

Abstract
Computational predictions have become indispensable for evaluating the disease-related impact of nonsynonymous single-nucleotide variants discovered in exome sequencing. Many such methods have their roots in molecular evolution, as they use information derived from multiple sequence alignments. We show that the performance of current methods (e.g., PolyPhen-2 and SIFT) is improved significantly by optimizing their statistical models on evolutionarily balanced training data, where equal numbers of positive and negative controls within each evolutionary conservation class are used. Evolutionary balancing significantly reduces the false-positive rates for variants observed at highly conserved sites and false-negative rates for variants observed at fast evolving sites. Use of these improved methods enables more accurate forecasting when concordant diagnosis from multiple methods is regarded as a more reliable indicator of the prediction. Applied to a large exome variation data set, we find that the current methods produce concordant predictions for less than half of the population variants. These advances are implemented in a web resource for use in practical applications (www.mypeg.info, last accessed March 13, 2013).
Keywords: evolutionary medicine, nonsynonymous single nucleotide variant, computational prediction
Powered by revolutionary sequencing technologies, an overwhelmingly large number of nonsynonymous single-nucleotide variants (nsSNVs) are being discovered in personal exomes and in the human population surveys. An assessment of the impact of these variants on human health and disease has now become a high priority. However, the lack of high-throughput assays to interrogate these nsSNVs in the laboratory has pushed computational predictions to the practical frontiers. A large number of methods have been developed in the past decade for predicting function-impacting nsSNVs, which are now routinely used by personal and population genomics researchers to help prioritize variants for further investigation (Zhu et al. 2008; Kumar et al. 2009; International HapMap Consortium 2010; Ng et al. 2010; Tennessen et al. 2012).
The most widely applied methods for computational diagnosis of nsSNVs of unknown health significance have based their predictions explicitly or implicitly on molecular evolutionary patterns, reviewed in Karchin (2009), Kumar et al. (2011), and Sunyaev (2012). They primarily use information derived from multiple sequence alignments to identify disease-related (non-neutral) variants. In many cases, a benchmark data set that includes positive controls (known disease-associated nsSNVs) and negative controls (neutral population nsSNVs) is used to build (train) a statistical model that produces an impact score for each variant. Then, a threshold impact score that provides optimal specificity and sensitivity of diagnosis is determined (e.g., Ng and Henikoff 2001; Adzhubei 2010; Kumar et al. 2012).
Despite many similarities and the use of the same training data set, different methods frequently produce contrasting diagnoses (e.g., Chun and Fay 2009; Karchin 2009). Therefore, many researchers now take a consensus approach, such that the concordance of diagnosis from multiple methods is considered more reliable (Zhu et al. 2008; International HapMap Consortium 2010; Tennessen et al. 2012). In addition, new hybrid approaches have been proposed that combine results from multiple methods statistically and produce a final diagnosis (Gonzalez-Perez and Lopez-Bigas 2011; Lopes et al. 2012; Olatubosun et al. 2012). An implicit requirement for the success of consensus and hybrid approaches is that individual methods are not biased in the same way, which would strengthen statistical signals and produce more reliable results. It has become clear that this implicit requirement is not fulfilled by some of the most widely used methods (Kumar et al. 2012). For example, PolyPhen-2 and SIFT individually, their consensus, and a hybrid approach (Condel) using results from these two methods show high false-positive rates (FPR; up to 89%) for nsSNVs at ultraconserved sites, which are positions that have not permitted amino acid change among vertebrates, that is, amino acid substitution rate per billion years, r, is close to 0 (Kumar et al. 2012). In addition, the false-negative rates (FNR) of these methods are high (up to 65%) for nsSNVs at less-conserved sites, where r > 1 amino acid substitutions per site per billion years (Kumar et al. 2012). We hypothesized that these problems result from evolutionary imbalance of the training and testing data used in PolyPhen-2, SIFT, and derived methods. If true, this would provide a solution to advance these two widely used methods, which is also necessary for using multimethod concordance approaches that are often used to produce more reliable inferences.
