

Summary
Initiation of translation involves recognition of the start codon by the initiator tRNA in the 30S subunit. To investigate the role of ribosomal RNA (rRNA) in this process, we isolated a number of 16S rRNA mutations that increase translation from the non-canonical start codon AUC. These mutations cluster to distinct regions that overlap remarkably well with previously identified class III protection sites and implicate both IF1 and IF3 in start codon selection. Two mutations map to the 790 loop and presumably act by inhibiting IF3 binding. Another cluster of mutations surrounds the conserved A1413○G1487 base pair of helix 44 in a region known to be distorted by IF1 and IF3. Site-directed mutagenesis in this region confirmed that this factor-induced rearrangement of helix 44 helps regulate initiation fidelity. A third cluster of mutations maps to the neck of the 30S subunit, suggesting that the dynamics of the head domain influences translation initiation. In addition to identifying mutations that decrease fidelity, we found that many P-site mutations increase the stringency of start codon selection. These data provide evidence that the interaction between the initiator tRNA and the 30S P site is tuned to balance efficiency and accuracy during initiation.
Introduction
Translation initiation involves recognition of the start codon by the initiator tRNA in the ribosomal P site. In bacteria, this process is kinetically controlled by three initiation factors (IF1, IF2 and IF3) and occurs in two major phases (Gualerzi and Pon, 1990; Gualerzi et al., 2001; Boelens and Gualerzi, 2002). The first phase is assembly of the 30S initiation complex (30SIC). During this phase, the initiator tRNA (fMet-tRNAfMet) binds the 30S P site and its anti-codon pairs with the start codon of mRNA. The second phase includes the docking of the 50S subunit onto the 30SIC, IF2-dependent GTP hydrolysis, and dissociation of the initiation factors. This generates a 70S initiation complex (70SIC) ready for the first round of elongation.
The efficiency of translation initiation is determined by cis-acting elements located in the translation initiation region of the mRNA. One important element is the start codon. In bacteria, AUG is the most common start codon, but GUG and UUG are also used quite often (Eduardo et al., 1999). Another important element is a purine-rich sequence (e.g. AGGA) termed the Shine–Dalgarno (SD) (Shine and Dalgarno, 1974), which lies 6–12 nucleotides upstream from the start codon (Shultzaberger et al., 2001). The SD pairs with the 3′ end of 16S ribosomal RNA (rRNA), termed anti-Shine–Dalgarno sequence (ASD), and this mRNA–rRNA interaction facilitates positioning of the start codon in the 30S P site (Steitz and Jakes, 1975; Kaminishi et al., 2007; Korostelev et al., 2007). The sequence of the SD, sequence of the start codon, number of nucleotides between the SD and start codon, and the secondary structure of the translation initiation region influence the efficiency of initiation (Ringquist et al., 1992; Vellanoweth and Rabinowitz, 1992; de Smit and van Duin, 2003; Studer and Joseph, 2006), indicating the importance of each of these determinants in the initiation process.
Initiation at the correct start codon is critical for establishing the translational reading frame. A number of mutations have been isolated in Escherichia coli that increase spurious initiation (i.e. translation from non-canonical start codons such as ACG, AUU, AUC and CUG). These mutations were mapped to infC, the gene encoding IF3, strongly implicating the factor in start codon selection (Sacerdot et al., 1996; Sussman et al., 1996; Haggerty and Lovett, 1997). Reducing the concentration of wild-type IF3 in the cell also increases spurious initiation, while overexpression of IF3 confers the opposite phenotype (Olsson et al., 1996; Sacerdot et al., 1996). Indeed, the ability of IF3 to repress translation from non-canonical start codons explains how the factor autoregulates translation of its own gene, which starts with AUU (Brombach and Pon, 1987; Butler et al., 1987; Sacerdot et al., 1996).
IF3 appears to act at both stages of initiation to ensure accuracy. IF3 destabilizes fMet-tRNAfMet during 30SIC formation, which presumably makes its binding more dependent on cognate codon–anti-codon interaction (La Teana et al., 1993; Antoun et al., 2006a,b). IF3 also inhibits formation of a stable 70SIC when the mRNA contains a non-canonical start codon such as AUU (Grigoriadou et al., 2007; Milon et al., 2008).
Although IF1 is an essential protein in the cell (Cummings and Hershey, 1994), its role in translation initiation remains elusive. IF1 is a small (~8 kDa) β-barrel protein that binds the 30S A site and enhances the activities of the other two factors (Stringer et al., 1977; Pon and Gualerzi, 1984; Moazed et al., 1995; Sette et al., 1997; Carter et al., 2001; Antoun et al., 2006a), which may stem in part from cooperative binding (Weiel and Hershey, 1982; Zucker and Hershey, 1986; Moazed et al., 1995). Recent biochemical studies suggest that IF1 negatively regulates 70SIC formation to ensure high fidelity (Milon et al., 2008). However, in vivo evidence that IF1 increases the accuracy of initiation is lacking.
Ribosomal RNA undoubtedly plays a role in initiation, but precisely how 16S rRNA participates in the process remains unclear. Here, we randomly mutagenized 16S rRNA and isolated a number of mutations that increase translation from the non-canonical start codon AUC. These mutations map to distinct regions including the neck, the 790 loop, and helix 44 (h44). The mutations in h44 map near the non-canonical A1413○G1487 pair in a region distorted by either IF1 or IF3. Site-directed mutagenesis in this region supports the idea that this factor-induced rearrangement of h44 helps control translation initiation. We also report that many P-site mutations increase the stringency of start codon selection. These data provide evidence that the affinity of fMet-tRNAfMet for the 30S P site is tuned to balance accuracy and efficiency during initiation.
Results
Identification of 16S rRNA mutations that increase translation from AUC
In previous work, a specialized ribosome system was used to identify two 16S rRNA mutations, G1338A and A790G, which increase translation from non-canonical start codons in vivo (Qin et al., 2007). Using the same genetic system, we sought to identify additional mutations with the same phenotype. Plasmid pKF207, encoding specialized 16S rRNA (ASD*: 5′-GGGGU-3′), was mutagenized by propagation in the mutator strain XL1-Red (Stratagene) and used to transform the indicator strain KLF2672. Strain KLF2672 expresses lacZ mRNA with the complementary SD sequence (SD*: 5′-AUCCC-3′) and the start codon AUC. Transformants were screened on Xgal plates for those expressing increased levels of β-galactosidase. Nineteen independent isolates carrying one or more mutations in 16S rRNA were identified (Table 1). With one exception (U13G), these mutations were transitions, a bias that appears to be due largely to the mutagenesis with XL1-Red (data not shown). Mutation G1539A mapped to the ASD* and is predicted to change the only wobble base pair in the SD*-ASD* helix to a Watson-Crick base pair. This mutation uniformly increased expression from SD*-AUC-lacZ and SD*-AUG-lacZ by about threefold (data not shown). The other 16S rRNA mutations cluster to distinct regions that overlap with class III protection sites (Fig. 1). Class III sites were previously implicated in ligand-induced conformational changes based on chemical protection experiments (Moazed and Noller, 1987). Two mutations, U789C and A790G, map to the IF3-binding site. Four mutations cluster in h44, just ‘down’ from the 30S A site, and surround A1413○G1487 (Fig. 1B and C), a non-canonical pair conserved in bacteria and perturbed by either IF1 or IF3 (Moazed et al., 1995; Dahlquist and Puglisi, 2000; Carter et al., 2001; Cannone et al., 2002). Another group of mutations cluster near the neck of the 30S subunit (Fig. 1D).
