

Abstract
Observations in real-time can provide insights into the timing of injury and the mechanisms of damage in neural ischemia-reperfusion. Continuous digital imaging of morphology and cell viability was applied in a novel model of simulated ischemia-reperfusion in cultured cortical neurons, consisting of exposure to severe hypoxia combined with glucose deprivation, mild acidosis, hypercapnia, and elevated potassium, followed by return of oxygenated, glucose-containing physiological saline. Substantial acute injury resulted following 1 hour of simulated ischemia, with 36 ± 8% neurons dying within 2 hours of reperfusion. Inclusion of moderate glutamate elevation (30 μM) in the simulation of ischemia increased the acute neuronal death to 51 ± 6% at 2 hours of reperfusion. While some swelling and neuritic breakdown occurred during ischemia, particularly with inclusion of glutamate, neuronal death, as marked by loss of somatic membrane integrity, was entirely restricted to the reperfusion phase. Morphological and cytoskeletal changes suggested a predominance of necrotic death in the acute phase of reperfusion, with more complete delayed death accompanied by some apoptotic features occurring over subsequent days. Prolonged simulated ischemia, without reperfusion, did not induce significant acute neuronal death even when extended to 3 hours. We conclude that while morphological changes suggesting initiation of neuronal injury appear during severe simulated ischemia, the irreversible injury signaled by membrane breakdown is accelerated by the events of reperfusion itself.
Keywords: Cortical neurons, ischemia, reperfusion, stroke, in vitro model, necrosis, excitotoxicity, neuroprotection
Introduction
Reperfusion of the ischemic brain is associated with the sudden return of blood flow to oxygen-starved, energetically compromised tissue. It is widely held that much of the injury produced by transient ischemia results from these events of reperfusion itself, giving rise to the hope that injury can be ameliorated by neuroprotective treatments during reperfusion (reviewed by Hess, 1984). However, direct testing of the timing of injury during neuronal ischemia-reperfusion has been limited. In vivo, comparisons of infarct volumes at 24 hours after transient or permanent middle cerebral artery occlusions have provided suggestive evidence for brain reperfusion injury (Yang and Betz, 1994, Aronowski, et al., 1997), but interpretation of these results is confounded by the increased tissue edema accompanying reperfusion. Detailed observations of the timing and mechanisms of ischemia-reperfusion injury are difficult to conduct in the intact brain, and assessment of injury without at least a brief re-exposure to oxygen during brain perfusion-fixation or surgical removal is technically challenging. Considerable evidence for reperfusion-associated injury has been presented in myocardial cells (Vanden Hoek, et al., 1996, Vanden Hoek, et al., 2003, Qin, et al., 2004) and cardiac tissues (Hearse, et al., 1973, Ambrosio and Flaherty, 1992). Similar experiments documenting specific reperfusion-phase injury in neurons with careful examination of the timing of initiation of injury are lacking. Nevertheless, understanding the timing and mechanisms of initiation of injury around the time of reperfusion is important for prioritizing attempts at neuroprotection versus reperfusion-directed thrombolysis in treatment of acute stroke.
In order to achieve the temporal resolution necessary to associate the timing of signs of injury with ischemia versus reperfusion, in vitro models of ischemic injury offer distinct advantages. Ischemia can be simulated in vitro simply by oxygen and glucose deprivation (`OGD') (Goldberg, et al., 1987), with many important features of cellular injury disclosed, including the preferential vulnerability of neurons over glia, the participation of secondary excitotoxic mechanisms in injury, and the early necrotic death followed by delayed apoptotic death of neurons. However, in addition to low oxygen and lack of glucose, the ischemic brain tissue milieu includes additional features that are known to be relevant to neuronal injury, including depolarizing elevations of extracellular [K+] (Abele, et al., 1990), elevated PCO2 associated with reduced pH (Tomlinson, et al., 1993), and elevated extracellular glutamate (Schneweis, et al., 2001). We have developed a model of neuronal ischemia-reperfusion that incorporates each of these features, and allows continuous real-time observation of cell morphology and viability. Here we report that this model of simulated ischemia-reperfusion injury demonstrates that neuronal death is associated temporally with reperfusion. In addition, neuronal death is substantially prevented during prolonged ischemia without reperfusion, showing that reperfusion leads to the accelerated appearance of acute neuronal necrosis following ischemia. The results suggest that this novel model of simulated ischemia, demonstrating reperfusion-associated neuronal injury, promises to provide a useful approach to the elucidation of the mechanisms important to neuronal ischemic injury.
Methods
Primary cultures of cortical neurons
All animal procedures were approved by the University of Chicago Institutional Animal Care and Use Committee. Dissociated cultures of cortical neurons were prepared from C57BL/10 mice, embryonic day E16.5, as previously described (Li, et al., 2005). Cells were plated on poly-L-lysine-coated 15 or 25-mm coverslips at a density of 8 × 104 cells/cm2 and maintained in serum-free medium (Neurobasal A/B-27; Invitrogen Corp., Grand Island, NY) supplemented with 0.5 mM L-glutamine and 5 μM 5-fluoro-2'-deoxyuridine. Under these conditions neuronal purity is > 95% (Li, et al., 2005).
