

Abstract
Eal is an EAL domain protein in Xylella fastidiosa homologous to one involved in resistance to tobramycin in Pseudomonas aeruginosa. EAL and HD-GYP domain proteins are implicated in the hydrolysis of the secondary messenger bis-(3′-5′)-cyclic dimeric GMP (cyclic di-GMP). Cell density-dependent communication mediated by a Diffusible Signal Factor (DSF) also modulates cyclic di-GMP levels in X. fastidiosa, thereby controlling the expression of virulence genes and genes involved in insect transmission. The possible linkage of Eal to both extrinsic factors such as antibiotics and intrinsic factors such as quorum sensing, and whether both affect virulence, was thus addressed. Expression of eal was induced by subinhibitory concentrations of tobramycin, and an eal deletion mutant was more susceptible to this antibiotic than the wild-type strain and exhibited phenotypes similar to those of an rpfF deletion mutant blocked in DSF production, such as hypermotility, reduced biofilm formation, and hypervirulence to grape. Consistent with that, the rpfF mutant was more susceptible than the wild-type strain to tobramycin. Therefore, we propose that cell-cell communication and antibiotic stress can apparently lead to similar modulations of cyclic di-GMP in X. fastidiosa, resulting in similar phenotypes. However, the effect of cell density is dominant compared to that of antibiotic stress, since eal is suppressed by RpfF, which may prevent inappropriate behavioral changes in response to antibiotic stress when DSF accumulates.
INTRODUCTION
The plant pathogen Xylella fastidiosa colonizes xylem vessels of many economically important crop plants, including grape, almond, and citrus, often blocking water flow and thus causing disease symptoms. The pathogen is transmitted from plant to plant by xylem-feeding leafhoppers (1), where it attaches to the precibarium in the mouthpart. In order to colonize both plant and insect hosts, X. fastidiosa has evolved a “bipolar” lifestyle, either movement within the plant or acquisition by insects, whereby different subpopulations within a plant express traits that allow bacterial behaviors that appear incompatible (2–4). In the plant, most cells exhibit an initial “exploratory” phase, enabling spread both within and between xylem vessels, in which genes involved in active movement and plant cell wall degradation are expressed while genes involved in attachment are suppressed. Only when the cell density in some colonized vessels becomes high do those cells increase the expression of genes for the production of adhesins and exopolyssacharide (EPS) and suppress the expression of gene products involved in active movement such as extracellular enzymes and type IV pili, thereby transitioning to an “insect acquisition phase,” in which biofilm-related features allow the adherence of the bacterium to the insect's precibarium upon acquisition (5).
The behavioral transitions of X. fastidiosa are regulated by signal transduction pathways involving bis-(3′-5′)-cyclic dimeric GMP (cyclic di-GMP)-modulating proteins. Cyclic di-GMP is an intracellular secondary messenger exclusively found in bacteria that is produced by enzymes containing GGDEF domains (diguanylate cyclases [DGCs]) and is hydrolyzed by those containing either EAL or HD-GYP domains (phosphodiesterases [PDEs]) (6–10). Cyclic di-GMP-mediated signaling is often involved in the transition between the motile and sessile bacterial lifestyles. X. fastidiosa harbors five genes encoding proteins with the capacity to modulate cyclic di-GMP levels; PD1671 and PD1994 encode GGDEF/EAL dual-domain proteins, cgsA (PD0279) encodes a GGDEF protein, rpfG (PD0405) encodes a HD-GYP protein, and PD1617 encodes an EAL protein. These proteins also have N-terminal sensory input domains; PD1671 and RpfG have a CheY-like REC domain typical of response regulators in phosphorelay systems (11), PD1994 has a Per-Arnt-Sim (PAS) domain that typically binds to a specific ligand (12), and CgsA and PD1617 have transmembrane helices that place their sensory domains in the periplasm.
RpfG is the response regulator of the RpfCG phosphor-relay system that receives and converts a quorum-sensing signal (Diffusible Signal Factor [DSF]) to the cyclic di-GMP currency. RpfC, a hybrid membrane sensor kinase, phosphorylates RpfG upon interacting with DSF, which is synthesized by RpfF (9, 13). The activated RpfG hydrolyzes cyclic di-GMP, thereby reducing its availability to Clp, a transcriptional regulator (14). An rpfF mutant (blocked in the production of DSF and therefore in the hydrolysis of cyclic di-GMP by RpfG) is hypervirulent to grape and poorly adherent and thus attenuated in biofilm formation (3, 15). Consistent with this, a cgsA mutant (attenuated in cyclic di-GMP synthesis) is hypovirulent to grape and hyperadhesive (16), suggesting that low levels of cyclic di-GMP in X. fastidiosa are associated with the “insect acquisition phase” whereas high levels of cyclic di-GMP are associated with the “plant exploratory phase.” This was unexpected, since the activity of diguanylate cyclases in other bacteria facilitates biofilm formation (reviewed in reference 17). RpfF was also reported to suppress the expression of cgsA (2), suggesting that signaling through the Rpf system reduces cyclic di-GMP levels in more than one manner.
In addition to the role of cyclic di-GMP-modulating proteins in modulating surface features of cells and their virulence, there is evidence that they control responses to a wide variety of environmental factors such as nutrient limitation (18) and antibiotic exposure (19–21). In Pseudomonas aeruginosa, subinhibitory concentrations of the aminoglycoside antibiotic tobramycin trigger biofilm formation. This trait requires Arr (for Aminoglycoside Response Regulator), a membrane-associated EAL domain protein (19). PD1617 encodes a protein with 43% identity to Arr and has similar EAL, transmembrane, and periplasmic domains (hereby designated Eal). In this work, we address the possible role of eal in drug resistance in X. fastidiosa and explore whether cross talk involving the common intracellular signal cyclic di-GMP could lead to common responses of this pathogen to the different input cues of cell density and antibiotics.
MATERIALS AND METHODS
Bacterial strains and culture media.
The strains and plasmids that were employed in this study are listed in Table 1. X. fastidiosa strains were routinely grown on PW broth (22), PWG plates (PW with 8 g/liter gelrite), or XFMP broth (XFM plus 1% pectin) (23, 24) at 28°C. Escherichia coli strains were grown on LB media (10 g/liter Bacto tryptone, 5 g/liter yeast extract, 5 g/liter NaCl) at 37°C. Antibiotics were used at the following concentrations: kanamycin at 50 μg/ml and gentamicin at 15 μg/ml.
Table 1.
Strains and plasmids
| Strain or plasmid | Genotype or description | Reference or source |
|---|---|---|
| X. fastidiosa Temecula1 strains | Wild type | ATCC 700964 |
| DifrpfF | X. fastidiosa Temecula1 ΔrpfF (markerless) | 2 |
| ADS1 | X. fastidiosa Temecula1 Δeal (kanR) | This study |
| MIX1 | X. fastidiosa Temecula1 Δclp (gentR) | This study |
| P. aeruginosa PAO1 cyclic di-GMP bioreporter | ΔpelA ΔpslBCD mini-Tn7-PcdrA-RBSII-gfp | 29 |
| Plasmid | ||
| pUC19 | Cloning vector, ampR (bla) | Invitrogen |
| pBBR1MCS-2 | kanR gene [aph(3′)II] | 25 |
| pBBR1MCS-5 | gentR gene (aaaC1) | 25 |
| pFXFkan | pUC19 kanR [aph(3′)II] | This study |
| pFXFgent | pUC19 gentR (aaaC1) | This study |
| pFXF1 | pFXFkan eal knockout vector | This study |
| pFXF2 | pBBR1MCS-5 eal complementation vector | This study |
| pFXF3 | pFXFgent clp knockout vector | This study |
| pEAL | pBBR1MCS-2 lacZ′::eal (aa 275–522)a | This study |
aa, amino acids.
