPC revealed that the apoenzyme and the SA U44069- or PA 12415-bound protein clearly show distinct interaction modes with the micelles. In light of our data we propose that, when bound to its product, L-PGDS would be able to transiently bind the membrane of ER allowing the release of PGD2 into the lipid bilayer. Similarly to PGIS and TXAS, L-PGDS would then be able to mediate both the synthesis and the transport of PGD2. The present work of combining X-ray crystallography and NMR allows us to better understand the mechanisms and dynamics of ligand binding to L-PGDS. Both methods support a model whereby ligand binding is accompanied by protein conformational changes mainly on the Ω loop and the E-F loop. Prostaglandin transport through the ER membrane into the cytoplasm was poorly understood, but our studies with membrane mimetics have provided evidence that differential protein-membrane interaction depends on ligand binding.
Supplementary Material
Acknowledgments
The authors would like to thank both the Protein Science Facility (PSF) in Karolinska Institutet and the Protein Production Platform (PPP) in Nanyang Technological University for doing initial cloning and screening of human L-PGDS constructs. The authors also thank Mr. Leo E. Wong who assisted in reviewing NMR resonance assignments.
Footnotes
Abbreviations:
- AA
- arachidonic acid
- c.m.c
- critical micelle concentration
- COX
- cyclooxygenase
- DPC
- dodecylphosphocholine
- HSQC
- heteronuclear single quantum correlation
- H-PGDS
- hematopoietic prostaglandin D synthase
- IPTG
- isopropyl-beta-D-1-thiogalactopyranoside
- L-PGDS
- lipocalin prostaglandin D synthase
- MOX
- methoxylamine
- NOESY
- nuclear Overhauser effect spectroscopy
- OD
- optical density
- PA
- product analog
- PGD2
- prostaglandin D2
- PGDS
- prostaglandin D synthase
- PGES
- prostaglandin E synthase
- PGH2
- prostaglandin H2
- PGI2
- prostaglandin I2
- PGIS
- prostaglandin I2 synthase
- PPAR
- peroxisome proliferator-activated receptor
- RA
- retinoic acid
- SA
- substrate analog
- TCEP
- tris-(2-carboxyethyl)phosphine
- TROSY
- transfer relaxation optimization spectroscopy
- TSA
- thermal shift assay
- TXAS
- thromboxane A2 synthase
This work was financially supported by the Singapore National Research Foundation Competitive Research Program (CRP) funding scheme, NRF2008NRF-CRP002-067.
The online version of this article (available at http://www.jlr.org) contains supplementary data in the form of four figures and two tables.
mPhe 83, mSer 81, mCys 65, mThr 67, and mSer 45 are the amino acids in mouse L-PGDS.
REFERENCES
- 1.Wymann M. P., Schneiter R. 2008. Lipid signalling in disease. Nat. Rev. Mol. Cell Biol. 9: 162–176 [DOI] [PubMed] [Google Scholar]
- 2.Simmons D. L., Botting R. M., Hla T. 2004. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol. Rev. 56: 387–437 [DOI] [PubMed] [Google Scholar]
- 3.Schneider C., Pozzi A. 2011. Cyclooxygenases and lipoxygenases in cancer. Cancer Metastasis Rev. 30: 277–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang D., Dubois R. N. 2010. Eicosanoids and cancer. Nat. Rev. Cancer. 10: 181–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith W. L. 1989. The eicosanoids and their biochemical mechanisms of action. Biochem. J. 259: 315–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Urade Y., Eguchi N. 2002. Lipocalin-type and hematopoietic prostaglandin D synthases as a novel example of functional convergence. Prostaglandins Other Lipid Mediat. 68–69: 375–382 [DOI] [PubMed] [Google Scholar]