Distributions of Impact Scores
To test the above hypothesis, we first examined the distributions of PolyPhen-2 scores using the HumVar (Adzhubei 2010) benchmark training and testing nsSNVs. Cumulative distributions of PolyPhen-2 impact scores of negative controls (neutral nsSNVs) at ultraconserved sites revealed clear biases, as they were concentrated at the high end (non-neutral) of the distribution (fig. 1a). Consequently, the use of the same default threshold (0.49) for all variants (shown by a vertical line), irrespective of the evolutionary conservation of their positions, produced high FPR at ultraconserved sites (89%, shaded area in fig. 1a). In contrast, FPRs were much lower for nsSNVs at well- and less-conserved sites, because the score distributions of neutral nsSNVs were concentrated toward the low end (neutral) of the distribution. Non-neutral variants also showed large differences across evolutionary classes (fig. 1b), which resulted in high FNR at fast evolving sites when the default threshold is used. Distributions of SIFT scores for neutral and non-neutral nsSNVs also showed similar trends comparable to PolyPhen-2 scores (fig. 1c and d), resulting in high FPRs at ultraconserved sites and FNRs at less-conserved sites.
Fig. 1.

Cumulative distributions of impact scores (S) for population polymorphisms (neutral nsSNVs) and disease-associated (non-neutral) nsSNVs. Distributions are shown for PolyPhen-2 (a and b), SIFT (c and d), and Condel (e and f) for nsSNVs found at ultraconserved sites (top line), well-conserved sites (middle line), and less-conserved sites (bottom line). Neutral nsSNVs are plotted in panels a, c, and e. Non-neutral nsSNVs are plotted in panels b, d, and f. Vertical lines mark the original threshold scores to designate non-neutral alleles: 0.49 for PolyPhen-2, 0.05 for SIFT, and 0.47 for Condel. A solid line marks neutral diagnosis and a broken line marks non-neutral diagnosis (left and right of the threshold scores, respectively). Shaded areas show incorrect predictions (false positives in panels a, c, and e and false negatives in panels b, d, and f).
We then examined the composition of the HumVar benchmark nsSNVs. The positive controls outnumbered negative controls by 10 to 1 at ultraconserved sites and negative controls outnumbered positive controls by 6 to 1 at less-conserved sites. Therefore, there exists an imbalance in the training data sets at ultraconserved and at less-conserved sites, even though the full HumVar data set has similar numbers of positive and negative controls over all sites when one disregards evolutionary conservation. This disparity leads to significant bias in the obtained models, which mirrored the high FPR at ultraconserved sites and high FNR at less-conserved sites. Overall, these patterns explain why PolyPhen-2 and SIFT, as well as a hybrid tool (Gonzalez-Perez and Lopez-Bigas 2011) that employs their impact scores, showed inconsistent performance for nsSNVs in different evolutionary categories (fig. 1e and f). Our finding is consistent with the expectation that the use of unequal numbers of positive and negative controls in the training data set (i.e., imbalanced training) may significantly bias the performance of statistical prediction methods in general (Valliant et al. 2000).
At the same time, we found that the impact scores for negative and positive controls produced by the statistical model trained using the whole data set show good differentiation within each evolutionary class (compare fig. 1a with fig. 1b for PolyPhen-2, and fig.1c with fig. 1d for SIFT). These patterns suggested that impact scores from PolyPhen-2 and SIFT have the power to diagnose function-impacting nsSNVs, as long as the thresholds are determined separately for each class by using balanced sampling. Therefore, we generated separate thresholds for ultra-, well-, and less- conserved site classes, such that the diagnosis accuracy was improved in a 10-fold cross-validation, as explained below.
