

SUMMARY
The molecular mechanisms involved in the development of obesity and related complications remain unclear. Here, we report that obese mice and human subjects have increased activity of neutrophil elastase (NE) and decreased serum levels of the NE inhibitor, α1-antitrypsin (A1AT, SerpinA1). NE null (Ela2−/−) mice and A1AT transgenic mice were resistant to high-fat diet (HFD)-induced bodyweight gain, insulin resistance, inflammation and fatty liver. NE inhibitor GW311616A reversed insulin resistance and bodyweight gain in HFD-fed mice. Compared with wild-type mice, Ela2−/− mice augmented circulating high molecular weight (HMW) adiponectin levels, phosphorylation of AMP-activated protein kinase (AMPK) and fatty acid oxidation (FAO) in the liver and brown adipose tissue (BAT), and uncoupling protein (UCP1) levels in the BAT. These data suggest that the A1AT-NE system regulates AMPK signaling, FAO and energy expenditure. The imbalance between A1AT and NE contributes to the development of obesity and related inflammation, insulin resistance and liver steatosis.
INTRODUCTION
Obesity is a major contributing factor to the worldwide prevalence of type II diabetes, non-alcoholic fatty liver disease, cardiovascular disease, and many other disorders. Leptin, a hormone secreted from adipose tissue, plays a key role in energy balance and feeding behavior through neuronal regulation, and both leptin deficiency and leptin resistance are associated with the development of obesity (Myers et al., 2010). Other factors implicated in the development of obesity-related diseases include adipokines, fatty acids, mitochondrial dysfunction, ER stress, and hypoxia (Sun et al., 2011). Despite great progress in the field, the molecular mechanisms that precede the development of obesity and related complications are not fully understood.
Many recent studies have suggested that obesity is associated with chronic adipose tissue inflammation, which results in increased levels of proinflammatory factors, such as tumor necrosis factor (TNFα) and monocyte chemoattractant protein (MCP-1), and decreased production of anti-inflammatory adipokines such as adiponectin (Hotamisligil, 2006; Kamei et al., 2006; Lumeng and Saltiel, 2011; Ouchi et al., 2011; Shoelson et al., 2006). Moreover, adipose tissue is infiltrated by proinflammatory cells such as lymphocytes, mast cells, NK cells, and neutrophils in the early stages of obesity, and macrophages accumulate at later times (Elgazar-Carmon et al., 2008; Feuerer et al., 2009; Liu et al., 2009; Nishimura et al., 2009; Weisberg et al., 2003; Winer et al., 2011; Xu et al., 2003). Ample evidence supports that adipose inflammation is related to the development of insulin resistance (Osborn and Olefsky, 2012; Ouchi et al., 2011; Sun et al., 2012). However, little is known about the molecular events that lead to immune cell infiltration and inflammatory cytokine production in adipose tissue, and the subsequent development of systemic insulin resistance.
In an effort to identify factors involved in the development of obesity-related metabolic complications, we compared the serum protein profiles of leptin-deficient obese (ob/ob) mice and lean wild-type (WT) mice, using a combination of glycoprotein enrichment and quantitative proteomic approaches (Tian et al., 2007). Several proteins showed differential expression, one of which, the serine protease inhibitor α1-antitrypsin (A1AT, also called SerpinA1), was dramatically reduced in the serum and liver of ob/ob mice. A1AT is produced in the liver and is an endogenous inhibitor of neutrophil elastase (NE), a proteolytic enzyme produced by neutrophils during inflammation (Korkmaz et al., 2010; Pham, 2006). Interestingly, leptin treatment increased A1AT expression both in cultured hepatocytes and in the liver of ob/ob mice. In contrast, we observed that NE activity was significantly elevated in serum of both ob/ob and high-fat diet (HFD) fed mice, suggesting that obesity was associated with a significant increase in the ratio of the NE protease over its natural inhibitor A1AT. We show here that genetic deletion of NE and overexpression of human A1AT (hA1AT) dramatically alleviated the adipose inflammation, insulin resistance, bodyweight gain and liver steatosis in mice fed with HFD. NE null mice also showed increased serum HMW adiponectin levels, AMPK signaling and fatty acid oxidation (FAO) in both the liver and BAT, and higher UCP1 protein levels in the BAT. We also confirmed that human obese subjects had significantly reduced serum A1AT levels and increased NE activities, which correlated with body mass index (BMI) and leptin resistance. Collectively, our data provides the first evidence that leptin regulates A1AT expression in the liver, and suggests that the imbalance between the activities of NE and its inhibitor A1AT may be an important contributing factor for the development of obesity, inflammation and insulin resistance.
RESULTS
Identification of α1-Antitrypsin as a Serum Protein Differentially Expressed in Obese Mice
We sought to identify differentially expressed serum proteins in the leptin-deficient obese B6.V-Lepob/J (ob/ob) mouse model using glycoprotein enrichment and quantitative proteomic approaches (Tian et al., 2007; Zhang et al., 2003) Serum proteins from 12-week-old obese ob/ob mice or age-matched lean C57BL/6 mice were purified by solid-phase extraction of N-linked glycopeptides, followed by stable isotope labeling and subsequent identification and quantification of glycopeptides using tandem mass spectrometry (Figure 1A). As shown in Table S1, we detected 11 glycoproteins with significantly different levels in the serum of obese mice compared with the lean mice. Among these proteins, multiple members of the serine protease inhibitor Serpin family, including SerpinA1 (α1-antitrypsin, A1AT), were significantly reduced in the obese mice. A1AT is produced and secreted mainly by the liver and plays a critical tissue protective role by inactivation of proteases, including NE, produced by tissue-resident inflammatory cells such as neutrophils. We confirmed by ELISA assay that serum A1AT levels were significantly lower (40%) in 13-week-old ob/ob mice than in lean control mice, whereas NE activity was almost 2-fold higher (Figures 1B & 1C). In addition, C57BL/6J lean control mice fed a HFD (60 kcal% fat) showed a decrease in serum A1AT levels and an increase in serum NE activity with time (Figures 1D & 1E). Thus, serum A1AT decreased and NE activity increased in a reciprocal manner over the period of HFD feeding in mice.
Figure 1. Changes in Serum A1AT Levels and NE Activity in Obese Mice and the Effect of Leptin on A1AT Expression.

(A) Schematic of the glycoprotein enrichment and quantitative serum proteomic approach for comparing serum protein profiles of male obese and lean mice. Serum α1-antitrypsin (A1AT) levels (B) or NE activity (C) in ob/ob mice (12-week-old male) and age-matched lean C57BL/6 controls were quantified by ELISA (A1AT) or a colorimetric assay (NE activity). Data are presented as mean ± SEM of 6 to 8 animals with triplicate samples. **P < 0.01. (D) Serum A1AT levels or (E) NE activity in male C67BL/6 mice at 7 weeks old fed 60% HFD for 2 to 17 weeks. Data are presented as mean ± SE of 5 to 9 animals with triplicate samples. *P < 0.05. (F and G) Leptin regulates A1AT expression in HepG2 cells. Cells were serum starved overnight and incubated with leptin (75 ng/ml) for the indicated times. (F) A1AT mRNA levels were determined by qRT-PCR and (G) A1AT protein levels were analyzed by Western blotting followed by quantification of band intensity. Results are presented as fold change relative to vehicle-treated control cells and are the mean ± SEM of 4 independent experiments. **P < 0.01. (H and I) Leptin stimulates A1AT expression in vivo. Alzet osmotic pumps were implanted subcutaneously into ob/ob male mice (10 weeks of age) and programmed to deliver vehicle or 10 μg/day of leptin for 7 days. Liver A1AT mRNA (H) and serum A1AT protein (I) were quantified by qRT-PCR and ELISA, respectively. Data are presented as mean ± SEM (n = 5). **P < 0.01. Also see Table S1 and Figure S1.