Table 1.
Mutations in 16S rRNA obtained in a screen for increased translation from AUC.
| 16S mutation(s) | Independent isolatesa |
Location |
|---|---|---|
| U13G | 1 | Central pseudoknot |
| A790G | 1 | 790 loop |
| G886A | 1 | h27 |
| U1083C | 1 | Neck region |
| A1092G | 1 | Neck region |
| G1094A | 1 | Neck region |
| A1179G | 1 | Junction of h38-h39-h40 |
| C1389U | 1 | Neck helix (h28) |
| A1410G | 4 | h44 |
| C1411U | 1 | h44 |
| U1414Cb | 2 | h44 |
| U1414Cb, G616A | 1 | h44, h21 |
| U789C, U1118C | 1 | 790 loop, junction of h38-h39-h40 |
| G1486Ab, C840U | 1 | h44, h26 |
| ASD sequence (GGGGU⇨GGGAU)c |
1 | 3′ end |
Isolates were only considered independent if they originated from separate preparations of mutagenized pKF207.
U1414 and G1486 form a wobble base pair.
This mutation changes the single U-G wobble base pair in the SD*-ASD* helix to U-A.
Fig. 1.

Nucleotides identified in this study as important for initiation fidelity cluster to h44, the 790 loop and the neck region. A. Mutation sites overlap with class III protection sites. Positions of mutations identified in this study (red triangles); class III protection sites attributed to ligand-induced conformation changes (black squares; Moazed and Noller, 1987). B. Positions of mutations in h44 and in the 790 loop (image based on Selmer et al., 2006; PDB 2J02). C. Binding of IF1 to the 30S subunit distorts h44 where our mutations map. Blue ribbon, with IF1; grey ribbon, without IF1 (image based on Carter et al., 2001; PDB 1HR0 and 1J5E). D. Positions of mutations in the neck region (image based on Selmer et al., 2006; PDB 2J02).
To determine which mutations recovered were responsible for the phenotype, each mutation was introduced into pKF207 de novo by site-directed mutagenesis. The resulting plasmids were transformed into an indicator strain containing SD*-AUC-lacZ. Mutations U13G,U789C, A790G, G886A, U1083C, A1092G, G1094A, C1389U, A1410G, C1411U, U1414C, G1486A were each confirmed to increase expression from SD*-AUC-lacZ. To quantify the phenotypes, the plasmids were also moved into strains containing SD*-ACG-lacZ or SD*-AUG-lacZ and β-galactosidase activity of all resulting strains was compared (Table S1). Translation from AUC and ACG was increased specifically and to various extents depending on the mutation. The level of β-galactosidase from SD*-AUC-lacZ or SD*-ACG-lacZ relative to SD*-AUG-lacZ was used to approximate the frequency of spurious initiation (Fig. 2). Mutations U789C and A790G appeared most defective in initiation fidelity (fivefold to sixfold decrease), similar to effects conferred by various infC alleles (Sacerdot et al., 1996; Sussman et al., 1996; Maar et al., 2008). The other mutations had modest but significant effects. We also determined the effects of the 16S rRNA mutations on initiation fidelity in an infC362 background. This mutation in the IF3 gene increased spurious initiation by about fivefold. The effects of infC362 and the 16S rRNA mutations were completely additive in some cases (U1083C, A1092G, G1094A, C1389U, C1411U and U1414C) and partially additive in the others (Fig. 2).
Fig. 2.
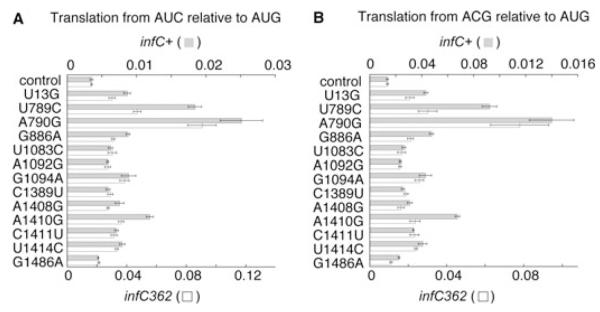
Effects of 16S rRNA mutations on the relative level of translation from AUC and ACG. Values correspond to the level of β-galactosidase expressed by specialized ribosomes from either SD*-AUC-lacZ (A) or SD*-ACG-lacZ (B) relative to SD*-AUG-lacZ. Grey bars, wild-type infC+ background; white bars, infC362 background. Values represent the quotient of two means ± standard error.
Mutant ribosomes can support the growth of E. coli
As a first step towards analysing the mutant 30S subunits in vitro, we moved a number of mutations (U13G, U789C, A790G, A790U, A790C, G886A, G1094A, G1338A, G1338U, A1408G, A1410G and U1414C) into a Δ7 prrn strain of E. coli, SQZ10. This strain lacks all chromosomal rRNA operons and relies on a single plasmid-borne rRNA operon, and hence a homogeneous population of ribosomes. Each mutation could be readily moved into the Δ7 prrn background, and the resulting strains did not exhibit obvious growth defects (data not shown). All ligand-binding experiments described below were performed using 30S subunits purified from these Δ7 prrn strains as described (Qin et al., 2007).
A790G and U789C confer a defect in IF3 binding
A790G was previously shown to decrease the affinity of IF3 for the 30S subunit by ~10-fold (Qin et al., 2007). We presumed that the adjacent mutation U789C similarly inhibited IF3 binding. To test this, we purified subunits containing U789C or each substitution at position 790 and compared their ability to bind IF3 (Qin et al., 2007). IF3 was labelled with amino-reactive fluorophore (Alexa Fluor 488) and sucrose gradient sedimentation analysis was used to detect the binding of labelled IF3 (IF3-AF) to the mutant 30S subunits (Fig. 3). Little or no IF3-AF co-migrated with 30S subunits containing A790G or U789C, indicating that both mutations conferred a substantial defect in IF3 binding. A790U reduced the level of co-migrating IF3-AF while A790C had no appreciable effect on the level of co-migrating IF3-AF.
Fig. 3.