New Balanced Approaches
We took 21,119 neutral nsSNVs and 22,196 non-neutral nsSNVs in the HumVar collection as the benchmark for training and testing, as it has been used by authors of PolyPhen-2 and SIFT previously. Precomputed impact scores by PolyPhen-2 and SIFT were retrieved from dbNSFP (Liu et al. 2011). We generated balanced training and testing data to select threshold impact score for PolyPhen-2 and SIFT (called balanced PolyPhen-2 and balanced SIFT) and then estimated the performance of the new approaches via 10-fold cross-validation. In this procedure, equal numbers of neutral and non-neutral nsSNVs were randomly sampled from within each evolutionary class that was then partitioned into two disjoint data sets for training and testing purposes. The sample size of training data and testing data was determined as 90% and 10% of the number of nsSNVs in the underrepresented class, respectively. For example, the ultraconserved class contained 1,101 negative controls and 10,559 positive controls, from which 991 randomly selected nsSNVs from each control group were included in the training data and 110 samples from each control group in the testing data. With an interval of 0.01, we generated a series of threshold values and measured the corresponding performances of the PolyPhen-2 and SIFT in the training data (see Materials and Methods). The optimal value was selected as the score that maximizes the accuracy of diagnosis. This process was repeated 1,000 times, and the average value of the threshold scores was used as the final value of threshold impact score.
After evolutionarily balanced training, the threshold impact scores for PolyPhen-2 were quite different from each other for diagnosing nsSNVs at ultra-, well-, and less-conserved sites (0.98, 0.61, and 0.03, respectively) when compared with the score of 0.49 used by original PolyPhen-2. For SIFT, the newly determined thresholds were 0.01, 0.05, and 0.19 for ultra, well, and less-conserved sites, respectively, when compared with the default score of 0.05. Projected onto the receiver operating characteristic curves (ROC), the new thresholds lead to better performance, as the original thresholds would clearly lead to higher FPRs at ultraconserved and lower true-positive rates (TPRs) at less-conserved sites (fig. 2a and b). By applying these new thresholds, the FPR at ultraconserved sites and FNR at less-conserved sites were reduced by as much as 30% (table 1). We retrieved the population allele frequency of HumVar nsSNVs from a 5,400-exome data set (Tennessen et al. 2012) and found consistent improvements across the spectrum of rare (<0.1%) to common (>5%) alleles (fig. 2c and d). Also, the performance of balanced PolyPhen-2 and balanced SIFT became more consistent across conservation classes and similar to another method (EvoD [Kumar et al. 2012]) that already uses balanced training and testing (fig. 3). We also examined the performance of balanced PolyPhen-2 and balanced SIFT on another data set (HumDiv [Adzhubei 2010]), which consists of 4,698 non-neutral variants associated with Mendelian diseases and 5,786 differences between human proteins and their closely related mammalian homologs, assumed to be neutral. The FPR at ultraconserved sites was reduced by 27% for PolyPhen-2 and 24% for SIFT, and the FNR at less-conserved sites was reduced by 31% for PolyPhen-2 and 22% for SIFT.
Fig. 2.

Performance improvement. Receiver operating characteristic (ROC) curves for PolyPhen-2 (a) and SIFT (b) in ultraconserved class (solid lines) and in less-conserved classes (broken lines). Arrows represent the direction of improvement by using the new thresholds (close circles) instead of the original thresholds (open circles). Area under curve (AUC) is shown for each curve. The diagonal lines represent random predictions. Reductions on FPR in ultraconserved class (c) and reductions on FNR in less-conserved class (d) achieved by balanced versions of PolyPhen-2 (solid bars) and SIFT (open bars) are depicted as percent improvement over the corresponding original versions for ncSNVs occurring with different population frequencies. Allele frequency data for HumVar variants were retrieved from the ESP5400 data set.
Table 1.
