

Abstract
Methylation of lysine 4 on histone H3 (H3K4) at promoters is tightly linked to transcriptional regulation in human cells. At least six different COMPASS-like multisubunit (SET1/MLL) complexes that contain methyltransferase activity for H3K4 have been described, but a comprehensive and quantitative analysis of these SET1/MLL complexes is lacking. We applied label-free quantitative mass spectrometry to determine the subunit composition and stoichiometry of the human SET1/MLL complexes. We identified both known and novel, unique and shared interactors and determined their distribution and stoichiometry over the different SET1/MLL complexes. In addition to being a core COMPASS subunit, the Dpy30 protein is a genuine subunit of the NURF chromatin remodeling complex. Furthermore, we identified the Bod1 protein as a discriminator between the SET1B and SET1A complexes, and we show that the H3K36me-interactor Psip1 preferentially binds to the MLL2 complex. Finally, absolute protein quantification in crude lysates mirrors many of the observed SET1/MLL complex stoichiometries. Our findings provide a molecular framework for understanding the diversity and abundance of the different SET1/MLL complexes, which together establish the H3K4 methylation landscape in human cells.
INTRODUCTION
The basic repeating unit of chromatin in eukaryotic cells constitutes of ∼147 bp of DNA wrapped around an octamer of histone proteins to form the nucleosome core particle (1). These histone proteins are subject to posttranslational modifications (PTMs), such as methylation, acetylation, phosphorylation, and ubiquitination (2, 3). In human cells, nucleosomes and their PTMs are involved in regulation of virtually all DNA-associated processes, such as transcription, replication, and response to DNA damage (4, 5). Methylated lysines and arginines are known to recruit effector proteins to specific genomic loci to impose their specific regulatory function upon the underlying DNA (2). Methylation on histone H3 at lysine 4 (H3K4) is conserved from Saccharomyces cerevisiae to humans and is tightly linked to the transcription of genes by RNA polymerase II (6). Whereas trimethylation of H3K4 (H3K4me3) primarily marks promoters of actively transcribed genes, monomethylation (H3K4me1) in combination with H3K27 acetylation has recently been established as a hallmark of active enhancers (7, 8). H3K4me3 can be recognized by PHD finger-containing proteins, such as the Bptf subunit of the NURF chromatin remodeling complex and the Taf3 subunit of the basal transcription factor TFIID, thereby recruiting the basal transcription machinery to activated promoters (9, 10).
In yeast cells, the Set domain-containing protein Set1p is the only methyltransferase for H3K4. Set1p together with other proteins (Cps25/Sdc1p, Cps30/Swd3p, Cps35/Swd2p, Cps40/Spp1p, Cps50/Swd1p, Csp15/Shg1p, and Cps60/Bre2p) assembles into the Set1/COMPASS complex (11). In contrast, higher eukaryotes contain at least six COMPASS-like complexes with H3K4 methyltransferase activity. These complexes are distinguished by six different catalytic Set domain proteins (Set1a, Set1b, Mll1, Mll2, Mll3, and Mll4, referred to here as HMTs [for histone methyltransferases]) (12). The SET1A and SET1B complexes are responsible for maintaining global levels of H3K4me3 (13), whereas complexes with the mixed-lineage leukemia proteins (Mll1 to Mll4) display gene specificity (for clarity, complex names are given in capitals to differentiate from the protein names). Interestingly, the Trr protein of fruit flies (and by analogy mammalian Mll3/4) has recently been found to be critical for H3K4me1 (14). SET1/MLL gene deletion studies in mice revealed diverse nonoverlapping phenotypes, which indicates that these genes perform nonredundant functions during development (15). The six distinct SET1/MLL complexes share a conserved core consisting of Wdr5, Rbbp5, Ash2l, and Dpy30 (named WRAD here). This WRAD module can associate with the catalytic subunit and has been implicated in regulating its enzymatic activity (16–18). Specific subunits, such as the menin subunit of MLL1/2 and the Ptip subunit of MLL3/4, however, have been shown to direct these distinct complexes to certain genomic loci (19).
The exact subunit composition and abundance of the SET1/MLL complexes in human cells is unknown at present, which complicates assessment of their contributions in establishing and maintaining methylation of H3K4. Recent developments in label-free quantitative mass spectrometry-based interaction proteomics (20) and the application of a novel method for quantifying the stoichiometry of these interactions (21) enabled a careful analysis of protein complex composition in a quantitative manner. Here we provide the first comprehensive and unbiased analysis of the six different SET1/MLL complexes in human cells. Single-step affinity purification of shared and unique subunits of the different complexes from nuclear extracts revealed a high degree of heterogeneity in the subunit composition. We found that the WRAD core subunits Dpy30 and Wdr5, but not Ash2l or Rbbp5, are present in other large protein complexes. Additionally, we propose that Bod1 is the human homolog of yeast Shg1p. Furthermore, Bod1 and Psip1 bind selectively to the SET1B and MLL2 complexes, respectively.
MATERIALS AND METHODS
Plasmids and cell culture.
The open reading frame (ORF) of the bait protein was amplified by PCR using the relevant human cDNA constructs and introduced into pDONR2.1. The DNA sequence of the amplified ORF was verified and introduced into a GATEWAY-compatible plasmid, pCDNA5/FRT/TO. All proteins except for menin were tagged with green fluorescent protein (GFP) at the N terminus. cDNA constructs for Wdr5, Dpy30, Rbbp5, and Ash2l (short isoform) were kindly provided by Ali Shilatifard, and Bod1 cDNA was a kind gift from Jason Swedlow. Human Pa1 and Cfp1 and mouse Wdr82 and Ptip cDNAs were obtained from Source Bioscience (Cfp1, catalog no. IRATp970F0412D; Wdr82, IRCKp5014J0617Q; Pa1, IRAUp969E1119D; Ptip1, IRAV9968G04124D). Stable doxycycline-inducible cell lines were created by transfecting pCDNA5/FRT/TO and pOG44 into HeLa FRT cells carrying the TET repressor using polyethyleneimine followed by antibiotic selection. Cells were grown in Dulbecco's modified Eagle medium (DMEM) with high glucose supplemented with penicillin-streptomycin and l-glutamine (all from Lonza) under blasticidin and hygromycin B selection. Protein expression was induced by addition of 0.5 μg/ml doxycycline to the culture medium 16 h prior to cell harvesting. Expression of the proper-sized GFP fusion protein was validated by immunoblotting and by fluorescence microscopy. Bap18-GFP bacterial artificial chromosomes were stably transfected in HeLa cells and selected using Geneticin (G418; Gibco).
GFP affinity purification and sample preparation.