Leptin Regulates A1AT Expression
To identify the factor(s) involved in the regulation of liver A1AT expression, we examined the effects of several obesity-related factors on A1AT gene expression in cultured hepatic cell lines. A1AT expression was unaffected in HepG2 cells treated with insulin, docosahexaenoic acid, eicosapentaenoic acid, or palmitate (data not shown). However, A1AT mRNA and protein levels were significantly increased by leptin treatment of human HepG2 cells, murine Hep1-6 cells, and primary murine hepatocytes (Figures 1F and G, Figures S1A–C). HepG2 cells treated with leptin also displayed increased phosphorylation of the transcription factor signal transducer and activator of transcription 3 (STAT3) (Figure S1D). Analysis of the serpina1 (A1AT) promoter (Kalsheker et al., 2002) revealed the presence of consensus sequences for binding of STAT3 and several other transcription factors, including hepatocyte nuclear factor-1/4 (HNF1/4) and specificity protein-1 (SP1). To determine the mechanism by which leptin increases expression of A1AT and phosphorylation of STAT3, hepatic cells were incubated with leptin in the presence of inhibitors of key signaling molecules, including Jak2 (AG490), HNF-1 (UCDA), SP1 (mithramycin), Akt1/2 (Akti-1/2) and MEK1/2 (U0126). Notably, the leptin-induced increase in A1AT expression and STAT3 phosphorylation was specifically abolished by AG490 (Figure S1A–D), demonstrating that leptin stimulates A1AT expression via the Jak2-Stat3 pathway in hepatocytes. Leptin had no effect on A1AT mRNA stability (Figure S1E). To examine the effects of leptin on A1AT expression in vivo, recombinant mouse leptin was administered to leptin-deficient ob/ob mice for 7 days through implanted osmotic infusion pumps. Leptin treatment significantly increased A1AT mRNA and protein expression in the liver and sera of ob/ob mice compared with vehicle-treated ob/ob mice (Figures 1H and 1I), besides its inhibitory effects on food intake and bodyweight (Figures S1F and S1G).
Imbalance of Serum A1AT Levels and NE Activities in Human Obese Subjects
To validate whether the expression of A1AT and NE is also altered in obese human subjects, we examined sera from 62 healthy South Asian males and 17 US Caucasian males with varying BMIs (Table S2). In the South Asian subjects, serum leptin levels were markedly increased in the obese subjects (BMI > 27, standard for South Asian populations) and positively correlated with BMI (Figures 2A & S2A). Interestingly, serum A1AT levels were negatively correlated with BMI and leptin (Figures 2B & 2C). Serum A1AT levels were significantly reduced by 27% in the obese group (BMI >27) compared with the non-obese group (BMI <27), and A1AT levels in subjects with BMI >30 were about 40% lower than in the lean subjects with BMI <23 (Figure S2B). We confirmed these findings in sera from 17 US Caucasian males (Figures 2D–2J) and found that the obese subjects (classified as BMI >30 for US population) had lower serum A1AT levels and higher leptin levels than their lean counterparts (BMI <26). We examined serum NE elastase activity in the US subjects and noted that, in contrast to A1AT, NE activity in the obese subjects was significantly higher than in the lean controls (Figure 2F). Interestingly, serum A1AT levels were negatively correlated to NE activity (Figure 2G). Furthermore, serum leptin levels were negatively correlated with A1AT levels (Figure 2H) and positively correlated with both NE activity and the ratio of NE activity to A1AT levels (Figures 2I & 2J). However, both serum A1AT levels and NE activity were not associated with blood glucose levels (Figure S2C & S2D). Together, these data suggest that obesity and leptin resistance are associated with an imbalance of serum NE activity and A1AT levels in those subjects.
Figure 2. Reduced A1AT Levels and Increased Elastase Activity Correlate with Body Mass Index in Obese Human Subjects.
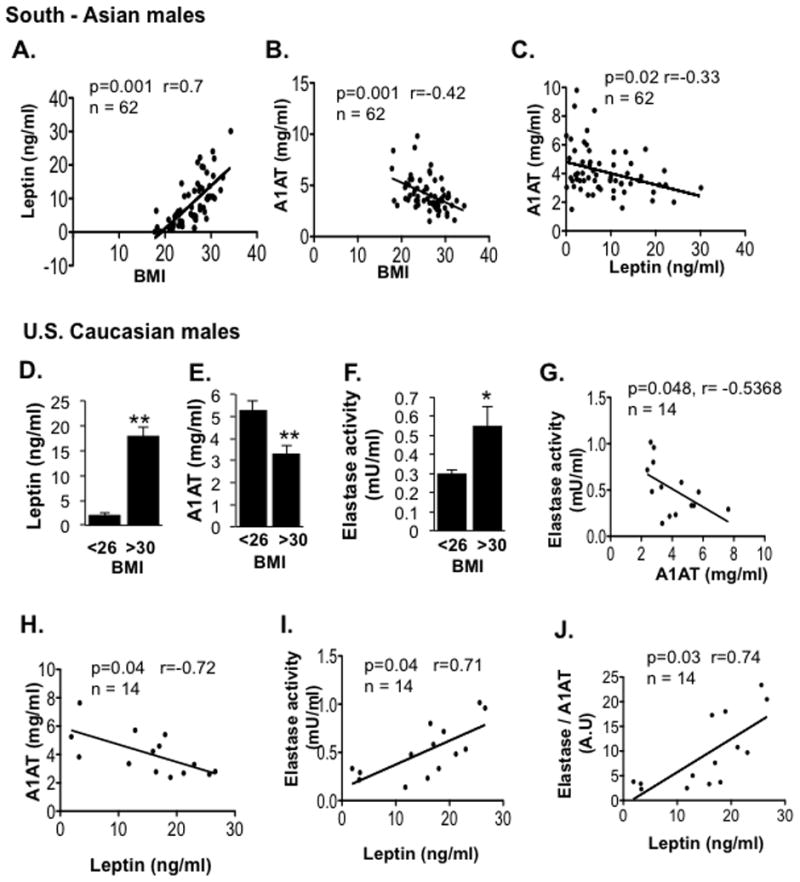
Serum samples were collected from 62 South Asian males (A–C) and 17 US Caucasian males (D–F). Leptin and A1AT were measured by ELISA, and serum NE activity was measured by a colorimetric assay. Prism software was used to analyze correlations between BMI and leptin, BMI and A1AT, or leptin and A1AT for South Asian men (A – C, n=62). Also displayed are correlations between leptin levels with serum A1AT levels (H), NE activity (I), and the ratio of NE activity verses A1AT (J), or NE activity verses A1AT (G) for the US subjects (n=14, BMI > 25). *P < 0.05, **P < 0.01. P values, regression coefficients and number of subjects (n) are indicated for the correlations. Also see Table S2 and Figure S2.
NE Knockout (Ela2−/−) Mice are Resistant to HFD-Induced Obesity, Liver Steatosis, and Insulin Resistance
We next determined whether alterations in the balance between serum NE activity and A1AT levels would affect basic metabolic functions in mice. For this, we used NE-deficient mice (B6.129X1-Elanetm1Sds/J, referred as Ela2−/−) (Belaaouaj et al., 1998). We found that Ela2−/− mice fed a 60% HFD showed reduced fat accumulation in the adipose tissue and were thus more resistant than WT controls to HFD-induced bodyweight gain (Figure 3A & 3B). Moreover, adipocytes in the visceral fat of Ela2−/− mice were significantly smaller than in WT control mice (Figures 3C – 3E). We also observed that the lipid content of the liver, muscle, and serum, but not the feces, were significantly reduced in HFD-fed Ela2−/− mice (Figures 3F & 3G), suggesting that Ela2−/− mice absorb fat normally but accumulate less fat in the tissues. However, there was only a small difference in bodyweight, but not lipid contents in tissues, between Ela2−/− and WT mice fed with a 10% low fat diet (Figures S3A–S3C). Metabolic cages were used to analyze energy expenditure and we found that Ela2−/− mice have significantly higher energy expenditure than the control mice during both light and dark cycles measured after 10 weeks of HFD feeding (Figure 3H – 3J), although there were no significant differences between the two groups in food intake or locomotory activity (Figures S4A – S4D). The rectal temperature was slightly increased in Ela2−/− mice challenged with an HFD (Figure 3K). Similarly, surface body temperature measured with a thermal camera was significantly higher in Ela2−/− mice than in WT mice fed with HFD for 6 weeks (Figures S4E & S4F), further supporting the notion of increased energy expenditure in the knockout mice.
Figure 3. Improved Metabolic Profiles in Neutrophil Elastase-Deficient Mice Fed with a HFD.