Performance of Original and Evolutionary Balanced Versions of PolyPhen-2 and SIFT and Triple Consensus Methods Using the HumVar Data Set.
| Method | Evolutionary Conservation | TN | FP | FN | TP | Diagnosis Rate (%) |
Accuracy (%) |
||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| TNR | FPR | FNR | TPR | BAcc | MCC | ||||||
| PolyPhen-2 | Ultra | 197 | 862 | 495 | 9,628 | 19 | 81 | 5 | 95 | 57 | 21 |
| Well | 2,958 | 1,778 | 1,204 | 5,207 | 62 | 38 | 19 | 81 | 72 | 44 | |
| Less | 11,403 | 1,130 | 1,265 | 818 | 91 | 9 | 61 | 39 | 65 | 35 | |
| Balanced Poly-Phen-2 | Ultra | 542 | 517 | 2,349 | 7,774 | 51 | 49 | 23 | 77 | 64 | 29 |
| Well | 3,227 | 1,509 | 1,492 | 4,919 | 68 | 32 | 23 | 77 | 72 | 45 | |
| Less | 8,896 | 3,637 | 639 | 1,444 | 71 | 29 | 31 | 69 | 70 | 40 | |
| SIFT | Ultra | 531 | 528 | 1,303 | 8,820 | 50 | 50 | 13 | 87 | 69 | 40 |
| Well | 3,451 | 1,285 | 1,682 | 4,729 | 73 | 27 | 26 | 74 | 73 | 47 | |
| Less | 10,956 | 1,577 | 1,187 | 896 | 87 | 13 | 57 | 43 | 65 | 34 | |
| Balanced SIFT | Ultra | 761 | 298 | 2,693 | 7,430 | 72 | 28 | 27 | 73 | 73 | 45 |
| Well | 3,451 | 1,285 | 1,682 | 4,729 | 73 | 27 | 26 | 74 | 73 | 47 | |
| Less | 8,797 | 3,736 | 730 | 1,353 | 70 | 30 | 35 | 65 | 68 | 35 | |
| Concordant | Ultra | 332 | 107 | 508 | 4,928 | 76 | 24 | 9 | 91 | 83 | 67 |
| Well | 2,281 | 476 | 437 | 3,489 | 83 | 17 | 11 | 89 | 86 | 72 | |
| Less | 5,973 | 1,271 | 239 | 986 | 82 | 18 | 20 | 80 | 81 | 63 | |
Note.—The diagnosis rates and accuracy were estimated using the full HumVar data set.
Fig. 3.
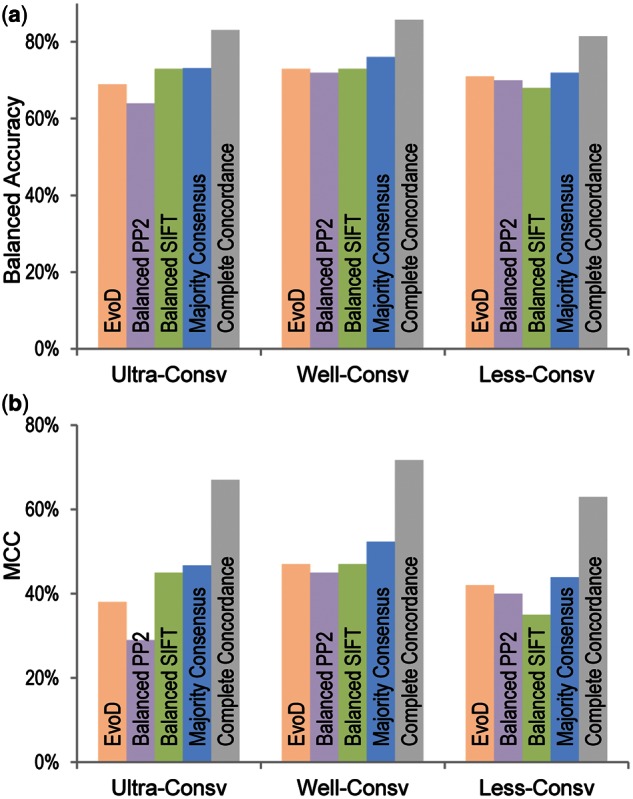
Diagnosis accuracy. (a) Balanced accuracy and (b) MCC for EvoD, balanced PolyPhen-2, balanced SIFT, majority consensus, and complete concordant diagnosis.