Deletion of eal (PD1617) and clp (PD0755).
Deletion of eal (Δeal) and clp (Δclp) was carried out by transforming X. fastidiosa strain Temecula1 with a pUC19-based suicide vector harboring a kanamycin [aph(3′)II] or a gentamicin (aaaC1) resistance gene, flanked by two 1,000-bp genomic fragments which adjoin the target gene in the X. fastidiosa genome. The kanamycin and gentamicin resistance genes were amplified from pBBR1MCS-2 and pBBR1MCS-5, respectively (25), using Phusion DNA polymerase (Finnzymes, Finland) and the primers listed in Table S1 in the supplemental material. The PCR products and pUC19 were digested with BamHI (NEB) and ligated with T4 DNA ligase (Roche, Switzerland) to generate pFXFkan and pFXFgent. The orientation of the resistance gene was determined by sequencing. Regions flanking the target genes were amplified and cloned into the pFXFkan vector using the primers listed in Table S1 in the supplemental material to generate pFXF1 (eal::kanR) and pFXF3 (clp::gentR). These suicide vectors were transformed into X. fastidiosa Temecula1 by exploiting its natural competence (26). Transformed cells were cultured on PWG plates supplemented with kanamycin or gentamicin and incubated for 2 to 3 weeks at 28°C. Colonies were screened for deletion of eal or clp by PCR using the relevant primers listed in Table S1 in the supplemental material. The presence of a single insertion of the kanamycin gene in the eal mutant genome (see Fig. S1 in the supplemental material) was confirmed by Southern blotting (27) using a PCR DIG (digoxigenin) probe synthesis kit (Roche, Switzerland) and the primers employed to amplify the kanR cassette from pBBR1MCS-2 (see Table S1 in the supplemental material).
Complementation of Δeal.
eal was cloned into pBBR1MCS-5 using the primers listed in Table S1 in the supplemental material and introduced into the eal mutant by using a previously described transformation protocol (28). The restoration of some phenotypes was verified (see Fig. S2 in the supplemental material).
Verification of Eal cyclic di-GMP phosphodiesterase activity.
The EAL domain of eal (amino acids 275 to 522) was amplified from the X. fastidiosa genome with the primers listed in Table S1 in the supplemental material. The forward primer had a tail harboring the ribosome binding site of P. aeruginosa PAO1 rpsO followed by an ATG sequence. The PCR product was cloned into pBBR1MCS-2 (25) at the HindII and EcoRI restriction sites as described above to generate pEAL (Table 1). pEAL was transformed by electroporation into the (nonaggregative) P. aeruginosa PAO1 Δpel Δpsl double mutant, which harbors a chromosomal cdrA′::gfp transcriptional fusion, previously shown to respond to cyclic d-GMP levels (29). The green fluorescent protein (GFP) fluorescence of this sensor harboring either pEAL or the empty vector pBBR1MCS-2 was assessed. Colonies were collected from LB plates supplemented with 150 μg/ml kanamycin and 60 μg/ml gentamicin and suspended in ABTG-casA broth (29) to an optical density at 600 (OD600) of 0.05. Aliquots (500 μl) were then incubated for 48 h in a clear 48-well microtiter plate at 37°C without shaking. GFP fluorescence (excitation wavelength of 485 nm and emission wavelength of 528 nm) and the OD600 were recorded using a Synergy 2 plate reader (BioTek). cdrA promoter activity was expressed as the fluorescence divided by the OD600.
Tobramycin MIC determination and biofilm induction.
The MIC of tobramycin for the wild-type X. fastidiosa strain was determined with cells collected from PWG plates and suspended in PW broth to OD600 = 0.1 in glass tubes. The tubes were shaken in a rotary shaker at 120 rpm and 28°C, and different concentrations of tobramycin (ranging from 0 to 30 μg/ml) were added at days 1, 4, 7, and 10 after inoculation. A 100-μl aliquot of undiluted suspension was plated onto fresh PWG plates after 24 h of exposure to the antibiotic. The MIC was determined as the lowest concentration of tobramycin that completely inhibited cell viability (fewer than 2 colonies obtained from triplicate plating) (30). Testing of sensitivity to subinhibitory doses was performed in a similar manner, using 0, 0.3, 0.5, and 1 μg/ml tobramycin. To assess the induction of biofilm formation by tobramycin, 0, 0.3, 0.5, or 1.0 μg/ml was added to cell suspensions after 4 days of incubation; the biofilm biomass at the liquid-air interface was stained and quantified with 1% crystal violet 24 h after antibiotic addition as described below.
Biofilm assay.
Cells were collected from PWG plates and suspended in PW broth to OD600 = 0.1 (approximately 108 CFU/ml), and 300-μl aliquots were transferred to 12-well polystyrene culture plates or glass tubes containing 3 ml of XFMP broth and incubated at 28°C without shaking (plates) or with shaking at 200 rpm (tubes). After 7 days of growth, the medium was discarded and the wells were gently washed with double-distilled water (DDW) to remove planktonic and loosely attached cells. The attached biofilm was quantified by staining the remaining cells with 1% crystal violet for 20 min at room temperature. Excess crystal violet was removed by washing the wells several times with DDW. Crystal violet bound to the attached cells was recovered in 1 ml of 90% ethanol and quantified by measuring absorbance at 595 nm.
Biofilm was also formed on coverslips at the bottom of 6-well polystyrene culture plates containing 1 ml of XFMP broth. To each well, 100 μl of cell suspension (OD600 = 0.1) was added, and the plate was incubated at 28°C without shaking. After 10 days, the medium was removed and the coverslips were washed with DDW. Cells were stained with Syto-9 (Invitrogen) in a 1:600 dilution in MilliQ autoclaved H2O (Millipore). Fluorescence images were captured using a Zeiss 510 confocal laser scanning microscope (Zeiss, Germany) (excitation wavelength of 488 nm and emission wavelengths of 505 to 550 nm) at the University of California College of Natural Resources Biological Imaging Facility.
Determination of the exopolysaccharides (EPS).
Cells were collected from PWG plates and resuspended in phosphate-buffered saline (PBS) to OD600 = 0.1, and 40-μl aliquots were spread on 6 plates of XFMP medium. The plates were incubated on 28°C for 10 days, and the cells were collected and resuspended in PBS. Two different methods were used to quantify EPS: (i) a protein A double-antibody sandwich enzyme-linked immunosorbent assay (ELISA) according to a protocol described before (31) and (ii) quantification of total carbohydrate content using Anthrone reagent (32). The protein A double-antibody sandwich ELISA was performed using an antibody raised against EPS of an X. campestris pv. campestris gumI mutant (31) (provided by B. Kirkpatrick, University of California, Davis). Briefly, the wells of a MaxiSorp microtiter plate (Nalge Nunc) were coated with 5 μg/ml anti-EPS F(ab)2 fragments in 0.015 M Na2CO3 (0.035 M)–NaHCO3 (pH 9.6). The wells were blocked with 1% nonfat milk for 1 h at room temperature and washed three times with phosphate-buffered saline–0.05% Tween 20 (PBST). Cell were suspended in PBS (OD600 = 0.1), added to the antibody-coated wells, and incubated for 1.5 h at 37°C, and then the wells were washed three times with PBST. Anti-EPS IgG was then added to the wells at a concentration of 5 μg/ml, and the plate was incubated for 1.5 h at 37°C followed by three washes with PBST. Protein A-alkaline phosphatase conjugate (Sigma-Aldrich) was diluted 1:1,500 in PBS, added to the wells, and incubated for 1 h at 37°C. Sigma Fast p-nitrophenyl phosphate (Sigma-Aldrich) was used as the substrate. The substrate color was allowed to develop for 1 h, and the absorbance was measured at 405 nm using a SpectraMax M2 Microplate Reader (Molecular Devices).