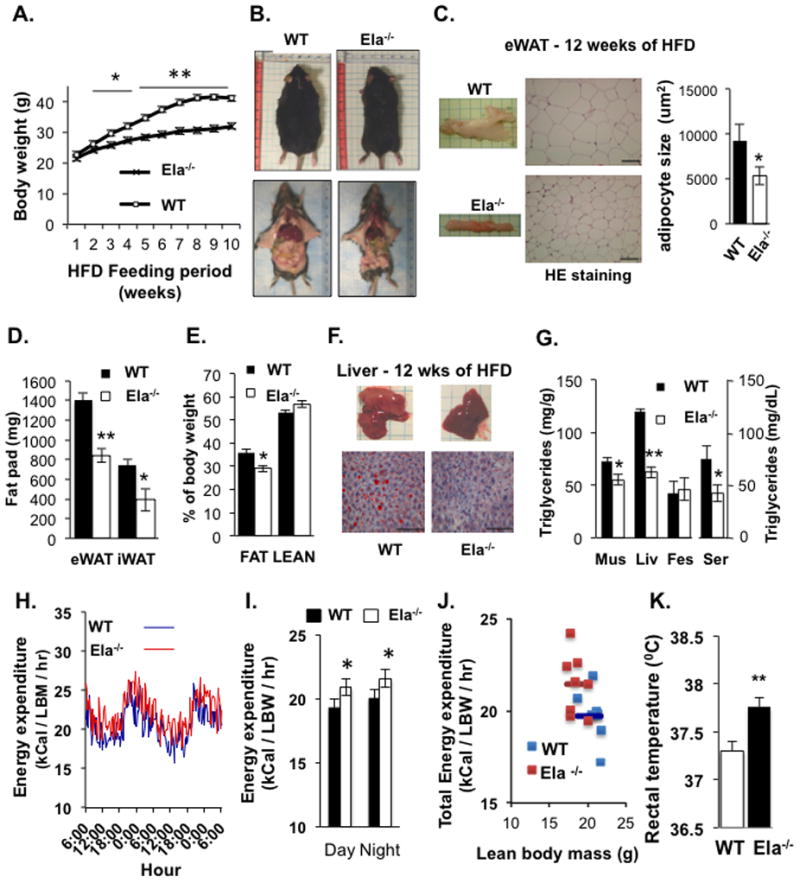
(A) Mean bodyweights of C57BL/6 (WT) and neutrophil elastase-deficient (Ela−/−) mice during 10 weeks on 60% HFD (n = 9). *P < 0.05, **P < 0.01, Ela2−/− vs WT. (B) Representative images of mice sacrificed after feeding the HFD for 12 weeks. (C) Images of epididymal white adipose tissue (eWAT), H&E staining, and mean area of the adipocytes calculated with ImageJ software. Scale bars = 100 μm. (D) Weight of epididymal (eWAT) and inguinal (iWAT) fat pads of mice fed a HFD for 9 weeks (n = 9). *P < 0.05, **P < 0.01. (E) Adipose tissue as a percentage of total bodyweight in mice fed the HFD for 9 weeks, measured by MRI (n = 9). *P < 0.05. (F) Representative images of livers and histologic sections (Oil Red O and methylene blue staining) of livers from WT and Ela2−/− mice fed a HFD for 12 weeks. Scale bars = 100 μm. (G) Triglyceride content of muscle (mus), liver (liv), feces, and serum (ser) of mice fed with the HFD for 12 weeks. Samples were collected after mice were fasted for 4 hrs. Energy expenditure (H, I. J) of mice fed with the HFD diet for 10 weeks and then monitored in metabolic cages for 48 hrs (n = 8 mice per group). Lean body weight: WT, 20.8 ± 0.76 (g); Ela2−/−, 18.5 ± 0.92 (g); Fat mass: WT, 15.1 ± 0.12 (g); Ela2−/−, 9 ± 0.13 (g). (K) Mouse rectal temperatures measured with a rectal probe (BAT-12 Microprobe Thermometer, Physitemp Instruments) between 4 to 5 pm after feeding with a HFD for 6 to 7 weeks. Data are presented as mean ± SEM from three experiments. **P < 0.01, Ela2−/− vs WT. Also see Figures S3 & S4.
To compare insulin sensitivity in WT and Ela2−/− mice, we measured fasting blood glucose and serum insulin levels in HFD-fed mice. Interestingly, Ela2−/− mice had significantly lower levels than WT mice of both fasting glucose and insulin (Figures 4A and 4B), suggesting these mice have a greater sensitivity to insulin. Consistent with this, glucose and insulin tolerance tests showed that Ela2−/− mice were more tolerant to glucose challenge and more sensitive to insulin stimulation (Figures 4C–4E). There were no differences in IPGTT, fasting blood glucose and serum insulin levels between WT and knockout mice fed with low fat diet (Figures S3D–S3F). In addition, the levels of HMW adiponectin, an insulin sensitizing adipokine, were increased by 2.5-fold in the serum of HFD-fed Ela2−/− mice compared with similarly treated WT mice (Figures 4F & G).
Figure 4. Recovery of Insulin Sensitivity and Increase in Serum HMW Adiponectin Levels in Elastase-Deficient Mice (A–G) and Pharmacological Inhibitor-Treated Mice (H–O).

Fasting blood glucose levels (A) and serum insulin levels (B) in WT and Ela2−/− mice fed a 60% HFD over for 9 weeks and 12 weeks, respectively (n = 9), *P < 0.05. (C) WT and Ela2−/− mice were given an intraperitoneal insulin tolerance test (IPITT, i.p. injection of insulin at the dose of 0.75 U/kg bodyweight) after feeding HFD for 9.5 weeks or (D) an intraperitoneal glucose tolerance test (IPGTT, i.p. injection of glucose at the dose of 0.9 g/kg bodyweight) after HFD for 10.5 weeks. (E) Area under the curve (AUC) for the IPGTT data. Data are presented as mean ± SEM (n = 9 in each group), **P < 0.01. (F) HMW adiponectin in serum samples (1.5 μl each) from WT and Ela2−/− mice was resolved by 2–15% gradient SDS-PAGE under non-reducing conditions (upper panel, different polymer adiponectin) or reducing and heat-denaturing conditions (lower panel, total adiponectin) and blotted with an antibody to adiponectin. (G) Quantification of serum HMW adiponectin in WT and Ela2−/− mice (mean ± SEM, n = 8, *P < 0.05, Ela2−/− vs WT). For the experiment with NE chemical inhibitor, C57BL/6 mice at 8 weeks of age were fed a 60% HFD for 19 weeks and then administered a NE inhibitor (GW311616A; inhib) (2 mg/kg bodyweight) or vehicle (H2O) by gavage every other day for up to 6 weeks. All data are mean ± SEM of 5 mice per group. (H) Serum NE activity after treatment for 1 or 3 weeks. **P < 0.01. (I) Change in bodyweight during 6 weeks of treatment. **P < 0.01 comparing before and after treatment. (J) Fasting glucose levels were measured after 6 weeks of treatment. *P < 0.05. IPGTT: (L and M) and IPITT (K) were performed after treatment with inhibitor for 5 or 6 weeks, respectively. *P < 0.05, **P < 0.01. (N and O) Serum samples from HFD-fed mice were taken after treatment with vehicle or GW311616A for 6 weeks and HMW adiponectin was quantified. *P < 0.05. Also see Figures S6.
A Small Molecule Inhibitor of NE Reverses HFD-Induced Insulin Resistance
We next asked if pharmacological inhibition of NE could reverse obesity and improve insulin sensitivity. For this, WT mice fed with an HFD for 19 weeks were orally administered by gavage with a long-term (4 days) effective, specific NE inhibitor GW311616A (inhib) (Macdonald et al., 2001) every other day for up to 6 weeks. GW311616A inhibits NE activity by covalently binding to the enzyme. As shown in Figure 4H, GW311616A treatment inhibited serum NE activity by 60% within 1 week of treatment, and this inhibitory effect was sustained during the treatment. GW311616A inhibition of NE activity resulted in significant reductions in HFD-induced bodyweight gain (Figure 4I) and fasting blood glucose levels (Figure 4J). Moreover, both insulin sensitivity and glucose tolerance were significantly improved in the obese WT mice treated with GW311616A for 5 to 6 weeks, compared with the vehicle-treated mice (Figures 4K–4M). Interestingly, GW311616A also significantly increased the level of HMW adiponectin and the ratio of HMW adiponectin to total adiponectin in the sera of HFD-induced obese mice (Figures 4N & 4O).