The observed differences between the original and balanced versions of PolyPhen-2 and SIFT suggested that the statistical P values reported by PolyPhen-2 and SIFT for hypothesis testing are unlikely to be appropriate. For example, the FPR is 43% for PolyPhen-2 at ultraconserved sites at a 5% overall error rate (P < 0.05). Therefore, to generate the statistical significance of a diagnosis made by balanced PolyPhen-2 or balanced SIFT, we calculated empirical P values based on the cumulative distributions of impact scores for neutral and non-neutral nsSNVs separately within each conservation class following Kumar et al. (2012). For a predicted neutral nsSNV with a score (S), the P value is the probability that this is a false-negative diagnosis, that is, observing a non-neutral variant in the training data set with a score lower than S. And for a non-neutral designations, it is the probability that this is a false-positive diagnosis, that is, observing a neutral variant with a score higher than S. The set of S scores that corresponded to various P values were provided as alternative cutoffs. One may consider diagnoses made with P < 0.05 to be significant and those with P < 0.01 to be highly significant.
Concordant Diagnosis
As mentioned earlier, balanced PolyPhen-2, balanced SIFT, and EvoD show similar overall performance in each evolutionary class. So, we examined the concordance of their diagnosis for the HumVar data set. All three produced the same diagnosis for only 57% of the nsSNVs, with 26% were designated neutral and 30% non-neutral. We found that the accuracy of diagnosis was much higher for these nsSNVs, as the neutral diagnoses were correct 88% of the times, whereas the non-neutral diagnoses were correct 84% of the time, which are significantly higher than the use of any one method alone (fig. 3, all comparisons have P value < 10−12). We also assessed the accuracy of predictions from the use of majority rule consensus, where two out of three methods produced the same diagnosis. The accuracy of diagnosis was only slightly better than that obtained by using each method separately (fig. 3). Therefore, complete concordance from three methods leads to more reliable inferences.
Analysis of Population Variation
We analyzed 240,625 nsSNVs from the ESP5400 Project, a large-scale population survey (Tennessen et al. 2012). Of these, 51,792 (22%) nsSNVs were predicted to be neutral and 52,802 (22%) nsSNVs were predicted to be non-neutral by all three methods. Rare nsSNVs (minor allele frequency [maf] < 0.5%) were twice as likely to be non-neutral as were the common nsSNVs (maf > 5%) (fig. 4); see also figure 3b of Tennessen et al. (2012). This trend is reasonable because the purifying selection acts against harmful variants, which also predicts that many more harmful rare variants will exist in heterozygous states where their negative effects are masked by wild-type alleles. Indeed, a higher proportion of heterozygous alleles was diagnosed to be non-neutral when compared with homozygous alleles for low frequency alleles (maf < 1%; P < 0.01). No significant difference was observed in nsSNVs with higher population frequency (maf > 1%), because they will be frequently exposed to purifying selection in homozygous states.
Fig. 4.

Analysis of population variation data. Proportions of nsSNVs diagnosed to be non-neutral by using complete concordance approach (solid lines) in different maf categories. A total of 240,625 nsSNVs from the ESP5400 Project were analyzed (Tennessen et al. 2012). Private nsSNVs are those observed only once in the population. The other maf categories contain nonprivate nsSNVs. A negative correlation was observed between maf and proportion of nsSNVs diagnosed to be non-neutral (percentages displayed above columns).