Continuous perfusion model of simulated ischemia-reperfusion
Coverslips with cultured cells were placed in a sealed flow-through chamber created by clamping together 2 coverslips separated by a stainless steel spacer ring (1.2 ml volume), as previously described (Vanden Hoek, et al., 1996, Levraut, et al., 2003). This chamber was mounted on a heated microscope stage with temperature monitored within the chamber by a thermocouple, and maintained at 37.0°C ± 0.5°C. The chamber was continuously perfused at 0.25 ml/min with saline equilibrated with O2-CO2 gas mixtures in a warmed water–jacketed column. Perfusate was conducted in tubing constructed of PharMed (Cole-Parmer Instruments, Vernon Hills, IL) or stainless steel to minimize oxygen leaks. Standard perfusion was with oxygenated, bicarbonate-buffered balanced salt solution (BSS) of composition (in mM): 104 NaCl, 18 NaHCO3, 4.0 KCl, 0.8 MgSO4, 1.0 NaH2PO4, 1.2 CaCl2, and 10 glucose, bubbled continuously with gas of composition 21%O2/5% CO2/74% N2 to produce a PO2 of ~150 torr, PCO2 of ~40 torr, and pH of 7.35. Ischemia was simulated with a solution containing (in mM): 0 glucose, 114 NaCl, 21.4 NaHCO3, 8.0 KCl, 0.8 MgSO4, 1.0 NaH2PO4, 1.2 CaCl2, and bubbled with 80% N2 and 20% CO2 for 30 min to produce conditions of severe hypoxia with PCO2 ~144 torr, and pH of 6.8. Monitoring of PO2 directly downstream from this perfusion chamber using an O2 electrode in a sealed flow-through chamber (Micro Flow-through O2 system, Lazar Research Laboratories, Los Angeles, CA) demonstrated that PO2 of < 5 torr, simulating that in the core of focal brain ischemia (Liu, et al., 2004), is attained in this system within ~10 minutes. Reperfusion was simulated by transition back to oxygenated BSS. All solutions contained 5 μM propidium iodide (Invitrogen, Grand Island, NY) to monitor cell viability.
Neurons were continuously imaged with a Nikon Eclipse TE 2000-U inverted phase/epifluorescent microscope. Prior to each experiment, a suitable microscopic field was selected and regions of interest were defined over somata of 30–60 healthy neurons in phase-contrast images. Phase-contrast and fluorescence images were acquired using a CoolSNAP ES PC-controlled camera (Photometrics, Tuscon, AZ) coupled with MetaMorph software, and were captured every 30 min during an experiment. Preselected neurons acquiring nuclear propidium fluorescence were counted at each interval as dead neurons.
Delayed survival following simulated ischemia-reperfusion injury
For assay of delayed survival following treatments, coverslips were pretreated with DNase solution, 15 μg/ml for 60 min, to eliminate nuclear `tombstones' of previously dead cells. Coverslips were removed from standard incubation conditions, rinsed in BSS, and then placed in dishes filled with standard BSS, or with ischemia solution, preequilibrated in an `ischemia chamber' containing a regulated atmosphere of 0.5% O2 and 20% CO2 at 37°C. These dishes were then kept in the standard incubator or in the ischemia chamber for 60 min, followed by transfer of coverslips back to dishes containing culture medium, in standard 20% O2 incubation conditions, for defined periods of simulated reperfusion. Survival after reperfusion was assessed using staining of living and dead neurons with fluorescein diacetate and propidium iodide, as previously described (Li, et al., 2005). Automated counts of living and dead neurons were generated using an algorithm based on CellProfiler cell image analysis software (Carpenter, et al., 2006) (available at www.cellprofiler.org). Counts from images of 3 – 4 randomly selected microscopic fields for each condition were generated and percent survival was calculated. To eliminate the contributions of neurons dying from attrition or manipulation, survival was normalized to that in parallel control coverslips treated with saline alone for 60 min (with cell survivals of 81 ± 2 at 24 hours and 71 ± 2 at 72 hours), to give relative survival.
Cell staining
For microtubule-associated protein-2 (MAP-2) immunostaining, coverslips were rapidly removed from the perfusion chamber during simulated ischemia or reperfusion and immediately submersed in 4% paraformaldehyde solution at 37°C for fixation (15 min), followed by immunostaining using MAP-2 monoclonal antibody (Chemicon; 1:200) and an Alexa-488-linked goat anti-mouse secondary antibody (Molecular Probes; 1:300), and imaging by epifluorescence microscopy. Visualization of nuclear morphology was accomplished by incubation with Hoechst 33342 dye (10μg/ml in phosphate buffered saline for 5 min), followed by epifluorescence microscopy. TUNEL staining was preformed as previously described (Li, et al., 2005), with blinded counts of stained and unstained cells or nuclei made in images of 3 fields from each condition in each independent experiment.
Data analysis
Pooled results are reported and graphed as mean values ± standard error (S.E.). Testing of significance was carried out by two-way repeated measures ANOVA testing, with time of reperfusion as the first factor and treatment condition as the second factor. ANOVA testing was followed by Student-Newman-Keuls post-hoc pairwise comparisons, with p< 0.05 considered significant (SigmaStat version 1.0, Jandel Scientific Corp.).