Extract preparation (22) and affinity purifications using GFP beads (20) were performed essentially as described before. Briefly, nuclei were isolated and nuclear extracts were prepared using hypotonic lysis. Purifications for GFP lines and WT HeLa cells were performed in triplicate using 1 mg of nuclear extract per purification and GFP binder beads (Chromotek) in 20 mM HEPES-KOH (pH 7.9), 20% glycerol, 300 mM NaCl, 2 mM MgCl2, 0.2 mM EDTA, 0.1% NP-40, 0.5 mM dithiothreitol (DTT), and complete protease inhibitors (Roche). All purifications included 50 μg/ml ethidium bromide to suppress DNA-mediated interactions. After 2.5 h incubation at 4°C, the beads were extensively washed, and on-bead digestion was performed using trypsin (Promega). After desalting and concentration on StageTips, the peptides were subjected to online nano-liquid chromatography-tandem mass spectrometry (LC-MS/MS), using a 120-min acetonitrile gradient (5.6 to 76%). Mass spectra were recorded on an LTQ-Orbitrap-Velos mass spectrometer (Thermo), selecting the 15 most intense precursor ions of every full scan for fragmentation.
Data analysis.
Raw data were analyzed using MaxQuant 1.3.0.5, with label-free quantification (LFQ), match between runs (between triplicates), and the iBAQ algorithm enabled (23). The identified proteins were filtered for known contaminants and reverse hits, as well as hits without unique peptides. Protein interactor identification was done as described previously (20). In short, the normalized mass spectrometric intensities (LFQ intensities) were compared between the GFP-tagged and control samples, using an adapted permutation-based false discovery rate (FDR) t test in Perseus (MaxQuant package). The threshold for significant identify interactors is based on both the FDR and the ratio between GFP and control sample. This threshold was empirically optimized for each experiment.
The stoichiometry was determined for significant interactors as previously described (21). Here, mass-spectrometric intensities were normalized for the theoretical number of observable peptides by the iBAQ algorithm. Thereby, the normalized intensities (iBAQ intensities) act as a measure of protein abundance and can be directly compared between proteins. The iBAQ intensities measured in the control sample indicate the amount of background binding and were therefore subtracted from the iBAQ intensities obtained in the GFP sample. Finally, the corrected iBAQ intensities were scaled to the total amount of Set1/Mll subunit, thereby allowing direct comparison of the different purifications. The average and standard deviation of the resulting stoichiometries were calculated per triplicate.
Absolute protein quantification in HeLa nuclear extracts.
Nuclear extracts from the HeLa Kyoto cell line were subjected to absolute protein quantification using iBAQ and a universal protein mix standard (UPS2), as described before (24). Briefly, 3 μg of UPS2 was added to 10 μg of HeLa nuclear extract, followed by FASP digestion (25). In parallel, 100 μg of HeLa nuclear extract was applied to FASP followed by strong anion exchange (SAX) chromatography, resulting in 8 fractions. After purification on stage tips, all 9 samples were separately measured by LC-MS/MS over a 4-h acetonitrile gradient (5.6% to 76%). Raw data were analyzed by MaxQuant 1.3.0.5 with iBAQ quantification enabled. A linear regression curve was made between the known UPS2 concentrations (log scale) and the measured iBAQ intensities for these proteins (log scale), which was used to extrapolate the absolute protein numbers of all measured proteins in this sample. Next, linear regression of these absolute protein abundances and their iBAQ intensities in the SAX data set allowed us to quantify the absolute amounts for all proteins found in the large SAX data set.
Proteomics data accession number.
The mass spectrometry proteomics data have been deposited in the Proteome Xchange Consortium database (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (26) with the data set identifier PXD000172.
RESULTS
In order to obtain quantitative information on the different human COMPASS-like complexes, individual subunits of these complexes were tagged with GFP, allowing single-step affinity purification and identification of complexes in a single liquid chromatography combined with tandem mass spectrometry (LC-MS/MS) run (20, 27). We made use of HeLa cell lines in which expression of the tagged protein can be induced upon doxycycline addition (28). Nuclear extracts obtained from these cells and from the parental cell line (wild type [WT]) were subjected to GFP affinity purification in triplicate as described previously (20), followed by direct on-bead digestion and mass spectrometry analysis on an LTQ-Orbitrap-Velos instrument. Once the interactors were identified, their relative stoichiometry was determined as described previously (21) (Fig. 1A). To allow comparison of stoichiometries between different experiments, all data were normalized to the total amount of catalytic subunit in that specific experiment, and consequently the stoichiometry is expressed relative to the total amount of HMT. We used this workflow to analyze the shared SET1/MLL complex subunits (Ash2l, Rbbp5, Wdr5, and Dpy30), as well as two specific subunits for MLL1/2 (menin and Psip1), MLL3/4 (Pa1 and Ptip), and SET1A/B (Wdr82 and Cfp1).
Fig 1.

(A) Experimental workflow. Nuclear extracts were prepared from HeLa wild-type-protein- or GFP-fusion protein-expressing cells. GFP pulldowns were performed in triplicate and analyzed separately by mass spectrometry. Raw data were analyzed by MaxQuant, and specific interactors were selected from background using label-free quantification in Perseus. iBAQ intensities were used to calculate the relative abundance of interaction partners. (B and C) Identification of interacting proteins for Ash2l (B) and Rbbp5 (C) by volcano plots (left) and the stoichiometry (>0.01) of these interactors presented by bar graphs (right). In the volcano plots, the ratio of GFP to WT in label-free quantification are plotted against the −log10 of the false discovery rate (FDR) calculated by a permutation-based FDR adapted t test. Significant outliers are labeled. Bar graphs indicate the stoichiometry of interacting proteins (indicated at the bottom) relative to Set1/Mll proteins. The dashed line indicates a ratio to the total Mll/Set1 protein of 1. Error bars indicate the standard deviations of the biochemical triplicate for each experiment.
SET1/MLL core subunit interactions.
Rbbp5 and Ash2l are part of the stable core of the human SET1/MLL complexes (Fig. 1B and C). Ash2l, Rbbp5, Wdr5, and Dpy30 were previously reported to form the WRAD complex independently of the catalytic HMT subunit (16–18, 29). Purification of Ash2l or Rbbp5 revealed that Ash2l, Rbbp5, and Dpy30 are more abundant than the combined Set1/Mll proteins (Fig. 1B and C). These observations suggest that a proportion of the WRAD module is not associated with HMT activity. The exception to this is the Wdr5 subunit, which is present in a 1:1 ratio with the HMTs (Fig. 1B and C). This is in agreement with previously reported direct interactions between Mll1 and Wdr5 (30). These observations suggest the existence of a subcomplex consisting of Rbbp5, Ash2l, and Dpy30. Alternatively, this putative RAD module is stabilized by overexpression of one of its components.