Colony morphology.
Differences in the morphologies of individual colonies of mutants of similar ages grown on PWG plates were assessed. The plates were incubated at 28°C for 10 days, and the morphology of the fringe of the colonies was examined using a Lumar dissecting microscope (Zeiss, Germany). Colonies with an obvious peripheral fringe were designated motile (34). These colonies were further examined by scanning electronic microscopy at the Electron Microscope Laboratory at the University of California, Berkeley. The samples were prepared as previously described (16).
Pathogenicity assays.
Cell suspensions (OD600 = 1.0) of various strains recovered after growth for 10 days on PWG were mechanically inoculated into 15 Thompson seedless grapevines using a droplet puncture method as described before (33). The number of symptomatic leaves was counted weekly starting 10 weeks after inoculation. Symptomatic leaves were scored for the presence of typical Pierce's disease symptoms (loss of chlorophyll and death of the leaf margins). The population size and migration distance were determined 15 weeks postinoculation in petioles collected at 7 different locations (0, 30, 60, 120, 150, 180, and 210 cm from the inoculation point). The petioles were surface sterilized and weighed, and cells were extracted as described before (35). Serial dilutions of tissue macerates were cultured on PWG plates, and the number of CFU obtained was normalized by the weight of the petiole.
RNA isolation, cDNA synthesis, and quantitative PCR (qPCR).
The abundance of transcripts of genes associated with biofilm formation and motility was measured for wild-type and mutant cells collected after 10 days of growth on XFMP plates. Cells were washed and collected by centrifugation at 8,000 × g for 5 min at 4°C with diethylpyrocarbonate-treated water. Total RNA was isolated using an RNeasy RNA extraction kit (Qiagen, Germany); DNA was eliminated using an on-column RNase-Free DNase apparatus (Qiagen, Germany). RNA samples were stored at −80°C, and new 1-μg aliquots were used for cDNA synthesis before each analysis, using 3 μg of random hexamers and Superscript II reverse transcriptase (Life Technologies) according to the manufacturer's instructions.
Quantitative PCR was performed in an ABI PRISM 7100 sequence detector system (Applied Biosystems). Detection of PCR products was done by measuring the increase in fluorescence produced upon binding of SYBR green dye (Qiagen, Germany) to double-stranded DNA. Both rpoD and rpsO were used as endogenous control genes to normalize gene expression. The specificity of primers (listed in Table S1 in the supplemental material) for the target genes was verified by DNA sequencing of PCR products at the University of California Biological Sequence Facility. To ensure that the threshold cycle (CT) values obtained were from a single PCR product, melting curve analysis was run at the end of each expression analysis. Relative expression (RQ) was calculated from the CT values (36) as follows: dCT = CT (target gene) − CT (endogenous control); ddCT = dCT (treatment) − dCT (reference); RQ = 2(−ddCT). To estimate the abundance of eal and rpfG transcripts in young (1-day-old) and old (7-day-old) wild-type cultures, a semiquantitative analysis was performed in which the transcript level was normalized to the total RNA level as follows: dCT = CT (7 days) − CT (1 day); RQ = 2(−dCT) (36).
RESULTS
X. fastidiosa eal is a phosphodiesterase involved in the response to tobramycin.
The phosphodiesterase activity of X. fastidiosa Eal (PD1617) was verified using a P. aeruginosa-based cyclic di-GMP bioreporter (29) in which GFP fluorescence is dependent upon cytoplasmic cyclic di-GMP levels. Heterologous expression of the EAL domain of Eal in this bioreporter resulted in a (ca. 30%) reduction in the GFP fluorescence of this bioreporter (see Fig. S3 in the supplemental material), suggesting that cyclic di-GMP levels had decreased due to the expression of the EAL domain.
Eal has an organization of transmembrane, periplasm, and EAL domains similar to that of Arr (Fig. 1), suggesting that it might also confer antibiotic resistance in X. fastidiosa. To test this conjecture, we assessed eal expression in cells exposed to subinhibitory concentrations of tobramycin. The wild-type strain exhibited considerable resistance to tobramycin, and survival (not shown) and growth (see Fig. S4 in the supplemental material) were reduced significantly at concentrations higher than 5 μg/ml. After incubation for 7 days in the presence of 0.5 and 1 μg/ml tobramycin, eal expression was 5- and 10-fold higher, respectively, than in control cells grown without this antibiotic (Fig. 2A). Expression of the rpfG gene, encoding a protein also having cyclic di-GMP hydrolysis activity such as Eal apparently does, was induced by only 1.5-fold following exposure to 1 μg/ml tobramycin (Fig. 2A). The induction of eal expression by tobramycin suggested that elevated cyclic di-GMP hydrolysis is linked to tobramycin resistance and that an eal deletion mutant would exhibit hypersensitivity to this antibiotic. The number of viable cells in cultures of the eal mutant and the wild-type strain to which various concentrations of tobramycin were added at various times was therefore assessed. The eal mutant was highly susceptible to concentrations of tobramycin that were only slightly inhibitory to the wild-type strain (Fig. 2B). For example, while the viable cell concentration in cultures of the mutant to which 0.3 μg/ml tobramycin was added after 4 days of growth was reduced 1,000-fold or more, such an addition had a modest effect on the culturability of the wild-type strain (Fig. 2B and C). Furthermore, the susceptibility increased with the age of the culture (Fig. 2C); no viable cells were recovered from cultures of the mutant in the presence of even as little as 0.3 μg/ml tobramycin when it was added after either 7 or 10 days of growth (Fig. 2C). This suggests that Eal, presumably through its cyclic di-GMP hydrolysis activity, is involved in the regulation of the cellular response to tobramycin stress. The increased antibiotic susceptibility of cells lacking Eal in older cultures suggests that another factor(s) is responsible for stress tolerance in young cultures and that Eal becomes essential only in later growth stages.
Fig 1.

Pairwise alignment comparison of Pseudomonas aeruginosa PAO1 Arr and Xylella fastidosa Eal. The EAL domain is boxed in a black frame, the signal peptide in a solid red frame, and the transmembrane (TM) helix in a dashed red frame. The comparison was performed using the EBI server, the image was generated using the BOXSHADE server, the EAL domain was located using the NCBI CDD database, and signal peptides and transmembrane helices were predicted using the TMpred server (56).
Fig 2.
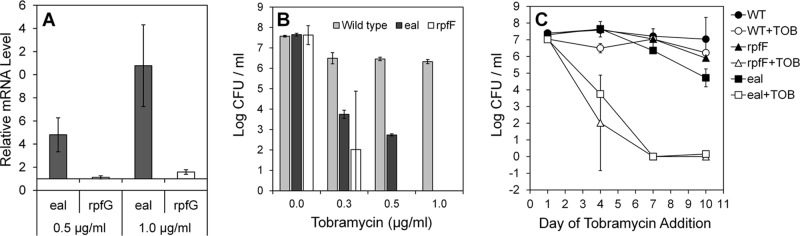
Eal is required for tobramycin resistance. (A) Relative expression of eal and rpfG in wild-type cells of Xylella fastidiosa grown for 7 days in the presence of 0.5 or 1 μg/ml tobramycin; expression values are relative to the expression of each gene in cells grown without tobramycin. (B) Loss of culturability of cells exposed to different concentrations of tobramycin added at day 4 of growth to cultures of the wild-type strain, an eal mutant, or an rpfF mutant. (C) Loss of culturability of cells exposed to 0.3 μg/ml added at day 1, 4, 7, or 10 of growth to cultures of the wild-type (WT) strain, an eal mutant, or an rpfF mutant. Viable cells were enumerated 24 h after the addition of the antibiotic. Vertical bars represent the standard errors of the means (n = 5 in panel A and n = 3 in panels B and C).
eal is involved in biofilm formation.