Results
To simulate ischemia in cortical neurons in vitro, we established a novel model of ischemia-reperfusion injury incorporating multiple features of the ischemic cellular milieu, including severe hypoxia, lack of metabolic substrate (glucose-free), elevated [K+] to 8 mM, and elevated PCO2 with resultant acidosis (pH 6.8). These conditions were applied to cortical neurons cultured for 10 days on glass coverslips and placed in a sealed flow-through chamber, allowing continuous observation by digital imaging techniques, to monitor morphology and survival of neurons during 1 hour simulated ischemia followed by 2 hours of simulated reperfusion (re-exposure to oxygen- and glucose-containing physiological saline). While neuronal morphology was monitored with phase-contrast imaging, the appearance of nuclear propidium fluorescence, indicating loss of somatic membrane integrity, was used as an unequivocal marker of cell death. Two distinct phases of cellular injury were observed (Figure 1): during ischemia itself, subtle morphological changes of cellular swelling and cytoplasmic lucency could be detected in some cells, but very few neurons underwent completion of necrosis as marked by propidium uptake. However, upon reperfusion with oxygenated, glucose-containing physiological saline buffer, somatic swelling and neuritic beading occurred more prominently, and completion of neuronal death proceeded, with the appearance of nuclear uptake of propidium in many of the neurons. At 2 hours of reperfusion after simulated ischemia, 36 ± 8 % of neurons were dead by this criterion, as compared to 5 ± 3 % dead in controls perfused continuously with physiological saline for the same duration (see Figure 1B).
Figure 1. Ischemia-reperfusion injury in cultured neurons.

A) Representative digital images (combined phase-contrast and propidium fluorescence imaging) of cortical neurons (10 days in vitro) subjected to ischemia-reperfusion. Left-hand column, cell images after 0.5 hour baseline equilibration by perfusion with physiological balanced salt solution (BSS). Middle column, images after an additional 1 hour perfusion with BSS (control), with glutamate, or with simulated ischemia, without or with glutamate (30 μM). Right-hand column, images after simulated reperfusion with oxygenated BSS for an additional 2 hours. In control experiments (`Cont', top row), neurons remained normal in morphology during the entire 3.5 hour protocol. Application of 30μM glutamate alone for 60 minutes (`Glu', second row) produced considerable cell swelling, but no significant increase in cell death within 3 hours. With application of simulated ischemia solution, without addition of glutamate (`IR', third row), only subtle morphologic changes occurred during ischemia (arrow), but a progressively increasing fraction of neurons died with reperfusion as marked by newly acquired nuclear propidium fluorescence (arrowhead). During application of simulated ischemia including glutamate (`IR/G', bottom row), dramatic cell swelling and neuritic breakdown occurred during ischemia when glutamate was included (arrow), but again cell death, marked by propidium uptake, took place only upon reperfusion (arrowhead). Scale bar in lower right panel represents 20 microns for all images. B) Cell death, defined by propidium uptake, in ischemia-reperfusion, comparing results from parallel experiments in controls, ischemia alone, and ischemia with glutamate (n = 6, 9, and 5 independent replicates respectively, with 25–53 neurons imaged per field; *significantly different from parallel control value, and †significantly different from parallel value in ischemia without glutamate condition, p< 0.05). C) Cell death in experiments comparing survival with 60 minute exposures to glutamate (30 μM) alone to simulated ischemia plus glutamate, in an independent set of replicates of the latter condition performed in parallel (n = 4 and 3 replicates, respectively, with 44–58 neurons imaged per field; *significantly different, p< 0.05).
As extracellular glutamate accumulation, with resulting excitotoxic neuronal injury, is also a well-known feature of brain ischemia (Rothman, 1983), we compared these effects of simulated ischemia alone to those of simulated ischemia with the addition of 30 μM glutamate, a value near the median of reported measured values of extracellular glutamate in ischemic human brain (Schneweis, et al., 2001). Exposing neurons to this combination produced markedly more severe cellular swelling within 30 minutes, with the widespread appearance at 1 hour of large, round, swollen cell bodies with ill-defined phase-lucent cytoplasm, and severe breakdown of neurites (Figure 1). Again, however, little cell death occurred during the 1 hour of ischemic exposure. Widespread appearance of propidium fluorescence, signaling membrane breakdown and cell death, instead occurred only during the subsequent 2 hours of reperfusion, with a significantly greater fraction of neurons dying by 2 hours (51 ± 6%) than during reperfusion after ischemia without glutamate (see Figure 1B). Exposure to glutamate alone, without accompanying ischemia, produced similar features of cytoplasmic swelling and neurite disruption, but no significant increase in cell death, in contrast to the significant death again produced by ischemia with glutamate and reperfusion, during the 3 hour period of observation (Figure 1C). Thus acute cell injury was specifically triggered by simulated ischemia, and exacerbated by accompanying glutamate excitotoxicity. However, the temporal association of ischemia-induced neuronal death with reperfusion suggested the acceleration of the necrotic destruction of neurons by reperfusion.
To further delineate the structural changes occurring in neurons during simulated ischemia-reperfusion, cells exposed to an identical protocol were removed and rapidly fixed at baseline, after 0.5 or 1 hour ischemia, or after 1 or 2 hours reperfusion, and then immunostained for the cytoskeletal protein MAP-2, a neuron-specific somato-dendritic marker (Figure 2). This more clearly demonstrated changes in neuronal dendrites associated with the damage of ischemia-reperfusion, as well as somatic swelling during ischemia and cellular breakdown during reperfusion. While some focal dendritic swelling could be seen during glutamate-free ischemia, this was substantially more severe during ischemia with glutamate, and rapidly progressed to full breakdown of dendrites to discontinuous beaded structures during reperfusion, with diminished MAP-2 immunoreactivity in cell bodies. These morphological changes during reperfusion signaled the rapid appearance of irreversible cellular injury by a necrotic mode of death.