Wdr5 plays an important role in self-renewal and reprogramming and many of these functions are attributed to its role in SET1/MLL (31, 32). In addition, Wdr5 has been found in other complexes, including the ATAC histone acetyltransferase complex (30, 33). We found Wdr5 to interact, either directly or indirectly, with almost 200 different proteins (Fig. 2A). As expected (30), among these are subunits of the ATAC, NSL, HBO1, and anaphase-promoting complexes. Additionally, INO80 and TFIID subunits were identified as interactors. These complexes were not identified using the other (Ash2l, Rbbp5, and Dpy30) core subunits as bait, indicating that they are exclusive for Wdr5 (34). Interestingly, intensities of the different identified complexes are comparable, indicating that Wdr5 is equally distributed over several chromatin-associated protein complexes. This suggests that Wdr5 is more abundant in cells than the other WRAD members. To investigate this, we performed intensity based absolute quantification (iBAQ) of HeLa nuclear extract (24) (Table 1). This analysis resulted in the identification and absolute quantification of ∼4800 proteins. As expected, Wdr5 is ∼10-fold more abundant than Rbbp5 and Ash2l (Table 1). This supports the idea that Wdr5 is a universal hub in chromatin and transcription regulatory pathways (30). Strikingly, the WRAD members are at least four times more abundant than the sum of all HMT subunits. This is in line with findings by others (17, 18) and by us (Fig. 1 and 2) that Wdr5, Rbbp5, Ash2l, and Dpy30 may form various subcomplexes.
Fig 2.
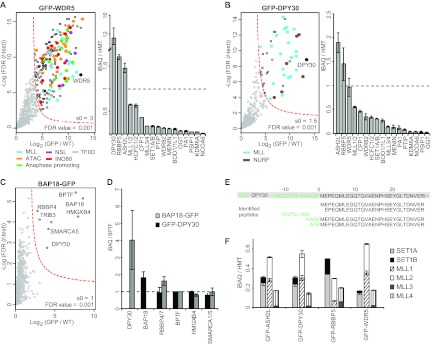
(A and B) (Left) Volcano plots of Wdr5- and Dpy30-interacting proteins. Coloring of significant outliers is based on GO classification (cellular compartment). (Right) Bar graphs of the stoichiometry (>0.01) of interacting proteins. The layout is as in Fig. 1. (C) Volcano plot of Bap18-interacting proteins. The layout is as in Fig. 1. (D) Stoichiometry of Dpy30 and Bap18-bound NURF subunits relative to Bptf. (E) Peptide sequences of Dpy30 N-terminal peptides identified in the Dpy30 pulldown. (F) Stoichiometry of HMTs in WRAD pulldowns.
Table 1.
Absolute protein quantification of HeLa cell nuclear extracts
| Protein ID | Protein name | Amta |
|---|---|---|
| O75475 | PSIP1 | 68,047.34 |
| P61964 | WDR5 | 8,870.15 |
| B4DIS3 | DPY30 | 5,799.25 |
| Q6UXN9 | WDR82 | 4,908.15 |
| Q15291 | RBBP5 | 894.73 |
| Q9UBL3 | ASH2L | 596.69 |
| Q8NFC6 | BOD1/1L1 | 201.70 |
| Q9P0U4-2 | CFP1 | 189.01 |
| O00255-2 | MEN1 | 137.71 |
| O15047/Q9UPS6 | SET1A/B | 133.68 |
| Q6ZW49 | PTIP | 58.14 |
| Q9BTK6 | PA1 | 7.31 |
| E9PQG7/Q9UMN6 | MLL1/2 | 1.91 |
| O14686-3/Q8NEZ4-3 | MLL3/4 | 1.62 |
| O15047 | SET1A | 132.68 ± 7.0 |
| O14686-3 | MLL4 | 1.44 ± 0.069 |
| E9PQG7 | MLL1 | 1.27 ± 0.070 |
| Q9UPS6 | SET1B | 1.00 ± 0.48 |
| Q9UMN6 | MLL2 | 0.64 ± 0.060 |
| Q8NEZ4-3 | MLL3 | 0.18 ± 0.0024 |
Values are given in fmol per mg of nuclear extract. For the individual HMTs, the abundance is estimated based on the intensity of unique peptides. The error indicates the relative abundance obtained from shared peptides, which cannot be assigned uniquely to a single HMT but are used in the abundance calculations.
Dpy30 isolation led to the copurification of SET1/MLL components, among which Ash2l showed the highest stoichiometry, which suggests a direct interaction between Ash2l and Dpy30 (Fig. 2B). Surprisingly, all known subunits of the NURF chromatin-remodeling complex copurified with Dpy30 (Fig. 2B). This indicates that Dpy30 is also an integral component of the NURF complex. Due to its small size (11 kDa), this protein may have been missed in previous gel-based analyses of the NURF complex (35). Roughly equal amounts of HMT and NURF complexes were present in the Dpy30 pulldowns. Bap18 is a core component of NURF (27) and was used to validate the Dpy30-NURF interaction (Fig. 2C). We determined the stoichiometry of the NURF subunits in the Bap18 and Dpy30 pulldowns and found that Bap18 is present in two more copies than the other subunits (Fig. 2D). By analogy, we propose that Bap18 might serve as the direct anchor for Dpy30 in NURF. In the GFP-Dpy30 purifications, we identified tryptic peptides from the junction of GFP-Dpy30 fusion protein and from the N terminus of endogenous Dpy30. This indicates copurification of GFP-tagged and endogenous Dpy30, which reveals the existence of Dpy30 multimers (Fig. 2E). Notably, Dpy30 is 6 to 10 times more abundant in HeLa nuclear extracts than Rbbp5 and Ash2l (Table 1).
Relative abundance of the SET1/MLL complexes.
Analysis of the shared subunits of the SET1/MLL complexes revealed that all six HMTs (Mll1 to -4 and Set1a and -1b) are present in a complex with the WRAD module (Fig. 1B and C and 2A and B). These homologous HMTs have evolved from a common ancestor and consequently share a number of tryptic peptides. To determine the exact stoichiometry of each HMT complex, HMTs that share tryptic peptides are collapsed into a single stoichiometry value. This analysis revealed that the MLL1/2 complexes are most abundant (on average 50% of all complexes), whereas SET1A/B and MLL3/4 account for 32% and 18% of the total pool of WRAD-bound HMT, respectively (Fig. 2F). Based on the intensity of the unique peptides for each HMT, we estimated their relative abundance (Fig. 2F). Whereas the Mll1 and Mll2 proteins are present in roughly equal amounts (both ∼25%), the amount of Mll3 is fairly low compared to that of Mll4 (∼3% versus ∼15%, respectively). The same holds true for Set1a, which is more abundant than Set1b (∼25% and ∼7%, respectively) (Fig. 2F).
The relative presence of the enzymatic components in the SET1/MLL complexes could be regulated posttranslationally or a direct effect of differential protein expression. Interestingly, Set1a is the most abundant HMT as indicated by its absolute protein abundance in HeLa nuclear extract (Table 1), which is not reflected in its degree of integration in SET1/MLL complexes (32% of all complexes). MLL1/2 and MLL3/4 complexes have similar abundances (1.9 and 1.6 fmol/mg, respectively), whereas their degrees of interaction with WRAD are not equal (50% versus 18%). Therefore, the integration of these enzymatic components is not strictly determined by their absolute abundance but probably due to the presence of certain HMTs (like Mll3/4 and Set1a) in other complexes lacking the WRAD proteins.