- 7.Hayaishi O., Urade Y. 2002. Prostaglandin D2 in sleep-wake regulation: recent progress and perspectives. Neuroscientist. 8: 12–15 [DOI] [PubMed] [Google Scholar]
- 8.Eguchi N., Minami T., Shirafuji N., Kanaoka Y., Tanaka T., Nagata A., Yoshida N., Urade Y., Ito S., Hayaishi O. 1999. Lack of tactile pain (allodynia) in lipocalin-type prostaglandin D synthase-deficient mice. Proc. Natl. Acad. Sci. USA. 96: 726–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ragolia L., Palaia T., Hall C. E., Maesaka J. K., Eguchi N., Urade Y. 2005. Accelerated glucose intolerance, nephropathy, and atherosclerosis in prostaglandin D2 synthase knock-out mice. J. Biol. Chem. 280: 29946–29955 [DOI] [PubMed] [Google Scholar]
- 10.Joo M., Kwon M., Sadikot R. T., Kingsley P. J., Marnett L. J., Blackwell T. S., Peebles R. S., Jr, Urade Y., Christman J. W. 2007. Induction and function of lipocalin prostaglandin D synthase in host immunity. J. Immunol. 179: 2565–2575 [DOI] [PubMed] [Google Scholar]
- 11.Virtue S., Masoodi M., Velagapudi V., Tan C. Y., Dale M., Suorti T., Slawik M., Blount M., Burling K., Campbell M., et al. 2012. Lipocalin prostaglandin D synthase and PPARgamma2 coordinate to regulate carbohydrate and lipid metabolism in vivo. PLoS ONE. 7: e39512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saso L., Leone M. G., Sorrentino C., Giacomelli S., Silvestrini B., Grima J., Li J. C., Samy E., Mruk D., Cheng C. Y. 1998. Quantification of prostaglandin D synthetase in cerebrospinal fluid: a potential marker for brain tumor. Biochem. Mol. Biol. Int. 46: 643–656 [DOI] [PubMed] [Google Scholar]
- 13.Kim J., Yang P., Suraokar M., Sabichi A. L., Llansa N. D., Mendoza G., Subbarayan V., Logothetis C. J., Newman R. A., Lippman S. M., et al. 2005. Suppression of prostate tumor cell growth by stromal cell prostaglandin D synthase-derived products. Cancer Res. 65: 6189–6198 [DOI] [PubMed] [Google Scholar]
- 14.Hirawa N., Uehara Y., Yamakado M., Toya Y., Gomi T., Ikeda T., Eguchi Y., Takagi M., Oda H., Seiki K., et al. 2002. Lipocalin-type prostaglandin d synthase in essential hypertension. Hypertension. 39: 449–454 [DOI] [PubMed] [Google Scholar]
- 15.Nakayama H., Echizen H., Gomi T., Shibuya Y., Nakamura Y., Nakano K., Arashi H., Itai T., Ohnishi S., Tanaka M., et al. 2009. Urinary lipocalin-type prostaglandin D synthase: a potential marker for early gentamicin-induced renal damage? Ther. Drug Monit. 31: 126–130 [DOI] [PubMed] [Google Scholar]
- 16.Evans J. F., Islam S., Urade Y., Eguchi N., Ragolia L. 2013. The lipocalin-type prostaglandin D2 synthase knockout mouse model of insulin resistance and obesity demonstrates early hypothalamic-pituitary-adrenal axis hyperactivity. J. Endocrinol. 216: 169–180 [DOI] [PubMed] [Google Scholar]
- 17.Heshmat S. M., Mullen J. B., Jarvi K. A., Soosaipillai A., Diamandis E. P., Hamilton R. J., Lo K. C. 2008. Seminal plasma lipocalin-type prostaglandin D synthase: a potential new marker for the diagnosis of obstructive azoospermia. J. Urol. 179: 1077–1080 [DOI] [PubMed] [Google Scholar]
- 18.Lescuyer P., Gandini A., Burkhard P. R., Hochstrasser D. F., Sanchez J. C. 2005. Prostaglandin D2 synthase and its post-translational modifications in neurological disorders. Electrophoresis. 26: 4563–4570 [DOI] [PubMed] [Google Scholar]