Figure 2. Morphological changes during ischemia-reperfusion in cortical neurons marked by MAP-2 immunostaining.

Representative digital images of MAP-2 immunofluorescence in cortical neurons subjected to simulated ischemia alone (`− Glu', left column) or simulated ischemia plus 30 μM glutamate (`+ Glu', right column) at baseline after 0.5 hour equilibration (`Equi 0.5 hr'), after 0.5 or 1 hour simulated ischemia (`IS'), and following 1 or 2 hours of simulated reperfusion (`RE'). Scale bar, 20 microns for all panels. In ischemia without glutamate, subtle neuronal swelling could be seen during ischemia (arrows), as well as scattered areas of breakdown of dendrites to a beaded appearance, becoming more prominent with reperfusion (arrowheads). In ischemia with glutamate, prominent somatic swelling and dendritic beading was seen during ischemia, followed by widespread dendritic breakdown and loss of cellular MAP-2 immunoreactivity during reperfusion.
While most neurons displayed morphological signs of injury during the acute period of observation, only half or fewer died, as marked by propidium uptake with 2 hours of reperfusion. It is important to also assess the morphology and survival rate of neurons for longer periods after simulated ischemia-reperfusion injury. To do so, we applied similar exposure conditions of simulated ischemia to neurons in sterile culture dishes, equilibrated with an atmosphere of regulated hypoxia and hypercapnia. In this model, following ischemia alone or with glutamate, survival at 2 hours reperfusion was modestly decreased relative to control conditions, with further death occurring over subsequent hours, so that survival at 24 or 72 hours was significantly less than that at 2 hours reperfusion (Figure 3B). At all time points, the addition of glutamate to simulated ischemia produced more severe injury, producing virtually complete neuronal death by 24 to 72 hours of reperfusion. Thus, both the protracted time course of neuronal death after reperfusion, and the significant additional injury triggered by glutamate during ischemia, were demonstrated.
Figure 3. Delayed injury to cortical neurons after simulated ischemia.

A) Hoechst staining for nuclear morphology showed pyknotic changes of most nuclei at 2 or 24 hours following ischemia with or without glutamate (arrowheads, examples). A fraction of nuclei showed severe swelling instead of condensation, especially after ischemia with glutamate (arrow). Only rare nuclei showed nuclear fragmentation characteristic of apoptosis (open arrow). B) Relative survival, determined by live/dead counts and normalized to parallel controls, showed significant declines in survival from 2 to 24 and 72 hours following ischemia-reperfusion (IR). At each time point, survival in reperfusion following ischemia plus glutamate (IR/G) was significantly less than that following ischemia alone (n = 3 independent replicates with 134–206 cells counted per condition; two-way ANOVA, effects of time and treatment both significant at p<0.05; post-hoc pairwise comparisons: *different than corresponding 2 hour survival, †different than 24 hour survival). C) At 2 hours of reperfusion, TUNEL staining revealed frequent TUNEL-positive pyknotic nuclei both in cells with apparently intact surrounding membranes and without evident surrounding cell membranes; few TUNEL-positive nuclei had a fragmented appearance. By 24 hours after IR/G, few intact cells could be seen but many TUNEL-positive as well as TUNEL-negative bare pyknotic nuclei remained. D) Quantitation of percentages of neurons staining by the TUNEL method. As this staining method does not provide a sensitive determination of preservation of intact somatic membrane, nuclei without evident surrounding cells were included in the counts. Proportions of cells staining for DNA strand breaks were increased at 2 and 24 hours following simulated ischemia alone and with glutamate (n = 3 independent replicates with 167–223 cells counted per condition; two-way ANOVA, effects of time and treatment both significant at p<0.05; post-hoc pairwise comparisons: *different than corresponding control; †different than ischemia alone condition).
The acute death within 2 hours of reperfusion bore signs of passive necrosis with cytoplasmic swelling and lucency, and nuclear propidium uptake, suggesting osmotic overload and membrane dissolution. However, various forms of programmed cell death are also known to contribute to ischemic neuronal damage (Bredesen, 2007). To more fully characterize the neuronal death following simulated ischemia in this model, we assessed nuclear morphological changes with DNA staining with the membrane permeable dye Hoechst 33342, and DNA strand breakage with terminal deoxynucleotidyl transferase-mediated biotinylated dUTP nick-end labeling (TUNEL) staining. Hoechst staining of neurons (Figure 3A) following 2 hours of reperfusion demonstrated severe swelling of nuclei in occasional neurons, and others with early pyknotic condensation of nuclear DNA. By 24 hours, most ischemia-exposed neurons showed clear signs of nuclear pyknosis. At either time point, only a minority showed nuclear fragmentation consistent with classical descriptions of apoptosis. Similarly, TUNEL staining revealed widespread induction of DNA strand breakage following ischemia, with most of the positive nuclei again showing a condensed pyknotic appearance at 2 hours or 24 hours. Both nuclei lacking any evident surrounding cytoplasmic membrane as well as nuclei contained within apparently intact cell bodies could be TUNEL-positive at 2 hours, and many bare nuclei remaining from necrotic cells were TUNEL-negative, emphasizing that TUNEL staining should not be considered a simple marker of living or dead cells. After ischemia with glutamate, a fraction of TUNEL-staining nuclei had a markedly swollen appearance, and most lacked evidence of an intact surrounding cell body, with only a rare minority showing classical nuclear fragmentation or karyorrhexis (Figure 3C). Quantitation of TUNEL staining, as a fraction of all cellular objects (including nuclei without evidence of surrounding cell membranes), showed significant increases at 2 or 24 hours following ischemia as compared with controls (Figure 3D). Thus ischemia-reperfusion injury produced substantial DNA strand breakage in the neurons, though sometimes this occurred in cells apparently undergoing rapid necrotic death as well as in some cells possibly dying by an apoptotic pathway.
