

1. Introduction
A simple, primary amine bearing a C10H15 alkyl residue was found to display potent anti-Influenza A properties in 1964.1 Soon thereafter, antiviral activity of this amine was found against Rubella viruses,2 and other viral targets soon followed. Antiviral drugs incorporating the adamantane moiety were quickly introduced to the market, and other targets for pharmaceuticals featuring this symmetrical “bullet” hydrocarbon were successfully identified. In other words, amantadine and the aminoadamantane antivirals constitute the birth of medicinal chemistry of adamantane derivatives. This happened just a few years after Schleyer's seminal synthesis3 of the parent hydrocarbon, adamantane (tricyclo[3.3.1.11,7]decane), through Lewis-acid induced rearrangement of the C10H16 precursor tetrahydrodicyclopentadiene in 1957. Albeit isolated in 1933 from crude oil4 and synthesized chemically for the first time in 1941,5 and some insecticidal properties of chloroadamantanes were reported in 1959,6 the study of adamantane and its functionalization was limited until adamantane became widely available through Schleyer's synthesis. Syntheses of adamantane derivatives started fueling pharmaceutical studies thereof, and they continue to do so to date.
This review concentrates on the numerous applications in medicinal chemistry and drug development of adamantane derivatives. No other singular hydrocarbon moiety (apart from the methyl group) has the success rate of adamantane in improving or providing pharmacological activity in and of best-selling pharmaceuticals. Having the “lipophilic bullet” (adamantane often is viewed as providing just the critical lipophilicity) readily available as an “add-on” for known pharmacophors, it was used in the modification of, e. g., hypoglycemic sulfonylureas,7 anabolic steroids,8 and nucleosides.9 The adamantane modifications were chosen to enhance lipophilicity and stability of the drugs, thereby improving their pharmacokinetics. Aminoadamantanes like amantadine,1 rimantadine,10 and tromantadine11,12 were among the first “hits” that successfully made it to the pharmaceutical market, and most of them are still being used to date. These remarkable bottom-of-the-line structures are not only efficiently being used as pharmaceuticals, but also led to an improved understanding of the molecular mechanisms underlying, e. g., the replication of Influenza A viruses. Research in this area is still highly fruitful as exemplified by numerous studies of the interactions of aminoadamantane anti-Influenza A agents with their target, the M2 ion channel. Electrophysiology,13 NMR techniques,14–16 and recently X-ray structures of the drug and models of its target14 are being used to understand the aminoadamantane's mechanism of action on a molecular level, and also address the increased emergence of “adamantane-resistant” strains of Influenza A in recent years.17
The aminoadamantanes are synthetic drugs that have not been inspired by natural products like numerous other drugs. There are, however, also isolates from Nature incorporating an adamantane motif, like “nature's marvel”, Tetrodotoxin.18 Like Amantadine, it also blocks an ion channel, the sodium channel. This potent pharmaceutical activity may be, at least in part, attributed to its rigid dioxaadamantane core. We will not elaborate on the development of pharmaceuticals from oxaadamantanes, azaadamantanes, or any other heteroadamantanes, that could be viewed as inspired by natural products. There are, however, also natural products that incorporate the adamantane skeleton,19–21 some of which also display interesting biological properties. This review focuses on the medicinal chemistry of the parent hydrocarbon, adamantane, and its derivatives. We will describe the development of adamantane-based chemotherapeutics for viral infections including Influenza A, Herpes simplex,22 Hepatitis C,23 and HIV24,25 in chapter four. Adamantane derivatives to be used as antimalarials26–29 will also be covered there. Emerging adamantane resistant strains of Influenza A may lead to an end of the use of aminoadamantanes against these infections, whereas the utilization of the pronounced lipophilic properties of adamantane derivatives in pharmaceuticals for neurological disorders still prospers. We focus on this field in chapters five and six. Frequently, the addition of adamantane moieties increases the permeability of the modified compound through the blood-brain-barrier.30,31 Therefore, targets of the CNS today are probably most promising both academically and commercially. With the fortuitous finding that amantadine gives symptomatic benefits in Parkinson disease32 and the approval of memantine by EMA and FDA for the treatment of moderate to severe stages of Alzheimer disease,33–36 two neurodegenerative diseases of increasing importance in aging societies are being addressed with (structurally again remarkably simple) adamantane derivatives. Memantine's pharmacological properties and its mechanism of action as a moderate, non-competitive NMDA-receptor antagonist have been studied extensively.37–41 Other targets for drug development of adamantane derivatives for CNS disorders are AMPA- and KATP channels42,43 and the GABAergic system.44,45 Incorporation of the adamantane moiety into neuropeptides and related signalling molecules frequently gives increased selectivities for receptor subtypes and enhanced stability in vivo, as may be exemplified by CCK derivatives equipped with an adamantane core.46,47
An emerging filed with respect to the application of adamantane derivatives is the inhibition of enzymes using adamantane based scaffolds. We cover this field in chapter seven. Most important are the DPP-IV inhibitors vildagliptin and saxagliptin,48–50 that currently enter the multi-billion dollar market of diabetes management. Other examples of enzyme targets for adamantane-based pharmaceuticals are soluble epoxide hydrolases,51,52 protein phosphatase 2A,53 or the hydroxysteroid dehydrogenases.54–56 The various enzymes targeted by functionalized adamantane-based pharmaceuticals demonstrate the trend towards an utilization of the adamantane scaffold to orientate (co)pharmacophors to positions beneficial for enhanced interaction with the target's active site. We will conclude this chapter with an antibiotic natural product modified with an adamantane core,57 re-iterating the concept of natural-product inspired pharmaceuticals. Chapter eight adds discussions on three classes of adamantane derivatives of relevance in cancer research. The “add-on” strategy is followed by adamantane derivatives of cisplatin, Adaphostin, and the adamantyl retinoids represent alternative routes to fight cancer cell proliferation. Adapalene, an anti-acne drug sold as hydrogel, is also covered here as it is also a retinoid originating from similar research. Some concluding remarks on the metabolism of adamantane based pharmaceuticals are given in chapter nine.
Though the present review has grown to considerable length during its making, it still is far from being complete: for reasons of brevity, we could not describe biological signaling cascades or life cycles of viruses and parasites as would certainly be necessary to present the whole picture of the targets hit by molecules incorporating adamantane; the reader is referred to review articles in these cases. Likewise, three reviews of smaller scope than the present one58–61 describing adamantane derivatives in medicinal chemistry had already been published before, one during the revision of the present manuscript62 to affirm our conviction that the topic deserves presentation in Chemical Reviews.
This latest review article gives clogP data on adamantyl derivatives comparing them with “non-adamantyl”-analogs. Clearly, having adamantane present in a test drug gives a molecule of significantly higher lipophilicity compared to a molecule with just a proton or a methyl group instead of the adamantane moiety. Therefore, we have chosen an alternative approach in selected cases: after identifying low-lying conformers of the respective molecules via molecular mechanics (MMFF force field63 and conformer search as implemented in Avogadro64) we have used ALOGPs65 to estimate logP of adamantane derivatives and analogues with other “bulky & lipophilic” groups. The ALOGPs values are given in square brackets in tables and schemes. Generally, these ALOGPs numbers support our conviction that in most cases there is more to adamantane's value in medicinal chemistry than just adding several logP units to a given pharmaceutical. This involves molecular properties beyond Lipinski's classical “rule of five”.66,67 Adamantane is not a “magic bullet”, but it certainly is more than just a lipophilic add-on, as we have tried to devise in this review.
2. Natural Products Incorporating an Adamantane Motif
As nature is the oldest source of biologically active substances, extracts from natural sources have traditionally fueled medicinal chemistry and continue to do so to date.68 One example for a natural product of remarkable biological activity is the potent toxic principle from puffer fish (Tora fugu), tetrodotoxin (Scheme 1, TTX, 1). Its “elaborate chemical architecture, crafted from a densely oxygenated cyclohexane framework (…), is matched only by its awesome potency as a selective blocker of voltage-gated Na+ ion channels.”18 The selectivity and potency of this compound certainly is warranted by several factors, one of which being the rigid central dioxaadamantane skeleton that orientates the multiple functional groups to fit tightly into the ion channel. TTX remains both a challenge for the synthetic organic chemist69–71 and a prototype tool for the elucidation of structure and functionality of its target ion channel.72 Muamvatin 2, the first trioxaadamantane natural product that could be extracted from limpets Siphonaria normalis73,74 and Caloundrin B 3 from Siphonaria zelandica,75 are further examples of natural products incorporating a heteroadamantane skeleton. Further naturally occuring “trioxaadamantanes” with sedative properties are Daigremontianin 4 and Bersaldegenin-1,3,5-orthoacetate 5, extracted from plants. Their orthoacetate structural motif is reflected in the synthetic class of the bananins. These compounds have been reported to inhibit the replication of the SARS corona virus.76,77 While being heteroadamantanes, (strikingly, Ansabananin 6 also incorporates an azaadamantane moiety), we will not cover them in detail here since, obviously, both chemical and medicinal properties of these compounds will differ markedly from adamantane derivatives.
Scheme 1.
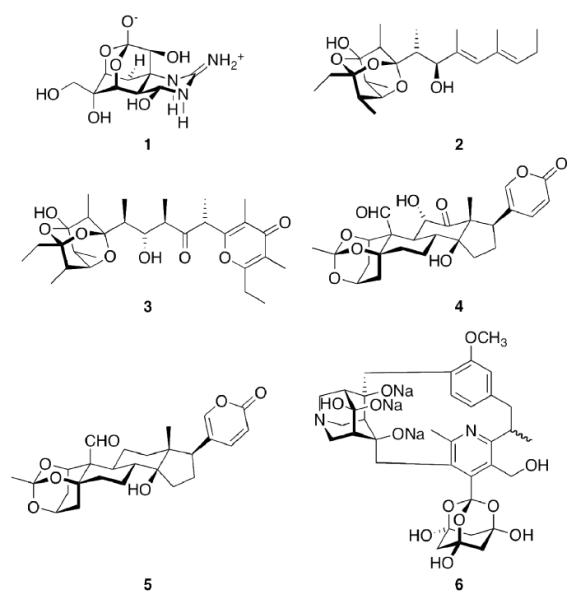
Naturally occuring and nature-inspired heteroadamantanes
There are, however, also compounds to be found in nature that incorporate the adamantane hydrocarbon scaffold itself (Scheme 2). Hypericum Sampsonii are guttiferae that have been used in traditional Chinese medicine for centuries to treat a multitude of disorders including snakebites, swellings, backache, and diarrhea.78 The active ingredients are not (yet) identified specifically, but some of the isolated compounds, the Sampsoniones and Plukenetiones, were isolated and their chemical structure was elucidated. Investigations of the constituents of extracts from various Guttiferae identified polyprenylated acylphloroglucinols as the source of the adamantane derivatives via secondary cyclizations.20 Substituted homoadamantanes (Sampsoniones A–H)78–80 were also isolated.
Scheme 2.
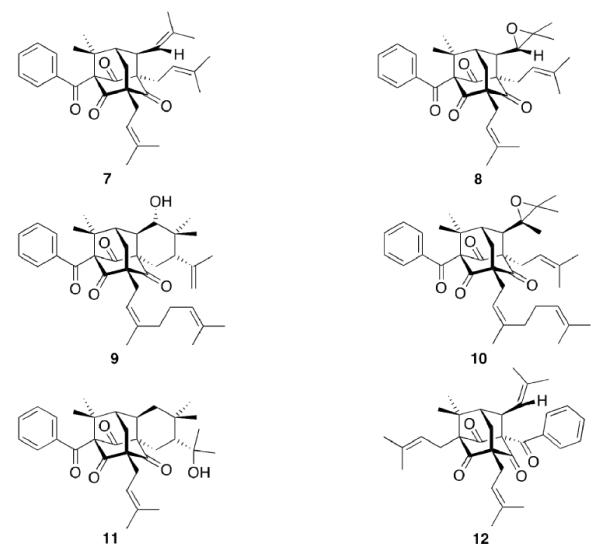
Natural products isolated from plants incorporating an adamantane scaffold
Plukenetione A (7) was first isolated from Clusia plukenetii in 1996,81 its 28,29-epoxy derivative 8 in 200182 from the fruit of Clusia havetiodes var. stenocarpa. Compound 7 as an ingredient of Cuban propolis was identified cytotoxic in a panel of cell lines for different cancer entities, the propolis itself being anti-metastatic in a mouse model.83 Meanwhile, antiretroviral activity of 7 has been reported84 and a total synthesis of the natural product has recently been published.85 The interesting pharmaceutical activities fueled synthetic efforts to the core scaffold.86 Sampsonione I 9 as well as Sampsonione J 10 have been isolated and tested for cytotoxicity towards a P388 cell line.87 While 9 displayed cytotoxicity against this cell line, 10 did not significantly act cytotoxic. Likewise, Hyperibone K 11 isolated from the Uzhbekistan medicinal plant Hypericum scrabum showed moderate cytotoxicity in two human cancer cell lines.88 The same authors could not find an anti-HIV activity of 11. 12 was isolated from Clusia obdeltifolia (interestingly from the Chapada Diamantina region in Brazil),89 but no bilogical tests of this compound have been disclosed yet.
The (hetero)adamantane scaffold might play a decisive role in the three-dimensional adjustment of the pharmacophors of the abovementioned natural products and natural-product inspired compounds. It is clear that the sheer activity of a drug, exerted by the fit into a receptor's binding pocket, the active site, is essentially triggered by this three-dimensional structure. This has historically been described as the “key-lock” principle and nowadays carries the moniker “induced fit”, expressing a more dynamic understanding of the receptor-ligand interactions.
More intriguing is the relationship between a drug's three-dimensional structure and its ADME characteristics. Both a drug's absorption, e. g., from the digestive tract into the bloodstream, and its distribution in the system, ideally enriching in the targeted tissue, depend (amongst others, e. g., lipophilicity) on its structure. In prodrug concepts, drug carriers designed for a guided distribution of the drug are frequently being utilized. Metabolism and excretion are mainly organic-chemical processes of the drug's action and, as such, also highly dependent of the three-dimensional structure.
Nature has designed the central scaffold of, e. g., Tetrodotoxin (1) to improve its efficiency, and the medicinal chemistry of adamantane derivatives can be regarded as an example for the design of primarily all-synthetic drugs to achieve the same ultimate goals, that is, high potency and selectivity. While the natural products hitherto mentioned stem from plant and animal sources, one must not forget that adamantane 134 and the higher diamondoids90 up to at least undecamantane are found in trace amounts in most raw petroleums and are, therefore, natural products as well (Scheme 3).
Scheme 3.
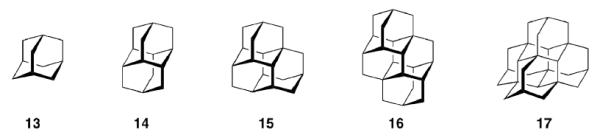
Polymantanes isolated frome crude oil
Diamondoids like diamantane 14, triamantane 15, [121]tetramantane 16, and [1(2,3)4]pentamantane 17 are a current focus of interest due to their ready accessibility; selective functionalizations91,92 yield building blocks of interest for, e. g., nanotechnology and electronics.93 Given this recent reincarnation of diamondoid chemistry, it remains to be seen whether one of these hydrocarbon scaffolds will start a career as building block in medicinal chemistry as fruitful as adamantane itself did.
3. “Add-On” to Known Pharmaceuticals
Fueled by the new widespread availability of synthetic adamantane and some of its derivatives after Schleyer's synthesis of the parent hydrocarbon, this almost spherical, lipophilic entity has been used mainly as an “add-on” to known pharmaceuticals or a replacement for other lipophilic groups. Gerzon et al.7 replaced the n-butyl sidechain in a popular sulfonylurea hypoglycemic drug that is sometimes still being prescribed today, Tolbutamide (18, Table 1), with various hydrophobic residues including several adamantane derivatives. When comparing the relative potencies of these compounds, a marked increase in hypoglycemic activity could be observed, along with a longer-lasting, constant activity. In humans, 19 was about five times more potent than tolbutamide on a weight basis. A marked decrease in potency when varying the adamantane substituent is most striking. When considering the mechanism of action of tolbutamide 18 and other sulfonylurea antidiabetics as an KATP channel blocker,94 a precise fit into the binding pocket (the sulfonylurea receptor or SUR subunit) is obviously a prerequisite for the induction of channel closure, which leads to stimulation of insulin secretion from pancreatic β-cells (see also Chapters 5.2 and 7.3). An increase in lipophilicity is beneficial, but there oviously is a “size limit” of the lipophilic add-on as exemplified by the sharp decrease in potency for the 3,5-dimethyladamantyl derivative 23.
Table 1.
Adamantanes as add-on to hypoglycemic sulfonylureas. 
| Substance | R1 | R2 | ALOGPs | relative potency (rat) |
|---|---|---|---|---|
| 18 | n-C4H9− | -CH3 | [2.04] | 1 |
| 19 | 1-Adamantyl- | -CH3 | [2.78] | 15.5 |
| 20 | Cyclohexyl- | -CH3 | [2.65] | 12.8 |
| 21 | 1-Adamantyl- | -C2H5 | [3.37] | 14.8 |
| 22 | 3-Methyl-1-adamantyl- | -CH3 | [2.81] | 2.8 |
| 23 | 3,5-Dimethyl-1-adamantyl- | -CH3 | [3.17] | 0 |
| 24 | Adamantyl-1-CH2− | -C2H5 | [4.09] | 0.2 |
Adding an adamantane moiety to anabolic steroids was reported by the same group of researchers at Eli Lilly.8 Since it was already known that esterification of the 17β-hyroxy group of steroids increased and prolonged the anabolic action (palmitic and stearic acid esters gave long duration of action at reduced intensity), adamantane-1-carboxylic acid as well as its methyl and dimethl derivates were studied here. As with the sulfonylureas above, the potency of the resulting drugs was strongly depending on the shape of the adamantane moiety and not on the overall lipophilicity, 3-methyl- and 3,5-dimethyl-substitution markedly decreasing the anabolic potency as measured by the weight gain of muscles in immature male castrate rats (Scheme 4).
Scheme 4.
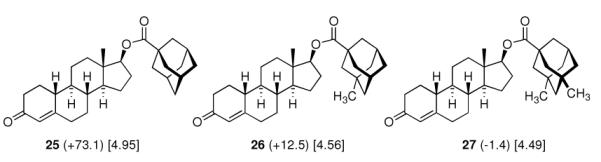
Anabolic activity of adamantoates of 19-nortestosterone (Nandrolone) as measured by the weight gain of the levator ani muscle over control (values in mg in brackets) in rats. Values in square brackets: ALOGPs data.
The easily available95,96 adamantane-1-carboxylic acid and some similarly straightforward derivatives have been popular “add-ons” therafter. Examples (Scheme 5) include adamantoylated nucleoside derivatives9 like 28, whose increased lipophilicity is crucial for medicinal applications. A series of these adamantoylated nucleosides was tested in different animal models of numerous disorders, e. g., suppression of antibody formation, antitumor activity, cytotoxicity, antiviral properties, and inhibition of adenosine deaminase.
Scheme 5.
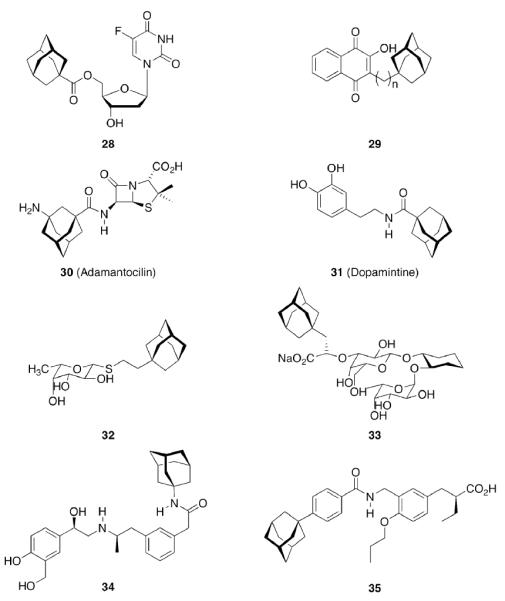
Add-on strategy to generate drug candidates incorporating an adamantane moiety
Upon this “search for an application” for an easily available class of compounds, again a crucial influence of the actual adamantyl moiety used on the potency of the test compound was observed. While the addition of an adamantane-1-carboxylic acid ester moiety generally improved the compounds' distribution in the host and enhanced metabolic stability, minor changes like additional 3-methyl- or 3,5-dimethyl substitution again resulted in markedly different potencies of the tested compounds. Naphthoquinones were a well-known class of antimalarials in the 1960's, and because of encouraging results using cyclohexyl substituted derivatives, 1-adamantyl substituted analogs 29 (n = 1–5) were prepared and tested in Plasmodium berghei infected mice – without success due to significant toxicity.26 Even more popular pharmaceuticals tested with adamantane “add-on” building blocks were Penicillin to give 30 (“Adamantocilin”) and Dopamine to furnish 31 (or “Dopamintine”).97 More recent examples for the “add-on” strategy of modifications of a primary pharmacophor with the adamantane “lipophilic bullet” acting as a functional subunit, sometimes more suitably described as a “secondary pharmacophor”, include glycolipids. Amongst other lipophilic residues, the 2-(1-adamantyl)ethyl group in the 1-thio-β-L-fucopyranosyllipid 32 was utilized to modify the potency of these compounds that can be used as immunologic adjuvants.98 Another example for carbohydrate-derived core structures modified with the lipophilic bullet are the E-selectin antagonists derived from the tetrasaccharide epitope Sialyl Lewis X99. In 33 (Scheme 5), the adamantane moiety was introduced to pre-organize the pharmacophors in solution, thereby lowering the entropic penalty upon receptor binding.100 For the treatment of asthma and chronic obstructive pulmonary disease, β2-adrenoceptor antagonists are currently being used clinically. Available preparations have to be inhaled twice daily. There is considerable interest to improve the pharmacokinetic properties of these drugs: longer acting compounds are desirable, as are compounds that are less likely to be taken up orally through swallowing during inhalation. Therefore, the pharmacokinetics of formeterol derivative 34 were studied.101 This compound displayed an excellent β1 selectivity. In a guinea pig tissue assay, it showed prolonged duration of action, and in vivo in guinea pigs, the results were promising. When compared to the parent compound not incorporating the adamantane moiety, the authors assigned “intrinsically poor cell permeability” to 34, it is being effluxed via transporter proteins and additionally showed a low microsomal stability.
A 4-adamantyl-phenyl group is described as a “lipophilic tail” that has recently been utilized to generate ligands for the peroxisome proliferator-activated receptors (PPARs). These are transcription factors that are activated by fatty acids and their metabolites. The PPARs heteodimerize with the retinoid X receptor upon ligand binding (we will discuss retinoid receptor binding adamantane derivatives in detail in chapter eight). These heterodimers regulate gene expression by binding to the peroxisome proliferator responsive element, a specific consensus DNA sequence. The PPARα, -γ, and -δ subtypes each play a pivotal role in lipid, lipoprotein, and glucose homeostasis. Simultaneously activating all three subtypes with one PPAR pan-agonist is an attractive target for treatment of, e. g., the metabolic syndrome. Steric bulkiness at the distal benzene ring in 35 helped to achieve this goal. The EC50 values for 35 in HEK-293 cell lines transiently transfected with PPAR-GAL4 chimeric receptors were 61, 120, and 43 nM for the PPARα, PPARδ, and PPARγ, respectively.102
Gaucher disease is a heritable lysosomal storage disease caused by the deficiency of lysosomal glucocerebrosidase. As a consequence, this enzyme's substrate, glucosylceramide, accumulates, ultimately leading to the pathogenesis of the disease. Hydrophobic adamantyl deoxynojirimycin derivatives have been designed as selective and potent inhibitors of the non-lysosomal glucosylceramidase103 while the active-site-directed “chemical chaperone”, N-nonyl-deoxynojirimycin, increases the activity of the Gaucher disease associated N370S mutant of glucocerebrosidase, presumably through its stabilization against proteasomal degradation.104 Combination of 2,5- anhydro-2,5-imino-D-glucitol 36 and isofagomine 38 moieties104 as active site binding primary pharmacophor with hydrophobic alkyl adamantyl amides and an appropriate linker gave small molecules 37 and 39 (Scheme 6) that increased the activity of N370S and G202R glucocerebrosidase mutants in patient-derived fibroblasts.105 Using 39, a 7.2-fold increase in glucocerebrosidase activity was observed, which represents the highest drug potency in cell lines reported to date. A docking study of one of these adamantane derivatives was also performed, based upon the X-ray crystal structure of glucocerebrosidase with an N-hexanoic acid adamantyl amide DNJ modeled into the active site (Figure 2). The crystal structure data indicated a lipophilic cleft in proximity to the active site, which could be “filled” with the adamantylamide moiety.
Scheme 6.
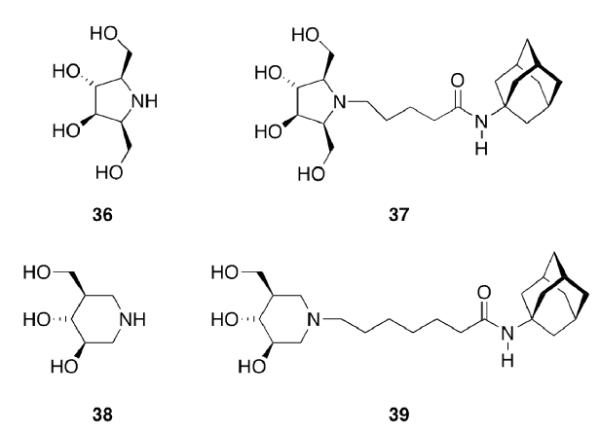
Design of activators for glucocerebrosidase
Figure 2.
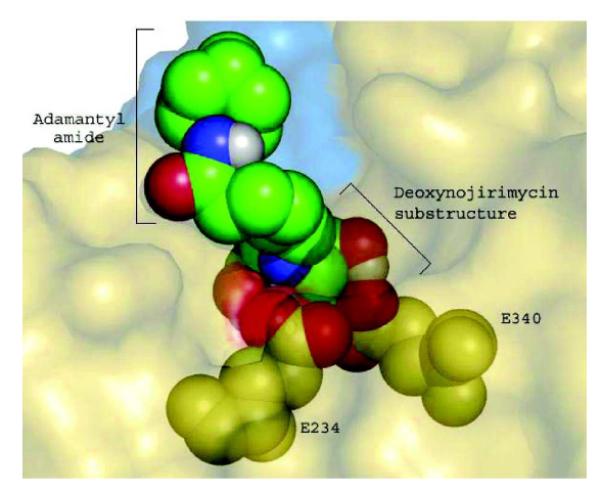
Docking of N-hexanoic acid adamantyl amide deoxynojirimycin into the active site and a nearby hydrophobic cleft of glucocerebrosidase.
(Yu, Z.; Sawkar, A. R.; Whalen, L. J.; Wong, C. H.; Kelly, J. W. J. Med. Chem. 2007, 50, 94-100.)
The combined pharmacophors facilitate glucocerebrosidase trafficking, increasing its activity in patient-derived cell lines. As the metabolism of glycosphingolipids is associated with atherogenesis, targeting glycosphingolipid metabolism with (adamantylated) pharmaceuticals might become useful also in the field of atherosclerosis, the underlying cause of ~ 50% of deaths in Western countries.106 “Click Chemistry”-introduced triazole based linkers of variable length between the deoxynojirimycin- and adamantyl- co-pharmacophores recently showed that these structures can be fine-tuned to act at a collection of glycosidases, and, as yet another target, they have been shown to act as correctors for defective mutants of the cystic fibrosis transmembrane conductance regulator (CFTR).107
The ever-increasing need for novel antibiotics fueled the synthesis of Gramicidin S analogues incorporating adamantyl amino acids. The adamantylated analog 41 (Scheme 7) “…emerges as the most promising compound because of its ability to distinguish, at a specific concentration, between bacteria and mammalian cells.”108
Scheme 7.
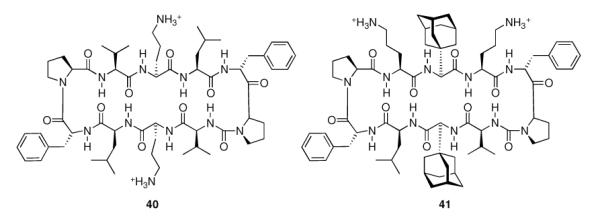
Gramicidin S (40) and adamantylated analog (41).
While several of the drugs and drug candidates we will discuss in the following chapters have been discovered following other design approaches, the above examples given for the “add-on” strategy illustrate nicely some of the most important aspects of adamantane's medicinal chemistry: Its size and approximately spherical shape obviously render it to interact favorably with many lipophilic binding sites, increasing the potency of the modified drugs and drug-like molecules, and its unique lipophilicity as well as relative chemical stability of esters and amides positively influence the drug's pharmacokinetic properties.
4. The First Hit: Adamantane Derivatives as Antivirals and against Infectious Diseases
Adamantane is easily functionalized at the tertiary positions, most conveniently by bromination using elemental bromine and subsequent substitutions with other functionalities. The first striking application of a simple, monofunctionalized adamantane derivative in medicinal chemistry originated in 1963, when in clinical trials the antiviral activity of 1-aminoadamantane hydrochloride, amantadine (42, Scheme 8), was reported.109 It had been shown previously that ammonium ions can inhibit Influenza virus growth in tissue culture,110 so the identification of amantadine as an antiviral can be regarded as a “hit” during a random screen.1,111 Subsequent research in tissue culture, chick embryos, and mouse models confirmed amantadine's strong activity against several strains of Influenza A viruses at low toxicity levels (LD50 in mice was 1080 mg/kg upon oral application).1,112 Two strains of Influenza B were not susceptive for inhibition through amantadine. These findings sparked the synthesis of a multitude of aminoadamantanes and related structures and their study as anti-Influenza agents as well as their application to other viral or infectious diseases, and their use as model substrates in the study of the mechanisms underlying the multiplication of Influenza viruses and other targets.
Scheme 8.
Early structural variations of anti-Influenza A drug candidates alongside the amantadine lead. AVI50 values118 in mg/kg (mouse) given in parentheses.
4.1 Influenza A – The Amantadine & Rimantadine Story
Not knowing the exact target, it was found that the virus' penetration of the host cell is blocked by amantadine, causing the virus to remain susceptible to antibody inactivation for a prolonged period of time.1 Having this antiviral “hit” at hand and in clinical trials, numerous structural variations alongside the “lipophilic, spheric cage hydrocarbon amine” lead structure followed.
4.1.1 Modifications Alongside the Aminoadamantane Motif
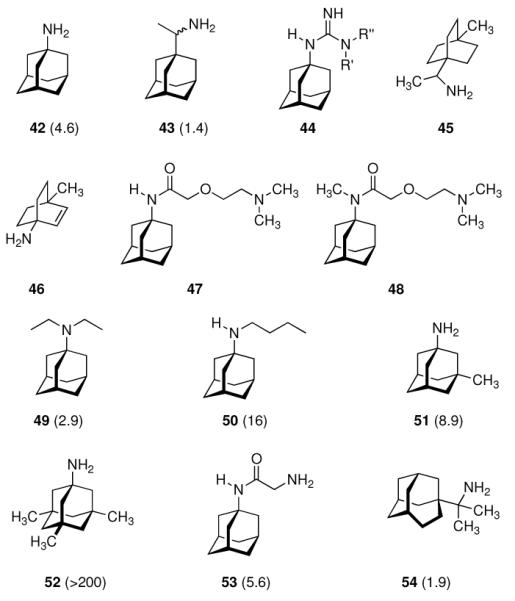
In 1965, α-methyl-1-adamantanemethylamine 43 was reported to display even stronger anti-Influenza A2 (Japan 305 strain) properties when compared to amantadine in a primary calf kidney cell culture assay as well as in vivo in mice and ferrets.10 1-Adamantyl guanidines like 44 were considered attractive because of their higher basicity, but out of a series of eight 1-adamantyl guanidines, prepared from amantadine via the 1-adamantyl cyanamide and bearing structural variations at the N-alkyation, only the non-alkylated parent structure (44, R' = R” = H) significantly inhibited the Influenza A multiplication in vitro and in vivo.113 The compounds were screened against the Influenza A (Swine) S15/1930 and Influenza A2 (Japan) 305/1957 strains in chick embryo fibroblast cultures as a testbed, but a quantitative comparison to amantadine showed the amine to be superior to the guanidine. Variations in the hydrocarbon moiety were also performed, and both for bicyclo[2.2.2]octane amines like, e. g., 45 and bicyclo[2.2.2]oct-2-enamines (46) in some cases comparable or slightly higher anti-Influenza A activities than for amantadine were observed.114 The screening of 156 structurally diverse biologically active compounds reported to have antiviral properties against Influenza A (PR8) and Influenza A2 (Japan/305) strains in mice confirmed an activity against these viruses only for amantadine and (DL)-noformicin.115 Alkylamines materialized as the lead structure. When compared to cyclooctylamine, amantadine proved to be more active against the Influenza A2 (Loma Linola/1/1969) strain at lower concentrations.116 Scherm and Peteri prepared four N-(2-aminoethoxy)acetyl-aminoadamantanes from amantadine via acetylation with chloroacetylchloride and subsequent etherification with aminoethanolates and measured their antiviral properties with a simple plaque test. 47 and 48 were strongly active against Influenza A viruses, while 47 also displayed activity against Herpes viruses (vide infra).117
Continuing the chase for antivirals, the screening of 87 cage amines including N- and C-alkylated adamantane amines (49 and 50), 1-adamantane methylamines, and homoadamantane amines, some consistent structure-activity-relationships (SAR) were concluded with the antiviral dose50 (AVI50) as a quantitative measure of activity of the screened compounds in virus-infected mice (Influenza A (swine) S15).118 Out of 40 N-substituted compounds screened, none had significantly higher potency than amantadine. With increasing size of the N-substituents the activity diminishes; the same holds true when functional groups are incorporated into the N-substituents. Substitution of the tertiary positions of the adamantane nucleus in amantadine (51, 52) was also found to be detrimental to the compound's anti-Influenza A activity. Inserting a bridge of one or more carbons between the 1-adamantyl- and the amino group led to compounds with generally high antiviral activity, with rimantadine outperforming amantadine in terms of activity as already had been reported before in a different test setup.10 Rimantadine incorporates a stereogenic center at the carbon bridge. Resolution of the enantiomers via the diamides from tartranil proved the enantiomers of rimantadine to be essentially equipotent with the racemic compound. Furthermore, expansion of the adamantane skeleton to homoadamantane (54) did not provide enhanced activity.
The glycinate 53 displayed an AVI50 comparable to that of amantadine. Replacing the amino group in amantadine with other functional groups like –OH, –SH, –CN, –CO2H, –Cl, or –Br gave inactive compounds. Taken together, these early findings supported a concept of the amino group acting as a primary pharmacophor that is assisted by an (preferably unsubstituted) adamantane cage in the appropriate distance, acting as a secondary pharmacophor – both leading to pronounced anti-Influenza A activity in a synergistic manner. The parent compound amantadine was approved by the FDA in 1966 for the prevention and treatment of Influenza A infections. It is being used to date (however, see below) and sold as amantadine, Symmetrel, and Symadine. Compound 43 was FDA-approved in 1993 and is marketed as rimantadine and Flumadine. Mostly being referred to as “adamantanes” in pharmacological studies, these drugs recently faced increasing resistances of the circulating Influenza A strains,119–121 and with the majority of strains (including the 2009 H1N1 epidemic) not responding to “adamantane” treatment any more, amantadine and rimantadine are no longer the chemotherapy of choice against Influenza A epidemics. For our survey, it remains noteworthy that the anti-Influenza-“adamantanes” have been introduced and successfully utilized clinically without knowing their target.
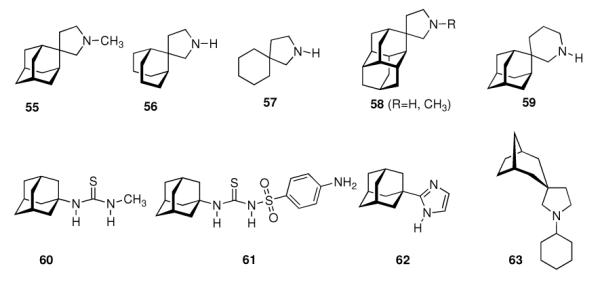
Ever since these days, newly synthesized (amino)adamantane derivatives are being tested with respect to their antiviral activities. Having adamantanone readily available in large scale, antiviral adamantane spiro-3'-pyrrolidines 55 (Scheme 9) were synthesized and their activity against several viruses in vitro (plaque assay) and against Influenza A2 (Japan) in mice was studied and compared with amantadine and structurally similar compounds.122,123 The N-methyl derivative 55 was “three times more active than amantadine” in vivo, also showing a broader antiviral spectrum in the in vitro tests. Small N-alkyl substituents were generally favorable. Of the other cages studied by these authors, the bicyclo[3.3.1]-derivative 56, structurally “most closely related to amantadine”, turned out to be about as active as amantadine in the testbed utilized here. Surprisingly, no antiviral activity at all could be measured for the cyclohexane derivative 57. As already deduced from alkylated adamantane derivatives, size and shape of the hydrocarbon co-pharmacophor does also matter in the spiro compounds, as shown by the diamantane spiro-3'-pyrrolidines 58 and spiro-3'-pyrrolidines incorporating the bornaneor perhydro-4,7-indane hydrocarbon moieties: all of these variations of the cage hydrocarbon gave compounds less active than amantadine in vivo. Of the six- and seven-membered ring analogues of these spiro compounds, the authors faced cytotoxicity problems in vitro except for two piperidine derivatives, of which the antiviral activity of 59in vivo was “comparable to that of 1-aminoadamantane or better”. Compound 55 (as the 1:1 maleate) was studied in greater detail, including a double-blind, placebo-controlled clinical prophylactic trial in 57 human volunteers.124 It was regarded more efficient than amantadine against Influenza A (Hong Kong)/1/68. When studying its potency against challenges of other virus strains, 55, like amantadine, was “quite ineffective” against Influenza B viruses but inhibited the growth of a number of Influenza A strains in vitro and Rhino viruses – albeit the anti-Rhino virus activity could not be confirmed in man.125 Another double-blind, placebo controlled trial of 55 in volunteers challenged with Influenza A (England/42/1972) examined its suitability as a therapeutic agent.126 No evidence of any toxic effect of the test drug was observed, but the generally observed reduction in severity of symptoms was not significant. New adamantane derivatives that were regarded not to be “adamantyl analogs” of corresponding drugs, e.g., halides, alcohols, ketones, carboxylic acids, esters, amides, nitriles, etc. were subsequently screened and compared to known adamantane derivatives in two in vitro assays using the Newcastle disease virus.127 Some of the novel compounds were reportedly “more active than amantadine, although most of them have no particular functional group with established biological activity”, but “cytotoxicieties of the compounds usually paralleled the antiviral activities, the activity and the cytotoxicity appearing at similar concentrations.” Essentially the same holds true for derivatives of 4-homotwistane, trimethylenenorbornane, and 1-tricyclo[4.3.1.12,5]undecane derivatives using the same in vitro testbed.128–130 A number of N-(1-adamantyl) thioureas (e.g., 60), were also prepared and screened against Influenza A2 (Bethesda) in mice; some of them significantly increasing the survival time of the animals challenged with the virus.131 Later on, the 2-adamantyl thioureas were also studied.132 In in vitro tests, some adamantyl thioureas furthermore showed activity against a number of other viruses.133 Since some of the adamantyl thioureas have proven active in vivo at low toxicity, a small library of nine 3-substituted 1-adamantylthioureas was synthesized and quantitative antiviral activities for three of them in mice challenged with the Influenza A2 (Asian)/J305 strain.134 Compound 61 was shown to compare favorably with amantadine with respect to both activity and toxicity. Furthermore, 61 caused a reduced level of CNS related side effects, producing mild tremors and ataxia in the animals only at very high doses (>600 mg/kg i.p.). Resembling the results mentioned above for the polycyclic spiro-3'-pyrrolidines, 1-adamantyl imidazoles like 62 and its N-methyl analog were identified to be active against Influenza A2 (Victoria) in chick embryos.135 In in vitro assays, the anti-Influenza A activities of even more cage amines were studied and compared to amantadine. The bicyclo[3.2.1]octane-3-spiro-3'-pyrrolidine 63 again was more potent than amantadine,136 while 4-amino-(D3)-trishomocubanes were about as active as amantadine at low levels of cytotoxicity.137
Scheme 9.
Development of adamantanes and other cage compounds as antiviral agents.
In a detailed review on laboratory and clinical data of amantadine's antiviral activity and the development of antiviral cage amines until 1980,111 the main achivements of the early days of chemotherapy in anti-Influenza A prophylaxis and treatment have been correlated to what was known about the life cycle of the virus on a molecular level. It had been known that amantadine and analogues inhibit an early step of the virus' reproduction, the uncoating step, but “it must be made clear that the precise point of action on Influenza virus replication of amantadine in molecular terms is not known. The details of the early stages of Influenza virus infection of cells are unclear and therefore the point of action of an inhibitor acting, as amantadine does, at this early stage remains unestablished”.
Starting in 1985,138 the mechanism of action of the aminoadamantanes against Influenza A has been elucidated on a molecular level. We will focus on that in chapter 4.1.2, but we should stress at this point that the knowledge of the drug target, the virus' M2 protein, and its function as a proton channel139,140 has driven the synthesis and screening of drug candidates to small molecules that could be accomodated by the target protein's binding site. At the same time, some of the aminoadamantanes, in particular the drugs amantadine and rimantadine, have also been highly useful as model substrates in the discovery of the M2 ion channel dynamics and its mechanism of proton conductivity.
With this concept in mind, the aminoadamantane motif was further refined focusing on the conformational propensities of the drug candidates. Using the strong anti-Influenza A potency of 55 as a starting point and varying the distance between the two pharmacophores, another series of 2-spiro adamantane derivatives was synthesized starting from adamantanone.141 While the spiro[cyclopropane-1,2'-adamantane]-2-amines (Scheme 10, 64a–c) were active at micromolar concentrations and were stronger antivirals than 42 was in the assay used, spiro[cyclopropane-1,2'-adamantane]-2-methanamine 65 was active at MIC50 = 0.8 μg/mL, or at a 125-times lower concentration than amantadine. Even more potent was 1-methylspiro[pyrrolidine-2,2'-adamantane] 66, being about 179-times more potent than amantadine. Expanding the nitrogen heterocycle gave spiro[piperidine-2,2'-adamantane] 67, which inhibited Influenza A2/Japan/305/1957 (H2N2) with MIC50 = 0.24 μg/mL, a concentration about 3.3 times lower than for amantadine in the control experiment using the MDCK cell line.142 The closely related spiro-[morpholine-3,2'-adamantane] 68 was notably less potent. Likewise, the conformationally somewhat more flexible “rimantadine analogs” 69 and 70 were also active at the same range of MIC50 like the “gold standard”, amantadine.
Scheme 10.
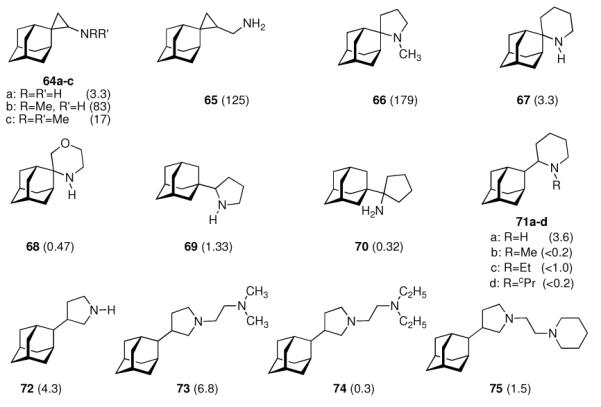
Refinement of adamantyl amines and -diamines targeting the Influenza A M2 ion channel. Numbers in parentheses are the relative potencies of the compounds measured as an x-fold increase or decrease in MIC50 compared to amantadine as positive control. 64 – 68: Influenza A (Ishikawa); 69 – 75: Influenza A2/Japan/305/1957 (H2N2). All compounds screened in MDCK cell culture assays.
These findings were not much of a surprise considering the very close structural relationships between the molecules and the identical mechanism of action. A range of other viruses was also studied, and some of the compounds displayed borderline anti-HIV activity (see chapter 4.3).143 Taking a closer look at 2-substituted adamantyl piperidines, the same authors subsequently studied 2-(2'-adamantyl)piperidines like 71a–d.144 While the unsubstituted parent compound 71a (R = H) was about 3.6 times more active against Influenza A2/Japan/305/1957 (H2N2) in the MDCK-cell based assay (MIC50 = 7.8 μM), the (cyclo)alkylated analogs 71b–d were significantly less active. From combined MD- and NMR studies the authors concluded that this is probably due to a change in the conformation of the piperidine moiety upon its N-alkylation. 3-(2-Adamantyl)pyrrolidines 72 – 74, screened using the same testbed,145 likewise were potent anti-Influenza A drug candidates.146 While the parent compound (72) with a MIC50 of 0.60 μM was about 4 times more active than amantadine, N-alkylation abates the activity significantly. However, introducing a second amino functionality via dialkylaminoethyl substitution gave diamines with strong anti-Influenza A activity (MIC50 (73) = 0.38 μM). Being more potent than the benchmark antivirals, amantadine and rimantadine, one major drawback (in addition to more laborious chemical syntheses when compared to, e. g., amantadine) of these compounds is their significantly higher cytotoxicity in cell based assays146 as well as in vivo.144
These issues are addressed, in part, with compounds 76a and 76b (Scheme 11) which combine peptidic fragments with known immunomodulatory activity and aminoadamantane derivatives with their well-established anti-Influenza A activity.147 They both displayed comparable potency to the benchmark, amantadine, at an approximately four-times lower cytotoxicity against the MDCK cells used. Notably, 76a, incorporating the D-2-Gly(2-Ada) residue, showed the same MIC to Influenza A H1N1 as does amantadine (MIC50 = 12.5 μg/200 μL), while 76b, incorporating the L-Gly(2-Ada) residue, displayed an MIC50 that was eight times higher. Testing the same drug candidates against the H3N2 subtype on the other hand showed 76b to be four-times more active while both modified peptides were significantly less active than amantadine. The effect of the size of the N-heterocyclic ring in “rimantadine analogues” was studied next. The work that had been begun with 2-(1-adamantyl)pyrrolidine (69) was subsequently extended by screening, amongst other aminoadamantanes, 2-(1-adamantyl)piperidine 77 and 2-(1-adamantyl)azepine 78.148 While 77 was, as the close relative 69, more potent than amantadine against Influenza A2/Japan/305/1957 (H2N2), the homolog incorporating a seven-membered heterocyclic ring 78 turned out to be significantly less potent at an increased cytotoxicity. Re-iterating the “rimantadine-derived” polycyclics, the same authors prepared 2-(1-adamantyl)-2-methyl pyrrolidine 79, the azetidine analog 80 and the aziridine derivative 81.149 In the MDCK cell based assay these compounds showed that reducing the heterocyclic ring size from a five-membered pyrrolidine to a three-membered aziridine gave test drugs with decreased potency. As noticed with numerous adamantane amines earlier, N-methylation also was detrimental for anti-Influenza A potency. While the test drugs' EC50 was in the low micromolar range for 79 and 80, meaning that they were more potent than amantadine and rimantadine in the same assay, their cytotoxicities were also much higher. For 81, finally the minimal cytotoxic concentration (55 μM) was below its EC50. As a reiteration of the bis-amino adamantane derivatives that had been studied before, a library of 2-adamantyl substituted (di)amino derivatives was reported.150
Scheme 11.
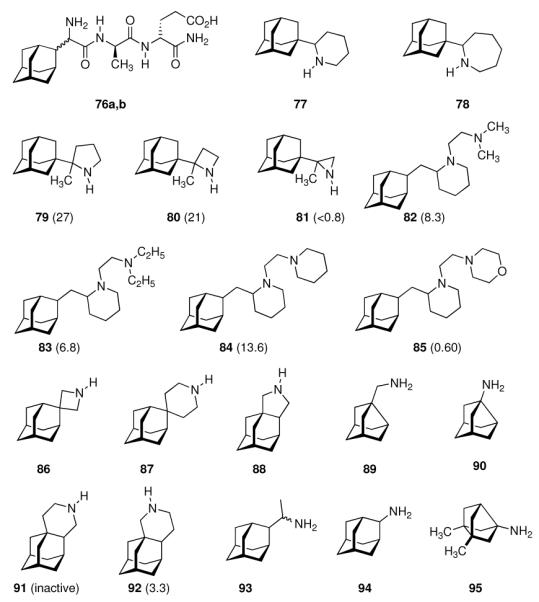
Recently reported aminoadamantanes with anti-Influenza A activity. Numbers in parentheses are potencies relative to amantadine in the same assays.
The piperidines 82 and 83 as well as bis-piperidine 84 were up to 14-times more potent than amantadine, but strikingly morpholine 85 was only about half as potent as the “gold standard” used as a benchmark in the cell-based assay. Moving the heterocyclic nitrogen atom one position (cf. 80), screening of the spiro-[azetidine-3,2'-adamantane] 86 along with spiro-[piperidine-4,2'-adamantane] 87 resembled closely the observations made before: Both test compounds were more potent than amantadine and rimantadine (albeit tested here in a slightly different testbed using Influenza A2/Hong Kong/7/1987 (H3N2) viruses in MDCK cells).151 Structure 84 furthermore displayed good activity against Trypanosoma brucei (see chapter 4.5). Again, N-methylation caused a dramatic reduction in these compounds' anti-Influenza A activity. Representing another recently studied class of aminoadamantanes designed and screened as antivirals, 1,2-annulated ring systems have also been studied.152 The potency of 88 along with a marked decrease in activity when N-alkylated (compared to amantadine, anti-Influenza A (H3N2) activity of 88 was 4.3-fold while the N-ethyl derivative was less potent than amantadine) confirmed the SAR from previous publications. Focusing on adamantyl piperidines, the differences in potency of 1,2-annulated piperidines 91 and 92 are striking.153 With 92 having a 3.3-fold higher potency against Influenza A/Hong Kong/7/1987 (H3N2) relative to amantadine, the isomer 91 was inactive in the concentration range studied. This is probably due to a conformation of the ammonium ion in which the NH- pharmacophore required for interaction with the M2 transmembrane domain is pushed into equatorial orientation, thereby distorting its possibility to interact favorably via hydrogen bonding. Not only the amino pharmacophore is, however, of importance for strong anti-Influenza A activity. As a recent example for studies focused on variations within the hydrocarbon “co-pharmacophore”, we conclude our survey with amino derivatives of noradamantane.154 The ones most closely resembling amantadine, 89 and 90, showed anti-Influenza A activities in about the same concentration range like the adamantane derivatives. The smaller bis-noradamantanes were less potent. The numerous amino derivatives of adamantane and some other polycyclic hydrocarbon cages (related in shape and size) screened to date, some of which we have presented herein to summarize four decades of SAR, demonstrate that the two co-pharmacophores (bulky hydrocarbon and amino group) have to be optimized to gain best potency. At the same time, minor stereoelectronic or conformational changes can strongly influence the drug's blocking of the M2 proton channel. This has also been demonstrated by comparing the antiviral potencies of “2-rimantadine” 93 and “2-amantadine” 94 with their respective 1-adamantyl brethren. 93 was 7.9 times more potent than amantadine and 3.7 times more potent than rimantadine, but 94 was significantly less potent than amantadine.155
Along with the increased occurence of resistances in most currently circulating strains of Influenza A (including H5N1 “avian Flu”156,157 as well as H1N1 “swine Flu”158,159), research in this field also seems to have reached a limit of maximum potency in the test compounds that, to date, has not justified the introduction of another aminoadamantane other than amantadine or rimantadine, also considering toxicity data as well as the more laborious synthesis for structurally somewhat more complex compounds. SAR of aminoadamantanes and related cage amines addressing resistances is, however, still a field of research actively pursued as exemplified by 95 (Scheme 11). As Influenza A strains resistant to the “adamantanes” carry mutations of the M2 transmembrane domain such as S31N or V27A, ring-contracted and ring-expanded amantadine derivatives have been envisaged to be valuable. Indeed, bisnoradamantane derivative 95 could be shown to inhibit the S31N mutant ion channel as expressed in Xenopus oocytes. Plaque reduction assays corroborated the electrophysiology measurements.160
Facing the paucity of anti-Influenza A drugs and targets, there still seems to be a role for the classic “adamantanes”, amantadine and rimantadine, in antiviral chemotherapy against Influenza A. (Dual) resistance develops also against the neuraminidase inhibitor, Oseltamivir.161 Furthermore, comparison of the actual clinical efficacy between amantadine and the neuraminidase inhibitors gave similar potency of the two classes of chemotherapeutics, at least against swine Flu.162 Monitoring genetic sequences obviously does not fully suffice to reflect the sensitivities of circulating as well as pandemic Influenza A strains to the aminoadamantanes. In H1N1 “swine Flu”, which was reported to be resistant to amantadine, yet later found to be sensitive to amantadine,162 the S31N mutation of the M2 protein modulates this ion channels' proton conductivity only mildly. Together with the findings that triple anti-Influenza A chemotherapy utilizing Ribavirin, the neuraminidase inhibitor Oseltamivir and the M2 blocker amantadine is synergistic and superior in vivo to the treatment with single drugs or any dual combination,163 and that this triple combination significantly suppresses breakthrough of resistant virus in vitro,164 there still might be clinical relevance of the “adamantanes” in the fight against Influenza A.
4.1.2 Insights Into the Mechanism of Action: Blocking the M2 Channel
In the early years following the discovery of amantadine's anti-Influenza A activity, structural variations and SAR were performed without even knowing the target of the drug. It was known then that the aminoadamantanes inhibit virus replication at the early steps of the infection, namely the penetration of the host cell by the virus particle and the release of the viral RNA. In 1985, Hay and coworkers analyzed the susceptibility of genetic reassortants of Influenza A obtained from the co-infection of amantadine-resistant isolates and sensitive ones.138 The gene encoding the M2 channel from the amantadine-sensitive parent strain is required for sensitivity of the reassortants and this gene alone was found to determine the differences in amantadine sensitivity. “The characterization of drug-resistant mutants by determining the nature and location of amino acid substitutions has now pinpointed more precisely the primary target of the drug action to the M2 protein”, the authors concluded from their studies. Infrequent occurence of changes in the sequence of the Influenza A haemagglutinine (HA) in amantadine-resistant strains of the virus and the lack of correlation of these mutations with the sequence changes in the M2 protein ruled out the possibility that HA mutations would play a role in determining the “adamantane” resistance. There is, however, an indirect effect of the M2 protein's mode of operation on HA maturation and function: While there was no direct evidence of the function of the M2 protein, the same group in 1990 attributed an indirect effect of the function of the M2 protein on HA: M2 was found to be capable of modulating the pH of compartments in the exocytic pathway. This contributes, together with protection of the integrity of the acid sensitive HA, to a promotion of the maturation of the active HA conformation.165 Shortly thereafter, it was discovered that alterations in the amino acid sequence of the transmembrane domain of the M2 protein abrogate the anti-Influenza A activity of amantadine.166 The M2 mRNA, when injected into oocytes of Xenopus laevis, induced expression of an amantadine-sensitive ion channel, as studied via two-electrode voltage clamp. Oocytes expressing the wild-type M2, when bathed in a solution containing amantadine at 100 μM, did not show this ion channel activity, whereas oocytes expressing mRNA of an amantadine-resistant strain of Influenza A were found to display ion channel activity with little influence of amantadine in the bathing solution. Furthermore, the amplitude of the conductance of the M2 channel in the oocytes expressing the wild-type M2 protein could be modulated by pH. “Thus, although final proof of the M2 channel activity will require purification and reconstitution of the protein into artificial bilayers, the data reported here, when taken together, provide strong evidence that the Influenza virus M2 protein is a bona fide ion channel,” the authors concluded. The role of this ion channel activity as a part of the replication cycle of the virus was identified at that time as being responsible for elevating the pH in the transgolgi network of virus infected cells.165,167 An elevation in luminal pH allows passage of the HA glycoprotein (which largely determines the infectivity of newly synthesized virus particles) without acid conversion. With the antiviral drugs amantadine or rimantadine present, the M2 channels would not be able to perform this function and the HA molecules would prematurely undergo a conformational change that prevents the formation of infective virus particles. Testing the hypothesis of M2 being a proton channel, the transmembrane domain (25 residues) of the M2 protein (a viral protein which, even in its full-length, functional form is very short, having 97 residues only) was synthesized and incorporated into voltage-clamped planar lipid bilayers.140 Amantadine(20 μM), applied on both sides of the artificial membrane, was able to block the channels formed by the M2 transmembrane peptide (M2TM) within 30 seconds. Again using oocytes as the model system, the M2 channel was found to be sensitive to amantadine-and rimantadine- block to about the same extent. However, from these electrophysiology studies it could not be ruled out that the adamantylamines act allosterically, that is, by not physically blocking the pore that is being formed by four helices of the M2 protein's transmembrane domain, but instead acting from the outside of the four-helix bundle that is forming the functional proton channel.168 Utilizing neutron diffraction, a deuterated 25-mer M2TM peptide as well as deuterated amantadine were used to study the location of the interaction between the two molecules in 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) multilayers as a membrane model.169 Three different M2TM peptides (residues 22 – 46 of full-length M2) were studied. Amantadine was found to reside about 0.5 nm from the center of the model lipid bilayer, which corresponded to the area around residues Val27 and Ser31 – consistent with the concept of a steric blockage within the ion channel. No such interaction was found when studying M2TM peptides corresponding to amantadine-resistant strains. These findings cannot be considered a clean-cut evidence of the mechanism of action of the aminoadamantane antivirals; they are, however, tempting (vide infra). The reconstitution of the Influenza A M2TMD in lipid bilayers, considered a milestone in the study of the M2 protein's significance and pharmacological targeting,166 was achieved in 1994.170 It had been found previously171,172 that amantadine reduces the rate of fusion of the Influenza A virion with liposomes of viral strains that are sensitive to the drug. A late effect (vide infra) of amantadine in the replication cycle is the premature conformational change of HA at the time when it is transported through the trans-golgi network (TGN), a low-pH conformation of HA being not as infectious as the high-pH conformation. Therefore, M2 ion channels within the TGN act to keep the pH of the TGN lumen above a threshold for the low-pH conformational change. Reconstituted M2 proteins in planar bilayer membranes were found to be regulated by pH (the channel is opened at low pH and closed at high pH), regulated by voltage, it can be blocked by aminoadamantane antivirals (when the source strain is amantadine sensitive), and it displays a substantial ion selectivity. We point out here that the mechanism of action on a precise molecular level has been (and still is to date) a subject of debate. Presumably, the mechanism of amantadine being a “cork plugging into a bottle”173 is not precise enough to explain the findings from studies using various models and experimental techniques. The demand for a more precise model of the channel is evident.
By co-expression of mixed oligomers of M2 from virus strains that were amantadine resistant and amantadine sensitive, the stoichiometry of the active M2 ion channel was studied more precisely.174 The whole surface current of the oocytes used, when measured electrophysiologically before and after treatment with amantadine, indicated that the functional channel is indeed a homotetramer. In the normal replication cycle of Influenza A, only 14–68 M2 proteins are being incorporated into the new virion, which corresponds to 3–17 functional M2 ion channels. Modeling and Cys scanning of the M2TM together with electrophysiology on Xenopus oocytes shed more light on the proton conductance needed for pH adjustment.175 The functional model generated utilizing these techniques suggested a four-helix bundle reminiscent of a four-stranded coiled coil as being the central structural feature of functional M2 proton channels, with His37 being most important for both pH-activation of the channel and its conductivity. While the native M2 channel is also being stabilized by intermolecular disulfide bonds, there is a pH-dependence of both the association and amantadine binding of peptides corresponding to the M2 transmembrane domain.176 Binding of amantadine shifts the monomer-tetramer equilibrium towards the tetrameric species when studied in dodecylphosphocholine micelles. Both the tetramerization and amantadine binding are more favorable at higher pH, with a pKa associated to a His side chain. Therefore, these authors suggested that the drug is competing with protons for binding to the His residues via hydogen bonds, thereby stabilizing the tetramer in a slightly altered conformation. These structural changes upon interaction with the drug can be monitored, amongst other techniques, by studying the pH dependence of Trp fluorescence when the drug is present or absent.177 Purified full-length M2 and M2 mutants were synthesized recombinantly in E. coli and used as tetramers. pH-Dependent structural changes could be observed. In particular, the fluorescence of Trp41, being an important part of the highly conserved HXXXW motif in the M2 protein, was shown to be quenched by His37 below pH 6, and this quenching could be reversed by rimantadine. The drug seemed to prevent protonation of His37 and other pH-dependent changes in the environment of Trp41. The specificity of the inhibition of the proton channel activity by rimantadine demonstrates that the early SAR calling for a large, hydrophobic cage amine118 could be confirmed here. Furthermore, titration of rimantadine's influence on Trp41 fluorescence in the amantadine-sensitive “wild-type” M2 protein at pH 5 clearly showed a 1 : 1 stoichiometry of the drug to functional M2 tetramer. The reduction in Trp41 flourescence below pH 6 could be blocked by rimantadine, which appears to be due to quenching of Trp41 fluorescence through His37. Busath and coworkers reported the single-channel proton currents using full-length M2 protein reconstituted in planar lipid bilayers,178 and the channel was found to form “a highly selective proton channel with a low probability of being open and partial block by amantadine.” The single-channel conductance was found to be about 6 pS, with a probability of the channel to be in its open state of ≤ 0.03. The function of the M2 ion channel was reviewed179 as displaying these main features (most of which have been confirmed utilizing aminoadamantanes and amantadine-sensitive as well as amantadine-resistant M2 model systems): One helix loop that is common to the Influenza A M2 channel and the Influenza B M2 channel is the conserved HXXXW motif which is critical for ion channel activity. His37 forms a selectivity filter, whereas the Trp41 tetrad forms a gate. The mechanism of channel inhibition by aminoadamantanes can be deduced from amantadine resistant mutants: A30T and G34E mutations both lead to drug resistance, presumably because they introduce less lipophilic residues into the aqueous pore of the channel. Inhibition of this channel occurs more readily at elevated pH, and the aminoadamantanes only inhibit when applied from the N-terminal ectodomain. Furthermore, by neutron diffraction it could be shown that amantadine lies in the outer region of artificial membranes. Together with the 1 : 1 ratio (rimantadine : M2-tetramer), these findings show that the aminoadamantanes act from the outside of the aqueous pore, the adamantane co-pharmacophore interacting favorably with lipophilic residues that line the inside of the channel. Perhaps the ammonium hydrogen is involved in an interaction with the His37 tetrad. But still, this is probably not the whole story, because the His37-Trp41 interactions and their fine-tuning through subtle conformational changes upon both drug–target interactions and mutations that render M2 drug resistant also seem to play a major role.
As the next milestone, experimentally detected high-resolution structures of functional models for the M2 proton channel came up. A structural model of Influenza A M2's backbone structure when blocked with amantadine was derived from ssNMR constraint data from the transmembrane domain peptide.180 Little, if any, direct interactions between His37-imidazole and the drug could be observed. The drug-resistant S31N mutation does not show a significant change in backbone structure when compared to the native, drug-sensitive sequence because upon drug binding the NMR data indicated a significant change in the structure of the drug sensitive model peptide, but no such change for the amantadine-resistant M2TM peptide mutant, so these authors concluded that amantadine does not bind to this mutant. Note that the tetrameric channel in this structural model was generated from structural data derived from one helix and a series of rigid-body transformations of this monomer subunit. The same group also studied the influence of amantadine binding to M2TM in terms of chemical and dynamical properties.181 Binding of the drug lowered the pKa values of His37 by about three orders of magnitude, which is strongly interfering with the channel's main function, proton conductivity. The structure of M2TM (residues 22–46) at high pH and in the absence of the drug is more dynamic and structurally heterogeneous when compared to the amantadine-bound structure. Furthermore, in addition to the reported change in His37-pKA, the binding of the drug molecule also seemed to prevent the formation of low-barrier hydrogen bonds between the His37 residues of different helices. This suggests that the amantadine-block is not based solely upon steric hindrance, but instead on a mechanism that interferes with the His-facilitation of proton transport through the channel's pore. 19F NMR was used to resolve for the first time resonances from a complex between fluorinated amantadine (96, Scheme 12) and a M2TM (residues 22–46, Udorn strain) peptide.182 The complex was studied in dodecylphosphocholine (DPC) micelles. In earlier work, the lineshape of 1H resonances of 96 had been utilized183 and found to give resonances too broad to be detected above pH 7.5, when the open-channel binding of amantadine to the M2 channel is expected to occur. When using 19F NMR, this could be circumvented and the authors found that the homotetramer of their fluorinated M2TM model peptide appears at pH 7, which is ~ 0.5 units below the pH threshold reported before using CD spectroscopy as the experimental method.176 They concluded that, above pH 7.0, the M2TM peptide adopts a neutral tertrameric form that is capable of binding the drug with high enough affinity to observe changes in the NMR spectra. Another ssNMR study of the M2 transmembrane peptides, aiming at elucidation of atomic resolution conformation and dynamics, was conducted in DLPC bilayers with and without amantadine being present.15 The focus lay on the conformational changes within the transmembrane domain while interaction with the drug takes place; this was monitored using magic angle spinning (MAS) 13C- and 15N NMR techniques. Again, comparable conclusions as before were drawn: Amantadine affects the M2 protein by changing the distribution and exchange rates between low-energetic conformers, wheras the average conformation and orientation seem to be only subtly modified. The most significant structural perturbations upon amantadine binding could be observed for the backbone of Gly34 and Ile35 as well as the Val27 sidechain, which gives the strongest change in chemical shift. Mutations of these residues are known to give increased amantadine resistance. While the apo-peptide backbone undergoes significant motions on a μs timescale, the binding of amantadine alters motional rates and reduces the conformer distribution. One decisive feature of the M2TM structure in this model environment, therefore, appears to be the presence of multiple low-lying conformations that are modified and selected via amantadine binding. The conformational plasticity seems to be essential for proton conduction through the M2 channel and the gating mechanism as well as, at least in part, amantadine's mechanism of action can be attributed to this modifying and selective influence on conformer distribution. Given the abovementioned NMR findings and the observance of fluorescence quenching of Trp41, it was attempted to screen a series of adamantane derivatives by a purely in vitro- assay.177,184 The series of 2-alkyl-2-aminoadamantanes that was studied amongst a number of other adamantane derivatives gave binding constants to a purified full-length M2 protein isolated from a Weybridge strain (Influenza A/chicken/germany/27 (H7N7)) virus that were comparable to the binding of amantadine (Scheme 12, compounds 94, 97–100), however, the relative binding affinities were not directly comparable to the relative antiviral potencies obtained in cell culture.
Scheme 12.
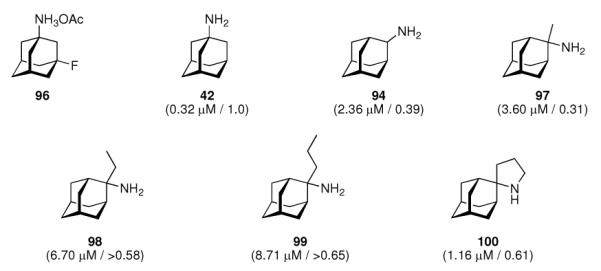
Fluoro-aminoadamantane 96 utilized in the 19F-NMR structure elucidation of M2TM peptide, aminoadamantanes 94 and 97–100 assayed in a Trp41 fluorescence-quenching assay. Values in parentheses are the binding constants to the model peptide and their potencies against Influenza A/Japan/305/57 (H2N2) as observed in an MDCK cell based assay, relative to amantadine
The authors concluded that the observed relative potencies in the cell-based assay are due to not only the pure interaction of the drug candidate with the target protein complex's binding site, but also the way of getting there by means of membrane diffusion.
The recent elucidation of high-resolution structures of M2 model systems both in solution via NMR methods and via X-ray crystallography, and both with and without an aminoadamantane drug being present, have fueled an intensive debate concerning the precise mechanism of action of the aminoadamantanes that, however, seems to be settled today. The X-ray structure of the transmembrane-spanning region of the M2 protein in its tetrameric form was reported by DeGrado and coworkers in 200814 (Figure 3, panels A – C). To obtain high resolution data, a series of peptides corresponding to the M2 transmembrane domain were used as the model system. A mutant with Selenomethionine instead of Ile33 could be used for crystallization with six molecules of the detergent octyl-β-glucopyranoside, forming a bilayer-like environment. For cocrystallization with amantadine, a lower pH of 5.3 and a different model peptide, incorporating the G34A mutation, had to be utilized. In the co-crystals, the drug (amantadine) is surrounded by residues that are mutated in clinical Influenza A isolates (Val27, Ala30, Ser31, Gly34) that are drug resistant. This drug-binding site within the tetramer of the M2TM peptide was found to be nearly identical between the two structures (with and without the drug molecule), these residues, however, do not include the residues responsible for the pH-dependent gating of the channel, His37 and Trp41. Amantadine's amino group is oriented towards, but does not directly touch via H-bonding, the His37 imidazol-residue. The authors furthermore modeled the most important mutation in Influenza A responsible for the widespread amantadine resistance, S31N,17 into their model of the M2 pore based upon X-ray data. The Asn residues in the amantadine-resistant mutant lead to a change in size and polarity of the amantadine binding site, while retarding H+ conductivity. The authors concluded: “The crystallographic structures are in excellent agreement with a wide body of functional and spectroscopic data and provide a basis for the design of new inhibitors that target amantadine-resistant mutants of M2. Inhibitors that target the cavity adjacent to the highly conserved His37 and Trp41 residues might reclaim the M2-blocking class of drugs both for the prophylaxis and for treatment of ongoing epidemic outbreaks and future pandemics of this deadly pathogen.” Back-to-back in the same issue of Nature, Schnell and Chou reported another high-resolution structure of a M model system.185 Their model is based upon a different construct of the M2 protein, residues 18–60 with a mutation (C50S). The peptide incorporates an unstructured N-terminus (18–23), the transmembrane-helix (25–46), a flexible loop region (47–50), and a C-terminal, amphiphatic helix (51–59). This model peptide formed stable tetramers in dihexanoylphosphatidylcholine (DHPC) detergent micelles. The tetramer in presence and without the “M2 channel-blocker” rimantadine was studied by means of NMR techniques at pH 7.5. By analyzing intermolecular M2–drug NOEs, they assigned a binding site of the rimantadine molecule between adjacent helices at the C-terminus of the transmembrane domain in proximity to the “gating” residue Trp41 (Figure 3, panels D – F). In total, four molecules of rimantadine bind at four equivalent sites on the side of the helices facing the lipid layer, thereby destabilizing the the closed conformation of the pore. These unexpected findings could explain some of the SAR observations made many years before.118 Like DeGrado and coworkers, Schnell and Chou also discussed the role of amantadine-resistant mutants. L26F, S31N, and L38F are mutations they assigned to the helix-helix-packing interface, whereas V27A, A30T, and G34E are mutations directly affecting the pore-lining sidechains. These drug-resistance mutations seem to counter the effect of drug binding in their model system by either providing more hydrophilic residues to the pore or by weakening the packing of the four transmembrane helices, thereby facilitating channel opening. “Having a cork-plugging-the-bottle model is not sufficient to explain all the results,” they concluded, and instead they proposed an “allosteric” mechanism of action of the aminoadamantane class of drugs, where drug-binding makes the M2 channel harder to be opened by pH changes.
Figure 3.
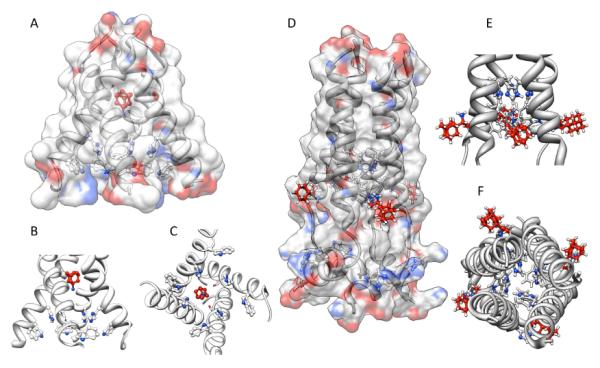
Binding modes of aminoadamantanes to Influenza A M2 model peptides. Panel A – C: Amantadine binding inside the pore of M2 transmembrane domain (X-ray diffraction data, pdb code 3c9j). Panel D – F: Rimantadine binding from the outside of the M2 model peptide (representative NMR data, pdf code 2rlf). The drug molecules are shown in red, His and Trp residues in the M2 model peptides are depictes as ball-and-stick models. See text for a discussion.
The mutations, in turn, destabilize the closed channel, making it easier to open – which counters the effect of the aminoadamantanes. This model could be advantageous for drug design, because drugs that are larger than hydrated ions do have to overcome a higher energetic barrier to find a binding site inside a pore than to bind to the channel complex from the membrane-facing “outside”. DeGrado's group commented on these findings by stating that Schnell and Chou's model structure represents a biologically relevant closed form of the M2 channel that is unable to bind amantadine in its central cavity. Both of these high-resolution structures are, however, models that necessarily are limited in describing the “truth” because they use fragments of the M2 protein only, are studied in model bilayers or micelles at different pH, the channels are not verified to be functional proton channels in the respective environment,186 and the two groups are using different antiviral drugs as model compounds. DeGrado's group was subsequently studying electrophysiologically the effect of the mutations leading to drug resistance in M2 ion channel activity.187 In this study, sightly longer model peptides for M2 were used (residues 19–46), because, in addition to being putatively a closer model for full-length M2 structure and function, these peptides also contain the Cys residue that plays a significant role in forming the functional tetrameric channel via intermolecular disulfide bonds. Furthermore, for functional assays, they were using full-length mutants. The sequence variants were shown to be thermodynamically more stable in lipids than they are in micelles, yet the effects of mutations on stability are correlated in both model environments. For the L26F and S31N mutations, the M2 channel stays active, but the sensitivity for amantadine is diminished. As an obvious contradiction to the “allosteric” model, they compared the ion channel activities of various mutants expressed in Xenopus laevis oocytes: The specific proton channel activities of both the S31N and the L26F mutant were found to be about 1.4 times the activity of the wild-type, amantadine-resistant channel. Additionally, these two mutants are about 100-fold more resistant to amantadine than the wild-type channel, but the “allosteric” model asks for a similar change in their conductance characteristics. Consequently, their electrophysiology results agree better with their own, X-ray structure-based model of amantadine binding to the pore of the channel, even more so because the NMR-based model from Schnell and Chou seems to have some weaknesses: Its stoichiometry of drug binding is not 1 : 1 (for aminoadamantane : M tetramer) as has been concluded from experimental data,168,177 the location of mutations which confer drug resistance are mostly close to the N-terminus of the transmembrane part, whereas in the NMR model the drug binds close to the C-terminus of the transmembrane domain. Furthermore, in the functional studies of DeGrado's group, mutations of residues that were predicted to be a part of the “allosteric” drug binding site did not interfere strongly with the amantadine blockage of the channel. Binding kinetics are very slow, which also does not favor the “allosteric” inhibition model, and even in the NMR structure itself, there appears to be an unfilled site within the channel that could accomodate amantadine (or rimantadine, for that matter). As a conclusion, the authors suggest that the “allosteric” sites from the NMR based structure model are secondary sites that come into play when certain conditions, like a closed channel, occur. Extensive molecular dynamics performed on the M2 transmembrane domain peptide, with and without amantadine as the prototype M2-active drug, shortly after suggested that the Val27 residues form a “secondary gate” of the channel, the primary gate being the one formed by the Trp41 sidechains along with the His37 residues.188 The role of these hydrophobic Val27 sidechains is to interrupt the water wire inside the channel, and upon amantadine binding, this water-free region is expanded, thereby blocking proton conductivity. The MD simulations confirmed the amantadine binding site found earlier from neutron diffraction data,169 being formed mainly by residues Ser31 and Ala30. Amantadine is predicted to be separated by three layers of water from the His37 proton relay. This primary proton gate, HXXXW, was recently studied via 19F ssNMR in a native lipid environment, but no aminoadamantane block was studied in this work.189 When studying the M2 transmembrane domain peptide in lipid bilayers by means of ssNMR techniques with and without amantadine being present, the drug did not seem to change the average structure of the channel very much, but spectral linewidths were narrower upon interaction with the drug, indicating a drug-induced change of the protein dynamics in the membrane.190 Among all residues, Ser31 showed the largest drug-induced changes in chemical shift, conformational dynamics, and conformational disorder. This residue, therefore, may be the amantadine binding site via H-bonds with the drug's amino group. MD simulations that were based upon ssNMR data180 of the M2 transmembrane domain peptide in POPC bilayers were aiming at a better understanding of amantadine resistance.191 These authors put their focus on a binding site of the drug inside the channel and not on the “allosteric” model of channel inhibition. Pielak, Schnell and Chou192 focused on their allosteric model, in particular on the binding site of the aminoadamantane, using NMR methods. The S31N mutated construct was analyzed and compared to the wild-type structure reported earlier, but this mutation had little effect on the pore structure. On the other hand, this mutation leading to drug-resistance in circulating viruses led to reduced binding as observed via NMR in this model system. Consequently, drug-resistant mutants seem to impair “allosteric” drug-binding by destabilizing the helix-helix-assembly. Ser31 did not directly interact with rimantadine (DeGrado and coworkers dicussed such an interaction for amantadine). A functional analysis of liposomal proton influx as measured for various M2 mutants was also performed.
The relevance of the “allosteric” binding site located outside the pore within the membrane, as discussed for rimantadine using the M2(18–60) peptide as a model at low pH-structure185 (vide supra) has recently been challenged by the findings reported by Cady et al.16 using 13C-labelled M2(22–46) peptide reconstituted in DMPC vesicles and ssNMR methods to study the binding of perdeuterated amantadine at high resolution and in different drug/peptide ratios. Only at very high concentrations of the drug, these authors could detect a binding of the aminoadamantane from the outside of the channel, while the “high-affinity” site inside the M2 pore is still being occupied by the drug, as it already is at an amantadine / M2-peptide ratio of 1 : 4, which reflects the previously reported binding stoichiometry. Furthermore, their 13C{2H}REDOR studies shed light on the primary site within the M2 lumen, indicating amantadine's hydrocarbon moiety to “fit snugly” into the N-terminal lumen. The amino group is most likely oriented towards the cavity near His37, as already postulated earlier. These authors, however, found that amantadine's structure is not perfectly optimized for the M2 channel pore, thus suggesting the chance of designing M2 channel blockers with enhanced affinity. Since this binding site involves the most conserved amino acids of Influenza A M2, these novel drugs193 could evade drug resistance. Recently, solid-state and solution NMR analyses on M2(18–60) in lipid bilayers and dodecylphosphocholine (DPC) micelles that also supported the “inside” binding mode of both amantadine and rimantadine have been reported.194 Rimantadine's methyl group was found to be in proximity to the Gly34 backbone, which results in better space-filling by the drug molecule and, consequently, its higher affinity to the pore compared to amantadine. Comparing amantadine and rimantadine binding, these NMR studies suggested rimantadine to have a slightly different equilibrium constant between the high-affinity (pore) binding site and the allosteric binding mode. Still, design of better space-filling M2 inhibitors could become feasible based upon these data, in particular, molecules that better fill the channel for both amantadine-sensitive and amantadine-resistant M2 channels. Support for the allosteric action of the aminoadamantanes through the “outside”, membrane-facing binding to the M2 tetramer further vanished based upon MD simulations using the crystal structure data as a starting point.195 In this study, all protonation states of the His37 tetrad of membrane-bound M2 transmembrane domain bound amantadine at the inside of the tetrameric bundle. No significant affinity of adamantane for the external binding site close to Asp44 was found. The external binding reported for very high concentrations of aminoadamantane drug correspond to secondary binding that are not pharmacologically relevant. A transporter-type mode of proton conductance through the channel, gated by Val27 and His37 tetrads, was suggested, at least in part explaining two slightly different binding modes inside the channel. Such a plasticity of the channel, required for action as a “proton pump”, is highly dependent not only on drug binding, but also on the environment of the model chosen as exemplified by, e. g., the membrane thickness.196 Likewise, structures of M2 detected in detergent micelles might have significant differences from those found in lipid bilayers.197 Validation of the models chosen to study M2 dynamics and its pharmacological blocking therefore requires scutiny. To this end, Influenza A M2 (22–62), a truncated model system that was studied via ssNMR techniques before, was subsequently functionally reconstituted in lipid bilayers.198 It displayed normal functionality in terms of proton flux activity, amantadine sensitivity, and proton selectivity. Detailed comparison of the available structural data and free-energy calculations based upon MD simulations indicated the pore-binding mode to be ~ 7 kcal/mol favored over the channel surface-binding “allosteric” mode.199 The aminoadamantanes' binding site in the pore is an aqueous cavity adjacent to the channel filter, but the channel blockers such as amantadine and rimantadine do not completely crowd out all of the water molecules from the inside of the channel. The role of the residual water molecules inside an M2 – aminoadamantane complex can be studied based upon high-resolution X-Ray data.200 The water cluster inside the channel was also studied via 2D-IR methods, showing its characteristics and proton transduction relevance. Water flows into the channel in its open state. Upon protonation of the His37 imidazoles, new conformations are being developed that allow for the water inside the channel to become “more liquid”, enabling diffusion of the protons into the virus interior.201 The precise mechanism of M2 proton transport into the virion can also be monitored by fluorescence correlation spectroscopy. One such “breath” of the transporter-like behavior of M2 channels takes about 500 μs, and bound drug molecules occlude the channel to block its “breathing”.202 Solid-state NMR studies later focused on how amantadine blocks the proton flux through the channel. Binding of amantadine suppresses proton exchange from His37 to the water within the channel upon reorientation of the His37 imidazole rings.203 Further ssNMR measurements showed once more the pharmacologically relevant binding site of the aminoadamantane drugs to be the pore of M2's transmembrane domain, this time using M2 (21–61) as a fully functional model system in DMPC bilayers.204 The drugs bind at Ser31 in this fully functional M2 channel model, and the additional fragment of the model peptide that constitutes amphipathic cytoplasmic helices does modulate the transmembrane domain's ability to bind channel inhibitors such as the aminoadamantanes. Combining computational methods and experimental verifications, a rationale how the M2 channel develops resistance to the aminoadamantanes on a molecular basis has been put forward.205 In this study, amantadine fits snugly into a binding pocket delimited by Val27 and Ser31 in the wild-type channel. The drug's positively charged amino group thereby is able to block proton conductance through electrostatic repulsion. Drug resistant channels either display mutations with larger pore-lining sidechains such as S31N to inhibit drug binding or mutations with smaller sidechains like V27A, leading to an larger binding pocket volume. Drug molecules bound in this “inflated” site remain mobile, the proton flux can not be blocked sufficiently by their amino group.
After performing a large-scale sequence analysis of the M-gene of more than 5,000 Influenza A virus isolates, Oshitani and coworkers concluded that there does not seem to be sufficient selectivity pressure through aminoadamantane antivirals on the Influenza A M2 gene to fully explain the widespread amantadine resistance observed in recent years.206 Strains with the S31N mutation appeared in human populations as early as 1930, decades before the discovery and introduction of amantadine and rimantadine. Clusters of M2 genes mutated at residue 31 have emerged separately in the late 1990s in different hosts and virus subtypes almost simultaneously, but this could not be related to amantadine use. M2 genes with the S31N mutation might have increased through “genetic drift” instead, and recent isolates show that mutations back to amantadine-sensitive might give the aminoadamantanes another chance in the future.207 As a conclusion for our survey of studies aiming at elucidation of the mechanism of action of the aminoadamantanes, we briefly summarize recent accounts on structure-activity relationships of novel compounds, now the mechanism of action of the aminoadamantanes blocking M2 has been elucidated by high-resolution structures. DeGrado and coworkers utilized their X-ray based model of inhibition of the M2 channel through aminoadamantanes by binding to a site inside the pore of the tertramer to design inhibitors for amantadine-resistant mutants of the M2 ion channel.208
Starting from the structure of BL1743 (101, scheme 13), a molecule known to bind inside the M2 pore, these authors have designed and tested a number of M2 channel blockers in amantadine resistant strains of Influenza A. The competition between BL1743 and amantadine further supported the binding mode of the aminoadamantanes to lie inside the outer portion of the channel's pore. Spiro[5,5]undecanes like 104 were found to be promising, approaching amantadine's potency even in this preliminary screening of a very small library of compounds (Scheme 13). For this reason, it seems to be possible to design antivirals specifically for amantadine-resistant “escape mutants” of Influenza A. As hydrophobicity is critical in improving antiviral activity of anti-Influenza A M2 channel blockers, replacement of the quaternary carbon in spirane amine inhibitors with silicon has been studied.209 Indeed, silanamine 105, sterically more demanding than is 104, was found to be able to fill in the extra space of the binding pocket generated by the V27A mutation that renders Influenza A amantadine resistant. The silaspiranes were as potent as their carbon analogues in blocking wild-type M2, but more potent targeting the drug resistant channels. Similar structure-guided considerations, based upon MD simulations, led to further development of spiranamines such as 102.210 Also targeting the previously “undruggable” V27A M2 channel (the major mutation emerging from drug pressure), “longer and more extended” drugs, compared to amantadine, have been envisioned to fill the additional volume in the binding site generated by this mutation and possibly reaching closer to the water cluster associated with Gly34 and His37, respectively. Molecules larger than amantadine are required that still are able to fit into the wild-type binding pocket. As a first step, 102 was found to inhibit M2 (V27A) with an IC50 = 84.9 ± 13.6 μM, while 104 was devoid of any blocking of this mutant. Homologs 106 and 107 displayed even higher M2 (V27A) blocking capabilities. Bis-cycloheptyl spiranamine 108 showed a two-fold higher potency than amantadine against wild-type M2, and an IC50 = 11.3 ± 0.7 μM against the V27A mutant. Diffuse densities of the upper ring in the spiranamine channel blockers led these authors to speculate that the binding affinities and, consequently, the channel block, might be further enhanced by modulating the bulk of this ring. To this end, they synthesized spiroadamantane 109 (Scheme 13). Indeed, 109's adamantane motif filled the extra space near Ala 27 in the amantadine-resistant mutant in MD simulations. In wild-type and Leu26Phe mutants the amino group was pushed closer to the His37 tetrad, resulting in water-mediated hydrogen bonding of its amino group with the His sidechains. The spiroadamantane 109 showed an IC50 = 0.3 μM, more than 280-fold lower than for 102. As MD computations and homology models suggest similar central cavities for the amantadine resistant M2 V27A and L26F mutants, repectively, it is no surprise that 109 also was found highly active against M2 (L26F). These findings were corroborated by plaque reduction assays. Strikingly, originating from amantadine resistance, structure-based design of novel cage amines to tackle this challenge at first led away from the adamantane motif, only to find novel adamantane derivatives such as 109 a few years later that are capable to inhibit Influenza A replication, at least in in vitro assays.
Scheme 13.
Recent channel blockers for amantadine-resistant M2 ion channels. The percentage of remaining M2 channel activity after application of 100 μM compound inhibition.208
In conclusion, amantadine and its derivatives have had (and still have) significant impact on the elucidation of the structure and dynamics of the M2 ion channel. Blocking this minimalistic ion channel has two effects on the replication cyle of Influenza viruses in infected cells: An early effect is concomitant with fusion between endosomal and viral membranes, which is mediated by haemagglutinin (HA). The M2 channel enables acidification of the viral interior inside the endosome, leading to uncoating, the release of ribonucleoprotein from the viral M1 matrix protein and finally nuclear infection.211 The late effect is associated with M2 channel function in post-golgi-vesicles, where acidification of these vesicles is undesirable, and the M2 channel counters any acidification here, so blocking the channel is detrimental for virus replication.165 The replication cycle is vastly similar when comparing Influenza A and Influenza B virus; moreover, Influenza B also encodes for a protein that features ion channel activity (BM2).212 The M2 channels of Influenza A (AM2) and Influenza B (BM2) have been compared in recent review articles based upon molecular structures and dynamics.213–215 They share the decisive HXXXW helix loop, but an alignment of the predicted transmembrane domains shows that AM2 and BM2 share almost no sequence homology. In particular, polar serine residues in BM2 are present in positions that presumably are pore-lining (Figure 4).216
Figure 4.

Sequence comparison between Influenza A and Influenza B M2 proteins. The transmembrane domain is underlined, the proton-gating HXXXW motif is in bold, the Ser residues presumably rendering the BM2 channel amantadine resistant by making the inside of the channel less hydrophobic are bold and in italics.
These pore-lining hydrophilic residues would reduce the affinity of the pore for binding to the lipophilic cage hydrocarbon moieties of the aminoadamantane antivirals amantadine and rimantadine, at the same time enhancing the proton channel activity. This is actually observed for the BM2 channel. Both AM2 and BM2 proton flux is gradient-driven and a “channel breathing” mechanism as outlined briefly above is the probable H+ translocating mechanism employed by both channels. Consequently, chimeric channels of AM2 and BM2 transmembrane domains, likewise, are being blocked by the aminoadamantanes through occluding the channel pore.217
While the approval of amantadine in 1966 and rimantadine in 1993 (note that the latter had been used in Eastern Europe before) were major achievements in combating the Influenza A virus, genetic drift rendered this class of drugs essentially futile. Still, they play a significant role in elucidating the precise molecular mechanism of their target, the M2 ion channel. While competing models in drug binding to the target have been discussed, extensive studies on the structural biology of M2 have led to the current consensus that the molecular mechanism of action of the aminoadamantanes primarily is a channel-occluding process. However, the picture is much more complex as model peptides, membrane environment, and other model setups have to be validated carefully. However, the structural and mechanistic insights gained for the M2 channel and its blocking through the aminoadamantanes has shown to offer opportunities to design new drugs that target M2, in particular, of “adamantane-resistant”218 strains. Oddly enough, this research –again– gave adamantane derivatives such as 109 as optimized lead structures. It remains to be seen whether adamantane derivatives will reclaim their pivotal role in this field of medicinal chemistry, but “…we have many reasons to believe that the new development in obtaining accurate picture of drug inhibition and drug resistance of the M2 channels will lead to development of more specific M2 inhibitors for treating influenza infections”.213
4.2 Herpes Simplex – The Tromantadine Story
While synthesizing and screening new aminoadamantane derivatives as antivirals, one of the earliest target viruses to be combated chemotherapeutically was the Herpes simplex virus (HSV). While one strategy was to synthesize nucleobase analogues that are being introduced into the viral DNA within the DNA replication through the DNA-polymerase of the respective virus, terminating and stopping DNA synthesis, another strategy is the inhibition of the viral polymerase.219 Drugs working through the former mechanism are Aciclovir (Zovirax) and Ganciclovir (110, Scheme 14). Following a prodrug approach and earlier findings on adamantane-modified nucleobase derivatives,9,220,221 Ganciclovir has been modified for topical application through esterification with lipophilic carboxylic acids, the prototype of which being adamantane-1-carboxylic acid.222 When compared to 110 and the bis-propionate 111, the bis-adamantanoylated prodrug 112 in addition to higher lipophilicity (see ALOGPs data in Scheme 14) also showed a markedly increased stability under acidic conditions, facilitating formulation and storage. Enzymatic saponification through esterases was studied in tissue homogenates and found to be in about the same range for the two bis-esters.223 An added benefit of the lipophilic bisadamantanoyl prodrug when considering its projected topical application is its low oral bioavailability, probably caused by its extremely low aqueous solubility (110: 3.7 mg/mL, 111: 2.1 mg/mL, 112: 2.4 × 10−5 g/mL).
Scheme 14.
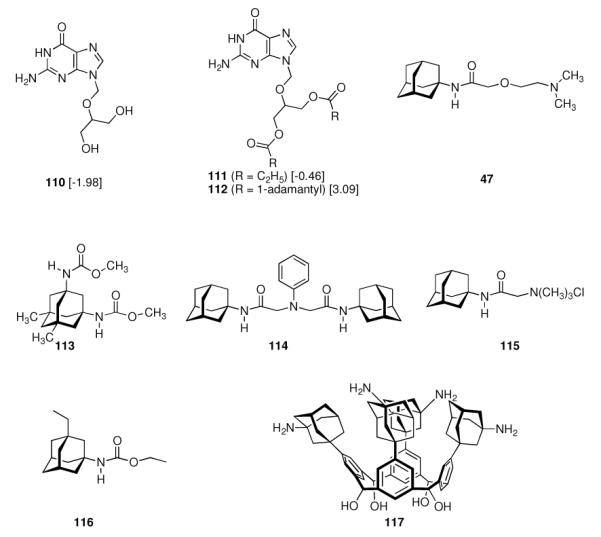
Anti-HSV agents. Values in square brackets: ALOGPs data.
Another class of compounds that have proven to be effective against Herpes simplex I and II are the aminoadamantanes – a result of the routine screening of a multitude of derivatives with respect to their activities against viruses other than Influenza A. Dialkyl esters of 5,7-dialkyladamantane-1,3-dicarboxylic acid like the dimethyl dicarbamate 113 (Scheme 14) were reported to show “some degree of protection” from Herpes simplex infections in mice.224 The di-n-propyl derivative was, however, inactive. 47 and 48 (Scheme 8), synthesized as a part of SAR modifications alongside the anti-Influenza A antiviral lead structure, were identified as active against Herpes simplex viruses in a plaque reduction assay117 where amantadine was inactive.225 Tromantadine (47) did not show a disadvantageous pharmacological profile in terms of acute and chronical toxicity, sensitization, and skin irritation in animals,226 so a small library of 37 compounds including 114 and 115 was synthesized and tested to establish an initial SAR,227 but no close structure-activity relationships could be found. Later, the adamantane carbamate motif was studied once more, identifying the ethyl carbamate 116 as an agent with antiviral activity that borders cytotoxicity.228 As mentioned before as well as in chapter 4.1.1, novel aminoadamantanes are frequently being screened in HSV assays as well, but no spectacular insights, other than mounting evidence for tromantadine's sidechain and the lipophilic adamantane moiety being equally important for anti-HSV potency, have been reported.141,142,229 An exotic strategy to combine the antiherpetic activity of some calixarenes and aminoadamantanes, the synthesis and antiherpetic activity of N-(3-amino-1-adamantyl)calix[4]arenes (e.g., 117) has been reported recently.230
Amantadine itself was identified showing some symptomatic benefit in a double-blind, placebo controlled clinical trial of a group of 100 patients infected with Herpes zoster, the virus that is causing shingles.231 Patients treated with amantadine had a significantly shorter duration of pain, the mode of action of the drug being unknown. Herpes zoster, however, is caused by the Varicella zoster virus; albeit belonging to the same family of herpes viridae, they constitute a different target presumably requiring different chemotherapeutics; however, to round off this chapter, we mention this study here.
Tromantadine (as the hydrochloride) is marketed as a 1% hydrogel (Viru-Merz®) for topical treatment of recurrent infections of Herpes simplex, to be applied before blisters appear. Its precise mechanism of action is still unknown, despite numerous studies addressing it. Comparative clinical trials of tromantadine and Aciclovir in the topical treatment of recurrent Herpes orofacialis232,233 did not show significant differences in the activity of the two drugs. Unlike Aciclovir, tromantadine does not require a viral kinase to become activated, so tromantadine is particularly interesting for the treatment of Aciclovir-resistant strains, which account for about 5 – 7% of HSV infections. In rare cases (< 1%), contact dermatitis occurs as an adverse effect of tromantadine.
Early attempts to elucidate tromantadine's MOA examined the timing of events required for cell fusion induced by HSV-1;234 adamantanone as “metabolic inhibitor” blocked excessive fusion of an infective HSV strain. When applied at different time points before and after the inoculation of two different tissue culture cell types with HSV-1 (KOS strain) virus, tromantadine was active well below its cytotoxic concentration, essentially not as a function of the timepoint the drug is added.12 As a conclusion, these authors believe the inhibitory properties of tromantadine on Herpes simplex virus multiplication to show at early and late events of the virus' reproductive cycle. Tromantadine's side chain seems to potentiate these effects. Studies of the metabolism of tromantadine showed more than 50% of the drug to be eliminated unmodified (also see chapter 9),235 confirming the drug itself as the active compound. The interaction of tromantadine and amantadine with model phospholipid membranes was examined in regard to the “working hypothesis” of tromantadine's MOA.236 The drug is believed to prevent membrane fusion of the HSV virion's membrane with the plasma membrane; therefore, balanced hydrophobic/hydrophilic group sizes is regarded crucial. Like other peptide-derived antivirals, Cyclosporin A stabilizes the bilayer phase of biomembranes; tromantadine had a similar effect, whereas amantadine was perturbing the model bilayers. Next to inhibiting cell-cell fusion, tromantadine also inhibits syncytium formation and seems to affect the synthesis of a protein required for fusion.22 In summary, tromantadine and other antiherpetic adamantane derivatives apparently, at least in part, exert their activities through influencing the fusion and assembly of lipid membranes, respectively.
An interesting aspect with respect to the MOA of tromantadine was reported recently, when adamantane substituted 2-phenylbenzimidazopyridines were found to display antiherpetic activity also. This class of compounds was initially identified during a project to identify small molecules that suppress excessive IgE response in allergy and asthma. To this end, a chemically diverse library of more than 300,000 compounds had been screened in a cell based HTS-assay, and the “hits” were 2-phenylbenzimidazoles like 119 (Scheme 15).237 These were subsequently modified, identifying derivatives modified with aliphatic cycloalkyl residues that showed an increased “block” of IgE-response. Considering potency as well as oral bioavailability, the authors selected the adamantyl amide 118. The library of adamantane derivatives studied was greatly expanded in subsequent work that was also directed towards identifying the mechanism of action of these compounds.238 The similar potency of the active compounds against multiple responses in different cell types suggests a highly conserved target integral to the cells that have been utilized, probably the Golgi or the endoplasmatic reticulum. The active structures had a strong preference for terminal large lipophilic groups, e. g., adamantane moieties.
Scheme 15.
2-phenylbenzimidazoles (118–121) and imidazopyridine 122 as anti-HSV 2 agents. Given in parentheses are IC50 values in a cell based assay.
Targeting the host cell rather than virus-encoded proteins offers significantly more target molecules and -processes, however, at the cost of a higher potential for cytotoxicity. Moreover, when fighting viral reproduction at the level of host cell processes, the pressure towards formation of resistant mutants of the virus would be lessened. The 2-phenylbenzimidazoles affect a group of proteins within the Golgi, while other organelles are apparently left unperturbed. These findings suggest that the impact of the drug candidates on the Golgi of eukaryotic cells may also affect the propagation of a number of viruses that exploit the Golgi for their intracellular maturation. Many viruses appear to rely on the Golgi apparatus to, for instance, contribute membranes for lipid envelopment or to participate in the processing of critical viral proteins. To this end, the influence of the “Golgi compounds” from the 2-phenylbenzimidazole class on nine viruses from eight families was studied.239 These viruses, including HSV-2, have been reported to either use membranes of the Golgi, the pre-Golgi compartment referred to as `endoplasmatic reticulum-Golgi intermediate compartment' (ERGIC), or the trans-Golgi network for their envelopment. Hence, these viruses rely on a functioning Golgi. Indeed, the 2-phenylbenzimidazoles (e.g., 118 – 121, Scheme 15) and 2-phenylimidazopyridine analogues like 122 were shown to be inhibitors of HSV-2 propagation in a cell-based assay in nanomolar concentrations. In a topical model for HSV infection in guinea pigs, the initial IgE-“blocker” 118 had no significant effect, while an ointment with 2% of 122 completely suppressed the appearance of vesicles. This dramatic difference was explained with the different lipid solubilities (which would translate into improved skin penetration) in addition to potence, both of which favor 122. Unfortunately, tromantadine was not included in this study, but these results together with the suggested MOA of tromantadine based upon the earlier studies mentioned above suggest a similar mode of action.
A recent report of novel antiherpetic adamantane derivatives might serve as another example how adamantane modification enhances potency and pharmacokinetics of known test drugs. Monoacyl derivatives of diaminopyridines were rendered less cytotoxic while maintaining antiviral activity when using adamantane-1-carboxylic acid as the acyl component (123, Scheme 16).240
Scheme 16.
Most recent adamantane derivatives with anti-HSV 1 properties. Given in parentheses is the EC50 (in μg/mL) as determined by plaque-reduction assay using Vero cells.
Among the novel compounds reported241 were 1-adamantyl-monothioureas derived from diaminocyclohexane (e. g., 124) and the bis-urea 125. These compounds inhibited HSV-1 reproduction as monitored by plaque reduction assays in Vero cells. These assays combined with a limited number of compounds screened do not offer additional insights into the MOA of aminoadamantanes against Herpes simplex. In conclusion, it is remarkable that tromantadine marks the third adamantane (more specifically: aminoadamantane) derivative that sucessfully was introduced to the market – even though its identification as an anti-Herpes simplex agent was not so much the result of research specifically addressing this use, but rather a finding from screenings of a group of aminoadamantanes with respect to their range of potency against various viruses.
4.3 Hepatitis C and HIV
Having two antiviral chemotherapeutics incorporating the 1-adamantyl residue reached the market, naturally researchers were intrigued by these virucidal effects combined with mostly straightforward chemical syntheses of the compounds and, consequently, adamantane derivatives are being considered and screened as chemotherapeutics active against most viruses that pose imminent threats on the human population. We will cover two of them, Hepatitis C (HCV), and human immunodeficiency virus (HIV) in this chapter.
The earliest report on adamantane derivatives displaying anti-hepatitis properties dates back to 1968 when pyridoxane adamantoates242 (e. g. 126, Scheme 17) were proposed as agents suitable for the treatment of viral hepatitis. Later, 2-adamantanone oxim esters like 127 were reported243 as having “exhibited antiviral activity against murine hepatitis virus.” Subsequently, paralleling in part the very early research dealing with utilizing lipophilic amines to combat Influenza A, ammonium chloride, amantadine, methylamine, and dansylcadaverine were examined in a monkey cell line infected with Hepatitis A.244 Lipophilic amines were known to accumulate and raise the pH in intracellular vesicles, thereby impeding the release of viral DNA into the cytoplasma, and, consequently, the lipophilic amines were the most potent inhibitors of viral antigen synthesis as monitored by immunofluorescence. A screening of forty known compounds with respect to an inhibitory effect on Hepatitis A virus in a human hepatoma cell line gave four active compounds as candidates for Hepatitis A chemotherapy: glycyrrhizin, pyrazofurin, ribavirin, and (of course) amantadine.245
Scheme 17.
Adamantane derivatives displaying anti-hepatitis properties and hexamethyleneamiloride (128)
In 1997, Smith reported the results of a clinical trial in 22 patients with chronic Hepatitis C infection who had previously failed the standard therapy (at that time) with interferon-α-2a.246 The patients received amantadine as a monotherapy in 2 × 100 mg doses each day, and 16 patients (64%) showed both a marked improvement in serum levels of alanine aminotransferase levels and HCV RNA titers during the six-month treatment period. This marked a significant improvement because the initial interferon chemotherapy of HCV infections only led to a sustained virological response in about one third of the patients. This initial report sparked off numerous follow-up trials trying to reproduce Smith's results and comparing amantadine's anti-HCV activity to various other antiviral chemotherapeutic regimens. The results of these trials remain elusive to date.
Several biochemical processes utilized by the HCV during its replication cycle were subsequently studied with regard to a possible effect of amantadine.247 These authors searched for homologies in the sequences of viral proteins between Influenza A and HCV, but no significant sequence or structural homologies, in particular with the Influenza A M2 protein, were identified. Furthermore, neither amantadine nor rimantadine displayed a dose-dependent inhibitory activity against HCV enzymes. Whether the aminoadamantanes are suitable anti-HCV agents – or, metaphorically speaking, whether amantadine is a “magic bullet or yet another dead duck”248 is a close call. Meta-analyses were conducted summarizing different aspects of clinical trials with amantadine following Smith's initial report. One of these,249 using data from a total of 972 patients out of six clinical trials, addressed an eventual advantage of interferon plus amantadine versus interferon alone, finding an absolute increase in sustained virological response after adding amantadine to interferon of around 6%. The authors concluded that their meta-analysis has proven the benefit of combining amantadine with interferon in treating naive patients with chronic hepatitis C. However, another meta-analysis, with a different frame of reference, raised more questions.250 Adding amantadine to an anti-HCV treatment using interferon alone showed promising results in some studies, while others did not confirm this. The authors reported that a triple therapy of interferon, Ribavirin, and amantadine seemed to improve sustained virological response overall, while in a subgroup analysis, non-responders (to previous chemotherapy) seemed to be the only subgroup of patients showing significant benefit when using amantadine. For other subgroups of HCV-infected patients, namely drug-naive patients and relapsers, combination therapy with amantadine is of no use when compared to the standard combination therapy of interferon and Ribavirin. More recent clinical trials compared amantadine in combination with PEGylated interferon and Ribavirin (triple therapy) with placebo and Ribavirin/PEGylated interferon,251 reporting a sustained virological response in 55.2% of patients infected with the difficult-to-treat genotype 1 HCV – the highest percentage of sustained response reported. Amantadine had an additive effect when combined with PEG-interferon and Ribavirin. Furthermore, the addition of amantadine to the regimen showed an initial additive effect of the aminoadamantane on the HCV DNA levels as detected by PCR at weeks 4 and 12 of the 48 week treatment period, confirming earlier reports of this effect of amantadine against HCV. However, a later clinical trial252 could not confirm these findings, concluding that amantadine addition to the PEG-interferon / Ribavirin combination therapy did not augment sustained virological response in drug-naive, genotype 1 patients. This effect is dependent on the HCV genotype, as a later trial in interferon-naive patients showed higher sustained virological response rates in the triple-therapy group (PEG-interferon, ribavirin, amantadine) compared to double therapy (PEG-interferon and Ribavirin), namely in genotype 1/4 patients, rendering amantadine beneficial (albeit the additional effect is small) in early-recognized, 'difficult-to-treat' patients.253 In another recent clinical trial of amantadine in HCV treatment of drug-naive patients, however, the addition of 400 mg / day amantadine to the regimen did not improve sustained virological response rates over the PEG-interferon / Ribavirin double therapy.254 There was a trend for some benefit of amantadine that did not reach statistical significance.
When studying re-treatment of non-responders to a previous anti-HCV chemotherapy, sustained virological response was observed in 20% of all re-treated patients overall, with an 8% difference between double therapy (PEG-interferon and Ribavirin, 16%) and triple therapy (PEG-interferon, ribavirin, and amantadine, 24%).255 The authors concluded that “…it is undisputable that a trend for beneficial effect of amantadine exists, especially in nonresponder patients.” The initial partial virological response when using amantadine is thereafter probably jeopardized by the emergence of escape mutants. A trend towards a beneficial effect of amantadine was also observed in another clinical trial with non-responders, concluding that “…the use of triple therapy would not be justified except in clinically controlled assays”.256
Treatment of post-transplantation HCV patients with amantadine monotherapy did not have an effect,257 while addition of amantadine to the PEG-interferon / Ribavirin therapy obviously reduced deterioration of depression, fatigue, and vigor during treatment.258 What one can conclude from these numerous trials is that there probably is an additional benefit when adding amantadine to the anti-HCV chemotherapy of PEG-interferon and Ribavirin, but this depends on the virus' genotype, the subgroup of patients,259 and the design of the trial. In spite of an obvious (small) beneficial effect of amantadine addition even to the current standard (PEGylated interferon and Ribavirin double therapy), the lack of in vitro assays and cell-based models for HCV infections limits further advance in the efficiency of anti-HCV chemotherapy because on one hand the selection of groups of patients where amantadine could be most effective is aggravated and the development of more efficient anti-HCV agents based upon the aminoadamantane motif is not possible. Both goals of drug development would benefit from the identification of the target of amantadine in HCV chemotherapy. The (at best) contradictory results from the amantadine/HCV trials on one hand and the prospect of protease-based antiviral therapies led Piccolo to devise an obituary: “Here lies amantadine hydrochloride (1997 – 2010), antiviral for HCV. For many years, through many clinical trials, it gave us hope for a better outcome in chronic hepatitis C. Unfortunately never fulfilled, as the evidence remained contradictory. Rest In Peace.”260
While the functions of several of the HCV proteins is poorly understood, the inability to culture the virus in vitro has retarded drug discovery. One small protein encoded by HCV is the p7 protein, a small, lipophilic member of the group of “viroporins” that is membrane-associated and mostly found within the endoplasmic reticulum. Notably, Influenza A M2 protein belongs to the same class of viral proteins. The HCV p7 protein is predicted to display two membrane-spanning helices linked by a small, charged loop. To determine whether HCV, like a number of other viroporins, also forms oligomers with a putative ion channel structure, the p7 protein was expressed in HepG2 cells and cross-linked chemically.261 Furthermore, these authors also synthesized recombinant p7 proteins in E. coli, and both GST-p7 fusion protein and His-tagged p7 formed ion channels in insulating artificial membranes, the His-tagged protein more efficiently than the GST fusion protein. Most notable in the context of the present review is the finding that the ion channel activity of the His-tagged p7 protein (but not the GST fusion protein) could be blocked by 1 μM amantadine. The demonstration that HCV p7 forms an amantadine-sensitive ion channel could explain amantadine's weak anti-HCV activity and enable further, target-directed drug development of HCV chemotheraputics once a HCV culture system has been established. In an artificial “black lipid membrane” bilayer system, reconstituted, chemically synthesized HCV p7 protein (N-terminally biotinylated) also displayed ion channel activity that could be blocked by iminosugars incorporating a long-chain alkyl sidechain; such iminosugars with shorter sidechains (e.g., n-butyl) were not as potent.262 The structure of the channel formed by homooligomers of HCV p7 in different model systems was proposed to be a hexamer261 (as derived from chemical cross-linking experiments as well as transmission electron microscopy), a heptamer (based upon TEM supplemented with computational analyses)263 or at least a pentamer (in black lipid membranes).262 These putative functional aggregates suggest that out of the two membrane-spanning helices of the full-length protein only one is contributing to the inner side of the ion channel, the conserved basic loop connecting these helices is important for the amantadine sensitivity of the channel.264 Using a model system that was known from studies of the Influenza A M2 channel, these authors were also able to demonstrate HCV p7 ion channel activity in living cells. Modeling such oligomeric HCV p7 channels with computational methods gave a first model of binding of the channel blocker, amantadine, to the target channel.265 The approximate dimensions of the channel were 1.5 nm for the inner diameter and 4.5 nm for the outer diameter of the pore, respectively. While being slightly smaller than the results from the TEM studies mentioned above, the most plausible oligomer is, therefore, the hexamer. Performing a docking analysis with amantadine shed light on the binding mode of the adamantylammonium ion inside the lumen of the model channel. The binding site lies in the vicinity of His17 while a hydrogen bond with Ser21 is probable. These models predict the His17 towards the interior of the channel, in analogy to the orientation and function of His37 of the Influenza A M2 tetramer. This suggests a role of the HCV p7 His17 in channel gating and proton conduction. NMR studies on recombinant HCV p7 model proteins266 in bicelles suggest a binding of amantadine to p7 via a lipophilic cluster of five transmembrane Leu residues.267 The drawback is, however, that at amantadine concentrations that are clinically achievable, the HCV p7 proton channel cannot be blocked efficiently.268 Results from molecular modeling269 studies suggest a binding of seven aminoadamantane molecules per HCV p7 channel, with the adamantanes bound at peripheral, membrane-exposed sites reminiscent of the low-affinity rimantadine binding site at the membrane-exposed outside of the Influenza A M2 channel discussed above. The location of this binding site corroborated earlier NMR studies. Mutations such as L20F that decrease the calculated affinity amantadine are observed as genuine resistance mutation in patients unresponsive to anti-HCV triple therapy (Interferon, Ribavirin, and amantadine) in response to selection driven by adamantane. Low loss of survival fitness at single mutations in the p7 protein that render it drug sensitive remain challenges for drug development in this field, however, screening of a small library of aminoadamantane derivatives through docking scores, corroborated by in vitro studies, do encourage drug development targeting the HCV p7 viroporin.
The lack of a small-animal model for HCV infections as well as a practicable, high-throuput assay to screen HCV p7 ion channel inhibitors is addressed by current research. Chimeric viruses were generated for the study of HCV p7 channels in a genuine cell culture system;270 amantadine reducing the virus titer to some extent. Subsequently, a different group reported a study of antivirals against a spectrum of HCV isolates and genotypes in cell culture.271 While these researchers could find an anti-HCV effect of iminosugars, amantadine was found to be inefficient in reducing RNA replication as well as release or infectivity of HCV particles. The lack of amantadine efficacy even at cytotoxic concentrations was attributed to the different p7 proteins used. Assaying the ion-channel activity of FLAG-labeled p7 in an in vitro system utilizing the release of a fluorescence indicator from liposomes, however, amantadine was found to be active, albeit, surprisingly, it did not block the carboxyfluorescein release completely even at 1 M.272 Additionally, in this assay rimantadine, hexamethyleneamiloride 128 (Scheme 17) and an experimental compound that was not disclosed were reported to be more active than amantadine itself. Since this liposome permeability assay allows for screening of both multiple HCV p7 sequences from various genotypes and libraries of drug candidates, screens on a medium or high-throughput scale may become feasible near-term. Refining assay development and combining rapid throughput in vitro assay with a HCV infectious culture system, the activity of some small molecule HCV p7 blockers was assessed against p7 channel activity as well as HCV assembly.273 The result indicated a genotype-dependent or subtype-dependent pattern of sensitivity that could imply the HCV p7 sequence determining whether there is an influence of small molecules, including amantadine and rimantadine, on HCV virus release. Release of infectious HCV in culture was reproducibly blocked by compounds that had been shown to inhibit p7 in vitro. The observed differential sensitivity of subtypes parallels reports from clinical trials of “escape mutants”. Lastly, the lack of a small animal model for HCV infections has been addressed recently. By replacing the p13 protein of GB virus B, a hepatotropic member of the flaviviridae family closely related to HCV (28% amino acid identity), with HCV p7, chimeric viruses were obtained that were infectious in marmosets.274 As amantadine inhibits spread of GB virus in primary marmoset hepatocytes, the animal model urgently needed in HCV drug development appears to be within reach.
Apart from the HCV p7 ion channel, other targets obviously have also been hit by the lipophilic adamantane bullet in the drug discovery process. A series of 1-boraadamantane complexes was identified that target the interaction between CD81, a membrane protein which signals for antiproliferation when bound by antibodies and the HCV-E2 glycoprotein, which is believed to be involved in mediating viral processes of cell entry and immune evasion.275 Out of a chemically diverse library of 500 compounds, boraadamantanes, e.g., 129 (Scheme 17) and related adamantoyl amides (130, 131) were the most promising compounds with 131 showing minimum cytotoxicity. These studies were assisted by molecular modeling based upon X-ray structural data of CD81-LEL. While few adamantane derivatives have been studied so far in HCV chemotherapy, these compounds obviously hit two targets and, while their efficiency remains under dispute, could at least serve as a starting point for target-directed drug discovery once the immanent problems in establishing suitable assays and animal models have been overcome.
The first chemotherapeutic against viral infections, amantadine has, as expected, also been studied in the context of human immunodeficciency virus (HIV). Amantadine was first suggested as a potential virustatic to combat HIV in 1987.276 The first effect to be studied was that of amantadine acting as a weak base.277 As, e. g., amantadine raises the pH of intracellular organelles like endosomes and lysosomes and the entry of HIV-1 following binding to the CD4-positive host cell was presumed to occur via membrane fusion, these authors have studied the pH-dependence of HIV-1 entry showing that HIV-1 infection is not inhibited by weak bases like amantadine, but rather the release of infectious virus from the cell was impaired by the bases. They also did not find evidence for an influence of bases on the synthesis of viral proteins. Using a different testbed of co-cultured human primary glial fibrilary acidic protein positive (GFAP+) cells with HIV-producing H9-cells, an anti-HIV effect of memantine could be found.278 While this effect was, at least in part, attributed to memantine's known NMDA-receptor antagonism (vide infra) that could ameliorate the effect of an HIV coat protein of increasing Calcium concentrations in culture, the authors also discussed an eventual participation of HIV-1 gp120 and its neurotoxicity as well as a pH-dependence of HIV-1 uptake by the GFAP+ cells as memantine's mode of action in this testbed. Using memantine in HIV-associated dementia was proposed. The neurotoxicity of HIV-1 coat protein gp120 was later also confirmed in rat cortical cell cultures; memantine (as well as an alternate NMDA-receptor antagonist, MK-801) dose-dependently prevented gp120 neurotoxicity at micromolar concentrations.279
A vast number of aminoadamantanes designed and synthesized as Influenza A M2 channel blockers (vide supra) were also screened with respect to a possible anti-HIV effect. While most of the compounds were inactive,141 borderline anti-HIV-1 activity in cell culture could be assigned to, e. g., compounds 67 and 68 (Scheme 10).142 Other adamantane derivatives reported by the same group include the 2-(1-adamantyl)piperidines 132–134 (Scheme 18),143,148 which had EC50 values in the low micromolar range and significantly higher cytotoxic concentrations (Scheme 18). In comparison to known anti-HIV agents, the potency of these adamantane derivatives was, however, markedly lower. Alongside the pharmacophors of known drugs Thalidomide, Phenytoin (an antiepileptic compound that also inhibits the binding of HIV to CD4 positive lymphocytes), and Ameltolide, a group of 24 substituted phtalimides, some of which incorporating the adamantane moiety, has later been synthesized and screened against HIV-1 and HIV-2 cytopathogenicity in CEM cells. Only the adamantylated Thalidomide derivative 135 displayed activity against HIV-1 (EC50 = 4.7 ± 0.1 μM) as well as HIV-2 (EC50 = 8 ± 0.0 μM) at concentrations below its cytotoxic concentration (CC50 = 10.3 ± 1.8 μM), the target of 135 being unknown. A follow-up study of related compounds280 including adamantylated benzamides (e.g. 136) found the analogues mostly being inactive below their cytotoxic concentrations. Reports of an ion-channel activity of a viral protein had prompted these workers to screen derivatives of known anticonvulsants against HIV. A different approach at anti-HIV compounds incorporating the adamantane motif was chosen due to the finding that leukocyte dialysate contains a low molecular weight immunoactive fraction, the major components of which are peptides like H-Tyr-Gly-OH or H-Tyr-Gly-Gly-OH, which resemble the N-terminal fragment of the enkephalins.281 Modifying enkephalins with an adamantane moiety gave hydrophobic peptides like 137, which was able to inhibit syncytium formation in T-lymphocytes. Because of the known tendency of aminoadamantanes to penetrate lipid bilayers,282 they assumed the anti-HIV activity of 137 to be possibly associated with processes on the surface of the host cell, e. g., interaction with opioid receptors. Polymeric compounds that incorporate aminoadamantane derivatives linked to polyanionic matrices (believed to enhance the membranotropic activity of the polymers) have also been described.283,284 HIV-1 infection could be inhibited in several cell lines using the polymer, whereas amantadine and rimantadine were ineffective in control experiments. Other compounds tested in regard to anti-HIV activity were hydrazides like 138 and 139. Compound 138 inhibited HIV at a level comparable to the anti-HIV drug Retrovir, whereas 139 did not. This cannot be ascribed to the 1-adamantyl pharmacophor alone, as the 2-norbornyl analogue of 138 also showed some activity. The combination of a heterocyclic pharmacophor with the “lipophilic bullet” adamantane was also studied in, amongst other heterocycles, a series of benzoxazinones,285 finding 140a and 140b to show moderate anti-HIV-1 activity in cell culture at micromolar concentrations. Finally, anti-HIV activity of 4-oxy-adamantane-4-one (“kemantane”) at low cytotoxicity has also been reported and, at least in part, was attributed to its membranotropic properties while being soluble in aqueous media.257
Scheme 18.
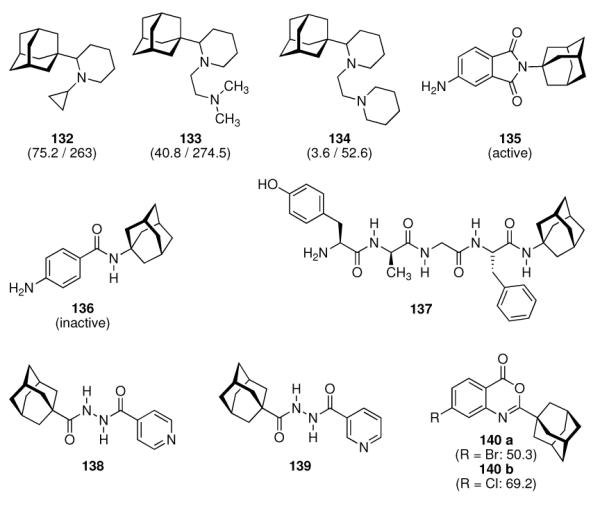
Outline of aminoadamantanes screened for anti-HIV-1 activity. Numbers in parentheses are the EC50 values in cell based assays / the CC50 values (in μM).
Another use of an adamantane derivative in pathologies associated with HIV infections should be mentioned here. Memantine, an uncompetitive, fast-on / fast-off, NMDA-recetor antagonist (see chapter 5.3), has successfully been studied in this context. Lipton tested in 1992 whether memantine can in in vitro setups ameliorate the neurotoxicity exerted by gp120, the HIV envelope glycoprotein that together with gp41 forms oligomers essential for host-cell receptor binding of the HI virus particle.286 gp120 can reduce certain neuronal populations by 20 to 50%, and NMDA-receptor mediated neuronal injury may also contribute to the pathologies summarized as “HIV-1 associated cognitive/motor complex”, a severe form of which is the so-called AIDS dementia complex. Several recombinant and native gp120 isoforms were studied, and memantine at doses as low as 2 μM completely protected the rat retinal ganglion cells used from gp120 neurotoxicity. Together with a “general effect on virus growth”, the neuroprotective effect of memantine has led to a nomination of memantine for the treatment of AIDS-related dementia.279 By cerebral expression of a fusion gene encoding gp120 in a transgenic mouse model, it could be demonstrated that the HIV glycoprotein induced an increase in the plasma concentrations of corticosterone as a result of activation of the hypothalamic-pituitary-adrenal (HPA-) axis. This is a Ca2+ dependent process that involves NMDA receptor stimulation, which can be ameliorated by memantine.287 Chronic treatment with memantine, initiated early, can antagonize gp120 induced neuronal damage in vivo; transgenic mice treated with memantine showed marked improvements in synaptic complexity as well as neuronal integrity when compared with untreated littermates.288 The role of gp120 in the neuropathogenesis of HIV-1 infection was further elucidated by directly detecting the glycoprotein in patients with HIV-1 encephalitis.289 These authors found that viral proteins, including gp120 and Tat, synergistically cause a massive derangement of neuronal function, in particular calcium dysregulation in neurons. A similar neuroprotective effect of memantine as discussed above was found in a mouse model for HIV-1 encephalitis.290 The memantine-treated mice displayed less severe impairment of long-term potentiation. Memantine went to clinical trials for the treatment of AIDS dementia.
In a first clinical trial of memantine, no trend toward clinical benefits could be observed in patients suffering from HIV-associated distal sensory neuropathy.291 Likewise, a different phase II clinical trial of memantine for the treatment of HIV-associated cognitive impairment did not confirm significant benefits in the drug treated group of patients.292 However, since MR examinations indicated that memantine may ameliorate neuronal metabolism, the beneficial effect of the drug might have been too weak to be significant within this relatively short (16 weeks treatment with the drug) trial. An overall positive result in a long-term study (60 weeks) has been, however, found recently as memantine was safe and tolearable, and (at least in an initial 12-week open-label phase) improvements in eight neuropsychological test scores were found.293
The “add-on” approach of attaching the adamantane moiety to known drugs or pharmacophors has also been undertaken in the field of HIV related drug discovery. AZT (Azidothymidine, Zidovudine), a nucleoside analogue reverse transcriptase inhibitor (NRTI), was the first approved treatment for HIV and remains to be used clinically to date, in particular in combination therapy regimens (Combivir, Trizivir). Since AZT is not able to cross the blood-brain-barrier (BBB), various lipophilic 5'-esters of AZT with 1-adamantyl substituted carboxylic acids have been synthesized and studied in vitro finding higher resistance against plasma esterases, as well as in vivo. Ester 141 (Scheme 19) could be detected in the brains of rats in 18-fold higher concentration than AZT itself.294 Combining the anti-HIV pharmacophores phthalimide (vide supra), adamantane, and AZT, the AZT prodrug candidate 142 was identified as showing considerably increased anti-HIV potency when compared to the analog without the adamantane modification (EC50 = 0.06 – 0.11 μM); this is, however, still an antiviral potency about 10- to 20-fold lower than that of AZT.295 That said, the authors still proposed this compound to be evaluated further because of its prospective in vivo pharmacological properties. Cosalane (143), a derivative of aurintricarboxylic acid (ATA) with improved anti-HIV-1 potency acting at the step of membrane fusion between the HI virion envelope and the host cell membrane via interference with the gp120-CD4 interaction as well as being an inhibitor for HIV-1 reverse transcriptase and protease, has also been modified with adamantanes. This time, the steroid pharmacophor has been replaced by adamantane. Cosalane and several derivatives, e.g. “Cosalog 25” (144) also displayed significant inhibitory activity against HIV-1 protease and HIV-1 integrase.296 While 144 (IC50 = 7 μM) was not as potent as the best analogues screened, it had a higher potency than cosalane itself. It also inhibited HIV-1 protease in vitro (IC50 = 0.37 μM) approximately four times stronger than cosalane. Unfortunately, however, the cosalane analogues were not as potent inhibitors of HIV-1 and HIV-2 cytopathicity as Cosalane is in cell based assays, at the same time showing more pronounced cytotoxic effects in the MT-4 and CEM-SS cells used.
Scheme 19.
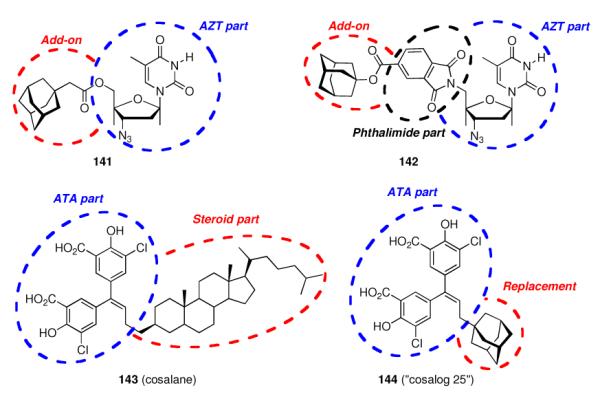
Modifications of known anti-HIV drugs by adamantane moieties
In 1999, as part of a screening of several thiourea derivatives, also adamantane substituted thioureas were examined with regard to their inhibition of HIV reverse transcriptase (RT).297 Later, Balzarini et al. have also screened their dideoxythymidine derivatives (some of which incorporating the adamantane cage) as potential inhibitors of recombinant HIV-1 RT (vide supra).295 However, these above examples did not hit this target in HIV drug discovery, being inactive at the concentrations tested. Again heterocyclic adamantane derivatives like the adamantyl-oxadiazoline thione 145 (Scheme 20) exhibited “borderline activity” against HIV in a cell-based assay.24
Scheme 20.
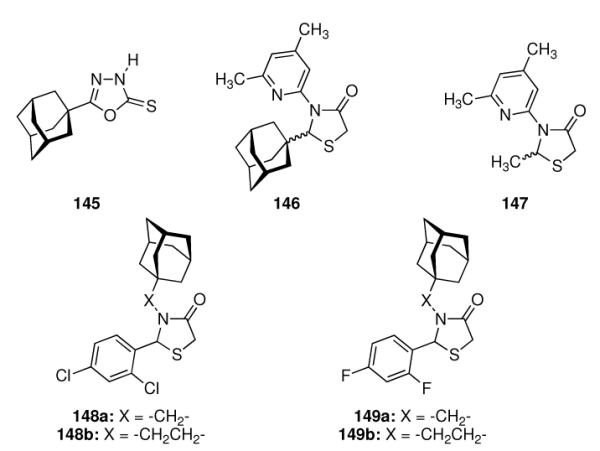
Experimental non-nucleoside inhibitors of HIV reverse transcriptase
Varying the heterocyclic moiety to structurally closely related thiazolidin-4-ones, a class of compounds utilized in many fields of medicinal chemistry including their use as non-nucleoside HIV reverse transcriptase inhibitors (NNRTIs), resulted in compounds with anti-HIV-1 potency in cell culture.298 The adamantane substituted thiazolidinone 146 displayed an EC50 of (0.35 ± 0.175) μM in CEM cells, while the methyl substituted analogue 147 was not active at all at concentrations as high as 450 μM. The (R)-enantiomer of 146 was about two times more potent than the (S)-enantiomer. The anti-HIV activity in cell culture was reflected, at least in part, by the inhibitory profile of the compounds against recombinant HIV reverse transcriptase (IC50 ((R)-146) = (19 ± 11) μM; IC50 (147) ≥ 900 μM. In other words, this novel class of NNRTIs obligatorily requires adamantane as a co-pharmacophore for activity. A follow-up study including a larger library of test compounds, e. g., pyrimidines, gave mostly active compounds, albeit with inferior potency compared to 146.299 Comparative measurements of some of the compounds' inhibitory activity against HIV-1 RT and HIV-2 RT clearly showed a selectivity of these NNRTIs for HIV-1 RT, the potency of HIV-1 RT inhibition paralleled the virustatic effect as measured in CEM cells (Table 2).
Table 2.
Comparison of NNRTI's incorporating 1-adamantane substituents. All EC50, CC50, and IC50 values in μM.
| (R)-146 | 148a | 148b | 149a | 149b | |
|---|---|---|---|---|---|
| HIV-1 inhibition (CEM cells), EC50 | 0.35 ± 0.18 | 1.0 ± 0.6 | 3.4 ± 1.2 | 11.0 ± 2.7 | 2.0 ± 0.36 |
| HIV-2 inhibition (CEM cells), EC50 | > 12.0 | ≥ 10.1 | ≥ 9.7 | 35.7 ± 4.4 | ≥ 4.0 |
| Cytotoxicity, CC50 | 42.8 ± 8.1 | 21.1 ± 2.3 | 19.0 ± 1.9 | > 275 | 7.9 ± 0.28 |
| HIV-1 RT, IC50 | 13.4 ± 13.6 | 42.9 ± 3.2 | 226 ± 58 | 495 ± 77 | 31.7 ± 17.7 |
| HIV-2 RT, IC50 | > 600 | ≥ 600 | ≥ 600 | ≥ 600 | ≥ 600 |
After hitting HIV targets that straightforwardly come into the reader's mind considering the previous chapters, either via classical SAR or via improving known pharmaceuticals by adding the “lipophilic bullet”, it also appears obvious to target biochemical functionalities harnessed by viruses that have been, at least in part, elucidated using pharmaceuticals derived from the adamantane motif. The prototype functional concept is the blockade of a pore or an ion channel which is formed by proteins encoded by the virus. Small viral proteins called viroporins300,301 oligomerize within the membrane and give rise to hydrophilic pores at the membranes of cells that are infected and “re-programmed” by the virus. Usually, these pores are not essential for viral replication, they do, however, enhance virus growth. We have already discussed in depth the Influenza A M2 ion channel (97 residues, tetramer), which at present is by far the best-studied viroporin. A great deal of insight into structure, dynamics, and function of M2 has been gained utilizing M2 blockers, namely aminoadamantane antivirals. The Influenza B M2 channel (82 residues, tetramer) may present a different sequence, but it shares structural motifs as well as residues crucial for its function (such as the HXXXW motif) with the Influenza A M2 channel.179 Likewise, we have also learned about the HCV p7 viroporin (63 residues, presumably a heptamer), whose validation as a target for chemotherapeutics also involves the aminoadamantanes. HIV-1 (but not HIV-2) encodes a 81-residue type 1 transmembrane phosphoprotein (Vpu) that plays a role in enhancement of HIV-1 virus release from infected cells as well as degradation of the CD4 receptor in the host cell. It shares some characteristics with the abovementioned viroporins, including a proton channel function.215
Early molecular modeling studies suggested homooligomers of HIV-1 Vpu to form weakly cation-selective ion channels.302 Since obviously the M2 viroporin from Influenza A and the HIV-1 Vpu protein share several similarities, an exchange of Vpu with M2 was considered. Replacing the transmembrane domain of Vpu in a simian-human immunodeficiency virus (SHIVKU1bMC33) with the transmembrane domain of (amantadine-sensitive) M2 gave a chimeric virus that did not change most of its properties:303 its maturation pattern was similar to the parent SHIV and it remained infectious causing severe loss of CD4+ T cells and AIDS in macaques, indicating that the modified VpuM2 protein was functional. The chimeric virus was furthermore found sensitive to the M2 channel blocker, rimantadine. The transmembrane domain of the HIV Vpu viroporin (subtype B) shows an analogy to the Influenza A M2TM: it incorporates an -AXXXW- motif in approximately the same position where the -HXXXW- subsequence is located in M2. Consequently, when substituting Ala19 with His in the Vpu TMD via site-directed mutagenesis, the resulting mutant virus also retained most of its functions unchanged, but its replication in culture could be almost completely abolished by 75–100 μM rimantadine.304 These findings validate, at least to some extent, the Vpu as a target for the development of anti-HIV chemotherapeutics. Subsequent solid-state NMR studies of a 36-residue polypeptide corresponding to the Vpu transmembrane domain in its native and mutated sequencee in DHPC micelles confirmed these findings on a molecular level.305 The newly introduced His residue of the rimantadine susceptible mutant displayed pronounced changes of its backbone amide resonance upon interaction with the drug, suggesting the binding site of the drug to be located here, as in Influenza A M2TM. No specific interaction of the drug with the unmutated Vpu polypeptide could be detected. The Vpu transmembrane domain is an ideal helix in phospholipid bicelles, and residues located on the same side of this helix like the His residue are also involved in the interaction with the drug. The mutation causes not only a local change of the Vpu TMD structure, but also influences secondary and tertiary structure of the protein, decisive for drug interaction with the homooligomeric Vpu channel.
The channel properties of the HIV-1 Vpu viroporin and the discovery of its molecular mechanism of action utilizing rimantadine-sensitive mutants are not the only parallel that can be drawn to the multiplication strategies of other viruses. In chapter 4.2 we have discussed a class of anti-Herpes simpex compounds targeting membranes instead of a clean-cut protein protein or oligomer, a development that was, at least in part, fueled by the known preferences of aminoadamantanes to interact with biomembranes.
In this context, it is of interest for us to note that it has been shown that amantadine (and other lysosomotropic agents that are able to diffuse across membranes) increases the infectivity of HIV-1SF-2 in HeLa cells dose-dependently above 25 μM.306 Next to the capability of amantadine to cross membranes, this effect is associated with its function as a weak base; thereby the drug is capable of elevating the pH of endosomes containing the virus after endocytosis. Endosome acidification raises the likelyhood of infectious particles to be deleted via lysosomal degradation, therefore, counteracting with a weak base like amantadine dramatically increases the overall infectivity of HIV-1 isolates.
As the fusion of HIV-1 with the host cell probably occurs in microdomains of the plasma membrane enriched with glycoshpingolipids (GSLs), also known as “lipid rafts” where the CD4 receptor is localized, and HIV gp120 as well as CD4 interact with several GSLs, synthetic analogs of GSLs mimicking these interactions could be anti-HIV agents.307 Indeed it has been shown that synthetic analogues of the GSL globotriaosylceramide (Gb3), which is also being referred to as a HIV-1 fusion cofactor, recognize HIV-1 gp120 and inhibit HIV-1 fusion.308 One such derivative is the adamantylated GSL adamantyl-globotriaosylceramide or ada-Gb3 (150, Scheme 21), a semisynthetic glycosphingolipid derivative with one of the fatty acid chains replaced by adamantane. This modification not only renders 150 soluble in aqueous media, but also changes the binding to ligands such as HIV-1 gp120, which recruits Gb3 in membranes during the formation of the HIV-1 fusion complex. Ada-Gb3 was found to specifically bind to gp120 far more rapidly than Gb3. Such adamantylated GSL derivatives therefore are considered invaluable tools for the study of the glycolipid receptor function. The effect of the adamantane substitution on ligand binding is considered an indirect one, with the adamantane rigidifying the monolayer (resembling a membrane microdomain or “lipid raft”), reducing its compressibility and, most importantly, modifying the organization of the carbohydrate moiety, thereby enhancing interaction with gp120. Presumably, the adamantane moiety in ada-Gb3 mimicks the effect of cholesterol, which is required for the gp-120 / Gb3- binding in microdomains. These authors subsequently studied the effect of ada-Gb3 on HIV-infection in cell culture.309 Infection of the Jurkat cells used was reduced dose-dependently with a reduction to background values at 300 μM ada-Gb3, and 50% inhibition was shown at about 150 μM. Via electron microscopy, the authors showed that the HI virions are not deleted, but rather their attachment to host cells is inhibited. Both HIV-1 and HIV-2 fusion is inhibited using ada-Gb3. The virus/ada-Gb3 binding is of central importance here, as a pre-incubation of the target cells with ada-Gb3 did not inhibit HIV multiplication. The authors also suggested that a reduction in lipophilicity of the modified GSL should further increase the specific activity. In conclusion, although the adamantane moiety is more lipophilic than the fatty acid chain it replaces, the adamantylated conjugates are remakably water-soluble and retain the functions in terms of ligand binding. Ada-Gb3 acts as a mimic of the Gb3/cholesterol complex, increases the receptor function of Gb3 about 1000-fold, and inhibits HIV-infection in cell culture.282 The modified GSLs could also offer an approach to the reversal of multi-drug resistance in cancer chemotherapy.310 Ada-Gb3 and other modified glycosphingolipids seem to offer the opportunity to investigate the GSL function in cellular physiology, and provide new approches to treat infectious diseases in which these membrane-associated functions are crucial, e. g., the recognition of and fusion with a host cell by a viral particle.
Scheme 21.
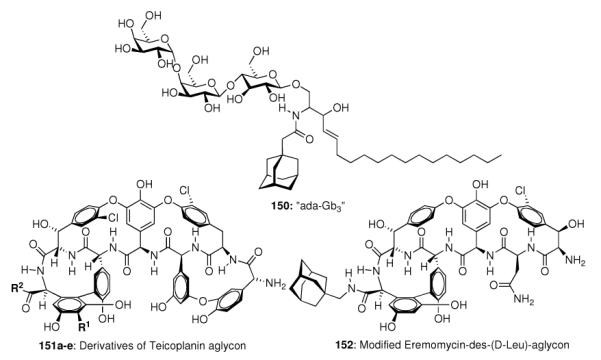
Semisynthetic anti-HIV compounds incorporating adamantane as lipophilic modifier.
Another group of pharmaceutically active compounds targeting the biosynthesis of bacterial cell walls are the glycopeptide antibiotics, the best-known example of which is Vancomycin. The search for glycopeptide derivatives able to combat glycopeptide-resistant bacteria led to lipophilically modified glycopeptide-derived compounds, some of which incorporate adamantane moieties. These compounds were also evaluated with regards to a potential use as anti-HIV compounds.311 While the unmodified antibiotics, e. g., Vancomycin and Eremomycin did not display anti-HIV activity, their lipophilically modified derivatives showed modest anti-HIV-1 activity, at increased cytotoxicity, however. Semisynthetic derivatives of Teicoplanin aglycon (151a–e, Scheme 21 and Table 3) were generally active against HIV-1 more than against HIV-2 in cell culture, and the lipophilically modified derivatives were about 10- to 20-fold more active than the unmodified parent (151a). However, since modifications other than the “lipophilic bullet” adamantane gave comparable results in this context (e.g., 151b vs. 151c, Scheme 21 and Table 3) and the most active compound screened (151e) does not incorporate the adamantane moiety, the role of the adamantane add-on here seems to be limited to just an increase in overall lipophilicity. While no details on the mechanism of action of these compounds were disclosed yet, they do inhibit an early step of the HIV-1 multiplication cycle, most likely viral entry to the host cell – a membrane-associated process which is the result of a specific interplay between the HIV glycoproteins gp120 and gp41 and cellular (co)receptors like CD4 (probably inside their lipophilic 'raft' neighborhood, as described above).
Table 3.
Details on the pharmacological properties of Teicoplanin aglycons 151 and the Eremomycin derivative 152 (see Scheme 21). EC50 values in cell culture are given in μM.
| R1 | R2 | HIV-1 | HIV-2 | |
|---|---|---|---|---|
| 151a | -H | -OH | 17 ± 3.5 | 20 ± 0.0 |
| 151b |
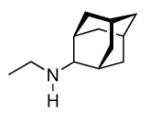
|
|
2.5 ± 0.7 | 8.0 ± 2.8 |
| 151c |
|
|
3 ± 0.0 | 5 ± 1.4 |
| 151d |
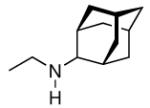
|
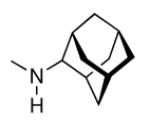
|
2.5 ± 0.7 | 3.5 ± 2.1 |
| 151e | -H |
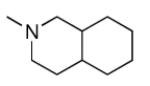
|
0.75 ± 0.07 | 4.5 ± 0.7 |
| 152 | - | - | 5.5 ± 0.0 | 3.5 ± 2.1 |
Having specific antiviral activity also implies that an antibacterial activity for compounds used to combat the HI virus should be minimized in order to prevent formation of glycopeptide resistant bacteria strains. This goal has been followed with the lipophilically modified glycopeptide derivatives described here by using the aglycons, which led to a significant decrease or loss of antibacterial activity.25 For that reason, we present 152 here as it was devoid of any antibacterial activity and, at the same time, about as potent against HIV as related glycopeptide aglycons (Table 3). However, SAR with a number of glycopeptide aglycons derived from Vancomycin, Eremomycin, and Dechloroeremomycin did not yield compounds with enhanced anti-HIV potency, as exemplified by the des-(N-Methyl-D-Leu) aglycon of Eremomycin 152 (Scheme 20, Table 3).
The authors concluded that “the investigation of antiretroviral activity demonstrated that amides with an (adamantyl-1) methyl substituent (…) are, as a rule, equally if not more active than compounds with adamantyl-2 substituents and ω-aminodecyl substituents”.25 As a result, the 'lipophilic bullet', adamantane, as of today may not have led to pharmaceuticals that are routinely being prescribed to combat HIV; the aminoadamantanes had, however, significant impact on the understanding and validation of the HIV-1 viroporin, Vpu, as a drug target. Moreover, as a recurring event in the medicinal chemistry of adamantane derivatives, the lipophilic add-on has successfully been utilized to enhance pharmacokinetics and/or pharmacodynamics of known pharmaceuticals. Several lines of evidence point towards the beneficial effect of adamantylation when membrane processes are targeted, as exemplified by adamantylated glycosphingolipids or glycopeptide aglycons derived from antibacterials.
4.4 Antimalarials Incorporating Adamantane
The “add-on” strategy of modifying known pharmaceuticals has also been followed in the context of antimalarials as early as 1967.26 Five adamantylated naphthoquinone antimalarials 29 (Scheme 22) were screened against plasmodium berghei, a protozoan that represents a classical model organism for human malaria, in mice along with other derivatives of the antimalarial drug, chloroquine. Adamantane substitution in this case showed no advantage over other alkyl and cycloalkyl naphthoquinones derived from Atovaquone (153). A recent example of this “add-on” approach is a report of enhanced activity of adamantylated derivatives of the early antimalarial drug, Chloroquine (154).312 The adamantylated 4-amino-7-chloroquinolines displayed excellent in vitro antimalarial activities, in particular also against chloroquine resistant strains of Plasmodium falciparum. Structure 155 was found active against the multidrug resistant P. falciparum TM91C235 strain (IC50 = 7.39 nM) as well as two other strains of P. falciparum; the IC50 of chloroquine itself was 124.24 nM in a parallel positive control using the same test system.
Scheme 22.
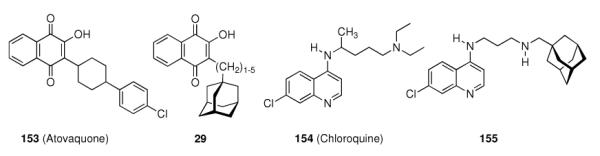
Modifications via adding adamantane moieties on known antimalarials
This class of compounds targets the heme polymerization inside the parasite's food vacuole, and the resistant strains of malaria parasites have found a way to efficiently remove drugs targeting this process outside their food vacuole, via the Plasmodium Falciparum Chloroquine Resistance Transporter (PfCRT), which, therefore, resembles one possible target to be hit in the development of antimalarials for chloroquine-resistant strains. Lastly, adamantaneethers, along with other lipophilic moieties, have been studied in the modification of the antimalarial natural compound isolated from Artemisia annua, artemisinin (vide infra).313 Some of the ether-derivatives were found to be 2- to 4-fold more active than arteether against multi-drug resistant P. yoelii nigeriensis in mice, but no special preference for adamantane can be observed here.314
The life cycle of malaria parasites along with a summary of possible and actual drug targets for antimalarial chemotherapy has been reviewed recently;315,316 it is almost an implicitness that adamantane-derived compounds also have been studied with regards to a possible antimalarial activity aiming at several molecular targets. Being reportedly a “membranotropic” compound, amantadine has been tested along with other membrane active drugs, Colchicine and Vinblastine, whether it was able to alter erythrocyte susceptibility for P. knowlesi in rehsus monkey erythrocytes.317 However, while Colchicine and Vinblastine did inhibit parasite invasion to some extent, amantadine did not. Being a weak base and an amphiphilic molecule, amantadine was hypothesized to be trapped in the acidic food vacuole via the same mechanism Chloroquine is taken up:318 After diffusion to this lysosome-like compartment, the weak bases become protonated and, as ionic species, do not diffuse out of the vacuole. Here, the drug molecules alter the local pH, thereby blocking the vacuolar digestion of host cell hemoglobin, ultimately causing parasite starvation (“lysosomotropic hypothesis” of antimalarial drug action319). Amantadine did show some activity against P. falciparum, in particular against strains that were Chloroquine resistant.318 Additionally, these authors also attributed amantadine's activity to its membrane-modulating properties, specifically in the context of modulating local charge density of the membrane. The same authors also studied possible interactions between amantadine and antimalarials like Chloroquine, Mefloquine, quinine, or Halofantrine using two malaria strains maintained in human red blood cells.320 In both strains, amantadine potentiated the effect of quinine and Chloroquine, respectively. The authors proposed that amantadine either increases the drug accumulation by acting on the resistance mechanism or somehow modifies the drug target, which was unknown at this time. Later, these researchers studied the effect of pH alterations on the potency of amantadine against P. falciparum, however, they found the pH not being the sole determinant of amantadine activity.321 In lieu thereof, it is presumably amantadine's interaction with membrane phospholipids that inhibits parasite maturation in the erythrocytes. Also in the context of studying the PfCRT as a drug target, it was studied more recently whether amantadine is capable of pressing for mutations in the PfCRT that render Chloroquine-resistant strains of Plasmodium parasites back to Chloroquine susceptibility.322 PfCRT incorporates ten putative transmembrane helices and is located in the food vacuole membrane within a parasitized erythrocyte. Data from cross-resistance studies indicated that PfCRT indeed constitutes a key determinant of Chloroquine- and amantadine-susceptibility. It remains to be shown whether amantadine acts in the context of synergistic action together with Chloroquine (vide supra – the lysosomotropic hypothesis) within the vacuole or by directly interacting with a lipophilic site within PfCRT. The widespread Chloroquine resistance of currently circulating strains of Plasmodium parasites has rendered Chloroquine (it nearly halved deaths from P. falciparum in children in sub-saharan africa) as a monotherapy essentially useless, and there is, as of today, still a need for replacement chemotherapeutics.323 A restoration of Chloroquine regimens via amantadine (or drugs developed based on this lead) and its presumable target, PfCRT, would, therefore, be most welcome. Amantadine itself does not qualify to fulfill these requirements since the concentrations of the drug needed to efficiently display the antimalarial effect are too high to be useful clinically.
Artemisinin (156, Scheme 23) and some semisynthetic derivatives, in particular Artemether or Artesunate, are currently part of combination therapies against multidrug resistant malaria. These natural product based therapies require plantation and processing of large amounts of Artemisia annua, imposing pressure towards the development of fully synthetic drugs with comparable or even better activities. The 1,2,4-trioxane pharmacophor accounts for artemisinin's antimalarial activity, and consequently the search for alternatives resulted in the synthesis and screening of trioxane derivatives. The first synthetic 1,2,4-trioxanes active against malaria in vivo (mice infected with P. berghei) were reported in 1992.324 The compounds, accessible through a straightforward acid-catalyzed condensation of adamantanone with β-hydroxyhydroperoxides, incorporate a spiroadamantane peroxide as the pharmacophor, were screened in a mouse model; 157a and 157b were active at doses as low as 30 mg/kg twice a day intraperitoneally. Spiroadamantane 1,2,4-trioxolanes have later also been identified as antimalarial drug candidates.325 Compounds 158 – 160 (Scheme 23) provided a foundational SAR: Trioxolanes 158 and 159 were completely inactive in tests against P. falciparum in vitro and P. berghei in mice, whereas 160 marked a “hit”, being about as potent as Artemether or Artesunate. Consistent with the “iron activation hypothesis” of antimalarial drug action,326,327 it could be shown by reacting the peroxides with Fe(II), mimicking the heme that is present in the food vacuole, and subsequent trapping of the reaction products with TEMPO, that this class of antimalarial peroxides indeed generates carbon-centered radicals that are believed to be the crucial alkylating species in Plasmodium chemotherapy through peroxide drugs. By Griesbaum co-ozonolysis328,329 utilizing O-methyl 2-adamantanone oxime as the key precursor along with suitable ketones, the synthesis of the ozonides in large scale (>100 mmol) gave satisfying yields without the need for chromatographic purifications. Ozonide 161 (OZ277, RBx11160, Arterolane) was no less potent than Artemether in in vitro assays against P. falciparum, and, more importantly, it was more active than the known peroxide antimalarials in vivo in mice inoculated with P. berghei. Consequently, 161 was chosen as a development candidate based upon its potency in vitro as well as in vivo, its toxicology, and its tissue ditribution, along with a simple and scaleable synthetic protocol. It has been advanced to “first in man” clinical trials in 2004.
Scheme 23.
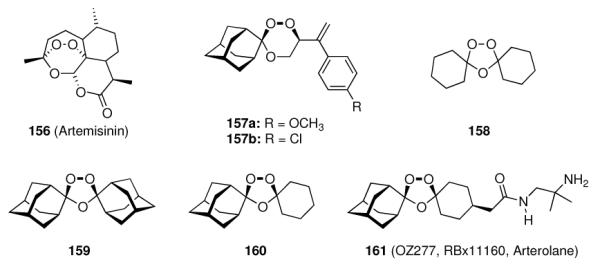
Artemisinin and spiroadamantane peroxide screened as antimalarials.
In the same year, spiroadamantane-1,2,4-trioxanes also re-gained attention. Some new 1,2,4-trioxanes were reported that showed good activity against multi-drug resistant P. yoelii in Swiss mice (Scheme 24).27 In particular, 163 suppressed the parasite completely when measured 4 days after the beginning of an oral treatment at 96 mg/kg/day, all five mice of the group survived past day 28.
Scheme 24.
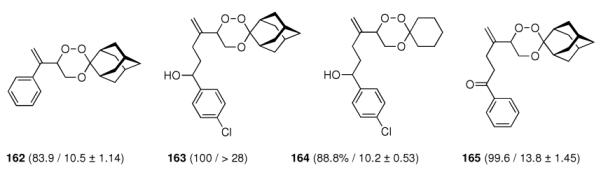
SAR of antimalarial spiroadamantane-1,2,4-trioxanes. Numbers in parentheses are the % suppression of P. yoelii parasites in Swiss mice on day 4 after receiving orally 96 mg/kg/day of the drug and the mean survival time in days.
Trioxanes 162, 164, and 165 also showed very promising antimalarial activity in the mouse model when given orally or i.m., however, the survival of the mice was not complete as with 163. Comparing the spiro-cyclohexyl trioxane 164 with the spiroadamantyl homolog 163, the striking importance of the adamantyl moiety is obvious: None of the mice treated with 164 survived past day 28.
The spiroannelated 1,2,4-trioxanes kept being studied. Out of several synthetic 1,2,4-trioxanes, synthesized via 1O2-ene-reaction of allylic alcohols and subsequent BF3-catalyzed peroxyacetalization and screened in vitro against the K1 strain of P. falciparum, also the ones incorporating the spiroadamantane-1,2,4-trioxane moiety (e.g., 166, Scheme 25; IC50 = 1.8 nM, cytotoxicity IC50 = 1.4 μg/mL) were the most potent ones.329 Compounds screened in this work with spirocyclopentane- or spirocyclohexanetrioxane groups were generally less active. Once more, the adamantane motif proved to enhance activity of the spiroannelated malaria chemotherapeutics.
Scheme 25.
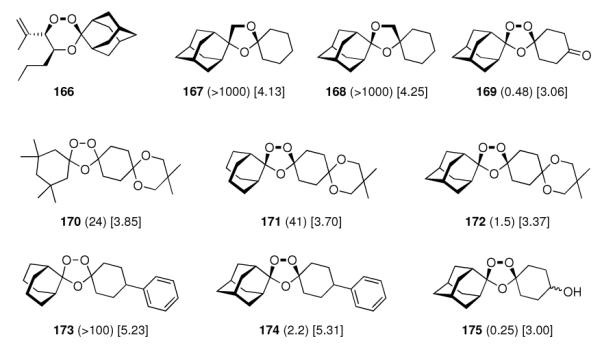
Antimalarial compounds for an SAR. Numbers in parentheses are the IC50 values against the Chloroquine-resistant K1 strain of P. falciparum in ng/mL measured in vitro, numbers in square brackets are ALOGPs data.
Trioxolane 161 was the first fully synthetic antimalarial peroxide that advanced to clinical trials. Since the antimalarial activity of the inspiring natural compound, Artemisinin, is largely determined by the peroxide group, a larger substrate library of compounds derived from the spiroadamantane-trioxolanes was synthesized via Griesbaum co-ozonolysis and subsequent conversions330 and screened to better describe the mechanism of action and to report a pharmacological and toxicological profiling of this class of compounds.28 What can be best described as the “lead structure”, 160 (Scheme 23) was chosen as a starting point for the SAR. The 1,3-dioxolane isosteres 167 and 168 (Scheme 25) were inactive in vitro as well as in vivo.
Likewise, the spiroadamantane motif is required for strong antimalarial activity or more generally, lipophilic antimalarial peroxides are potent. Moreover, most synthetic trioxolanes showed limited oral bioavailabilities, but the more lipophilic derivatives were better active orally than the polar counterparts. As mentioned above and in accordance with the iron activation hypothesis of antimalarial activity, with a peroxide group that is too exposed, the compounds largely lose activity due to high metabolism. Too inaccessible peroxide groups are equally detrimental since the Fe(II) species cannot attack the peroxide favorably to enable electron transfer. Consequently, compounds with balanced properties in terms of steric congestion, e. g., 169, display antimalarial activities comparable to artemisinin control experiments (IC50 (169) = (0.48 ± 0.37) ng/mL; IC50 (156) = 0.74 ng/mL). Lipophilic moieties other than adamantane were not as active, 170 and 171 were only weakly potent in vitro and completely inactive in vivo, while similarly lipophilic spiroadamantane 172 was significantly more active. This finding, along with the pronounced decrease in potency when comparing the spiroadamantane 174 with the spirobicyclo[3.3.1]nonane 173 proved the “…unique contribution of the spiroadamantane ring system to the antimalarial activity in these trioxolanes.” However, poor solubility seems to severely restrict the oral absorption of the trioxolanes studied. Compound 174 had the highest bioavailability at around 10% in rats and also the lowest plasma clearance. With regards to toxicology, the new spiroadamantane trioxolanes were generally uncomlicated, in particular their neurotoxicities against the NB2a neuroblastoma cell line was significantly lower than for Artemisinin.
The same authors later studied in detail the influence of functional group polarity on the physicochemical properties of 43 diversely functionalized spiroadamantane trioxolanes.331 Most of the compounds screened were quite potent against P. falciparum (Chloroquine-resistant K1 strain) with IC50s below 5 ng/mL. Moderately high lipophilicity is required for good antimalarial potency. Carboxylic acids were considerably less potent than were weak bases, which generally had good potency. In the in vivo model using P. berghei infected mice, the derivatives with good oral activities were lipophilic ones. As a conclusion, it became evident that in vitro potency alone is not a reliable predictor of in vivo activity. Mechanistic studies of the fragmentation process of various examples for antimalarial trioxolanes reacted with Fe(II) species showed a small preference for the attack of Fe(II) on the nonketal-peroxide oxygen in artemisinin, and less exposed and presumably more biologically stable peroxide bonds in related 1,2,4-trioxanes can at least in part explain the latter compounds' much weaker activity compared to artemisinin and the 1,2,4-trioxolanes.332 The electron transfer leading to carbon-centered radicals seems to constitute the required activation step of the artemisinins and other peroxidic antimalarials. With the pharmacokinetics being heavily influenced by the physicochemical properties of the peroxide antimalarials and formulation playing a significant role as well, alternative approaches to enhance the pharmacokinetics of the synthetic ozonides are being pursued. The binding of spiroadamantane antimalarials to cyclodextrins as vehicles for intravenous administration is one such example.333 Since cyclodextrins are also used for the solubilization of highly lipophilic drugs in aqueous media, these findings also could have impact on orally administered formulations.
Studies of the kinetics of Fe(II)-mediated degradation of the dispiro-1,2,4-trioxolane antimalarials represented another attempt to elucidate the MOA of these compouds, which remains controversial.334 Targets for alkylation after Fe(II)-mediated generation of C-centered radicals are, amongst others, the Plasmodium calcium ATPase (pfATP6), translationally controlled tumor protein (TCTP), haem, and glutathione. Alkylation studies for all of these substrates conducted with peroxide antimalarials require the presence of Fe(II). Studying the degradation kinetics of the compounds with FeSO4 via LC-MS analysis using 4-oxo-TEMPO as a radical trap to isolate intermediates, these authors found 158 (Scheme 23) essentially unreactive, whereas 160 and 169 (Scheme 25) were degraded fast. These observations essentially paralleled the antimalarial activity pattern. The mechanism of reductive degradation of the trioxolanes involves Fe(II)-induced homolytic peroxide-scission with subsequent rearrangement to C-centered radicals. The authors concluded: “The spiroadamantane substituent appears to impart some unique physicochemical, pharmacokinetic, or pharmacodynamic properties on either the radical intermediate, or the intact molecule, which are necessary for antimalarial activity of these trioxolanes, but not directly related to iron-mediated reactivity.” Fe(II) mediated reactivity appears as a necessery, but insufficient prerequisite for active antimalarials of the peroxide class. When comparing 1,2-dioxolanes to the corresponding 1,2,4-trioxolanes, the former were all either inactive or orders of magnitude less active than the latter against P. falciparum in in vitro screens or P. berghei in in vivo tests, respectively.29 The reason behind these findings is that reaction with Fe(II) gives two-electron reductions of the dioxolanes instead of one-electron reduction for the trioxolanes, so that much less C-centered radicals are being formed. Cytochrome P450 in liver microsomes has later been found to selectively hydroxylate one or two of the tertiary positions in the spiroadamantane framework.335 The resulting metabolites (see also chapter 8) are devoid of any antimalarial activity, demonstrating the essential contribution the unsubstituted spiroadamantane motif has to antimalarial activity. Most recent studies on the SAR of arterolane (161) and related spiroadamantane antimalarials therefore focused on modifications of the spirocyclohexane moiety.336 After the maleate of 161 in a combination with the antimalarial drug, Piperaquine has entered phase III clinical trials,337 still libraries of compounds with substituted cyclohexane moieties are being studied, bearing in mind that higher lipophilicity had been shown to generally enhance oral bioavailabilities and basic, but not acidic, peroxides are good antimalarials. The IC50s of these compound libraries are generally between 0.2 – 3.9 ng/mL, which is quite similar to Artesunate controls, in in vitro screens using different strains of the malaria parasite. The most active ozonides in vivo (P. berghei infected mice) were all weak bases, which are matabolically more stable than neutral ozonides, including the promising new ozonides 176 – 178 depicted in Scheme 26.
Scheme 26.
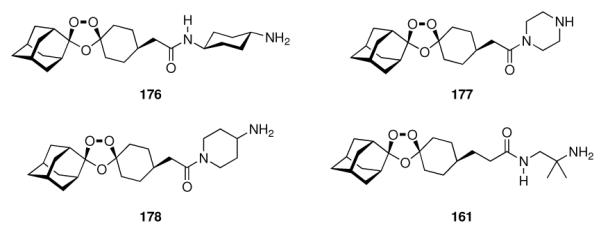
Further developments based upon the spiroadamantane-1,2,4-trioxolane motif.
Only 178 was found to be curative in the P. berghei infected mice when given at 3 mg/kg/day (three times post-infection): three out of five mice of the treated group survived with no detectable parasites at day 30 post infection. All the other ozonides screened here also increased mean survival time of the mice compared to control (no treatment) or, more importantly, compared to known drugs Artemisinine and Chloroquine, respectively. Aminopiperidine 178 also had the highest oral bioavailability in rats, being minimally toxic only when given orally to the rats in doses as high as 300 mg/kg for five days – overall, this is quite a simiar toxicity profile as for arterolane (161) itself. As a conclusion and in line with the mechanistic findings, the variations at the cyclohexane moiety do not influence the in vitro potency of the trioxolanes to large extent and can, therefore, be utilized for a fine-tuning of the compound's ADME properties. As a result, follow-up development candidates for 161 are available while the development of 161 continues. Recent reports include studies of the stage sensitivity of the malaria parasite against arterolane (161),338 finding radiolabelled 161 accumulated in red blood cells infected with P. falciparum. Arterolane (161) also has been shown to be active against P. vivax, a strain of malaria parasites that is more frequent in more temperate climates and also is beginning to develop resistances against known drugs.339 It is also excellently active against freshly obtained, Chloroquine-resistant isolates of P. falciparum in vitro.340 Antiplasmodial specificity has also been studied for 161.341 It inhibits P. falciparum ATPase 6 (PfATP6), albeit at a somewhat lower potency than Artemisinin.342 Iron chelators abrogate its activity, and 161 as well as artemisinin are concentrated 200- to 300-fold in intraerythrocytic parasites compared with extracellular concentrations. Via fluorescence labeling, these authors also studied the sub-cellular concentrations, again finding similar behavior of artemisinin and 161. Testing possible combination therapies, a combination of 161 with Piperaquine has been suggested as a “legitimate choice for development.”343 Arterolane 161 seems to similarly affect all stages of P. falciparum, at least in vitro.344 Addressing the MOA of 161, it was recently studied along with its non-peroxidic isosteres (vide supra) and nitroxyl radicals.345 The IC50s of Artemisinin and 161 were about 10,000-fold lower than those of the non-peroxide isosteres, and the antagonism of TEMPO to the action of both peroxide antimalarials again suggests formation of C-centered radicals as an essential prerequisite for activity in this class of antimalarials.
While the spiroadamantane-1,2,4-trioxolanes are actively being developed today and a drug candidate is in clinical trials, the closely related spiroadamantane-1,2,4-trioxanes also did receive renewed interest. Screening of a larger library of spiroadamantane trioxanes, synthesized via photooxygenation of allylic alcohols to β-hydroxy hydroperoxides which react readily with adamantanone, gave, amongst others, novel 6-arylvinyl- and 6-adamantylvinyl-substituted 1,2,4-trioxanes, e. g. 179 (Scheme 27).346
Scheme 27.
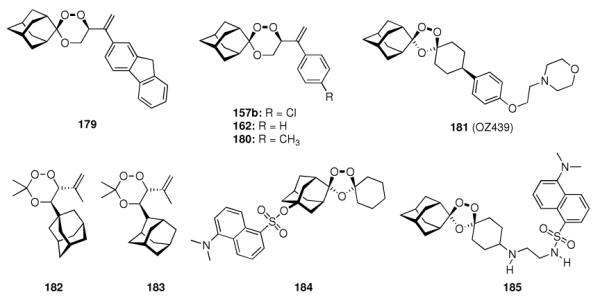
Recent developments of antimalarial 1,2,4-trioxanes.
This 6-fluorenylvinyl substituted 1,2,4-trioxane gave 100% suppression of the parasitaemia of P. yoelii in Swiss mice at 48 mg/kg × 4 days, with all the treated mice surviving beyond day 28 post-infection. However, when studied in rhesus moneys infected with P. cynomolgi, 179 was active when given orally at 10 or 20 mg/kg × 4 days, clearing 100% of the parasitaemia, but it did not provide long-term protection and was poorly active when given by the intramuscular route. As an additional requirement for activity, the adamantane has to be spiroannelated to position 3 of the 1,2,4-trioxane skeleton. Compounds 157b, 162, and 180 (Scheme 27) were also studied by the same authors,347 finding 157b and 180 100% curative in rhesus monkeys infected with P. knowlesii when given at 80 mg/kg × 5 days via the intramuscular route. The authors state that these compounds can be considered promising for development for the treatment of severe malaria. Currently in Phase II clinical trials is another synthetic spiroadamantane endoperoxide named OZ439 (181) that has been developed in an attempt at extended in vivo half life of only 1.3 h for Arterolane (161) in rats.348 In vitro Fe(II) induced degradation of 181 was > 50 times slower than that of 161. Likewise, 181 was 15 times more stable in healthy rat blood, and > 20 times more stable in healthy human blood samples. In rats, 181's half life following oral dodosing was > 20 h. Drug candidate 181 is highly active with fast onset of action in vivo in P. berghei infected mice, at the same time displaying significant prophylactic activity. The authors expressed their hope that this second generation spiroadamantane endoperoxide could end up as a single-dose cure. SAR efforts are also ongoing on the field of 5-adamantyl substituted 1,2,4-trioxanes such as 182 and 183, both of which are active against P. falciparum.349
Additionally, efforts are being undertaken to study the mechanism of action of the spiroadamantane endoperoxides and artemisinin derivatives. To this end, biotinylated derivatives of both classes of antimalarials have been reported350 that are envisioned to label the proteins that are being alkylated by the endoperoxides with a (cleavable) biotin moiety. Streptavidin-biotin “pull-down” assays could subsequently be utilized to identify the target protein(s) that is/are being alkylated by the antimalarial drug. Having said that, fluorescent-labeled spiroadamantane 1,2,4-trioxolane analogues such as 184 and 185 have been synthesized. Utilizing fluorescent microscopy clearly showed accumulation of fluorescence label in the food vacuole of the parasite for 184, carrying the label at the adamantane moiety, whereas for 185, with the label at the cyclohexane moiety, showed no clear localization pattern. This clearly shows that the C-centered radicals that act as alkylating agents must be adamantane-derived.351
In view of the far more than 1,000 endoperoxides that have been prepared and screened, the adamantane-annelated spirotrioxanes and spirotrioxolanes seem to be the principal pharmacophores.352 Arterolane (161) marked a breakthrough that is now in late clinical trials, and possible alternatives have been identified and also advanced to the clinic. It remains to be seen if the spiroadamantane endoperoxides will successfully reach the pharmaceutical market.
4.5. Aminoadamantanes With Trypanocidal Activity
Other protozoans having significant impact, mainly in sub-saharan africa, are the parasites of the Trypanosoma genus, more specifically, of Trypanosoma brucei gambiense and Trypanosoma rhodesiense. These parasites use the Tsetse fly as their insect vector and mammals, including humans, as a secondary hosts. T. brucei lives in the bloodstream of the mammalian host, ready to re-infect the fly vector upon biting, and during later stages of the infection in mammals, other areas of the host are affected, including the CNS. At this stage of the infection, the parasites cause loss of neuronal populations resulting in, amongst other symptoms, disturbance of the sleepcycle that gave the human african trypanosomiasis (HAT) its more familiar name, “african sleeping sickness”. The disease is invariably fatal if not treated. Since the parasites are repeatedly changing their surface coat which is presented to the host's immune system (“antigenic variation”), the prospects of vaccination are poor, rendering chemotherapeutics essential. The affected human population is, however, mostly poor, rendering drug development economically unattractive. This is reflected, at least in part, by the small number of available drugs to combat T. brucei, in particular in the neurological phase of the infection. Available drugs require medical supervision and the drug of choice for advanced stages of the disease, Melarsoprol, an arsenic compound, causes severe side-effects leading to reactive encephalopathy and death in about 5–10% of the treated patients. Other chemotherapeutics are, therefore, urgently needed, but only one drug, Eflornithine, has been registered in the last 50 years.353,354
In 1999, Kelly et al. reported that upon studying the expression of Influenza A M2 protein into trypanosomes, they discovered an in vitro anti-trypanosome effect of the antiviral drug, rimantadine (43, vide supra) against the bloodstream form of the parasite.355 The authors found the trypanocidal effect of rimantadine and amantadine to be pH-dependent (at higher pH, the drugs become more potent), with IC50 values in the μg/mL range. Rimantadine was somewhat more active than amantadine. Rimantadine was also found to be active against other strains of the parasite, namely T. cruzi and the related organism Leishmania major. The mechanism of action of the aminoadamantanes remained unclear, the authors assumed, by analogy with the anti-Influenza activity, interference of the drug with pH-maintenance of the parasite, possibly by blocking of a transmembrane transporter or proton pump. In a follow-up study, a larger compound library of 62 aminoadamantane- and aminocyclohexane derivatives was screened against T. brucei in an in vitro assay.356 Of the screened compounds, 17 were more active than rimantadine, some of which are depicted in Scheme 28. Increased hyrophobicity obviously enhances trypanocidal activity, and compound 188 was the most potent screened, being about 20–25 times more effective than rimantadine. In an in vivo model using immunosuppressed CD1 mice infected with T. brucei (strain 427), this most potent compound had to be discontinued due to significant toxicity. This was also the case with 186, as well as possibly with a number of other trypanocidal compounds screened. As for the mechanism of action, other than the rudimentary “lipophilicity enhances potency” SAR nothing new was discovered, but the authors discussed the well-known enrichment of the aminoadamantanes in lysosomes in mammalian cells, where they elevate the pH. The adamantane scaffold is not a prerequisite for activity, as a number of aminocyclohexanes also were trypanocidal, the most potent one being 190. Still, aminoadamantane 188 which is about as lipophilic displays significantly stonger trypanocidal activity (Scheme 28). Since the most critical stage of T. brucei infections is the invasion of the parasites of the CNS, the aminoadamantanes as CNS-active drugs (vide infra) readily distribute into the CNS, too, and for this reason should be ideal drug candidates for the treatment of advanced stages of HAT. Subsequent research gave compounds with trypanocidal activities comparable to or better than rimantadine (Scheme 28). Some spiroadamantane heterocycles, primarily synthesized as anti-Influenza A compounds, also displayed trypanocidal activity, like the spiro barbituric analog 191 or piperidine 87.151 Cage hydrocarbons other than adamantane have also been utilized as scaffolds for anti-T. brucei compounds.
Scheme 28.
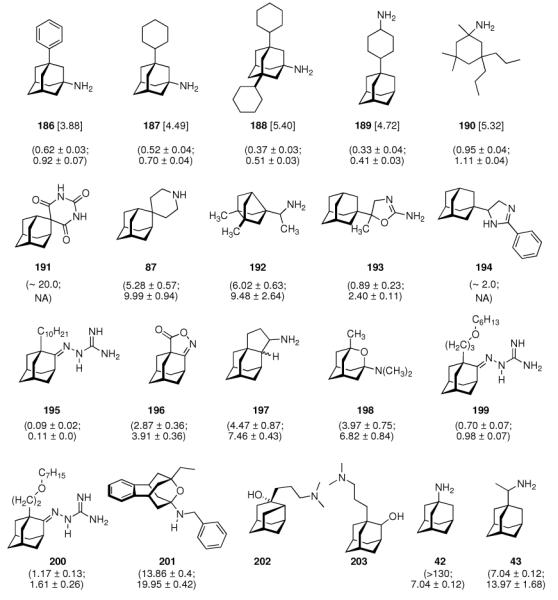
Aminoadamantanes and related compounds screened as tryptanocidals. Numbers in square brackets are ALOGPs data. Values in parentheses represent IC50 and IC90 data in μM. All values detected in vitro using T. brucei (strain 427) cultured in Iscove's medium at pH = 7.4 and 37 °C.
(±)-Bisnoradamantyl methylamine 192, structurally closely related to rimantadine, gave very similar IC50- and IC90 values, respectively.154 Out of a series of adamantane-2-oxazolines tested, only 193 displayed significant activity, and since this class of adamantyl-heterocycles has an effect on microtubule association that does not correlate with their trypanocidal potency, the effect on tubulin polymerization obviously is not contributing to anti-trypanosomal activity.357 4,5-Dihydroimidazole 194,358 adamantane oxazolone 196, and adamantane cyclopentane amine 197152 also are examples for adamantyl substituted heterocyles with trypanocidal activity at comparable potency in the in vitro test. Combining the guanylhydrazone pharmacophore, which is known to be a S-adenosylmethionine decarboxylase inhibitor and a trypanocidal motif by itself, with lipophilically substituted adamantane moieties, a synergistic effect of lipophilicity at C1 of the adamantane scaffold and the C2 functionality could be observed in compounds 195,359 199, and 200.360 For this class of trypanocides, a decane- (or “oxadecane” substitution, for that matter) substituent at C1 was shown to be ideal in terms of in vitro trypanocidal potency. Lastly, oxaheterocyclic amines like oxaadamantane 198361 and the secondary amine 201362 have also been shown to display anti-T. brucei activity in vitro; furthermore, the latter oxapolycyclic amine is devoid of any NMDA-receptor antagonism, which could be useful to avoid CNS-related side effects when using these compounds to combat T. brucei and other trypanosomes clinically. Recent additions to the library of adamantane-derived trypanocidal compounds are amino alcohols such as protoadamantane 202 and 2-hydroxyderivative 203363 as well as spiro carboxyclic adamantylidene diketopiperazines.364
While these findings are interesting and to some extent encouraging for the development of (low cost) chemotherapeutics for these kinds of “neglected diseases”, the data provided in this chapter mostly only provide in vitro potencies for various cage amines generated using a simple assay of cultures of T. brucei in its bloodstream form. Important tests, like routine toxicity screens in vitro or better in vivo have mostly not been performed, and physicochemical or pharmacokinetic data are also absent, so that the fortuitous finding that aminoadamantanes and related compounds are anti-trypanosomal agents has to be taken with a grain of salt. Much more data, in particular with regards to the mechanism of action of these molecules, are required to judge whether the aminoadamantanes could become clinically useful in the disease control of HAT.
5. Adamantanes Against Diseases of the Central Nervous System: Blocking Channels
5.1 The Dopaminergic System and Parkinson Disease – Another Amantadine Story
“In early April 1968, a 58-year-old woman with moderately severe bilateral Parkinson's disease recounted to us that three months before, while taking amantadine hydrochloride 100 mg twice daily, to prevent the flu, she experienced a remarkable remission in her symptoms of rigidity, tremor, and akinesia. These promptly returned on stopping the drug after six weeks.” This fortuitous finding reported by Schwab et al.32 in May 1969 marks the beginning of medicinal chemistry of adamantane derivatives in the context of diseases affecting the central nervous system, which, accounting for its lipophilicity and BBB-penetration enhancing properties, has since then become the el dorado for the utilization of adamantane derivatives in medicinal chemistry. When the findings of the above case report were reassessed in a group of 163 patients suffering from Parkinson disease (PD), 107 or 66% showed some improvement when treated with amantadine. A majority of patients kept sustained benefits after three to eight months duration of amantadine treatment, but a slow and steady reduction of benefit could be seen in one third of the patients who had previously improved. When a group of patients was shifted from amantadine to a placebo without their knowledge, they became aware of the change within one day. The group of patients in this initial trial was heterogeneous with respect to, amongst others, treatment regimen, previous treatment history, age, and duration of the disease, however the benefit of amantadine treatment remains remarkable. Schwab et al. conceded: “How amantadine hydrochloride lessens rigidity, akinesia, and even Parkinson's tremor is not clear from its pharmacology.” Nevertheless, a second clinical application of amantadine had been discovered, and it is being used as an adjunct therapy of PD, in particular L-DOPA induced dyskinesia, until today.
Subsequent clinical trials in general corroborated the above findings.365–368 The effect of amantadine was proven to be beneficial, in particular when given together with the standard therapy, L-DOPA. In general, typical dosing regimens used were between 100 and 300 mg per day, but even higher doses were regarded safe as notable side effects were weak and reversible. One outpatient took 100 capsules, 100 mg amantadine hydrochloride each, attempting suicide – and survived.368 The other aminoadamantane antiviral marketed, rimantadine was also studied in 17 patients, giving essentially no beneficial effect in an early clinical trial,368 however, more recently369 it was stated that some symptomatic benefit could be achieved in a group of 14 patients when taking rimantadine, in particular, rigidity improved. As the first-line PD treatment L-DOPA/Carbidopa induces well-known side-effects like motor fluctuations, ameliorating these is also needed. Amantadine (100 – 200 mg/day) can be added to a stable anti-PD drug regimen and decreases the severity of end-of-dose deterioration (“wearing-off”) in chronically treated PD patients.370 Amantadine is effective irrespective of the duration of the disease and duration of a previous treatment.371 Another simple aminoadamantane initially studied for its anti-Influenza A activity, memantine (190, Scheme 28), was identified as being of symptomatic benefit in PD – but the small structral differences (methyl substitution at two of the three remaining tertiary positions of the adamantane scaffold) also lead to intriguing differences in the pharmacological profile that became particularly obvious in drug research in the field of PD.
After the identification of amantadine as an agent useful clinically in the management of PD, a multitude of derivatives have been synthesized and studied. An obvious approach (vide supra) was the combination of pharmacophores. The cholinolytic activity of phosphoramidates had been found earlier, so this class of compounds was combined with aminoadamantane to give phosphoamidates like 204 (Scheme 29) that also would display amantadine's dopamine-enhancing effect and possibly an improvement in pharmacokinetics for CNS-targeting compounds.372 In reserpine-induced catalepsy in rats, an experimental model for toxin-induced PD,373 several 2-substituted adamantanealkanamines, prepared using for instance Pb(OAc)4-mediated intrameolecular C–H- functionalization of the secondary position, have been found to be about as potent as amantadine, while diol 206 showed the best activity under this paradigm.374 Several aminoadamantanes, including amantadine and memantine, were reported to be active in chemically induced (via spiroperidol) catalepsy PD in Wistar rats. While memantine (20 mg/kg, i.p.) reduced the catalepsy by 98.7%, amantadine at the same dosage only gave a reduction by 37.0%.375 These workers concluded that memantine has a stronger action on the CNS than amantadine. Protoadamantanamines as well as newly synthesized substituted adamantanealkanamines screened as anti-PD agents in the reversal of reserpine-induced catalepsy in mice and rats showed that 207 gave complete reversal of the catalepsy at 0.14 mmol/kg at an LD50 of about 1.0 mmol/kg in mice.376 A systematic study of the effect of bridgehead-substitution of 1-aminoadamantanes with a small library of seven alkyl-substituted adamantane amines utilized three models of PD: the stimulation of spontaneous locomotor activity in mice, the modification of circling behavior of mice with an unilateral striatal lesion from 6-hydroxydopamine injections, and the reversal of reserpine-induced akinesia in mice.377 The induction of locomotor activity saw striking differences even within this small compound library. While amantadine was the least active, memantine was as active as amphetamine under this paradigm. The authors attributed these differences to two separate dopaminergic mechanisms of action: indirect agonism for memantine and the methyl-substituted aminoadamantanes and a direct dopamine agonist component in ethyl derivatives like 208 and 209. While lipophilicity was found to contribute positively to the antiparkinsonian activity of the aminoadamantanes, it clearly is not the sole activity-determining factor, at least in the class of the aminoadamantanes. The workers stated that a factor best describing the observed activity trends would be “molecular shape”. Similar results were reported for N-alkyl substituted amantadine and memantine derivatives like 210, compounds with increased lipophilicity and somewhat enhanced activity; however, the reasons underlying the beneficial effect of N-alkyl substitution remained obscure.378 2-Aminoadamantane 94 was also found to increase the dopamine-release from dopaminergic nerve terminals.31,379 Within a series of nine adamantyl-substituted aminoalcohols studied in a multiparameter screen in mice, 211 and 212 were found to diplay effective anti-PD activities.31 The authors attributed these findings to the CNS-penetration capabilities of the compounds, which they found most pronounced in compounds bearing C4 – C8- as well as C14 – C18- alkyl residues. Comparison of the binding of twelve aminoadamantanes to the MK-801 binding site of the NMDA-receptor (memantine binds to this site, vide infra) via displacement of [3H] MK-801 in membrane homogenates of post-mortem human frontal cortex) found 212 was the strongest binding aminoadamantane studied (Ki = 0.19 ± 0.06 μM), while N-methylaminoadamantane 213 was the weakest binding compound.380 Memantine was nearly as strongly binding as the diethyl analog (Ki = 0.54 ± 0.23 μM), while amantadine's ~20-times higher Ki (10.5 ± 6.1 μM) reflects the 5 – 10 times higher doses of the latter over memantine used clinically in PD. Structure 214 is another aminoadamantane derivative that has been studied as an antiparkinsonian agent, e. g., in chemically induced parkinsonism in mice.381 More recent adamantane derivatives studied in the context of PD are TEMPO-derivatives like 215382 or the fullerene-derivative 216.383 Both compounds have been shown to be active against chemically induced parkinsonism in rodents. Recent developments also include the synthesis of subtype-specific NMDA-receptor antagonists like 217 or 218.384,385 Compound 217 was found to possess selectivity for the NR2B subunit of the NMDA-receptor and it did not show an effect on dopamine-concentrations in 6-hydroxydopamine-lesioned rats, while it increased the release of noradrenaline and dopamine from hippocampal and striatal slices at low micromolar concentrations.
Scheme 29.
Antiparkinsonian agents incorporating the adamantane motif.
How do the aminoadamantanes improve PD symptoms, what is their mechanism of action? It has been known that degeneration of dopaminergic pathways in the basal ganglia is the main pathology in PD,386 so affecting dopamine levels and those of its metabolites was the first potential pharmacological activity diuscussed for the aminoadamantanes' potential in PD. Most of the eary studies were based upon indirect in vivo evidence, however, a direct dopaminomimetic activity of the aminoadamantanes had been proposed, but remained controversial – probably also due to the lack of methodology to study other processes in the 1970s.387 In dogs, Grelak et al. found that amantadine increases dopamine concentrations; the authors assumed that the drug has a dopamine release-enhancing effect.388 Experimental observations in various model systems had been reported to corroborate amantadine's anti-PD activity, including enhancement of the synthesis and release of dopamine,389,390 or the blockage of dopamine reuptake through transporter systems by neurons.391 Both of these activities would end up in additional dopamine available for neuronal transmission in the synaptic cleft. Direct action as an agonist on the dopamine receptors has also been discussed for amantadine,392 but according to other studies,393–395 this inhibitory activity of amantadine is too weak of an action to fully explain its symptomatic benefit in PD. The increase in dopamine synthesis ex vivo by 40 mg/kg amantadine389 could never be reproduced.396 Even more controversially, Vaastra et al. reported on both an enhancing effect of adamantane on dopamine release and an inhibiting effect on dopamine-reuptake in the same study,397 but even these two potential actions of amantadine in PD models were considered not sufficient to fully explain why amantadine is so useful in PD. Cox et al.387 studied the tremor amantadine causes in mice when given in higher doses (ED50 = 140 mg/kg; at 200 mg/kg, all animals displayed tremor), which they assume to be of central origin. While p-chloro phenylalanine was somewhat protective against the amantadine-induced tremor, depletors of brain amines, e. g., diethyldithiocarbamate (which depletes in particular brain noradrenaline), cause significant potentiation of amantadine's tremor-inducing effect. Obviously, the loss of catecholamine stores increases the susceptibility of the mice to this effect of high doses of amantadine. Since p-chloro phenylalanine selectively depletes brain 5-hydroxytryptamine (serotonin) and pre-treatment with this drug almost completely abolished the tremor induced by amantadine, this effect of amantadine was attributed to the serotonin system. An effect on the activity of brain monoamine oxidase (MAO), an enzyme crucial for the biosynthesis of catecholamines, could not be shown; likewise, no effect on serotonin reuptake was observed. As a result, these authors concluded that amantadine sensitizes serotonin receptors – and in rat fundus as a model, they could observe an enhancement of serotonin activity through amantadine. Since in PD a decrease in serotonin is observed in addition to the well-known loss of dopamine, amantadine's additional effect could lie in the serotonin system. Wesemann et al.398 studied this serotonergic activity in more detail using isolated structures like synaptic vesicles, membranes, and synaptosomes. The uptake, binding and release of serotonin were studied. The results were in favor of the hypothesis that serotonin (re)uptake into synaptic vesicles is inhibited by amantadine, however, a high Ki and the low inhibitory effect make it unlikely that this is amantadine's only effect. Memantine's effect in this respect is somewhat stronger. Both the inhibition of serotonin release from and serotonin reuptake to synaptosomes by amantadine and memantine suggest an interaction of the aminoadamantanes with the nerve ending membranes or with the intracellular compartmentation of serotonin. However, even these findings cannot explain how the 1-aminoadamantanes “interfere on the molecular level with 5-HT and DA transport and distribution.”398 When studying the effect of memantine on membrane potentials as measured in isolated nerve fibre bundles, some effect of the drug on the permeability of the membranes for K+, Na+, and Cl− was found and used as an explanation for the aminoadamantanes' activity in PD.399 Memantine also was found to inhibit the activity of several cation channels, leading to suppression of neuronal activity, in cell culture experiments.400 The NMDA-receptor has subsequently been intensively studied as a potential target for the aminoadamantanes' activity in PD;401,402 experiments studying the binding of aminoadamantanes to NMDA-receptors showed an effect of memantine not only at the MK-801 site of the NMDA-receptor, but also a much higher selectivity for memantine (over amantadine) for this receptor: no interactions with the dopamine-, opioid-, GABA-, or adrenergic receptors were found in these studies.380,403,404 Electrophysiology showed that memantine, in concentrations obtained clinically, indeed prevents NMDA-receptor mediated neurotoxicity in rat retinal ganglion cells as well as in cortical neurons.38 The concentrations of memantine obtained clinically in humans in the cerebrospinal fluid (CSF) correlate to the serum concentrations (0.025 – 0.529 μM when given in doses of 5 – 30 mg/day) and could well be within the range required to bind to the NMDA receptor (Ki = 0.5 μM in human frontal cortex).405 Likewise, amantadine concentrations obtained clinically are also within the range of its Ki at the PCP-binding site in NMDA receptors as well as being high enough for σ-receptor stimulation:406 Post mortem concentrations of amantadine in brain slices of patients that were under an amantadine regimen gave a homogeneous distribution of this drug in the brain in concentrations ranging from 48.2 to 386 μM;406 much lower concentrations were detected in CSF and serum. Six weeks treatment of mice with amantadine resulted in a reduced effectiveness of stimulants of the CNS which act pre-synaptically, e. g., amphetamine.407 Their proposal of amantadine's effect is an interaction at the postsynaptic side within the membrane adjacent to the actual recognition site, thereby increasing the dopamine receptor affinity for its agonist. Amantadine has also been studied in a more reliable model for PD, that is, 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)- induced parkinsonism in mice.408 The drug was found to cause an 34% increase of striatal dopamine turnover in the animals. An excellent summary of research conducted pre-clinically in the field of aminoadamantanes to treat PD was published in 1997 by Danysz et al.396 summarizing in particular the change of the scientific community's attention towards NMDA-receptor antagonism as a major activity of the aminoadamantanes in the late 1980s. Summarizing the early in vitro studies, amantadine and memantine inhibit NMDA-stimulated acetylcholine release, an indirect action that is probably pivotal in the antiparkinsonian effects exerted by these drugs. Aminoadamantanes, in particular amantadine at the higher concentrations obtained clinically, may also act at σ1-receptors and nicotinic receptors,409–412 or enhance noradrenergic neurotransmission.413,414 Biochemically, a relatively high concentration of the aminoadamantanes would be required for a clear antagonism at the NMDA-receptor (depending on subtype), but memantine consistently gave higher affinities for this ion channel.40 Even high doses of amantadine (150 mg/kg) did not affect the dopamine levels in whole brain; instead, a decrease of noradrenaline and serotonin turnover was observed.415 Brown et al.416 as well as Maj et al.413 also questioned a direct dopaminomimetic effect of amantadine at high doses of up to 80 mg/kg. Likewise, memantine was found to change the levels of serotonin, dopamine, and their metabolites in various brain regions when studied ex vivo, but these results were highly inconsistent and of small magnitude and cannot satisfactorily explain memantine's potency.417 Moving on to in vivo studies, also high-dosage experiments were discussed. In cats perfused with amantadine,418 an increase in preloaded dopamine was observed; other in vivo studies to show a modest and highly variable effect of Aminoadamantanes on the dopaminergic system likewise use extremely high, clinically irrelevant concentrations of the drugs and are, therefore, considered not reliable for the elucidation of the primary MOA of these drugs.419–421 The NMDA-receptor antagonism was more and more considered to be a result of open-channel block of the receptor's pore, as the aminoadamantanes are strongly voltage-dependent antagonists.38,422,423 The difference in activities between amantadine and memantine found in various brain regions strikingly parallels the NMDA-receptor induced acetylcholine release in these areas (here: Amantadine has higher activity in striatal slices when compared to hippocampal slices).424–426 Clinically relevant concentrations of amantadine are more active in the striatum, whereas memantine at (lower!) clinically relevant concentrations seems to be more active in non-striatal parts of the brain, which may explain why amantadine is better suited to treat PD in humans.421,427,428 The low affinity and fast unblocking characteristics of the aminoadamantanes are particularly advantageous clinically, since this allows the receptors to stay functional, unlike with high-affinity NMDAR antagonists, e. g., MK-801. The distribution of NMDAR-subtypes in the brain and memantine's higher selectivity may also explain the alternate blocking mechanisms of the two drugs that have been observed.429
Summarizing the preclinical studies performed with amantadine and memantine, particulary emphasizing NMDAR antagonism, the aminoadamantanes obviously do not directly act on the dopaminergic system as was the consensus earlier, but rather manifest their antiparkinsonian effect through an attenuation of the imbalance of dopaminergic and glutamatergic pathways – at least to large extent mediated by NMDA receptor antagonism. The reason why amantadine seems to be better suited for PD treatment could lie, in addition to abovementioned NMDAR subtypes and their occurence in brain regions primarily affected by the pathology, in amantadine's binding to σ1-sites,37 its blocking of neuronal nicotinic acetylcholine receptors,430 and its capability to increase noradrenaline-release.390 These three latter properties could not be attributed to memantine. Another concept particularly interesting in the context of neurodegenerative diseases like PD are “neuroprotective” properties that more and more became en vogue in the 1990s. Excessive concentrations of the excitatory neurotransmitter glutamate are, for instance, neurotoxic in in vitro stuties, and glutamate-mediated “excitotoxicity” is, amongst other insults, attenuated by amantadine and memantine.39,428 Consequently, there have been reports that amantadine increases life expectancy in PD patients,431 an effect that could also be, at least in part, explained by a direct trophic effect of the drug.432 To summarize, the mid-1990s saw a consensus in the scientific community that the antiparkinsonian properties of the aminoadamantanes can be explained by their NMDAR-antagonism at clinically relevant concentrations. Additional effects observed for amantadine could explain its better applicability in PD in comparison to memantine (e. g., interactions with σ1-receptors, etc.), and low concentrations of the aminoadamantanes could also exert neuroprotective activity under excitotoxic conditions. The latter potential effects are clearly interesting for other neurodegenerative diseases (e.g., dementia, Huntington chorea) as well, and we will continue this discussion in chapter 5.3. More recent studies to explain amantadine's antiparkinsonian activity include a possible modulatory effect on the production of reactive oxygen species (ROS),433 an effect that must be indirect since aminoadamantanes per se did not act as scavengers or quenchers of •OH. Amantadine has furthermore been identified to be a non-competitive antagonist of α7, α4b2, and α3b4 nicotinic acetylcholine receptors at doses that did not alter the function of other ligand-gated ion channels on rat hippocampal neurons. With regards to the α7 nAChR, amantadine's IC50 (≈ 6.5 μM) again was close to the clinically obtained concentrations of the drug.430 At doses up to 100 μM in mice, memantine was also found to exert effects beyond NMDA-antagonism, in particular, stimulating effects on cholinergic signalling via muscarinic receptors could be detected in the mouse hippocampus.434 An amine transporter, hOCT2, could be responsible for monoamine reuptake and was found to be blocked competitively by amantadine and memantine.435 Furthermore, in rats, amantadine and memantine (like other glutamate antagonists) caused a pronounced increase in aromatic L-amino acid decarboxylase (AADC, also known as DOPA-decarboxylase), an effect which also would contribute to enhanced dopamine turnover upon aminoadamantane treatment.436,437 The gene-expression of AADC, which is of special importance during L-DOPA therapy in PD, has been found to be induced by 10 – 100 μM amantadine in cell culture.438 The effect of amantadine on dopamine (re)uptake into synaptosomes (30% increase after seven days treatment of rats at 40 mg/day, i.p., before using the brains of the animals for the preparation of synaptosome fractions) was not an effect of altered expression of the dopamine transporter (DAT), but rather an indirect effect via glutamate receptor antagonism. These receptors also regulate DAT phosphorylation, thereby regulating dopamine reuptake.439 Memantine also inhibits KATP channels in dopaminergic neurons at 30 and 100 μM,440 corroborating findings from studies performed 30 years earlier.399 Recent studies on mechanisms of action of amantadine showed that the drug's capability to ameliorate L-DOPA induced dyskensia, a side effect of the primary PD treatment that still is far from being understood fully,441 is associated with modulation of the striato-nigral pathway in mice and rats.442 Amantadine reduces the activation of microglia, ameliorating inflammatory processes and, consequently, neuronal loss.443,444 In astroglia, increased expression of neurotrophic factors such as glia-derived neurotrophic factor (GDNF) was effected by amantadine, another mechanism of action to be summarized under the term “neuroprotection”.444 Lastly, noradrenaline transporters are also blocked by amantadine, an effect also seen with amphetamin, but to a lesser extent with the aminoadamantane.445
In conclusion, usage of memantine and, even more so, amantadine, against PD symptoms most certainly involves interaction of the simple aminoadamantanes not only with a host of receptor systems, but also, as we have learned in the previous chapters, indirect modes of action via the pronounced membrane modifying capabilities of the drugs. This “promiscuous” receptor binding of amantadine and memantine are probably best summarized by the term “dirty drugs”.446 These might help in the drug discovery process, as an alternative route to drugs for complex diseases447 – and PD is, most certainly, one such complex disease.
As an example, A-77636 ((R,S)-219, Scheme 30) can be briefly mentioned here. This adamantane derivative, also resembling catecholamines structurally, has been found to be a long-acting, selective dopamine D1 receptor agonist at nanomolar affinity in rats.448 In MPTP-treated marmosets, it reverses the parkinsonian symptoms, whereas the enantiomer, (S,R)-219, does not. These data substantiated the hypothesis that D1-receptor agonists may be of use clinically in PD. A follow-up study showed, however, rapid de-sensitization of the D1-receptor; dose escalation was not suited to fully restore the anti-PD symptom benefit in the MPTP-treated animals.449 Cell culture studies later showed that (R,S)-219 obviously is too good of an agonist for D1 receptors: it dissociates extremely slowly from the receptor, causing long duration of receptor activation with subsequent destabilization, which finally gives rise to behavioral tolerance in vivo.450 Further supporting the principle suitability of D1 receptors as drug targets in PD, behavioral studies in the MPTP-treated marmoset model, more recent studies focused on the effect of D1 receptor stimulation during treatment of the animals with L-DOPA / Carbidopa. D1 agonists, amongst others, (R,S)-219, showed symptomatic benefit in this model for PD.451 The dopaminergic system is depending on intact D1- and D2-receptors, however, as the antiparkinsonian effect of (R,S)-219 in the abovementioned marmoset model is strongly diminished upon addition of selective D2 antagonists.452 Structure (R,S)-219 subsequently played a role in elucidating the negative feedback regulation of dopamine release in mammalian brain, knowledge of which may also be of significance in PD research.453
Scheme 30.
Selective dopamine-D1 receptor agonist (R,S)-219 and its significantly less active enantiomer, (S,R)-219
Compound (R,S)-219 appears to be binding to the receptor complex to such an extent as to stabilize the D1 subtype during and after internalization into the neuron.454 In this study, the D1 receptor recovered to the cell surface later – demonstrating activities of potent, long-acting D1 agonists beyond immediate activation of the receptor, probably caused by allosteric interactions.
From this chapter on the utilization of adamantane derivatives clinically to combat PD symptoms as well as to elucidate experimentally the molecular dysfunctions underlying PD and related disorders, we could learn that amantadine is safe and efficient clinically, in particular when given as an adjunct to L-DOPA regimens.455,456 At least in part, amantadine's and, even more so, memantine's benefits in PD can be attributed to a non-competitive NMDA receptor antagonism457–459 which also acts neuroprotective and leads to a later onset of dementia in PD patients treated with amantadine,460 an effect that directly points towards the next major application of adamantane derivatives (see chapter 5.3). Likewise, with the aminoadamantanes being “dirty drugs”, a host of other sites of interaction of the compounds, mostly located in the CNS, has been identified that could give rise to drug development for these targets in the future.
5.2 KATP Channels and AMPA receptor channels
In the previous chapter, we have encountered several times effects of adamantane derivatives on ion permeabilities, which can be ascribed to interactions with ion channels other than the NMDA-receptor.
Potassium channels are found in most cell types and play important roles in, e. g., the regulation of blood pressure and maintaining and shaping of membrane potential. Therefore, these ion channels represent rewarding targets for drug development. ATP sensitive potassium (KATP) channels are controlled by the intracellular concentration of ATP an their opening can be caused by a decrease in intracellular ATP. Over the last decade or so, there has been a steady drumbeat of studies via X-ray crystallography and electrophysiology to elucidate structure as well as kinetics and pharmacological interference of this class of ion channel and its subtypes.461,462 Control of pancreatic KATP channels through pharmaceuticals like, e. g., Tolbutamide, results in an increase of insulin excretion. We have already heard of hypoglycemic properties of adamantane-derived sulfonylureas,7 and starting in the 1980s, other adamantane derivatives have been found to exhibit insulin releasing activities in vitro. When studied in mouse islets as the model system, amantadine was reported to be as potent as 2-aminoadamantane in this respect, while adamantane-1-carboxylic acid displayed a markedly weaker effect, and 1-adamantanol was devoid of any insulin-releasing potency.463 The authors concluded that aminoadamantanes decrease the K+ permeability of β-cell membranes, this causes depolarization of voltage-dependent Ca2+ channels, followed by Ca2+ influx and subsequent insulin release. However, the authors did not expect aminoadamantanes per se to become useful as drugs in the treatment of diabetes, but “…derivatives of this molecule or addition of this molecule to already active compounds may perhaps provide useful drugs.” (see also chapter 7.3). Nevertheless, the aminoadamantanes were by far the simplest molecules to inhibit potassium channels in β-cells. Effects on potassium channels were reported for a number of adamantane derivatives,464 including 220 (Scheme 31), which has been found to selectively block myocardial K+ channels and to antagonize the effects of acetylcholine, which is a potassium channel activator.465 Since openers of potassium channels are vasodilators and antihypertensives, their antagonism is highly useful in the drug development in this arena. There has been previously only one sulfonylurea derivative that consistently blocks the actions of potassium channel openers; the first non-sulfonylurea exhibiting this profile beneficial in the target validation process was the guanidine derivative 221, which is a compound capable of a consistent and selective blocking of the pharmacological responses to K+ channel openers in vitro and in vivo.466 A synergism of 221 and the sulfonylurea glyburide in their action as antagonists of vasodilation by KATP channel openers has been, for example, utilized for the elucidation of the regulation of vascular KATP activity.467 By studying the effect of 221 and its analogues 222 and 223 in follicle-enclosed oocytes of Xenopus, these derivatives' capabilities as potassium channel blockers were studied in more detail.42 While 222, like 221, was able to inhibit KATP currents and to displace [3H]-221 from its binding site, 223 was not, indicating a decisive effect of the adamantane cage for receptor binding. Another major feature of 221 is its selectivity, because, unlike amantadine (vide supra), it does not stimulate insulin excretion. More recently, 221 was used to study various subtyes of cloned KATP channes in Xenopus oocytes43 and KATP channels in rat middle meningeal arteries, since dilating such intracranial arteries may be causing migraine.468 Having reached the brain area once more, we also point out here that part of memantine's anti-parkinsonian activity may be, in addition to its NMDA-receptor antagonism, related with an effect on KATP channels in the brain. As KATP conductances play a role in the degeneration of dopaminergic neurons (KATP channels are opened in conditions of metabolic stress), pharmacologically blocking these channels may, at least in part, be responsible for memantine's antiparkinsonian effect.440
Scheme 31.
Adamantane derivatives used to study KATP channels
Another ion channel of medicinal importance is the so-called AMPA receptor, which is gated by α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (224, Scheme 32). Together with the glutamate receptors opened by NMDA and Kainate, these ionotropic glutamate receptors play crucial roles in processes of long-term potentiation (LTP) or long-term depression underlying learning and memory and are, therefore, valuable targets for pharmacological research. Because the only X-ray crystal structure of ionotropic glutamate receptors available then was of a truncated amino terminal domain from the AMPA receptor subtype GluR2 (more recently referred to as GluA2),469 the molecular machinery of the gating, ion conductivity, and pharmacological manipulation remains to be fully understood. AMPARs are tetrameric complexes, variably composed of a “dimer of dimers” recruited from the receptor subtypes GluR1 – GluR4. The ion channel assemblies have four sites to bind the agonist, and are (depending on the subtype composition) permeable for a variety of cations including Ca2+, Na+, and K+. Notably, GluR2-lacking AMPARs have an impaired conductivity for Ca2+, which is of importance under “excitotoxic” conditions known from a variety of neurological disorders. Regulation through phosphorylation (at four sites of GluR1) as well as other intracellular processes leading to an increase in AMPARs is believed to take part in LTP. AMPARs as well as the other ionotropic glutamate receptor ion channels mediate the vast majority of fast excitatory interneural communication. Studies on the distributon of the AMPAR subtypes in the different neuronal populations and their role in neuronal network activity as well as their role in disease states depend on the availability of subtype-selective ligands of these receptors (which additionally have to be distinguished from NMDA and kainate receptors),470 and in this field, some adamantane derivatives (initially considered as blockers for NMDA or nicotinic acetylcholine receptors471) have been developed and successfully utilized pre-clinically.
Scheme 32.
Compounds used to elucidate the pharmacology of AMPA receptors and a homology model of the pore built around IEM-1460.473
The dicationic adamantane derivatives 225 and 226 (Scheme 32) were used to study electrophysiologically the block of recombinant AMPARs as expressed in oocytes of Xenopus laevis as well as in in vitro experiments using neurons from rat hippocampus.472 Both compounds non-competitively blocked the conductivity of the channel in a use- and voltage-dependent manner. They also displayed subtype-selective antagonism, as the blockage was cell-type specific in the rat neurons studied. The presence of “edited” GluR2 subunits diminishes the blocking capabilities of the adamantane containing dications, indicating a use of these compounds for selectively studying the function of subtypes of AMPARs. As the edited GluR2 subunits incorporate an Arg residue instead of a Gln residue in their M2 segment (the location of this mutation is being referred to as the “Q/R-site”), an additional positive charge within the pore naturally inhibits binding of positively charged molecules simply by electrostatic repulsion. Compound 225 was found to be several times more potent than 226 in these assays of AMPARs while it is 5.6-times less potent as an NMDA receptor inhibitor. In all cases, these compounds are open channel blockers, which means that agonist has to be present to open the channel pore, allowing the dication to access the pore region.
This basic functional mode of the AMPAR is corroborated by several modeling studies.474,475 The Q/R site within the M2 part of the AMPAR that forms a membrane re-entry loop incorporates sidechains that presumably do not completely extend into the pore but rather form a macrocycle stabilized by intersegment H-bonding of the tetrameric receptor complex. This rationale shows some three-dimensional similarity with a bacterial K+ channel for which X-ray data are available. To this end, other than adamantyl dications, polyamine toxin-derived compounds have also been utilized to study AMPARs. The main problem of these polyamines is their lack of selectivity; consequently, libraries of derivatives have been synthesized. Notably, the combination of adamantane derivatives in the headgroup of AMPAR channel blockers derived from polyamine toxins have been found to be the most potent open-channel blockers of AMPARs to date.476 While the 37 compounds studied in this account via two-electron voltage clamp techniques in X. laevis oocytes expressing homomeric GluR1 AMPARs all incorporate the same polyamine motif (227 a–c, Scheme 32), their headgroup was varied using various amino acid side chains (R1) as well as small and large N-acyl sidechains (R2). Surprisingly, adamantylacetamides were the two most potent AMPA open-channel blockers (227a, 227b), with the ones incorporating charged amino acid side chains (His and Arg) being preferred. Compound 227a, the adamantane derivative that is the most potent AMPAR channel blocker, displays about 100-fold higher activity (Ki ≈ 2 nM) when compared to the n-butanoyl derivative 227c (Ki ≈ 200 nM). Thus, the adamantane derivatives reported in this context, considered “hybrid molecules” resembling features derived from polyamine toxins and the semipotent AMPAR channel blockers like 225 and 226 surprisingly are significantly more potent than either of their parent structures.
Studying the determinants of selectivity for the dicationic adamantane derivatives, Magazanik and coworkers found that the maximum activity for AMPAR channel block is oserved with a -(CH2)5- chain connecting the ammonium groups, as can be seen in, e. g., 225, 226, 228, and 229.477 The dications of varying length have been used to gain insight into the topography of the ion channel. The adamantane and the terminal ammonium group were postulated to interact with a lipophilic and a nucleophilic site, which were assumed to be separated by ~ 10 Å in the AMPAR, but close to one another in the NMDAR, thereby explaining, at least in part, the selectivity of the dications for the AMPAR. Mechanistically, the blocking rate constant was considered responsible to determine the subtype selectivity of 225, with the subunit composition of the actual AMPAR channel influencing accessibility of the binding site within the pore.478 Later, based upon homology modeling of the AMPA receptor ion channel “around” bound dication 225 (see Figure in Scheme 32), the binding sites of spider and wasp toxin polycations was elucidated.473 This model was derived from the K+ channel of Methanobacterium autotrophicum, which belongs to the same receptor superfamily. More recently, further detailed electrophysiology studies found that the adamantane containing dications including 228 and 229 permeate into the cytoplasm through the open and the closed AMPAR channel.479 When applied intracellularly, 229 was able to block Ca2+ permeable AMPARs (that do not incorporate the GluR2 subunit) both with and without the agonist present, that is, it acted as an open channel blocker and a closed channel blocker. The extracellular blocking of 229 could be decreased by intracellular 229, showing that the binding site in both cases is the same or at least overlapping significantly.480 While a clinical use of the AMPAR channel blockers reported here has not been disclosed, they found several applications in pre-clinical research. Populations of AMPARs in various rat brain cell populations have been characterized using 225,481 finding that 225 is a convenient marker for the absence of the GluR2 subunit in AMPARs. Some cell types seemed to express both types of AMPARs, sensitive and unsensitive for blockage through 225. This compound also has been found to decrease excitatory postsynaptic currents in rat hippocampus.482 Finally, 225 along with other mono- and dicationic derivatives of adamantane and phenylcyclohexane was used to distinguish between NMDA and AMPA mediated processes underlying the prevention of pentylenetetrazol-induced kindling in experimental mice.483
5.3 The NMDA Receptor and Alzheimer Disease – The memantine Story
The other main ionotropic glutamate receptor in the CNS has been classified as the NMDA receptor after N-methyl-D-aspartate, its selective agonist. Its channel is permeable for several cations, but characteristically, it is both ligand-gated and voltage dependent and requires two excitatory amino acids, glutamate and glycine, to become activated. Its involvement in neural transmission as well as several neurological disorders is well-known and this glutamate receptor subtype is generally the best studied drug target in this field.484 NMDA-receptor mediated neurotransmission is the primary interneuronal communication underlying synaptic plasticity. Consequently, the NMDA receptor has also been targeted by adamantane derivatives.
5.3.1 Discovery of Memantine
3,5-Dimethylaminoadamantane (memantine, 230, Table 4) was reported for the first time in a medicinal chemistry context as an intermediate for the preparation of hypoglycemic sulfonylureas,7 but this class of compounds was not followed in the field of diabetes research. Instead, Merz filed a patent application in 1972485 stating that memantine and other aminoadamantanes are suited to combat symptoms of Parkinson Disease because they release dopamine in the brain. No examples, details, or studies were disclosed at this point. In another patent application, Merz disclosed the preparation of several aminoadamantanes via the reaction of 1-haloadamantanes with ureas and subsequent hydrolysis.375 Additionally, the authors stated that these aminoadamantane derivatives display valuable pharmacological properties to influence the CNS in man and animal, which is particularly useful in PD. They assumed an effect of the test compounds on the catecholamine-metabolism, in line with the consensus of PD research at the time (see Chapter 5.1). Some basic experiments in drug-induced parkinsonism in mice were disclosed to confirm the superiority of memantine over amantadine. Other earlier aminoadamantanes reported valuable in this context were mostly alkylated aminoadamantanes.486 Later, Svensson et al. also reported on the differences between memantine and amantadine.487 A summary of early studies on the CNS effects of memantine was published in 1982 by Maj et al.,413 again stressing a very pronounced stimulation of the dopaminergic system, but also a stimulation of the serotoninergic and GABAergic systems. Pharmacodynamical and pharmacokinetical data on memantine as well as clinical observations corroborated these findings.488 The first clinical trial of memantine in dementia was reported in 1986.489 Ten patients suffering severe AD were treated with 20 – 30 mg/day memantine (i.v.), but no significant differences to a placebo group could be demonstrated. However, a mild amelioration of sleep-wakefulness disturbances was found in the drug-treated group.
Table 4.
Summary of preclinical data on various NMDA receptor antagonists.
| No. | Structure | Anticonvulsive ED50 [mg/kg]491 | MK-801-extrusion Ki [μM] | Affinity at σ-sites Ki [μM] | Binding Ki [μM] | Patch Clamp IC50 [μM] |
|---|---|---|---|---|---|---|
| 230 |
|
16 | 0.54 ± 0.23 | 19.98 ± 3.08 | 0.70 ± 0.11 | 2.3 ± 0.3 |
| 208 |
|
30 | 0.60 ± 0.27 | - | - | - |
| 209 |
|
24 | 0.19 ± 0.06 | - | 0.32 ± 0.09 | 3.0 ± 0.2 |
| 231 |
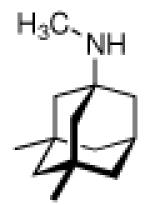
|
13 | 1.61 ± 1.17 | 0.341 ± 0.055 | 1.34 ± 0.17 | 4.4 ± 1.2 |
| 232 |
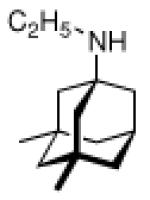
|
- | 1.72 ± 0.43 | 1.34 ± 0.23 | - | - |
| 233 |
|
- | 4.08 ± 0.73 | - | - | - |
| 234 |
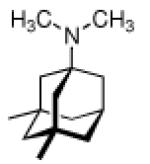
|
- | 4.40 ± 2.18 | 0.273 ± 0.019 | 2.01 ± 0.20 | 28.4 ± 1.4 |
| 235 |
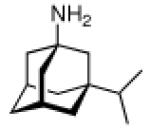
|
- | 7.21 ± 9.54 | - | 0.71 ± 0.18 | 4.2 ± 0.2 |
| 42 |
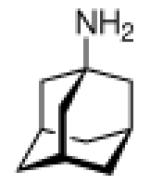
|
- | 10.50 ± 6.10 | 20.25 ± 16.84 | 20.42 ± 5.43 | 71.0 ± 11.1 |
| 186 |
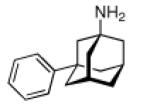
|
- | 15.16 ± 0.87 | 2.60 ± 0.43 | - | - |
| 236 |
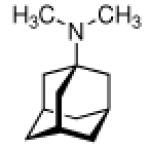
|
- | 20.77 ± 2.14 | 2.54 ± 0.24 | - | - |
| 213 |
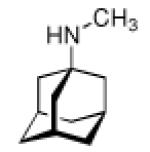
|
- | 21.72 ± 1.63 | 1.56 ± 0.19 | - | - |
| 237 |
|
- | - | - | 1.96 ± 0.31 | 6.9 ± 1.0 |
| 238 |
|
- | - | - | 0.41 ± 0.17 | 4.3 ± 0.9 |
| 239 |
|
- | - | - | 0.89 ± 0.23 | 6.9 ± 0.1 |
| 240 |
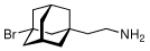
|
- | - | - | 1.0 ± 0.14 | 6.2 ± 0.1 |
| 241 |
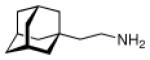
|
- | - | - | 0.49 ± 0.02 | 8.0 ± 1.2 |
| 242 |
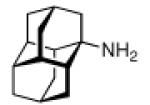
|
- | - | - | 2.17 ± 0.28 | 9.6 ± 1.3 |
| 243 |
|
- | - | - | 0.89 ± 0.20 | 7.3 ± 1.9 |
| 244 |
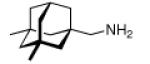
|
- | - | - | 3.88 ± 1.96 | 14.0 ± 1.1 |
| 245 |
|
- | - | - | 3.04 ± 0.60 | 1.5 ± 0.7 |
| 246 |
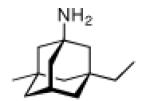
|
- | - | - | 0.60 ± 0.12 | 3.1 ± 0.07 |
|
| ||||||
| 247 |
|
9 | - | - | 0.50 ± 0.10 | 1.0 ± 0.20 |
| 248 |
|
< 1 | - | - | 0.0026 ± 0.0002 | 0.14 ± 0.10 |
5.3.2. Other Adamantane Derivatives Studied as NMDAR Antagonists
A number of aminoadamantanes that were synthesized for the prevention and treatment of cerebral ischemia were reported in 1989.490 As one of several pathologies causing ischemia, AD was briefly mentioned here. The focus of this patent application lay on the preparation and pre-clinical screening of a small library of aminoadamantanes. As their anticonvulsive action correlates with the binding to NMDAR, this was studied using the supermaximal electroshock method in mice, 40 min after the test compounds were given i.p. to the animals in various concentrations (Table 4). In addition, memantine was shown to act neuroprotectively in a model for ischemia (closure of the carotid artery in rats). More preclinical data on these aminoadamantanes were disclosed in 1991, including a comparison of the displacement of [3H]-(+)-MK-801 (248) from membrane homogenates of human post-mortem frontal cortex.380 Likewise, post-mortem human frontal cortex homogenate was also utilized to detect the affinity of the aminoadamantanes to the σ-site; here, competition experiments of the aminoadamantanes with [3H]-(+)-pentazocine were performed.37
At concentrations resembling those obtained therapeutically (10 – 30 μM428), amantadine probably binds to the NMDAR (PCP-site) as well as to the σ1-site (Table 4). In contrast, memantine at therapeutic concentrations (< 2 μM492) probably does not bind to σ1-sites, rendering it a more selective NMDAR antagonist and contributing to its clinically safe profile. Further studies, including whole-cell patch clamp studies, receptor binding assays, three independent in vivo convulsion models as well as models of motor impairment in vivo showed that the aminoadamantanes in general had a poor therapeutic index in the epilepsy models.40 Summarizing these data, it is obvious that memantine has a higher affinity and selectivity to the PCP-site of the NMDAR than amantadine and most of the other aminoadamantanes. However, within the relatively small library of aminoadamantanes summarized in Table 4, there are molecules that display higher affinities (e. g., 209). The reason why development of memantine for this indication was continued lies in its favorably fast receptor blocking/unblocking kinetics that has been studied extensively, in particular comparing amantadine and memantine to compounds like 247 and 248. We will discuss this topic later (see Chapter 5.3.4). Most recent reports on NMDAR blockers incorporating adamantane or related cages include oxacyclic compounds like 249 and 250 (Scheme 33).361 The most active channel blockers have blocking abilities against Ca2+-influx in NMDA-challenged primary neuronal cultures between those of amantadine and memantine, however, electrophysiological measurements of these new compounds have not been reported to date.
Scheme 33.
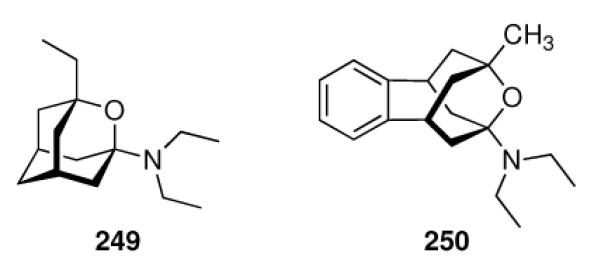
Recent cage compounds studied as NMDAR blockers.
5.3.3. Clinical Trials
The first clinical trial studying memantine in 20 patients with severe AD gave inconclusive results (vide supra); however, given the short treatment period in this study (20 days) and the small sample size together with an obvious bias due to increased attention to the patients participating required additional trials.489 Renewed interest in this class of drugs due to lack of other pharmacological treatment options, along with the change in the consensus of the mechanism of action (from dopaminergic to NMDAR-blocking) warranted numerous trials, some of which we will briefly mention here.493
The 12-week trial reported by Winblad et al. in 1999494 comprising 166 severely demented patients (51% vascular dementia, 49% Alzheimer disease) gave encouraging results. In this group of patients, treatment with memantine (10 mg/day) led to significant functional improvement and reduced care dependency of the treated group over placebo controls. Together with the finding that treatment with amantadine led to a delayed onset of dementia in PD patients,460 a host of clinical trials495 followed that have been reviewed and summarized elsewhere:496 “Memantine has a small beneficial, clinically detectable effect on cognitive function and functional decline measured at 6 months in patients with moderate to severe Alzheimer's Disease (AD). In patients with mild to moderate dementia, the small beneficial effect on cognition was not clinically detectable in those with vascular dementia and barely detectable in those with AD. It is well tolerated.” A recent meta-analysis, summarizing data from placebo-controlled phase III clinical trials of 1,826 patients with moderate to severe AD also supports efficacy and safety of memantine at these stages of the disease:497 “Memantine treatment resulted in a statistically significant benefit in four efficacy domains: the cognitive, functional, global, and behavioural endpoint. These data were the basis for the extension of the memantine indication to comprise moderate and severe AD in Europe.” Meanwhile, after having been used clinically in Germany for some time in cognitive disorders, memantine was approved for moderate to severe AD by the EMA in 2002 and by the FDA in October, 2003.498 It has gained “blockbuster” status since then. Memantine's efficacy, in particular in mild to moderate stages of AD, has since been questioned by some trials,499 while others found it to improve communication skills in patients with moderate AD.500
5.3.4. The Mechanism of Action: Moderate Affinity, Noncompetitive, Fast
How does it work? Memantine had been found relatively early by patch-clamp measurements in spinal neurons from mouse embryos to be an equally effective NMDAR antagonist like MK-801 (dizolcipine, 248).404 It binds to the receptor only when it is opened by endogenous agonists (Glycine, Glutamate) or NMDA (“open channel blocker”) in a non-competitive fashion. Hence, memantine displaces [3H]-(+)-MK-801 from its binding site within the NMDAR channel in post-mortem human frontal cortex at therapeutically obtained concentrations (Ki = (0.536 ± 0.035) μM), which is comparable to the potency of Ketamine, the dissociative anesthetic (ab)used as a “recreational drug” which is known to bind to the NMDAR's PCP-site.403 However, even though it binds to the same site at the same receptor with comparable potency, memantine obviously lacks the psychotomimetic side effects of, e. g., PCP (247). To this end, the behavioral and neurochemical effects of competitive and noncompetitive NMDAR antagonists (including memantine) were studied, showing differences in the behaviour of the treated animals.417 The non-competitive NMDAR-antagonist memantine resembled the stimulatory effects of amphetamine in the animal model. In addition, the concept of excitotoxicity, excessive Ca2+ influx into neurons mediated by overly activated NMDAR through high levels of glutamate, gained popularity.501–503 Insults causing these neurotoxic processes include ischemia, trauma, epilepsy, and neurodegeneration, and a selective block of NMDAR channels could, at least in part, decrease Ca2+ influx. However, none of the NMDAR antagonists had been proven effective and safe clinically – their strong and selective binding to the receptor causes considerable side effects. Whole-cell patch-clamp measurement and single channel recordings in various retinal and cortex cultures of the rat showed that memantine indeed acts via an open-channel block similar to dizolcipine (that is, it only acts as antagonist when the agonists open the NMDAR ion channel). However, compared to 248, memantine displays faster kinetics of action with faster blocking and unblocking rates. Being an uncompetitive antagonist, the presence of clinically obtained concentrations of memantine would allow for near normal physiological function of NMDAR-mediated signalling, unlike NMDAR blockers with slower kinetics.38 The effects of higher concentrations of the agonist NMDA were blocked by memantine to a higher degree than at lower concentrations, allowing near normal function of the NMDAR channel in processes like LTP. These authors also reported that low micromolar concentrations of memantine prevent the neurotoxic effects mediated by NMDAR in rat neurons, and that it also acts in a neuroprotective fashion in a reliable animal model of stroke. In in vitro measurements on rat retinal ganglion cells, the delayed application of 12 μM memantine protected the cells from neurotoxic insults exerted by potentially toxic exposure to glutamate.504 Corroborating these findings, a subsequent study showed that memantine dose-dependently antagonizes the responses to NMDA (100 μM) in patch-clamp measurements on cultured neurons with an IC50 of (2.92 ± 0.05) μM. Application of 8 μM memantine did not affect the current responses to the neurotransmitters, AMPA or GABA. The uncompetitive, use- and voltage-dependent mode of action was demonstrated once more. Moreover, memantine has no affinity to the NMDAR Gly-site, as even 100 μM glycine did not reverse memantine's effects on the current responses.422 The concentrations of the drug in serum and cerebrospinal fluid (CSF) were measured in patients under a dose regimen used clinically.405 Patients taking 5 – 30 mg memantine per day displayed serum concentrations of 0.025 – 0.529 μM; CSF concentrations were about half of that. These concentrations are well within range of the Ki value at the PCP site. However, the concentrations detected in CSF are more akin to the extracellular concentrations, not so much to the intracellular concentrations as detected in, e. g., homogenates that are considerably higher than measured for, e. g., amantadine.406 In cerebellar and cortical rat neurons, both amantadine and memantine protected against glutamate toxicity, memantine in this model even more potently than MK-801; however, this was not true in mesencephalic cultures, probably because this cell line contains a relatively large percentage of dopaminergic neurons.39 These findings indicate a relatively higher usefulness of amantadine in diseases like PD (vide supra). Chronic (20 months) treatment with memantine in aged rats (22 months old) has been reported to exert an effect on the polyamine and glycine binding sites of the NMDAR complex, because in the rats treated with memantine, a larger number of MK-801 sites was found, along with higher affinity for glycine.505 These authors postulated that some of memantine's reported effects in vitro and in vivo may be due to its indirect modification of the ion channel at sites different from the MK-801/PCP binding site. Other effects discussed included the interaction of memantine with muscarinic cholinoceptors and metabotropic glutamate receptors.419 Effects on the phosphoinositide turnover have also been suggested here.
Quantitative receptor autoradiography has been used to study the regional variations in the binding of NMDAR channel blockers amantadine and memantine, along with eight non-adamantane test drugs, in the brain. The rule of thumb that lower affinity and faster on/off kinetics means better tolerability was supplemented with the subtype specificity of the drugs.429 The compounds displaced [3H]-MK-801 from six different regions in rat brain slices to different extent. In general, clinically tolerable NMDAR antagonists had a higher affinity in the cerebellum and lower affinity in the forebrain whereas the test drugs with the highest overall affinity (including the troublesome NMDAR antagonists, MK-801 and PCP) showed no clear regional preferences. Obviously, cerebellar and forebrain NMDARs have different pharmacological profiles, which is probably due to differences in the NMDAR2 subunits of the receptor complex. These authors also discussed the issue of the drastically lower “behavioral toxicity” (that is, clinical tolerability) of memantine compared to 248: the affinities of these two drugs are roughly similar, but they differ in their kinetics and in their regional preferences. Memantine has a higher affinity in the cerebellum than in the forebrain. This could be due to the NMDAR2C subunit, which is expressed almost exclusively in the cerebellum. To this end, freshly dissociated rat hippocampal and striatal neurons were studied using patch-clamp- and voltage clamp techniques.423 The inward current responses of hippocampal neurons in the presence of 500 μM NMDA and 5 μM glycine were selectively antagonized by 5 μM ketamine, 10 μM memantine and 100 μM amantadine. When studying the striatal neurons, 248, ketamine and memantine were about three to four times less potent, whereas amantadine was more potent in this neuronal population than it was in the striatal neurons. Along with the favorable, fast blocking kinetics of memantine, the authors used these findings to substantiate the intriguing differences in the aminoadamantane series of drugs, where amantadine seems better suited for PD, whereas memantine is more adequate to treat AD. The shift to NMDAR antagonism as the major pharmaceutical mode of action of the aminoadamantanes in the CNS has been reviewed,396,506,507 and overwhelming accumulated preclinical evidence shows that memantine attenuates the disruption of neuronal plasticity due to an overstimulation of NMDARs. This symptomatological improvement is probably best described by a “reduction of signaling noise” (that is, excitotoxicity), to increase the signal/noise ratio because normal NMDAR mediated signaling is not fully suppressed by memantine. No undesired interactions of memantine with the approved first-line AD drugs, the reversible AChE-inhibitors, were found in vitro; the drugs were reported to act synergistically instead.508,509 The mechanism of action of memantine is still an object of intense studies. MK-801 (248) displays pronounced trapping block in the NMDAR channel, memantine shows intermediate, “partial trapping” properties in electrophysiological studies.510 These authors ascribed this to the NMDAR having a “deep” and a “shallow” binding site. Mutational analyses on NR1/NR2A-NMDARs expressed in oocytes of X. laevis also hint to a high-affinity site near the Mg2+ binding site of the channel's specificity filter, and another, low-affinity site at the vestibule of the channel.511 Adamantane derivatives equipped with fluorescence labels block NMDARs, while being a tool for assay development or sucellular localization studies.512 A recent study finally suggested that Memantine's ameliorating effect on cognitive decline in AD patients could, at least in part, be mediated by activation of histamine neurons, which still can be activated in AD.513
The abovementioned findings and models briefly describe the current consensus of the mechanism of action of memantine as a neuroprotective treatment in AD. After the drugs approval by EMA and FDA in 2002/2003, numerous detailed reviews have been published;33,34,459,514–518 we advise readers to them for an in-depth look at memantine's MOA. Though the cost-benefit ratio of memantine in AD is a matter of discussion,518,519 it already has gained “blockbuster” status.520
5.3.5. Uses of Memantine other than AD and PD
We learned that memantine's career as a blockbuster drug for the treatment of AD is somewhat an example for an “off-label” use itself.490 Together with the multitude of pharmacological effects discussed for the aminoadamantanes, in particular the host of psychiatric disorders attributed, at least in part, to a glutamatergic dysfunction, it does not come as a surprise that numerous other applications for memantine have been proposed and studied. Clinical trials of memantine in the treatment of narcotic and opiate dependence,521 schizophrenia,522 depression, obsessive-compulsive disorder, bipolar disorder, AIDS dementia, epilepsy, glaucoma, hepathic encephalopathy, multiple sclerosis, stroke, tardive dyskinesia and so forth have been reported and reviewed,506,523 however, therapeutic use of memantine in these disorders has not been recommended.
5.3.6 Recent Developments
As one of the major pathological hallmarks in AD is the formation of amyloid plaques formed by APP metabolites such as Aβ1–40 and Aβ1–42, the effects of these protein fragments in AD has been studied intensively. Aβ1–40 also acts neurotoxic via an excitotoxic mechanism, so it has been studied whether memantine could exert neuroprotective action under an Aβ insult.41 Rats treated for nine days with memantine at a plasma concentration of (2.34 ± 0.23) μM showed significantly less neuronal damage after an Aβ1–40 insult as detected immunohistochemically. The histological findings were reflected in behavioural tests.
5.3.6.1 NMDAR Subtypes
Next to various neurotoxic challenges whose consequences are ameliorated upon treatment with memantine, we have also already learned about subtype preferences of the aminoadamantanes and other NMDAR antagonists. CR-3394 (217) and CR-3391 (218, Scheme 29) are NMDAR-subtype selective agents that have been studied using NMDA-evoked currents as the measure for their binding affinity towards NMDARs expressed in cortical neuron cultures.384,385 Compound 217 furthermore was protective in an in vitro model of neurotoxicity and it increases the basal catecholamine release in vitro and in vivo, it is more potent on the NR1a/NR2B than on the NR1a/NR2A NMDAR subtype. Physiological concentrations of extracellular Mg2+ (1 mM) were found to decrease the inhibition of NMDAR mediated currents through memantine near 20-fold for the NR1/2A- and the NR1/2B subtype, whereas the decrease through Mg2+ at this concentration is only about three-fold for the NR1/2C and NR1/2D subtypes.524 This is indicative of a primary therapeutic neuroprotective effect of memantine at the NR1/2C and NR1/2D subtypes.
5.3.6.2 Enhanced Neurogenesis
The neuroprotection observed for memantine and related compounds has also been ascribed to secondary effects. Memantine restored the levels of substance P (a neuropeptide that has been associated with neurogenesis) and somatostatin to control levels525 and the usual activation of glia, frequently observed in AD and probably a reason for excitotoxicity, was not observed in rats treated with memantine. Additionally, amantadine and memantine have been found to dose-dependently increase the level of glial-derived neurotrophic factor (GDNF) in C6 glioma cell cultures at submicromolar concentrations.526 The drugs modulate the expression of the trophic factor at the gene level. Later, another study of memantine in primary midbrain neuron-glia cultures and other cell lines also showed the neurotrophic and neuroprotective effects of the drug; the authors found these effects to be glia-dependent, and postulated that probably memantine enhances GDNF-production in astroglia.527 In vivo, memantine promotes proliferation of progenitor cells in the hippocampus of adult mice.528 As in AD the level of adult neurogenesis is increased, probably as part of a compensatory mechanism to counteract neuronal loss, modulation of the levels of endogenous neurotrophic agents through small molecules is an attractive therapeutical target, as usually neurotrophic factors themselves do not cross the blood-brain-barrier and are rapidly degraded through proteolytic enzymes. Memantine, like tacrine and galantamine, has shown to enhance neurogenesis in vitro as well as in vivo as evidenced by the incorporation of BrdU.529 An increase in neurogenesis of up to 45% over controls could be observed in vivo. Furthermore, memantine upregulates brain-derived neurotrophic factor (BDNF) in rhesus monkeys infected with simian HIV at an early, non-symptomatic stage of the immunodeficiency.530 mRNA and protein expression of BDNF has been shown to be upregulated by memantine. The authors assumed that this may be the reason for the protective effect of memantine on dopaminergic neurons. Latest research on this field combines active regions of trophic factors with aminoadamantane derivatives, as exemplified by the ciliary neurotrophic factor (CNTF)-derived compounds 251 and 252 (Scheme 34).531
Scheme 34.
CNTF-derived, adamantane-modified peptides that enhance neurogenesis in vitro as well as in vivo and improve learning and memory normal mice.
5.3.6.3 Follow-up Compounds
As patent protection of memantine expires in 2013 (Europe) / 2015 (USA), follow-up compounds are being developed. Using memantine as the template, a number of aminoalkyl cyclohexanes are being studied (253 – 256, Scheme 35) and developed as NMDA receptor antagonists.35 Neramexane (253) binds to the same binding site in the NMDAR channel with comparable affinity, it displays very similar blocking kinetics and bioavailability in vivo to those of memantine. Neramexane went to clinical trials for four indications including AD, and 254 – 256 also were found to displace [3H]-MK-801 (248) binding to rat cortical membranes and antagonized inward current responses of cultured hippocampal neurons with moderate affinities, comparable to those of Neramexane and memantine.
Scheme 35.
Aminoalkylcyclohexanes as NMDAR antagonists with Memantine as the “template”.
5.3.6.4 Miscellaneous Other Targets
Studying the effects of memantine on the Ca2+ conductance through α7* nicotinic acetylcholine receptors (nAChR) in cultured rat hippocampal neurons, Aracava et al. found that at 60 mV membrane potential, the IC50 of memantine for the nAChR was 0.34 μM, whereas IC50 for the NMDAR was 5.14 μM.532 The authors stated that this could be counteracting memantine's beneficial effect, in particular at early stages of AD. These findings were, however, subject of debate as there are, for instance, clinical trials that have shown clinical benefit of memantine also in early stages of AD.533 According to these authors, it is not clear whether nAChRs are actually involved here, and work has been published that both nAChR antagonists and agonists displayed neuroprotection. Moreover, there could be species differences between the nAChRs as studies of human nAChRs expressed in X. laevis oocytes showed a much lower affinity of memantine for this receptor (IC50 (memantine) = 5 μM; cf. the clinically obtained concentrations of ~1 μM). However, Aracava et al. insisted upon their concept, asking for more preclinical studies of memantine before it can be approved for mild-to-moderate AD.533
A possible “downstream effect” of memantine was reported in 2006 by de Sarno et al., who found an increase in serine-phosphorylation of glycogen synthase kinase 3 (GSK-3), an enzyme playing a decisive role in AD pathogenesis, in particular, phosphorylation of the microtubule-associated protein Tau (vide infra) whose abnormal hyperphosphorylation lies at the heart of AD's second major pathological hallmark, the formation of neurofibrillary tangles.534 Phosphorylation decreases GSK-3's activity, so phosphorylation of this kinase would be beneficial in AD.
As the NMDAR is regulated via nitrosylation, some authors have suggested using the aminoadamantane as a target-directed shuttle to bring NO close to a site within the NMDAR where it can nitrosylate and thereby regulate the NMDARs ion channel conductivity.535 Other terapeutic targets that might be hit by memantine and other adamantane derivatives are oxidative stress,536 possibly in combination with vitamin D as an additional antioxidant,537 acetylcholine esterase inhibitors modified by addition of adamantane moieties,538 and, returning to the early postulated effects, a direct agonist activity at dopamine D2high receptors at an affinity similar to the affinity to the NMDAR.539
In conclusion, aminoadamantanes hit several targets involved in the pathogenesis of AD, enabling unique treatment options in severe stages of AD.540 In addition, follow-ups directly derived from aminoadamantane template help to fill the pipeline. We will once more return to AD below.
5.3.6.5 Protein Phosphatase 2A (PP-2A)
Another recent finding in the field of Alzheimer disease involves an enzyme as the target to be hit by memantine. In addition to its effect as an NMDAR antagonist, memantine has also been found to be able to inhibit and reverse abnormal hyperphosphorylation of the microtubule-assiciated protein tau.541 In its abnormally hyperphosphorylated form, this protein is the major constituent of the neurofibrillary tangles that are, next to amyloid plaques, the second major hallmark in AD pathogenesis.541,542 The major enzyme responsible for dephosphorylation of tau is protein phosphatase 2A (PP-2A), which is intracellularly regulated by endogenous inhibitors named I1PP2A and I2PP2A. Up-regulation of these inhibitors led to hyperphosphorylation of tau, a process that is probably a consequence of the inhibition of PP-2A and an increase in the phosphorylation of tau by kinases that are regulated by PP-2A.543 Down-regulation of PP-2A inhibitors, therefore, represents a target in medicinal chemistry. While memantine had no effect on the activity of PP-2A, it obviously interacts with I2PP2A within the cell, thereby reducing tau phosphorylation.53,544 This effect of memantine has not only been observed in vitro, but also in vivo in individuals suffering from AD.545 In order for memantine to inhibit I2PP2A activity and the consequent abnormal hyperphosphorylation of tau, it must gain entry into neurons in the brain. Memantine is positively charged and probably enters neurons primarily during excitotoxicity when the NMDAR Ca2+ channels are open and it binds to the Mg2+ site or the NR-1 subunit of the NMDAR.511 Probably because of this limitation, mostly patients with moderate to severe stages of AD, whose brains experience persistent excitotoxicity, have been found to benefit from memantine. Thus, development of derivatives of memantine that can enter neurons and inhibit I2PP2A, even in the absence of excitotoxicity, is promising for the treatment of AD as well as other neurodegenerative disorders called “tauopathies” which are also characterized by neurofibrillary tangles formed by abnormally hyperphosphorylated tau.
5.4 The GABAergic System
After tackling targets in excitatory neurotransmitter systems, showing a multitude of interactions of aminoadamantanes and other adamantane derivatives, some of which can be beneficially utilized clinically, one would also like to know about inhibitory neurotransmitters.
γ-Amino butyric acid (GABA), the main inhibitory neurotransmitter in the CNS, plays a decisive role in signal transmission and, from a medicinal point of view, in pathological changes in the CNS underlying diseases like epilepsy. GABA exerts its inhibitory activity via different subtypes of GABA receptors and transporters,546–548 For CNS activity of pharmaceuticals, analogues with enhanced lipophilicity are desirable. By “adding lipophilicity and conformational rigidity” to GABA as the lead structure the medicinal chemist is guided to Gabapentin (257, Scheme 36), initially developed to treat epilepsy but now marketed mainly to relieve neuropathic pain. However, this drug does neither target GABA receptors and transporters, nor enzymes in the GABA metabolic pathway.549 Today's consensus is that Gabapentin interacts with the α2-δ subunit of voltage-gated Ca2+ channels, modulating them to reduce Ca2+ influx into neurons, which finally reduces the release of neurotransmitters and modulators like glutamate, norepinephrine, and substance P. Reduced release of excitatory neurotransmitters leads to control of, e. g., epileptic seizures.550 Alkylated analogues of gabapentin have been developed, including 258 and the adamantane-based γ-amino acid “AdGABA” (259).44,551 Several of these alkylated Gabapentin derivatives have been reported to display higher affinity to the α2-δ site (40 ± 8 nM for 258, 140 nM for 257),551 at a comparable anticonvulsive activity in vivo in an animal model. The adamantane-derived γ-amino acid 259 was shown to bind to the same site and to reduce neuronal Ca2+ current as measured electrophysiologically.44 In addition to its anticonvulsive properties, it also has been shown to act as an analgesic in mice. Notably, conformationally rigid analogues like 3-aminoadamantane-1-carboxylic acid 260 were not yet studied as analogues of GABA. However, 259 has been used recently to study the pharmacology of L-type Ca2+ channels with an α2-δ subunit.552
Scheme 36.
Lipophilic GABA analogues
5.5 Neuropeptides and Serotonin Analogues
After elaborating some of the numerous applications of aminoadamantane derivatives of remarkably simple structures, in particular in drug research targeting pathologies of the nervous system, strikingly the aminoadamantanes have been reported frequently as an “add-on” to modify neuropeptides and neuropeptide-derived compounds, attempting to render these peptide-derived compounds more “druggable.”
5.5.1 Cholecystokinin
The cholecystokinins (CCK) are a family of six neuropeptides varying in length from eight to 83 residues. However, CCK8 (H-Asp-Tyr(SO3−)-Met-Gly-Trp-Met-Asp-Phe-NH2) represents the C-terminal sequence of all members of the CCK family. CCK mediates satiety by acting on CCK receptors that are distributed widely in the CNS. The CCK receptors are also involved in memory and anxiety and are, therefore, a valuable target for the development of pharmaceuticals.
Memantine has been reported to increase the CCK binding in mouse brain.553 Directly using CCK-peptides as the templates, drug development in this field involved compound libraries of adamantane-modified molecules we will briefly discuss here. Based upon CCK4 (Trp-Met-Asp-Phe), Davey et al. reported on the design of CCK-B receptor antagonists bearing a 2-adamantyloxycarbonyl (2-Adoc) protective group at the N-terminus.554 In mouse cerebral cortex, 261 (Scheme 37) displaced [125I] labeled CCK8 from CCK-B sites at an IC50 of 78 nM. Next to potency, pharmacokinetics needs to be addressed. A larger library of compounds based upon this lead was screened subsequently, with 262 as the most potent member (Ki = 6.1 ± 0.3 nM) as measured at guinea pig cortex.46 For binding, obviously the adamantane moiety is essential as the compounds not incorporating this motif displayed Ki values over 3,000 nM. An estimated 0.2% of 262 crossed the blood-brain-barrier after i.p. administration in mice. As an example for a future application, the authors mentioned anxiolytic compounds, since selective agonists for the G-protein coupled CCK-B receptors cause anxiety and memory perturbation. First selective agonists for the CCK-A receptor are structurally very similar, as they incorporated the 2-Adoc-α-MeTrp motif. Derivative 263 reportedly was the smallest biologically active CCK-A agonist reported at the time.555 Further development of these compounds yielded selective CCK-A agonists like 264 which next to being a full agonist at CCK-A receptors in pancreatic cells from rats also was longer acting than the octapeptide CCK8.556 Starting from peptoids like 262, conformationally constrained peptides were screened in an attempt to identify the conformational requirements for potent binding at CCK-B receptors.47 Antagonists with favorable selectivities for the CCK-B receptor required both R stereochemistry at the Trp Cα and cis configuration at the pyrrolidine moiety. Compound 265 had an Ki of 26.3 nM at CCK-B sites, an affinity 80-fold higher than for CCK-A sites. Due to problems with the oral bioavailability of some of the peptoids reported hitherto, smaller compounds were sought as CCK-B antagonists.557 Notably, the 2-Adoc-αMe-Trp motif was maintained in these compounds. Structure 266 displayed favorable selectivity (Ki (CCK-B) = 3.0 nM; Ki (CCK-A) = 2,900 nM), improved oral bioavailability in rats as well as better penetration of the BBB. Therefore, this compound was selected as a development candidate for an anxiolytic drug. The significance of medicinal chemistry in this field is reflected also in laborious syntheses, including 14C-radiolabelled analogues for the study of pharmacokinetics of 266.558 Structural modifications lead to compounds like the 3H labelled 267 that was utilized in the identification of two distinct CCK-B sites in rat cortex.559 It also proved to be stable upon incubation with the membranes for 150 min. Returning to the search for a selective CCK1 receptor agonist, 268 has been reported as a full agonist at CCK1 high affinity sites while being an antagonist at low affinity sites and a CCK2 receptor antagonist.560 Incorporation of a type II β-turn mimetic into 268 again gave conformationally rigidified peptoids; 269 maintained nanomolar affinity at the CCK1 receptor at an increased selectivity, while not relying on the 2-adamantyloxycarbonyl motif.561
Scheme 37.
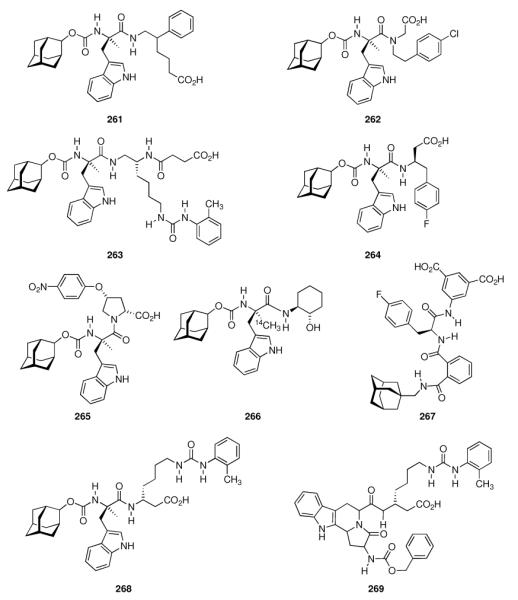
Adamantylated peptoids derived from CCK4.
A reversal of selectivities for CCK1 and CCK2 receptors has been observed by, amongst other modifications, replacing the 2-adamantyloxycarbonyl group with N-Boc (Scheme 38).562 Albeit having an overall low potency, these compounds (270, 271) represented the first selective CCK2 antagonists in the field of peptoids. Measured as the ratio of Ki values upon inhibition of specific binding of [3H]-pCCK8 to CCK1 receptors in rat pancreas and CCK2 in rat cortex homogenates, the K1 (CCK1) / Ki (CCK2) ratio was > 83 with the adamantane derived peptoid displaying significantly higher affinity towards the CCK2 receptors ((121 ± 17) nM for 270, > 10,000 nM for 271). Fine-tuning of the hydrophilicities of even smaller CCK2 antagonists led to substituted imidazoles displaying nanomolar receptor affinity (e. g., 272).563 A 3D-QSAR method called “scaffold hopping” that involves the in silico screening of compounds with unrelated scaffolds but similar pharmacophores led to compounds like 273, which were then synthesized and found to be not as potent as the indoles and pyrazoles described above, and suffered from reduced bilary excretion, at lower molecular weights.
Scheme 38.
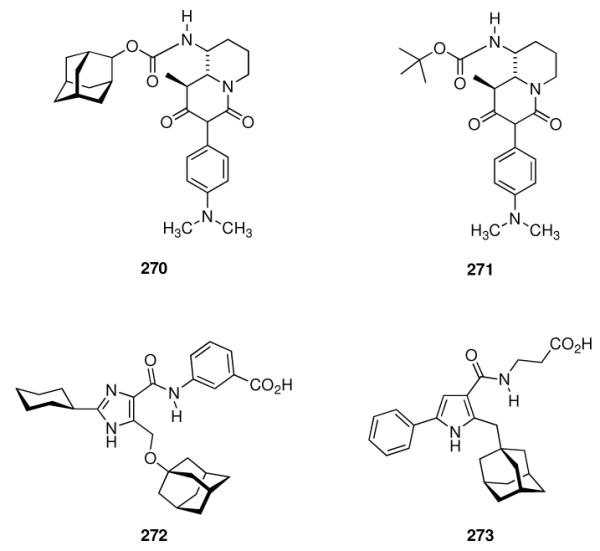
Recent developments of small-molecule CCK receptor ligands.
Notably, an adamantane substituent stays present in these ligands.564 Amongst other compounds, the adamantylated CCK receptor ligands have been used to improve these theoretical methods, as the standard 3D-QSAR screenings are sometimes troublesome when screening compound libraries with vast differences in their backbone scaffolds.565 While to the best of our knowledge none of the adamantylated CCK receptor ligands are routinely used clinically, several are being used to elucidate structure and function of the various CCK receptors (“target validation”).566
5.5.2 Neurotensin
The endogenous tridecapeptide neurotensin (NT) plays a dual role as (i) a peptidic hormone in the periphery and (ii) a neuromoduator or neurotransmitter in the dopaminergic system of the CNS. It exhibits hypothermic, psychotropic, and analgesic properties, but only its C-terminal pentameric subsequence is necessary for the analgesic effect. Modulating this pentamer led to the discovery of adamantanecarbonyl-NT(9–13) (274, Scheme 39) with longer duration of action compared to the parent peptide.567 Leaving the path of peptidic backbones, 275 was identified which acts as an antagonist at neurotensin receptors in nanomolar affinity in vitro and displays better bioavailability than 274.568 A 3H labeled isotopomer has been used to elucidate the binding properties and distribution of neurotensin receptors NTR1 and NTR2 in rat brain569 and 274 also found utilization in the development of antipsychotic drugs.570 Closely related derivative 275 found application in molecular modeling studies to identify the binding sites of NTR1 antagonists and -agonists.571 Blocking NTRs with 275 attenuates amphetamine-induced locomotor sensitization; therefore, this compound has been proposed as a clinically useful agent for neuropsychiatric disorders.572 Recently, 275 was used to study the actions of neurotensin at midbrain neurons in descending analgesic pathways.573
Scheme 39.
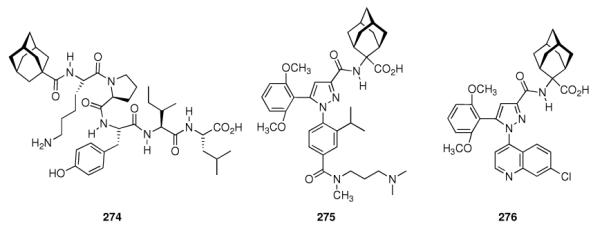
Neurotensin receptor antagonists incorporating an adamantane motif.
5.5.3 Bradykinin
The nonapeptide bradykinin (H-Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg-OH) acts as a vasodilator as well as playing a role in pain. It increases the calcium levels in neurons co-cultured with astroglia, but not in neuronal cultures, by stimulating glutamate excretion from astroglia.574 In rats, the N-terminally adamantane-1-acetic acid-acylated bradykinin antagonist D-Arg-Arg-Pro-Hyp-Gly-Thi-Ser-D-Phe-Thi-Arg resulted in a more than tenfold increase in bradykinin-antagonistic potency.575 Acylating shorter peptidic bradykinin antagonists with adamantane-1-carboxylic acid at the N-terminus gave bradykinin antagonists with at least 33-fold activity.576 Adamantane derivatives of different substructures and modifications of the bradykinin sequence have been synthesized and utilized to gain insight into the pharmacology of bradykinin and its receptors,577,578 e. g., in the characterization of bradykinin receptors in the human nasal airway,579 the elucidation of the thrombin-bradykinin interplay in inflammatory response,580 in studies dealing with the stimulation of the production of vasodilators mediated by bradykinin,578 and recently in the regulation of blood pressure, possibly through bradykinin receptor subtypes, in rats.581
5.5.4 Vasopressins and Oxytocin
The vasopressins are nonameric peptide hormones that act at three receptor systems, one of which, the Arginine vasopressin receptor 1B (AVPR1B), is located in the brain and plays a role in hormone secretion under stress. A concept reminiscent of the modification of neurotensin's C-terminus to obtain 274 has also been utilized to design vasopressin analogues. Simply adding adamantane-1-acetic acid at the N-terminus of Argvasopressin(4–9) gave 277 (Scheme 40), which acts as a selective antagonist of V2 receptors. This derivative was used to study functions of vasopressin (4–9) in the cholinergic system. Arginine-vasopressin (4–9) is believed to stimulate ACh release in rat hippocampus, thereby facilitating learning and memory in rodents.582 Compound 277 acts as an antagonist at the vasopressin receptor and consequently has been used to localize intravascular vasopressin receptors that play important roles in the regulation of blood pressure.580
Scheme 40.
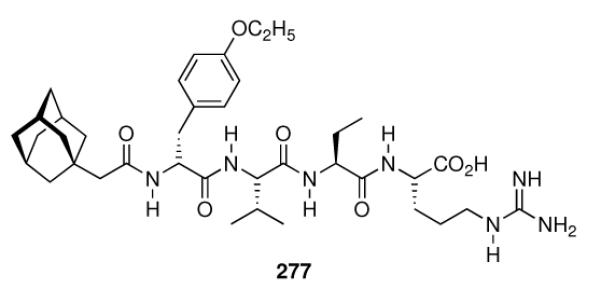
Adamantaneacetyl-D-Tyr(OEt)-Val-Abu-Arg-H, a vasopressin receptor antagonist.
Very early, amantadine was studied in the rat uterus and in vitro and in vivo oxytocic effects of the aminoadamantane were reported.583 Lastly, there is also one report of a modified oxytocin derivative incorporating 2,2-substituted adamantane as a building block.584
5.5.5 Enkephalins
The enkephalins are two pentapeptides acting as ligands at the opioid receptor in the brain. They differ in their sequence at the C-terminal amino acid only: [Met]enkephalin, H-Tyr-Gly-Gly-Phe-Met-OH, and [Leu]enkephalin, H-Tyr-Gly-Gly-Phe-Leu-OH. They act as neurotransmitters and neuromodulators in the brain and due to their opioid receptor activities, they were used as the lead structures in the development of peptide-derived analgesics with fewer side-effects like, e. g., propensity for addiction.585,586 These include peptides incorporating β- and γ-amino acids based upon the adamantane scaffold, as exemplified by 278 and 279 (Scheme 41).587 Likewise, the adamantylated α-amino acid “adamantyl glycine” (Ada) has also been used, as in 280.588 Structure 280 was about three times more potent than an analogue incorporating Leu instead of Ada. Added benefit of the adamantane derivatives is their markedly increased stability towards proteolytic degradation in human plasma589 and their enhanced penetration through the BBB due to higher lipophilicity.590
Scheme 41.
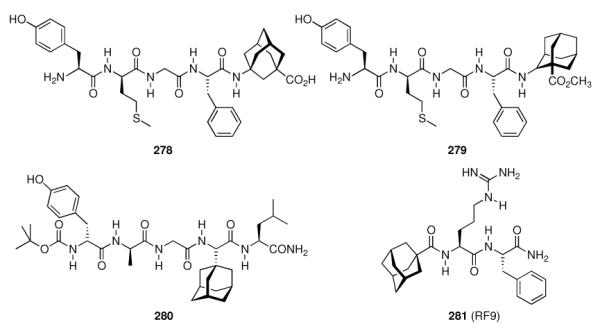
Adamantane-based enkephalin-derived peptides and the NPFF receptor antagonist RF9 (281).
5.5.6 Neuropeptide FF (NPFF)
The octapeptide H-Phe-Leu-Phe-Gln-Pro-Gln-Arg-Phe-NH2 or neuropeptide FF (NPFF), a member of the larger family of the RF peptide amides, plays roles in pain modulation, insulin release, food intake, memory, blood pressure, nociception and modulation of opioid-induced analgesia, in particular, opioid tolerance.591 Although two G-protein coupled receptors, NPFFR1 and NPFFR2, had been cloned, a lack of pharmacological tools to study the pharmacology and effects of this messaging system hampered further research. Dansylated peptide amides of the C-terminal fragment from NPFF were among the compounds studied for this purpose, and Dansyl-Pro-Gln-Arg-Phe-NH2 displaced the radiolabeled NPFF analogue H-[125I]Tyr-Leu-Phe-Gln-Pro-Gln-Arg-Phe-NH2 in rat spinal cord membrane preparations (Ki = 6.1 nM).592 Starting from N-terminally benzoylated RF amide, the synthesis and screening of a small library of about 100 acylated RF peptide amides later identified few small compounds with favorable affinity to the NPFF receptors, one of which, RF9 (281, Scheme 41) showed good affinities at both human NPFF receptors (Ki = 75 ± 9 nM at hNPFFR2; Ki = 58 ± 5 nM at hNPFFR1).591 This adamantanoylated dipeptide derivative was found to block the effects of NPFF, namely the increase in blood pressure and heart rate in rats, and, when co-administered with heroin, prevents hyperalgesia from daily heroin administration. An added benefit is that RF9 exerts these effects when used systemically. In recent years, this compound helped to elucidate the function and (clinical) relevance of the NPFF system. Next to the abovementioned effect in the prevention of heroin-induced hyperalgesia,591 it was found to have no direct agonist activity when administered intraventricularly, but to antagonize NPFF-induced hypothermia593 and to attenuate lipopolysaccharide-induced fever.594 RF9 helped to prove the idea that NPFF receptors mediate the process of opioid-induced hypothermia in conscious rats.595 The RF peptide amides not only share their C-terminal dipeptide motif, but also act at each other's receptors as the prolactin-releasing peptide (PrRP) interacts with the NPFF receptors – as found out utilizing RF9 as a selective pharmacological tool.596 Latest benefits from having available RF9 as selective NPFF receptor antagonist include the finding that there are marked species variations in the NPFF system, as RF9 is about 10-fold more active at the mouse NPFFR2 than at the human NPFFR2,597 and the finding that RF9 in rodents has a gonadotropic effect.598
5.5.7 Serotonin
Next to its role in the regulation of blood pressure, 5-hydroxytryptamin (5-HT) or serotonin is also a neurotransmitter in the CNS involved in depression and anxiety. The serotoninergic system is diffuse and involves numerous receptor subtypes. Selective agonists/antagonists are, therfore, needed to study pathologies involving serotoninergic transmission and possible therapeutic options. Numerous effects of aminoadamantanes on the serotoninergic system have been reported (see Chapters 5.1 and 5.3), and we will discuss here several programs focused on the identification of selective ligands for serotonin receptor subtypes that led to adamantane derived compounds as most promising drug candidates.
NAN-190 (282, Scheme 42), an early postsynaptic antagonist at 5-HT1A sites, displayed good affinity to the target receptor (Ki = 0.6 nM), but also significant affinity to α1 adrenergic receptors (Ki = 0.8 nM). Consequently, more selective ligands were sought thus eliminating the intrinsic risk of unwanted side effects.599 Amide 283 retained postsynaptic 5-HT1A antagonistic activity (Ki = (0.4 ± 0.03) nM) at drastically reduced affinity at α1 sites (Ki = (64 ± 6) nM). The adamantane-derived benzodioxane (−)HT-90B (284) later has been described as an agonist for the 5-HT1A receptor and antagonist for the 5-HT2A subtype.600 In in vitro radioligand binding assays, this compound displays high affinities at both serotonin receptor subtypes (Ki = 0.18 nM for the 5-HT1A receptor, Ki = 9.2 nM for the 5-HT2 subtype). To other serotonin receptor subtypes (5-HT1C, 5-HT3, 5-HT4), it has virtually no affinity. Furthermore, 284 is also active in vivo in rodents and shows good bioavailability. The behavioral effects of 284 in rats and mice qualified it as a potent antidepressant and anxiolytic compound.601
Scheme 42.
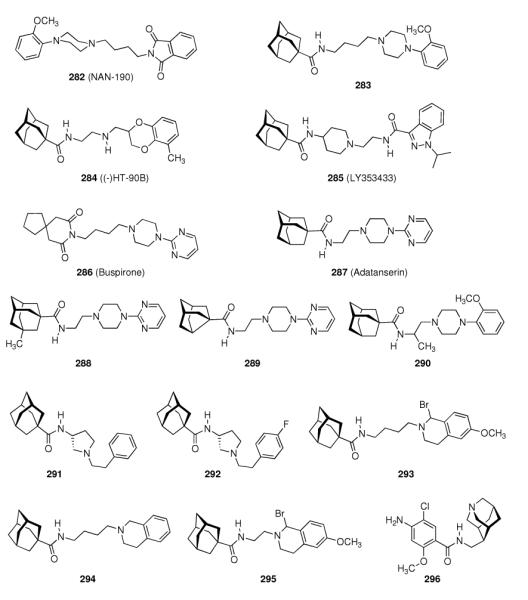
High-affinity serotonin receptor ligands
A screening of libraries of substituted indazoles and benzimidazoles identified LY353433 (285) as a potent, orally active 5-HT4 antagonist whose selectivity profile and duration of action602 led to further development of this indazole.603 This 5-HT4 receptor selective compound has no affinity for adrenergic, dopaminergic, histaminergic, muscarinergic, and GABAergic receptors. The p.o. / i.v. dose ratio of about 1 indicates its good oral bioavailability in rats. Another screening program at Wyeth was based upon substituted aryl- or heteroaryl piperazines.604 Buspirone (286), a drug used against anxiety and depression, served as the template. Since the 5-HT1A receptor serves as the primary target for these antidepressants, the screening involving variations at the imide functionality attempted at improving potency and selectivity at this receptor. Adatanserin (287) displayed high affinity (Ki = 1 nM) to the 5-HT1A receptor, which is significantly higher than to the 5-HT2 subtype (Ki = 73 nM).
Furthermore, affinity to the D2 receptor was also lower (Ki = 166 nM). Adatanserin (287) is a 5-HT1A agonist and a 5-HT2 antagonist in vivo. Variations at the cage hydrocarbon moiety have also been studied. While 3-methyladamantane-1-carboxylic acid derivative 288 displayed significantly lower affinity to the 5-HT1A receptor (Ki = 30 nM), the noradamantane derivative 289 (Ki = 2.4 nM) showed good affinity, meaning that the hydrocarbon moiety probably is directly relevant for receptor binding. The 2-methoxyphenyl piperazine 290 displayed the highest affinity to the target receptor (Ki = 0.22 nM). In rats and pigeons, 287 indeed showed anxiolytic properties at less strong side effects (sedative action) than known drugs. Along with a strong antidepressive activity, these findings led to the development of 287, which was advanced to phase II clinical trials.605 1-Adamantane carboxamides have been used to study peripherally active 5-HT2 receptors. Aminopyrrolidine 291 has been shown to possess high affinity and selectivity for 5-HT2 receptors (Ki = 0.24 nM for the 5-HT2 receptor; Ki = 26 nM for the 5-HT1A receptor, Ki = 6.4 nM for the α1 receptor), and it exerted also an anti-platelet effect in vitro and in vivo.606 Fine-tuning led to the 4-fluoro-derivative 292 (affinities for the abovementioned receptors 0.09 nM, 79 nM, and 3.0 nM, respectively). The in vitro anti-platelet aggregation effects were measured as IC50 = 3.0 nM for 291 and 1.9 nM for 292, respectively. Substituted tetrahydroisoquinolines were also screened as selective 5-HT2A receptor ligands, again finding high-affinity compounds incorporating the adamantane motif.607 The prototype structure, 294 (Ki = 0.95 ± 0.004 nM at the 5-HT1A subtype; Ki = 452 ± 7 nM at the 5-HT2A subtype), showed high selectivity, while 293 (6.2 ± 0.4 nM and 40 ± 1 nM) and 295 (15 ± 0.2 nM and 640 ± 80 nM), respectively, did not show any higher selectivities. These compounds are better described as “dual affinity” derivatives. Subsequent studies included the solid-phase synthesis of a 72-member library of derivatives alongside the arylpiperazine core pharmacophore.608 SAR on the field of serotonin receptor ligands has been continued, also finding 1-azaadamantane derivatives like 296 as 5-HT4 selective agonists. Its 5-HT4 affinity is 51 nM, it is an agonist in rats, but it is also a 5-HT3 ligand at an affinity of 25.4 nM.609
5.5.8 Somatostatin
Next to its role in the digestive system, somatostatins also are produced in several brain cell populations, and somatostatin receptors are expressed at many different sites of the brain, e. g., the arcuate nucleus and the hippocampus. As somatostatins, along with other neuropeptides like neuropeptide Y, are reduced in organic mental disorders and neurodegenerative diseases, the fact that amantadine610 and memantine525 along with other dopaminergic drugs increase somatostatin levels in brains of normal rats is one of the numerous effects of the aminoadamantanes in neurochemistry. Among all neuropeptides, somatostatin is the one most significantly reduced in post-mortem brains of AD patients.611 Five receptor subtypes have been identified in the late 1990s (sst1–5), and the somatostatin peptides act as ligands for all of them. Therapeutically, the use of somatostatin peptides is hampered by the short half-life of less than three minutes in rat plasma.612 The pharmacophore within the tetradecameric, disulfide bound cyclopeptide has been identified to be represented by a -Tyr-Trp-Lys- motif, and peptidic somatostatin derivatives with enhanced stability and bioavailability are being sought.613 The strategy has been to maintain the decisive residues, truncate the peptide chain, add conformational rigidity through N-methyl amino acids or cyclization, and to introduce unnatural amino acids. As inadequate pharmacokinetics and selectivity for the sst1–5 receptor subtypes pose challenges on drug development in this field, numerous peptidic ligands for somatostatin receptors have been studied.614
One such ligand is the sst2 selective ultrashort somatostatin 14 analogue 297 (Scheme 43), incorporating a rigid diaminoadamantane moiety and D-Trp to stabilize β-turn conformation. This analogue has high affinity (KD = 63 nM) and inhibits the release of growth hormone in vitro and in vivo in rats.614 Taking the pharmacophore region of somatostatin to synthesize linear, lipophilic peptides has been another strategy to obtain somatostatin derivatives with enhanced stability. Boc-Tyr-D-Trp-1-adamantylamide (298) has been found to be the most potent derivative of a number of test compounds in the inhibition of cellular proliferation in A431 cells, a process that is believed to be mediated by sst receptors.615 While 10 μM 298 inhibited cellular proliferation by 72 ± 1.4%, the 2-adamantylamide 299 only gave (26 ± 5.8)% inhibition, and the tert.-butylamide 300 showed almost no antiproliferative activity at this concentration ((3 ± 7.2)%).
Scheme 43.
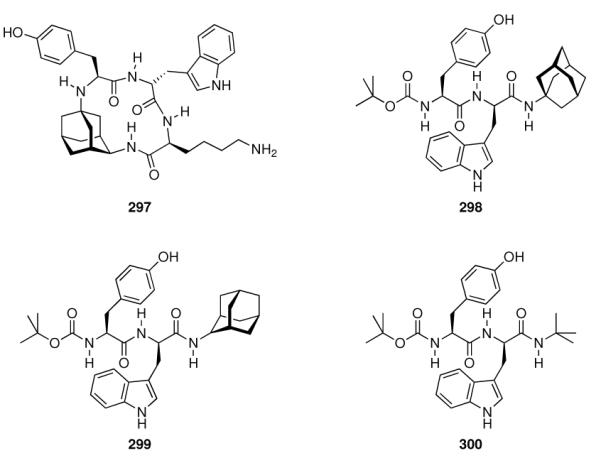
Lipophilically modified somatostatin analogues
6. Other targets in the CNS
Adamantane-modified neuropeptides can be considered a typical example of adamantane's impact on medicinal chemistry: it is used as a lipophilic building block that directly influences a compound's stability and systemic distribution, in particular, BBB penetration, while being biocompatible. Added benefit for peptidic structures is the enhanced stability against proteolytic degradation, enhancing the half-life of peptides and peptide-derived compounds significantly.589 Some of these biophysical properties also play decisive roles in several other CNS-targets hit by adamantane derivatives.
6.1 Opioid Receptors
While we have already discussed adamantylated derivatives of the enkephalins, which are sometimes being referred to as endogenous opioids, we will discuss adamantane derivatives targeting the opioid receptors, G-protein coupled transmembrane proteins of three main subtypes (δ, κ, μ, ORL1) separately in brief. These opioid receptors are ~40% identical to the somatostatin receptors. Together with the opioid action of enkephalins and the knowledge of other naturally occuring short peptides with opioid activities,616 modified short peptides were the starting point for research to develop selective opioid receptor ligands – and adamantane modification, once more, has been proven to be a highly useful tool to influence both affinity and selectivity as well as pharmacokinetics of the resulting compounds.
C-terminally modified truncated analogues of dermorphin (H-Tyr-D-Ala-Phe-Gly-Tyr-Pro-Ser-NH2), a frog heptapeptide that binds selectively to μ opioid receptors,617 maintained their opioid activity. In particular, 1-adamantanamine derivatives like 301 (Scheme 44) were found to produce central and peripheral opioid activities.616 Another study focusing on the development of peptidic inhibitors for human renin618 reported adamantane substituted peptides that also displayed high affinities to opiate receptors. The 2-adamantylamino derivative 302 displayed an IC50 of 1.9 · 10−10 M in a radioligand binding assay using rat brain homogenate. This was a higher affinity than morphine had in this assay (IC50 = 4 · 10−9 M). Deltorphin A (H-Tyr-D-Met-Phe-His-Leu-Met-Asp-NH2) and deltorphin C (H-Tyr-D-Ala-Phe-Asp-Val-Val-Gly-NH2) are two more opioid peptides with high affinities and selectivities for δ receptors. Truncated derivatives like 303 were studied but found to lose their selectivity.619 C-terminally adamantane modified tri- and dipeptide fragments derived from enkephalins (vide supra) like 304 and 305 on the other hand were identified as functional opioid receptor agonists.620 In electrically stimulated longitudinal muscle stripes of guinea pig ileum, an assay for μ-opioid receptors, 304 has an IC50 = 413 ± 10 nM while 305 was more active at IC50 = 4 ± 0.3 nM. In mouse vas defens muscle strips, a model to study δ opioid receptors, 304 was markedly less active (IC50 = 1510 ± 340 nM), while 305 also showed nanomolar activity (Ki = 42 ± 3 nM). In general, an adamantane unit at the C-terminus of the short enkephalin derivatives led to a decrease in the δ-opioid receptor activity, leading to increased μ selectivity. These authors postulated that membrane incorporation of the lipophilic peptides precedes interaction with the receptors. An example for a very popular peptide-derived pharmacophor with high affinity to opioid receptors is the 2,6-dimethyltyrosine-tetrahydroisoquinoline or “Dmt-Tic” group. Enhancing its affinity and selectivity for opioid receptors has intensively been studied. H-Dmt-Tic-OH displays very high affinity for δ-opioid receptors (Ki = 0.02 nM),621 along with in vitro δ-receptor antagonism. As has been established using knockout mice, the μ receptor subtype is the primary target for analgesic action of morphines, and non-addictive opioids primarily exert their analgesic effects through δ-receptor agonism. More potent δ-opioid receptor antagonists were obtained through enhancement of the lipophilicity of the respective parent peptides; 306 and 307 displayed very high δ receptor binding (Ki below 0.2 nM), 306 also acted as a μ-receptor agonist. The enhanced lipophilicity of these compounds were beneficial in the crossing of the BBB when compared to H-Dmt-Tic-OH. BBB penetration also depends on interactions of the brain-directed pharmaceutical with human multidrug resistant 1 glycoprotein that is abundant within the BBB; 308 has been found to interact here, which should be a favorable property for a chemosensitizing agent in cancer chemotherapy regimens.622 A comparison of 306 (Kiδ = (0.16 ± 0.02) nM; Kiμ = (1.12 ± 0.10) nM), 308 (Kiδ = (0.26 ± 0.05) nM; Kiμ = (0.76 ± 0.05) nM), and 309 (Kiδ = (0.12 ± 0.02) nM; Kiμ = (2,435 ± 462) nM) reveals that for μ-receptor affinity the C-terminal adamantane modification is beneficial while δ-receptor affinity is largely maintained.623 Other peptidic opioid receptor ligands were inspired by the endogenous peptidic opioids called endomorphins. Once more, truncated, C-terminally adamantane modified analogues were studied, finding mixed μ/δ agonists and δ antagonistic derivatives.624 Adamantane derivatives of Endomorphin-2 analogues were among the few compounds studied that displayed nanomolar affinity for the μ-receptor. Structure 310 displayed relatively high affinity for the μ receptor (Ki = (6.59 ± 0.06) nM); and good selectivity (Kiδ = (5,560 ± 1,770) nM).
Scheme 44.

Peptide-derived, lipophilic opioid receptor ligands
6.2 Sigma receptors
Once thought to be another subtype of opioid receptors, the pharmacology of σ1 and σ2 receptors is still poorly understood.625 Only in recent years, with the development of selective opioid ligands, it became clear that these receptors are involved in, e. g., psychotic disorders, neuroprotection, and depression.625–627 They are also involved in the regulation of NMDARs, and the σ1 receptor has been found to be involved in the modulation of dopaminergic transmission through amantadine.628 Consequently, selective σ receptor ligands need to be developed – and this is yet another CNS target hit by adamantane derivatives.
An early SAR on selective ligands has been reported in 1990 by Scherz et al. who systematically studied derivatives of N,N'-di-2-tolylguanidine (DTG).629 While the adamantane derivatives studied were amongst the highest-affinity ligands in this study (e.g., 311, Scheme 45, IC50 = 5.2 ± 0.4 nM), the most active compound screened was the closely related exo-norbornane guanidine 312 (IC50 = 4 ± 1 nM). The aminoadamantanes' binding affinity to σ sites was also studied (as mentioned above, see chapter 5.3), finding affinities in the low micromolecular range for the 1-aminoadamantane derivatives as measured in a radioligand binding competition assay using human post-mortem frontal cortex homogenates. Both amantadine and memantine had affinities of ~20 μM to the σ1 receptor studied, while others, in particular dimethylamino derivatives like 234 (Ki = (0.237 ± 0.019) μM) exhibited much higher affinities.37 Among the most potent σ1 ligands were cyclopropyl derivatives modified with aminoadamantanes (313, Ki = (0.6 ± 0.04) nM),630 other adamantane derivatives of this series furthermore displayed σ receptor subtype selectivities. As σ1 receptors are also involved in the Ca2+ signaling via an intracellular site of action and a site at the plasma membrane and they also have been found in retinal neurons, 315 has been successfully used as a neuroprotective agent in an ischemia model using retinal degradation in rats.631 The σ1 and σ2 receptor ligand SR125329A (314) has been found to possess other highly interesting pharmacological properties. Yeast sterol isomerase, an enzyme involved in inflammatory processes of, e. g., rheumathoid arthritis, has been found to be structurally homologous to some extent to the 26 kDa σ1 receptor, and as human sterol isomerase also shows pharmacological similarities to σ1 receptors, ligands for the latter have been studied here. A cyclohexane derivative of 314 had been developed as a σ-receptor ligand with strong inhibition of TNF-α and, at the same time, this compound also stimulated the excretion of the anti-inflammatory interleukin-10.632 The problem of this compound was, however, rapid metabolic degradation through Cytochrome 450, but 314, the adamantane-derived “backup compound”, showed even higher affinities for both σ1 and σ2 sites and also higher affinity to human sterol isomerase. Compound 314 has been characterized in vitro and in vivo; it was proven to trigger anti-inflammatory pathways as expected, which could make such adamantane-derivatives useful as pharmaceuticals in the treatment of rheumathoid arthritis and other inflammatory diseases.
Scheme 45.
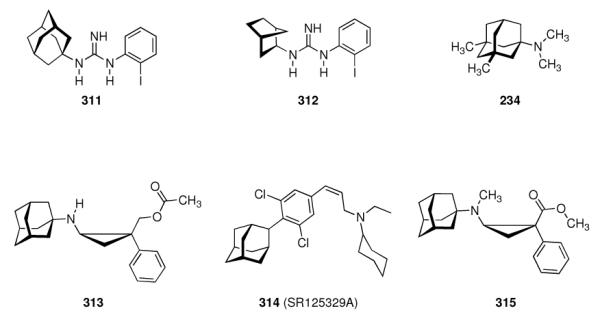
High-affinity ligands for σ-receptors
6.3 Estrogen Receptors
The steroid hormone estrogen influences growth and differentiation of many target tissues. Its receptors, ERα and ERβ, are activated by the hormone ligands that induce a conformational change, whereupon the receptors dimerize. Then, the ER dimer binds to chromatin to modulate transcription of target genes.633 Due to the tissue selectivity of many estrogens, differentiation between agonistic or antagonistic action as well as subtype selectivity for pharmocological studies of the ERs is delicate. To this end, ERα knockout mice have been bred. These animals have reduced fertility, reflecting a primary distribution of ERα in breast and uterus. By contrast, ERβ knockout mice show impairment of cognitive functions; ERβ being located mainly in the brain. Compounds displaying ER binding at micromolar concentrations include phenols like the 4-adamantyl substituted 316 or the diamantyl phenol 317 (Scheme 46). Even though these compounds bind at the ER at 200- to 300-fold lower affinities compared to 17β-estradiol, the number of lipophilic phenols and their ubiquity in human environment possibly pose health risks.634 The ER modulating properties of this class of compounds was studied further by fluorescence polarization.635 Also via competition experiments, the binding affinities have been measured: Ad-DP (318) bound to human ERα at somewhat lower affinity (IC50 = (200 ± 1) nM) than to human ERβ (IC50 = (80 ± 0.5) nM). Affinities for AdP are somewhat lower (IC50 (ERα) = 1000 nM; IC50 (ERβ) = 200 nM). Based upon these and other binding studies, adamantyl phenols were proposed as novel, non-steroid ER ligands, potentially useful as a starting point for the design of ER modulators. The adamantylated estradiol 320 is a non-receptor binding estrogen analogue that has been found to display neuroprotective properties through a decrease of glutamate-induced excitotoxicity – for therapeutic applications, this seems interesting as androgen effects of neuroprotective compounds are certainly not desirable.636 Neuroprotection in cortical neurons has also been found for 321, which acts neither through ER pathways nor as an NMDAR antagonist, so an activity as a radical scavenger was postulated.637 Neuroprotection of rat retinal ganglion RGC-5 cells under glutamate toxicity, a model for glaucoma, by 321 has later been studied in detail – 321 was the most active compound in this study.638 Since 321 had been shown not to bind to the ERa predominantly expressed in the retinal ganglion cells, it is obvious that the neuroprotection must be ascribed to other characteristics like, e. g., antioxidant activity.637 Since protein phosphatases are involved in 17β-estradiol mediated neuroprotection, influence on phosphatases and kinases could also be involved in 321's neuroprotective effect. Indeed, 321 was not protective against insults with the phosphatase inhibitors, okadaic acid and calyculin A, in primary cortical neurons. Furthermore, co-administration of 321 and glutamate prevented the reduction in phosphatase levels observed under glutamate treatment without the neuroprotectant.639 An example for compounds developed as subtype selective ER ligands is 322, which displayed high affinities and some subtype selectivity; however, other hydrocarbon cages instead of the 2-adamantylidene residue appeared better suited.640,641
Scheme 46.
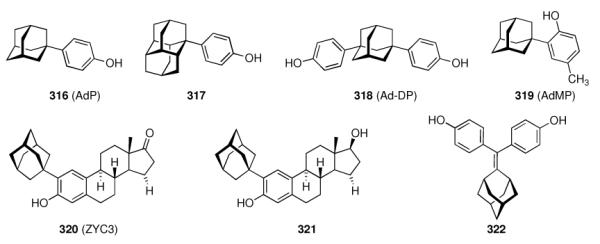
Adamantane modified ligands for estrogen receptors
7. Miscellaneous non-CNS Targets
While in the above chapters, in particular in chapters 5 and 6, the strong propensity of adamantane derived test pharmaceuticals to display activity in the CNS, owing to the pronounced lipophilicity of the added adamantane building block, has been repeatedly elaborated on, we have also encountered targets that play roles both in the CNS and outside. Therefore, we will discuss non-CNS targets hit by the lipophilic bullet in the following chapters.
7.1 P2X7 Receptors
ATP-activated, ligand gated ion channels are being referred to as P2X receptors. Seven subtypes are known, homomeric and heteromeric trimers that are selectively permeable for cations (Ca2, Na+, K+). Though significant progress has been made in elucidating the channels' function, still there is paucity of potent, selective pharmacological tools.642 Homomeric P2X7 receptors can be found on immune cells and glia, and they allow for passage of larger molecular weight compounds upon extended interaction with an agonist. They mediate cytokine release, important in inflammatory processes, and cell proliferation as well as apoptosis. Series of druggable compounds have been developed as P2X7 antagonists, including adamantane derivatives.643–646
AstraZeneca initiated a high-throughput screening to identify P2X7 antagonists for the use in chronic inflammatory diseases where activation of P2X7 receptors mediates the release of the proinflammatory cytokine interleukin-1β (IL-1β). As a chronic disease, long-term treatment promises a large market. Based on a cell-based high-throughput-screening in 96-well format, adamantane bisamide 323 (Scheme 47) was identified as a “hit”. However, this molecule was considered too large and too lipophilic, so “hit-to-lead-studies” have been performed.643 Notably, no replacement for the adamantane moiety was found. Instead, 324 was identified as fulfilling the requirement in molecular weight. The 2-chloro-substitution was essential due to requirements on the conformational preferences, while 4-chloro-substitution is detrimental for potency. Further screening of compound libraries identified 5-indazolyl amide 325 as a lead structure. This structure was further developed. Since in blood from P2X7 knockout mice ATP-stimulated extracellular IL-1β was lower than in the blood from normal mice and the P2X7-ko mice also suffered less severe arthritis in an anti-collagen antibody arthritis model, the target receptor seemed valid to justify development. Compound 326 was discovered as a highly selective, metabolically stable antagonist for human P2X7 receptors, while 327 was less active at hP2X7, but strongly active at rat P2X7 receptors in vitro as well as in vivo.647 This latter compound also shows good oral bioavailability. Phase II clinical trials of P2X7 inhibitors for the treatment of rheumatoid arthritis have been initiated since then, however, neither AstraZeneca nor Pfizer have disclosed the structure of the respective drugs.648
Scheme 47.
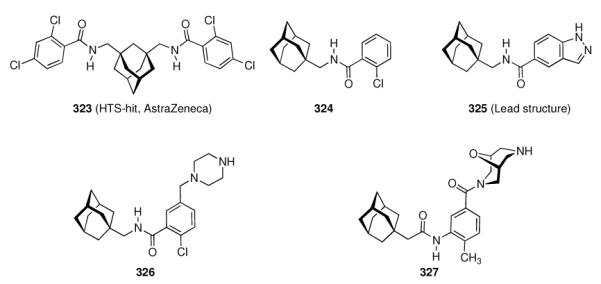
Adamantane amides in the development of P2X7 antagonists
7.2 Soluble Epoxide Hydrolase
Adamantane derivatives have also successfully hit enzyme targets. An early example is the discovery of N-adamantylurea derivatives as inhibitors for soluble epoxide hydrolase (sEH). Next to hepatic microsomal epoxide hydrolase, sEH is present in mammals; the two hydrolases complement each other in detoxifying activity through hydrolysis of various mutagenic, toxic, and carcinogenic epoxides. Furthermore, several diols that are being produced via sEH catalysis cause inflammation. Therefore, inhibition of sEH represents a valuable target for a number of diseases where such compounds are involved. Recombinant human and murine sEHs are available, as are crystal structures,649 rendering the design and study of sEH inhibitors a promising field in medicinal chemistry. Adamantyl ureas have been reported as sEH inhibitors in 1999,650 N-1-adamantyl-N'-cyclohexylurea 328 inhibited recombinant murine sEH (MsEH) with an IC50 = (0.06 ± 0.01) μM, and recombinant human sEH (HsEH) with IC50 = (0.12 ± 0.01) μM. A potent lead structure had been identified, however, the pronounced lipophilicity caused solubility issues and, therefore, more soluble, “druggable” compounds were sought.
Addition of a polar functional group at the end of a long aliphatic chain on one side of the urea pharmacophor retained inhibitory activity but increased solubility. Short aliphatic chains (less than seven carbon atoms) can be modified with a carboxylic acid group to enhance solubility; however, this will reduce inhibition. Ester groups are tolerated, and the resulting urea esters retain activity. Notably, in this class of inhibitors, adamantyl substitution as in 330 gives a markedly stronger sEH inhibitor (IC50 = 0.17 ± 0.01 μM) than the cyclohexyl derivative 329 (IC50 = 1.02 ± 0.05 μM).51 The dodecanoic acid derivative 331 (AUDA) has been used as sEH inhibitor in an animal model of hypertension: manipulation of endogenous lipids by orally active sEH inhibitors like 331 is beneficial in angiotensin-induced hypertension.651 Further studies in vivo showed no effect of 331 on blood pressure in normal mice, but it reduced angiotensin-induced hypertension. However, the adamantylurea was inactive in other mouse models of hypertension.652
sEH inhibition through 331 obviously inhibited the hypertensive effects of angiotensin II in mice by decreasing renal excretion of salt and water. Subsequent development of the sEH inhibitors focused on the synthesis of compounds that warrant simpler formulation.653 Replacing the urea motif by an amide in 332 resulted in a drop in activity to human sEH (IC50 = (0.43 ± 0.02) μM, however, inhibitory activity for murine sEH was retained (IC50 = (0.05 ± 0.01) μM; compare to 330). At the same time, solubilities of the amides in aqueous buffer were 10- to 30-fold higher than for the corresponding urea derivatives. Solid-phase methods have subsequently been utilized to synthesize libraries of sEH inhibitors in a combinatorial approach.654 Screening 192 inhibitors, it was found that in some cases the adamantane moiety can be replaced with no significant drop in sEH inhibition: both 333 and 334 inhibited human sEH with the same potency (IC50 = (0.05 ± 0.1) nM). Further variation of the 1,4-disubstituted cyclohexyl motif in 335 gave compounds like 336 (human sEH IC50 = 1.3 ± 0.05 nM) with an excellent oral bioavailability of 98%.52 In addition, a prodrug approach was followed to enhance the duration of action of AUDA (331). Esters of 331, once more, facilitated formulation, and glycosylated derivatives like 337 (human sEH IC50 = (0.1 ± 0.01) μM) significantly inceased water solubilities (38 μg/mL for 337).655 N-Acetyl piperidine urea 338 has recently been selected as a development candidate based upon its potency in in vitro and in vivo preclinical tests.656 While compounds with higher potencies such as 339 and 340 have been reported, they suffered from decreased oral exposure.
The adamantane based sEH inhibitors have been used to establish sEH as a target in inflammatory diseases, as inhibition of sEH decreases the plasma concentrations of proinflammatory cytokines. Furthermore, sEH inhibitors also led to enhanced formation of lipoxins. As epoxyeicosatrienoic acids (EETs) possess anti-inflammatory activity, and sEH catalyzes their hydration, sEH inhibition should result in enhanced levels of EETs and, therefore, alleviate inflammatory processes. In mice challenged with lipopolysaccharides, sEH inhibitors indeed accelerated inflammatory resolution.657 The use of AUDA-n-butyl ester in cisplatin-nephrotoxicity658 and a neuroprotective effect of AUDA in a rat model of cerebral ischemia659 support the concept that adamantane derived sEH inhibitors are multifactorial and multitarget agents that, amongst other effects, modulate acute inflammatory responses in vivo, at excellent oral bioavailabilities for some examples. As the N-adamantylurea sEH inhibitors' prime effects are in the fields of regulation of blood pressure660 and endothelial dysfunction;661 adamantane-derived 338 represents a candidate for clinical development in the field of “metabolic syndrome” - a multi-billion market.656
7.3 Dipeptidyl Peptidase IV (DPP-IV)
We have already described very early studies on the hypoglycemic effect of adamantane-modified sulfonylureas.7 An adamantane-modified, truncated B-chain of insulin has later also been reported,662 but the real breakthrough of adamantane derivatives in the development of antidiabetes drugs was published in 2003 with the discovery of 346 (LAF-237, vildagliptin, Scheme 49) by researchers from Novartis as a potent, orally active inhibitor of dipeptidyl peptidase IV (DPP-IV) for the treatment of type 2 diabetes mellitus (T2DM).48 DPP-IV is an ubiquitous, yet highly specific exopeptidase that cleaves a dipeptide fragment from the N-terminus of its substrates, which all incorporate either L-Pro or L-Ala at the penultimate position. Through this modification, several circulating regulatory peptides are regulated, amongst others, the incretin hormone glucagon-like peptide 1 (GLP-1). This peptide is the most potent insulinotropic hormone. Triggered by elevated glucose levels, GLP-1 is excreted and activates β-cells to release insulin as a response to glucose intake. Action of DPP-IV inactivates GLP-1 by truncation, consequently, inhibition of DPP-IV is a promising target for the treatment of T2DM.663 Known DPP-IV inhibitors were analogues of the dipeptide unit to be cleaved by DPP-IV. Similarly, the discovery of vildagliptin commenced from peptide-derived structures.
Scheme 49.
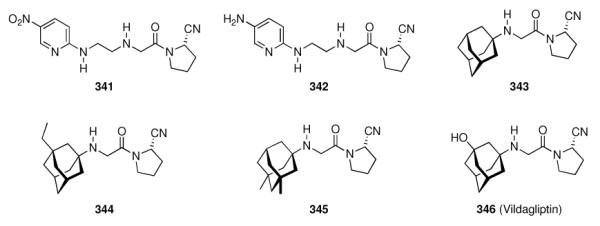
Discovery of the dipeptidyl peptidase IV inhibitor, vildagliptin
Assaying various 2-cyanopyrrolidine derivatives for their inhibition of human DPP-IV in Caco cells, 341 was identified as “hit”, with an IC50 = 8 ± 3 nM. From this structure as a starting point, a first candidate for clinical develompent was derived (342, IC50 = (22 ± 4) nM). As the X-ray crystal structure of DPP-IV in complex with an inhibitor revealed a large enough cavity for the so-called “P2-site”, the workers studied N-substituted glycine derivatives at this position of the inhibitors, e. g., 1-adamantyl derivative 343 (IC50 = (3 ± 2) nM), ((3-ethyl)-1-adamantyl) derivative 344 (IC50 = (7 ± 1) nM), and the ((3,5-dimethyl)-1-adamantyl) derivative 345 (IC50 = 17 ± 0 nM). Clearly, bulky, bi- and tricyclic aliphatic residues at the glycine's amino group like the 1-adamantyl substituent were beneficial for potency, but subtle modifications of the lipophilic bullet caused marked decreases in potency. As studies of primary metabolites indicated a 3-hydroxylation of the adamantane moiety, the workers synthesized and screened hydroxylated adamantane derivatives as well. Vildagliptin (346) was identified (IC50 = (3.5 ± 1.5) nM) as a potent, orally active DPP-IV inhibitor that led to increased insulin levels in a rat model for T2DM. Furthermore, in monkeys, its oral bioavailability was found to be >90%, and its longer half-life in vivo was indicative of a possible once-daily dosing regimen in humans. Clinical trials proved, amongst other aspects studied, activity in man,664 sustained insulin levels upon treatment with vildagliptin,665 and improvement of meal-related β-cell function.666 As T2DM is becoming an epidemic in industrialized countries and pharmacological treatment necessarily needs to be chronic, T2DM drugs are highly interesting for “big pharma”. Therefore, not surprisingly, Bristol-Myers-Squibb in 2005 disclosed the discovery of a closely related DPP-IV inhibitor.49 Similar to the discovery process reported above for vildagliptin, these workers also started from a previously identified β-quaternary amino acid linked to their primary pharmacophore, L-cis-4,5-methanoprolinenitrile. These DPP-IV inhibitors had been found to strongly inhibit the formation of inactive GLP-1 (9–36) from active GLP-1 (7–36) via N-terminal truncation by two residues, the process that is known to represent the major degradtion route for GLP-1 in vivo. As a “logical extension” of their earlier findings, the prototype scaffold 347 (Scheme 50) was further modified. Finding the cyclopentyl substituted 348 to yield a potent inhibitor (Ki = (3.9 ± 0.6) nM) with favorable duration of action, but very low oral bioavailability (oral bioavailability in rats is 5.3%), they also found that the turnover of 348 in rat liver microsomes is too high. Putative metabolites of 348, the hydroxymethyl derivative 349 (Ki = (7.4 ± 1.1) nM) and the bishydroxylated 350 (Ki = (143 ± 15) nM) displayed significantly better oral bioavailabilities.
Scheme 50.
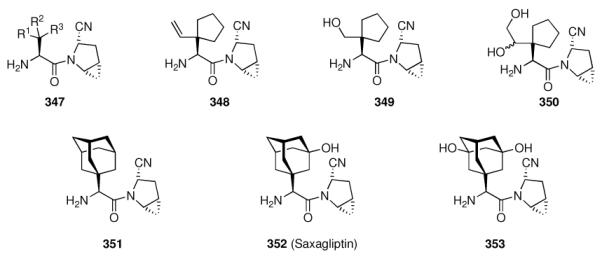
Discovery of Saxagliptin
The L-adamantylglycine derivative 351 was very potent (Ki = (0.9 ± 0.32) nM), at low bioavailability; conveying the rationale that hydroxylated derivatives were constantly equipped with markedly higher bioavailabilities, they synthesized and screened saxagliptin (352, Ki = (0.6 ± 0.06) nM) which also displayed the expected oral bioavailability in animal models as well as a long duration of action in rat plasma (87% inhibition of DPP-IV after 30 min and after 4 h). As the bishydroxylated putative metabolite 353 (Ki = (2.1 ± 0.3) nM) had markedly worse absorption, saxagliptin was selected as a candidate for clinical development. In a rat model for T2DM (Zuckerfa/fa rats), saxagliptin was proven to be suitable in vivo and the long duration of action appeared promising for a once-daily regimen in man.
Vildagliptin in recent years has been intensively studied pre-clinically in vitro as well as in vivo, and a number of clinical trials have been performed.667 The drug is a slowly binding transition-state mimetic displaying two-step inhibition kinetics. It is selectively inhibiting DPP-IV, as no significant inhibitory activity was shown in screenings using, e. g., DPP-II and DPP-VIII.668 In brief, vildagliptin reduces the concentration of active DPP-IV, thereby increasing GLP-1 levels. Furthermore, in two rodent models, vildagliptin was shown to be orally bioavailable and to augment levels of intact GLP-1.669 In these animals, the effect of vildagliptin is antihyperglycemic, not hypoglycemic, thereby bearing intrisically an improved safety profile for clinical applications.
With vildagliptin and saxagliptin entering clinical trials, large-scale syntheses have been worked out. These included enzymatically catalyzed transformations as shown in Scheme 51.
Scheme 51.
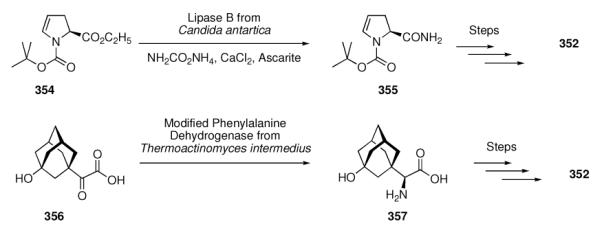
Enzymatic steps in the large-scale synthesis of Saxagliptin (352)
Lipase B was utilized to transform the ester group in 354 to the amide in 356;670 genetically engineered dehydrogenase from Thermoactinomyces intermedius was utilized for the stereospecific transformation of 3-hydroxyadamantane derivative 356 to the adamantylglycine 357.671 This transformation has been scaled up to batches up to several hundred kg at close to 100% conversion. In their development of DPP-IV inhibitors, Abbott also studied adamantane derivatives like 358 (Ki = 62 nM, Scheme 52).672 Focus of this research was on the modification of the cyanopyrrolidin motif, thereby enhancing selectivity for DPP-IV inhibiton over other enzymes. However, their candidate for clinical development is piperidine derivative 359 (ABT-279). Pfizer reported on cis-2,5-dicyanopyrrolidines, e. g. 360 (Ki = 74 nM), as these are achiral, selective, orally bioavailable, and bioactive in vivo. The mechanism of inhibition could be corroborated experimentally by cocrystallization of DPP-IV with an inhibitor, showing the inhibitor to covalently bind with the nitrile group to Ser.673 Proton NMR resonances at low field also supported this binding mode.674 These workers suggested 361 as a development candidate. Chemical efforts in the course of the development of the two adamantane-derived dipeptidyl peptidase IV inhibitors, vildagliptin and saxagliptin also included the synthesis of labelled compounds required to support animal ADME studies.675,676 For the synthesis of 14C labelled derivatives en route to saxagliptin, 13-step and ten-step syntheses of 362 have been reported (Scheme 52). Potent non-nitrile DPP-IV inhibitors have also been reported. Research at Bristol-Myers-Squibb elucidated that “…the unique structural features imparted to inhibitors by the hydroxyadamantylglycine P2 group appear to be capable of conferring significant potency to non-nitrile compounds…” The added benefit of such compounds is increased stability as one possible degradation reaction would be cyclization via the cyano group. Methanopyrrolidine 363 (BMS-538305) was reported as one such compound (Ki = (10 ± 3) nM).677 Mechanistic studies suggest a two-step binding mode of the cyanopyrrolidines by first forming an initial encounter complex, whereupon a covalently bound intermediate with strong enzyme-inhibitor H-bonds forms.678 Active site mutants of recombinant DPP-IV co-crystallized with inhibitors further supported this mechanism of action (Figure 5). Inhibitors, e. g., saxagliptin, bind covalently but reversibly to Ser630, a process that is assisted by the typical catalytic triad of serine proteases, involving His740 acting as a general base to enhance nucleophilicity of Ser630 for the formation of a covalently bound adduct.674
Scheme 52.
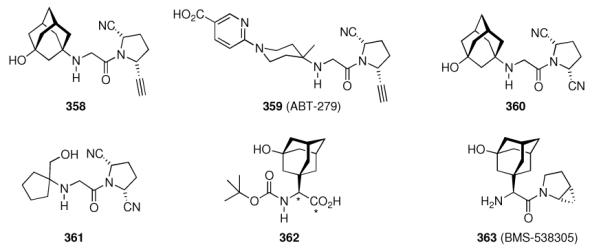
further developments en route to inhibitors of DPP-IV. Asterisks indicate 14C labels
Figure 5.
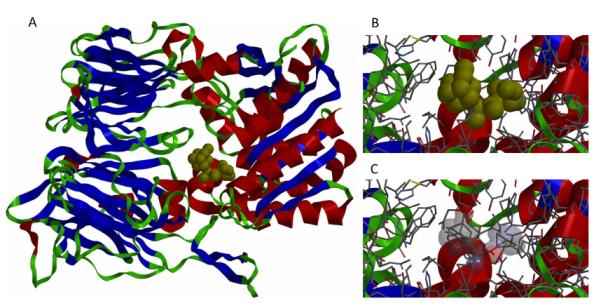
Saxagliptin (352) comlexed with DPP-IV, pdb code 3bmj. A: Binding location in the Enzyme; B: closeup with Saxagliptin in spacefill; C: closeup showing Saxagliptin as a tube model. See text for discussion.
In the multi-billion dollar market of T2DM, big pharma has initiated drug development programs towards the development of DPP-IV, a new target. This target was successfully hit by adamantane compounds; vildagliptin and saxagliptin have been approved in recent years.679 Clinical results are generally favorable (as is mostly the case with any new drug).50,680,681 Adamantane derivatives hit yet another target.
7.4 Hydroxysteroid Dehydrogenases
A strategy to pharmaceutically block the formation of androgens, e. g., in the treatment of prostate cancers, involves blockade of the formation of androgens T and dihydrotestosterone. To this end, a potent inhibitor of type 3 17β-hydroxysteroid dehydrogenase (17β-HSD-3) would be useful.
Hitting this target, adamantane derivatives have been reported to successfuly modify the lead, androsterone (ADT), to enhance inhibition of 17β-HSD-3.682 ADT inhibited this enzyme at an IC50 = 330 nM, while the adamantane derivative 364 (Scheme 53) showed about ten-times enhanced potency (IC50 = 35 nM). However, these authors also found adamantane substitution not essential, as compounds like 365 were equally potent. Another target enzyme is 11β-HSD type 1 (HSD1), which catalyzes the reaction from cortisone 366 to cortisol 367 (Scheme 54). Selectively inhibiting this enzyme over the 11β-HSD type 2 (HSD2) is a valuable target in the pharmacological treatment of metabolic syndrome and atherosclerotic diseases. Adamantane tetrazoles like 368 were non-steroid hits for this target.683
Scheme 53.
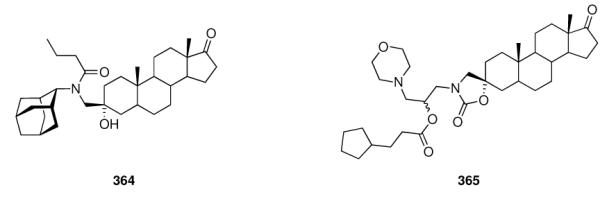
Inhibitors for type 3 17 β-hydroxysteroid dehydrogenase
Scheme 54.
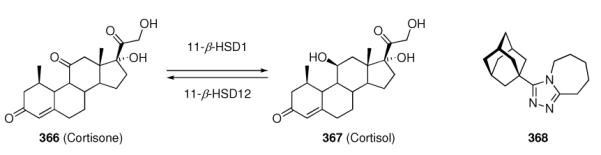
Substrates for HSD1 and HSD2 and 349, an HSD1 inhibitor.
However, establishing a clean SAR was difficult as the potency of the screening library members was not directly influenced by structural modifications. Compound 368 has also shown to be active in vivo in a mouse model. It inhibited both hHSD1 over hHSD2 (IC50 = 7.5 ± 0.5 nM vs. >3,300 nM) and murine HSD1 over mHSD2 (IC50 = 97 ± 5.1 nM vs. >10,000 nM).684 A host of studies dealing with design, synthesis, and screening of 11β-HSD inhibitors incorporating adamantane based building blocks have been published since. Dual human and mouse 11β-HSD1 inhibitors gave 4-amino-3-carboxamidoadmantanes like 369 (Scheme 55) as hits (hHSD1: Ki = 8 nM, hHSD2: IC50 > 10,000 nM; mHSD1: Ki = 15 nM, mHSD2: IC50 > 100,000 nM.685 Non-adamantane derivatives like the bicyclo[2.2.2]octane derivative 370 were about as potent as these (370: hHSD1: IC50 = 13 nM, hHSD2: IC50 = > 100,000 nM; mHSD1: IC50 = 28 nM; mHSD2: IC50 > 100,000 nM).56
Scheme 55.
Various 11 β-hydroxysteroid dehydrogenase inhibitors
Further refinement of the adamantylcarboxamides gave butyrolactams like 371 (hHSD1: IC50 = 3 nM, hHSD2: IC50 = 23,000 nM; mHSD1: IC50 = 2 nM, mHSD2: IC50 = 10,000 nM).55 This compound, in addition to potency and selectivity, also showed a good pharmacodynamic profile and was selected for in vivo efficacy evaluation in obese mice, an animal model for metabolic syndrome. Therefore, scalable and high-yielding syntheses had to be worked out.686 Related derivatives like 372 were somewhat less potent at similar selectivities for HSD1 over HSD2 (hHSD1: IC50 = 39 nM, hHSD2: IC50 = 100,000 nM; mHSD1: IC50 = 26 nM, mHSD2: IC50 > 100,000 nM).687 These were further refined in a goal to push potency below 30 nM at high HSD1 selectivities in rodent models, 373 is one such candidate inhibitor (hHSD1: IC50 = 6 nM, hHSD2: IC50 = 18,000 nM; mHSD1: IC50 = 4 nM, mHSD2: IC50 = 55,000 nM).688 This inhibitor further displayed good potency in a hHSD1 assay from HEK-293 cells (IC50 = 21 nM) as well as reasonable stability as 57% of the compound were present after 30 min incubation with mouse liver microsomes. The introduction of metabolically stable HSD1 inhibitors was the goal of another report, finding 5-hydroxy-2-adamantanamines like 374 as promising compounds (rHSD1: Ki = 6 nM, rHSD2: IC50 > 10,000 nM). Its in vivo half life in rats was 1.4 h.689 Guided by the X-ray crystal structure of the enzyme in complex with an inhibitor, SAR of adamantane based HSD inhibitors was advanced further, sulfones like 375 also displayed low nanomolar affinities at good selectivity (hHSD1: Ki = 7 nM, hHSD2: IC50 = 26,000 nM; mHSD1: Ki = 4 nM, mHSD2: IC50 > 100,000 nM).690 Its stability in the mouse liver microsome assay was as good as for 373. Addressing tissue selectivity is another important aspect when it comes to clinical applications. 376 was found to display full inhibitory activities in tissues like liver, fat, and brain, but not in the kidney. In these smaller compounds, the adamantane moiety could not be replaced without drastic loss in potency.691 Lastly, utilizing HTS hit 377 co-crystallized inside the HSD binding pocket led to the design of protoadamantane 378 (hHSD1: Ki = 3 ± 1 nM, hHSD2: IC50 > 10,000 nM)692 and a docking approach was utilized to identify 379 and 380 as small-molecule inhibitors with sub-micromolar potency for human 11β-hydroxysteroid dehydrogenase type 1 in HEK-293 cells.693 Compound 375 could be co-crystallized with hHSD-1 (Figure 6).690 This substituted adamantanesulfone binds to the steroid binding site, its amide close to the enzyme's residues responsible for substrate ketone reduction.
Figure 6.
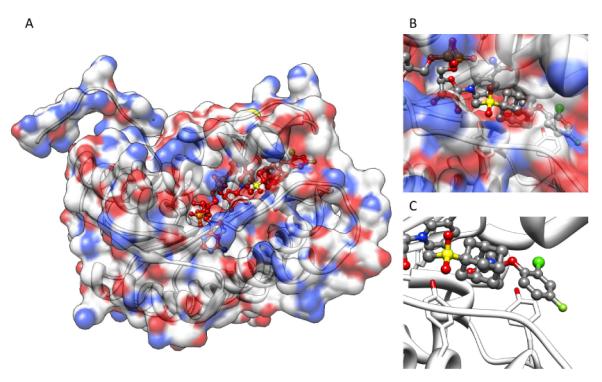
Crystal structure of hydroxysteroid dehydrogenase inhibitor 375 bound to hHSD-1 (pdb code: 2ilt). A: Full protein with the Ligand depicted in red; B: closeup showing the location of inhibitor binding and residues Tyr177 and Tyr 183 as a tube model; C: closeup with the protein surface omitted for clarity. Yellow: sulfur; red: oxygen, blue: nitrogen, light green: fluorine, deep green: chlorine.
While, to the best of our knowledge, clinical applications of adamantane-derived inhibitors for hydroxysteroid dehydrogenases have not been reported, the importance of an adamantane framework in most of these structures to maintain potency, selectivity, and favorable absortion and metabolism in vivo is impressive. Unlike in most of the examples of adamantane modifications of pharmacophores by just adding 1-adamantylor alkyladamantyl residues to a test drug to influence its properties, the 11β-hydroxysteroid dehydrogenase inhibitors described above also utilize the rigid adamantane framework as a scaffold that is capable of orientating pharmacophores into a three-dimensional structure with few degrees of conformational flexibility, which is favorable for enzyme-inhibitor interactions.
7.5 Adamantaplatensimycin
This concept outlined in the previous chapter was also followed in the synthesis of both isomers of Adamantaplatensimycin. The ever-increasing need for new antibiotics, in particular against multi-drug resistant bacteria, challenges both drug discovery, in this field strongly influenced by the isolation of natural products, and synthetic chemists.694 One such class of natural products with potent antibiotic properties targets the fatty acid synthesis of the bacteria, and, via natural product screening, Platensimycin (381, Scheme 56) was identified as a novel antibiotic.695 Numerous publications dealing with its biosynthesis696,697 signify its relevance. Nicolaou et al. reasoned that by replacing Platensimycin's central cagelike domain by approximately isosteric adamantane would facilitate the synthesis and maintain platensimycin's bioactivity. Synthesis of “Adamantaplatensimycin” 384 from 1-bromoadamantane included Rhodium acetate catalyzed C–H bond insertion reaction of diazoketone 382 to decompose to tetracyclic intermediate (±)-383. The following steps, including separation of enantiomers utilizing (−)-menthol as an auxiliary furnished (+)- and (−)-adamantaplatensimycin, and, indeed, the adamantane scaffold obviously maintained appropriate functional group orientations as (–)-384 showed comparable bioactivity to the parent compound.
Scheme 56.
Adamantaplatensimycin
8. Adamantane Derivatives in Cancer Research
With cancer research making up a significant chunk in medicinal chemistry research conducted in both academia and pharmaceutical companies, it does not come as a surprise that adamantane derivatives have also been successfully utilized here. In this chapter, we will discuss some important examples of them.
8.1 Cisplatin derivatives
To selectively eradicate cancer cells while sparing “normal” ones is the ultimate goal of chemotherapeutics. A classic chemotherapeutic, the DNS-crosslinking agent cisplatin (385, Scheme 57) represents a cornerstone in present-day chemotherapy.698 Still, tumor resistance to cisplatin and its less nephrotoxic analogue, carboplatin (386), leads to a significant number of relapses where cisplatin and carboplatin cannot be used any more. In addition, oral bioavailability of cisplatin is limited, hence, more effective and less toxic platinum-based cancer chemotherapeutics are desirable that should be active even against cisplatin-resistant cancer cell lines. To this end, adamantane derivatives have also been studied in a drug discovery program focused on trans-platinum complexes. Trans Pt(IV) complex 387 indeed displayed cytotoxicity against a number of human tumor cell lines in vitro contrary to the earlier established structure-activity rules of platinum complexes. Of particular interest seemed the finding that 387 in vivo was found to be amongst the very few trans Pt complexes that retained activity against a cisplatin-resistant tumor, displaying a significantly higher therapeutic index when compared to the cyclohexyl- and cycloheptyl analogues, respectively.699 In 2004, the closely related cis Pt(IV) complex 388 was synthesized and screened in vitro against a series of cisplatin-resistant tumor cell lines, finding no cross-resistance with cisplatin.700
Scheme 57.
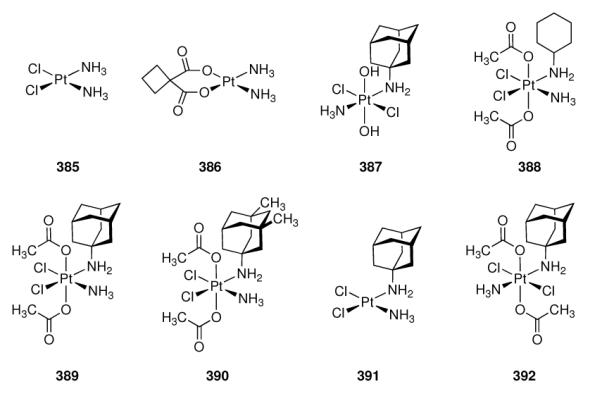
Development of cisplatin analogues incorporating aminoadamantane ligands.
Since a DNA-crosslinking mechanism of action of the Platinum drugs requires entry into the cells, the capability of the Pt-complexes to cross cell membranes is of utmost importance. To this end, the same group of researchers also studied some physicochemical data of, amongst others, 388 – 390, all of which feature one lipophilic amino ligand.701 Unfortunately, in this study the in vitro cytotoxicities of 390 have not been studied. HPLC-based studies of the compounds' rates of ligand exchange of Cl− through H2O in a number of buffers showed no significant influence of the lipophilic amine ligand. Though a correlation of cell uptake with lipophilicity as the main reason for enhanced cytotoxicities seems obvious, 389 displayed cytotoxicity against cancer cell lines displaying differing mechanisms of cisplatin resistance, suggesting another effect of the bulky, lipophilic adamantane ligand not directly related with cell uptake. Complex 389 was also found to display a higher degree of cytotoxicity against both cisplatin-sensitive and cisplatin-resistant ovarian cancer cells compared to 385, presumably due to an additional activity adding to the known apoptosis-inducing capabilities of the latter.702 The 1-aminoadamantane complex 389 reached phase I clinical trials; its oral dose regimens were studied in mouse models,703,704 and in intrinsically cisplatin-resistant ovarian adenocarcinoma, cytokinetic parameters differed markedly when compared with the Pt(II) complex 391.705 Subtle differences in the cell cycle modulation between Cisplatin and 389 have also been found in ovarian carcinoma. While cisplatin treatment led to a G2/M cell cycle arrest, an equitoxic 24 h treatment with 389 resulted in accumulation of S-phase cells.706 The authors attributed this accumulation to the increased uptake and accumulation of the aminoadamantane complex, leading to an inhibition of DNA-polymerization, slowed-down DNA-repair, and an increase of DNA/protein crosslinking. Both Pt complexes led to an increase of p53-expression; however, 389 exerts its cytotoxicity presumably via both DNA-dependent and DNA-independent pathways. In rat liver epithel cells (WB-F344), 389 also displayed enhanced induction of apoptosis compared with cisplatin, as a result of rapid penetration of the adamantane-derived drug into the cell and an attack on DNA, whereby p53, the “guardian of the genome”,707 is activated.708 Complex 389 obviously shifts gene expression of the cancer cells through both p53-mediated and p53-independent pathways, as 389 disrupts proliferation of the cells regardless of their p53-status; however, functional p53 did amplify this effect, as found in a panel of human carcinoma cell lines possessing distinct p53 statuses.709 Most pronounced pro-apoptotic effects of 389 have been found in p53-deficient cells or in cells where p53 is inactive.710 The changes in transcription that are induced by 389 differ from those that cisplatin induces, which may be part of the cytotoxic effect of 389 in cisplatin-resistant cancer cell lines. In other words, the spectrum of genes expressed through the action of platinum complexes on cancer cells differs when comparing 389 with cisplatin. A direct interaction of 389 with chaperone Hsp90 was found via immunoprecipitation of Hsp90 from cells treated with 389.711 Pt could be detected in the immunoprecipitate via atomic absorption spectroscopy. Inhibition of the Hsp90-chaperoning of p53 through 389 could therefore also be contributing to its mechanism of action. Lastly, a recent report on the effect of cisplatin and 389 adresses combinations of the platinum complexes with TRAIL, the TNF-related apoptosis inducing ligand.712 As cisplatin had been found before to enhance the “cell-killing capacities” of TRAIL, 389 has been studied in colon and prostate cancer cell lines and compared with cisplatin. Aminoadamantane complex 389 displayed similar enhancements of TRAIL, but at an ~20 times lower dose. Both platinum complexes obviously modulate parts of the upstream TRAIL signaling, an extrinsic apoptotic pathway in line with the conclusions of earlier reports asking for a DNA-independent additional effect. As for trans Pt complexes, both trans Pt(II) and trans Pt(IV) complexes incorporating an alkylamino ligand such as 392 remain cytotoxic in a selection of two cisplatin-sensitive and two cisplatin-resistant cell lines - while transplatin does not.713 Corroborating the above findings, these authors also found a more than thirty-fold cellular uptake of the adamantylamine complexes compared to cisplatin. Obviously, introduction of the adamantane ligand simply enhances uptake into the cell, expanding the therapeutic window, but this is not the full picture as a number of modifications in the mechanism of action of the Pt complexes accompanies their modification with the lipophilic bullet.
8.2 Generating ROS: Adaphostin
When screening a library of derivatives of the Protein Tyrosine Kinase (PTK) inhibitor Lavendustin (393, Scheme 58) in a panel of about sixty different cancer cell lines, esters incorporating the primary pharmacophore of the starting compound, Lavendustin, as seen in the methyl ester 394 or the 1-adamantyl and 1-adamantylmethyl esters (395 – 397) have been studied. Not surprising in the context of this review, 395 and 396 were reported as “especially promising” in terms of a broad-band antiproliferative effect and in vivo biological activity (hollow fiber assay in mice, subcutaneous xenografts in mice).714–716 The in vitro structure-activity-relationship confirmed the superiority of lipophilic, sterically hindered esters compared with the methyl ester in 394 that is probably being hydrolyzed much faster than the adamantane analogues, contributing to its fast biological half-life of three minutes after i.v. administration in mice. Adamantyl ester 395 in contrast displayed a plasma clearance rate that was 50% lower, leading to a mean residence time of 36 min. Phenol ethers (in the hydroquinone moiety of the test compounds) abrogated their antiproliferative activity, and quinones (e. g., 397 and 398) showed reduced anti-kinase activity as well as decreased growth inhibition. As a potential mechanism of action, the authors considered an intracellular oxidation of the drug to the corresponding quinone that subsequently modifies target proteins covalently via 1,4-addition reactions. Maximum activity in the hollow fiber assay using a panel of leukemia cells was also found for 395. It showed maximum cell growth inhibition both in vitro and in vivo while maintaining the autokinase inhibitory activity it initially had been synthesized for; therefore, 395 was selected for further development and referred to in later studies as NSC680410 or Adaphostin, respectively.
Scheme 58.
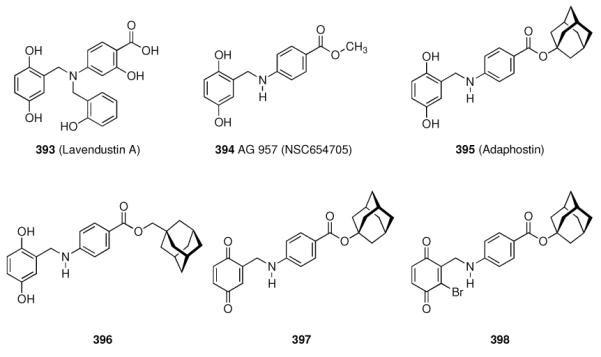
Adaphostin and some of its derivatives
How does it work? Comparing 394 and 395 with one another, Svingen et al.715 also found that Adaphostin was inhibiting the kinase responsible for a majority of chronic myelogenous leukemia (CML), p210bcr/abl, more potently than the methyl ester 394. Adaphostin downregulated the expression of this kinase three-fold stronger than 394, and it also induced apoptosis in K562 cells stronger than 394 did. By comparison with an inhibitor for the tyrosine kinase that alters the ATP binding site of p210bcr/abl, the inhibitory activity of Adaphostin instead alters the binding site of the peptide substrate.717 Tests in ten human leukemia and glioblastoma cells subsequently showed that Adaphostin had a broader applicability than just leukemias, as it consistenly showed lower IC50 values than a structurally unrelated antileucemic compound via a mechanism that is p53-independent.718 Later reports supplemented these findings by studying Adaphostin in three different glioblastoma cell lines.719 The simple oxidant tert.-butyl hydroperoxide was found predictive for the Adaphostin-sensitivities found in these cell lines, clearly supporting a mechanism of action of the latter through generation of reactive oxygen species (ROS) in the cell. In prostate cancer cells, Adaphostin induced G1 phase cell cycle arrest, and the drug also inhibits the growth of many adherent and suspension cell lines with IC50 values between 79 ± 1.8 nM and 9.2 ± 0.2 μM.720 Recently, Adaphostin was also found active against a non-small cell lung-cancer cell line (NCI-H 522).721
At least two different modes of action are important for Adaphostin's selective722 cytotoxicities: “Tyrphostin” effect (that is, inhibition of a tyrosine kinase), and a ROS-mediated mechanism. Generation of ROS preceded DNA-damage in heuman leukemia cells exposed to Adaphostin,723 and it also acts upstream of perturbations in stress-, survival-, and cell-cycle-associated proteins, as well as mitochondrial injury.724 Monitoring the Adaphostin-induced changes in transcription, the release of “free iron”, leading to redox perturbation, was also found as alternative or additional mode of action.725 Kinase inhibition by Adaphostin was also found in cell lines resistant to the tyrosine kinase inhibitor, Gleevec due to several point mutations.726,727 As N-acetyl cystein, a radical scavenger, triggers a partial reduction in ROS generation in leukemia cells with wild-type and mutated p210 tyrosine kinase, ROS-generation and subsequent pathways probably confer the vast majority of Adaphostin's anti-leukemia activity.726 However, induction of oxidative stress still is not sufficient to fully explain Adaphostin's actions.728 As Adaphostin is concentrated up to 300-fold in cells and 3,000-fold in mitochondria compared with the extracellular medium, the respiration of the mitochondria has been suspected as the prime origin of Adaphostin-induced rises in the levels of ROS.729 Within the mitochondria, Adaphostin probably binds to and acts at the respiratory complex III, as corroborated also by in silico docking studies. In human multiple myeloma cells, Adaphostin causes an upregulation of c-Jun, activating caspases, cell-growth arrest, and finally apoptosis.730 Proteomic profiling of Adaphostin-treated leukemia cells revealed similar effects of the Tyrphostin as seen when treating the cells with oxidizers such as H2O2 or hydroquinone.731 In this study, two Adaphostin resistance genes have been identified. Adaphostin has also been identified as an active agent in a screening for agents that increase the levels of hypoxia-inducible factor 1-α in multiple myeloma.732
A number of studies focused on combinations of Adaphostin with other anticancer drugs, for instance, STI571 (Gleevec),733 acted synergistically with Adaphostin in leukemia cells; Fludarabine displayed synergy in chronic lymphotic leukemia;734 the multikinase inhibitor Sorafenib enhanced Adaphostin-induced apoptosis in K562 leukemia cells.735 Combinations of Adaphostin and Bortezomib, an inhibitor of the proteasome, have been found highly efficient in leukemia cells bearing point mutations in the tyrosine phosphatase.726 In three different leukemia cell lines, combination of Adaphostin and the peptidic aldehyde proteasome inhibitor MG-132 acted synergistically in the induction of apoptosis, shifting the balance towards stress-related signal transduction pathways and increasing ROS-dependent cell death while normal hematopoietic cells were spared relative to transformed ones.736 Recently, caffeine was also found beneficial in leukemia therapy when co-administered with Adaphostin.737
Even though Adaphostin obviously acts via a number of different modes, one of which is enhanced stability and target cell penetration through the lipophilic bullet, Adaphostin and its analogs have been characterized as “CSA compounds”, that is, a class of drugs with consistent structure activity relationship.738 This could warrant further studies on novel derivatives such as compounds with a modified adamantane moiety to enhance Adaphostin's low oral bioavailability.739 Such compounds have not been reported in the literature to date.
8.3 Adamantyl Retinoids: Adapalene, CD437 and ST1926
Retinoids are structures related to the vitamin A metabolite, (all-trans) retinoic acid, ATRA (399, Table 5) that binds to and activates the retinoic acid receptors, a class of nuclear receptors capable of DNA-binding and regulation of gene expression. Consequently, retinoids such as ATRA and its analogues are capable of direct modulation of, e. g., cellular proliferation, differentiation, and apoptosis. Therefore, together with their natural and synthetic ligands, the retinoic acid receptors represent targets in a diverse series of pathologies.740
Table 5.
Development of Adamantane-substituted retinoids
| No. | Structure | Receptor Affinity Kd [nM] |
ALOGPs | Ref. | ||
|---|---|---|---|---|---|---|
| RARα | RARβ | RARγ | ||||
| 399 |
|
15.5 | 4.5 | 3.0 | [5.66] | 747 |
| 400 |
|
1100 ± 70 | 34 ± 4 | 130 ± 25 | [6.06] | 747 |
| 401 |
|
6500 ± 2500 | 36 ± 8 | 426 ± 16 | [6.17] | 747 |
| 402 |
|
920.0 | 26.0 | 160.0 | [6.71] | 747 |
| 403 |
|
6500 ± 570 | 2480 ± 620 | 77 ± 18 | [5.89] | 747 |
| 404 |
|
2750.0 | 1500.0 | 150.0 | [5.93] | 747 |
| 405 |
|
2240.0 | 2300.0 | 68.0 | [5.79] | 747 |
| 406 |
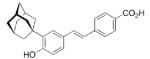
|
610 ± 29 | 70 ± 29 | 20 ± 6 | [5.12] | 741 |
| 407 |
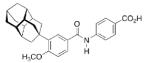
|
93.0 | 870.0 | N.D. | [4.16] | 748 |
| 408 |
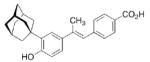
|
1144 ± 67 | 1245 ± 30 | 53 ± 7 | [5.04] | 741 |
| 409 |
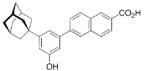
|
- | - | - | [5.98] | 748 |
| 410 |
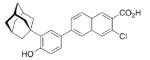
|
- | - | - | [6.44] | 749 |
| 411 |
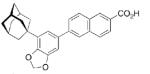
|
- | - | - | [5.68] | 750 |
| 412 |
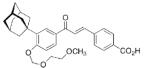
|
- | - | - | [5.15] | 750,751 |
Acne vulgaris is a multifactorial disease leading to the well-known lesions and was treated with ATRA for decades. As ATRA interacts with retinoic acid receptors (RAR) and retinoid X receptors (RXR) without pronounced selectivity, RAR subtype-specific analogues have been sought to reduce side effects.741 Adapalene (400, Table 5) was identified in this screening as a mixed RARβ-γ agonist with low RARa affinity742 that was selected for development as topical treatment of Acne vulgaris; its pharmacological and chemical properties include photochemical stability, local activity, high stability and - as expected - low local side-effect profile. It has subsequently been proven equally active than ATRA at lower concentrations in the topical treatment of Acne vulgaris,743,744 and it is used until today as hydrogel containing 0.1% or 0.3% of the adamantyl retinoid as the active pharmaceutical ingredient (brand names are Differin,® Pimpal,® and Gallet,® respectively). Combination of adapalene (0.1%) with antimicrobial benzoyl peroxide (2.5%) offers enhanced efficiency while maintaining adapalene's favorable safety profile.745 Recently, adapalene has been studied more closely via molecular modeling and UV-Vis spectroscopy with respect to its direct binding to DNA.746 Docking revealed that retinoids without the adamantane moiety might conveniently contact the DNA minor groove, however, the presence of the “lipophilic bullet”, adamantane, was shown to clearly contribute to a stronger DNA binding of adamantyl-substituted 8–16, which is mainly based upon lipophilic interactions.
The successful introduction of yet another adamantane derivative, adapalene, to the pharmaceutical market was, however, not the end of this story. While 401 and 402, analogs of adapalene, are of similar RARβ-specificity, the simple change from a 4'-methoxy- to a 4'-hydroxy group as found in 403 modifies the test drugs selective for RARγ, rendering them interesting tools to study the retinoic acid receptors' relevance in, e. g., dermatology.752 RARγ-selective 403 has been found to upregulate the expression of alkaline phosphatase in teratocarcinoma cells,753 and, more importantly, it induces G0/G1 cell cycle arrest and apoptosis in human breast carcinoma and human leukemia cell lines in a p53-independent fashion.754 These effects are obviously RAR independent, involving enhanced expression of p21waf1/cip1, a component of cyclin-CDK complexes that also inhibit DNA polymerase activity.755,756 Additionally, substituted stilbenes such as 406 bearing the same 4'-hydroxy- / 3'-(1-adamantyl) substitution pattern have also found to be RARγ selective.741 Comparison of the growth-inhibitory activities of 37 natural and synthetic retinoids on eight non-small cell lung cancer (NSCLC) lines gave striking proof for the importance of the adamantane moiety for induction of growth inhibition.748 Independent of their RAR subtype selectivities, the adamantane-substituted retinoids were the most successful growth inhibitors in this study: the one RARα-selective retinoid to display NSCLC growth inhibition in more than just one cell line was 407, incorporating a diamantyl moiety. This compound with the bulkier diamondoid residue, however, is not as potent a growth inhibitor as is 403, which displayed IC50 values in the range of <0.13 – 0.53 μM. Moreover, all of the three RARγ-selective retinoids that showed significant growth inhibition in the NSCLC panel incorporated the adamantane moiety. To complete the list, adapalene was the one (out of four tested) RARβ/γ selective retinoid found active in this study. 405, lacking the adamantane group, did not inhibit NSCLC growth via this RAR-independent mechanism.757
A wealth of studies aiming at demonstration of the scope and elucidation of the mechanism by which 403, also referred to as CD437, induces cell-cycle arrest and apoptosis followed. CD437 (403) activates and upregulates the transcription factor AP-1 in human melanoma cells;758 in human lung cancer cell lines, it was shown to act via a non-retinoid acid receptor (RAR) or retinoid X receptor (RXR)-mediated pathway.759 In mice bearing Xenografts of human ovarian cancer cells, 403 was found active after i.p. and oral application.760 Unlike adaphostin (cf. chapter 8.2), 403 does not seem to increase ROS in the cancer cells.761 The wealth of studies covering the scope of cellular model systems sensitive to the cell cycle-modifying and apoptosis-inducing actions of 403 cover human lung cancer cells,762–768 leukemias,769–774 hepatoma cell lines,775,776 prostate cancer cells,777–780 breast cancer cell cultures,781–784 ovarian carcinoma,785–792 myeloma,793,794 human bronchial epithel cells,795,796 neuroblastoma,797 glioblastoma,798,799 head and neck squamous carcinoma,800 normal mammary epithelial cells,801 malignant human epidermal keratinocytes,802 non-tumorigenic keratinocytes,803 cutaneous squamous cell carcinoma,804 melanoma,805–807 and primary microglia from rats,808 showing 403's capability to regulate cellular differentiation in neuroinflammatory disease. These extensive studies have been reviewed elsewhere; consensus exists in the finding that the vast majority of CD437's pro-apoptotic activities is mediated without direct interaction of the molecule with the receptor(s) it has initially been developed for, RARβ and RARγ, respectively.809–813
Clearly, the evident cytotoxic effect that adapalene's parent structure, 403 (CD437, AHPN) displayed in multiple experimental settings encouraged the synthesis and screening of libraries of derivatives aiming at enhanced potencies, selectivities, and improved pharmacokinetics. Both 403 (RARβ/γ- selective, Table 5) and the adamantyl stilbene 408 (RARγ-selective) inhibited the differentiation of human head and neck squamous myeloma cells via processes that have been linked to RAR interaction, while also inducing apoptosis via RAR-independent pathways.800 Binding of 403 to an unidentified 95 kD protein from nuclear extracts of human breast carcinoma, as shown by size-exclusion chromatographic analysis, has linked the activities of the adamantyl retinoids to an “orphan receptor” at the nucleus.782 The question whether 400 also binds to this orphan receptor and, more specifically, to what extent in comparison to 403, has been deemed essential in the elucidation of the non-RAR/RXR-mediated mechanism of action of the retinoids, at least in human prostate carcinoma.778 Out of 38 natural and synthetic retinoids screened in a panel of head and neck squamous carcinoma, 403 emerged as the most promising derivative, showing lower IC50 values in most cell lines compared to non-adamantylated retinoids and adamantyl derivatives such as 405, 407, or 400.814 Minimizing RAR- or RXR- transactivation has been a goal in the design of novel adamantyl retinoids. 410 has been reported as an equally good inducer of apoptosis as 403 with a further reduced transactivation of RARs.749 Molecular modeling in this study suggested that for reduced interaction with the RAR, the adamantane moiety should extrude from the plane of the naphthalene moiety. Data on 403, 411, 412 and nonadamantylated retinoids have been used to derive a model for the pro-apoptotic activities of some retinoids involving both RAR-dependent (for 412) and RAR-independent (411, 412) pathways;750 adamantyl arotinoids such as 412 are being considered as retinoids that exert their apoptotic activity “completely independent” from the RARs.751 These retinoid-related molecules, including the RAR antagonist 415 (Scheme 59), target kinases and phosphatases such as IκBα kinase (IKK), for which RAR antagonist 412 is a potent inhibitor, triggering signal transduction pathways ultimately leading to apoptosis. These intracellular pathways appear to converge at the mitochondria.815 Further support for the non-RAR-mediated apoptosis-inducing properties in B-cell chronic leukemia and acute lymphoblastic leukemia cells resulted from comparing 403 to 413 in these cell lines.816 The non-chlorinated analog, 414 (ST1926, scheme 59), emerged as a lead structure as it displayed potent antiproliferative activity on a panel of 13 human tumor cell lines, being almost completely devoid of RAR transactivation. The 3-(1-adamantyl)-4-hydroxyphenyl- moiety is regarded as the co-pharmacophor together with the carboxylic acid group that is about 11.4 Å away in active adamantyl retinoids.817 To study in detail binding to and activation of RARs, the “twist” in the central biaryl bond has been further examined.818 In compounds 403 and 414 the adamantyl group resides in the plane of the aromatic scaffold, while in chloro derivatives 417 and 413 and in the acetamidopropoxy derivative 416, the adamantyl group is forced out of the plane, as shown by DFT computations. Docking studies with a RARγ model showed that 403 and 414 on one hand and 417 and 413 on the other bound to the ligand binding domain of the RARγ model, but the latter two derivatives bearing the ortho-chloro-substitution cause altered binding of the RARγ-drug complex with coactivator or repressor proteins, reducing RAR transactivation. As 416 antagonizes apoptosis induced in a panel of various cancer cells through compounds 403 and 413, mechanisms of action proposed earlier were questioned, namely direct free-radical generation through the phenol groups (present in both apoptotic derivatives and their antagonist) or nonspecific mitochondrial permeation (expected to be similar in apoptotic derivatives and their antagonist), narrowing down the molecular target to events upstream of direct effects on the mitochondrial membrane. These could involve enhanced proteolysis of epidermal growth factor receptor (EGFR) and the Ser/Thr kinase AKT.819 An extensive screening revealed the relatively strict requirements for apoptotic activity of the retinoids: they require presence of the carboxylic acid group, E-configuration of the acyclic double bond, this double bond must not incorporate large substituents, substituents at the aromatic rings do not per se enhance apoptotic activity, and a bulky, lipophilic substituent in 3'-position is necessary, “…adamantan-1-yl being so far the best.”820 As also shown by the ALOGPs data summarized in Table 5, lipophilicity alone is not the prerequisite for activity. The ALOGPs of these synthetic retinoids are mostly above logP = 5, the value that has been considered the upper limit of drug-like molecules in Lipinski's classic rule of five - but this also holds true for ATRA itself. Precise orientation of pharmacophores, including the 1-adamantyl substituent, instead shapes up as main determinant not only for RAR subtype specificity, but also for the cell-cycle modifying activities of the retinoids. Compound 414 has been selected for further evaluation. The optimal distance between carboxylic acid group and the lipophilic add-on was also mentioned as critical by these authors; as a consequence, the aromatic scaffold should be a rigid one. Furthermore, they also observed a qualitative correlation between logP and the anti-proliferative activity of the retinoid related molecules. Cinnamic acid derivative 414 exerts its apoptotic effects, at least in NB4 leukemia cells, by the same mechanism of action as 403, at a significantly higher apoptotic index. The effects on gene expression of the cinnamic acid derivative and the naphthoic acid derivative are similar.821 Furthermore, the cinnamic acid derivative 414 is orally active in SCID mice bearing NB4 xenografts. Notably, it increases the influx of Ca2+. Retinoid related molecules bearing the adamantane moiety tend to spare untransformed cells while a large variety of leukemias and solid tumor cell lines are being inhibited; the two probably best-studied adamantyl retinoids, 403 and 414, are of low toxicities in vivo, 414 being more potent in vivo.822 “Studies on the molecular mechanism of action of this class of compounds must continue,” these authors concluded, considering at least 403, 413, and 414 clinically interesting. Combination therapies are appraised particularly feasible.823
Scheme 59.
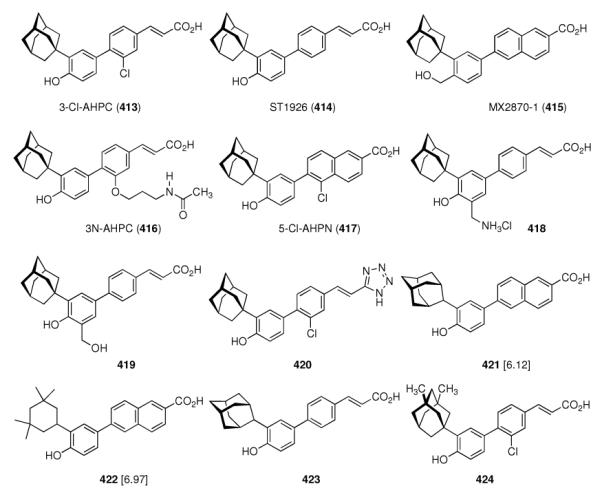
Adamantane-derivatives as retinoids. ALOGPs data in square brackets.
This request for more studies to elucidate the MOA has subsequently been satisfied. In ovarian carcinoma, 414 was found to induce apoptosis stronger in p53-wt cells.824 Combination of 414 with an EGFR inhibitor in human ovarian-, lung-, and breast carcinoma was found synergistic825 and 414 sensitizes ovarian tumors in vivo in mice for the cytotoxic actions of cisplatin.826 Activation of nuclear factor κB in the presence of 413 (a similar mechanism was observed with 403 in prostate cancer cells780) results in an enhanced expression of pro-apoptotic mediators and reduced expression of inhibitors of apoptosis.827 Cross-resistance with 403 was observed in H460 lung cancer cells that developed resistance to 413, further supporting a common MOA.828 Gene expression profiling, likewise, gave similar results.829 Further development of adamantyl retinoids exerting “genotoxic stress” on cancer cells gave, amongst others, 418 and 419 (Scheme 59).830 In NB4 human promyelotic leukemia cells, still 414 was the most potent inhibitor of proliferation (IC50 (414) = 0.082 ± 0.005 μM; IC50 (419) = 0.19 ± 0.01 μM; IC50 (418) = 0.18 ± 0.08 μM). Structure 414 also is more potent than “mother compounds” 403 and ATRA in preclinical models of neuroblastoma in vitro as well as in xenografted mice.831 With respect to the elucidation of one precise molecular target, probably the most convincing findings relate to binding of the retinoids to the nuclear orphan receptor “small heterodimer partner” (SHP, NROB2).832 The nuclear receptor SHP binds to, amongst others, the RAR and RXR receptors to form heterodimeric complexes. The carboxylic acid acid group in the retinoids is essential for formation of these complexes, as 413 is highly apoptotic whereas carboxylic acid isosteres such as tetrazole 420 are largely inactive.833
Subsequent structure-anticancer-relationship studies once more highlighted the importance of both the 1-adamantyl group and the phenolic hydroxy group for apoptotic activity. The binding pocket of SHP requires interaction with both of these pharmacophores, as replacing the 1-adamantyl group and/or the hydroxy group with isosteric groups impacts the induction of apoptosis.834 At a concentration of 0.1 μM in HL-60R acute myelotic leukemia cells, the well-known 3'-(1-adamantyl) retinoid 403 (ALOGPs = 5.89, Table 5) induced apoptosis in 73% of the cells. Strikingly, the almost equally lipophilic 3'-(2-adamantyl)- analog 421 (ALOGPs = 6.12, scheme 59) did not induce apoptosis (0% apoptotic cells) under identical conditions. The tetramethylcyclohexyl derivative 422 (ALOGPs = 6.97) gave 26% apoptotic HL-60R AML cells. Likewise as expected, the 3'(1-adamatyl)-cinnamic acid analog 414 induced apoptosis more potently (90% apoptotic cells in this experimental setting), while its 3'-(2-adamantyl)- analog 423 gave only 2% apoptotic cells. Comparable effects of attenuated induction of apoptosis upon deviations from the “lipophilic bullet” substituent were observed in other cell culture settings as well; with hydrohobic parameters such as symmetry and chain length also playing a role in SHP binding. In H292 non small cell lung cancer cells, the 3'-(3,5-dimethyl)adamantyl- derivative 424 only gave IC50 = 3,6 μM, parent compound 413 being significantly more potent (IC50 = 0.4 μM). The importance of the shape of the lipophilic 3'-substituent was underscored by binding studies of a series of (adamantyl) retinoids to recombinant SHP. Lacking X-ray structural data, homology models of SHP, a class I orphan nuclear receptor (NR) transcription factor, are being utilized to dissect the molecular MOA of the adamantyl retinoids. The binding pocket identified was examined via in silico docking studies. It features H-bonding of the retinoid's carboxylic acid, “edge-to-face”- binding of the phenolic hydroxy group to a Phe sidechain, and a lipophilic pocket for the adamantyl substituent incorporating residues such as Ala145, Trp148, Leu231, and Leu235. SHP binds to heterodimer partners - amongst others, those can be the RARs or RXRs, respectively - to form heterodimeric receptor complexes that do not act as classical NR transcription factors, but rather as bridging proteins between NR and repressor proteins. It is this formation of oligomeric NR complexes that is probably the molecular target of adamantyl-retinoid mediated apoptosis and cell-cycle regulation, respectively. SHP now is considered an “adopted” orphan nuclear receptor, as several ligands modulating its interactions with other heterooligomeric partners forming a multiprotein repressor complex have been identified, such as 413 or 414. This formation of repressor protein complexes is probably the major process directly targeted by the adamantyl retinoids, as most of the changes in protein expression leading to cell cycle arrest and/or apoptosis in a cell-type specific manner can be explained with this MOA. To summarize, the prototype “retinoid related molecules” (RRMs), 403 and 414, are similar to (natural) retinoids, but display a MOA that is not depending on RARs and RXRs, their major advantage being absence of cross-resistance with most current anticancer chemotherapies.835
The multimolecular repressor complex formed by SHP and Sin 3A represents the target of the adamantyl retinoids in human breast carcinoma and leukemia cells, as in such cells with lost SHP/Sin3A expression, senstitivity towards induction of apoptotis by 413 is absent. Still, inhibition of proliferation is observed.836 Together with the finding that SHP ist responsible for binding of 413 to nuclear extracts, further emphasizes SHP as the primary target of the adamantyl retinoids.
Improved synthetic protocols utilizing microwave-assisted Suzuki-couplings to assemble the central biphenyls were reported in an effort to screen ortho-substituted retinoids. In a panel of six cell lines using the MTT assay to monitor inhibition of cell proliferation, adamantyl derivatives 425 and 426 (Scheme 60) were the strongest inducers of apoptosis.
Scheme 60.
Recent modifications for retinoid-related molecules (RRMs) incorporating an adamantane substituent.
As can also be found summarized in a recent review by researchers heading leading groups in the field,837 development candidate 414 has been selected for combination therapy regimens based upon the fact that nontoxic concentrations in mice were found too low for monotherapy; synergism with cisplatin warrants further development of 414 as combination chemotherapies; however, the relatively high toxicity in vivo together with rapid glucuronidation and excretion of the drug in humans (vide infra) also led these authors to conclude that still structural modifications of lead compound 414 may be useful. One such modified derivative of 414 is 427 (Scheme 60), a derivative that is equally active in vitro and in vivo in mice. In cancer cells with knocked-down SHP, both 413 and 427 lost their apoptotic activities, supporting an MOA mediated by SHP.838 Xenographed severe combined immunodeficient (SCID) mice carrying TF(v-SRC) cells, an ATRA-resistant leukemia cell line, when treated with 427, showed marked increase in length of survival with no evidence of leukemia in 87% of the animals. The higher solubilities in aqueous media of these heteroatom derivatives of 413 could, at least in part, account for their equal or even higher biological activities.839 In this recent screening effort, strategies to further enhance pharmacological properties such as toxicity, solubility, and bioavailability have been pursued: next to the introduction of heteroatoms into the cinnamic acid ring (yielding, e. g., substituted pyridines or pyrimidines), replacement of the aromatic cinnamic acid ring with saturated heterocycles and replacement of the cinnamic acid double bond with several heteroatom functionalities has been reported. However, only the heteroaromatic derivatives maintained strong apoptotic effects in a panel of acute myeloid leukemia cells and solid tumor cells. Binding of the screening candidates to recombinant GST-SHP as studied by proton NMR revealed binding of both 427 and 413 to the SHP fusion protein; docking showed that the test drugs displaying apoptotic activity bound in a mode very similar to 413. Pyrimidine 427 may have potential for treatment of acute myelotic leukemia.
Finally, the MOA of the prototype adamantyl retinoid, 403, was also shown to be mediated by SHP: retinoid 403 acts by regulating SHP gene expression and by promoting SHP-translocation to the mitochondria. As a result, by using various in vitro and in vivo cellular models, these authors demonstrate a role of SHP in the regulation of mitochondrial activity;840 next to a MOA of 403 involving kinases and phosphatases841 and further transcription factors,842 this could account for 403's apoptotic activity.
Other molecular mechanisms of action of adamantyl retinoids discussed in the recent literature include genotoxic stress, that is, DNA-damage843 and nuclear factor κB (NFκB), which is activated by phosphorylation through kinases IKKα and IKKβ who are, in turn, being activated by 413.844 In the human ovarian cancer cell line IGROV-1, retinoids 428 – 430 have been studied, identifying them as growth inhibitors for the cancer cells (IC50 (428) = 0.21 ± 0.06 μM; IC50 (429) = 1.31 ± 1.31 ± 0.2 μM; IC50 (430) = 0.62 ± 0.2 μM).845 They share mechanism of action and their influence on gene expression as shown by proteomics; a majority of the proteins whose expression is influenced by these latter three retinoids have a role in Ca2+ homeostasis regulation. SHP, the probable target protein of the adamantyl retinoids, also is of significance in metabolic anormalities such as diabetes, fatty liver, and insulin resistance; therefore, SHP regulation is a valuable goal in these fields also.846 Development of further, selective SHP ligands, therefore, surely is warranted; together with advancements in overcoming the intrinsically low solubilities of most of the adamantyl retinoids in aqueous media via, e. g., formation of β-cyclodextrin complexes as has been successfully shown for 403,847 further pre-clinical and clinical drug development in the field of adamantyl retinoids is to be expected.
9. Metabolism of Pharmaceuticals Incorporating Adamantane
What happens to the adamantane-derived pharmaceuticals once they have been administered? In this final chapter, we will briefly discuss metabolic degradation of some drugs incorporating adamantane as a building block.
In mice, most of the amantadine dose given has been found to be excreted unchanged with urine within 12 hours after oral application.848 About 2% of the drug can be found in feces, so there is good oral absorption. Some metabolism does occur as still part of the dose cannot be collected in the animal's urine and feces. In rats, recovery of amantadine was lower; likewise, in dogs only 19% of the drug could be recovered from urine. N-methylation occurs in dog, as in this species, ~10% of the excreted amantadine is actually metabolized to 213 (Scheme 61). In monkeys, the half-life of the drug is ~5 h, and some metabolic conversions do also occur. This report also stated that in man ~86% of the drug can be recovered from urine, and there was no evidence of acylated, methylated, or otherwise modified amantadine metabolites in any of the urine samples from humans that were analyzed by GLC.
Scheme 61.
Metabolites of amantadine
However, after analyzing samples from an individual attempting suicide by taking very large amounts of the drug indicating that metabolic conversions do occur in humans, the metabolism of amantadine under therapeutic dose regimens was revisited.849 When taking 200 mg orally, still the vast majority of amantadine (65–85%) can be detected in urine. The major metabolite in urine was acetylated 432, 5–15% were found in urine. A total of eight metabolites could be identified; all of which incorporated an unmodified adamantane moiety.
Such modifications of the tricyclic hydrocarbon moiety were, however, found for memantine (Scheme 62) in tissue and urine samples from rats after i.p. injection.850 In rats, the hydroxymethyl derivative 438 was found in tissue and urine as the main metabolite. Likewise in rats, ~ 4% of the memantine administered metabolizes, 17% to secondary alcohol 439, 2% to the substituted carbinol 437, and 35% to 441;488 cage hydroxylation as the main metabolic degradation takes place for memantine, in marked contrast to amantadine.
Scheme 62.
Memantine and its metabolites
The anti-herpes drug tromantadine (47, Scheme 63) bears an amide functionality along with a tertiary amine, and therefore is being metabolized somewhat differently. Still, most of the drug (>50%) can be detected unmodified in urine of humans after taking a single dose of 120 mg of the drug.235 Metabolites identified include a cleavage product of the amide to form 1-aminoadamantane (amantadine) and of the ether to give 445 as the two major metabolites; modifications at the tertiary amino group account for the other metabolites (442 – 444, 446). As with amantadine, the adamantyl cage is not modified.
Scheme 63.
Metabolites of Tromantadine
In contrast, metabolism of the antiviral rimantadine (Scheme 64) mainly takes place at the adamantane cage.851 After a single dose of 200 mg p.o., still unmodified drug is the major compound isolated in urine in man, but cage hydroxylated metabolites 447 – 449 account for a combined 29% of the drug recovered in the urine samples from 0 – 72 h post-dose. In another study, traces of a glucuronid derivative could also be detected.852
Scheme 64.
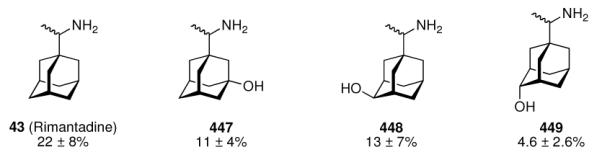
Rimantadine and metabolites detected in human urine
Using 14C-labeled drug in rats and dogs, the major metabolic pathways have been established to be (i) oxidation of the adamantane moiety, (ii) oxidation of the amine, giving hydroxylamine, and (iii) abovementioned glucuronidation and sulfate ester formation.853 In humans, a total of 42% of the recovered drug has been found to be hydroxylated in this study.
The CCK-B receptor antagonist 450 (Scheme 65) also contains an 1-adamantyl moiety not bound to an electronegative atom. This compound has been considered for development of an anti-anxiety drug, so its metabolism was studied.854 A total of eight metabolites have been identified, but even though this compound bears numerous functional groups that could be modified metabolically, the major metabolite other than unmodified compound (450) is modified at the “paraffine” residue to give 451. These findings corroborated results from in vitro studies using liver microsomes.855
Scheme 65.
CCK-B antagonist 385 and its major metabolite
The DPP-IV inhibitor vildagiptin (346, Scheme 66) has been studied after orally giving 100 mg of 14C-labeled test drug to healthy human volunteers.856 In plasma samples, 25.7% of the radioactivity was detected as unmodified drug, whereas 55% were present as a carboxylic acid metabolite (452).
Scheme 66.
Vildagliptin and some of its metabolites.
In urine samples, cleavage of the amide accounts for 3.4% of the dose as isolated as 453, glucuronidation gives 4.4% 454, and oxidative modifications of the pyrrolidine moiety lead to less than 1% each of 455 and 456. The primary elimination route is renal excretion (22.6% of radioactivity), 4.5 % of the initial radioactivity can be found in feces. Furthermore, it has been shown that the hydrolysis of the nitrile in vildagliptin is actually catalyzed by the target enzyme, DPP-IV (vide supra). Similar results have been reported for other species, including rats and dogs.857 The second approved adamantane-based DPP-IV inhibitor, saxagliptin (Scheme 67) incorporates a 3-hydroxyadamantyl residue; its primary metabolic clearance route has been shown to involve bis-hydroxylated 457.858
Scheme 67.
Saxagliptin and major metabolite 457
The antimalarial ozonide OZ277 (161, Scheme 68) has been studied on human liver microsomes335 and in rat plasma.859 In these experiments, two metabolites with an hydroxylated spiroadamantane moiety have been isolated.335 Both major (“cis”, 458) and minor (“trans”, 459) metabolite are devoid of any antimalarial activity.
Scheme 68.
Antimalarial spiroadamantane ozonide OZ277 and its metabolites
Metabolism of pro-apoptotic adamantyl retinoid 414 (Scheme 69) has been studied during early phase I clinical trials. In the ovarian cancer patients, only low and variable absorption of the drug was observed when applied orally for five consecutive days.860 While showing excellent plasma stabilities, the elimination half-lives varied between two and seven hours, because more than 80% of the drug underwent glucuronidation at three different sites, dramatically lowering its bioavailability. The glucuronidated metabolites could be detected in patient blood samples via LC/MS methods; two of them (460, 461) are shown in Scheme 69; the site for glururonidation for the third identified metabolite probably is the cinnamic acid double bond via C-glucuronidation.
Scheme 69.
Adamantyl retinoid 414 (ST1926, AHPC) and two of its metabolites.
The rationale behind the metabolization of adamantane based drugs is clear: Compounds incorporating an electron-rich adamantane core are mostly being modified via hydroxylation involving cytochromes P450.91 As can be deduced from the crystal structure of CYP450CAM complexed with adamantane,861 this process is generally not selective and, in absence of further functional groups in the drug that could orientate the cage hydrocarbon, several sites are being hydroxylated – as can be seen with, e. g., rimantadine and memantine.
9. Conclusions & Outlook
In the above chapters, we summarized the major fields in medicinal chemistry and drug development where the tricyclic C10H16 isomer adamantane has been having striking impact. Remarkably simple aminoadamantane structures hit targets like the viroporins to combat Influenza A as well as the NMDA receptor for symptomatic relief in Parkinson Disease and Alzheimer Disease. This “lipophilic bullet” building block can favorably modify known pharmacophors. We have also learned about drugs using adamantane as an “add-on” to interact with a lipophilic pocket in proximity to the active site of the target, giving rise to enhanced selectivities. Lastly, in the DPP-IV inhibitors approved recently, the adamantane moiety bears a functional group different from short akyl groups; we can consider this as an orientating effect the rigid adamantane scaffold exerts on the functional groups capable of forming hydrogen bonds or even covalent attachments to the enzyme's active site. Several of the drugs introduced in the above chapters, including adamantane-based experimental drugs derived from the neuropeptides (Chapter 5.5), but also the DPP-IV inhibitors, saxagliptin and vildagliptin, exemplify the process of utilizing endogenous peptides or subsequences thereof as lead structures for further drug development.
Seven adamantane-based drugs have been approved so far, and while the aminoadamantanes are no more recommended as anti-Influenza A agents because of resistances in the circulating strains of the virus, memantine has become a blockbuster drug, and probably the DPP-IV inhibitors will also become successful in a multi-billion market. Notably, none of the approved drugs has been discovered directly via modern HTS strategies; the DPP-IV inhibitors were, however, an indirect result of HTS efforts after validation of dipeptidyl peptidase IV as a drug target, as in both cases the initial HTS-hit did not contain the “lipophilic bullet”. This is another notable story behind adamantane based pharmaceuticals: They are derived from a very small library of adamantane derivatives, mostly simple aminoadamantanes or adamantane carboxylic acids, probably because these are commercially available and were, therefore, more frequently considered as building blocks in test drug library syntheses. With the advent of a large variety of selective C–H bond functionalization methods on the adamantane core and the availability of even more lipophilic diamondoid building blocks (Scheme 3),90 much is to be expected to use these unique hydrocarbons as modifiers or enhancers of active pharmacophors.92,93,862–868 This uncharted territory certainly holds great promise for the pharmaceutical industry.
We consider this a major arena for future drug development. As can be seen in the DPPIV inhibitors, the development of selective inhibitors for 11β-hydroxysteroid dehydrogenase (Chapter 7.4) and also the somatostatin analogue 281, bis-functionalized adamantane derivatives have already hit several targets. These “oligofunctionalized” adamantane derivatives are also available,869–871 which should encourage medicinal chemists to consider these building blocks at an early stage of the drug discovery process, in particular when utilizing today's powerful methods of combinatorial chemistry.
What makes this globular paraffine so special? First of all, size matters: To hit targets like ion channels, adamantane and some methyl substituted derivatives obviously have the appropriate molecular dimensions to functionally block the conductance of viroporins or calcium channels. Secondly, lipophilicity has a direct effect on affinity in several cases, but also an indirect one, enhancing CNS-access through facilitated penetration of the blood-brain-barrier. Lipophilicity is also beneficial for test drugs to hit targets in membranes, in particular, in lipid rafts. In addition, the rigid hydrocarbon protects functional groups in its proximity from metabolic cleavage, enhancing duration of action of, amongst others, peptide-derived drugs. On the other hand, it is “biocompatible”, as metabolism can take place in the liver, so that toxic effects by accumulation upon chronic treatment are not to be expected. Most adamantane-based drugs for which metabolites have been studied are, in fact, being excreted largely unmodified, which has the added benefit that possible side effects arising from bioactive metabolites are intrinsically improbable. Lastly, the trend of larger molecules being exploited as pharmaceuticals (this trend also is reflected by the adamantane derivatives described in this article), control over the functional group orientation has an ever-increasing relevance to optimize potency and selectivity. We expect adamantane-based scaffolds to become highly relevant especially in this respect. Adamantane may not be a “magic bullet”248 and adamantane-based pharmaceuticals certainly are not “silver bullets”, yet the lipophilic bullet can be expected to hit even more targets in medicinal chemistry in the years to come.
Request for copyright permission
-
(A)
The Photo in Figure 1 has been taken by Dr. L. Wanka. As he is coauthor of the present manuscript, he agrees to transfer copyright of the photo.
Please send a copyright transfer form to: Dr. L. Wanka Institute for Organic Chemistry Heinrich Buff Ring 58 D-35392 Giessen, Germany Fax: +49(641)9934309 Lukas.Wanka@org.chemie.uni-giessen.de
-
(B)
For Figure 2 of the present manuscript, taken from Yu, Z.; Sawkar, A. R.; Whalen, L. J.; Wong, C. H.; Kelly, J. W. J. Med. Chem. 2007, 50, 94), we need copyright permission.
In addition, in Scheme 32 of the present manuscript we used a graphic from: Andersen, T. F.; Tikhonov, D. B.; Bolcho, U.; Bolshakov, K.; Nelson, J. K.; Pluteanu, F.; Mellor, I. R.; Egebjerg, J.; Stromgaard, K. J. Med. Chem. 2006, 49, 5414. As this is an ACS journal, we are positive that the Editorial Office might handle these cases.
Figure 1.
This silver sculpture, “cloud gate” in Chicago, Ill reflects the seven drugs on the market today incorporating the adamantane motif. Chicagoans refer to cloud gate as “the bean”, and reserachers at universities near “the bean” have contributed to the elucidation of the mechanisms of action of some of these drugs. Photograph taken by L. Wanka.
Scheme 48.
Adamantane derived sEH inhibitors.
Acknowledgement
Our work on adamantane and diomondoid chemistry was supported for many years by the Deutsche Forschungsgemeinschaft and the Fonds der Chemischen Industrie. Khalid Iqbal's lab is supported by the New York State Office of People with Developmental Disabilities, NIH grants AG019158, AG028538 from the United States Department of Public Health, and by a research grant from Ever NeuroPharma GmbH, Unterach, Austria. Finally, we thank the referees for their contribution to enhance the quality of this review.
11. Acronyms
- 2-Adoc
2-adamantyloxycarbonyl
- 5-HT
5-hydroxytryptamine (serotonin)
- 17β-HSD-3 type 3
17β-hydroxysteroid dehydrogenase
- AADC
aromatic L-amino acid decarboxylase
- AChE
acetylcholine esterase
- AD
Alzheimer disease
- Ada
adamantyl glycine
- ADME
absorption, distribution, metabolism, excretion
- ADT
androsterone
- AIDS
acquired immunodeficiency syndrome
- AKT
Protein Kinase B
- AMPA
α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate
- ATP
adenosine triphosphate
- ATRA
all-trans retinoic acid
- AVI50
50% antiviral dose
- AVPR1B
Vasopressin receptor 1B
- AZT
Azidothymidine (Zidovudine)
- BBB
blood-brain-barrier
- BDNF
brain-derived neurotrophic factor
- BrDU
bromodeoxyuridine
- CC50
half maximal cytotoxic concentration
- CCK
cholecystokinin
- CD4
cluster of differentiation 4surface glycoprotein
- CFTR
cystic fibrosis transmembrane conductance regulator
- CML
chronic myelogenous leukemia
- CNS
central nervous system
- CNTF
cilliary neurotrophic factor
- CSA
consistent structure-activity relationship
- CSF
cerebrospinal fluid
- CYP450
cytochrome P450
- DAT
dopamine transporter
- DHPC
dihexanoyl phosphatidylcholine
- DLPC
dilauroyl phosphatidyl choline
- DMPC
dimyristoyl phosphatidylcoline
- Dmt-Tic
2,6-dimethyltyrosine-tetrahydroisoquinoline
- DNA
deoxyribonucleic acid
- DNJ
deoxynojirimycin
- DOPC
1,2-dioleoyl-sn-glycero-3-phosphocholine
- DPC
dodecyl phosphocholine
- DPP-IV
dipeptidyl peptidase IV
- DTG
N, N'-di-2-tolylguanidine
- EC50
half maximal effective concentration
- EET
epoxyecosatrienoic acid
- EGFR
epidermal growth factor receptor
- EMA
European Medicines Agency
- ER
estrogen receptor
- ERGIC
endoplasmatic reticulum-golgi intermediate compartment
- FDA
United States food and drug administration
- FLAG
polypeptide protein tag (DYKDDDDK)
- GABA
γ-amino butyric acid
- Gb3 GSL
globotriaosylceramide
- GDNF
glial derived neurotrophic factor
- GFAP
glial fibrillary acid protein
- GLP-1
glucagon-like peptide 1
- GSK-3
glycogen synthase kinase 3
- GSL
glycosphingolipid
- GST
glutathion-S-transferase
- HA
Influenza hemagglutinin surface protein
- HAT
human african trypanosomiasis (“african sleeping sickness”)
- HCV
hepatitis C virus
- HIV
human immunodeficiency virus
- hOCT2
human organic cation transporter 2
- HPA
hypothalamic-pituitary-adrenal-
- HSD
hydroxysteroid dehydrogenase
- HSD1
11β-HSD type 1
- HSD2
11β-HSD type 2
- HSV
Herpes simplex virus
- HTS
high throughput screening
- i.m.
intramuscular
- i.p.
intraperitoneally
- i.v.
intravenous
- I2PP2A
inhibitor 2 for protein phosphatase 2A
- IC50
half maximal inhibitory concentration
- IL-1β
interleukin-1β
- KD
dissociation constant
- Ki
binding affinity
- LD50
median lethal dose
- LTP
long term potentiation
- M2
Influenza ion channel forming surface protein
- M2TM
transmembrane domain of Influenza A M2 surface protein
- MAS
magic angle spinning
- MDCK
Madin Darby canine kidney
- MIC50
half minimum inhibitory concentration
- MOA
mechanism of action
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- mRNA
messenger ribonucleic acid
- MTT
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- nAChR
nicotinic acetylcholine receptor
- NFκB
nuclear factor κB
- NMDA
N-methyl-D-aspartate
- NNRTI
non-nucleoside (HIV) reverse transcriptase inhibitor
- NOE
nuclear Overhauser effect
- NPFF
neuroeptide FF
- NR
nuclear receptor
- NRTI
nucleoside analog reverse transcriptase inhibitor
- NSCLC
non-small cell lung cancer
- NT
neurotensin
- NTR
neurotensin receptor
- PCP
phencyclidine
- PCR
polymerase chain reaction
- PD
Parkinson disease
- PEG
polyethylene glycol
- pfATP6
Plasmodium falciparum ATPase 6
- PfCRT
Plasmodium falciparum chloroquine resistance transporter
- p.o.
per os (by mouth)
- POPC
palmitoyl oleoyl phosphatidylcholine
- PP-2A
protein phosphatase 2A
- PPAR
peroxisome proliferator-activated receptor
- PrRP
prolactin-releasing peptide
- PTK
protein tyrosine kinase
- QSAR
quantitative structure-activity relationship
- RAR
retinoic acid receptor
- REDOR
rotational echo double resonance
- RNA
ribonucleic acid
- ROS
reactive oxygen species
- RRMs
retinoid-related molecules
- RT
reverse transcriptase
- RXR
retinoid X receptor
- SAR
structure-activity-relationship
- SARS
severe acute respiratory syndrome
- SCID
severe combined immunodeficient
- sEH
soluble epoxide hydrolase
- SHIV
simian-human immunodeficiency virus
- SHP
small heterodimer partner
- ssNMR
solid state nuclear magnetic resonance spectroscopy
- sst
somatostatin
- SUR
sulfonylurea receptor
- T2DM
type 2 diabetes mellitus
- TCTP
translationally controlled tumor protein
- TEM
transmission electron microscopy
- TEMPO
2,2,6,6-Tetramethylpiperidine-1-oxyl
- TGN
trans-golgi network
- TNF-α
tumor necrosis factor alpha
- TRAIL
TNF-related apoptosis inducing ligand
- TTX
Tetrodotoxin
Biographies
Photographs and short biographies of each author
(A) Dr. Lukas Wanka

L. Wanka (b. 1974) currently serves as a deputy professor of Organic Chemistry at the Justus-Liebig University Giessen. He studied Chemistry in Giessen, where he worked on conformational analyses of enamines to earn his Diploma with Prof. H. Ahlbrecht. Subsequently, he joined Prof. Peter R. Schreiner's group, working on the synthesis of adamantane-based amino acids and their incorporation into novel peptidic structures to receive his Dr. rer. nat. (2007) in Organic Chemistry. He then received post-doctoral training in Prof. Khalid Iqbal's group at the Department of Neurochemistry of NYS's Institute for Basic Research (IBR), working on the fields of phosphatase regulation as well as the enhancement of neurogenesis with small peptidic molecules. After returning to the University of Giessen, his current research deals with peptide-derived binding pockets for Organocatalysis as well as the aggregation behavior of post-translationally modified protein fragments.
(B) Prof. Dr. Khalid Iqbal, Ph. D

Khalid Iqbal (b. 1943) is Professor and Chairman, Department of Neurochemistry at the New York State Institute for Basic Research in Developmental Disabilities, Staten Island, New York. He received his Ph.D. in Biochemistry in 1969 from the University of Edinburgh, Edinburgh, U.K. His pioneering studies on neuronal protein pathology and discoveries of tau protein and its abnormal hyperphosphorylation in Alzheimer disease have won him many prestigious honors and awards, including the Potamkin Prize for Alzheimer Disease research from the American Academy of Neurology, and the Zenith Award from the Alzheimer's Association, U.S.A. In 2007, Alzheimer's Association, USA, established a Khalid Iqbal Life Time Achievement Award for Alzheimer's Disease Research, which is given out annually at the International Conference on Alzheimer's Disease (ICAD) to a senior established Alzheimer disease researcher. Khalid has authored over 200 scientific papers in prestigious American and international scientific journals and edited seven books on research advances in Alzheimer disease and related neurodegenerative disorders. He currently serves on the editorial boards of several journals and scientific advisory committees of biotechnology companies.
(C) Prof. Dr. Peter Schreiner, Ph. D

P. R. Schreiner (b. 1965) is professor of Organic Chemistry at the Justus-Liebig University Giessen. He studied chemistry in his native city at the University of Erlangen-Nürnberg, Germany, where he received his Dr. rer. nat. (1994) in Organic Chemistry. Simultaneously, he obtained a PhD (1995) in Computational Chemistry from the University of Georgia, USA. He completed his habilitation at the University of Göttingen (1999). P. R. Schreiner is the 2003 recipient of the Dirac Medal and received the ADUC-Prize for Assistant Professors in 1999 and was a Liebig-Fellow (1997–1999). He currently serves as an Editor for the Journal of Computational Chemistry, as Editor-in-Chief for WIRES – Computational Molecular Sciences, Associate Editor for the Beilstein Journal of Organic Chemistry, and as an international advisory board member of several organic chemistry journals.
14. References
- (1).Davies WL, Grunert RR, Haff RF, McGahen JW, Neumayer EM, Paulshock M, Watts JC, Wood TR, Hermann EC, Hoffmann CE. Science. 1964;144:862. doi: 10.1126/science.144.3620.862. [DOI] [PubMed] [Google Scholar]
- (2).Maassab HF, Cochran KW. Science. 1964;145:1443. doi: 10.1126/science.145.3639.1443. [DOI] [PubMed] [Google Scholar]
- (3).Schleyer P. v. R. J. Am. Chem. Soc. 1957;79:3292. [Google Scholar]
- (4).Landa S, Machacek V. Collect. Czech. Chem. C. 1933;5:1. [Google Scholar]
- (5).Prelog V, Seiwerth R. Berichte der Deutschen Chemischen Gesellschaft [Abteilung] B: Abhandlungen. 1941;74B:1644. [Google Scholar]
- (6).Webber WC, Harthoorn PA. Chlorinated and brominated polycyclic hydrocarbons. Br. Pat. Appl. 819240. 1959 Sep 02; [Google Scholar]
- (7).Gerzon K, Krumalns EV, Brindle RL, Marshall FJ, Root MA. J. Med. Chem. 1963;6:760. doi: 10.1021/jm00342a029. [DOI] [PubMed] [Google Scholar]
- (8).Rapala RT, Kraay RJ, Gerzon K. J. Med. Chem. 1965;8:580. doi: 10.1021/jm00329a007. [DOI] [PubMed] [Google Scholar]
- (9).Gerzon K, Kau D. J. Med. Chem. 1967;10:189. doi: 10.1021/jm00314a014. [DOI] [PubMed] [Google Scholar]
- (10).Tsunoda A, Maassab HF, Cochran KW, Eveland WC. Antimicrob. Agents Ch. 1965;553 doi: 10.1128/AAC.5.6.553. [DOI] [PubMed] [Google Scholar]
- (11).Fanta D. Wien. Med. Wochenschr. 1976;126:315. [PubMed] [Google Scholar]
- (12).Rosenthal KS, Sokol MS, Ingram RL, Subramanian R, Fort RC. Antimicrob. Agents Ch. 1982;22:1031. doi: 10.1128/aac.22.6.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Mould JA, Li HC, Dudlak CS, Lear JD, Pekosz A, Lamb RA, Pinto LH. J. Biol. Chem. 2000;275:8592. doi: 10.1074/jbc.275.12.8592. [DOI] [PubMed] [Google Scholar]
- (14).Stouffer AL, Acharya R, Salom D, Levine AS, Di Costanzo L, Soto CS, Tereshko V, Nanda V, Stayrook S, DeGrado WF. Nature. 2008;451:596. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Cady SD, Hong M. Proc. Natl. Acad. Sci. U.S.A. 2008;105:1483. doi: 10.1073/pnas.0711500105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Cady SD, Schmidt-Rohr K, Wang J, Soto CS, Degrado WF, Hong M. Nature. 2010;463:689. doi: 10.1038/nature08722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Bright RA, Medina M.-j., Xu X, Perez-Oronoz G, Wallis TR, Davis XM, Povinelli L, Cox NJ, Klimov AI. Lancet. 2005;366:1175. doi: 10.1016/S0140-6736(05)67338-2. [DOI] [PubMed] [Google Scholar]
- (18).Hinman A, Du Bois J. J. Am. Chem. Soc. 2003;125:11510. doi: 10.1021/ja0368305. [DOI] [PubMed] [Google Scholar]
- (19).Hu L-H, Sim K-Y. Tetrahedron. 2000;56:1379. [Google Scholar]
- (20).Ciochina R, Grossman RB. Chem. Rev. 2006;106:3963. doi: 10.1021/cr0500582. [DOI] [PubMed] [Google Scholar]
- (21).Rezanka T, Sigler K. Phytochemistry. 2007;68:1272. doi: 10.1016/j.phytochem.2007.02.029. [DOI] [PubMed] [Google Scholar]
- (22).Ickes DE, Venetta TM, Phonphok Y, Rosenthal KS. Antivir. Res. 1990;14:75. doi: 10.1016/0166-3542(90)90045-9. [DOI] [PubMed] [Google Scholar]
- (23).Berg T, Kronenberger B, Hinrichsen H, Gerlach T, Buggisch P, Herrmann E, Spengler U, Goeser T, Nasser S, Wursthorn K, Pape GR, Hopf U, Zeuzem S. Hepatology. 2003;37:1359. doi: 10.1053/jhep.2003.50219. [DOI] [PubMed] [Google Scholar]
- (24).El-Emam AA, Al-Deeb OA, Al-Omar M, Lehmann J. Bioorgan. Med. Chem. 2004;12:5107. doi: 10.1016/j.bmc.2004.07.033. [DOI] [PubMed] [Google Scholar]
- (25).Printsevskaya SS, Solovieva SE, Olsufyeva EN, Mirchink EP, Isakova EB, De Clercq E, Balzarini J, Preobrazhenskaya MN. J. Med. Chem. 2005;48:3885. doi: 10.1021/jm0500774. [DOI] [PubMed] [Google Scholar]
- (26).Fieser LF, Nazer MZ, Archer S, Berberian DA, Slighter RG. J. Med. Chem. 1967;10:517. doi: 10.1021/jm00316a002. [DOI] [PubMed] [Google Scholar]
- (27).Singh C, Gupta N, Puri SK. Bioorgan. Med. Chem. 2004;12:5553. doi: 10.1016/j.bmc.2004.08.005. [DOI] [PubMed] [Google Scholar]
- (28).Dong Y, Chollet J, Matile H, Charman SA, Chiu FC, Charman WN, Scorneaux B, Urwyler H, Santo Tomas J, Scheurer C, Snyder C, Dorn A, Wang X, Karle JM, Tang Y, Wittlin S, Brun R, Vennerstrom JL. J. Med. Chem. 2005;48:4953. doi: 10.1021/jm049040u. [DOI] [PubMed] [Google Scholar]
- (29).Wang X, Dong Y, Wittlin S, Creek D, Chollet J, Charman SA, SantoTomas J, Scheurer C, Snyder C, Vennerstrom JL. J. Med. Chem. 2007;50:5840. doi: 10.1021/jm0707673. [DOI] [PubMed] [Google Scholar]
- (30).Gerzon K, Tobias DJ, Holmes RE, Rathbun RE, Kattau RW. J. Med. Chem. 1967;10:603. doi: 10.1021/jm00316a018. [DOI] [PubMed] [Google Scholar]
- (31).Swift PA, Stagnito ML, Mullen GB, Palmer GC, Georgiev VS. Eur. J. Med. Chem. 1988;23:465. [Google Scholar]
- (32).Schwab RS, England AC, Jr., Poskanzer DC, Young RR. J. Am. Med. Assoc. 1969;208:1168. [PubMed] [Google Scholar]
- (33).Sonkusare SK, Kaul CL, Ramarao P. Pharmacol. Res. 2005;51:1. doi: 10.1016/j.phrs.2004.05.005. [DOI] [PubMed] [Google Scholar]
- (34).Lipton SA. Nat. Rev. Drug. Discov. 2006;5:160. doi: 10.1038/nrd1958. [DOI] [PubMed] [Google Scholar]
- (35).Parsons CG, Danysz W, Quack G. Amino Acids. 2000;19:157. doi: 10.1007/s007260070044. [DOI] [PubMed] [Google Scholar]
- (36).Danysz W, Parsons CG, Quack G. Amino Acids. 2000;19:167. [PubMed] [Google Scholar]
- (37).Kornhuber J, Schoppmeyer K, Riederer P. Neurosci. Lett. 1993;163:129. doi: 10.1016/0304-3940(93)90362-o. [DOI] [PubMed] [Google Scholar]
- (38).Chen HS, Pellegrini JW, Aggarwal SK, Lei SZ, Warach S, Jensen FE, Lipton SA. J. Neurosci. 1992;12:4427. doi: 10.1523/JNEUROSCI.12-11-04427.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Weller M, Finiels-Marlier F, Paul SM. Brain Res. 1993;613:143. doi: 10.1016/0006-8993(93)90464-x. [DOI] [PubMed] [Google Scholar]
- (40).Parsons CG, Quack G, Bresink I, Baran L, Przegalinski E, Kostowski W, Krzascik P, Hartmann S, Danysz W. Neuropharmacology. 1995;34:1239. doi: 10.1016/0028-3908(95)00092-k. [DOI] [PubMed] [Google Scholar]
- (41).Miguel-Hidalgo JJ, Alvarez XA, Cacabelos R, Quack G. Brain Res. 2002;958:210. doi: 10.1016/s0006-8993(02)03731-9. [DOI] [PubMed] [Google Scholar]
- (42).Guillemare E, Honore E, De Weille J, Fosset M, Lazdunski M, Meisheri K. Mol. Pharmacol. 1994;46:139. [PubMed] [Google Scholar]
- (43).Kovalev H, Quayle JM, Kamishima T, Lodwick D. Brit. J. Pharmacol. 2004;141:867. doi: 10.1038/sj.bjp.0705670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Zoidis G, Papanastasiou I, Dotsikas I, Sandoval A, Dos Santos RG, Papadopoulou-Daifoti Z, Vamvakides A, Kolocouris N, Felix R. Bioorgan. Med. Chem. 2005;13:2791. doi: 10.1016/j.bmc.2005.02.030. [DOI] [PubMed] [Google Scholar]
- (45).Schreiner PR, Wanka L. New aminoadamantane derivatives useful for treating e.g. viral diseases, Parkinson's disease, Huntington's chorea, Alzheimer's disease, autism, tinnitus, Tourette syndrome, hypertension, sleep disorders and psychosis. Ger. Offen. DE13042006010362. 2006 Mar 16; [Google Scholar]
- (46).Blommaert AGS, Weng JH, Dorville A, McCort I, Ducos B, Durieux C, Roques BP. J. Med. Chem. 1993;36:2868. doi: 10.1021/jm00072a005. [DOI] [PubMed] [Google Scholar]
- (47).Bellier B, McCort-Tranchepain I, Ducos B, Danascimento S, Meudal H, Noble F, Garbay C, Roques BP. J. Med. Chem. 1997;40:3947. doi: 10.1021/jm970439a. [DOI] [PubMed] [Google Scholar]
- (48).Villhauer EB, Brinkman JA, Naderi GB, Burkey BF, Dunning BE, Prasad K, Mangold BL, Russell ME, Hughes TE. J. Med. Chem. 2003;46:2774. doi: 10.1021/jm030091l. [DOI] [PubMed] [Google Scholar]
- (49).Augeri DJ, Robl JA, Betebenner DA, Magnin DR, Khanna A, Robertson JG, Wang A, Simpkins LM, Taunk P, Huang Q, Han S-P, Abboa-Offei B, Cap M, Xin L, Tao L, Tozzo E, Welzel GE, Egan DM, Marcinkeviciene J, Chang SY, Biller SA, Kirby MS, Parker RA, Hamann LG. J. Med. Chem. 2005;48:5025. doi: 10.1021/jm050261p. [DOI] [PubMed] [Google Scholar]
- (50).Barnett A. Int. J. Clin. Pract. 2006;60:1454. doi: 10.1111/j.1742-1241.2006.01178.x. [DOI] [PubMed] [Google Scholar]
- (51).Kim I-H, Morisseau C, Watanabe T, Hammock BD. J. Med. Chem. 2004;47:2110. doi: 10.1021/jm030514j. [DOI] [PubMed] [Google Scholar]
- (52).Hwang SH, Tsai HJ, Liu JY, Morisseau C, Hammock BD. J. Med. Chem. 2007;50:3825. doi: 10.1021/jm070270t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Chohan MO, Khatoon S, Iqbal I-G, Iqbal K. FEBS Lett. 2006;580:3973. doi: 10.1016/j.febslet.2006.06.021. [DOI] [PubMed] [Google Scholar]
- (54).Tsu H, Chen X, Chen C-T, Lee S-J, Chang C-N, Kao K-H, Coumar MS, Yeh Y-T, Chien C-H, Wang H-S, Lin K-T, Chang Y-Y, Wu S-H, Chen Y-S, Lu I-L, Wu S-Y, Tsai T-Y, Chen W-C, Hsieh H-P, Chao Y-S, Jiaang W-T. J. Med. Chem. 2006;49:373. doi: 10.1021/jm0507781. [DOI] [PubMed] [Google Scholar]
- (55).Yeh VSC, Kurukulasuriya R, Fung S, Monzon K, Chiou W, Wang J, Stolarik D, Imade H, Shapiro R, Knourek-Segel V, Bush E, Wilcox D, Nguyen PT, Brune M, Jacobson P, Link JT. Bioorg. Med. Chem. Lett. 2006;16:5555. doi: 10.1016/j.bmcl.2006.08.034. [DOI] [PubMed] [Google Scholar]
- (56).Yeh VSC, Kurukulasuriya R, Madar D, Patel JR, Fung S, Monzon K, Chiou W, Wang J, Jacobson P, Sham HL, Link JT. Bioorg. Med. Chem. Lett. 2006;16:5408. doi: 10.1016/j.bmcl.2006.07.062. [DOI] [PubMed] [Google Scholar]
- (57).Nicolaou KC, T. L., Denton Ross M., Montero Ana, Edmonds David J. Angew. Chem. Int. Edit. 2007;46:4712. doi: 10.1002/anie.200701548. [DOI] [PubMed] [Google Scholar]
- (58).Spasov A, Khamidova T, Bugaeva L, Morozov I. Pharm. Chem. J. 2000;34:1. [PubMed] [Google Scholar]
- (59).Lamoureux G, Artavia G. Curr. Med. Chem. 2010;17:2967. doi: 10.2174/092986710792065027. [DOI] [PubMed] [Google Scholar]
- (60).Trommer H. PZ Prisma. 2010;17:43. [Google Scholar]
- (61).Trommer H. PZ Prisma. 2010;17:109. [Google Scholar]
- (62).Liu J, Obando D, Liao V, Lifa T, Codd R. Eur. J. Med. Chem. 2011;46:1949. doi: 10.1016/j.ejmech.2011.01.047. [DOI] [PubMed] [Google Scholar]
- (63).Halgren TA. J. Comput. Chem. 1996;17:490. [Google Scholar]
- (64).Avogadro: an open-source molecular builder and visualization tool. (Version 1.03) http://avogadro.openmolecules.net/
- (65).Tetko IV, Gasteiger J, Todeschini R, Mauri A, Livingstone D, Ertl P, Palyulin VA, Radchenko EV, Zefirov NS, Makarenko AS, Tanchuk VY, Prokopenko VV. J. Comput. Aid. Mol. Des. 2005;19:453. doi: 10.1007/s10822-005-8694-y. [DOI] [PubMed] [Google Scholar]
- (66).Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv. Drug Deliver. Rev. 1997;23:3. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- (67).Lipinski C, Hopkins A. Nature. 2004;432:855. doi: 10.1038/nature03193. [DOI] [PubMed] [Google Scholar]
- (68).Butler MS. J. Nat. Prod. 2004;67:2141. doi: 10.1021/np040106y. [DOI] [PubMed] [Google Scholar]
- (69).Koert U. Angew. Chem. Int. Edit. 2004;43:5572. doi: 10.1002/anie.200461097. [DOI] [PubMed] [Google Scholar]
- (70).Sato K, Akai S, Sugita N, Ohsawa T, Kogure T, Shoji H, Yoshimura J. J. Org. Chem. 2005;70:7496. doi: 10.1021/jo050342t. [DOI] [PubMed] [Google Scholar]
- (71).Urabe D, Nishikawa T, Isobe M. Chemistry-an Asian Journal. 2006;1:125. doi: 10.1002/asia.200600038. [DOI] [PubMed] [Google Scholar]
- (72).Hucho F. Angew. Chem. Int. Edit. 1995;34:39. [Google Scholar]
- (73).Roll DM, Biskupiak JE, Mayne CL, Ireland CM. J. Am. Chem. Soc. 1986;108:6680. [Google Scholar]
- (74).Hoffman RW, Dahmann G. Tetrahedron Lett. 1993;34:1115. [Google Scholar]
- (75).Blanchfield JT, Brecknell DJ, Brereton IM, Garson MJ, Jones DD. Aust. J. Chem. 1994;47:2255. [Google Scholar]
- (76).Tanner JA, Zheng B-J, Zhou J, Watt RM, Jiang J-Q, Wong K-L, Lin Y-P, Lu L-Y, He M-L, Kung H-F, Kesel AJ, Huang J-D. Chem. Biol. 2005;12:303. doi: 10.1016/j.chembiol.2005.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Kesel AJ. Anti-Inf. Agents Med. Chem. 2006;5:161. [Google Scholar]
- (78).Hu L-H, Sim K-Y. Tetrahedron Lett. 1998;39:7999. [Google Scholar]
- (79).Hu L-H, Sim K-Y. Tetrahedron Lett. 1999;40:759. [Google Scholar]
- (80).Lin Y-L, Wu Y-S. Helv. Chim. Acta. 2003;86:2156. [Google Scholar]
- (81).Henry GE, Jacobs H, Carrington CMS, McLean S, Reynolds WF. Tetrahedron Lett. 1996;37:8663. [Google Scholar]
- (82).Christian OE, Henry GE, Jacobs H, McLean S, Reynolds WF. J. Nat. Prod. 2001;64:23. doi: 10.1021/np000321o. [DOI] [PubMed] [Google Scholar]
- (83).Diaz-Carballo D, Malak S, Bardenheuer W, Freistuehler M, Peter Reusch H. Bioorgan. Med. Chem. 2008;16:9635. doi: 10.1016/j.bmc.2008.10.019. [DOI] [PubMed] [Google Scholar]
- (84).Diaz-Carballo D, Ueberla K, Kleff V, Ergun S, Malak S, Freistuehler M, Somogyi S, Kucherer C, Bardenheuer W, Strumberg D. Int. J. Clin. Pharm. Th. 2010;48:670. doi: 10.5414/cpp48670. [DOI] [PubMed] [Google Scholar]
- (85).Zhang Q, Mitasev B, Qi J, Porco JA., Jr. J. Am. Chem. Soc. 2010;132:14212. doi: 10.1021/ja105784s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Takagi R, Inoue Y, Ohkata K. J. Org. Chem. 2008;73:9320. doi: 10.1021/jo801595y. [DOI] [PubMed] [Google Scholar]
- (87).Hu LH, Sim KY. Org. Lett. 1999;1:879. doi: 10.1021/ol9907825. [DOI] [PubMed] [Google Scholar]
- (88).Tanaka N, Takaishi Y, Shikishima Y, Nakanishi Y, Bastow K, Lee K-H, Honda G, Ito M, Takeda Y, Kodzhimatov OK, Ashurmetov O. J. Nat. Prod. 2004;67:1870. doi: 10.1021/np040024+. [DOI] [PubMed] [Google Scholar]
- (89).Teixeira JSAR, Cruz FG. Tetrahedron Lett. 2005;46:2813. [Google Scholar]
- (90).Dahl JE, Liu SG, Carlson RMK. Science. 2003;299:96. doi: 10.1126/science.1078239. [DOI] [PubMed] [Google Scholar]
- (91).Fokin AA, Schreiner PR. Chem. Rev. 2002;102:1551. doi: 10.1021/cr000453m. [DOI] [PubMed] [Google Scholar]
- (92).Schwertfeger H, Fokin AA, Schreiner PR. Angew. Chem. Int. Edit. 2008;47:1022. doi: 10.1002/anie.200701684. [DOI] [PubMed] [Google Scholar]
- (93).Schreiner PR, Fokina NA, Tkachenko BA, Hausmann H, Serafin M, Dahl JEP, Liu S, Carlson RMK, Fokin AA. J. Org. Chem. 2006;71:6709. doi: 10.1021/jo052646l. [DOI] [PubMed] [Google Scholar]
- (94).Proks P, Reimann F, Green N, Gribble F, Ashcroft F. Diabetes. 2002;51:S368. doi: 10.2337/diabetes.51.2007.s368. [DOI] [PubMed] [Google Scholar]
- (95).Koch H, Haaf W. Angew. Chem. 1960;72:628. [Google Scholar]
- (96).Koch H, Haaf W. Liebigs Ann. 1958;618:251. [Google Scholar]
- (97).Kovtun VY, Plakhotnik VM. Pharm. Chem. J. 1987;21:555. [Google Scholar]
- (98).Ponpipom MM, Bugianesi RL, Shen TY, Friedman A. J. Med. Chem. 1980;23:1184. doi: 10.1021/jm00185a007. [DOI] [PubMed] [Google Scholar]
- (99).Kurokawa K, Kumihara H, Kondo H. Bioorg. Med. Chem. Lett. 2000;10:1827. doi: 10.1016/s0960-894x(00)00358-9. [DOI] [PubMed] [Google Scholar]
- (100).Titz A, Patton J, Alker AM, Porro M, Schwardt O, Hennig M, Francotte E, Magnani J, Ernst B. Bioorgan. Med. Chem. 2008;16:1046. doi: 10.1016/j.bmc.2007.07.025. [DOI] [PubMed] [Google Scholar]
- (101).Brown AD, Bunnage ME, Glossop PA, James K, Jones R, Lane CA, Lewthwaite RA, Mantell S, Perros-Huguet C, Price DA, Trevethick M, Webster R. Bioorg. Med. Chem. Lett. 2008;18:1280. doi: 10.1016/j.bmcl.2008.01.034. [DOI] [PubMed] [Google Scholar]
- (102).Kasuga J, Yamasaki D, Ogura K, Shimizu M, Sato M, Makishima M, Doi T, Hashimoto Y, Miyachi H. Bioorg. Med. Chem. Lett. 2008;18:1110. doi: 10.1016/j.bmcl.2007.12.001. [DOI] [PubMed] [Google Scholar]
- (103).Overkleeft HS, Renkema GH, Neele J, Vianello P, Hung IO, Strijland A, van der Burg AM, Koomen G-J, Pandit UK, Aerts JMFG. J. Biol. Chem. 1998;273:26522. doi: 10.1074/jbc.273.41.26522. [DOI] [PubMed] [Google Scholar]
- (104).Sawkar AR, Adamski-Werner SL, Cheng WC, Wong CH, Beutler E, Zimmer KP, Kelly JW. Chem. Biol. 2005;12:1235. doi: 10.1016/j.chembiol.2005.09.007. [DOI] [PubMed] [Google Scholar]
- (105).Yu Z, Sawkar AR, Whalen LJ, Wong C-H, Kelly JW. J. Med. Chem. 2007;50:94. doi: 10.1021/jm060677i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Bietrix F, Lombardo E, van Roomen CPAA, Ottenhoff R, Vos M, Rensen PCN, Verhoeven AJ, Aerts JM, Groen AK. Arterioscl. Throm. Vas. 2010;30:931. doi: 10.1161/ATVBAHA.109.201673. [DOI] [PubMed] [Google Scholar]
- (107).Ardes-Guisot N, Alonzi DS, Reinkensmeier G, Butters TD, Norez C, Becq F, Shimada Y, Nakagawa S, Kato A, Bleriot Y, Sollogoub M, Vauzeilles B. Org. Biomol. Chem. 2011;9:5373. doi: 10.1039/c1ob05119a. [DOI] [PubMed] [Google Scholar]
- (108).Kapoerchan VV, Knijnenburg AD, Niamat M, Spalburg E, de Neeling AJ, Nibbering PH, Mars-Groenendijk RH, Noort D, Otero JM, Llamas-Saiz AL, van Raaij MJ, van der Marel GA, Overkleeft HS, Overhand M. Chem.-Eur. J. 2010;16:12174. doi: 10.1002/chem.201001686. [DOI] [PubMed] [Google Scholar]
- (109).Jackson GG, Muldoon RL, Akers LW. Antimicrob. Agents Ch. 1963;161:703. [PubMed] [Google Scholar]
- (110).Eaton MD, Low IE, Scala AR, Uretsky S. Virology. 1962;18:102. doi: 10.1016/0042-6822(62)90182-4. [DOI] [PubMed] [Google Scholar]
- (111).Oxford JS, Galbraith A. Pharmacol. Therapeut. 1980;11:181. doi: 10.1016/0163-7258(80)90072-8. [DOI] [PubMed] [Google Scholar]
- (112).Grunert RR, McGahen JW, Davies WL. Virology. 1965;26:262. doi: 10.1016/0042-6822(65)90273-4. [DOI] [PubMed] [Google Scholar]
- (113).Geluk HW, Schut J, Schlatmann JLMA. J. Med. Chem. 1969;12:712. doi: 10.1021/jm00304a045. [DOI] [PubMed] [Google Scholar]
- (114).Whitney JG, Gregory WA, Kauer JC, Roland JR, Snyder JA, Benson RE, Hermann EC. J. Med. Chem. 1970;13:254. doi: 10.1021/jm00296a021. [DOI] [PubMed] [Google Scholar]
- (115).Sidwell RW, Dixon GJ, Sellers SM, Schabel FM., Jr. Appl. Microbiol. 1968;16:370. doi: 10.1128/am.16.2.370-392.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Greene JF, Jr., Tilton BE, Quilligan JJ., Jr. Appl. Microbiol. 1969;18:533. doi: 10.1128/am.18.4.533-534.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Scherm A, Peteri D. Antiviral 1-(aminoethoxyacetylamino)adamantanes. Ger. Offen. DE1941218. 1971 Feb 25; [Google Scholar]
- (118).Aldrich PE, Hermann EC, Meier WE, Paulshock M, Prichard WW, Synder JA, Watts JC. J. Med. Chem. 1971;14:535. doi: 10.1021/jm00288a019. [DOI] [PubMed] [Google Scholar]
- (119).Schmidtke M, Zell R, Bauer K, Krumbholz A, Schrader C, Suess J, Wutzler P. Intervirology. 2006;49:286. doi: 10.1159/000094244. [DOI] [PubMed] [Google Scholar]
- (120).Nelson MI, Simonsen L, Viboud C, Miller MA, Holmes EC. Virology. 2009;388:270. doi: 10.1016/j.virol.2009.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Weinstock DM, Zuccotti G. J. Am. Med. Assoc. 2009;301:1066. doi: 10.1001/jama.2009.324. [DOI] [PubMed] [Google Scholar]
- (122).Lundahl K, Schut J, Schlatmann JLMA, Paerels GB, Peters A. J. Med. Chem. 1972;15:129. doi: 10.1021/jm00272a003. [DOI] [PubMed] [Google Scholar]
- (123).Van Hes R, Smit A, Kralt T, Peters A. J. Med. Chem. 1972;15:132. doi: 10.1021/jm00272a004. [DOI] [PubMed] [Google Scholar]
- (124).Beare AS, Hall TS, Tyrrell DA. Lancet. 1972;1:1039. doi: 10.1016/s0140-6736(72)91220-2. [DOI] [PubMed] [Google Scholar]
- (125).Mathur A, Beare AS, Reed SE. Antimicrob. Agents Ch. 1973;4:421. doi: 10.1128/aac.4.4.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Arroyo M, Beare AS, Reed SE, Craig JW. J. Antimicrob. Chemoth. 1975;1:87. doi: 10.1093/jac/1.suppl_4.87. [DOI] [PubMed] [Google Scholar]
- (127).Aigami K, Inamoto Y, Takaishi N, Hattori K, Takatsuki A. J. Med. Chem. 1975;18:713. doi: 10.1021/jm00241a015. [DOI] [PubMed] [Google Scholar]
- (128).Aigami K, Inamoto Y, Takaishi N, Fujikura Y. J. Med. Chem. 1976;19:536. doi: 10.1021/jm00226a018. [DOI] [PubMed] [Google Scholar]
- (129).Inamoto Y, Aigami K, Kadono T, Nakayama H, takatsuki A, Tamura G. J. Med. Chem. 1977;20:1371. doi: 10.1021/jm00221a003. [DOI] [PubMed] [Google Scholar]
- (130).Inamoto Y, Aigami K, Takaishi N, Fujikura Y, Ohsugi M, Ikeda H, Tsuchihashi K, Takatsuki A, Tamura G. J. Med. Chem. 1979;22:1206. doi: 10.1021/jm00196a011. [DOI] [PubMed] [Google Scholar]
- (131).Kreutzberger A, Schroeders HH. Tetrahedron Lett. 1969:5101. doi: 10.1016/s0040-4039(00)99744-2. [DOI] [PubMed] [Google Scholar]
- (132).Kreutzberger A, Tantawy A. Arzneimittel-Forsch. 1983;33:512. [PubMed] [Google Scholar]
- (133).Kreutzberger A, Schroders H-H. Tetrahedron Lett. 1973;14:687. [Google Scholar]
- (134).Tilley JW, Levitan P, Kramer MJ. J. Med. Chem. 1979;22:1009. doi: 10.1021/jm00194a025. [DOI] [PubMed] [Google Scholar]
- (135).Pellicciari R, Fioretti MC, Cogolli P, Tiecco M. Arzneimittel-Forsch. 1980;30:2103. [PubMed] [Google Scholar]
- (136).de la Cuesta E, Ballesteros P, Trigo GG. J. Pharm. Sci. 1984;73:1307. doi: 10.1002/jps.2600730933. [DOI] [PubMed] [Google Scholar]
- (137).Oliver DW, Dekker TG, Snyckers FO. Arznei-Forschung. 1991;41:549. [PubMed] [Google Scholar]
- (138).Hay AJ, Wolstenholme AJ, Skehel JJ, Smith MH. 1985. EMBO J. 4:3021. doi: 10.1002/j.1460-2075.1985.tb04038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Kempf C, Michel MR, Kohler U, Koblet H. Bioscience Rep. 1987;7:761. doi: 10.1007/BF01116748. [DOI] [PubMed] [Google Scholar]
- (140).Duff KC, Ashley RH. Virology. 1992;190:485. doi: 10.1016/0042-6822(92)91239-q. [DOI] [PubMed] [Google Scholar]
- (141).Kolocouris N, Foscolos GB, Kolocouris A, Marakos P, Pouli N, Fytas G, Ikeda S, De Clercq E. J. Med. Chem. 1994;37:2896. doi: 10.1021/jm00044a010. [DOI] [PubMed] [Google Scholar]
- (142).Kolocouris N, Kolocouris A, Foscolos GB, Fytas G, Neyts J, Padalko E, Balzarini J, Snoeck R, Andrei G, De Clercq E. J. Med. Chem. 1996;39:3307. doi: 10.1021/jm950891z. [DOI] [PubMed] [Google Scholar]
- (143).Fytas G, Stamatiou G, Foscolos GB, Kolocouris A, Kolocouris N, Witvrouw M, Pannecouque C, De Clercq E. Bioorg. Med. Chem. Lett. 1997;7:1887. [Google Scholar]
- (144).Kolocouris A, Tataridis D, Fytas G, Mavromoustakos T, Foscolos GB, Kolocouris N, De Clercq E. Bioorg. Med. Chem. Lett. 1999;9:3465. doi: 10.1016/s0960-894x(99)00631-9. [DOI] [PubMed] [Google Scholar]
- (145).Shigeta S, Konno K, Yokota T, Nakamura K, De Clercq E. Antimicrob. Agents Ch. 1988;32:906. doi: 10.1128/aac.32.6.906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Stamatiou G, Kolocouris A, Kolocouris N, Fytas G, Foscolos GB, Neyts J, De Clercq E. Bioorg. Med. Chem. Lett. 2001;11:2137. doi: 10.1016/s0960-894x(01)00388-2. [DOI] [PubMed] [Google Scholar]
- (147).Vranesic B, Tomasic J, Smerdel S, Kantoci D, Benedetti F. Helv. Chim. Acta. 1993;76:1752. [Google Scholar]
- (148).Stamatiou G, Foscolos GB, Fytas G, Kolocouris A, Kolocouris N, Pannecouque C, Witvrouw M, Padalko E, Neyts J, De Clercq E. Bioorgan. Med. Chem. 2003;11:5485. doi: 10.1016/j.bmc.2003.09.024. [DOI] [PubMed] [Google Scholar]
- (149).Zoidis G, Fytas C, Papanastasiou I, Foscolos GB, Fytas G, Padalko E, De Clercq E, Naesens L, Neyts J, Kolocouris N. Bioorgan. Med. Chem. 2006;14:3341. doi: 10.1016/j.bmc.2005.12.056. [DOI] [PubMed] [Google Scholar]
- (150).Setaki D, Tataridis D, Stamatiou G, Kolocouris A, Foscolos GB, Fytas G, Kolocouris N, Padalko E, Neyts J, De Clercq E. Bioorg. Chem. 2006;34:248. doi: 10.1016/j.bioorg.2006.05.004. [DOI] [PubMed] [Google Scholar]
- (151).Kolocouris N, Zoidis G, Foscolos GB, Fytas G, Prathalingham SR, Kelly JM, Naesens L, De Clercq E. Bioorg. Med. Chem. Lett. 2007;17:4358. doi: 10.1016/j.bmcl.2007.04.108. [DOI] [PubMed] [Google Scholar]
- (152).Zoidis G, Tsotinis A, Kolocouris N, Kelly JM, Prathalingam SR, Naesens L, De Clercq E. Org. Biomol. Chem. 2008;6:3177. doi: 10.1039/b804907f. [DOI] [PubMed] [Google Scholar]
- (153).Zoidis G, Kolocouris N, Naesens L, Clercq ED. Bioorgan. Med. Chem. 2009;17:1534. doi: 10.1016/j.bmc.2009.01.009. [DOI] [PubMed] [Google Scholar]
- (154).Camps P, Duque MD, Vazquez S, Naesens L, De Clercq E, Sureda FX, Lopez-Querol M, Camins A, Pallas M, Prathalingam SR, Kelly JM, Romero V, Ivorra D, Cortes D. Bioorgan. Med. Chem. 2008;16:9925. doi: 10.1016/j.bmc.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (155).Zoidis G, Kolocouris N, Foscolos GB, Kolocouris A, Fytas G, Karayannis P, Padalko E, Neyts J, De Clercq E. Antivir. Chem. Chemoth. 2003;14:153. doi: 10.1177/095632020301400305. [DOI] [PubMed] [Google Scholar]
- (156).Hurt AC, Selleck P, Komadina N, Shaw R, Brown L, Barr IG. Antivir. Res. 2007;73:228. doi: 10.1016/j.antiviral.2006.10.004. [DOI] [PubMed] [Google Scholar]
- (157).De Clercq E, Neyts J. Trends Pharmacol. Sci. 2007;28:280. doi: 10.1016/j.tips.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (158).Wang JF, Wei DQ, Chou KC. Biochem. Bioph. Res. Co. 2009;388:413. doi: 10.1016/j.bbrc.2009.08.026. [DOI] [PubMed] [Google Scholar]
- (159).Rungrotmongkol T, Intharathep P, Malaisree M, Nunthaboot N, Kaiyawet N, Sompornpisut P, Payungporn S, Poovorawan Y, Hannongbua S. Biochem. Bioph. Res. Co. 2009;385:390. doi: 10.1016/j.bbrc.2009.05.066. [DOI] [PubMed] [Google Scholar]
- (160).Duque MD, Ma CL, Torres E, Wang J, Naesens L, Juarez-Jimenez J, Camps P, Luque FJ, DeGrado WF, Lamb RA, Pinto LH, Vazquez S. J. Med. Chem. 2011;54:2646. doi: 10.1021/jm101334y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Sheu TG, Fry AM, Garten RJ, Deyde VM, Shwe T, Bullion L, Peebles PJ, Li Y, Klimov AI, Gubareva LV. J. Infect. Dis. 2011;203:13. doi: 10.1093/infdis/jiq005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (162).Miyachi K, Watanabe A, Iida H, Hattori H, Ukai H, Takano T, Hankins RW. J. Infect. Chemother. 2011;17:524. doi: 10.1007/s10156-011-0217-2. [DOI] [PubMed] [Google Scholar]
- (163).Nguyen JT, Smee DF, Barnard DL, Julander JG, Gross M, de Jong MD, Went GT. PLoS One. 2012;7:e31006. doi: 10.1371/journal.pone.0031006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Hoopes JD, Driebe EM, Kelley E, Engelthaler DM, Keim PS, Perelson AS, Rong L, Went GT, Nguyen JT. PLoS One. 2012;6:e29778. doi: 10.1371/journal.pone.0029778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (165).Sugrue RJ, Bahadur G, Zambon MC, Hall-Smith M, Douglas AR, Hay AJ. EMBO J. 1990;9:3469. doi: 10.1002/j.1460-2075.1990.tb07555.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Pinto LH, Holsinger LJ, Lamb RA. Cell. 1992;69:517. doi: 10.1016/0092-8674(92)90452-i. [DOI] [PubMed] [Google Scholar]
- (167).Helenius A. Cell. 1992;69:577. doi: 10.1016/0092-8674(92)90219-3. [DOI] [PubMed] [Google Scholar]
- (168).Wang C, Takeuchi K, Pinto LH, Lamb RA. J. Virol. 1993;67:5585. doi: 10.1128/jvi.67.9.5585-5594.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Duff KC, Gilchrist PJ, Saxena AM, Bradshaw JP. Virology. 1994;202:287. doi: 10.1006/viro.1994.1345. [DOI] [PubMed] [Google Scholar]
- (170).Tosteson MT, Pinto LH, Holsinger LJ, Lamb RA. J. Membrane Biol. 1994;142:117. doi: 10.1007/BF00233389. [DOI] [PubMed] [Google Scholar]
- (171).Bron R, Kendal AP, Klenk HD, Wilschut J. Virology. 1993;195:808. doi: 10.1006/viro.1993.1435. [DOI] [PubMed] [Google Scholar]
- (172).Wharton SA, Belshe RB, Skehel JJ, Hay AJ. J. Gen. Virol. 1994;75:945. doi: 10.1099/0022-1317-75-4-945. [DOI] [PubMed] [Google Scholar]
- (173).Pinto LH, Lamb RA. Trends Microbiol. 1995;3:271. doi: 10.1016/s0966-842x(00)88942-8. [DOI] [PubMed] [Google Scholar]
- (174).Sakaguchi T, Tu QA, Pinto LH, Lamb RA. Proc. Natl. Acad. Sci. U.S.A. 1997;94:5000. doi: 10.1073/pnas.94.10.5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (175).Pinto LH, Dieckmann GR, Gandhi CS, Papworth CG, Braman J, Shaughnessy MA, Lear JD, Lamb RA, DeGrado WF. Proc. Natl. Acad. Sci. U.S.A. 1997;94:11301. doi: 10.1073/pnas.94.21.11301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (176).Salom D, Hill BR, Lear JD, DeGrado WF. Biochemistry. 2000;39:14160. doi: 10.1021/bi001799u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (177).Czabotar PE, Martin SR, Hay AJ. Virus Res. 2004;99:57. doi: 10.1016/j.virusres.2003.10.004. [DOI] [PubMed] [Google Scholar]
- (178).Vijayvergiya V, Wilson R, Chorak A, Gao PF, Cross TA, Busath DD. Biophys. J. 2004;87:1697. doi: 10.1529/biophysj.104.043018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (179).Pinto LH, Lamb RA. J. Biol. Chem. 2006;281:8997. doi: 10.1074/jbc.R500020200. [DOI] [PubMed] [Google Scholar]
- (180).Hu J, Asbury T, Achuthan S, Li C, Bertram R, Quine JR, Fu R, Cross TA. Biophys. J. 2007;92:4335. doi: 10.1529/biophysj.106.090183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (181).Hu J, Fu R, Cross TA. Biophys. J. 2007;93:276. doi: 10.1529/biophysj.106.102103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (182).Kolocouris A, Zikos C, Broadhurst RW. Bioorg. Med. Chem. Lett. 2007;17:3947. doi: 10.1016/j.bmcl.2007.04.100. [DOI] [PubMed] [Google Scholar]
- (183).Kolocouris A, Hansen RK, Broadhurst RW. J. Med. Chem. 2004;47:4975. doi: 10.1021/jm0496685. [DOI] [PubMed] [Google Scholar]
- (184).Kolocouris A, Spearpoint P, Martin SR, Hay AJ, Lopez-Querol M, Sureda FX, Padalko E, Neyts J, De Clercq E. Bioorg. Med. Chem. Lett. 2008;18:6156. doi: 10.1016/j.bmcl.2008.10.003. [DOI] [PubMed] [Google Scholar]
- (185).Schnell JR, Chou JJ. Nature. 2008;451:591. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (186).Miller C. Nature. 2008;451:532. doi: 10.1038/451532a. [DOI] [PubMed] [Google Scholar]
- (187).Stouffer AL, Ma CL, Cristian L, Ohigashi Y, Lamb RA, Lear JD, Pinto LH, DeGrado WF. Structure. 2008;16:1067. doi: 10.1016/j.str.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (188).Yi M, Cross TA, Zhou HX. J. Phys. Chem. B. 2008;112:7977. doi: 10.1021/jp800171m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (189).Witter R, Nozirov F, Sternberg U, Cross TA, Ulrich AS, Fu R. J. Am. Chem. Soc. 2008;130:918. doi: 10.1021/ja0754305. [DOI] [PubMed] [Google Scholar]
- (190).Cady SD, Mishanina TV, Hong M. J. Mol. Biol. 2009;385:1127. doi: 10.1016/j.jmb.2008.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (191).Laohpongspaisan C, Rungrotmongkol T, Intharathep P, Malaisree M, Decha P, Aruksakunwong O, Sompornpisut P, Hannongbua S. J. Chem. Inf. Model. 2009;49:847. doi: 10.1021/ci800267a. [DOI] [PubMed] [Google Scholar]
- (192).Pielak RM, Schnell JR, Chou JJ. Proc. Natl. Acad. Sci. U.S.A. 2009;106:7379. doi: 10.1073/pnas.0902548106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (193).Wang J, Cady SD, Balannik V, Pinto LH, DeGrado WF, Hong M. J. Am. Chem. Soc. 2009;131:8066. doi: 10.1021/ja900063s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (194).Cady SD, Wang J, Wu Y, DeGrado WF, Hong M. J. Am. Chem. Soc. 2011;133:4274. doi: 10.1021/ja102581n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (195).Khurana E, DeVane RH, Dal Peraro M, Klein ML. BBA-Biomembranes. 2011;1808:530. doi: 10.1016/j.bbamem.2010.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (196).Hu F, Luo W, Cady SD, Hong M. Biochim. Biophysica Acta. 2011;1808:415. doi: 10.1016/j.bbamem.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (197).Cross TA, Sharma M, Yi M, Zhou HX. Trends Biochem. Sci. 2011;36:117. doi: 10.1016/j.tibs.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (198).Peterson E, Ryser T, Funk S, Inouye D, Sharma M, Qin HJ, Cross TA, Busath DD. BBA-Biomembranes. 2011;1808:516. doi: 10.1016/j.bbamem.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (199).Gu RX, Liu LA, Wei DQ, Du JG, Liu L, Liu H. J. Am. Chem. Soc. 2011;133:10817. doi: 10.1021/ja1114198. [DOI] [PubMed] [Google Scholar]
- (200).Wang J, Qiu JXY, Soto C, DeGrado WF. Curr. Opin. Struc. Biol. 2011;21:68. doi: 10.1016/j.sbi.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (201).Ghosh A, Qiu J, DeGrado WF, Hochstrasser RM. Proc. Natl. Acad. Sci. U.S.A. 2011;108:6115. doi: 10.1073/pnas.1103027108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (202).Rogers JMG, Poishchuk AL, Guo L, Wang J, DeGrado WF, Gai F. Langmuir. 2011;27:3815. doi: 10.1021/la200480d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (203).Hu F, Schmidt-Rohr K, Hong M. J. Am. Chem. Soc. 2011 doi: 10.1021/ja2081185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (204).Cady S, Wang T, Hong M. J. Am. Chem. Soc. 2011;133:11572. doi: 10.1021/ja202051n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (205).Leonov H, Astrahan P, Krugliak M, Arkin IT. J. Am. Chem. Soc. 2011;133:9903. doi: 10.1021/ja202288m. [DOI] [PubMed] [Google Scholar]
- (206).Furuse Y, Suzuki A, Oshitani H. Antimicrob. Agents Ch. 2009;53:4457. doi: 10.1128/AAC.00650-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (207).Furuse Y, Suzuki A, Kamigaki T, Shimizu M, Fuji N, Oshitani H. J. Clin. Microbiol. 2009;47:841. doi: 10.1128/JCM.01622-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (208).Balannik V, Wang J, Ohigashi Y, Jing X, Magavern E, Lamb RA, Degrado WF, Pinto LH. Biochemistry. 2009;48:11872. doi: 10.1021/bi9014488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (209).Wang J, Ma CL, Wu YB, Lamb RA, Pinto LH, DeGrado WF. J. Am. Chem. Soc. 2011;133:13844. doi: 10.1021/ja2050666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (210).Wang J, Ma C, Fiorin G, Carnevale V, Wang T, Hu F, Lamb RA, Pinto LH, Hong M, Klein ML, DeGrado WF. J. Am. Chem. Soc. 2011;133:12834. doi: 10.1021/ja204969m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (211).Martin K, Helenius A. Cell. 1991;67:117. doi: 10.1016/0092-8674(91)90576-k. [DOI] [PubMed] [Google Scholar]
- (212).Mould JA, Paterson RG, Takeda M, Ohigashi Y, Venkataraman P, Lamb RA, Pinto LH. Develop. Cell. 2003;5:175. doi: 10.1016/s1534-5807(03)00190-4. [DOI] [PubMed] [Google Scholar]
- (213).Pielak R, Chou J. Prot. Cell. 2010;1:246. doi: 10.1007/s13238-010-0025-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (214).Pielak RM, Chou JJ. BBA-Biomembranes. 2011;1808:522. doi: 10.1016/j.bbamem.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (215).Sharma M, Li CG, Busath DD, Zhou HX, Cross TA. BBA-Biomembranes. 2011;1808:538. doi: 10.1016/j.bbamem.2010.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (216).Pinto LH, Lamb RA. Mol. Biosyst. 2007;3:18. doi: 10.1039/b611613m. [DOI] [PubMed] [Google Scholar]
- (217).Pielak RM, Oxenoid K, Chou JJ. Structure. 2011;19:1655. doi: 10.1016/j.str.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (218).Hayden FG, de Jong MD. J. Infect. Dis. 2011;203:6. doi: 10.1093/infdis/jiq012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (219).Keppeler K, Kiefer G, De Clercq E. Arch. Pharm. 1984;317:867. doi: 10.1002/ardp.19843171011. [DOI] [PubMed] [Google Scholar]
- (220).Gish DT, Kelly RC, Camiener GW, Wechter WJ. J. Med. Chem. 1971;14:1159. doi: 10.1021/jm00294a004. [DOI] [PubMed] [Google Scholar]
- (221).Wechter WJ, Gish DT, Greig ME, Gray GD, Moxley TE, Kuentzel SL, Gray LG, Gibbons AJ, Griffin RL, Neil GL. J. Med. Chem. 1976;19:1013. doi: 10.1021/jm00230a007. [DOI] [PubMed] [Google Scholar]
- (222).Martin JC, Tippie MA, McGee DPC, Verheyden JPH. J. Pharm. Sci. 1987;76:180. doi: 10.1002/jps.2600760221. [DOI] [PubMed] [Google Scholar]
- (223).Powell MF, Magill A, Chu N, Hama K, Mau CI, Foster L, Bergstrom R. Pharmaceut. Res. 1991;8:1418. doi: 10.1023/a:1015809408908. [DOI] [PubMed] [Google Scholar]
- (224).Moore RE. Dicarbamate Derivatives of Alkyladamantanes. U. S. Patent 3450744. 1969 Jun 17;
- (225).Scherm A, Peteri D. Virucidal N-(1-adamantyl)-2-(aminoethoxy)acetamides. Ger. Offen. DE2316839. 1974 Oct 17; [Google Scholar]
- (226).Peteri D, Sterner W. Arznei-Forschung. 1973;23:577. [PubMed] [Google Scholar]
- (227).May G, Peteri D. Arzneimittel-Forsch. 1973;23:718. [PubMed] [Google Scholar]
- (228).Klimochkin YN, Moiseev IK, Boreko EI, Vladyko GV, Korobchenko LV. Pharm. Chem. J. 1989;23:304. [Google Scholar]
- (229).Makarova NV, Boreko EI, Moiseev IK, Pavlova NI, Nikolaeva SN, Zemtsova MN, Vladyko GV. Pharm. Chem. J. 2002;36:3. [Google Scholar]
- (230).Motornaya A, Alimbarova L, Shokova É, Kovalev V. Pharm. Chem. J. 2006;40:68. [Google Scholar]
- (231).Galbraith AW. Brit. Med. J. 1973;4:693. doi: 10.1136/bmj.4.5894.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (232).Diezel W, Michel G, Gortelmeyer R, Ostheimer KE. Arznei-Forschung. 1993;43:491. [PubMed] [Google Scholar]
- (233).Ostheimer KE, Busch T, Gortelmeyer R, Hahn KD. Arznei-Forschung. 1989;39:1152. [PubMed] [Google Scholar]
- (234).Kousoulas KG, Person S, Holland TC. J. Virol. 1978;27:505. doi: 10.1128/jvi.27.3.505-512.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (235).Koppel C, Tenczer J, Rutten E, Klaschka F. Biomed. Mass. Spectrom. 1985;12:487. [PubMed] [Google Scholar]
- (236).Cheetham JJ, Epand RM. Bioscience Rep. 1987;7:225. doi: 10.1007/BF01124793. [DOI] [PubMed] [Google Scholar]
- (237).Richards ML, Lio SC, Sinha A, Tieu KK, Sircar JC. J. Med. Chem. 2004;47:6451. doi: 10.1021/jm049288j. [DOI] [PubMed] [Google Scholar]
- (238).Richards ML, Lio SC, Sinha A, Banie H, Thomas RJ, Major M, Tanji M, Sircar JC. Eur. J. Med. Chem. 2006;41:950. doi: 10.1016/j.ejmech.2006.03.014. [DOI] [PubMed] [Google Scholar]
- (239).Banie H, Sinha A, Thomas RJ, Sircar JC, Richards ML. J. Med. Chem. 2007;50:5984. doi: 10.1021/jm0704907. [DOI] [PubMed] [Google Scholar]
- (240).Mibu N, Yokomizo K, Miyata T, Sumoto K. Chem. Pharm. Bull. 2007;55:1406. doi: 10.1248/cpb.55.1406. [DOI] [PubMed] [Google Scholar]
- (241).Mibu N, Yokomizo K, Oishi M, Miyata T, Sumoto K. Chem. Pharm. Bull. 2008;56:1052. doi: 10.1248/cpb.56.1052. [DOI] [PubMed] [Google Scholar]
- (242).Sarbach R, Mizoule J, Ricci C, Yavordios D. Cholagogically-active Adamantanecarboxylic Acids of Pyridoxines, Pyridoxamines, or Pyridoxals. Ger. Offen. 1920673B1. 1971 Apr 8; [Google Scholar]
- (243).Kinsolving CR, Georgiev VS. Esters of 2-adamantanone oxime. U. S. Patent 4486601. 1984 Dec 4;
- (244).Superti F, Seganti L, Orsi N, Divizia M, Gabrieli R, Pana A. Arch. Virol. 1987;96:289. doi: 10.1007/BF01320970. [DOI] [PubMed] [Google Scholar]
- (245).Crance JM, Biziagos E, Passagot J, van Cuyck-Gandre H, Deloince R. J. Med. Virol. 1990;31:155. doi: 10.1002/jmv.1890310214. [DOI] [PubMed] [Google Scholar]
- (246).Smith JP. Digest. Dis. Sci. 1997;42:1681. doi: 10.1023/a:1018857314351. [DOI] [PubMed] [Google Scholar]
- (247).Jubin R, Murray MG, Howe AY, Butkiewicz N, Hong Z, Lau JY. J. Infect. Dis. 2000;181:331. doi: 10.1086/315175. [DOI] [PubMed] [Google Scholar]
- (248).Craxi A, Lo Lacono O. J. Hepatol. 2001;35:527. doi: 10.1016/S0168-8278(01)00184-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (249).Mangia A, Leandro G, Helbling B, Renner EL, Tabone M, Sidoli L, Caronia S, Foster GR, Zeuzem S, Berg T, Di Marco V, Cino N, Andriulli A. J. Hepatol. 2004;40:478. doi: 10.1016/j.jhep.2003.11.002. [DOI] [PubMed] [Google Scholar]
- (250).Deltenre P, Henrion J, Canva V, Dharancy S, Texier F, Louvet A, De Maeght S, Paris JC, Mathurin P. J. Hepatol. 2004;41:462. doi: 10.1016/j.jhep.2004.05.019. [DOI] [PubMed] [Google Scholar]
- (251).Mangia A, Ricci GL, Persico M, Minerva N, Carretta V, Bacca D, Cela M, Piattelli M, Annese M, Maio G, Conte D, Guadagnino V, Pazienza V, Festi D, Spirito F, Andriulli A. J. Viral Hepatitis. 2005;12:292. doi: 10.1111/j.1365-2893.2005.00591.x. [DOI] [PubMed] [Google Scholar]
- (252).Ferenci P, Formann E, Laferl H, Gschwantler M, Hackl F, Brunner H, Hubmann R, Datz C, Stauber R, Steindl-Munda P, Kessler HH, Klingler A, Gangl A. J. Hepatol. 2006;44:275. doi: 10.1016/j.jhep.2005.09.015. [DOI] [PubMed] [Google Scholar]
- (253).Angelico M, Koehler-Horst B, Piccolo P, Angelico F, Gentile S, Francioso S, Tarquini P, Vecchia RD, Ponti L, Pilleri G, Barlattani A, Grieco A, Soccorsi F, Guarascio P, Demelia L, Sorbello O, Rossi Z, Forlini G, Zaru S, Bandiera F. Eur. J. Gastroen. Hepatol. 2008;20:680. doi: 10.1097/MEG.0b013e3282f5196c. [DOI] [PubMed] [Google Scholar]
- (254).von Wagner M, Hofmann WP, Teuber G, Berg T, Goeser T, Spengler U, Hinrichsen H, Weidenbach H, Gerken G, Manns M, Buggisch P, Herrmann E, Zeuzem S. Hepatology. 2008;48:1404. doi: 10.1002/hep.22483. [DOI] [PubMed] [Google Scholar]
- (255).Maynard M, Pradat P, Bailly F, Rozier F, Nemoz C, Si Ahmed SN, Adeleine P, Trepo C. J. Hepatol. 2006;44:484. doi: 10.1016/j.jhep.2005.11.038. [DOI] [PubMed] [Google Scholar]
- (256).Salmeron J, Diago M, Andrade R, Perez R, Sola R, Romero M, de la Mata M, Granados R, Ruiz-Extremera A, Munoz de Rueda P. J. Viral Hepatitis. 2007;14:89. doi: 10.1111/j.1365-2893.2006.00771.x. [DOI] [PubMed] [Google Scholar]
- (257).Muzi F, Orlando G, Ielpo B, Anselmo A, Sabato Ceraldi S, de Liguori Carino N, Manzia T, D'Andria D, Tariciotti L, Angelico M, Tisone G. Transplant. P. 2005;37:1705. doi: 10.1016/j.transproceed.2005.03.077. [DOI] [PubMed] [Google Scholar]
- (258).Kronenberger B, Berg T, Herrmann E, Hinrichsen H, Gerlach T, Buggisch P, Spengler U, Goeser T, Nasser S, Wursthorn K, Pape GR, Hopf U, Zeuzem S. Eur. J. Gastroen. Hepatol. 2007;19:639. doi: 10.1097/MEG.0b013e3281ac20ca. [DOI] [PubMed] [Google Scholar]
- (259).Mendez-Navarro J, Chirino RA, Corey KE, Gorospe EC, Zheng H, Moran S, Juarez JA, Chung RT, Dehesa-Violante M. Digest. Dis. Sci. 2010;55:2629. doi: 10.1007/s10620-009-1062-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (260).Piccolo P. Digest. Liver Dis. 2010;42:468. doi: 10.1016/j.dld.2010.04.009. [DOI] [PubMed] [Google Scholar]
- (261).Griffin SD, Beales LP, Clarke DS, Worsfold O, Evans SD, Jaeger J, Harris MP, Rowlands DJ. FEBS Lett. 2003;535:34. doi: 10.1016/s0014-5793(02)03851-6. [DOI] [PubMed] [Google Scholar]
- (262).Pavlovic D, Neville DCA, Argaud O, Blumberg B, Dwek RA, Fischer WB, Zitzmann N. Proc. Natl. Acad. Sci. U.S.A. 2003;100:6104. doi: 10.1073/pnas.1031527100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (263).Clarke D, Griffin S, Beales L, Gelais CS, Burgess S, Harris M, Rowlands D. J. Biol. Chem. 2006;281:37057. doi: 10.1074/jbc.M602434200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (264).Griffin SD, Harvey R, Clarke DS, Barclay WS, Harris M, Rowlands DJ. J. Gen. Virol. 2004;85:451. doi: 10.1099/vir.0.19634-0. [DOI] [PubMed] [Google Scholar]
- (265).Patargias G, Zitzmann N, Dwek R, Fischer WB. J. Med. Chem. 2006;49:648. doi: 10.1021/jm050721e. [DOI] [PubMed] [Google Scholar]
- (266).Cook GA, Stefer S, Opella SJ. Biopolymers. 2011;96:32. doi: 10.1002/bip.21453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (267).Cook GA, Opella SJ. Eur. Biophys. J. 2010;39:1097. doi: 10.1007/s00249-009-0533-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (268).Wozniak AL, Griffin S, Rowlands D, Harris M, Yi M, Lemon SM, Weinman SA. PLoS Pathog. 2010;6:e1001087. doi: 10.1371/journal.ppat.1001087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (269).Foster TL, Verow M, Wozniak AL, Bentham MJ, Thompson J, Atkins E, Weinman SA, Fishwick C, Foster R, Harris M, Griffin S. Hepatology. 2011;54:79. doi: 10.1002/hep.24371. [DOI] [PubMed] [Google Scholar]
- (270).Haqshenas G, Dong X, Ewart G, Bowden S, Gowans EJ. Virology. 2007;360:17. doi: 10.1016/j.virol.2006.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (271).Steinmann E, Whitfield T, Kallis S, Dwek RA, Zitzmann N, Pietschmann T, Bartenschlager R. Hepatology. 2007;46:330. doi: 10.1002/hep.21686. [DOI] [PubMed] [Google Scholar]
- (272).StGelais C, Tuthill TJ, Clarke DS, Rowlands DJ, Harris M, Griffin S. Antivir. Res. 2007;76:48. doi: 10.1016/j.antiviral.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (273).Griffin S, Stgelais C, Owsianka AM, Patel AH, Rowlands D, Harris M. Hepatology. 2008;48:1779. doi: 10.1002/hep.22555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (274).Griffin S, Trowbridge R, Thommes P, Parry N, Rowlands D, Harris M, Bright H. J. Hepatol. 2008;49:908. doi: 10.1016/j.jhep.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (275).Wagner CE, Mohler ML, Kang GS, Miller DD, Geisert EE, Chang YA, Fleischer EB, Shea KJ. J. Med. Chem. 2003;46:2823. doi: 10.1021/jm020326d. [DOI] [PubMed] [Google Scholar]
- (276).Kagan BL. Western J. Med. 1987;146:234. [PMC free article] [PubMed] [Google Scholar]
- (277).McClure MO, Marsh M, Weiss RA. EMBO J. 1988;7:513. doi: 10.1002/j.1460-2075.1988.tb02839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (278).Rytik PG, Eremin VF, Kvacheva ZB, Poleschuk NN, Popov SA, Schroder HC, Bachmann M, Weiler BE, Muller WE. AIDS Res. Hum. Retroviruses. 1991;7:89. doi: 10.1089/aid.1991.7.89. [DOI] [PubMed] [Google Scholar]
- (279).Muller WEG, Schroder HC, Ushijima H, Dapper J, Bormann J. Eur. J. Pharm.-Molec. Ph. 1992;226:209. doi: 10.1016/0922-4106(92)90063-2. [DOI] [PubMed] [Google Scholar]
- (280).Vamecq J, Van derpoorten K, Poupaert JH, Balzarini J, De Clercq E, Stables JP. Life Sci. 1998;63:PL267. doi: 10.1016/s0024-3205(98)00445-7. [DOI] [PubMed] [Google Scholar]
- (281).Sinha SK, Sizemore RC, Gottlieb AA. Nat. Biotechnol. 1988;6:810. [Google Scholar]
- (282).Epand RM, Epand RF, McKenzie RC. J. Biol. Chem. 1987;262:1526. [PubMed] [Google Scholar]
- (283).Burstein ME, Serbin AV, Khakhulina TV, Alymova IV, Stotskaya LL, Bogdan OP, Manukchina EE, Jdanov VV, Sharova NK, Bukrinskaya AG. Antivir. Res. 1999;41:135. doi: 10.1016/s0166-3542(99)00006-6. [DOI] [PubMed] [Google Scholar]
- (284).Rybalko S, Nesterova N, Diadiun S, Danylenko G, Danylenko V, Guzhova S, Maksimov Y, Arkadiev V, Ivans'ka N, Maksymenok O, Vrnycianu N, Zhtrebtsova E, Grygoreva T. Acta Biochim. Pol. 2001;48:241. [PubMed] [Google Scholar]
- (285).El-Sherbeny MA. Arch. Pharm. 2000;333:323. doi: 10.1002/1521-4184(200010)333:10<323::aid-ardp323>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- (286).Lipton SA. Neurology. 1992;42:1403. doi: 10.1212/wnl.42.7.1403. [DOI] [PubMed] [Google Scholar]
- (287).Raber J, Toggas SM, Lee S, Bloom FE, Epstein CJ, Mucke L. Virology. 1996;226:362. doi: 10.1006/viro.1996.0664. [DOI] [PubMed] [Google Scholar]
- (288).Toggas SM, Masliah E, Mucke L. Brain Res. 1996;706:303. doi: 10.1016/0006-8993(95)01197-8. [DOI] [PubMed] [Google Scholar]
- (289).Nath A, Haughey NJ, Jones M, Anderson C, Bell JE, Geiger JD. Ann. Neurol. 2000;47:186. [PubMed] [Google Scholar]
- (290).Anderson ER, Gendelman HE, Xiong HG. J. Neurosci. 2004;24:7194. doi: 10.1523/JNEUROSCI.1933-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (291).Schifitto G, Yiannoutsos CT, Simpson DM, Marra CM, Singer EJ, Kolson DL, Nath A, Berger JR, Navia B. J. Neurovirol. 2006;12:328. doi: 10.1080/13550280600873835. [DOI] [PubMed] [Google Scholar]
- (292).Schifitto G, Navia BA, Yiannoutsos CT, Marra CM, Chang L, Ernst T, Jarvik JG, Miller EN, Singer EJ, Ellis RJ, Kolson DL, Simpson D, Nath A, Berger J, Shriver SL, Millar LL, Colquhoun D, Lenkinski R, Gonzalez RG, Lipton SA. AIDS. 2007;21:1877. doi: 10.1097/QAD.0b013e32813384e8. [DOI] [PubMed] [Google Scholar]
- (293).Zhao Y, Navia BA, Marra CM, Singer EJ, Chang L, Berger J, Ellis RJ, Kolson DL, Simpson D, Miller EN, