

Abstract
Corneal angiogenesis and lymphangiogenesis are induced by vascular endothelial growth factors (VEGFs) signaling through its receptors VEGFR-1, -2, and -3. Endostatin is a peptide antagonist of these receptors that causes inhibition of bFGF-induced corneal angiogenesis and lymphangiogenesis. Here we show that binding of VEGF-C and endostatin to recombinant VEGFR-3 is competitive. Alignments of the primary amino acid sequences of VEGF-C and the C-terminal endostatin peptide (mEP: LEQKAASCHNSYIVLCIENSFMTSFSK) identified two conserved cysteine residues separated by seven amino acids. Peptides of VEGF-C and mEP containing these conserved residues bound toVEGFR-3. However, substitution of alanine for either of the cysteines in the mEP peptide perturbed the secondary structure, and this mutated peptide was unable to bind to VEGFR-3. Analysis by surface plasmon resonance demonstrated that the binding of the mEP peptide for recombinant VEGFR-3 had a Ka of 1.41 × 107M−1s−1, Kd of 0.6718 s−1, and a KD of 4.78 10−8M. Characterization of the mechanism of endostatin binding to VEGFR-3 may lead to the development of novel therapies for lymphangiogenesis-related disorders, such as transplant rejection, lymphedema, and cancer metastasis.
Keywords: Endostatin, lymphangiogenesis, peptide fragment, surface plasmon resonance, VEGF, VEGFR
INTRODUCTION
Endostatin is a 20-kDa proteolytic fragment of the C-terminal non-collagenous domain of Collagen XVIII [1,2]. Endostatin is a tightly packed globular protein, and its structure is maintained by two disulfide bonds in a unique nested pattern [3]. These disulfide bonds are intimately related to the native conformation, stability, and activity of endostatin. In endothelial or tumor cell lines, endostatin inhibits the angiogenic signaling cascade by binding to specific cell surface receptors and interfering with growth factor signaling [4]. Putative endostatin receptors include vascular endothelial growth factor receptors (VEGFR) -2 and -3, integrins α5 and αV, and glypican-1 and -4 [5–9]. Activation of these receptors by a ligand induces endothelial cell apoptosis and inhibits endothelial cell proliferation and migration. Although endostatin does not bind to VEGF or basic fibroblast growth factor (bFGF) [7], endostatin suppresses VEGF activity by competition for the growth factor receptor [10]. The motifs in endostatin that are involved in binding to growth factor receptors are not well characterized.
We have previously found that the 28-kDa endostatin-containing fragment of collagen XVIII binds to recombinant VEGFR-3 (VEGFR-3-Fc) and inhibits b FGF-induced corneal lymphangiogenesis [8]. Here we have begun to characterize the interaction in vitro between an endostatin short peptide and VEGFR-3, using a combination of structure-function analysis and surface plasmon resonance (SPR) analysis of the binding kinetics. Our results have refined our understanding of the VEGFR 3-binding domain of endostatin and will be important in the development of peptide antagonists that specifically target lymphatic endothelial cells to treat lymphatic-related diseases.
MATERIALS AND METHODS
Cell Culture and Endostatin Peptides
Primary human lung lymphatic endothelial cells (h-LECs, Lonza, Allendale, NJ) were cultured for 24 h in serum-free medium and then stimulated for 24 h with VEGF-C with or without endostatin peptide. The following peptides were used: Four 27-amino acid endostatin peptides (mEP: LEQKAASCHNSYIVLCIENSFMTSFSK; mEP-CA: LEQKAASCHNSYIVLAIENSFMTSFSK; mEP-AC: LEQKAASAHNSYIVLCIENSFMTSFSK; and mEP-AA: LEQKAASAHNSYIVLAIENSFMTSFSK) based on the C-terminal endostatin sequence, a 25-amino acid control peptide based on the internal endostatin sequence (FDGRDVLRHPAWPQKSVWHGSDPSG), and a computer-generated, scrambled peptide (CVCYMEAEKHIFSNILTSSALKLFQNSS). These short endostatin peptides are used instead of the entire peptide because it is difficult to synthesize large quantities of 40–50-amino acid peptides. Additionally, the longer lengths may affect peptide folding and secondary structure [10]. A total of 10 mg of each peptide was coupled to Sepharose CL4B beads in 10 ml of slurry resin. Pull-down assays were performed using 50 μg of sepharose-conjugated peptide.