Depletion of NE Increases Insulin and AMP-Activated Protein Kinase (AMPK) Signaling, Fatty Acid Oxidation in Periphery Tissues, and UCP1 Protein Levels in the BAT
To explore the molecular mechanisms whereby the altered ratio of NE to A1AT might regulate metabolic functions, we further analyzed biochemical parameters involved in the regulation of glucose and fatty acid metabolisms. Ela2−/− and WT mice were fed 60% HFD for 12 weeks and muscle, liver, and epididymal white adipose tissue (eWAT) were then used for analyzing AMPK signaling and expression of genes important for fatty acid metabolism (Kahn et al., 2005). For detection of insulin-induced Akt phosphorylation, mice were fasted before insulin injection (i.p.). As shown in Figures 5A, B and C, treatment of mice with insulin increased the phosphorylation of Akt in the liver, skeletal muscle, and eWAT of both Ela2−/− and WT mice, but the effect was significantly higher in Ela2−/− mice. We also observed that the protein levels of IRS1 in eWAT and of IRS2 in both liver and eWAT, but not skeletal muscle, were significantly increased in Ela2−/− mice. Interestingly, basal phosphorylation of AMPK and acetyl-CoA carb oxylase 2 (ACC2), two enzymes involved in the regulation of fatty acid oxidation, were increased 3-fold and 2-fold, respectively, in the liver of HFD-fed Ela2−/− mice compared with WT mice (Figure 5A). Of note, the total protein levels of ACC2 were significantly lower in the liver of Ela2−/− mice. Increased AMPK phosphorylation was also observed in the liver from control low fat diet-fed Ela2−/− mice (Figure S3G & S3H). We also examined the expression of genes involved in glucose and lipid metabolism in the HFD-fed Ela2−/− and WT mice. Expression levels of genes involved in gluconeogenesis such as phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6P) were significantly decreased in the liver of Ela2−/− mice (Figure S5A). However, there were no differences in the phosphorylation of AMPK and ACC (Figure 5B) or the expression of G6P, PEPCK, SREBP1c and PGC1α (Figure S5B) in skeletal muscle from Ela2−/− and WT mice.
Figure 5. Alternation of Signaling Proteins and Fatty Acid Oxidation (FAO) in Tissues of Elastase-Deficient Mice.
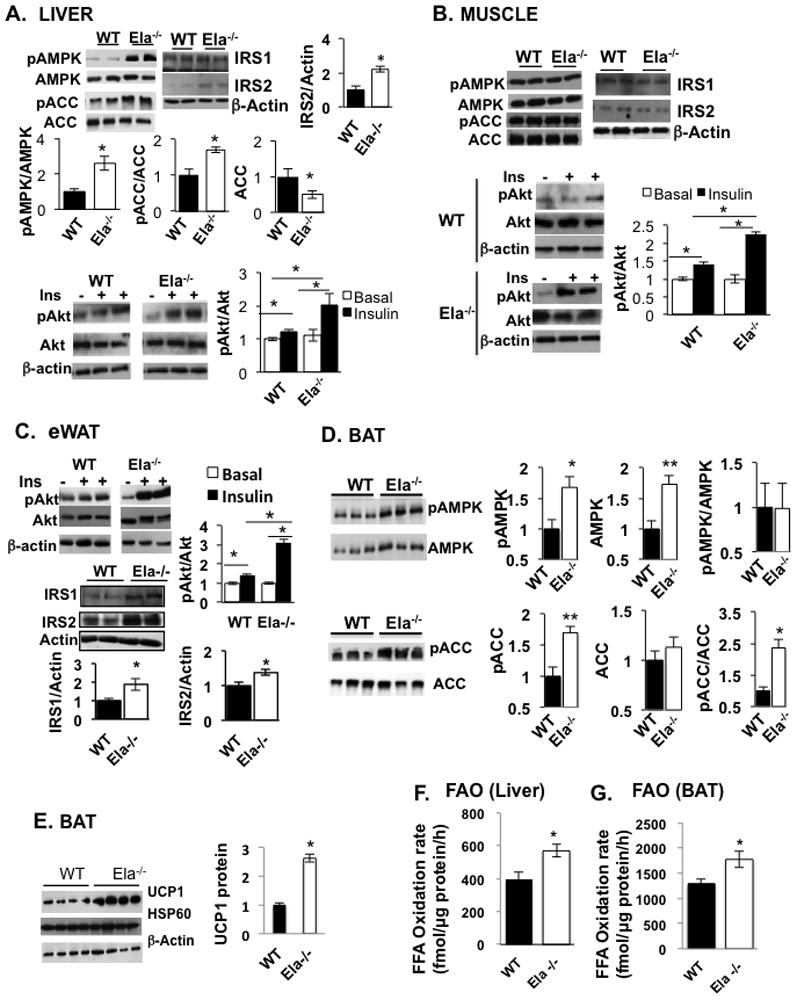
Ela2−/− and WT mice were fed an HFD for 12 weeks, fasted for 6 hrs, and injected with vehicle or insulin (i.p. 5 U/kg bodyweight), and then sacrificed 15 min later. Tissue homogenates from liver (A), skeletal muscle (B), eWAT (C) were BAT (E) resolved by SDS-PAGE and immunoblotted with antibodies specific for pAkt, Akt, pACC (phospho-Ser79), ACC, pAMPK (phospho-Thr172), AMPK, UCP1, or β-Actin, as indicated. For analyzing the AMPK pathway in BAT (D) and FAO rates in liver (F) and BAT (G) homogenates, 9 weeks old WT and Ela2−/− mice were fed with an HFD for only 2 weeks (D) and 5 days (F & G). Data are presented as mean ± SEM with 8 animals in each group. *P < 0.05, **P < 0.01 as indicated or Ela2−/− vs WT mice. Also see Figure S5 for gene expression data.
Interestingly, both AMPK protein levels and phosphorylation of AMPK and ACC2 in the BAT from Ela2−/− mice were significantly higher than WT mice fed with an HFD for 2 weeks (Figure 5D). In addition, BAT from Ela2−/− mice expressed significantly higher levels of UCP1 protein than WT mice fed with HFD for 12-weeks (Figure 5E), although there was no significant difference in the expression of ucp1, ucp3 or other mitochondrial genes, except ucp2 and cox7a, in the BAT between the two groups of mice (Figure S5D). Since the AMPK-ACC2 pathway is involved in the regulation of FAO, we also measured palmitate oxidation rates in the liver and BAT homogenates. Interestingly, palmitate oxidation rates were 43% increased in the liver and 38% higher in the BAT from Ela2−/− mice when compared with WT mice both fed with an HFD for only 5 days (Figures 5F & 5G), consistent with the increase of the AMPK signaling in both tissues.
These data suggest that the insulin/Akt signaling is significantly increased in the periphery tissues while the activity of the AMPK pathway is increased in the liver and BAT of Ela2−/− mice, leading to the decreased expression of genes involved gluconeogenesis in the liver and the increase of FAO rates in both liver and BAT. Together, the increase of FAO and UCP1 protein levels contribute, at least partially, to the resistance of HFD-induced bodyweight gain and liver steatosis in Ela2−/− mice.
We did not observe significant differences between the two groups of mice in expression of genes involved in the regulation of adipose differentiation or in expression of adiponectin (Figure S5C). However, HMW adiponectin levels were increased in the serum of HFD-fed Ela2−/− mice (Figures 4F & 4G). HMW adiponectin is the active form of the adipokine involved in the regulation of insulin sensitivity and fatty acid metabolism through activation of the AMPK pathway (Kadowaki et al., 2006; Turer and Scherer, 2012). This finding is consistent with the observation that the AMPK pathway is more active in the liver, but not muscle, of Ela2−/− mice, because HMW adiponectin has a greater effect on liver (Kadowaki et al., 2006; Shetty et al., 2009). The observed increase in serum HMW adiponectin in Ela2−/− mice could be due to increased secretion from adipose tissues or depletion of NE that cleaves adiponectin (Waki et al., 2005). Consistent with the latter notion, we demonstrated that incubation of purified leukocyte elastase with adiponectin-containing conditioned medium from 3T3-L1 adipocytes reduced the level of HMW adiponectin and increased the production of small fragments of adiponectin (Figure S6). Our data suggest that Ela2−/− mice express increased levels of HMW adiponectin, which is involved in regulating lipid metabolism and insulin sensitivity via activation of the AMPK pathway in the liver.