- 19.Garza L. A., Liu Y., Yang Z., Alagesan B., Lawson J. A., Norberg S. M., Loy D. E., Zhao T., Blatt H. B., Stanton D. C., et al. 2012. Prostaglandin D2 inhibits hair growth and is elevated in bald scalp of men with androgenetic alopecia. Sci. Transl. Med. 4: 126–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujimori K., Inui T., Uodome N., Kadoyama K., Aritake K., Urade Y. 2006. Zebrafish and chicken lipocalin-type prostaglandin D synthase homologues: conservation of mammalian gene structure and binding ability for lipophilic molecules, and difference in expression profile and enzyme activity. Gene. 375: 14–25 [DOI] [PubMed] [Google Scholar]
- 21.Kume S., Lee Y. H., Miyamoto Y., Fukada H., Goto Y., Inui T. 2012. Systematic interaction analysis of human lipocalin-type prostaglandin D synthase with small lipophilic ligands. Biochem. J. 446: 279–289 [DOI] [PubMed] [Google Scholar]
- 22.Beuckmann C. T., Aoyagi M., Okazaki I., Hiroike T., Toh H., Hayaishi O., Urade Y. 1999. Binding of biliverdin, bilirubin, and thyroid hormones to lipocalin-type prostaglandin D synthase. Biochemistry. 38: 8006–8013 [DOI] [PubMed] [Google Scholar]
- 23.Tanaka T., Urade Y., Kimura H., Eguchi N., Nishikawa A., Hayaishi O. 1997. Lipocalin-type prostaglandin D synthase (beta-trace) is a newly recognized type of retinoid transporter. J. Biol. Chem. 272: 15789–15795 [DOI] [PubMed] [Google Scholar]
- 24.Flower D. R. 1996. The lipocalin protein family: structure and function. Biochem. J. 318: 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fukuhara A., Nakajima H., Miyamoto Y., Inoue K., Kume S., Lee Y. H., Noda M., Uchiyama S., Shimamoto S., Nishimura S., et al. 2012. Drug delivery system for poorly water-soluble compounds using lipocalin-type prostaglandin D synthase. J. Control. Release. 159: 143–150 [DOI] [PubMed] [Google Scholar]
- 26.Urade Y., Hayaishi O. 2000. Biochemical, structural, genetic, physiological, and pathophysiological features of lipocalin-type prostaglandin D synthase. Biochim. Biophys. Acta. 1482: 259–271 [DOI] [PubMed] [Google Scholar]
- 27.Kumasaka T., Aritake K., Ago H., Irikura D., Tsurumura T., Yamamoto M., Miyano M., Urade Y., Hayaishi O. 2009. Structural basis of the catalytic mechanism operating in open-closed conformers of lipocalin type prostaglandin D synthase. J. Biol. Chem. 284: 22344–22352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miyamoto Y., Nishimura S., Inoue K., Shimamoto S., Yoshida T., Fukuhara A., Yamada M., Urade Y., Yagi N., Ohkubo T., et al. 2010. Structural analysis of lipocalin-type prostaglandin D synthase complexed with biliverdin by small-angle X-ray scattering and multi-dimensional NMR. J. Struct. Biol. 169: 209–218 [DOI] [PubMed] [Google Scholar]
- 29.Shimamoto S., Yoshida T., Inui T., Gohda K., Kobayashi Y., Fujimori K., Tsurumura T., Aritake K., Urade Y., Ohkubo T. 2007. NMR solution structure of lipocalin-type prostaglandin D synthase: evidence for partial overlapping of catalytic pocket and retinoic acid-binding pocket within the central cavity. J. Biol. Chem. 282: 31373–31379 [DOI] [PubMed] [Google Scholar]
- 30.Zhou Y., Shaw N., Li Y., Zhao Y., Zhang R., Liu Z. J. 2010. Structure-function analysis of human l-prostaglandin D synthase bound with fatty acid molecules. FASEB J. 24: 4668–4677 [DOI] [PubMed] [Google Scholar]
- 31.Otwinowski Z., Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. In Macromolecular Crystallography, Part A. C. W. Carter, Jr. and R. M. Sweet, editors. Academic Press, New York. 307–326. [DOI] [PubMed]
- 32.Leslie A. G. W., Powell H. R. 2007. Processing diffraction data with Mosflm. In Evolving Methods for Macromolecular Crystallography. R. J. Read and J. L. Sussman, editors. Springer, Dordrecht, The Netherlands. 41–51.