Endostatin-derived and Mutated Peptide Synthesis and Analysis by Mass Spectrometry
The peptides were synthesized by solid-phase synthesis on a Symphony Peptide Synthesizer (Protein Technologies, Tucson, AZ) using Fmoc-Rink Amide-MBHA resin (AnaS-pec, San Jose, CA; Novabiochem, Gibbstown, NJ), and purified by reversed-phase C18 HPLC [11]. Peptides were spotted and dried onto a matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) target for analysis in positive-ion reflector mode with delayed extraction over the m/z range 700–4000 using a Voyager DE-PRO Mass Spectrometer (Applied Biosystems, Foster City, CA) equipped with a nitrogen laser.
Circular Dichroismanalysis
The mEP, mEP-CA, -AC, and –AA peptides were dissolved in a 50:50 solution of acetonitrile and water to a final concentration of 200 μg/ml and scanned with a J-710 spec-tropolarimeter (Jasco, Easton, MD) at 25 °C calibrated with d10 camphor sulfonic acid in 1-mm path-length fused-quartz cuvette. The resulting spectra were corrected with scans of the solvent mixture and smoothed.
Pull-down Assays
Recombinant mouse VEGFR-3-Fc proteins (R&D Systems, Minneapolis, MN) were preabsorbed against Sepha-rose 4B in RIPA buffer for 1 h at 4°C. Unconjugated sepha-rose beads bound to VEGFR-3-Fc was removed by centrifugation, and then 50 ng VEGFR-3-Fc in RIPA buffer (50 mM Tris-HCl, pH 7.4, and 150 mM NaCl, 1 mM EDTA, 0.25% Na-deoxycholate, and 1% NP-40) was incubated with 50 μg mEP-conjugated Sepharose 4B beads for 2 h at 4°C. Pull-down complexes were washed three times with PBS and analyzed by western blotting with rat anti-mouse VEGFR-3 antibody (1:1,000, eBiosciences, San Diego, CA) and horse-radish peroxidase-conjugated anti-rat IgG antibody (1:20,000, Cell Signaling Technology, Danvers, MA). The ECL detection system (Amersham, Piscataway, NJ) detected bound VEGFR-3-Fc.
SPR binding Assays
Carboxymethylated dextran biosensor CM5 chips were prepared by amine-coupling performed on a Biacore T100 instrument at 25°C at a flow rate of 10 μL/min in 10 mM HEPES, 150 mM NaCl, pH 7.4, and with the surface activated by injecting both 1-ethyl-3-(3-dimethylaminopropyl) carbodimide and N-hydroxysuccinimide for 15 minutes. A total of 25 μg of mEP was dissolved in immobilization solution (10 mM sodium acetate, pH 5.0) and injected for 10 minutes over the CM5 chip. Ethanolamine was injected for 7 minutes to block residual activated groups. After immobilization, the instruments were primed with the analysis running buffer (50 mM Tris-HCl, 150 mM NaCl, 10 mM MgCl2, 0.1% Tween 20, 0.1% Brij-35, 5% dimethyl sulfoxide, pH 8.0). Binding affinities were determined from experiments in which VEGFR-3-Fc at a range of concentrations (0, 0.01, 0.05, 0.25, 0.5, 1.0, 2.5, 5.0, and 10 μM) was passed over the mEP peptide-immobilized CM5 chip. The sensorgrams from the different concentrations of VEGFR-3-Fc were simultaneously fitted and constrained the kinetic rate constants to a single value for each set of curves by the BIA evaluation 3.1 software.
Bromodeoxyuridine (BrdU) Assays
h-LECs were seeded at 5 × 104 cells/well in 96-well plates coated with collagen, starved of serum overnight, and then incubated with 100 ng/mL VEGF-C and 0, 20, or 100 μg/mL mEP for 24 h at 37°C. Cells were then incubated in fresh media containing 10 μmol/L BrdU for 120 min and analyzed by ELISA, according to the manufacturer's instructions (Roche Molecular Biochemicals, Mannheim, Germany).