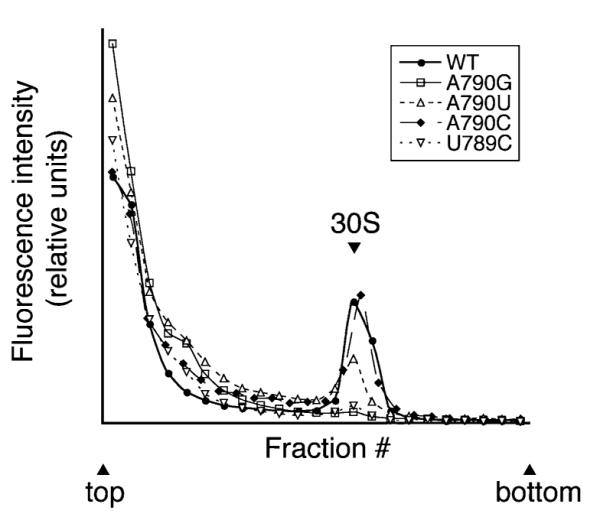
Effect of mutations in the 790 loop on the binding of IF3-AF to the 30S subunit. Fluorescently labelled IF3 (IF3-AF; 1 μM) was incubated with each preparation of 30S subunits (1 μM) at 37°C for 20 min and then subjected to sucrose gradient sedimentation analysis. Fractions (0.5 ml) were collected and the fluorescence intensity (excitation 495 nm; emission 520 nm) of each was quantified to determine the distribution of IF3-AF in the gradient. The position of 30S subunits in the gradient, as identified by A260, is indicated.
A1410G and G886A confer a defect in IF1 binding
A group of mutations mapped to h44 of 16S rRNA (Fig. 1B and C). Structural and biochemical studies have shown that IF1 binds the 30S A site, close to where our h44 mutations cluster (Moazed et al., 1995; Dahlquist and Puglisi, 2000; Carter et al., 2001). Thus, we considered the possibility that some of our mutations affected IF1 binding. 30S subunits containing U13G, G886A, A1408G, A1410G or U1414C were purified and the affinities of [ 35S]-IF1 for these subunits were measured using membrane filtration devices (Microcon YM100, MWCO 100K; Millipore Corporation) to separate free and bound [35S]-IF1 (Fig. 4). The dissociation constant (KD) for IF1 binding to control 30S subunits was estimated at ~0.6 μM, consistent with earlier studies using true equilibrium methods (Zucker and Hershey, 1986; Dahlquist and Puglisi, 2000). G886A and A1410G reduced the affinity for IF1 by twofold and threefold respectively, while the other mutations tested did not affect IF1 binding (Fig. 4A, Table 2). 50S subunits failed to bind [35S]-IF1, indicating that interaction between [35S]-IF1 and the 30S subunit was specific. As an additional control, we tested A1408G, a mutation shown previously to confer a defect in IF1 binding (Dahlquist and Puglisi, 2000). A1408G conferred a defect in our hands as well, but of lesser magnitude than that reported previously.
Fig. 4.
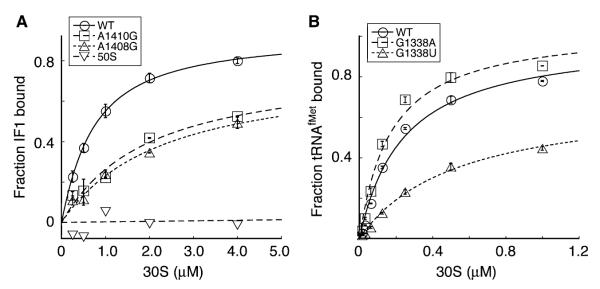
Effects of 16S rRNA mutations on the binding of IF1 or tRNAfMet to the 30S subunit. A. Examples of experiments used to estimate the affinity of IF1 for mutant 30S subunits. [35S]-IF1 (0.25 μM) was incubated with 30S subunits (various concentration) in buffer C [10 mM Tris-HCl (pH 7.5), 100 mM NH4Cl, 5 mM Mg(OAc)2, 6 mM β-ME and 0.03% Nikkol] at room temperature for 20 min. Microfiltration devices were used to separate free and bound [35S]-IF1. The fraction of [35S]-IF1 bound, averaged from at least three independent experiments, was plotted as a function of the concentration of 30S subunits, and the data were fit using KaleidaGraph. B. Examples of experiments to estimate the affinity of tRNAfMet for mutant 30S subunits. 3′-[32P] -tRNAfMet (5 nM) was incubated with 30S subunits (various concentration) with a model mRNA containing an AUG start codon (m701; 3 μM) in buffer D [50 mM Tris-HCl (pH 7.5), 100 mM NH4Cl, 20 mM MgCl2, 6 mM β-ME] for 20 min at 37°C. The free and bound 3′-[32P]-tRNAfMet were separated by filtration through a bilayer of nitrocellulose and nylon membranes. The fraction of 3′-[32P]-tRNAfMet bound was plotted as a function of 30S subunit concentration, and the data were fit using KaleidaGraph. Equilibrium binding constants (KD) listed in Table 2 represent the mean ± SEM from three independent experiments.
Table 2.
Effects of 16S rRNA mutations on ligand binding.
| 16S allelea |
IF1 bindingb |
tRNA bindingc |
IF3 bindingd |
|
|---|---|---|---|---|
| −IF3 | +IF3 | |||
| Control | 600 ± 80 | 30 ± 6 | 240 ± 10 | ++ |
| U13G | 700 ± 100 | ND | ND | ND |
| U789C | ND | ND | 340 ± 10 | − |
| A790G | 700 ± 100 | ND | 300 ± 10 | − |
| A790U | ND | ND | 510 ± 30 | + |
| A790C | ND | ND | 520 ± 10 | ++ |
| G886A | 1100 ± 200 | 60 ± 9 | 240 ± 10 | ++ |
| G1094A | ND | ND | 350 ± 30 | ND |
| G1338A | 700 ± 200 | ND | 170 ± 10 | ++ |
| G1338U | ND | ND | 540 ± 50 | ND |
| A1408G | 2100 ± 600 | ND | ND | ND |
| A1410G | 2200 ± 500 | 90 ± 20 | 210 ± 20 | ++ |
| U1414C | 600 ± 100 | 40 ± 20 | 240 ± 10 | ++ |
ND, not determined.
Mutant 30S subunits were purified from a Δ7 prrn strain of E. coli (see text).
Values correspond to the estimated KD ± standard error (nM) derived from curve fitting. When present, the concentration of IF3 was 350 nM.
Values correspond to KD (nM; mean ± SEM) obtained from three independent experiments.
Binding of IF3-AF to 30S was assessed by the level of co-migration after sucrose gradient sedimentation assay. ++, extent of co-migration indistinguishable from control; +, extent of co-migration substantially reduced; −, little or no co-migration.
As IF1 and IF3 bind cooperatively to the 30S subunit, we also measured the binding of [35S]-IF1 to mutant 30S subunits in the presence of saturating IF3 (Table 2). IF3 (350 nM) increased the affinity of IF1 for control 30S subunits by ~30-fold, consistent with previous studies (Zucker and Hershey, 1986). G886A or A1410G decreased the affinity for IF1 by twofold to threefold in the presence of IF3, as was observed in its absence (Table 2). The fact that these mutations similarly affected IF1 binding in the absence and presence of IF3 indicates that they do not disrupt the mechanism of cooperativity between the two factors.