MLL1/2 specific interactors.
Menin acts as a tumor suppressor in the neuroendocrine MEN1 tumor syndrome but is an essential cofactor for the oncogenic activity of rearranged MLL (36, 37). Menin binds to Mll1/2 and the transcription factor JunD in a mutually exclusive manner (37–39). Purification of the core MLL subunits revealed a 4- to 5-fold-lower abundance of menin than Mll1/2, indicating that not all MLL1/2 complexes contain menin (Fig. 1B and C and 2A and B). On the other hand the fraction of menin, which is associated with Mll1/2, binds to the WRAD complex with a stoichiometry similar to 1:1/1/1/6 (menin:Wdr5/Rbbp5/Ash2l/Dpy30) (Fig. 3A). Among the other menin-interacting proteins, JunD was highly enriched but in a lower stoichiometry than MLL1/2 complex subunits. Intriguingly, the JunD dimerization partners c-Fos, Fos1l, Atf2/7, and Atf3 were also identified in this experiment (Fig. 3A and C). This suggests that menin interacts with dimers of JunD and other AP-1 transcription factors. Additionally, menin was previously shown to localize to chromatin after UV radiation, which may depend on these AP-1 transcription factors (40). We also identified the nucleotide excision repair proteins Rad23a/b as novel menin interactors, which may hint at a function for menin in this pathway. Notably, the RNA-binding protein Taf15 was also identified, which might link menin to other non-MLL-related functions.
Fig 3.

(A and B) (Left) Volcano plots of menin- and Psip1-interacting proteins. (Right) Bar graphs of the stoichiometry (>0.01) of interacting SET1/MLL complex subunits. The layout is as in Fig. 1. (C and D) Stoichiometry of specific interactors for menin (C) and Psip1 (D). (E) Relative amounts of Mll1 and Mll2 proteins associating with menin or Psip1.
Psip1 (Ledgf/p75) is well studied as the interaction partner for the HIV integrase (41) and was more recently also implicated in MLL-linked leukemia (42). Like menin, Psip1 does not interact with the Set1a/b or Mll3/4 methyltransferases. Analysis of the stoichiometry of Psip1-bound MLL subunits resulted in the same picture as when menin was used as bait (Fig. 3A and B). As the menin protein has a 1:1 stoichiometry relative to Mll1/2 in the GFP-Psip1 purifications, it is likely that menin and Psip1 interact only in the context of Mll1/2. On the other hand, menin pulldown indicated that Psip1 is present in only ∼25% of the menin-MLL1/2 complexes. This low abundance is supported by the analysis of WRAD subunits as bait, in which Psip1 was not identified as an interactor (Fig. 1B and C and 2A and B), together indicating that Psip1 is a substoichiometric component in this complex. Interestingly, Psip1 enriches mostly Mll2, whereas menin binds equal amounts of the Mll1 and Mll2 proteins (Fig. 3E). This indicates preferential binding of Psip1 to the MLL2 complex.
Our analyses confirmed Cdca7l/Jpo2 as a Psip1 interactor (43). We found that Psip-Cdca7l is 18-fold more abundant than the Psip-MLL1/2 complexes (Fig. 3D). This is confirmed by the absolute protein quantification data set, which shows that Psip1 is the most abundant SET1/MLL interactor, with a 100-fold higher abundance than Rbbp5 and Ash2l (Table 1). As reported previously, Psip1 interacts with Dbf4 and Cdc7, which likely form a complex, and with the Pogz transcription factor (44, 45). These interactions are of a lower abundance than Mll1/2. Together, these observations reveal that only ∼5% of Psip1 is present in MLL complexes, and that the bulk of Psip1 performs other functions.
Hcfc1 and -2 were previously identified as members of the MLL1 complex, but they are also linked to the SET1 complexes (46). Our experiments validate these proteins as substoichiometric interactors for SET1A/B and MLL1/2 and provide no evidence for an interaction of Hcfc1/2 with the MLL3/4 complexes (Fig. 3A and B and 4A and B).
Fig 4.

(A and B) (Left) Volcano plots of Ptip- and Pa1-interacting proteins. (Right) Bar graphs of the stoichiometry (>0.01) of interacting SET1/MLL complex subunits. The layout is as in Fig. 1. (C) Stoichiometry of specific interactors for Ptip. (D) Relative amounts of Mll3 and Mll4 proteins associating with Ptip or Pa1.
MLL3/4-specific interactors.
Ptip was previously identified as a bona fide subunit of the MLL3/4 complexes and found to be associated with the 53bp1 protein, which is involved in the DNA damage response pathway (47, 48). The interaction with Mll3/4 was confirmed (Fig. 4A), and Ptip-bound MLL3/4 complexes seem to have a 1:1/1/1/3 (Ptip:Wdr5/Rbbp5/Ash2l/Dpy30) stoichiometry. Interestingly, Dpy30 is probably present as a trimer, compared to the hexamer in MLL1/2. Other known MLL3/4 subunits, the histone H3K27 demethylase Kdm6a/Utx and the nuclear receptor coactivator NcoA6, are present in only ∼19% and ∼30% of Ptip pulldowns relative to Mll3/4, respectively (Fig. 4A and B). This indicates that these subunits are genuine substoichiometric components of the Ptip-containing MLL3/4 complexes.
Notably, Ptip-53bp1 complexes are more abundant than Ptip-HMT complexes (Fig. 4C). The GFP-Ptip purification (Fig. 4C) also identified Dclre1C, Mdc1, and Blm, which are all implicated in the DNA damage response pathway (49). These proteins were not identified previously as Ptip interactors, which may indicate that they interact via 53bp1. The zinc finger transcription factor Zbtb2 has been implicated in the p53 pathway (50), and it was found to be a strong Ptip interactor. The Zbtb2-Ptip interaction seems to be independent of the SET1/MLL complexes, because Zbtb2 was not identified in WRAD purifications (Fig. 4C). Furthermore, the Smad2/3 proteins interacted with Ptip. Interestingly, a role in TGFβ signaling via interaction with Smad proteins was previously described for the Xenopus Ptip homolog Swift (51). The novel interaction found with the zinc finger Znf639 may hint at a role for this protein in localizing Ptip and its associated proteins to specific genomic loci. It is important to note that these interactions were not found with Pa1 or other SET1/MLL proteins and could therefore also be linked to the 53bp1 DNA damage pathway or to novel functions of the Ptip protein.
Ptip-associated protein (Pa1) interacts with the MLL3/4 complexes with stoichiometry similar to that of Ptip, 1:1/1/1/3 (Pa1:Wdr5/Rbbp5/Ash2l/Dpy30) (Fig. 4B). Moreover, a larger proportion (∼2-fold) of Ptip compared to Mll3/4 is found in these experiments. Conversely, Pa1 was 2-fold more abundant relative to Mll3/4 in the GFP-Ptip analysis (Fig. 4A). Ptip and Pa1 are found with a lower stoichiometry than Mll3/4 in the pulldowns of the WRAD components, which makes a Ptip/Pa1 dimer within the MLL3/4 complex unlikely (Fig. 1B and C and 2A and B). Instead, these data suggest that Ptip and Pa1 form a complex outside the MLL3/4 complex. Analysis of the distribution of Mll3 and Mll4 proteins in the Ptip and Pa1 pulldowns showed Mll3 and Mll4 distributions similar to those found in the WRAD purifications, indicating no preference of Ptip and Pa1 for either of these HMTs (Fig. 4D).
SET1A/B-specific interactors.
The beta-propeller protein Wdr82 is part of the SET1A/B complexes and directly involved in regulation global levels of H3K4me3 (13, 52). Wdr82 has also been found to interact with the PP1 phosphatase complex (53). Wdr82 is a stoichiometric component of the SET1A/B complexes based on pulldowns of Ash2l, Rbbp5, and Dpy30 (Fig. 1A and B and 2B). Purification of Wdr82 indicated stoichiometric amounts of Cfp1 and Set1a/b and slightly smaller amounts of the WRAD components (Fig. 5A). Again, Dpy30 is present as a trimer. The Wdr82 purification also identified the Bod1 and Bod1L1 proteins as novel interactors, which we studied further (see below). As expected (53), we also identified the whole PP1 protein phosphatase complex (Tox4, Pp1a/b/c/, Ppp1R10), which was present at an ∼10-fold higher abundance than the Set1a/b proteins (Fig. 5C). In addition, we identified several novel Wdr82-interacting proteins, which are in much lower abundances than PP1 and SET1A/B complexes (Fig. 5C).
Fig 5.
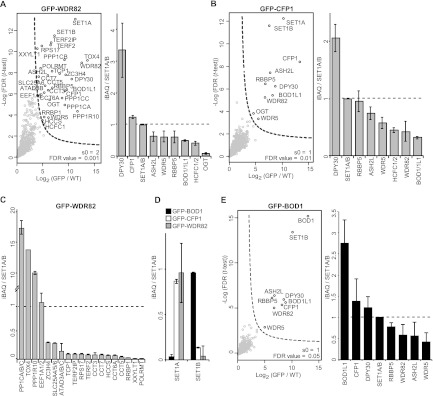
(A and B) (Left) Volcano plots of Wdr82- and Cfp1-interacting proteins. (Right) Bar graphs of the stoichiometry (>0.01) of interacting SET1/MLL complex subunits. The layout is as in Fig. 1. (C) Stoichiometry of specific interactors for Wdr82. (D) Relative amounts of Set1a and Set1b proteins associating with Ptip, Pa1, or Bod1. (E) (Left) Volcano plot of Bod1-interacting proteins with significant outliers indicated. (Right) Bar graphs of the stoichiometry (>0.01) of interacting proteins. The layout is as in Fig. 1.
Cfp1 purification resulted in SET1A/B complex members, and Cfp1 appears to be an exclusive and specific SET1A/B subunit (Fig. 5B). Similar to Wdr82 purification, the Bod1 and Bod1L1 proteins were identified as interactors, which suggests that they are genuine interactors of the SET1A/B complexes. Analysis of Cfp1 stoichiometry in WRAD pulldowns indicated that Cfp1 binds with a stoichiometry of approximately 1.6 relative to the total HMT subunit, 1.6:1/1/1/2 (Cfp1:Wdr5/Rbbp5/Ash2l/Dpy30). This may indicate a potential dimerization of Cfp1 in the SET1A/B complexes.
Bod1 and Bod1L1 are human paralogs of yShg1p and exclusive to SET1B.
The bi-orientation defected 1 protein (Bod1) is important for proper chromosome segregation during mitosis (54). Bod1 and the highly similar Bod1L1 were identified in our experiments as interacting with all members of the WRAD complex (Fig. 1B and C and 2A and B). Purification of the specific SET1A/B subunits Cfp1 and Wdr82 revealed that Bod1 and Bod1L1 are exclusively present in SET1A/B complexes, as we found specific and shared peptides for both proteins (Fig. 5A and B).
To investigate this further, we constructed a GFP-Bod1-expressing cell line and analyzed its interactors. Interestingly, Set1b was the only HMT interactor (Fig. 5E). Estimation of the relative abundance of Set1a and Set1b in the Bod1, Cfp1, and Wdr82 pulldowns confirmed that Bod1 interacts exclusively with the SET1B complex (Fig. 5D). In contrast, Cfp1 and Wdr82 interact mostly, but not exclusively, with Set1a, and the ratio of Set1a to Set1b is similar to the ratios in the WRAD purifications (Fig. 5D and 2F).
The Bod1 pulldown identified unique peptides for Bod1L1 with higher iBAQ intensity than the other SET1B complex members, which may indicate that Bod1 and Bod1L1 could interact outside the SET1B complex. Alignment of the Bod1 homologous sequences from human, fruit fly, and zebrafish revealed a high degree of similarity with the Saccharomyces cerevisiae protein Shg1p (data not shown). Intriguingly, Shg1p (a.k.a. Csp15) is part of the yeast COMPASS complex. Shg1p interacts with RNA, which affects yeast SET1 complex assembly (55, 56). Based on the identification of Bod1 in SET1B and the high sequence similarity, we propose that Bod1 and Bod1L1 are the higher eukaryotic paralogs of yeast Shg1p.
DISCUSSION
Recent developments in quantitative mass spectrometry allowed an unbiased, quantitative, and comprehensive interaction study on the six COMPASS-like H3K4 methyltransferase complexes in humans (21). We identified many known interactors, illustrating the high quality of our data sets, and calculated their stoichiometries. The 10 SET1/MLL pulldowns were clustered based on the identified interactors and presented as a heat map reflecting their stoichiometry (Fig. 6A; a summary is presented in Fig. 6B). Our approach indicates a high degree of diversity in the peripheral subunits of the different HMT complexes.
Fig 6.
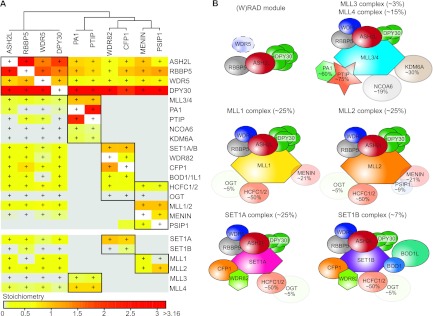
SET1/MLL interactome. (A) Hierarchical clustering of overlapping interactors identified for Wdr5, Rbbp5, Ash2L, and Dpy30 pulldowns. Bait proteins are indicated on top and identified interactors on the right. Coloring (ascending from gray to red via yellow and orange) is based on the stoichiometry of the interactor in that pulldown, relative to the total amount of Set1a/b and Mll1 to Mll4 proteins found for that bait. Significant outliers are indicated (+) for each bait protein. (B) Summary of the composition and relative amounts of the SET1/MLL complexes determined in this study. Dpy30 is present as a multimer dependent on the complex context. Proteins that have a stoichiometry of <1 are displayed as transparent, and their relative abundance is indicated.