Cell Migration Assays
Cell migration was measured by monolayer scratch wounding. Confluent cell cultures in 12-wel plates were cultured in serum-free medium for 24 h and then rinsed with Dulbecco's modified Eagle's medium. Each well was scratched with a sterile pipette tip, and cells were then washed twice with PBS, incubated in experimental medium for 24 h, rinsed twice with PBS, and fixed with 4% paraformaldehyde. The number of cells that migrated beyond the scratch was counted in at least three fields per well, and three wells were examined for each experiment and condition.
RESULTS
VEGF-C interferes with the Binding of Endostatin-containing Fragments toVEGFR-3
Both endostatin and VEGF-C can bind to VEGFR-3, but whether this binding is competitive is unknown. We tested for competitiveness between the ligands by analyzing the binding of endostatin-containing fragments to recombinant VEGFR-3-Fc in the presence of various concentrations of VEGF-C. GST-endostatin-containing fragments, VEGFR-3, and a range of concentrations of VEGF-C were incubated and the receptor-ligand complexes were analyzed using a pull-down assay. We found that VEGF-C competed with GST-endostatin-containing fragments in binding to recombinant VEGFR-3-Fc (Fig. 1). GST-endostatin containing fragment was able to bind VEGFR-3-Fc in the absence of VEGF-C and at low concentration of VEGF-C ((Fig. 1), lanes 1 and 2). However, this binding was diminished by VEGF-C at a higher concentration ((Fig. 1), lane 3). This result shows that VEGF-C competes with endostatin in binding to VEGFR-3.
Figure 1.
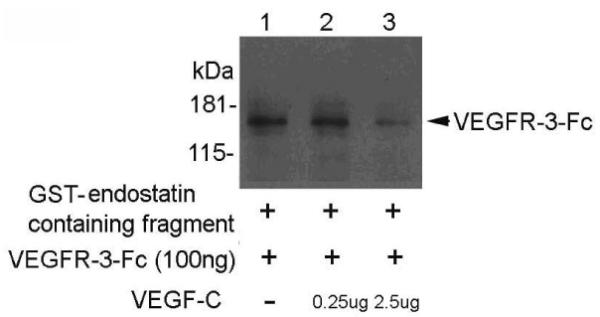
VEGF-C competes with endostatin-containing fragments to bind recombinant VEGFR-3-Fc. Western blot analysis of pull-down assays using an anti-VEGFR-3 antibody and the indicated concentrations of recombinant VEGF-C incubated with VEGFR-3-Fc and GST-endostatin-containing fragments.
A Cysteine-rich Conserved Motif in Endostatin-peptide and VEGF-C
To understand the basis of the competitive binding of endostatin and VEGF-C to VEGFR-3, we compared the primary sequences of the 28-kDa endostatin-containing fragment of collagen XVIII, VEGF-C, and VEGF-D using EMBOSS Needle-Pairwise Sequence Alignment (http://www.ebi.ac.uk/Tools/psa/emboss_needle/). This alignment identified a conserved motif [(R/K)xxxCxNSxx(V/L)xCxxxS] between residues 8–16 of the C-terminal end of endostatin and VEGF-C (Fig. 2A). Western blot analysis confirmed that both synthesized mEP and VEGF-C peptides bound recombinant VEGFR-3 (Fig. 2B).
Figure 2.