Depletion of NE Blocks Inflammation in Mouse White Adipose Tissues at Early and Late Stages of HFD Feeding
Because low-grade chronic inflammation plays an important role in obesity-related insulin resistance, we examined HFD-induced macrophage infiltration into visceral eWAT in the NE-deficient mice. The eWAT of WT mice fed a HFD for 12 weeks contained about 6% macrophage crown-like cells (CLC), as revealed by positive F4/80 immunohistochemical staining (Figure 6A/B). Remarkably, CLC were reduced to less than 0.6% in the eWAT from Ela2−/− mice fed an HFD for 12 weeks. Using Masson’s trichrome stain, we observed the accumulation of connective tissues (blue staining) between adipocytes in eWAT of WT mice, but not Ela2−/− mice, fed with the HFD for 12 weeks (Figure 6C). These data suggest that Ela2−/− mice are resistant to HFD-induced macrophage infiltration, tissue damage and fibrosis. NE expression in eWAT was also examined with immunohistochemical staining. As shown in Figure 6D, similar as F4/80 and connective tissue staining, NE accumulated around small adipocytes in eWAT from WT mice fed with the HFD for 12 weeks, suggesting that NE may contribute to tissue damage and consequent fibrosis in eWAT due to HFD feeding.
Figure 6. Reduction of Adipose Inflammation and Fibrosis in Elastase-Deficient Mice at Early and Late Stages of HFD Feeding.

(A and B) Representative images and quantification of immunostaining of the macrophage marker F4/80 in eWAT sections of WT and Ela2−/− mice fed a HFD for 12 weeks. The graph shows the percentage of macrophage-infiltrated cells with a characteristic crown-like structure in total adipocytes, counted on whole tissue sections scanned with an Aperio ScanScope system (Aperio, Vista, CA). (C) Representative images of Masson’s trichrome stain in eWAT from WT and Ela2−/− mice fed with normal chow diet (NCD) or HFD for 12 weeks. The blue stain indicates connective tissue fibrosis. (D) Representative images of immunohistochemical stain of NE in eWAT of WT mice fed with NCD or HFD for 12 weeks. Red arrows indicate positive staining of F4/80 (A), fibrosis (C), and NE (D). Scale bar = 50 μm. (E and F) Percentage of neutrophils (CD11b+/LY6G+/F480−/CD11c− cells) in vascular stromal fractions and representative neutrophil profiles analyzed with FACS. (G) qRT-PCR analysis of mRNA extracted from eWAT of WT and Ela2−/− mice fed with NCD for 3 days (Day3 NCD), HFD for 3 days (Day3 HFD), or HFD for 12 weeks (Week12 HFD). The indicated inflammation-associated genes were analyzed to profile the inflammatory state of the adipose tissue. All graphs are mean ± SEM, n = 4 and 6 animals for (E) and (G), respectively. *P < 0.05 or **P < 0.01, WT vs Ela2−/− mice fed with HFD for 12 weeks; § P < 0.01, WT vs Ela2−/− mice fed with HFD for 3 days. Also see Figure S7.
We next determined whether short-term feeding of 60% HFD affected metabolic function in Ela2−/− mice. After feeding with HFD for 3 days, WT and Ela2−/− mice showed no significant change in total bodyweight or epididymal fat pad weight (Figures S7A & S7B), but blood glucose levels following a 6-hr fast were significantly lower in Ela2−/− mice than in WT mice (Figure S7C). Surprisingly, after 3 days on the HFD, WT mice showed about 2-fold increase in serum NE activity, although serum A1AT levels were unchanged (Figures S7D & S7E).
Neutrophils are among the first immune cells to be activated during tissue inflammation (Elgazar-Carmon et al., 2008). Using a FACS analysis approach, we detected neutrophils (1.6%) positively expressing both cell surface markers Ly6G and CD11b in the stromal vascular fractions isolated from eWAT of WT mice fed an HFD for 2 weeks, but not in HFD-fed Ela2−/− mice or WT mice fed normal chow (Figures 6E & 6F). This data suggests neutrophil infiltration in eWAT at the early stage of HFD feeding in WT mice, but not Ela2−/− mice. We further compared the expression of several inflammation markers in eWAT from WT and Ela2−/− mice fed with HFD for either 3 days or 12 weeks. Ela2−/− mice fed a HFD for 3 days showed no increase in expression of other early inflammation makers such as the chemoattractants MCP-1 and IL-8 and the leukocyte adhesion molecule ICAM-1 as observed in WT mice (Figure 6G). In comparison with WT mice, eWAT from Ela2−/− mice fed with the HFD for 12 weeks showed significant lower levels of the chemoattractants MCP-1, IL-8, and markers of “classically activated” M1 macrophages such as F4/80, CD68, TNFα, and CD11c; in contrast, there was no difference in the expression of CD163, a marker of the alternatively activated M2 macrophages. There were no significant changes in these macrophage markers in eWAT from either mouse strain after only 3 days of HFD feeding. These data therefore suggest that Ela2−/− mice are resistant to adipose inflammation induced by both short-term and long-term feeding with a HFD. Thus, NE is an important factor involved in HFD-induced adipose inflammation and tissue damage.
Mice Overexpressing Human a1-Antitrypsin (hA1AT) are Resistant to HFD-Induced Obesity, Fatty Liver, Insulin Resistance and Inflammation
To evaluate the effect of over-expression of hA1AT on HFD-induced insulin resistance, obesity and inflammation, transgenic mice expressing hA1AT on C57BL/6J × CBA background (Kelsey et al., 1987) were back-crossed with C57BL/6J mice for 6 generations before feeding them with 60% HFD. As shown in Figure 7A, mice overexpressing hA1AT had high-level expression of hA1AT in the serum. Worth noting, transgenic mice were resistant to HFD-induced bodyweight gain, fat mass and blood glucose increase (Figures 7B–7D), and were more insulin sensitive and glucose tolerant than WT littermates (Figures 7E–7G). The transgenic mice showed increased AMPK protein phosphorylation and palmitate oxidation rates in the liver, and reduced HFD-induced liver steatosis after 11 weeks on 60% HFD (Figures 7H–7J). Overexpression of hA1AT also protected HFD-induced decrease of mouse endogenous A1AT expression in the liver (data not shown). In addition, HFD-induced infiltration of F4/80 positive CLC and mRNA expression of inflammatory markers, F4/80, CD11c and MCP1, was significantly decreased in the eWAT from hA1AT mice (Figures 7K & 7L). These data suggest that hA1AT has multiple protective effects on obesity-related complications.
Figure 7. Overexpression of hA1AT in Mice Ameliorates HFD-Induced Insulin Resistance, Liver steatosis, Bodyweight Gain and Adipose Inflammation.