A1408G causes spurious initiation in vivo
A1408G was not isolated in the genetic screen but conferred a defect in IF1 binding. Thus, it was of interest to determine whether A1408G also increased spurious initiation. We engineered A1408G into pKF207, transformed the appropriate indicator strains, and found that, indeed, A1408G increased translation from AUC and ACG to the same degree as other h44 mutations (Fig. 2, Table S1). These data further implicate IF1 in the mechanism of initiation fidelity.
Watson-Crick base pairs at 1413–1487 and 1414–1486 decrease initiation fidelity
In contrast to mutation A1410G, U1414C did not affect IF1 binding despite that it conferred a similar decrease in initiation fidelity (Fig. 2). Structural studies have shown that bound IF1 locally displaces one strand of h44 along the helical axis, disrupting several base pairs including A1413○G1487 and U1414•G1486 (Carter et al., 2001). Both U1414C and G1486A were isolated and are predicted to change the wobble U1414•G1486 base pair to C-G and U-A respectively, suggesting that they may decrease initiation fidelity by stabilizing the paired conformation of h44. With this in mind, we engineered mutations to generate alternative base pairs at positions 1413–1487 and 1414–1486 and tested their effects on initiation fidelity (Fig. 5, Table S2). Substitutions predicted to stabilize base-pairing at either position conferred increased levels of spurious initiation. In contrast, mismatch A1413 × A1487 decreased spurious initiation, while G1413 × A1487 and U1414 × C1486 had no appreciable effect. The only exceptional case was G1414 × G1486, which increased spurious initiation somewhatdespite that it would be predicted to destabilize the paired conformation of h44.
Fig. 5.

Effects of alternative base pairs at 1410–1490, 1413–1487 and 1414–1486 on the relative level of translation from AUC and ACG. Values correspond to the level of β-galactosidase expressed by specialized ribosomes from either SD*-AUC-lacZ (grey bars) or SD*-ACG-lacZ (white bars) relative to SD*-AUG-lacZ. Values represent the quotient of two means ± standard error.
A1410 forms a Watson-Crick base pair with U1490, which remains paired upon IF1 binding (Carter et al., 2001). A1410G was isolated and is predicted to change the Watson-Crick base pair to a wobble G•U pair. We engineered alternative base pairs at this position and found that U1490C increased spurious initiation by about fourfold. A1410G–U1490C double mutation, which generates an alternative Watson-Crick base pair at 1410–1490, restored initiation fidelity (Fig. 5, Table S2). These data suggest that a Watson-Crick base pair at 1410–1490 is important for initiation fidelity, consistent with 95% conservation of a Watson-Crick pair at this position (Cannone et al., 2002).
Most 30S P-site mutations increase the stringency of start codon selection
It has been proposed that IF3 acts to increase fidelity in part by destabilizing fMet-tRNAfMet in the 30S P site (La Teana et al., 1993; Antoun et al., 2006a,b). To explore this possibility, we analysed a number of P-site mutations for their effects on initiation fidelity. Most P-site mutations decreased translation from both SD*-AUC-lacZ and SD*-AUG-lacZ, but decreased translation from AUC to a larger degree (Table 3). In other words, these P-site mutations effectively increase the stringency of start codon selection.
Table 3.
Effects of P-site mutations on translation from AUC and AUG.
| β-Galactosidase activityb |
Relative translation levelc |
||
|---|---|---|---|
| 16S allelea |
AUC | AUG | AUC/AUG |
| Control | 8.0 ± 0.4 | 2300 ± 80 | 0.0035 ± 0.0002 |
| G926A | < 0.12 | 490 ± 90 | < 0.0002 |
| G926U | < 0.12 | 470 ± 100 | < 0.0003 |
| G926C | < 0.12 | 330 ± 90 | < 0.0004 |
| G1338A | 11 ± 0.3 | 2400 ± 60 | 0.0046 ± 0.0001 |
| G1338U | 0.5 ± 0.1 | 510 ± 30 | 0.001 ± 0.0002 |
| G1338C | < 0.12 | 110 ± 20 | < 0.001 |
| C1400A | 1.5 ± 0.5 | 450 ± 20 | 0.0032 ± 0.001 |
| C1400U | 1.9 ± 0.4 | 1200 ± 300 | 0.0016 ± 0.0004 |
| C1400G | 0.2 ± 0.05 | 190 ± 30 | 0.0012 ± 0.0003 |
| A790G | 13 ± 0.1 | 500 ± 60 | 0.025 ± 0.003 |
| A790C | 1.5 ± 0.2 | 1300 ± 300 | 0.0012 ± 0.0003 |
| A790U | 4.3 ± 0.6 | 1500 ± 200 | 0.0027 ± 0.0005 |
| G966A | 1.6 ± 0.3 | 800 ± 100 | 0.0019 ± 0.0005 |
| G966U | 0.5 ± 0.1 | 700 ± 80 | 0.0008 ± 0.0002 |
| G966C | 0.2 ± 0.01 | 500 ± 50 | 0.0004 ± 0.00005 |
Each 16S allele contains the specialized SD* sequence to allow specific translation of the lacZ reporter gene.
Specialized 16S rRNA (without or with mutations as indicated) was expressed in indicator strains KLF2526 and KLF2528 (harbouring SD*-AUC-lacZ or SD*-AUG-lacZ respectively) and specific β-galactosidase activity (based on cleavage of chlorophenol red-β-d-galactopyranoside) was measured. Values represent the mean ± SEM (arbitrary units) of at least three independent experiments.
Values represent the quotient of two means ± standard error.
Effect of 30S mutations on tRNAfMet binding
Most P-site mutations would be expected to destabilize fMet-tRNAfMet in the 30SIC. A notable exception is G1338A, which stabilizes tRNAfMet in the P site (Qin et al., 2007). To determine how other 16S rRNA mutations affected tRNAfMet binding, we estimated the affinity of 3′-[32P]-tRNAfMet for the 30S P site in various mutant 30S subunits using filter binding as described (Qin et al., 2007) (Fig. 4B, Table 2). Of the mutations tested, G1338U, A790C and A790U were the most defective in tRNAfMet binding. A790G, U789C and G1094A conferred a smaller defect, while A1410G, U1414C and G886A showed no apparent defect.
Discussion
In this study, a number of mutations in 16S rRNA that increase translation from AUC were isolated. These mutations cluster to distinct regions that overlap remarkably well with previously identified class III protection sites. Class III sites were defined as those nucleotides protected from chemical probes by multiple ligands (e.g. 50S subunits, tRNA, various antibiotics). Because these ligands can bind simultaneously to the 30S subunit, these class III protections were attributed to ligand-induced conformational changes, rather than to direct protections (Moazed and Noller, 1987). The fact that many of our mutations map near class III sites suggests that they perturb rRNA rearrangements important for initiation.