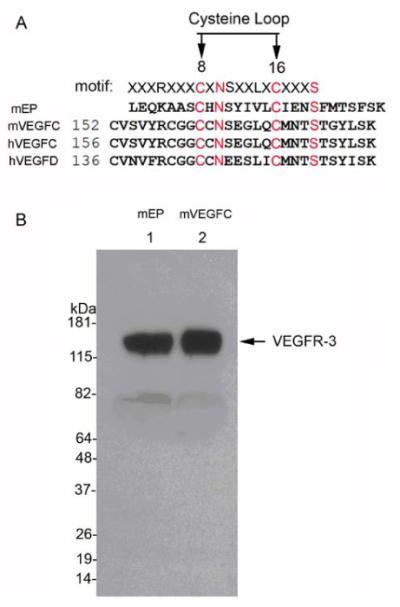
(A) Comparison of the amino acid sequences from the C-terminus of endostatin and the cysteine loops of VEGF-C and -D (identical amino acid sequences indicated in red). (B) Western blot analysis of pull-down assays; mEP (lane 1) and mVEGF-C (lane 2) bind to recombinant VEGFR-3.
The Conserved Cysteines of mEP are Required for Binding to VEGFR-3
To address whether this motif was required for binding to VEGFR-3, we tested the binding of peptides containing alanine substituted for either one or both conserved cysteine residues (peptides mEP-CA, -AC, and -AA; (Fig. 3A)). Analysis of these peptides using the receptor pull-down assay indicated that binding of the mEP to VEGFR-3 was abolished when either Cys was substituted with alanine (Fig. 3D). Analysis of the native and substituted peptides using circular dichroism showed that the Ellipticity between 205 and 225 nm of the native peptide was diminished by either of the Cys-to-Ala substitutions of the peptides (Fig. 3B). MALDI-TOF analysis of mEP showed a single relative absorbance peak with a m/z of 3163.52 (Fig. 3C).
Figure 3.
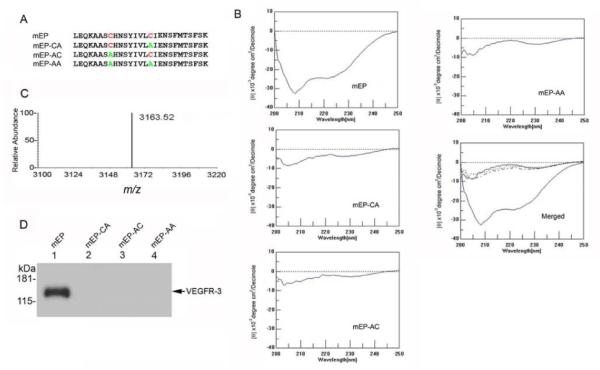
Substitutions within the mEP cysteine loop abolish binding to recombinant VEGFR-3. (A) Sequences of synthetic peptides containing Cys-to-Ala substitutions as indicated (B) Analysis of endostatin peptides using circular dichroism. The pronounced trough between −205 nm and −225 nm indicates secondary structure in the mEP peptide, whereas the substitution peptides mEP-CA, -AC and –AA lack this feature. (C) MALDI-TOF of mEP showed a single peak with a m/z ratio of 3163.52. (D) Protein pull-down assays with 50 μg mEP (lane 1), mEP-CA (lane 2), mEP-AC (lane 3), or mEP-AA (lane 4) found binding to recombinant VEGFR-3 by mEP via western blot using anti-VEGFR-3 antibody.
Binding of VEGFR-3-Fc to mEP analyzed by SPR
We characterized the binding of VEGFR3-Fc to mEP using SPR. mEP or a control endostatin peptide was injected and immobilized on a CM5 chip, and then, VEGFR3-Fc at a range of concentrations (0, 0.01, 0.05, 0.25, 0.5, 1.0, 2.5, 5.0, and 10 μM) was passed over the immobilized peptides. VEGFR3-Fc bound mEP at all concentrations (Fig. 4A). From this analysis, we determined that the binding parameters were as follows: Ka = 1.41× 107M−1s−1, Kd = 0.6718 s−1, and KD = 4.78×10−8 M (Fig. 4B).
Figure 4.