The smallest core subunit of the SET1/MLL complexes, Dpy30, shows an interesting behavior. It was shown to interact with Ash2l as a dimer (29). However, we observe a stoichiometry of ∼6 in Mll1/2, whereas Mll3/4 and Set1A/B bind ∼3 molecules of Dpy30 per complex (Fig. 6B). This argues for additional binding sites on the Mll and Set proteins or on the peripheral subunits. Dpy30 is only 11 kDa; therefore, the quantification is based on a limited number of tryptic peptides (11) relative to the other interactors (81 peptides on average), which might explain the high standard deviation for this protein in the stoichiometry determinations. We also identified all members of the NURF complex (35) in the GFP-Dpy30 pulldown, and we identified Dpy30 in the pulldown with the Bap18 subunit of NURF (Fig. 2C). These findings imply that Dpy30 is a genuine subunit of NURF and that its function should not be interpreted solely in the context of HMT complexes (57).
The same holds true for Wdr5, which was identified as a central hub in chromatin-associated complexes. Since Wdr5 was described as a SET1/MLL complex member, many Wdr5 interactors have been linked to the SET1/MLL complexes, such as the INO80, TFIID, and NSL complexes (30). This annotation is still present in gene ontology databases. These interactions are, however, not found in the other core SET1/MLL subunit pulldowns (Rbbp5, Ash2l, or Dpy30). This indicates that rather than SET1/MLL complex interactors, these protein complexes should be annotated as exclusive Wdr5 interactors. It was shown that Wdr5 binds to the tail of histone H3 and that this may facilitate methylation by HMTs (34, 58). Similarly, H3 tail presentation by Wdr5 could be relevant for the binding or modification by the complexes identified as Wdr5 interactors here.
Our results address the question of the limiting components for the assembly of SET1/MLL complexes in cycling HeLa cells. With the exception of Set1a, the HMTs are present in small amounts in crude nuclear extracts. The WRAD subunits and specific interactors, like Wdr82, Psip, menin, and Bod1, are more abundant. These observations support the existence of (W)RAD subcomplexes lacking the HMT subunit (17). Furthermore, our unbiased approach shows that the complex-specific subunits can also be part of other complexes, like Dpy30 in NURF, Wdr82 in the PP1 phosphatase (53), and Psip in the Cdc7/Dbf4/Cdca7l complexes (43, 44). These observations raise questions regarding the regulation of these different interactions during the cell cycle or upon cellular stress. Interestingly, different components of the MLL1 complex display differential chromatin binding during mitosis (59).
Whereas menin binds Mll1 and Mll2 with equal affinity, Psip1 shows a striking preference for Mll2, under the conditions used for HeLa cells (Fig. 6B). This conflicts with an earlier protein-protein interaction study using fragments of these proteins (38). Although Psip1 and menin are substoichiometric MLL1/2 subunits, both proteins are essential for oncogenic transformation by Mll1 fusions (42, 60). Our observation that Psip is mainly a MLL2 complex member may imply that the function of Psip during Mll1-fusion mediated oncogenesis depends on MLL2 function as much as it depends on the wild-type MLL1 complex (61).
Bod1 was linked previously to chromosome segregation during mitosis (54). The phenotype of Bod1 depletion and the association of the other SET1B complex member, Wdr82, with PP1, whose inactivation leads to a mitotic arrest (62), hints at a link between the SET1B complex, histone methylation, and mitotic progression. In yeast, Set1p was reported to inhibit aurora kinase function during chromosome segregation, which involves methylation of the Dam1p kinetochore protein (63). Based on these observations, we speculate that the SET1B complex plays a similar role during mitosis in mammalian cells.
The absolute protein quantification data of the nuclear extract are in good (but not perfect) agreement with the observed stoichiometries of SET1/MLL complex subunits. The higher abundance of WRAD subunits supports existence of free (W)RAD subcomplexes (17). Additionally, the finding that Wdr5 and Dpy30 are subunits of other chromatin-regulatory complexes is mirrored by their higher abundance in nuclear extracts. This suggests that absolute protein quantifications could be predictive for multimerization and/or association with multiple complexes. This concept could be applied to predict composition of protein complexes in tissue samples for which insufficient material is available for biochemical purification.
Taken together, the results of our quantitative mass spectrometry analysis of the major H3K4 methyltransferase complexes in human cells revealed both known and novel, unique and shared interactors and determined their distribution and stoichiometry over the different SET1/MLL complexes. The comprehensive and quantitative mapping of subunit composition and abundance provides a molecular framework for understanding the diversity and contributions of the SET1/MLL complexes in establishing H3K4 methylation patterns.
ACKNOWLEDGMENTS
We thank Steven Taylor for providing the HeLa FRT cells, Ali Shilatifard for the Ash2l, Rbbp5, Dpy30, and Wdr5 cDNA constructs, and Jason Swedlow for the Bod1 cDNA construct.
This work was supported by the Netherlands Organization for Scientific Research (NWO) through a TOP grant (700.57.302) to H.T.M.T. and by the Netherlands Proteomics Centre. Work in the laboratory of M.V. is supported by a VIDI grant (NWO) and a project grant from the Dutch Cancer Society (KWF).