In the continuous perfusion model of ischemia, the temporal association of the onset of acute loss of neuronal viability with reperfusion suggested the possibility of reperfusion-triggered injury. However, the timing might be coincidental. To explore whether reperfusion specifically accelerated neuronal injury, we compared the results described above in Figure 1 to those occurring without reperfusion, prolonging ischemia for a full 3 hour period, alone and in combination with glutamate (Figure 4). During prolonged ischemia alone, only minimal changes of neuronal swelling and lucency appeared in a fraction of neurons. With the inclusion of added glutamate with ischemia, widespread cellular morphological changes of swelling and neuritic beading were seen, suggesting the initiation of significant neuronal injury, despite maintenance of membrane integrity, as determined by exclusion of propidium. Nevertheless, through durations of continuous ischemia of up to 3 hours, either without or with glutamate, there was a remarkable prevention of cellular death (3 ± 1% without glutamate, or 7 ± 3% with glutamate). The contrast of these results to the rapid death of neurons occurring with reperfusion over the same time frame (Figure 1) suggests that reperfusion itself is necessary to produce the rapid development of loss of membrane integrity, marking the demise of neurons.
Figure 4. Survival of cortical neurons after prolonged simulated ischemia.
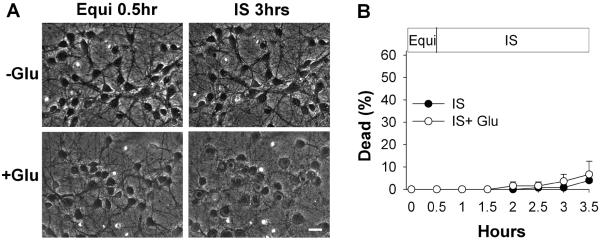
A) Representative digital images (combined phase-contrast and fluorescence) of cortical neurons (10 days in vitro) at baseline after equilibration (`Equi') and after continuous simulated ischemia without or with glutamate for 3 hours. Scale bar, 20 microns for all panels. B) Cell death during continuous simulated ischemia, without (filled circles) or with 30 μM glutamate (open circles); n = 4 replicates, with 21–36 neurons imaged per field (error bars are hidden by symbols).
Discussion
While the acute sensitivity of neurons to transient ischemia is well known, specific evidence linking initiation of the injury to the events of reperfusion itself is scant. Here, in an in vitro model of reperfusion following severe ischemia, we provide direct evidence for the acceleration of neuronal injury by reperfusion.
Numerous previous studies of cultured neurons or brain slices have simulated ischemia using a simple OGD model (Goldberg, et al., 1987, Gwag, et al., 1995, Perez-Velazquez, et al., 1997, Grabb and Choi, 1999). The present studies characterize a novel model of in vitro ischemia, incorporating multiple relevant features of ischemic brain tissue including oxygen and glucose deprivation combined with hypercapnia, acidosis, partially depolarizing concentrations of K+, and elevated glutamate. This insult is predictably more severe than simple OGD, in that several of the additional factors, including acidosis (Kraig, et al., 1987), elevated K+ concentration (Abele, et al., 1990), and glutamate (Choi, et al., 1987) can themselves produce lethal injury to neurons. In particular, the addition of glutamate produces much more striking morphological injury, and nearly doubles the acute death occurring during 2 hours reperfusion, as compared to simulated ischemia alone. Nevertheless, the net injury from simulated ischemia is not likely to be simply the sum of its parts, as interactions between the individual features may either worsen or lessen the net injury. For example, while the effects of adding glutamate to simulated ischemia include increased energy demand exacerbating cellular energy failure (Sibson, et al., 1998), and formation of damaging reactive oxygen species (Dugan, et al., 1995), the moderate acidosis used here is known to lessen the effects of glutamate excitotoxicity by modulating N-methyl-D-aspartate receptor activity (Tombaugh and Sapolsky, 1990, Giffard, et al., 1990). On the other hand, even in severe hypoxia, the presence of trace amounts of molecular O2 can be expected to hasten excitotoxic death through oxygen radical production (Dubinsky, et al., 1995), as compared to protective effects of true functional anoxia used in some models of OGD (Goldberg and Choi, 1993). The combined effects of these several elements in addition to oxygen and glucose deprivation produce a model simulating the milieu experienced by neurons during severe ischemia. The severity of this combined insult may explain why the injury is acutely lethal to approximately half of the neurons within 2 hours of reperfusion, and to greater than 90% within 24 hours, contrasting with the relative resistance of cortical neurons to acute death following OGD in some examples, with 1 hour OGD producing ~ 20% death at 2 hours reperfusion (Lim, et al., 2006) and 2 hours of OGD producing death of only 30% of neurons at 24 hours (Furuichi, et al., 2005). In this regard, the present model of simulated ischemia, including moderate glutamate, may better approximate the severity of cortical transient ischemia in vivo, which produces substantial neuronal death after as short as 20 min ischemia (Heiss and Rosner, 1983, Sicard, et al., 2006). The severity of the injury in the present model may also limit the potential reversibility of damage at the time of reperfusion. Nevertheless, the acute manifestation of injury by breakdown of somatic membrane integrity, measured here as propidium uptake, can be prevented at least for a time by extending ischemia, avoiding reperfusion.