Tobramycin exposure induces biofilm formation in P. aeruginosa PAO1 whereas an arr mutant is attenuated in this process (19). We did not observe such an increase in biofilm formation in the wild-type strain of X. fastidiosa in response to any concentration of tobramycin (Fig. 3B; see also Fig. S2 in the supplemental material). In fact, a slight decrease in biofilm formation was observed. Similarly, it was reported (37) that the biofilm formed in the presence of the aminoglycoside antibiotic streptomycin by X. fastidiosa 9a5c did not differ from that formed by nontreated cells, suggesting that responses to antibiotic stress in X. fastidiosa do not necessarily depend upon traits linked with biofilm formation. It was clear, however, that both eal and rpfF deletion mutants formed less biofilm than the wild-type strain even in the absence of the antibiotic (Fig. 3A and B) as measured by three different methods: crystal violet staining of cells attached at the liquid-air interface in glass culture tubes (Fig. 3A), crystal violet staining of cells attached on the bottom of polystyrene microtiter plates (Fig. 3B), and examination of biofilms that had formed on glass coverslips immersed within cultures using confocal microscopy (Fig. 3C). Crystal violet staining revealed that the eal mutant exhibited only 25% of the attachment of the wild-type strain (Fig. 3A and B). eal mutants were mostly attached as single cells or as small aggregates, while the wild-type strain formed a thicker and more structured biofilm (Fig. 3C). In repeated experiments, it was also observed that the eal mutant was more impaired in biofilm formation than the rpfF mutant (P < 0.05), suggesting either that Eal and RpfF might both control the same traits in quantitatively different manners or that they control separate traits that each contribute to this process. These results clearly indicate that Eal is involved in the regulation of fundamental processes such as attachment to surfaces in X. fastidiosa. Since Eal can control both biofilm formation and antibiotic resistance whereas tobramycin-dependent elevation of its expression did not confer an increase in biofilm formation, we posit that it controls a tobramycin-dependent set of traits contributing to antibiotic resistance independently of traits contributing to biofilm formation.
Fig 3.

eal and rpfF contribute to biofilm formation. (A) Crystal violet staining of biofilm formed at the liquid-air interface of cultures of various strains of Xylella fastidiosa in glass culture tubes. (B) Quantification of crystal violet staining of cells attached to the bottom of wells of polystyrene microtiter plates after 10 days of growth. (C) Confocal fluorescence imaging of Syto 9-stained biofilms formed on glass coverslips placed in the bottom of 6-well microtiter plates in which various strains of Xylella fastidiosa were grown for 10 days. Vertical bars represent the standard deviations of the means (n = 6).
eal and rpfF mutants exhibit similar in vitro and in planta phenotypes.
Given that expression of both Eal and RpfF might be expected to lead to decreases in cyclic di-GMP levels, the phenotypes of the eal and rpfF mutants were compared to determine the extent to which they overlapped. The rpfF mutant was more susceptible to tobramycin than the wild-type strain and even slightly more susceptible than the eal mutant (Fig. 2B). As with the eal mutant, its susceptibility increased with the age of the culture (Fig. 2C), indicating that accumulation of DSF in late growth stages regulates traits involved in tobramycin resistance that seem to be different from those regulated by Eal. Both mutants were also attenuated in EPS production as measured with either a protein A double-antibody sandwich ELISA using antibodies raised against EPS produced by the Xanthomonas campestris gumI mutant (31) or a colorimetric carbohydrate assay using Anthrone reagent. The amounts of EPS produced by the rpfF and eal mutants were significantly (P < 0.01) lower than that produced by the wild-type strain (Fig. 4A and B). Since EPS accumulation contributes to biofilm formation (39), this deficiency may partly account for the reduced levels of biofilm produced by these mutants. Both mutants were also hypermotile, having fringed colony margins not seen in the wild-type strain on a medium which tends to suppress motility (Fig. 4C; see also Fig. S2 in the supplemental material). The eal and rpfF mutants both also expressed more polar pili that might have been type IV pili that were observable with scanning electron microscopy than the wild-type strain (Fig. 4D). Type IV pili have been associated with the ability of X. fastidiosa to move along xylem vessels by twitching (34, 40). Taken together, these observations suggest that RpfF and Eal independently negatively control the expression of traits associated with movement but induce traits involved in attachment, biofilm formation, and resistance to subinhibitory concentrations of tobramycin.
Fig 4.

eal and rpfF mutants of Xylella fastidiosa exhibit similar phenotypes. (A) EPS production of different strains assessed with protein A double-antibody sandwich ELISA with anti-EPS antibodies. (B) Quantification of total carbohydrates with Anthrone reagent. (C) Colony morphology of colonies grown for 10 days. (D) Scanning electron microscopy of cells collected from the colonies shown in panel C. White arrows point toward pilus-like structures. Vertical bars represent the standard deviations of the means (n = 3).
We further compared the abilities of the wild-type strain and the eal and rpfF mutants of X. fastidiosa strains to cause Pierce's disease in grapevine after mechanical inoculation into stems. The virulence of the strains was assessed by determining the rate of change of the incidence of symptomatic leaves in vines inoculated with a given strain with time after inoculation. The incidence of symptomatic leaves increased linearly with time in all treatments. Disease progression was significantly more rapid in plants inoculated with the eal mutant (1.36 leaves per plant/week) than in plants inoculated with the wild-type strain (0.69 leaves per plant/week) (P < 0.0001). Likewise, disease progression in plants inoculated with the rpfF mutant (1.43 leaves per plant/week) was higher than that in plants infected with the wild-type strain (P < 0.0002). The levels of virulence of eal and rpfF mutants did not differ significantly (P = 0.68). Enumeration of bacterial population size in petioles at different distances from the point of inoculation revealed that the eal mutant moved further than the wild-type strain; by 15 weeks after inoculation, both the wild-type strain and the rpfF mutant were detected in all petioles at up to 180 cm away from the point of inoculation whereas the eal mutant was detected also in petioles at a distance of up to 210 cm (Fig. 5A). The population sizes of both mutants were either similar to or larger than that of the wild-type strain in all petioles at the distances at which all of the strains were present (Fig. 5A).
Fig 5.
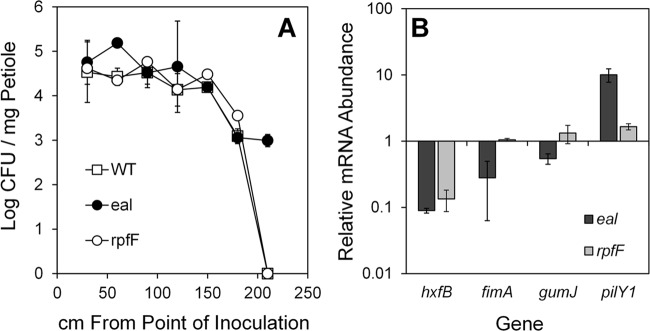
eal mutants of Xylella fastidiosa are hypermotile. (A) Population sizes of cells of various strains in petioles collected 11 weeks after inoculation at various distances from the point of inoculation. (B) Transcript abundance of various genes influencing biofilm formation, movement, and virulence determined by quantitative reverse transcriptase PCR in cells after 10 days of growth on XFMP plates. Vertical bars represent the standard deviations of the means (n = 3).
Eal controls the expression of virulence genes.