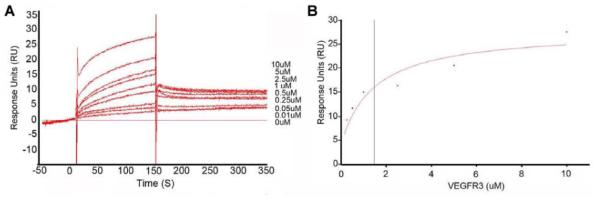
(A) Interaction of mEP with various concentrations of VEGFR-3-Fc, as shown by a representative sensorgram of fitted kinetic data. (B) The fitted curve is the solid red line super-imposed on the responses, and the VEGFR-3 concentrations are 0, 0.01, 0.05, 0.25, 0.5, 1.0, 2.5, 5.0, and 10 μM.
mEP Inhibits proliferation and Migration of Endothelial Cells
We predicted that mEP binding to VEGFR-3 is inhibitory and would therefore decrease VEGF-dependent lymphatic endothelial cell proliferation and migration. To test this hypothesis, we used BrdU incorporation to assess the proliferation of hLECs in the presence of VEGF-C and 0, 20, or 100 μg/mL mEP. VEGF-C-stimulated cell proliferation in the presence of either 20 or 100 μg/mL mEP was reduced by one-third and more than 3-fold, respectively, compared to VEGF-C alone (Fig. 5A). The scratch migration assay showed that mEP suppressed lymphatic cell migration. In the absence of either ligand, the cells did not migrate into the denuded area (Fig. 5B). However, VEGF-C stimulation led to approximately 50% repopulation of the scratch surface. In the presence of both mEP and VEGF-C, only a few cells migrated beyond the borders of the scratch. These results indicate that mEP can inhibit VEGF-C-induced proliferation and migration of endothelial cells.
Figure 5.
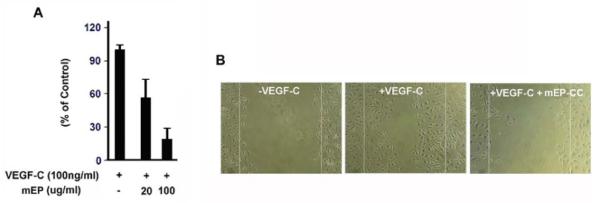
mEP inhibits proliferation (A) and migration (B) of hLECs stimulated with 100 ng/mL VEGF-C.
DISCUSSION
Our goal is to use surface plasmon resonance to characterize endostatin short peptide bound to VEGFR-3. We have demonstrated that VEGF-C competes in a concentration-dependent manner with an endostatin-containing fragment for binding to VEGFR-3. We also determined that the endostatin short peptide binds to recombinant VEGFR-3-Fc with a binding affinity (KD = 4.78×10−8M) that is considerably less than the 1 nM KD of fully processed VEGF-C binding to VEGFR3-AP (a VEGFR-3 fusion protein) [12]. Preincubation of endostatin with VEGFR-2-Fc before endostatin treatment of endothelial cells was suggested to block VEGF binding because endostatin directly blocks the interaction between VEGF-A and VEGFR-2 to inhibit VEGF-A-induced signaling. It has been shown that a 10-min preincubation of endostatin with VEGFR-2-Fc blocks the interaction of VEGF-A and VEGFR-2-Fc in a dose-dependent manner, and a high concentration of endostatin (10 μg/mL) does not affect the dissociation rate of VEGFR-2-Fc from VEGF and its receptor complex [7].
Our structure-function analysis showed that the binding of endostatin short peptide to VEGFR-3 depends upon two cysteine residues that are conserved in VEGF ligands. The C-terminal domain of endostatin contains a secondary structure [14] that is consistent with our circular dichroism data. Substitution of either conserved cysteine residue abolished mEP binding to recombinant VEGFR-3. Therefore, it is possible that these cysteine residues stabilize the secondary structure. These cysteines are likely to be important for the activity of VEGF ligands. Human VEGF-C is an antiparallel homodimer covalently linked by two disulfide bridges between Cys156 and Cys165 [13,14], and the binding of VEGF-C to VEGFR-2 is structurally determined by Cys165 of VEGF-C that is localized to loop 2 and beta 4 of VEGF-C and regulates VEGFR-2 binding [13].
The endostatin peptide was capable of inhibiting VEGF-induced lymphatic cell proliferation and migration in a concentration-dependent manner. The N-terminal, internal, and C-terminal fragments of endostatin are anti-angiogenic [3,15–20], and the N- and C-terminal fragments have full biological activity in angiogenesis assays and may have more potency and efficacy than full-length human endostatin [18].
We have shown that the binding of endostatin to VEGFR-3 depends upon a cysteine motif that is conserved in VEGF ligands. This structural similarity may underlie the competition between these VEGFR ligands, which are likely to bind the receptor in a similar manner [21]. We anticipate that additional characterization of the endostatin domains involved in binding VEGFR-3 will facilitate the design of small peptides that can specifically target lymphatic endothelial cells to treat lymphangiogenesis-related disorders such as transplant rejection, lymphedema, and cancer metastasis.
ACKNOWLEDGEMENTS
This study was supported by NIH grants EY021886 to J.H.C., EY10101 to D.T.A., and EY01792 to D.T.A., a Midwest Eye Bank Research grant (Ann Arbor, Michigan), and an unrestricted grant from Research to Prevent Blindness (New York, NY).
ABBREVIATIONS
- h-LECs
Human lung lymphatic endothelial cells
- mEP
Murine endostatin peptide
- SPR
Surface plasmon resonance
Footnotes
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.
REFERENCES
- [1].O'Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR, Folkman J. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88:277–85. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- [2].Veillard F, Saidi A, Burden RE, Scott CJ, Gillet L, Lecaille F, Lalmanach G. Cysteine cathepsins S and L modulate anti-angiogenic activities of human endostatin. J. Biol. Chem. 2011;286:37158–67. doi: 10.1074/jbc.M111.284869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Becker CM, Sampson DA, Short SM, Javaherian K, Folkman J, D'Amato RJ. Short synthetic endostatin peptides inhibit endothelial migration in vitro and endometriosis in a mouse model. Fertil Steril. 2006;85:71–7. doi: 10.1016/j.fertnstert.2005.07.1290. [DOI] [PubMed] [Google Scholar]
- [4].A. Abdollahi A, Hahnfeldt P, Maercker C, Gröne HJ, Debus J, Ansorge W, Folkman J, Hlatky L, Huber PE. Endostatin's antiangiogenic signaling network. Mol. Cell. 2004;13:649–63. doi: 10.1016/s1097-2765(04)00102-9. [DOI] [PubMed] [Google Scholar]
- [5].Faye C, Moreau C, Chautard E, Jetne R, Fukai N, Ruggiero F, Humphries MJ, Olsen BR, Ricard-Blum S. Molecular interplay between endostatin, integrins, and heparan sulfate. J. Biol. Chem. 2009;284:22029–40. doi: 10.1074/jbc.M109.002840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Rehn M, Veikkola T, Kukk-Valdre E, Nakamura H, Ilmonen M, Lombardo C, Pihlajaniemi T, Alitalo K, Vuori K. Interaction of endostatin with integrins implicated in angiogenesis. Proc. Natl. Acad. Sci. USA. 2001;98:1024–9. doi: 10.1073/pnas.031564998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kim YM, Hwang S, Kim YM, Pyun BJ, Kim TY, Lee ST, Gho YS, Kwon YG. Endostatin blocks vascular endothelial growth factor-mediated signaling via direct interaction with KDR/Flk-1. J. Biol. Chem. 2002;277:27872–9. doi: 10.1074/jbc.M202771200. [DOI] [PubMed] [Google Scholar]
- [8].Kojima T, Azar DT, Chang JH. Neostatin-7 regulates bFGF-induced corneal lymphangiogenesis. FEBS Lett. 2008;582:2515–20. doi: 10.1016/j.febslet.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Karihalo A, Karumanchi SA, Barasch J, Jha V, Nickel CH, Yang J, Grisaru S, Bush KT, Nigam S, Rosenblum ND, Sukhatme VP, Cantley LG. Endostatin regulates branching morphogenesis of renal epithelial cells and ureteric bud. Proc. Natl. Acad. Sci. USA. 2001;98:12509–14. doi: 10.1073/pnas.221205198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Xu HL, Tan HN, Wang FS, Tang W. Research advances of endostatin and its short internal fragments. Curr. Protein Pept. Sci. 2008;9:275–83. doi: 10.2174/138920308784534050. [DOI] [PubMed] [Google Scholar]
- [11].McKern NM, Edskes HK, Shukla DD. Purification of hydrophilic and hydrophobic peptide fragments on a single reversed phase high performance liquid chromatographic column. Biomed. Chromatogr. 1993;7:15–9. doi: 10.1002/bmc.1130070105. [DOI] [PubMed] [Google Scholar]
- [12].Pytowski B, Goldman J, Persaud K, Wu Y, Witte L, Hicklin DJ, Skobe M, Boardman KC, Swartz MA. Complete and specific inhibition of adult lymphatic regeneration by a novel VEGFR-3 neutralizing antibody. J. Natl. Cancer Inst. 2005;97:14–21. doi: 10.1093/jnci/dji003. [DOI] [PubMed] [Google Scholar]
- [13].Leppänen VM, Prota AE, Jeltsch M, Anisimov A, Kalkkinen N, Strandin T, Lankinen H, Goldman A, Ballmer-Hofer K, Alitalo K. Structural determinants of growth factor binding and specificity by VEGF receptor 2. Proc. Natl. Acad. Sci. USA. 2010;107:2425–30. doi: 10.1073/pnas.0914318107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yang Y, Xie P, Opatowsky Y, Schlessinger J. Direct contacts between extracellular membrane-proximal domains are required for VEGF receptor activation and cell signaling. Proc. Natl. Acad. Sci. USA. 2010;107:1906–11. doi: 10.1073/pnas.0914052107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Tjin Tham Sjin RM, Satchi-Fainaro R, Birsner AE, Ramanujam VM, Folkman J, Javaherian K. A 27-amino-acid synthetic peptide corresponding to the NH2-terminal zinc-binding domain of endostatin is responsible for its antitumor activity. Cancer Res. 2005;65:3656–63. doi: 10.1158/0008-5472.CAN-04-1833. [DOI] [PubMed] [Google Scholar]
- [16].Yamaguchi N, Anand-Apte B, Lee M, Sasaki T, Fukai N, Shapiro R, Que I, Lowik C, Timpl R, Olsen BR. Endostatin inhibits VEGF-induced endothelial cell migration and tumor growth independently of zinc binding. Embo. J. 1999;18:4414–23. doi: 10.1093/emboj/18.16.4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Boehm T, O'Reilly MS, Keough K, Shiloach J, Shapiro R, Folkman J. Zinc-binding of endostatin is essential for its antiangiogenic activity. Biochem. Biophys. Res. Commun. 1998;252:190–4. doi: 10.1006/bbrc.1998.9617. [DOI] [PubMed] [Google Scholar]
- [18].Cattaneo MG, Pola S, Francescato P, Chillemi F, Vicentini LM. Human endostatin-derived synthetic peptides possess potent antiangiogenic properties in vitro and in vivo. Exp. Cell Res. 2003;283:230–6. doi: 10.1016/s0014-4827(02)00057-5. [DOI] [PubMed] [Google Scholar]
- [19].Zhang HT, Li HC, Li ZW, Guo CH, Chen YJ. An RGD-Modified Endostatin Peptide Expressed at E. coli Shows Anti-Tumor Activity In vivo. Protein Pept. Lett. 2011;79(9):2684–92. [PubMed] [Google Scholar]
- [20].Ou J, Li J, Pan F, Xie G, Zhou Q, Huang H, Liang H. Endostatin suppresses colorectal tumor-induced lymphangiogenesis by inhibiting expression of fibronectin extra domain A and integrin alpha9. J. Cell Biochem. 2011;112:2106–14. doi: 10.1002/jcb.23130. [DOI] [PubMed] [Google Scholar]
- [21].Jeltsch M, Karpanen T, Strandin T, Aho K, Lankinen H, Alitalo K. Vascular endothelial growth factor (VEGF)/VEGF-C mosaic molecules reveal specificity determinants and feature novel receptor binding patterns. J. Biol. Chem. 2006;281:12187–95. doi: 10.1074/jbc.M511593200. [DOI] [PubMed] [Google Scholar]