In summary, the approach of evolutionary balancing of training data sets leads to better predictive models aimed at identifying function-impacting nsSNVs. It is, however, clear that the current methods can reliably predict functional impacts of less than half (44% in our case) of population variants. This is because all current computational tools are designed to identify disease-associated alleles that have relatively large effects, owing to the dependence of the statistical models on training data implicated in Mendelian diseases. Many nsSNVs showing medium to low impact scores and, thus, insignificant P values may be involved in simple and complex heritable diseases (Thomas and Kejariwal 2004). Furthermore, when one interprets the diagnoses from predictive models that use disease-associated variants as positive controls, it is important to consider that the disease association of variants and their biochemical functional impact do not have a one-to-one mapping. Although an nsSNV may disrupt biochemical functions, it is not a necessary or sufficient condition for it to lead to a disease, which is an ultimate organismal phenotype determined by multiple factors. Similarly, an nsSNV showing no functional impact in experimental assays may in fact be involved in disease, because not all protein functions are known and can be assayed. Therefore, the use of forecasting methods discussed in this work as guides for including or excluding nsSNVs in further experimental and clinical analysis should depend on the objective of individual studies, where the improvements described here will now provide significantly better predictions for thousands of existing and novel variants. We have implemented these advances in the EvoD webserver (www.mypeg.info, last accessed March 13, 2013), which reports predictions and P values produced by EvoD, PolyPhen-2, and SIFT using both original and new thresholds, together with the concordance diagnosis. Batch processing is supported and can be used to analyze small- and exome-scale data sets.
Materials and Methods
We used the HumVar data set for training and testing the predictive models (Adzhubei 2010; Kumar et al. 2012). This data set consisted of 22,196 non-neutral nsSNVs associated with human diseases and 21,119 neutral nsSNVs commonly found in the human population. We also analyzed a population variation data set that contains exome sequencing data (269,277 nsSNVs) for approximately 5,400 individuals available from the ESP5400 Project at University of Washington (Tennessen et al. 2012). Precomputed PolyPhen-2 and SIFT scores for HumVar and ESP5400 variants were retrieved from dbNSFP (Liu et al. 2011). Variants with missing PolyPhen-2 or SIFT scores were removed (1,332 from HumVar data and 27,460 from ES5400 data). EvoD predictions were obtained using the EvoD online server (www.mypeg.info, last accessed March 13, 2013). To cross-reference data from different resources and methods, we mapped all variants to chromosomal locations and imposed a requirement for perfect matches on protein IDs, protein positions, wild-type amino acids, and variant amino acids. Unresolved and mismatching variants were excluded from subsequent analysis (5,038 from HumVar data and 192 from ESP5400 data). This resulted in a total of 36,945 and 240,625 nsSNVs in the final HumVar and ESP5400 data sets, respectively.
We employed several parameters to measure the performances of predictive models, including TPR (sensitivity), true-negative rate (TNR, specificity), FPR, FNR, overall accuracy, balanced accuracy, and Matthews correlation coefficient (MCC). We defined true positive (TP) as the number of correctly predicted disease-associated nsSNVs, true negative (TN) as the number of correctly predicted nsSNVs not associated with any disease (neutral), false positive (FP) as the number of neutral nsSNVs incorrectly predicted to be function impacting (non-neutral), and false negative (FN) as the number of disease-associated nsSNVs incorrectly predicted to be neutral. The aforementioned performance parameters are as follows: TPR = TP/(TP + FN); TNR =TN/(TN + FP); FPR = FP/(TN + FP); FNR = FN/(TP + FN). Overall accuracy = (TP + TN)/(TP + FN + TN + FP); balanced accuracy = (TPR + TNR)/2; MCC = (TPR × TNR – FPR ×FNR)/√([TPR + FNR] × [TNR + FPR] × [TPR + FPR] ×[TNR + FNR]). Because the extreme imbalance in the HumVar data in each evolutionary class affects the overall accuracy, we used the balanced accuracy to measure the performance of various methods. MCC, an alternative measurement that accounts for moderate imbalance in the data, is also inadequate in these extreme cases (Obayashi and Kinoshita 2009; Eiland et al., submitted). Therefore, we used values in a normalized joint probability table that are equivalent to replacing the TP, TN, FP, and FN in the MCC equation with TPR, TNR, FPR, and FNR, respectively (Kumar et al. 2012; Eiland et al. submitted). To test the null hypothesis of equal accuracy, we employed the two-proportion z test (one tailed) (McAfee 2010).