Nucleotides 783–793 constitute a major portion of the IF3-binding site (Moazed et al., 1995; Dallas and Noller, 2001; Fabbretti et al., 2007). Two mutations (U789C and A790G) lie in this region and confer the strongest phenotypes (Fig. 2). A790G and the adjacent G791A were shown previously to decrease the affinity of IF3 for the 30S subunit by ~10-fold (Tapprich et al., 1989; Qin et al., 2007), and U789C appears similarly defective in IF3 binding (Fig. 3). It is well known that IF3 regulates initiation complex formation to prevent initiation from non-canonical start codons (Sacerdot et al., 1996; Sussman et al., 1996; Haggerty and Lovett, 1997). Chromosomal mutations that increase spurious initiation events have been mapped almost exclusively to the IF3 gene (infC), in line with our observation that 16S rRNA mutations that target the IF3-binding site are the most defective in initiation fidelity. When mutations in IF3 (infC362) and its binding site (A790G) are present in the same cell, AUG is discriminated from AUC by a factor of 10 rather than 300 (Fig. 2), underscoring the importance of IF3 in regulating initiation.
A second cluster of mutations surrounds the conserved A1413○G1487 base pair in h44. Structural studies reveal that IF1 binds the 30S A site and induces a distortion of h44, involving displacement of one strand away from the helical axis and consequential disruption of base pairs A1413○G1487 and U1414•G1486 (Carter et al., 2001). This rearrangement of h44 is centred right where our mutations cluster, strongly implicating IF1 in the mechanism of initiation fidelity. While certain mutations (i.e. A1410G, A1408G, G886A) decrease the affinity of IF1 for the subunit, U1414C does not. These data suggest that defects in fidelity might stem from inhibition of IF1 binding, the factor-induced rRNA rearrangement, or both. With this in mind, we tested a number of substitutions predicted to stabilize base pairs 1413–1487 and 1414–1486. In each case, an increase in spurious initiation was observed, consistent with the idea that initiation is regulated by the factor-induced rearrangement of h44. By contrast, engineered mismatches G1413 × G1487, A1413 × A1487 and U1414 × C1486 did not decrease initiation fidelity. The only anomalous result came from the G × G mismatch at 1414–1486, which conferred a slight increase in spurious initiation. It is possible this purine–purine pair generates a unique conformation responsible for its phenotype, which fails to follow the trend.
A recent biochemical study suggests that IF1 helps IF3 negatively regulate 70SIC formation to enhance start codon selection (Milon et al., 2008). Changing the start codon from AUG to AUU slowed both IF3 dissociation and subunit joining, and omission of IF1 from the reaction increased the rates of these events in the presence of AUU. Interestingly, the ability of IF1 to inhibit IF3 release and subunit joining was lost in the presence of streptomycin. It was proposed that IF1 induces an ‘initiation-unfavourable’ conformation of the 30S subunit, which could be reversed by cognate codon–anti-codon pairing in the 30SIC or by streptomycin. Our data are consistent with this model and further suggest that the structural corollary of the ‘initiation-unfavourable’ conformation is the distorted state of h44, in which base pairs 1413–1487 and 1414–1486 are unpaired, as observed in the cocrystal structure of 30S-IF1 (Carter et al., 2001). Binding of either IF1 or IF3 to the 30S subunit causes G1487 to become hyper-reactive to kethoxal, suggesting that both factors stabilize the unpaired state of h44 (Moazed et al., 1995; Dahlquist and Puglisi, 2000). Streptomycin, on the other hand, protects G1487 from kethoxal modification, as does tRNA (presumably P-tRNA) (Moazed and Noller, 1987). These ligands do not directly interact with G1487; instead, they most probably stabilize the paired state of h44. Clues of how the unpaired state of h44 might inhibit 70SIC formation come from structural studies. The centre of the IF1-induced distortion lies between intersubunit bridges B2a and B3 (Carter et al., 2000; Korostelev et al., 2006; Selmer et al., 2006), suggesting that docking of the 50S subunit might be inhibitedby displacement of the 16S rRNA nucleotides that contribute to these bridges. The idea that IF1 helps regulate 50S docking is consistent with evidence that IF1 and IF3 work together to facilitate subunit splitting (Noll and Noll, 1972; Godefroy-Colburn et al., 1975; Pavlov et al., 2008).
A previous report showed that several mutations in infA failed to increase translation from GUG and UUG (Croitoru et al., 2005), and those data have been taken by some investigators as evidence that IF1 plays no role in start codon selection. However, mutations in infC also fail to increase translation from GUG and UUG (Sacerdot et al., 1996; Sussman et al., 1996), indicating that IF3 does not discriminate against these naturally occurring non-AUG start codons. In our view, there is no reason to suspect that IF1 would discriminate against these start codons either. For technical reasons, Croitoru et al. were unable to measure the effects of infA alleles on spurious initiation (i.e. translation from Class IIa codons such as AUU, AUC and ACG). Whether these infA alleles increase spurious initiation remains an open question worth addressing.
One of our mutations, U13G, maps to the central pseudoknot and has been isolated previously in a selection for streptomycin resistance (Harris et al., 1989). Streptomycin binds adjacent to U13 (Carter et al., 2000), and other base substitutions at this position have been shown to inhibit streptomycin binding (Pinard et al., 1993). Although not yet understood, this connection between streptomycin and initiation fidelity in vivo is noteworthy, particularly in light of the effects of streptomycin on the ability of IF1 to regulate initiation in vitro (Milon et al., 2008).
A third cluster of mutations localizes to the neck region, and these mutations may act by altering the dynamics of the head domain. Structural and biochemical studies have revealed that the head can assume various positions, and its orientation depends on which ligands (e.g. 50S subunit, translation factors, tRNA, antibiotics) are bound to the 30S subunit (Carter et al., 2001; Ogle et al., 2002; Hickerson et al., 2005; Schuwirth et al., 2005; Berk et al., 2006). Because P-site tRNA influences the orientation of the head domain (Berk et al., 2006), we tested whether G1094A increases the intrinsic stability of the initiator tRNA in the 30S subunit. On the contrary, G1094A decreases the affinity of tRNAfMet for the 30S P site, arguing against the idea that G1094A acts in a manner analogous to G1338A. Upon subunit joining, the head domain of the 30S subunit tilts towards the 50S subunit (Hickerson et al., 2005), which raises the possibility that these neck mutations might promote premature 50S docking. While we have not yet investigated the effects of any of our mutations on subunit association (in the absence or presence of factors), such experiments may prove quite informative.
A number of P-site mutations (e.g. G926A, G926C, G926U, m2G966C, G1338U) increase the stringency of start codon selection (Table 3). These mutations are likely to destabilize fMet-tRNAfMet in the 30SIC, consistent with the fact that G1338U destabilizes tRNAfMet in the 30S P site (Fig. 4B, Table 2). A notable exception is G1338A, which decreases the stringency of start codon selection and stabilizes tRNAfMet in the 30S P site (Qin et al., 2007). These data support to a model in which discrimination against non-canonical start codons relies, at least in part, on the mutual stabilization of mRNA and fMet-tRNAfMet in the 30SIC that comes from codon–anti-codon interaction (La Teana et al., 1993; Antoun et al., 2006a). In G1338U subunits, fMet-tRNAfMet is less stable in the 30SIC, and consequently the probability of productive initiation depends more heavily on cognate codon–anti-codon pairing. In G1338A subunits, the lifetime of 30SIC with fMet-tRNAfMet paired to AUC is lengthened, allowing a higher level of spurious initiation. Indeed, our data are consistent with the proposal that IF3 can repress translation from non-canonical start codons by simply destabilizing fMet-tRNAfMet in the 30SIC (Antoun et al., 2006a).
A790 interacts with both IF3 and fMet-tRNAfMet (Moazed et al., 1995; Dallas and Noller, 2001; Selmer et al., 2006), making it potentially more difficult to predict the effects of mutations at this position on 30SIC stability. However, of the three substitutions, A790G destabilizes IF3 the most and tRNAfMet the least, and it is the only mutation that increases spurious initiation. A790C has no apparent effect on IF3 binding, destabilizes tRNAfMet to the greatest degree, and decreases spurious initiation. A790U has an intermediate effect on IF3 binding, destabilizes tRNAfMet to the same degree as A790C, and confers the least effect on spurious initiation. All these results are in line with the model proposed by Ehrenberg in which IF3 and fMet-tRNAfMet bind the 30S subunit in an anti-cooperative manner, and their interplay during initiation determines the stringency of start codon selection (Antoun et al., 2006a). Clearly, it will be of interest in the future to directly test the effects of these mutations on fMet-tRNAfMet stability in the 30SIC.
Although ribosomes with certain P-site mutations can discriminate AUG from AUC very well, these mutant ribosomes translate from AUG with reduced efficiency (Table 3). Thus, it seems that the affinity of fMet-tRNAfMet for the 30S P site is tuned to balance efficiency and accuracy during initiation.
Experimental procedures
Bacterial strains
Strain KLF2511 is CSH142 [F-ara Δ(gpt-lac)5] containing λ(Pant-SD*-AUC-lacZ) and was constructed as described previously (Qin et al., 2007). Strain KLF2672 is KLF2511 con-taining Δ(recA-srl)306 srl301::Tn10. Other specialized ribosome strains were described previously (Qin et al., 2007). All E. coli Δ7 prrn strains were made using strain SQZ10 as described previously (Qin et al., 2007).
Genetic screen
Mutations were generated in the 16S rRNA gene by propagation of pKF207 in the mutator strain XL1-Red (Stratagene). Twelve transformants were independently grown to saturation iteratively for 7 days. Mutagenized plasmids were then purified from each culture and used to transform the reporter strain KLF2672. The transformed cells were plated on Luria– Bertani plates containing ampicillin (100 μg ml−1), kanamycin (30 μg ml−1), L-arabinose (5 mM) and Xgal (20 μg ml−1), and the resulting colonies were screened for those that exhibited a bluer colour than their neighbours. Plasmids were purified from the positive isolates, and the 16S rRNA gene was sequenced.
β-Galactosidase assay
Specific β-galactosidase activity was measured as described (Qin et al., 2007), except that cells were grown overnight on plates rather than in liquid culture.
Purification of 35S-labelled IF1
IF1 was overexpressed in strain BL21/DE3 (pLysS) from a pET24b-based construct (kindly provided by A. Dallas and H. Noller). The overproducer cells were grown at 37°C in 250 ml M9 medium supplemented with casamino acids (0.05%), thiamine (10 μg ml−1), proline, tryptophan, leucine (80 μg ml−1 each) and kanamycin (30 μg ml−1). At an OD600 of 0.5, IPTG (1 mM) and [35S] L-methionine (2 μCi ml−1, MP Biomedical) were added to the culture. The incubation was continued for 2 h, after which the cells were collected by centrifugation at 2800 g for 15 min, washed with buffer A [20 mM Tris-HCl (pH 7.6), 20 mM NaCl, 10 mM MgCl2, 6 mM β-ME, 0.5 mM EDTA]. The cells were lysed by freezing and thawing them twice, and the lysate was clarified by centrifugation at 1600 g for 10 min. The supernatant was passed through a 0.22 μm filter and loaded onto a Resource S column (GE Corporation). The bound protein was washed with 10 column volumes of buffer A, a NaCl gradient was applied, and IF1 was collected as a single peak (at ~125 mM NaCl). The IF1 preparation was dialysed against buffer B [20 mM Tris-HCl (pH 7.5), 100 mM NH4Cl, 10 mM Mg(OAc)2, 1 mM DTT and 10% glycerol] and stored at −70°C. The purity of IF1 was > 95%, as judged by SDS-PAGE.
Binding assays
Control and mutant 30S subunits were purified from Δ7 prrn strains of E. coli as described previously (Qin et al., 2007). To determine the affinity of IF1 for the 30S subunit in the absence of IF3, [35S]-IF1 (250 nM) was incubated with activated 30S subunits (various concentration) at room temperature for 20 min in buffer C [10 mM Tris-HCl (pH 7.5), 100 mM NH4Cl, 5 mM Mg(OAc)2, 6 mM β-ME and 0.03% Nikkol]. Binding reactions were loaded onto Microcon YM100 filter units (Millipore) and spun at 1600 g for 5 min to separate the bound and free [35S]-IF1. The flow through was collected and the signal was measured by liquid scintillation assay. A no-subunit control was included to estimate the total free [35S]-IF1. The concentration of bound [35S]-IF1, averaged from at least three independent experiments, was plotted versus the concentration of 30S subunits. To determine KD, the data were fit to the equation: [30S·IF1] = {[30S]input + [IF1]input + KD − (([30S]input + [IF1]input + KD)2−4 × [IF1]input × [30S]input)1/2}/2, using the program KaleidaGraph. The standard error reported in Table 2 derived from the curve-fitting. The same procedure was used to measure [35S]-IF1 binding in the presence of IF3 (350 nM), except that the concentration of [35S]-IF1 was 20 nM.
Assays used to measure binding of 3′-[32P]-tRNAfMet and IF3-AF to the 30S subunit were described previously (Qin et al., 2007).
Supplementary Material
Acknowledgements
We thank A. Dallas and H. Noller for the strain to overexpress IF1, C. Squires and S. Quan for the Δ7 prrn strain SQZ10, A. Smith for helpful suggestions regarding the IF1 binding assay, and S. Shoji for comments on the manuscript. This work was supported by NIH Grant GM072528.
Footnotes
Supporting information Additional supporting information may be found in the online version of this article.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Antoun A, Pavlov MY, Lovmar M, Ehrenberg M. How initiation factors maximize the accuracy of tRNA selection in initiation of bacterial protein synthesis. Mol Cell. 2006a;23:183–193. doi: 10.1016/j.molcel.2006.05.030. [DOI] [PubMed] [Google Scholar]
- Antoun A, Pavlov MY, Lovmar M, Ehrenberg M. How initiation factors tune the rate of initiation of protein synthesis in bacteria. EMBO J. 2006b;25:2539–2550. doi: 10.1038/sj.emboj.7601140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berk V, Zhang W, Pai RD, Cate JHD. Structural basis for mRNA and tRNA positioning on the ribosome. Proc Natl Acad Sci USA. 2006;103:15830–15834. doi: 10.1073/pnas.0607541103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boelens R, Gualerzi CO. Structure and function of bacterial initiation factors. Curr Protein Pept Sci. 2002;3:107–119. doi: 10.2174/1389203023380765. [DOI] [PubMed] [Google Scholar]
- Brombach M, Pon CL. The unusual translational initiation codon AUU limits the expression of the infC (initiation factor IF3) gene of Escherichia coli. Mol Gen Genet. 1987;208:94–100. doi: 10.1007/BF00330428. [DOI] [PubMed] [Google Scholar]
- Butler JS, Springer M, Grunberg-Manago M. AUU-to-AUG mutation in the initiator codon of the translation initiation factor IF3 abolishes translational autocontrol of its own gene (infC) in vivo. Proc Natl Acad Sci USA. 1987;84:4022–4025. doi: 10.1073/pnas.84.12.4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannone JJ, Subramanian S, Schnare MN, Collett JR, Du D’Souza LM, Y., et al. The Comparative RNA Web (CRW) Site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics. 2002;3:15. doi: 10.1186/1471-2105-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AP, Clemons WM, Brodersen DE, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. Functional insights from the structure of the 30S ribosomalsubunit and its interactions with antibiotics. Nature. 2000;407:340–348. doi: 10.1038/35030019. [DOI] [PubMed] [Google Scholar]
- Carter AP, Clemons WM, Jr, Brodersen DE, Morgan-Warren RJ, Hartsch T, Wimberly BT, Ramakrishnan V. Crystal structure of an initiation factor bound to the 30S ribosomal subunit. Science. 2001;291:498–501. doi: 10.1126/science.1057766. [DOI] [PubMed] [Google Scholar]
- Croitoru VV, Bucheli-Witschel M, Isaksson LA. In vivo involvement of mutated initiation factor IF1 in gene expression control at the translational level. FEBS Lett. 2005;579:995–1000. doi: 10.1016/j.febslet.2004.12.072. [DOI] [PubMed] [Google Scholar]
- Cummings HS, Hershey JWB. Translation initiation factor IF1 is essential for cell viability in Escherichia coli. J Bacteriol. 1994;176:198–205. doi: 10.1128/jb.176.1.198-205.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlquist KD, Puglisi JD. Interaction of translation initiation factor IF1 with the E. coli ribosomal A site. J Mol Biol. 2000;299:1–15. doi: 10.1006/jmbi.2000.3672. [DOI] [PubMed] [Google Scholar]
- Dallas A, Noller HF. Interaction of translation initiation factor 3 with the 30S ribosomal subunit. Mol Cell. 2001;8:855–864. doi: 10.1016/s1097-2765(01)00356-2. [DOI] [PubMed] [Google Scholar]
- Eduardo PCR, Antoine D, Alain V. Translation in Bacillus subtilis: roles and trends of initiation and termination, insights from a genome analysis. Nucleic Acids Res. 1999;27:3567–3576. doi: 10.1093/nar/27.17.3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbretti A, Pon CL, Hennelly SP, Hill WE, Lodmell JS, Gualerzi C. The real-time path of translation factor IF3 onto and off the ribosome. Mol Cell. 2007;25:285–296. doi: 10.1016/j.molcel.2006.12.011. [DOI] [PubMed] [Google Scholar]
- Godefroy-Colburn T, Wolfe AD, Dondon J, Grunberg-Manago M, Dessen P, Pantaloni D. Lightscattering studies showing the effect of initiation factors on the reversible dissociation of Escherichia coli ribosomes. J Mol Biol. 1975;94:461–478. doi: 10.1016/0022-2836(75)90215-6. [DOI] [PubMed] [Google Scholar]
- Grigoriadou C, Marzi S, Pan D, Gualerzi CO, Cooperman BS. The translational fidelity function of IF3 during transition from the 30S initiation complex to the 70S initiation complex. J Mol Biol. 2007;373:551–561. doi: 10.1016/j.jmb.2007.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gualerzi CO, Pon CL. Initiation of mRNA translation in prokaryotes. Biochemistry. 1990;29:5881–5889. doi: 10.1021/bi00477a001. [DOI] [PubMed] [Google Scholar]
- Gualerzi CO, Brandi L, Caserta E, Garofalo C, Lammi M, La Teana A, et al. Initiation factors in the early events of mRNA translation in bacteria. In: Stillman B, editor. The Ribosome. Vol. LXVI. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2001. pp. 363–376. [DOI] [PubMed] [Google Scholar]
- Haggerty TJ, Lovett ST. IF3-mediated suppression of a GUA initiation codon mutation in the recJ gene of Escherichia coli. J Bacteriol. 1997;179:6705–6713. doi: 10.1128/jb.179.21.6705-6713.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris EH, Burkhart BD, Gillham NW, Boynton JE. Antibiotic resistance mutations in the chloroplast 16S and 23S rRNA genes of Chlamydomonas reinhardtii: correlation of genetic and physical maps of the chloroplast genome. Genetics. 1989;123:281–292. doi: 10.1093/genetics/123.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickerson RP, Majumdar ZK, Baucom A, Clegg RM, Noller HF. Measurement of internal movements within the 30 S ribosomal subunit using Förster resonance energy transfer. J Mol Biol. 2005;354:459–472. doi: 10.1016/j.jmb.2005.09.010. [DOI] [PubMed] [Google Scholar]
- Kaminishi T, Wilson DN, Takemoto C, Harms JM, Kawazoe M, Schluenzen F, et al. A snapshot of the 30S ribosomal subunit capturing mRNA via the Shine– Dalgarno interaction. Structure. 2007;15:289–297. doi: 10.1016/j.str.2006.12.008. [DOI] [PubMed] [Google Scholar]
- Korostelev A, Trakhanov S, Laurberg M, Noller HF. Crystal structure of a 70S ribosome-tRNA complex reveals functional interactions and rearrangements. Cell. 2006;126:1–13. doi: 10.1016/j.cell.2006.08.032. [DOI] [PubMed] [Google Scholar]
- Korostelev A, Trakhanov S, Asahara H, Laurberg M, Lancaster L, Noller HF. Interactions and dynamics of the Shine–Dalgarno helix in the 70S ribosome. Proc Natl Acad Sci USA. 2007;104:16840–16843. doi: 10.1073/pnas.0707850104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Teana A, Pon CL, Gualerzi CO. Translation of mRNAs with degenerate initiation triplet AUU displays high initiation factor 2 dependence and is subject to initiation factor 3 repression. Proc Natl Acad Sci USA. 1993;90:4161–4165. doi: 10.1073/pnas.90.9.4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maar D, Liveris D, Sussman JK, Ringquist S, Moll I, Heredia N, et al. A Single Mutation in the IF3 N-Terminal Domain Perturbs the Fidelity of Translation Initiation at Three Levels. J Mol Biol. 2008;383:937–944. doi: 10.1016/j.jmb.2008.09.012. [DOI] [PubMed] [Google Scholar]