The present model produces neuronal injury most consistent with passive necrosis, featuring cellular swelling and membrane rupture, with only a minority of neurons exhibiting typical morphological changes of apoptosis. Numerous dying cells showed evidence of nuclear strand breakage by TUNEL staining. While often considered a marker of apoptotic death, TUNEL-detected strand breakage is well known to also occur in necrotic death induced by excitotoxic or ischemic injury (Bonfoco, et al., 1995, Didier, et al., 1996, de Torres, et al., 1997). Our results, showing rapid appearance of TUNEL staining within 2 hours of reperfusion in cells bearing morphological features suggestive of passive necrosis, support this conclusion that TUNEL staining is not inconsistent with necrosis. Rather than the nuclear fragmentation characteristic for apoptosis, most of the dying neurons in these experiments displayed nuclear pyknosis (Figure 3). Such nuclear pyknosis can also be consistent with either passive necrosis or apoptosis (de Torres, et al., 1997). Other non-apoptotic forms of programmed cell death may also contribute to ischemic neuronal death (Bredesen, 2007). The timing and morphological features of cell death in this model suggest that one effect of the addition of glutamate to simulated ischemia in this model may in fact be to push more neurons into a rapid necrotic mode of death from a more protracted cell death program.
The neuronal injury, while initiated during ischemia based on the onset of morphological changes, is clearly accelerated by reperfusion, suggesting the involvement of pathophysiological mechanisms of reperfusion in production of the observable features of necrosis. These reperfusion effects likely include activation of oxidant production by cytoplasmic and mitochondrial O2-dependent enzymes, and resulting destruction of cellular proteins and membranes (Hall and Braughler, 1989, Fiskum, et al., 1999, Chan, 2001, Abramov, et al., 2007). While avoiding reperfusion postpones necrotic neuronal death (Figure 3), without restoration of substrate for energy production the neurons will go on to die over subsequent hours (Goldberg and Choi, 1993), so that eventual reoxygenation must occur for survival. Even if the acute necrotic changes during reperfusion can somehow be averted, apoptotic cell death driven by oxidant-related mitochondrial-dependent pathways are likely to supervene (Chan, 2005).
Such oxidant-mediated reperfusion injury, with initiation of mitochondrial death pathways, has been characterized in cardiomyocytes using a comparable perfusion model of simulated ischemia (Vanden Hoek, et al., 1996, Vanden Hoek, et al., 2003), revealing important features shared with the current results. In particular, in both neurons and cardiomyocytes, shorter periods of ischemia with reperfusion produce more immediate cell death than does prolonged ischemia alone. The question of clinical relevance is whether, prior to the damage accompanying reperfusion, the neuronal injury induced by ischemia may still be reversible, by therapies such as those proven effective against cardiomyocyte injury in vitro (Vanden Hoek, et al., 2003, Shao, et al., 2006), or against heart and brain injury in a mouse cardiac arrest model in vivo (Abella, et al., 2004). If the events of reperfusion itself cause cellular destruction, and effective measures against these can be found, then such treatments may become a priority during resuscitation of cardiac arrest patients, who often succumb to post-reperfusion heart and brain injury despite initial return of spontaneous circulation, or during treatment of acute stroke by induction of arterial reperfusion with fibrinolytic or mechanical thrombolysis.
Acknowledgements
This work was supported by a Grant-in-Aid from the American Heart Association (J.R.B.), by NIH grants NS036260 (J.R.B.) and HL-068951 (T.L.V.), and by a gift from the Valenti Family Foundation. We thank Eleanor Mogongwa for assistance in performing neuronal cultures.
References
- Abele AE, Scholz KP, Scholz WK, Miller RJ. Excitotoxicity induced by enhanced excitatory neurotransmission in cultured hippocampal pyramidal neurons. Neuron. 1990;4:413–419. doi: 10.1016/0896-6273(90)90053-i. [DOI] [PubMed] [Google Scholar]
- Abella BS, Zhao D, Alvarado J, Hamann K, Vanden Hoek TL, Becker LB. Intra-arrest cooling improves outcomes in a murine cardiac arrest model. Circulation. 2004;109:2786–2791. doi: 10.1161/01.CIR.0000131940.19833.85. [DOI] [PubMed] [Google Scholar]
- Abramov AY, Scorziello A, Duchen MR. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J. Neurosc. 2007;27:1129–1138. doi: 10.1523/JNEUROSCI.4468-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosio G, Flaherty JT. Effects of the superoxide radical scavenger superoxide dismutase, and of the hydroxyl radical scavenger mannitol, on reperfusion injury in isolated rabbit hearts. Cardiovasc. Drugs Ther. 1992;6:623–632. doi: 10.1007/BF00052564. [DOI] [PubMed] [Google Scholar]