Acknowledgments
The authors thank Maxwell Sanderford for database cross-reference, Nevin Gerek for comments, and Carol Williams for editorial support. This research was supported by research grants from the National Institutes of Health (LM010834-03) and Mayo/Arizona State University seed grant to S.K.
References
- Adzhubei I, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun S, Fay JC. Identification of deleterious mutations within three human genomes. Genome Res. 2009;19:1553–1561. doi: 10.1101/gr.092619.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Perez A, Lopez-Bigas N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet. 2011;88:440–449. doi: 10.1016/j.ajhg.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International HapMap Consortium. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karchin R. Next generation tools for the annotation of human SNPs. Brief Bioinform. 2009;10:35–52. doi: 10.1093/bib/bbn047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Dudley JT, Filipski A, Liu L. Phylomedicine: an evolutionary telescope to explore and diagnose the universe of disease mutations. Trends Genet. 2011;27:377–386. doi: 10.1016/j.tig.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Sanderford M, Gray VE, Ye J, Liu L. Evolutionary diagnosis method for variants in personal exomes. Nat Methods. 2012;9:855–856. doi: 10.1038/nmeth.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Suleski MP, Markov GJ, Lawrence S, Marco A, Filipski AJ. Positional conservation and amino acids shape the correct diagnosis and population frequencies of benign and damaging personal amino acid mutations. Genome Res. 2009;19:1562–1569. doi: 10.1101/gr.091991.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Jian X, Boerwinkle E. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum Mutat. 2011;32:894–899. doi: 10.1002/humu.21517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes MC, Joyce C, Ritchie GR, John SL, Cunningham F, Asimit J, Zeggini E. A combined functional annotation score for non-synonymous variants. Hum Hered. 2012;73:47–51. doi: 10.1159/000334984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAfee G. Boston: Course Technology PTR; 2010. Master math: AP statistics. [Google Scholar]
- Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SB, Buckingham KJ, Lee C, et al. (12 co-authors) Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obayashi T, Kinoshita K. Rank of correlation coefficient as a comparable measure for biological significance of gene coexpression. DNA Res. 2009;16:249–260. doi: 10.1093/dnares/dsp016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olatubosun A, Valiaho J, Harkonen J, Thusberg J, Vihinen M. PON-P: integrated predictor for pathogenicity of missense variants. Hum Mutat. 2012;33:1166–1174. doi: 10.1002/humu.22102. [DOI] [PubMed] [Google Scholar]
- Sunyaev SR. Inferring causality and functional significance of human coding DNA variants. Hum Mol Genet. 2012;21:R10–R17. doi: 10.1093/hmg/dds385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennessen JA, Bigham AW, O'Connor TD, et al. (26 co-authors) Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas PD, Kejariwal A. Coding single-nucleotide polymorphisms associated with complex vs. Mendelian disease: evolutionary evidence for differences in molecular effects. Proc Natl Acad Sci U S A. 2004;101:15398–15403. doi: 10.1073/pnas.0404380101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valliant R, Dorfman AH, Royall RM. Finite population sampling and inference: a prediction approach. New York: John Wiley; 2000. [Google Scholar]
- Zhu Y, Stevens RG, Leaderer D, Hoffman A, Holford T, Zhang Y, Brown HN, Zheng T. Non-synonymous polymorphisms in the circadian gene NPAS2 and breast cancer risk. Breast Cancer Res Treat. 2008;107:421–425. doi: 10.1007/s10549-007-9565-0. [DOI] [PMC free article] [PubMed] [Google Scholar]