The levels of expression of four key genes involved in movement, biofilm formation, and virulence were compared in the X. fastidiosa wild-type strain and the eal and rpfF mutants to determine if they could be associated with the higher virulence of the mutants. The expression of hxfB, encoding a hemagglutinin-like protein that is an antivirulence factor that reduces cell movement within plants (38), was 11- and 7-fold lower in the eal and rpfF mutants, respectively, than in the wild-type strain (Fig. 5B). The expression of fimA, an attachment factor encoding type I pili, a mutant of which was reported to move longer distances in the plant than the wild-type strain (33), was 3.5-fold lower in the eal mutant than in the wild-type strain, while its expression in the rpfF mutant was similar to that in the parental strain (Fig. 5B). Likewise, the expression of gumJ, involved in exopolysaccharide production, was unchanged in the rpfF mutant but 2-fold lower in the eal mutant than in the parental strain (Fig. 5B). Transcripts of pilY1, encoding the adhesion protein of the type IV pilus enabling twitching motility (40), were 10-fold more abundant in the eal mutant than in the wild-type strain. This gene was also more highly expressed in the rpfF mutant, although its expression level was less than that in the eal mutant (Fig. 5B). The lower expression of the adhesins HxfB and FimA and the higher expression of PilY1, all of which would aid active movement, is consistent with the hypermotile phenotype exhibited by both eal and rpfF mutants in vitro and in planta, despite the fact that the two mutants differ quantitatively in expression of these genes.
eal is negatively controlled by RpfF and DSF.
Our results suggest that X. fastidiosa employs two different cyclic di-GMP hydrolyzing pathways to control similar traits but in response to different cues. RpfF was reported to repress cgsA and thus to suppress cyclic di-GMP synthesis (16). We thus hypothesized that RpfF might also induce the expression of eal, thus increasing cyclic di-GMP degradation, to confer a large and consistent effect of cell density on the abundance of this second messenger. Surprisingly, the expression of eal, like that of cgsA, was higher in the rpfF mutant than in the wild-type strain when assessed in older cultures. The levels of expression of cgsA and eal were 5 ± 2.2- and 8.6 ± 5.9-fold higher in the rpfF mutant than in the wild-type strain, respectively, while the levels of expression of a control gene, rpsO, did not differ (0.99 ± 0.48). This suggested that DSF represses eal (Fig. 6A).
Fig 6.
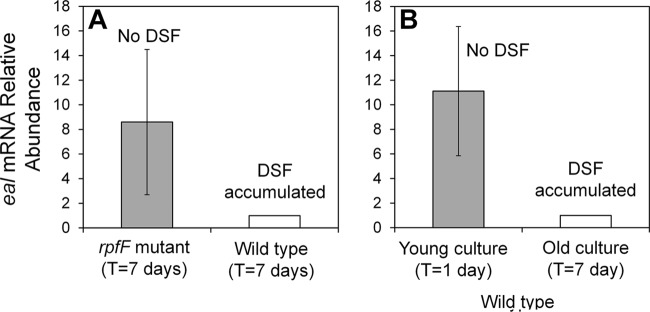
Regulation of eal by RpfF and DSF. (A) Relative expression of eal in the wild-type strain and the rpfF mutant of X. fastidiosa after 7 days of growth, during which DSF is expected to accumulate. (B) Relative expression of eal in wild-type cells after 1 day of growth (young culture), during which DSF had not yet accumulated, and after 7 days of growth (old culture), during which DSF is expected to have accumulated. The vertical bars represent the standard deviations of the means (n = 7 for panel A and n = 5 for panel B).
To test this model, the levels of expression of eal were compared in young cultures of the wild-type strain that had not accumulated DSF (1-day-old cultures) and older cultures in which DSF had accumulated (7 days). The abundance of eal transcript in older cells was about 10-fold lower than that in younger cells (ratio, 0.09 ± 0.19), supporting the conjecture that DSF-mediated signaling suppresses eal (Fig. 6B). In contrast, rpfG transcript abundance in the old cells was similar to that of the young cells (ratio, 1.07 ± 0.08). It is expected that rpfG is regulated by DSF at the protein level, and not at the transcriptional level, via phosphorylation by RpfC upon DSF sensing.
DISCUSSION
We demonstrate here that whereas eal, encoding a cyclic di-GMP PDE, plays a significant role in antibiotic resistance and biofilm formation in X. fastidiosa, it also controls motility and virulence, traits that are also controlled by the Rpf signaling system which regulates another cyclic di-GMP PDE, RpfG. A large number of different cyclic di-GMP-modifying enzymes containing GGDEF, EAL, and HD-GYP domains are found in many bacteria (6–8). The multiplicity of cyclic di-GMP-modulating proteins permits the integration of many signals, enabling several cellular processes to be regulated by separate pathways in parallel, but raises the issue of how they can exhibit specificity in outputs (41). It is intriguing that X. fastidiosa possesses only 5 such enzymes, many fewer than P. aeruginosa PAO1, which contains 38 (19), E. coli K-12, which contains 29 (41), or even the close relative X. campestris pv. campestris 8004, which contains 37 (42). The small number of such players in X. fastidiosa should facilitate understanding of how such a redundant system is buffered against stochastic noise and prevents excessive cross talk in regulation of target traits. However, to date, we have not observed substantial differences in the traits controlled by the different cyclic di-GMP modules tested in X. fastidiosa; all of the phenotypes tested (biofilm formation, motility, and virulence) were generally consistent with the cyclic di-GMP levels predicted to occur in the rpfF, cgsA, and eal mutants; and the similar canonical EAL/GGDEF domains found in PD1671 and PD1994 make it difficult to discern distinct roles of these two enzymes.
In other bacteria, DGCs and PDEs that harbor a single domain and thus have similar effects on cyclic-di-GMP levels can either similarly regulate a given trait or, unexpectedly, differ in such regulation. Of 19 predicted DGCs in P. fluorescens Pf0-1, mutations in only four genes caused significant reductions in biofilm formation, indicating that, despite their common enzymatic function, they have different roles (43). Mutants with mutations in the PDE-encoding genes rpfG and XC2161 of X. campestris pv. campestris 8004, which have a single HD-GYP and EAL domain, respectively, were attenuated in biofilm formation, whereas a mutant with a mutation in XC0362, also having a single HD-GYP domain, formed more biofilm than the wild-type strain (42). Likewise, it was previously reported (44) that two different DGC proteins, when abolished, conferred equally reduced levels of cyclic di-GMP and similar levels of impairment in biofilm formation in P. aeruginosa PA14 but had distinct regulatory targets, one controlling EPS production and the other flagellar motility. These discrete outputs at the same level of cyclic di-GMP were suggested to be due to distinct subcellular localizations of these proteins. Consistent with this model, other cyclic di-GMP-modulating proteins have been shown to be localized to the cell poles upon activation by phosphorylation (45). In fact, RpfG has been suggested to be spatially localized in X. campestrisis pv. campestris 8004 (46). In addition, enzymes such as HD-GYP proteins that are capable of modulating cyclic di-GMP levels regulate certain traits via direct interactions with other proteins (47). There are thus many possible reasons why rpfF and eal mutants of X. fastidiosa might have been expected to differ somewhat in their phenotypes. Indeed, while these mutants had similar defects in biofilm formation and antibiotic resistance and both were hypermotile and hypervirulent to plants, they differed quantitatively in many phenotypes and in the expression of certain target genes. For example, while the eal mutant was more impaired in biofilm formation than the rpfF mutant (Fig. 3), the rpfF mutant was more susceptible to tobramycin (Fig. 2); the decrease in expression of hxfB, fimA, and gumJ, which contribute to biofilm formation, was greater in the eal mutant (Fig. 5B), consistent with its lower biofilm formation ability. These results suggest that gene expression in the eal mutant was somewhat different from that of rpfF. Therefore, it is possible that eal and the Rpf/DSF system could differentially contribute to biofilm formation and tobramycin resistance by regulating different sets of genes or by differentially regulating a common set of genes. RpfG and Eal might differ in enzymatic efficiencies or be present at different levels or in different locations in the cell, thus affecting discrete cyclic di-GMP pools, or might confer different total cyclic di-GMP levels in the cell, thereby expressing subsets of common or distinct target genes.