- Aronowski J, Strong R, Grotta J. Reperfusion injury: Demonstration of brain damage produced by reperfusion after transient focal ischemia in rats. J. Cereb. Blood Flow Metab. 1997;17:1048–1056. doi: 10.1097/00004647-199710000-00006. [DOI] [PubMed] [Google Scholar]
- Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Apoptosis and necrosis: Two distinct events induced, respectively, by mild and intense insults with N-Methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc. Natl. Acad. Sci. USA. 1995;92:7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredesen DE. Toward a mechanistic taxonomy for cell death programs. Stroke. 2007;38:652–660. doi: 10.1161/01.STR.0000257802.82826.a7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biology. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J. Cereb. Blood Flow Metab. 2001;21:2–14. doi: 10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- Chan PH. Mitochondrial dysfunction and oxidative stress as determinants of cell death/survival in stroke. Annals New York Academy of Sciences. 2005;1042:203–209. doi: 10.1196/annals.1338.022. [DOI] [PubMed] [Google Scholar]
- Choi DW, Maulucci-Gedde M, Kriegstein AR. Glutamate neurotoxicity in cortical cell culture. J. Neurosci. 1987;7:357–368. doi: 10.1523/JNEUROSCI.07-02-00357.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Torres C, Munell F, Ferrer I, Reventós J, Macaya A. Identification of necrotic cell death by the TUNEL assay in the hypoxic-ischemic neonatal rat brain. Neurosci. Lett. 1997;230:1–4. doi: 10.1016/s0304-3940(97)00445-x. [DOI] [PubMed] [Google Scholar]
- Didier M, Bursztajn S, Adamec E, Passani L, Nixon RA, Coyle JT, Wei JY, Berman SA. DNA strand breaks induced by sustained glutamate excitotoxicity in primary neuronal cultures. J. Neurosci. 1996;16:2238–2250. doi: 10.1523/JNEUROSCI.16-07-02238.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubinsky J, Kristal B, Elizondo-Fournier M. An obligate role for oxygen in the early stages of glutamate-induced, delayed neuronal death. J. Neurosci. 1995;15:7071–7078. doi: 10.1523/JNEUROSCI.15-11-07071.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugan LL, Sensi SL, Canzoniero LMT, Handran SD, Rothman SM, Lin T-S, Goldberg MP, Choi DW. Mitochondrial production of reactive oxygen species in cortical neurons following exposure to N-methyl-D-aspartate. J. Neurosci. 1995;15:6377–6388. doi: 10.1523/JNEUROSCI.15-10-06377.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiskum G, Murphy AN, Beal MF. Mitochondria in neurodegeneration: Acute ischemia and chronic neurodegenerative diseases. J. Cereb. Blood Flow Metab. 1999;19:351–369. doi: 10.1097/00004647-199904000-00001. [DOI] [PubMed] [Google Scholar]
- Furuichi T, Liu W, Shi H, Miyake M, Liu KJ. Generation of hydrogen peroxide during brief oxygen-glucose deprivation induces preconditioning neuronal protection in primary cultured neurons. J. Neurosci. Res. 2005;79:816–824. doi: 10.1002/jnr.20402. [DOI] [PubMed] [Google Scholar]
- Giffard RG, Monyer H, Christine CW, Choi DW. Acidosis reduces NMDA receptor activation, glutamate neurotoxicity, and oxygen-glucose deprivation neuronal injury in cortical cultures. Brain Res. 1990;506:339–342. doi: 10.1016/0006-8993(90)91276-m. [DOI] [PubMed] [Google Scholar]
- Goldberg MP, Choi DW. Combined oxygen and glucose deprivation in cortical cell culture: Calcium-dependent and calcium-independent mechanisms of neuronal injury. J. Neurosci. 1993;13:3510–3524. doi: 10.1523/JNEUROSCI.13-08-03510.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MP, Weiss JW, Pham PC, Choi DW. N-methyl-D-aspartate receptors mediate hypoxic neuronal injury in cortical cultures. J. Pharmacol. Exp. Ther. 1987;243:784–791. [PubMed] [Google Scholar]
- Grabb MC, Choi DW. Ischemic tolerance in murine cortical cell culture: Critical role for NMDA receptors. J. Neurosci. 1999;19:1657–1662. doi: 10.1523/JNEUROSCI.19-05-01657.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwag BJ, Lobner D, Koh JY, Wie MB, Choi DW. Blockade of glutamate receptors unmasks neuronal apoptosis after oxygen-glucose deprivation in vitro. Neurosci. 1995;68:615–619. doi: 10.1016/0306-4522(95)00232-8. [DOI] [PubMed] [Google Scholar]
- Hall ED, Braughler JM. Central nervous system trauma and stroke. II. Physiological and pharmacological evidence for involvement of oxygen radicals and lipid peroxidation. Free Radic. Biol. Med. 1989;6:303–313. doi: 10.1016/0891-5849(89)90057-9. [DOI] [PubMed] [Google Scholar]
- Hearse DJ, Humphrey SM, Chain EB. Abrupt reoxygenation of the anoxic potassium-arrested perfused rat heart: A study of myocardial enzyme release. J. Mol. Cell. Cardiol. 1973;5:395–407. doi: 10.1016/0022-2828(73)90030-8. [DOI] [PubMed] [Google Scholar]
- Heiss W-D, Rosner G. Functional recovery of cortical neurons as related to degree and duration of ischemia. Ann. Neurol. 1983;14:294–301. doi: 10.1002/ana.410140307. [DOI] [PubMed] [Google Scholar]