- Milon P, Konevega AL, Gualerzi C, Rodnina M. Kinetic checkpoint at a late step in translation initiation. Mol Cell. 2008;30:712–720. doi: 10.1016/j.molcel.2008.04.014. [DOI] [PubMed] [Google Scholar]
- Moazed D, Noller HF. Interaction of antibiotics with functional sites in 16S ribosomal RNA. Nature. 1987;327:389–394. doi: 10.1038/327389a0. [DOI] [PubMed] [Google Scholar]
- Moazed D, Samaha RR, Gualerzi C, Noller HF. Specific protection of 16 S rRNA by translational initiation factors. J Mol Biol. 1995;248:207–210. doi: 10.1016/s0022-2836(95)80042-5. [DOI] [PubMed] [Google Scholar]
- Noll M, Noll H. Mechanism and control of initiation in the translation of R17 RNA. Nature New Biology. 1972;238:225–228. doi: 10.1038/newbio238225a0. [DOI] [PubMed] [Google Scholar]
- Ogle JM, Murphy FV, Tarry MJ, Ramakrishnan V. Selection of tRNA by the ribosome requires a transition from an open to a closed form. Cell. 2002;111:721–732. doi: 10.1016/s0092-8674(02)01086-3. [DOI] [PubMed] [Google Scholar]
- Olsson CL, Graffe M, Springer M, Hershey JWB. Physiological effects of translation initiation factor IF3 and ribosomal protein L20 in Escherichia coli. Mol Gen Genet. 1996;250:705–714. doi: 10.1007/BF02172982. [DOI] [PubMed] [Google Scholar]
- Pavlov MY, Antoun A, Lovmar M, Ehrenberg M. Complementary roles of initiation factor 1 and ribosome recycling factor in 70S ribosome splitting. EMBO J. 2008;27:1706–1717. doi: 10.1038/emboj.2008.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinard R, Payant C, Melançon P, Brakier-Gingras L. The 5′ proximal helix of 16S rRNA is involved in the binding of streptomycin to the ribosome. FASEB J. 1993;7:173–176. doi: 10.1096/fasebj.7.1.7678560. [DOI] [PubMed] [Google Scholar]
- Pon CL, Gualerzi CO. Mechanism of protein biosynthesis in prokaryotic cells. Effect of initiation factor IF1 on the initial rate of 30 S initiation complex formation. FEBS Lett. 1984;175:203–207. doi: 10.1016/0014-5793(84)80737-1. [DOI] [PubMed] [Google Scholar]
- Qin D, Abdi NM, Fredrick K. Characterization of 16S rRNA mutations that decrease the fidelity of translation initiation. RNA. 2007;13:2348–2355. doi: 10.1261/rna.715307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringquist S, Shinedling S, Barrick D, Green L, Binkley J, Stormo G, Gold L. Translation initiation in Escherichia coli: sequences within the ribosome-binding site. Mol Microbiol. 1992;6:1219–1229. doi: 10.1111/j.1365-2958.1992.tb01561.x. [DOI] [PubMed] [Google Scholar]
- Sacerdot C, Chiaruttini C, Engst K, Graffe M, Milet M, Mathy N, et al. The role of the AUU initiation codon in the negative feedback regulation of the gene for translation initiation factor IF3 in Escherichia coli. Mol Microbiol. 1996;21:331–346. doi: 10.1046/j.1365-2958.1996.6361359.x. [DOI] [PubMed] [Google Scholar]
- Schuwirth BS, Borovinskaya MA, Hau CW, Zhang W, Vila-Sanjurjo A, Holton JM, Cate JHD. Structures of the bacterial ribosome at 3.5 Å resolution. Science. 2005;310:827–834. doi: 10.1126/science.1117230. [DOI] [PubMed] [Google Scholar]
- Selmer M, Dunham CM, Murphy FV, Weixlbaumer A, Petry S, Kelley AC, et al. Structure of the 70S ribosome complexed with mRNA and tRNA. Science. 2006;313:1935–1942. doi: 10.1126/science.1131127. [DOI] [PubMed] [Google Scholar]
- Sette MPVT, Spurio R, Kaptein R, Paci M, Gualerzi CO, Boelens R. The structure of the translational initiation factor IF1 from E. coli contains an oligomer-binding motif. EMBO J. 1997;16:1436–1443. doi: 10.1093/emboj/16.6.1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shine J, Dalgarno L. The 3′-terminal sequence of Escherichia coli 16S ribosomal RNA: complementarity to nonsense triplets and ribosome binding sites. Proc Natl Acad Sci USA. 1974;71:1342–1346. doi: 10.1073/pnas.71.4.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shultzaberger RK, Bucheimer RE, Rudd KE, Schneider TD. Anatomy of Escherichia coli ribosome binding sites. J Mol Biol. 2001;313:215–228. doi: 10.1006/jmbi.2001.5040. [DOI] [PubMed] [Google Scholar]
- de Smit MH, van Duin J. Translational standby sites: how ribosomes may deal with the rapid folding kinetics of mRNA. J Mol Biol. 2003;331:737–743. doi: 10.1016/s0022-2836(03)00809-x. [DOI] [PubMed] [Google Scholar]
- Steitz JA, Jakes K. How ribosomes select initiator regions in mRNA: base pair formation between the 3′ terminus of 16S rRNA and the mRNA during initiation of protein synthesis in Escherichia coli. Proc Natl Acad Sci USA. 1975;72:4734–4738. doi: 10.1073/pnas.72.12.4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringer EA, Sarkar P, Maitra U. Function of initiation factor 1 in the binding and release of initiation factor 2 from ribosomal initiation complexes in Escherichia coli. J Biol Chem. 1977;252:1739–1744. [PubMed] [Google Scholar]
- Studer SM, Joseph S. Unfolding of mRNA secondary structure by the bacterial translation initiation complex. Mol Cell. 2006;22:105–115. doi: 10.1016/j.molcel.2006.02.014. [DOI] [PubMed] [Google Scholar]
- Sussman JK, Simons EL, Simons RW. Escherichia coli translation initiation factor 3 discriminates the initiation codon in vivo. Mol Microbiol. 1996;21:347–360. doi: 10.1046/j.1365-2958.1996.6371354.x. [DOI] [PubMed] [Google Scholar]
- Tapprich WE, Goss DJ, Dahlberg AE. Mutation at position 791 in Escherichia coli 16S ribosomal RNA affects processes involved in the initiation of protein synthesis. Proc Natl Acad Sci USA. 1989;86:4927–4931. doi: 10.1073/pnas.86.13.4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vellanoweth RL, Rabinowitz JC. The influence of ribosome-binding-site elements on translational efficiency in Bacillus subtilis and Escherichia coli in vivo. Mol Microbiol. 1992;6:1105–1114. doi: 10.1111/j.1365-2958.1992.tb01548.x. [DOI] [PubMed] [Google Scholar]
- Weiel J, Hershey JWB. The binding of fluorescein-labeled protein synthesis initiation factor 2 to Escherichia coli 30S ribosomal subunits determined by fluorescence polarization. J Biol Chem. 1982;257:1215–1220. [PubMed] [Google Scholar]
- Zucker FH, Hershey JWB. Binding of Escherichia coli protein synthesis initiation factor IF1–30S ribosomal subunits measured by fluorescence polarization. Biochemistry. 1986;25:3682–3690. doi: 10.1021/bi00360a031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.