There are apparently two main behavioral traits that are controlled by cyclic di-GMP in X. fastidiosa: traits contributing to the plant-exploratory phase which are induced by high levels of cyclic di-GMP and the converse phenotypes that enable insect acquisition which are induced by low levels of cyclic di-GMP (Fig. 7A). X. fastidiosa harbors at least four genes encoding proteins predicted to be directly regulated by cyclic di-GMP levels (cyclic di-GMP effector proteins): clp, PD1311, PD1497, and PD0726. Those proteins mediate the regulation of the opposing phenotypes, and it is expected that at least one would also control the response to antibiotic stress. We can eliminate Clp from that list, since a clp mutant assayed for tobramycin susceptibility did not show increased susceptibility (data not shown) such as the eal and rpfF mutants did. Tobramycin would not have been expected to induce biofilm formation if Eal controls biofilm formation and antibiotic resistance via two different cyclic di-GMP effector proteins. An increase in expression of eal upon tobramycin exposure might also increase the expression of a particular cyclic di-GMP effector protein, establishing an alternative signaling cascade that favors traits for antibiotic resistance rather than biofilm formation. Such a scenario can explain the reduction in biofilm formation seen in wild-type cells subjected to tobramycin exposure, if the signaling cascade favoring biofilm formation would then be weakened.
Fig 7.

(A) A mechanistic model of the cyclic di-GMP regulatory network in Xylella fastidiosa: X. fastidiosa possesses 5 proteins (RpfG, CgsA, Eal, PD1671, and PD1994) implicated in cyclic di-GMP metabolism and 4 proteins (Clp, PD1313, PD1497, and PD0276) predicted to be cyclic di-GMP effectors. RpfB produces a DSF precursor (57) that is modified by RpfF to produce DSF. DSF accumulates outside the cell and, upon reaching a threshold level, interacts with RpfC, which in turn phosphorylates RpfG. High levels of cyclic di-GMP induce motility and virulence, and low levels induce biofilm formation and antibiotic resistance. (B) Hierarchical regulatory model for DSF and tobramycin signaling: tobramycin induces eal expression and Eal and CgsA expression is suppressed by RpfF, presumably through RpfG. Both the Eal and the Rpf systems contribute to cyclic di-GMP reduction and thus to an increase in antibiotic resistance and biofilm formation. However, their discrete contributions are expected to overlap only partially.
Although tobramycin did not induce biofilm formation and since both mutants with mutations in eal and rpfF, like a P. aeruginosa arr mutant (19), were attenuated in biofilm formation, it is likely that their susceptibility to subinhibitory concentrations of tobramycin is at least partially due to their impairment in biofilm formation. It is well established that cells in biofilms are more resistant to antibiotics than planktonic cells (48, 49), but the basis for this resistance is not fully understood. In fact, an ndvB (encoding a glucosyltransferase involved in the formation of cyclic glucans) mutant of P. aeruginosa that formed biofilms with a normal architecture was still sensitive to tobramycin (50), indicating that physical features of biofilms do not confer resistance but rather that the physiological state of cells in biofilms (having altered patterns of gene expression) leads to resistance. Furthermore, planktonic aggregates of P. aeruginosa, but not planktonic solitary cells, exhibited an enhanced capacity to survive otherwise lethal antibiotic exposure (51), further indicating that surface attachment is not essential for resistance. We presume that one or more of the many genes regulated by DSF signaling (52) contribute to antibiotic resistance in X. fastidiosa. Likewise, Eal presumably also affects the expression of a large number of genes, some of which are involved in antibiotic resistance, and some (but not all) are also regulated by the Rpf/DSF system. Several traits such as multidrug transporters (53) or factors that prevent the antibiotics from reaching their site of action such as sequestration of them in the periplasm with glucose polymers (50) that are distinctly controlled by Eal and/or RpfF, and are expressed in a biofilm, may contribute to resistance.
It might be expected, given the apparent role of the Rpf/DSF signaling system in resistance to antibiotics and the fact that DSF signaling is more prominent in denser cultures, that the rpfF mutant would be more susceptible than the wild-type strain to antibiotics as the cells age. However, it was surprising that the eal mutant also exhibited such a pattern. The age-dependent susceptibility of both mutants indicates that both Eal and RpfF are functional in batch cultures with a high cell density in which DSF has already accumulated. Since each could not compensate for the loss of the other, each is probably regulating different traits. The considerable resistance seen in young cultures (1 day of growth) is likely due to factors that are expressed in early stages of growth. It is noteworthy that the rpfF mutant was more susceptible to antibiotics at all ages than the eal mutant. While Eal clearly controls traits that contribute to antibiotic resistance, additional traits that are controlled by RpfF but not Eal might contribute to the higher resistance of the eal mutant.
The attachment to surfaces and thus biofilm formation is a major life strategy required for dissemination of X. fastidiosa by insects (54, 55). Once inside xylem vessels of host plants, X. fastidiosa proliferates and spreads, apparently mostly as planktonic cells, although some can attach to the plant. X. fastidiosa reaches high cell densities in only a small proportion of vessels where it forms a biofilm along the xylem wall (reviewed in reference 3). Such cells are presumably more readily acquired by sharpshooter vectors due to their higher adhesiveness, and they form a biofilm on the cuticular lining of the insect foregut (5). Induction of biofilms and of cell-cell aggregation is thus a common behavior of bacteria unrelated to antibiotic exposure, although the changes in gene expression associated with biofilm formation often lead to higher antibiotic tolerance, as discussed above. Many of the cells that experience toxic compounds in planta are thus likely to be in a biofilm or an aggregated form that is more tolerant of the toxic compounds to which they may be exposed.
Given that DSF signaling suppresses Eal expression, the innate antibiotic resistance seen in wild-type cells might be due predominantly to those traits that are under RpfF but not Eal control. As cells become more abundant with age, DSF is expected to accumulate and reduce eal expression, thus enabling RpfG to function in a cellular environment without the influence of cyclic di-GMP degradation that would occur because of external events such as antibiotic-induced expression of Eal. Nevertheless, when cells experience tobramycin, eal expression seems to be maintained at a high level despite the presence of DSF, since both DSF-regulated traits and Eal-regulated traits are required for tobramycin resistance to be achieved. DSF-mediated suppression of Eal may occur to prevent excessive depletion of cyclic di-GMP and to avoid unnecessary expression of Eal in the absence of antibiotics. It might be expected that antimicrobial factors from which X. fastidiosa might need protection occur mostly in plants that are heavily colonized by this pathogen and thus are most symptomatic. This would explain why the RpfF system influences tolerance of antibiotics (and other toxic compounds) in addition to the more specific contextual regulator Eal. A common process that would link to both antibiotic resistance and biofilm formation is the modulation of the level of cyclic di-GMP. Cyclic di-GMP levels might be different in subcellular compartments, depending on the total cellular DGC and PDE activities which, in turn, depend on the levels and specific activities of each of the GGDEF, EAL, and HD-GYP domain proteins. These parameters are apparently dynamic and interconnected. RpfF temporally regulates at least 3 of the 5 cyclic di-GMP control modules: RpfG at the protein activity level (9) and both CgsA (16) and Eal at the transcriptional level (Fig. 6A and 7). Our results strongly suggest that the cell density-dependent Rpf cell-cell communication system of X. fastidiosa is dominant over the more contextual CgsA and Eal signaling systems and that intracellular signaling mediated by cyclic di-GMP is coordinated in a way to maximize pathogen fitness. Such complex contextual coordination of motility and biofilm formation is needed to enable the sequential colonization of host plants and insect vectors while at the same time allowing appropriate responses to toxic compounds and other stresses to which the cell might be exposed during any life stage.
Supplementary Material
ACKNOWLEDGMENTS
Funding for A.A.D.S. and A.M.D.S. was provided by the State of São Paulo Research Foundation/FAPESP (Proc. N 2008/03626-0 and 2010/16409-7). M.I. was supported by Vaadia-BARD Postdoctoral Fellowship Award No. FI-427-09 from BARD, The United States-Israel Binational Agricultural Research and Development Fund. Additional funding was provided by the California Department of Food and Agriculture, Pierce's Disease and Glassy-winged Sharpshooter Program.
We thank N. Killiny for assistance in ELISAs.
Footnotes
Published ahead of print 29 March 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03834-12.
REFERENCES
- 1. Hopkins DL, Purcell AH. 2002. Xylella fastidiosa: cause of Pierce's disease of grapevine and other emergent diseases. Plant Dis. 86: 1056– 1066 [DOI] [PubMed] [Google Scholar]
- 2. Chatterjee S, Wistrom C, Lindow SE. 2008. A cell-cell signaling sensor is required for virulence and insect transmission of Xylella fastidiosa. Proc. Natl. Acad. Sci. U. S. A. 105: 2670– 2675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chatterjee S, Almeida RPP, Lindow SE. 2008. Living in two worlds: the plant and insect lifestyles of Xylella fastidiosa. Annu. Rev. Phytopathol. 46: 243– 271 [DOI] [PubMed] [Google Scholar]
- 4. Chatterjee S, Newman KL, Lindow SE. 2008. Cell-cell signaling in Xylella fastidiosa suppresses movement and xylem vessel colonization in grape. Mol. Plant Microbe Interact. 21: 1309– 1315 [DOI] [PubMed] [Google Scholar]
- 5. Killiny N, Almeida RP. 2009. Xylella fastidiosa afimbrial adhesins mediate cell transmission to plants by leafhopper vectors. Appl. Environ. Microbiol. 75: 521– 528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. D'Argenio DA, Miller SI. 2004. Cyclic di-GMP as a bacterial second messenger. Microbiology 150: 2497– 2502 [DOI] [PubMed] [Google Scholar]
- 7. Jenal U. 2004. Cyclic di-guanosine-monophosphate comes of age: a novel secondary messenger involved in modulating cell surface structures in bacteria? Curr. Opin. Microbiol. 7: 185– 191 [DOI] [PubMed] [Google Scholar]
- 8. Römling U, Gomelsky M, Galperin MY. 2005. C-di-GMP: the dawning of a novel bacterial signaling system. Mol. Microbiol. 57: 629– 639 [DOI] [PubMed] [Google Scholar]
- 9. Ryan RP, Fouhy Y, Lucey JF, Crossman LC, Spiro S, He YW, Zhnag LH, Heeb S, Cámara M, Williams P, Dow JM. 2006. Cell-cell signaling in Xanthomonas campestris involves an HD-GYP domain protein that functions in cyclic di-GMP turnover. Proc. Natl. Acad. Sci. U. S. A. 103: 6712– 6717 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10. Cotter PA, Stibitz S. 2007. c-di-GMP-mediated regulation of virulence and biofilm formation. Curr. Opin. Microbiol. 10: 17– 23 [DOI] [PubMed] [Google Scholar]
- 11. Galperin MY. 2006. Structural classification of bacterial response regulators: diversity of output domains and domain combinations. J. Bacteriol. 188: 4169– 4182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ulrich LE, Koonin EV, Zhulin IB. 2005. One-component systems dominate signal transduction in prokaryotes. Trends Microbiol. 13: 52– 56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. He YW, Ng AY, Xu M, Lin K, Wang LH, Dong YH, Zhang LH. 2007. Xanthomonas campestris cell-cell communication involves a putative nucleotide receptor protein Clp and a hierarchical signaling network. Mol. Microbiol. 64: 281– 292 [DOI] [PubMed] [Google Scholar]
- 14. Tao F, He YW, Wu DH, Swarup S, Zhang LH. 2010. The cyclic nucleotide monophosphate domain of Xanthomonas campestris global regulator Clp defines a new class of cyclic di-GMP effectors. J. Bacteriol. 192: 1020– 1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Newman KL, Almeida RPP, Purcell AH, Lindow SE. 2004. Cell-cell signaling controls Xylella fastidiosa interaction with both insects and plants. Proc. Natl. Acad. Sci. U. S. A. 101: 1737– 1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chatterjee S, Killiny N, Almeida RP, Lindow SE. 2010. Role of cyclic di-GMP in Xylella fastidiosa biofilm formation, plant virulence, and insect transmission. Mol. Plant Microbe Interact. 23: 1356– 13563 [DOI] [PubMed] [Google Scholar]
- 17. Römling U. 2012. Cyclic di-GMP, an established secondary messenger still speeding up. Environ. Microbiol. 14: 1817– 1829 [DOI] [PubMed] [Google Scholar]
- 18. Boehm A, Kaiser M, Li H, Spangler C, Kasper CA, Ackermann M, Kaever V, Sourjik V, Roth V, Jenal U. 2010. Second messenger-mediated adjustment of bacterial swimming velocity. Cell 141: 107– 116 [DOI] [PubMed] [Google Scholar]
- 19. Hoffman LR, D'Argenio DA, MacCoss MJ, Zhang Z, Jones RA, Miller SI. 2005. Aminoglycoside antibiotics induce bacterial biofilm formation. Nature 436: 1171– 1175 [DOI] [PubMed] [Google Scholar]
- 20. Jenal U, Malone J. 2006. Mechanisms of cyclic-di-GMP signaling in bacteria. Annu. Rev. Genet. 40: 385– 407 [DOI] [PubMed] [Google Scholar]
- 21. Harmsen M, Yang L, Pamp SJ, Tolker-Nielsen T. 2010. An update on Pseudomonas aeruginosa biofilm formation, tolerance, and dispersal. FEMS Immunol. Med. Microbiol. 59: 253– 268 [DOI] [PubMed] [Google Scholar]
- 22. Davis MJ, French WJ, Schaad NW. 1981. Axenic culture of the bacteria associated with phony disease of peach and plum leaf scald. Curr. Microbiol. 6: 309– 314 [Google Scholar]
- 23. Almeida RP, Mann R, Purcell AH. 2004. Xylella fastidiosa cultivation on a minimal solid defined medium. Curr. Microbiol. 48: 368– 372 [DOI] [PubMed] [Google Scholar]
- 24. Killiny N, Almeida RP. 2009. Host structural carbohydrate induces vector transmission of a bacterial plant pathogen. Proc. Natl. Acad. Sci. U. S. A. 106: 22416– 22420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RMII, Peterson KM. 1995. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166: 175– 176 [DOI] [PubMed] [Google Scholar]
- 26. Kung SH, Almeida RP. 2011. Natural competence and recombination in the plant pathogen Xylella fastidiosa. Appl. Environ. Microbiol. 77: 5278– 5284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Southern EM. 1975. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J. Mol. Biol. 98: 503– 517 [DOI] [PubMed] [Google Scholar]