- Hess ML, Manson NH. Molecular oxygen: Friend and foe. The role of the oxygen free radical system in the calcium paradox, the oxygen paradox, and ischemia/reperfusion injury. J. Mol. Cell. Cardiol. 1984;16:969–985. doi: 10.1016/s0022-2828(84)80011-5. [DOI] [PubMed] [Google Scholar]
- Kraig RP, Petito CK, Plum F, Pulsinelli WA. Hydrogen ions kill brain at concentrations reached in ischemia. J. Cereb. Blood Flow Metab. 1987;7:379–386. doi: 10.1038/jcbfm.1987.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levraut J, Iwase H, Shao Z-H, Vanden Hoek T, Schumacker P. Cell death during ischemia: relationship to mitochondrial depolarization and ROS generation. Am. J. Physio..Heart Circ. Physiol. 2003;284:H549–H558. doi: 10.1152/ajpheart.00708.2002. [DOI] [PubMed] [Google Scholar]
- Li D, Marks JD, Schumacker PT, Young RM, Brorson JR. Physiological hypoxia promotes survival of cultured cortical neurons. Eur. J. Neurosci. 2005;22:1319–1326. doi: 10.1111/j.1460-9568.2005.04335.x. [DOI] [PubMed] [Google Scholar]
- Lim JH, Lee J-C, Lee YH, Choi IY, Oh Y-K, Kim H-S, Park J-S, Kim W-K. Simvastatin prevents oxygen and glucose deprivation/reoxygenation-induced death of cortical neurons by reducing the production and toxicity of 4-hydroxy-2E-nonenal. J. Neurochem. 2006;97:140–150. doi: 10.1111/j.1471-4159.2006.03715.x. [DOI] [PubMed] [Google Scholar]
- Liu S, Shi H, Liu W, Furuichi T, Timmins GS, Liu KJ. Interstitial pO2 in ischemic penumbra and core are differentially affected following transient focal cerebral ischemia in rats. J. Cereb. Blood Flow Metab. 2004;24:343–349. doi: 10.1097/01.WCB.0000110047.43905.01. [DOI] [PubMed] [Google Scholar]
- Perez-Velazquez J, Frantseva M, Carlen P. In vitro ischemia promotes glutamate-mediated free radical generation and intracellular calcium accumulation in hippocampal pyramidal neurons. J. Neurosci. 1997;17:9085–9094. doi: 10.1523/JNEUROSCI.17-23-09085.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y, Vanden Hoek TL, Wojcik K, Anderson T, Li C-Q, Shao Z-H, Becker LB, Hamann KJ. Caspase-dependent cytochrome c release and cell death in chick cardiomyocytes after simulate ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2004;286:H2280–H2286. doi: 10.1152/ajpheart.01063.2003. [DOI] [PubMed] [Google Scholar]
- Rothman SM. Synaptic activity mediates death of hypoxic neurons. Science. 1983;220:536–537. doi: 10.1126/science.6836300. [DOI] [PubMed] [Google Scholar]
- Schneweis S, Grond M, Staub F, Brinker G, Neveling M, Dohmen C, Graf R, Heiss W-D. Predictive value of neurochemical monitoring in large middle cerebral artery infarction. Stroke. 2001;32:1863–1867. doi: 10.1161/01.str.32.8.1863. [DOI] [PubMed] [Google Scholar]
- Shao Z-H, Chang W-T, Chan KC, Wojcik KR, Hsu C-W, Li C-Q, Li J, Anderson T, Qin Y, Becker LB, Hamann KJ, Vanden Hoek TL. Hypothermia-induced cardioprotection using extended ischemia and early reperfusion cooling. Am. J. Physiol. Heart Circ. Physiol. 2006 doi: 10.1152/ajpheart.01312.2005. [DOI] [PubMed] [Google Scholar]
- Sibson NR, Dhankhar A, Mason GF, Rothman DL, Behar KL, Shulman RG. Stoichiometric coupling of brain glucose metabolism and glutamatergic neuronal activity. Proc. Natl. Acad. Sci. USA. 1998;95:316–321. doi: 10.1073/pnas.95.1.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicard KM, Henniger N, Fisher M, Duong TQ, Ferris CF. Long-term changes of functional MRI-based brain function, behavioral status, and histopathology after transient focal cerebral ischemia in rats. Stroke. 2006;37:2593–2600. doi: 10.1161/01.STR.0000239667.15532.c1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tombaugh GC, Sapolsky RM. Mechanistic distinctions between excitotoxic and acidotic hippocampal damage in an in vitro model of ischemia. J. Cereb. Blood Flow Metab. 1990;10:527–535. doi: 10.1038/jcbfm.1990.94. [DOI] [PubMed] [Google Scholar]
- Tomlinson FH, Anderson RE, Meyer FB. Brain pHi, cerebral blood flow, and NADH fluorescence during severe incomplete global ischemia in rabbits. Stroke. 1993;24:435–443. doi: 10.1161/01.str.24.3.435. [DOI] [PubMed] [Google Scholar]
- Vanden Hoek T, Shao Z, Li C, Zak R, Schumacker P, Becker L. Reperfusion injury in cardiac myocytes after simulated ischemia. Am. J. Physiol. 1996;270:H1334–H1341. doi: 10.1152/ajpheart.1996.270.4.H1334. [DOI] [PubMed] [Google Scholar]
- Vanden Hoek TL, Qin Y, Wojcik K, Li C-Q, Shao Z-H, Anderson T, Becker LB, Hamann KJ. Reperfusion, not simulated ischemia, initiates intrinsic apoptosis injury in chick cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2003;284:H141–H150. doi: 10.1152/ajpheart.00132.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G-Y, Betz AL. Reperfusion-induced injury to the blood-brain barrier after middle cerebral artery occlusion in rats. Stroke. 1994;25:1658–1664. doi: 10.1161/01.str.25.8.1658. [DOI] [PubMed] [Google Scholar]