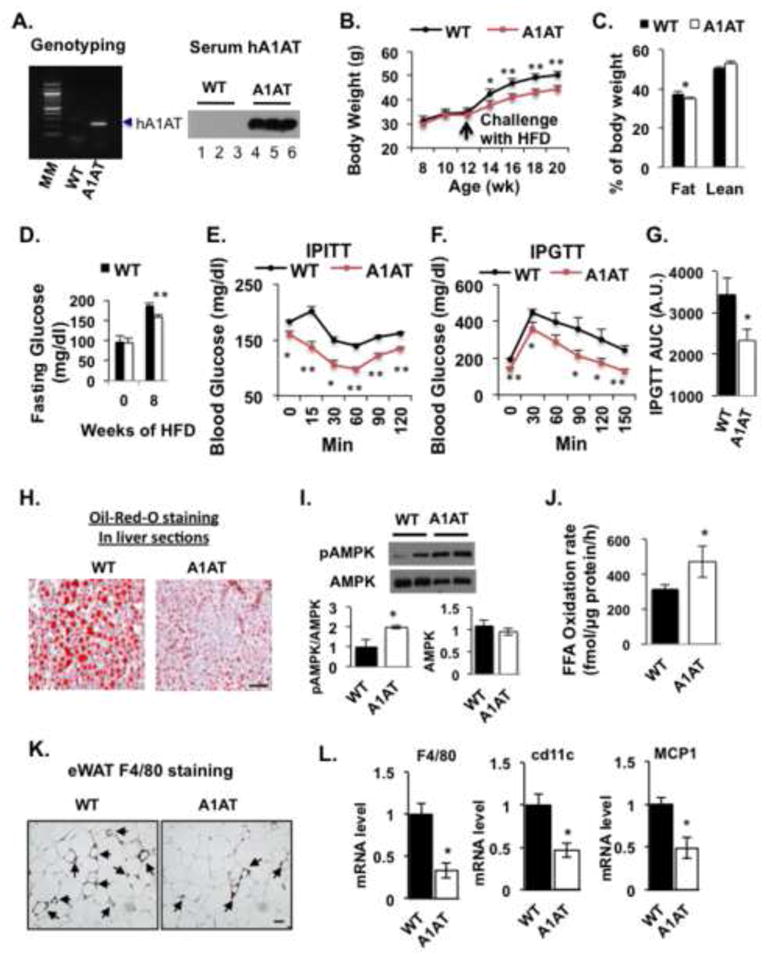
Mice expressing hA1AT were confirmed by both DNA genotyping and serum A1AT detection with specific antibody against human α-1-antitrypsin (A). Mean bodyweights (B) of hA1AT mice and WT C57BL/6 littermates during 8 weeks on 60% HFD (n = 8). (C) Percentage of lean and fat mass measured by MRI in mice fed the HFD for 9 weeks (n = 8). Data are presented as mean ± SEM from 8 mice in each group. *P < 0.05, ** P< 0.01, Ela2−/− vs WT. During HFD feeding, fasting blood glucose levels (D) were measured at week 9 while IPITT (E) and IPGTT (F, G) were examined at week 8 and 8.5, respectively. AMPK activation (I) and FAO rate (J) in liver homogenates after HFD feeding. (L) Inflammatory markers in eWAT from mice fed with a HFD for 9 weeks. Data are presented as mean ± SEM (n = 8 in each group), **P < 0.01. WT vs A1AT mice. Representative images of Oil Red O staining of liver histologic sections (H) and immunostaining of the macrophage marker F4/80 in eWAT sections (K) from mice fed a HFD for 9 weeks. Scale bars = 50 μm.
DISCUSSION
We applied a quantitative proteomic approach to detect serum proteins that might be differentially expressed during obesity, and identified A1AT as a major serum protein that is significantly reduced in both leptin-deficient ob/ob and HFD-induced obese mice. We showed that leptin stimulated A1AT expression through the Jak2-Stat3 pathway in cultured hepatocytes. This finding is consistent with previous reports that a Stat3 consensus sequence is present in the 3′ enhancer region of the serpina1 gene (Kalsheker et al., 2002). Moreover, leptin infusion into ob/ob mice increased serum A1AT levels, validating the significance of the in vitro findings. Importantly, studies with clinical samples from lean and obese South Asian and U.S. Caucasian subjects revealed that serum A1AT levels were significantly reduced in obese leptin-resistant subjects. Thus, it is possible that leptin is a physiological regulator of A1AT expression and function. Our data suggests for the first time that leptin deficiency and leptin resistance are both related to the observed obesity-related reduction in A1AT expression and secretion.
NE plays an important role in host defense and inflammation (Pham, 2006). NE also breaks down a range of matrix substrates including elastin and other circulating proteins, such as adiponectin (Waki et al., 2005). The catalytic function of NE is blocked by A1AT through covalent binding to each other; thus, A1AT protects tissues from serine protease-induced damage. Here, we report that serum NE activity was significantly increased in leptin-deficient ob/ob mice, HFD-induced obese mice, and obese human subjects, consistent with the reciprocal reductions in the level of the NE inhibitor A1AT. Of noted, we also observed that NE activity increase in the sera of mice fed with a HFD for only three days (Figure S7D) before the development of leptin resistance. Therefore, the imbalance between NE and A1AT occurs in HFD-induced obesity both at the early stage due to the increase of NE activity and at the later stage due to the decrease of A1AT expression. It is possible that increased NE activity is related to neutrophil activation in the early stage (Elgazar-Carmon et al., 2008) and the decrease A1AT expression is related to the development of leptin resistance and fatty liver at the later stage of HFD feeding. We hypothesized that this imbalance might be related to the pathological changes observed in obesity. This hypothesis was tested using NE null mice, hA1AT transgenic mice and NE chemical inhibitor. Both NE null mice and hA1AT transgenic mice were resistant to multiple HFD-induced metabolic disturbances, including weight gain, glucose intolerance, insulin resistance, fatty liver and macrophage infiltration in adipose tissues. Our data support the concept that the imbalance between NE and A1AT is a key factor contributing to the development of HFD-induced obesity, insulin resistance, and liver steatosis. This is particularly important because increased NE activity is observed at an early stage of over-nutrition in WT mice. Our data suggest that NE and A1AT are potential drug targets for treatment of obesity and insulin resistance. Thus, reversing the ratio of NE to A1AT could be beneficial for obesity and insulin resistance due to over-nutrition. This notion is further supported by the benefits of overexpression of hA1AT and treatment with a NE inhibitor on metabolic profiles in HFD-fed mice.
It is well established that obesity is accompanied by low-grade inflammation of adipose tissue, which manifests as infiltration of inflammatory cells such as macrophages, lymphocytes, mast cells, and neutrophils, as well as production of inflammatory cytokines and chemokines that contribute to leukocyte infiltration and insulin resistance (Lumeng and Saltiel, 2011; Osborn and Olefsky, 2012). Notably, we observed that visceral fat of Ela2−/− mice fed an HFD contained less than 10% of the number of macrophages present in fat of WT mice. The expression of proinflammatory M1 macrophage markers, but not M2 macrophage markers, was significantly lower in adipose tissue of Ela2−/− mice. This is consistent with the observation of classically activated M1 macrophages in inflamed adipose tissue in obesity (Lumeng et al., 2007). In addition, we observed no increase in early inflammation markers such as chemoattractant MCP-1, and adhesion molecule ICAM-1, after 3 days of HFD feeding of Ela2−/− mice. Furthermore, our data demonstrated that NE accumulated in eWAT in a similar pattern as macrophage infiltration and fibrosis. These data therefore suggest that NE is a key regulator of HFD-induced inflammation and tissue damage in eWAT throughout the development of obesity. It is possible that activation of NE by HFD feeding may induce vascular damage, which allows entry of neutrophils and M1 macrophages. Thus, this study supports the notion that the imbalance of NE and A1AT is a key pathological mechanism linking over-nutrition to leukocyte infiltration, adipose fibrosis and subsequent insulin resistance in obesity.
Adiponectin is a cytokine secreted by adipose tissue that exists in high, medium, and low molecular weight forms in the circulation. Compelling evidence suggests that adiponectin plays an important role in the regulation of insulin sensitivity, lipid and glucose metabolism, and inflammation (Kadowaki et al., 2006; Ohashi et al., 2010; Shetty et al., 2009). Multiple studies have reported that circulating adiponectin levels, particularly the HMW adipokine, are significantly reduced in obese and type II diabetic patients compared with lean subjects (Pajvani et al., 2003; Semple et al., 2007; Waki et al., 2003). HMW adiponectin has been shown to regulate expression of IRS2 and phosphorylation of AMPK and ACC2 in the liver, which are thought to be important for HMW adiponectin-mediated insulin sensitization and fatty acid oxidation (Awazawa et al., 2011; Kadowaki et al., 2006; Shetty et al., 2012; Yamauchi et al., 2002). In this study, Ela2−/− mice were resistant to the HFD-induced decrease in serum HMW adiponectin, suggesting that NE is a regulator of HMW adiponectin homeostasis. Interestingly, Ela2−/− mice showed increased phosphorylation of AMPK and FAO in the liver. These results are consistent not only with the observation that circulating HMW adiponectin levels are increased in Ela2−/− mice, but also with the functional analyses showing that those mice have improved insulin sensitivity and are resistant to HFD-induced liver steatosis. The reduced expression of PEPCK and glucose-6-phosphatase in the liver of Ela2−/− mice further supports these observations. Thus, our data suggest an additional mechanism by which NE regulates insulin sensitivity and obesity by increasing HMW adiponectin expression and AMPK activation in the liver. It is possible that NE directly cleaves adiponectin resulting in lower levels of HMW adiponectin. Alternatively, the inflammatory damage of adipose tissue may lead to less HMW adiponectin secretion and therefore, less serum HMW adiponectin in WT mice than in Ela2−/− mice.
Our data also provides a potential molecular mechanism whereby the imbalance of NE and A1AT affects HFD-induced bodyweight gain. We observed that both AMPK protein expression and phosphorylation were significantly increased in the BAT of Ela2−/− mice. The increase of the inhibitory phosphorylation of ACC2 by the activated AMPK may contribute to the increase in FAO in the BAT of NE null mice. Therefore, our data for the first time provides the link of the NE-A1AT system to AMPK signaling – FAO – energy expenditure axis. In addition, UCP1 protein levels were significantly increased in the BAT of NE null mice after HFD feeding. These data are consistent with the observation that Ela2−/− mice had higher body temperature, more energy expenditure and less bodyweight gain than WT mice fed with a HFD. It is possible that increased UCP1 expression in the BAT from NE null mice may be due to the decrease of inflammation and chemokine production in the BAT (data not shown). However, it remains to be evaluated how depletion of NE leads to the activation of the AMPK pathway and increase of UCP1 protein levels in the BAT.
Taken together, our data suggest that elevation of serum and tissue NE and reduction of A1AT are key events in obesity. Furthermore, the imbalance between NE and A1AT is associated with leptin resistance and it also precedes the decrease of HMW adiponectin. This study also suggests that the imbalance of NE and A1AT affects energy expenditure through regulating the AMPK pathway and contributes to the development obese-related inflammation, adipose tissue remodeling, insulin resistance and liver steatosis. Hence, both NE and A1AT are important molecular markers for obesity and may be potential drug targets for treatment of obesity and related diseases. Our proteomic profiling-based study is validated with clinical samples and is consistent with an independent recent report suggesting that elastase is involved in the development of inflammation and insulin resistance (Talukdar et al., 2012).
EXPERIMENTAL PROCEDURES
Experimental Animals
NE-deficient (Ela2−/−, B6.129X1-Elanetm1Sds/J) mice, B6.V-Lepob/J (ob/ob) mice, and C57BL/6 control mice were initially purchased from The Jackson Laboratory (Bar Harbor, ME). hA1AT transgenic mice were generated and maintained on a mixed C57BL/6J x CBA background (Kelsey et al., 1987) and were then backcrossed with C57BL/6J mice. All mouse work was performed according to the Institutional Animal Care and Use Committee guidelines.
Insulin and Glucose Tolerance Tests and Leptin Infusion
For insulin and glucose tolerance tests, mice were fasted for 6 hrs and overnight, respectively, before intraperitoneally injection with insulin (0.75 U/kg body weight) or glucose (0.9 g/kg body weight). Glucose levels in tail-vein blood samples were measured with a blood glucometer (Bayer) at different time points post-injection. To test leptin’s function in vivo, mouse recombinant leptin was delivered at 10 μg/day for 7 days via an Alzet pump (model 1007D, Alza Corp., Palo Alto, CA) implanted subcutaneously in ob/ob mice.
Body Composition Analysis and Comprehensive Laboratory Animal Monitoring System (CLAMS)
Body composition of live mice was measured using Minispec LF90II time domain NMR analyzer (Bruker Optics, Inc, Billerica, MA). Energy expenditure, food intake, and locomotory activity were measured using a CLAMS system from Columbus Instruments (Columbus, OH) as described in the Supplemental Experimental Procedures (S.E.P.).
Human Blood Samples
Sera were collected from both South Asian and U.S. Caucasian men with different BMI according to the guidelines of the Institutional Review Board. Serum A1AT and leptin levels were measured with ELISA kits according to the instructions provided by Immunology Consultants (Newberg, OR) and Millipore (Danvers, MA), respectively.
Measurement of Elastase Activity
Both human and mouse serum NE activities were measured with N-methoxysuccinyl-Ala-Ala-Pro-Val-p-nitroanilide, a highly specific synthetic substrate for NE, according to the methods described previously (Labow et al., 1996; Yoshimura et al., 1982).
Serum Proteomics Analysis
Glycosylated secreted proteins in serum samples from 12–week-old ob/ob and C57BL/6 control mice were analyzed using glycoprotein enrichment and quantitative proteomic approaches, as previously described (Tian et al., 2007; Zhang et al., 2003)
Cell Culture, Immunoblotting and Histological Analysis
Hepatic cell lines HepG2 and Hep1-6 were obtained from ATCC. Proteins were extracted from cells or tissues and immunoblotted with different antibodies as described in the S.E.P. Histological analyses of macrophage infiltration, neutrophil elastase deposition, lipid accumulation and fibrosis in different tissue sections are described in the S.E.P.
Gene Expression
Total RNAs were extracted from tissues or cultured cells, reverse transcribed into first-strand cDNA and then quantified with Real-time PCR using methods and PCR primers described in the S.E.P.
Fatty Acid Oxidation
Crude homogenates from freshly collected liver (500 μg protein) or brown fat tissues (100 μg protein) were used to induce fatty acid oxidation in a buffer containing [1-14C]-palmitate for 30 min at 37°C. Palmitate oxidation rate was quantified by measuring 14C-labeled carbon dioxide production as described in the S.E.P.
Isolation of the SVF and FACS Analysis
Anesthetized mice were perfused with PBS via left ventricles before perigonadal fat pads were excised and used for isolation of SVF as described in the S.E.P. The cells in SVF were blocked with 1 μg Fc receptor for 15 min at 4°C before incubation with 0.5 μg fluorochrome-conjugated primary antibodies for 30 min at 4°C in PBS containing 1 mM CaCl2 and 0.5% BSA. After staining with APC-conjugated anti-Ly-6G antibody, PE-conjugated anti-CD11b antibody, FITC-conjugated anti-F4/80 antibody, PerCP-conjugated anti-CD11c antibody and respective isotype controls, the cells were analyzed by BD Canto flow cytometer and data were analyzed using FlowJo 7.5.5 software. Neutrophils were identified as CD11c−F4/80−CD11b+Ly-6G+ cells.
Analysis of Adiponectin Complexes
Mouse sera were diluted into non-reducing sample buffer (3% SDS, 50 mM Tris-HCl pH 6.8, and 10% glycerol) before subjecting to SDS-PAGE for detecting adiponectin complexes under non-reducing and non-heat-denaturing conditions. For detecting total denatured adiponectin, sera diluted in sample buffer containing 5% 2-mercaptoethanol and 10 mM dithiothreitol were heated at 95°C for 10 min before dissolving in SDS-PAGE by electrophoresis and then immunoblotting with primary anti-adiponectin antibody.
Statistics
For correlation studies with human samples, P value and Pearson regression coefficient (r) were generated by Prism 5.0 (GraphPad, La Jolla, CA). Group-wise differences were assessed by two-tailed unpaired t-test with Welch correction. For mouse studies, evaluations were performed by two-way ANOVA and unpaired Student t-test. Error bar represents standard error of the mean (SEM).
Supplementary Material
Highlights.
Serum A1AT levels are decreased and neutrophil elastase (NE) activity is increased in obese men and mice
Neutrophil infiltration and NE deposition occur in adipose tissue from HFD-fed mice
NE-null and A1AT mice alleviate HFD-induced obesity, insulin resistance and inflammation
NE-null mice increase AMPK signaling and fatty acid oxidation in the liver and BAT
Acknowledgments
We wish to thank Julio Ayala, John Shelley, and SBMRI Cardiometabolic Phenotyping Core for excellent help in the CLAMS study and histology, Annette Khaled, Rebecca Boohaker, Mangala Soundarapandian, Ling Lai, Rita Luther and Darrin Kuystermans for technical help. We appreciate Philipp Scherer, Tim Osborne, Guy Salvesen, Dan Kelly and Dev Sikder for comments, and Shonna Small for administrative support. Z.Y.J. is supported by research grants from the American Diabetes Association (7-11-BS-72) and NIH grant R01 DK094025. X.X. is supported by a James and Esther King Postdoctoral Fellowship. R.L-B is funded by the UK Medical Research Council (U117512772). Z.Y.J. wrote the manuscript.