- 28. Matsumoto A, Young GM, Igo M. 2009. Chromosome-based genetic complementation system for Xylella fastidiosa. Appl. Environ. Microbiol. 75: 1679– 1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rybtkea MT, Borleeb BR, Murakamib K, Irieb Y, Hentzerc M, Nielsend TE, Givskova M, Parsekb MR, Tolker-Nielsena T. 2012. Fluorescence-based reporter for gauging cyclic di-GMP levels in Pseudomonas aeruginosa. Appl. Environ. Microbiol. 78: 5060– 5069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brooun A, Liu S, Lewis K. 2000. A dose-response study of antibiotic resistance in Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 44: 640– 646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Roper MC, Greve LC, Labavitch JM, Kirkpatrick BC. 2007. Detection and visualization of an exopolysaccharide produced by Xylella fastidiosa in vitro and in planta. Appl. Environ. Microbiol. 73: 7252– 7258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ionescu M, Belkin S. 2009. Overproduction of exopolysaccharides by an Escherichia coli K-12 rpoS mutant in response to osmotic stress. Appl. Environ. Microbiol. 75: 483– 492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hill BL, Purcell AH. 1995. Multiplication and movement of Xylella fastidiosa within grapevine and four other plants. Phytopathology 85: 1368– 1372 [Google Scholar]
- 34. Meng Y, Li Y, Galvani CD, Hao G, Turner JN, Burr TJ, Hoch HC. 2005. Upstream migration of Xylella fastidiosa via pilus-driven twitching motility. J. Bacteriol. 187: 5560– 5567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Baccari C, Lindow SE. 2011. Assessment of the process of movement of Xylella fastidiosa within susceptible and resistant grape cultivars. Phytopathology 101: 77– 84 [DOI] [PubMed] [Google Scholar]
- 36. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 25: 402– 408 [DOI] [PubMed] [Google Scholar]
- 37. Fogaça AC, Zaini PA, Wulff NA, Da Silva PIP, Fázio MA, Miranda A, Daffre S, Da Silva AM. 2010. Effects of the antimicrobial peptide gomesin on the global gene expression profile, virulence and biofilm formation of Xylella fastidiosa. FEMS Microbiol. Lett. 306: 152– 159 [DOI] [PubMed] [Google Scholar]
- 38. Danese PN, Pratt LA, Kolter R. 2000. Exopolysaccharide production is required for development of Escherichia coli K-12 biofilm architecture. J. Bacteriol. 182: 3593– 3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Guilhabert MR, Kirkpatrick BC. 2005. Identification of Xylella fastidiosa antivirulence genes: hemagglutinin adhesins contribute to biofilm maturation to X. fastidiosa and colonization and attenuate virulence. Mol. Plant Microbe Interact. 18: 856– 868 [DOI] [PubMed] [Google Scholar]
- 40. De La Fuente L, Burr TJ, Hoch HC. 2007. Mutations in type I and type IV pilus biosynthetic genes affect twitching motility rates in Xylella fastidiosa. J. Bacteriol. 189: 7507– 7510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hengge R. 2009. Principles of c-di-GMP signaling in bacteria. Nat. Rev. Microbiol. 7: 263– 273 [DOI] [PubMed] [Google Scholar]
- 42. Ryan RP, Fouhy Y, Lucey JF, Jiang BL, He YQ, Feng JX, Tang JL, Dow JM. 2007. Cyclic di-GMP signaling in the virulence and environmental adaptation of Xanthomonas campestris. Mol. Microbiol. 63: 429– 442 [DOI] [PubMed] [Google Scholar]
- 43. Newell PD, Yosiko S, Hvorecny KL, Monds RD, O'Toole GA. 2011. Systematic analysis of diguanylate cyclases that promote biofilm formation by Pseudomonas fluorescens Pf0-1. J. Bacteriol. 193: 4685– 4698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Merritt JH, Ha DG, Cowles KN, Lu W, Morales DK, Rabinowitz J, Gitai Z, O'Toole GA. 2010. Specific control of Pseudomonas aeruginosa surface-associated behaviors by two c-di-GMP diguanylate cyclases. mBio 1: e00183– 10 doi:10.1128/mBio.00183-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Paul R, Weiser S, Amiot NC, Chan C, Schirmer T, Giese B, Jenal U. 2004. Cell cycle-dependent dynamic localization of a bacterial response regulator with a novel di-guanylate cyclase output domain. Genes Dev. 18: 715– 727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ryan PR, McCarthy Y, Andrade M, Farah CS, Armitage JP, Dow JM. 2010. Cell-cell signal-dependent dynamic interactions between HD-GYP and GGDEF domain proteins mediate virulence in Xanthomonas campestris. Proc. Natl. Acad. Sci. U. S. A. 107: 5989– 5994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Andrade MO, Alegria MC, Guzzo CR, Docena C, Rosa MCP, Ramos CHI, Farah CS. 2006. The HD-GYP domain of RpfG mediates a direct linkage between the Rpf quorum-sensing pathway and a subset of diguanylate cyclase proteins in the phytopathogen Xanthomonas axonopodis pv citri. Mol. Microbiol. 62: 537– 551 [DOI] [PubMed] [Google Scholar]
- 48. Nickel JR, Ruseska I, Wright JB, Costerton JW. 1985. Tobramycin resistance of Pseudomonas aeruginosa cells growing as a biofilm on urinary tract catheter. Antimicrob. Agents Chemother. 27: 619– 624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Whiteley M, Ott JR, Weaver EA, McLean RJC. 2001. Effects of community composition and growth rate on aquifer biofilm bacteria and their susceptibility to betadine disinfection. Environ. Microbiol. 3: 43– 52 [DOI] [PubMed] [Google Scholar]
- 50. Mah TF, Pitts B, Pellock B, Walker GC, Stewart PS, O'Toole GA. 2003. A genetic basis for Pseudomonas aeruginosa biofilm antibiotic resistance. Nature 426: 306– 310 [DOI] [PubMed] [Google Scholar]
- 51. Alhede M, Kragh KN, Qvortrup K, Allesen-Holm M, van Gennip M, Christensen LD, Jensen PØ, Nielsen AK, Parsek M, Wozniak D, Molin M, Tolker-Nielsen T, Høiby N, Givskov M, Bjarnsholt T. 2011. Phenotypes of non-attached Pseudomonas aeruginosa aggregates resemble surface attached biofilm. PLoS One 6: e27943 doi:10.1371/journal.pone.0027943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang N, Li JL, Lindow SE. 2012. RpfF-dependent regulon of Xylella fastidiosa. Phytopathology 102:1045– 1053 [DOI] [PubMed] [Google Scholar]
- 53. Zgurskaya HI. 2009. Multicomponent drug efflux complexes: architecture and mechanism of assembly. Future Microbiol. 4: 919– 932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Marques LLR, Ceri H, Manfio GP, Reid DM, Olson ME. 2002. Characterization of biofilm formation by Xylella fastidiosa in vitro. Plant Dis. 86: 633– 638 [DOI] [PubMed] [Google Scholar]
- 55. Shi XY, Dumenyo CK, Hernandez-Martinez R, Azad H, Cooksey DA. 2009. Characterization of regulatory pathways in Xylella fastidiosa: genes and phenotypes controlled by gacA. Appl. Environ. Microbiol. 75: 2275– 2283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hofmann K, Stoffel W. 1993. TMBASE—a database of membrane spanning protein segments. Biol. Chem. Hoppe-Seyler 374: 166 [Google Scholar]
- 57. Almeida APP, Killiny N, Newman KL, Chatterjee S, Ionescu M, Lindow SE. 2012. Contribution of rpfB to cell-cell signal synthesis, virulence, and vector transmission of Xylella fastidiosa. Mol. Plant Microbe Interact. 25:453– 462 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.