- 33.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Stroroni L. C., Read R. J. 2007. Phaser crystalligraphic software. J. Appl. Crystallogr. 40: 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langer G., Cohen S. X., Lamzin V. S., Perrakis A. 2008. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3: 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Emsley P., Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- 36.Murshudov G. N., Vagin A. A., Dodson E. J. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53: 240–255 [DOI] [PubMed] [Google Scholar]
- 37.Collaborative Computational Project Number 4 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50: 760–763 [DOI] [PubMed] [Google Scholar]
- 38.Takeuchi K., Ng E., Malia T. J., Wagner G. 2007. 1–13C amino acid selective labeling in a 2H15N background for NMR studies of large proteins. J. Biomol. NMR. 38: 89–98 [DOI] [PubMed] [Google Scholar]
- 39.Keller R. L. J. 2004. The Computer Aided Resonance Assignment Tutorial ISBN 3-85600-112-3, first edition. Accessed September 30, 2011, at http://cara.nmr-software.org/downloads/3-85600-112-3.pdf [Google Scholar]
- 40.Senisterra G. A., Markin E., Yamazaki K., Hui R., Vedadi M., Awrey D. E. 2006. Screening for ligands using a generic and high-throughput light-scattering-based assay. J. Biomol. Screen. 11: 940–948 [DOI] [PubMed] [Google Scholar]
- 41.Vedadi M., Niesen F. H., Allali-Hassani A., Fedorov O. Y., Finerty P. J., Jr, Wasney G. A., Yeung R., Arrowsmith C., Ball L. J., Berglund H., et al. 2006. Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc. Natl. Acad. Sci. USA. 103: 15835–15840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ericsson U. B., Hallberg B. M., Detitta G. T., Dekker N., Nordlund P. 2006. Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal. Biochem. 357: 289–298 [DOI] [PubMed] [Google Scholar]
- 43.Inoue T., Irikura D., Okazaki N., Kinugasa S., Matsumura H., Uodome N., Yamamoto M., Kumasaka T., Miyano M., Kai Y., et al. 2003. Mechanism of metal activation of human hematopoietic prostaglandin D synthase. Nat. Struct. Biol. 10: 291–296 [DOI] [PubMed] [Google Scholar]
- 44.Byrne L. J., Sidhu A., Wallis A. K., Ruddock L. W., Freedman R. B., Howard M. J., Williamson R. A. 2009. Mapping of the ligand-binding site on the b′ domain of human PDI: interaction with peptide ligands and the x-linker region. Biochem. J. 423: 209–217 [DOI] [PubMed] [Google Scholar]
- 45.Marintchev A., Frueh D., Wagner G. 2007. NMR methods for studying protein-protein interactions involved in translation initiation. Methods Enzymol. 430: 283–331 [DOI] [PubMed] [Google Scholar]
- 46.McKay R. T., Tripet B. P., Hodges R. S., Sykes B. D. 1997. Interaction of the second binding region of troponin I with the regulatory domain of skeletal muscle troponin C as determined by NMR spectroscopy. J. Biol. Chem. 272: 28494–28500 [DOI] [PubMed] [Google Scholar]
- 47.Kallick D. A., Tessmer M. R., Watts C. R., Li C. Y. 1995. The use of dodecylphosphocholine micelles in solution NMR. J. Magn. Reson. B. 109: 60–65 [DOI] [PubMed] [Google Scholar]
- 48.Tham L. K. editor 2005. Protein-lipid interactions: from membrane domains to cellular networks. Wiley-VCH.
- 49.Forneris F., Mattevi A. 2008. Enzymes without borders: mobilizing substrates, delivering products. Science. 321: 213–216 [DOI] [PubMed] [Google Scholar]
- 50.Böcskei Z., Groom C. R., Flower D. R., Wright C. E., Phillips S. E., Cavaggioni A., Findlay J. B., North A. C. 1992. Pheromone binding to two rodent urinary proteins revealed by X-ray crystallography. Nature. 360: 186–188 [DOI] [PubMed] [Google Scholar]
- 51.Ahn V. E., Faull K. F., Whitelegge J. P., Fluharty A. L., Prive G. G. 2003. Crystal structure of saposin B reveals a dimeric shell for lipid binding. Proc. Natl. Acad. Sci. USA. 100: 38–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gilbert N. C., Rui Z., Neau D. B., Waight M. T., Bartlett S. G., Boeglin W. E., Brash A. R., Newcomer M. E. 2012. Conversion of human 5-lipoxygenase to a 15-lipoxygenase by a point mutation to mimic phosphorylation at Serine-663. FASEB J. 26: 3222–3229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.de Waart D. R., Paulusma C. C., Kunne C., Oude Elferink R. P. 2006. Multidrug resistance associated protein 2 mediates transport of prostaglandin E2. Liver Int. 26: 362–368 [DOI] [PubMed] [Google Scholar]
- 54.Deng H., Huang A., So S. P., Lin Y. Z., Ruan K. H. 2002. Substrate access channel topology in membrane-bound prostacyclin synthase. Biochem. J. 362: 545–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.