Footnotes
Supplemental Information includes 2 tables, 7 figures and the Supplemental Experimental Procedures can be found with this article online
All authors have no conflict of interest regarding this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Awazawa M, Ueki K, Inabe K, Yamauchi T, Kubota N, Kaneko K, Kobayashi M, Iwane A, Sasako T, Okazaki Y, et al. Adiponectin enhances insulin sensitivity by increasing hepatic IRS-2 expression via a macrophage-derived IL-6-dependent pathway. Cell Metab. 2011;13:401–412. doi: 10.1016/j.cmet.2011.02.010. [DOI] [PubMed] [Google Scholar]
- Belaaouaj A, McCarthy R, Baumann M, Gao Z, Ley TJ, Abraham SN, Shapiro SD. Mice lacking neutrophil elastase reveal impaired host defense against gram negative bacterial sepsis. Nat Med. 1998;4:615–618. doi: 10.1038/nm0598-615. [DOI] [PubMed] [Google Scholar]
- Elgazar-Carmon V, Rudich A, Hadad N, Levy R. Neutrophils transiently infiltrate intra-abdominal fat early in the course of high-fat feeding. Journal of lipid research. 2008;49:1894–1903. doi: 10.1194/jlr.M800132-JLR200. [DOI] [PubMed] [Google Scholar]
- Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, Lee J, Goldfine AB, Benoist C, Shoelson S, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 2009;15:930–939. doi: 10.1038/nm.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest. 2006;116:1784–1792. doi: 10.1172/JCI29126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1:15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Kalsheker N, Morley S, Morgan K. Gene regulation of the serine proteinase inhibitors alpha1-antitrypsin and alpha1-antichymotrypsin. Biochem Soc Trans. 2002;30:93–98. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- Kamei N, Tobe K, Suzuki R, Ohsugi M, Watanabe T, Kubota N, Ohtsuka-Kowatari N, Kumagai K, Sakamoto K, Kobayashi M, et al. Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J Biol Chem. 2006;281:26602–26614. doi: 10.1074/jbc.M601284200. [DOI] [PubMed] [Google Scholar]
- Kelsey GD, Povey S, Bygrave AE, Lovell-Badge RH. Species- and tissue-specific expression of human alpha 1-antitrypsin in transgenic mice. Genes Dev. 1987;1:161–171. doi: 10.1101/gad.1.2.161. [DOI] [PubMed] [Google Scholar]
- Korkmaz B, Horwitz MS, Jenne DE, Gauthier F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacological reviews. 2010;62:726–759. doi: 10.1124/pr.110.002733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labow RS, Erfle DJ, Santerre JP. Elastase-induced hydrolysis of synthetic solid substrates: poly(ester-urea-urethane) and poly(ether-urea-urethane) Biomaterials. 1996;17:2381–2388. doi: 10.1016/s0142-9612(96)00088-9. [DOI] [PubMed] [Google Scholar]
- Liu J, Divoux A, Sun J, Zhang J, Clement K, Glickman JN, Sukhova GK, Wolters PJ, Du J, Gorgun CZ, et al. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat Med. 2009;15:940–945. doi: 10.1038/nm.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117:175–184. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. 2011;121:2111–2117. doi: 10.1172/JCI57132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald SJ, Dowle MD, Harrison LA, Shah P, Johnson MR, Inglis GG, Clarke GD, Smith RA, Humphreys D, Molloy CR, et al. The discovery of a potent, intracellular, orally bioavailable, long duration inhibitor of human neutrophil elastase--GW311616A a development candidate. Bioorg Med Chem Lett. 2001;11:895–898. doi: 10.1016/s0960-894x(01)00078-6. [DOI] [PubMed] [Google Scholar]
- Myers MG, Jr, Leibel RL, Seeley RJ, Schwartz MW. Obesity and leptin resistance: distinguishing cause from effect. Trends Endocrinol Metab. 2010;21:643–651. doi: 10.1016/j.tem.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, Otsu M, Hara K, Ueki K, Sugiura S, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. 2009;15:914–920. doi: 10.1038/nm.1964. [DOI] [PubMed] [Google Scholar]
- Ohashi K, Parker JL, Ouchi N, Higuchi A, Vita JA, Gokce N, Pedersen AA, Kalthoff C, Tullin S, Sams A, et al. Adiponectin promotes macrophage polarization toward an anti-inflammatory phenotype. J Biol Chem. 2010;285:6153–6160. doi: 10.1074/jbc.M109.088708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn O, Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med. 2012;18:363–374. doi: 10.1038/nm.2627. [DOI] [PubMed] [Google Scholar]
- Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. 2011;11:85–97. doi: 10.1038/nri2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajvani UB, Du X, Combs TP, Berg AH, Rajala MW, Schulthess T, Engel J, Brownlee M, Scherer PE. Structure-function studies of the adipocyte-secreted hormone Acrp30/adiponectin. Implications fpr metabolic regulation and bioactivity. J Biol Chem. 2003;278:9073–9085. doi: 10.1074/jbc.M207198200. [DOI] [PubMed] [Google Scholar]
- Pham CT. Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol. 2006;6:541–550. doi: 10.1038/nri1841. [DOI] [PubMed] [Google Scholar]
- Semple RK, Halberg NH, Burling K, Soos MA, Schraw T, Luan J, Cochran EK, Dunger DB, Wareham NJ, Scherer PE, et al. Paradoxical elevation of high-molecular weight adiponectin in acquired extreme insulin resistance due to insulin receptor antibodies. Diabetes. 2007;56:1712–1717. doi: 10.2337/db06-1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty S, Kusminski CM, Scherer PE. Adiponectin in health and disease: evaluation of adiponectin-targeted drug development strategies. Trends Pharmacol Sci. 2009;30:234–239. doi: 10.1016/j.tips.2009.02.004. [DOI] [PubMed] [Google Scholar]
- Shetty S, Ramos-Roman MA, Cho YR, Brown J, Plutzky J, Muise ES, Horton JD, Scherer PE, Parks EJ. Enhanced fatty acid flux triggered by adiponectin overexpression. Endocrinology. 2012;153:113–122. doi: 10.1210/en.2011-1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Kusminski CM, Scherer PE. Adipose tissue remodeling and obesity. J Clin Invest. 2011;121:2094–2101. doi: 10.1172/JCI45887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Ji Y, Kersten S, Qi L. Mechanisms of inflammatory responses in obese adipose tissue. Annual review of nutrition. 2012;32:261–286. doi: 10.1146/annurev-nutr-071811-150623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, McNelis J, Lu M, Li P, Yan Q, Zhu Y, et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med. 2012 doi: 10.1038/nm.2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Zhou Y, Elliott S, Aebersold R, Zhang H. Solid-phase extraction of N-linked glycopeptides. Nature protocols. 2007;2:334–339. doi: 10.1038/nprot.2007.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turer AT, Scherer PE. Adiponectin: mechanistic insights and clinical implications. Diabetologia. 2012;55:2319–2326. doi: 10.1007/s00125-012-2598-x. [DOI] [PubMed] [Google Scholar]
- Waki H, Yamauchi T, Kamon J, Ito Y, Uchida S, Kita S, Hara K, Hada Y, Vasseur F, Froguel P, et al. Impaired multimerization of human adiponectin mutants associated with diabetes. Molecular structure and multimer formation of adiponectin. J Biol Chem. 2003;278:40352–40363. doi: 10.1074/jbc.M300365200. [DOI] [PubMed] [Google Scholar]
- Waki H, Yamauchi T, Kamon J, Kita S, Ito Y, Hada Y, Uchida S, Tsuchida A, Takekawa S, Kadowaki T. Generation of globular fragment of adiponectin by leukocyte elastase secreted by monocytic cell line THP-1. Endocrinology. 2005;146:790–796. doi: 10.1210/en.2004-1096. [DOI] [PubMed] [Google Scholar]
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G, Tsui H, Wu P, Davidson MG, Alonso MN, et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med. 2011;17:610–617. doi: 10.1038/nm.2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8:1288–1295. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- Yoshimura T, Barker LN, Powers JC. Specificity and reactivity of human leukocyte elastase, porcine pancreatic elastase, human granulocyte cathepsin G, and bovine pancreatic chymotrypsin with arylsulfonyl fluorides. Discovery of a new series of potent and specific irreversible elastase inhibitors. J Biol Chem. 1982;257:5077–5084. [PubMed] [Google Scholar]
- Zhang H, Li XJ, Martin DB, Aebersold R. Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nature biotechnology. 2003;21:660–666. doi: 10.1038/nbt827. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.