Footnotes
Published ahead of print 18 March 2013
REFERENCES
- 1. Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ. 1997. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389:251–260 [DOI] [PubMed] [Google Scholar]
- 2. Jenuwein T, Allis CD. 2001. Translating the histone code. Science 293:1074–1080 [DOI] [PubMed] [Google Scholar]
- 3. Berger SL. 2007. The complex language of chromatin regulation during transcription. Nature 447:407–412 [DOI] [PubMed] [Google Scholar]
- 4. Fischle W, Wang Y, Allis CD. 2003. Histone and chromatin cross-talk. Curr. Opin. Cell Biol. 15:172–183 [DOI] [PubMed] [Google Scholar]
- 5. Kouzarides T. 2007. Chromatin modifications and their function. Cell 128:693–705 [DOI] [PubMed] [Google Scholar]
- 6. Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, McMahon S, Karlsson EK, Kulbokas EJ, Gingeras TR, Schreiber SL, Lander ES. 2005. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120:169–181 [DOI] [PubMed] [Google Scholar]
- 7. Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, Wang W, Weng Z, Green RD, Crawford GE, Ren B. 2007. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 39:311–318 [DOI] [PubMed] [Google Scholar]
- 8. Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, Ching KA, Antosiewicz-Bourget JE, Liu H, Zhang X, Green RD, Lobanenkov VV, Stewart R, Thomson JA, Crawford GE, Kellis M, Ren B. 2009. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459:108–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vermeulen M, Mulder KW, Denissov S, Pijnappel WWMP, van Schaik FMA, Varier RA, Baltissen MPA, Stunnenberg HG, Mann M, Timmers HTM. 2007. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell 131:58–69 [DOI] [PubMed] [Google Scholar]
- 10. Wysocka J, Swigut T, Xiao H, Milne TA, Kwon SY, Landry J, Kauer M, Tackett AJ, Chait BT, Badenhorst P, Wu C, Allis CD. 2006. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature 442:86–90 [DOI] [PubMed] [Google Scholar]
- 11. Miller T, Krogan NJ, Dover J, Erdjument-Bromage H, Tempst P, Johnston M, Greenblatt JF, Shilatifard A. 2001. COMPASS: a complex of proteins associated with a trithorax-related SET domain protein. Proc. Natl. Acad. Sci. U. S. A. 98:12902–12907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shilatifard A. 2012. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu. Rev. Biochem. 81:65–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wu M, Wang PF, Lee JS, Martin-Brown S, Florens L, Washburn M, Shilatifard A. 2008. Molecular regulation of H3K4 trimethylation by Wdr82, a component of human Set1/COMPASS. Mol. Cell. Biol. 28:7337–7344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Herz H-M, Mohan M, Garruss AS, Liang K, Takahashi Y-H, Mickey K, Voets O, Verrijzer CP, Shilatifard A. 2012. Enhancer-associated H3K4 monomethylation by Trithorax-related, the Drosophila homolog of mammalian Mll3/Mll4. Genes Dev. 26:2604–2620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eissenberg JC, Shilatifard A. 2010. Histone H3 lysine 4 (H3K4) methylation in development and differentiation. Dev. Biol. 339:240–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Patel A, Vought VE, Dharmarajan V, Cosgrove MS. 2011. A novel non-SET domain multi-subunit methyltransferase required for sequential nucleosomal histone H3 methylation by the mixed lineage leukemia protein-1 (MLL1) core complex. J. Biol. Chem. 286:3359–3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Steward MM, Lee JS, O'Donovan A, Wyatt M, Bernstein BE, Shilatifard A. 2006. Molecular regulation of H3K4 trimethylation by ASH2L, a shared subunit of MLL complexes. Nat. Struct. Mol. Biol. 13:852–854 [DOI] [PubMed] [Google Scholar]
- 18. Dou Y, Milne TA, Ruthenburg AJ, Lee S, Lee JW, Verdine GL, Allis CD, Roeder RG. 2006. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat. Struct. Mol. Biol. 13:713–719 [DOI] [PubMed] [Google Scholar]
- 19. Wang P, Lin C, Smith ER, Guo H, Sanderson BW, Wu M, Gogol M, Alexander T, Seidel C, Wiedemann LM, Ge K, Krumlauf R, Shilatifard A. 2009. Global analysis of H3K4 methylation defines MLL family member targets and points to a role for MLL1-mediated H3K4 methylation in the regulation of transcriptional initiation by RNA polymerase II. Mol. Cell. Biol. 29:6074–6085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hubner NC, Bird AW, Cox J, Splettstoesser B, Bandilla P, Poser I, Hyman A, Mann M. 2010. Quantitative proteomics combined with BAC TransgeneOmics reveals in vivo protein interactions. J. Cell Biol. 189:739–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Smits AH, Jansen PWTC, Poser I, Hyman AA, Vermeulen M. 2012. Stoichiometry of chromatin-associated protein complexes revealed by label-free quantitative mass spectrometry-based proteomics. Nucleic Acids Res. 41:e28 doi:10.1093/nar/gks941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dignam JD, Lebovitz RM, Roeder RG. 1983. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11:1475–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cox J, Mann M. 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26:1367–1372 [DOI] [PubMed] [Google Scholar]
- 24. Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. 2011. Global quantification of mammalian gene expression control. Nature 473:337–342 [DOI] [PubMed] [Google Scholar]
- 25. Wiśniewski JR, Zougman A, Nagaraj N, Mann M. 2009. Universal sample preparation method for proteome analysis. Nat. Methods 6:359–362 [DOI] [PubMed] [Google Scholar]
- 26. Vizcaíno JA, Côté RG, Csordas A, Dianes JA, Fabregat A, Foster JM, Griss J, Alpi E, Birim M, Contell J, O'Kelly G, Schoenegger A, Ovelleiro D, Pérez-Riverol Y, Reisinger F, Ríos D, Wang R, Hermjakob H. 2013. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 41:D1063–D1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vermeulen M, Eberl HC, Matarese F, Marks H, Denissov S, Butter F, Lee KK, Olsen JV, Hyman AA, Stunnenberg HG, Mann M. 2010. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell 142:967–980 [DOI] [PubMed] [Google Scholar]
- 28. Tighe A, Staples O, Taylor S. 2008. Mps1 kinase activity restrains anaphase during an unperturbed mitosis and targets Mad2 to kinetochores. J. Cell Biol. 181:893–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Patel A, Dharmarajan V, Vought VE, Cosgrove MS. 2009. On the mechanism of multiple lysine methylation by the human mixed lineage leukemia protein-1 (MLL1) core complex. J. Biol. Chem. 284:24242–24256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Trievel RC, Shilatifard A. 2009. WDR5, a complexed protein. Nat. Struct. Mol. Biol. 16:678–680 [DOI] [PubMed] [Google Scholar]
- 31. Wysocka J, Swigut T, Milne TA, Dou Y, Zhang X, Burlingame AL, Roeder RG, Brivanlou AH, Allis CD. 2005. WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development. Cell 121:859–872 [DOI] [PubMed] [Google Scholar]
- 32. Ang Y-S, Tsai S-Y, Lee D-F, Monk J, Su J, Ratnakumar K, Ding J, Ge Y, Darr H, Chang B, Wang J, Rendl M, Bernstein E, Schaniel C, Lemischka IR. 2011. Wdr5 mediates self-renewal and reprogramming via the embryonic stem cell core transcriptional network. Cell 145:183–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Spedale G, Timmers HTM, Pijnappel WWMP. 2012. ATAC-king the complexity of SAGA during evolution. Genes Dev. 26:527–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Song J-J, Kingston RE. 2008. WDR5 interacts with mixed lineage leukemia (MLL) protein via the histone H3-binding pocket. J. Biol. Chem. 283:35258–35264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Barak O, Lazzaro MA, Lane WS, Speicher DW, Picketts DJ, Shiekhattar R. 2003. Isolation of human NURF: a regulator of Engrailed gene expression. EMBO J. 22:6089–6100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Karnik SK, Hughes CM, Gu X, Rozenblatt-Rosen O, McLean GW, Xiong Y, Meyerson M, Kim SK. 2005. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc. Natl. Acad. Sci. U. S. A. 102:14659–14664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hughes CM, Rozenblatt-Rosen O, Milne TA, Copeland TD, Levine SS, Lee JC, Hayes DN, Shanmugam KS, Bhattacharjee A, Biondi CA, Kay GF, Hayward NK, Hess JL, Meyerson M. 2004. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol. Cell 13:587–597 [DOI] [PubMed] [Google Scholar]
- 38. Huang J, Gurung B, Wan B, Matkar S, Veniaminova NA, Wan K, Merchant JL, Hua X, Lei M. 2012. The same pocket in menin binds both MLL and JUND but has opposite effects on transcription. Nature 482:542–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Agarwal SK, Guru SC, Heppner C, Erdos MR, Collins RM, Park SY, Saggar S, Chandrasekharappa SC, Collins FS, Spiegel AM, Marx SJ, Burns AL. 1999. Menin interacts with the AP1 transcription factor JunD and represses JunD-activated transcription. Cell 96:143–152 [DOI] [PubMed] [Google Scholar]
- 40. Farley SM, Chen G, Guo S, Wang M, Lee A JF, Lee F, Sawicki M. 2006. Menin localizes to chromatin through an ATR-CHK1 mediated pathway after UV-induced DNA damage. J. Surg. Res. 133:29–37 [DOI] [PubMed] [Google Scholar]
- 41. Marshall HM, Ronen K, Berry C, Llano M, Sutherland H, Saenz D, Bickmore W, Poeschla E, Bushman FD. 2007. Role of PSIP1/LEDGF/p75 in lentiviral infectivity and integration targeting. PLoS One 2:e1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yokoyama A, Cleary ML. 2008. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell 14:36–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Maertens GN, Cherepanov P, Engelman A. 2006. Transcriptional co-activator p75 binds and tethers the Myc-interacting protein JPO2 to chromatin. J. Cell Sci. 119:2563–2571 [DOI] [PubMed] [Google Scholar]
- 44. Hughes S, Jenkins V, Dar MJ, Engelman A, Cherepanov P. 2010. Transcriptional co-activator LEDGF interacts with Cdc7-activator of S-phase kinase (ASK) and stimulates its enzymatic activity. J. Biol. Chem. 285:541–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bartholomeeusen K, Christ F, Hendrix J, Rain J-C, Emiliani S, Benarous R, Debyser Z, Gijsbers R, De Rijck J. 2009. Lens epithelium-derived growth factor/p75 interacts with the transposase-derived DDE domain of PogZ. J. Biol. Chem. 284:11467–11477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wysocka J, Myers MP, Laherty CD, Eisenman RN, Herr W. 2003. Human Sin3 deacetylase and trithorax-related Set1/Ash2 histone H3-K4 methyltransferase are tethered together selectively by the cell-proliferation factor HCF-1. Genes Dev. 17:896–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Manke IA, Lowery DM, Nguyen A, Yaffe MB. 2003. BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science 302:636–639 [DOI] [PubMed] [Google Scholar]
- 48. Cho Y-W, Hong T, Hong S, Guo H, Yu H, Kim D, Guszczynski T, Dressler GR, Copeland TD, Kalkum M, Ge K. 2007. PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. J. Biol. Chem. 282:20395–20406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Thompson LH. 2012. Recognition, signaling, and repair of DNA double-strand breaks produced by ionizing radiation in mammalian cells: the molecular choreography. Mutat. Res. 751:158–246 [DOI] [PubMed] [Google Scholar]
- 50. Jeon B-N, Choi W-I, Yu M-Y, Yoon A-R, Kim M-H, Yun C-O, Hur M-W. 2009. ZBTB2, a novel master regulator of the p53 pathway. J. Biol. Chem. 284:17935–17946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shimizu K, Bourillot PY, Nielsen SJ, Zorn AM, Gurdon JB. 2001. Swift is a novel BRCT domain coactivator of Smad2 in transforming growth factor beta signaling. Mol. Cell. Biol. 21:3901–3912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lee J-H, Tate CM, You J-S, Skalnik DG. 2007. Identification and characterization of the human Set1B histone H3-Lys4 methyltransferase complex. J. Biol. Chem. 282:13419–13428 [DOI] [PubMed] [Google Scholar]
- 53. Lee J-H, You J, Dobrota E, Skalnik DG. 2010. Identification and characterization of a novel human PP1 phosphatase complex. J. Biol. Chem. 285:24466–24476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Porter IM, McClelland SE, Khoudoli GA, Hunter CJ, Andersen JS, McAinsh AD, Blow JJ, Swedlow JR. 2007. Bod1, a novel kinetochore protein required for chromosome biorientation. J. Cell Biol. 179:187–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Halbach A, Zhang H, Wengi A, Jablonska Z, Gruber IML, Halbeisen RE, Dehé Kemmeren P-MP, Holstege F, Géli V, Gerber AP, Dichtl B. 2009. Cotranslational assembly of the yeast SET1C histone methyltransferase complex. EMBO J. 28:2959–2970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dehé P-M, Dichtl B, Schaft D, Roguev A, Pamblanco M, Lebrun R, Rodríguez-Gil A, Mkandawire M, Landsberg K, Shevchenko A, Shevchenko A, Rosaleny LE, Tordera V, Chávez S, Stewart AF, Géli V. 2006. Protein interactions within the Set1 complex and their roles in the regulation of histone 3 lysine 4 methylation. J. Biol. Chem. 281:35404–35412 [DOI] [PubMed] [Google Scholar]
- 57. Jiang H, Shukla A, Wang X, Chen W-Y, Bernstein BE, Roeder RG. 2011. Role for Dpy-30 in ES cell-fate specification by regulation of H3K4 methylation within bivalent domains. Cell 144:513–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dharmarajan V, Lee J-H, Patel A, Skalnik DG, Cosgrove MS. 2012. Structural basis for WDR5 interaction (Win) motif recognition in human SET1 family histone methyltransferases. J. Biol. Chem. 287:27275–27289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Blobel GA, Kadauke S, Wang E, Lau AW, Zuber J, Chou MM, Vakoc CR. 2009. A reconfigured pattern of MLL occupancy within mitotic chromatin promotes rapid transcriptional reactivation following mitotic exit. Mol. Cell 36:970–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yokoyama A, Somervaille TCP, Smith KS, Rozenblatt-Rosen O, Meyerson M, Cleary ML. 2005. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell 123:207–218 [DOI] [PubMed] [Google Scholar]
- 61. Thiel AT, Blessington P, Zou T, Feather D, Wu X, Yan J, Zhang H, Liu Z, Ernst P, Koretzky GA, Hua X. 2010. MLL-AF9-induced leukemogenesis requires coexpression of the wild-type Mll allele. Cancer Cell 17:148–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fernandez A, Brautigan DL, Lamb NJ. 1992. Protein phosphatase type 1 in mammalian cell mitosis: chromosomal localization and involvement in mitotic exit. J. Cell Biol. 116:1421–1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhang K, Lin W, Latham JA, Riefler GM, Schumacher JM, Chan C, Tatchell K, Hawke DH, Kobayashi R, Dent SYR. 2005. The Set1 methyltransferase opposes Ipl1 aurora kinase functions in chromosome segregation. Cell 122:723–734 [DOI] [PMC free article] [PubMed] [Google Scholar]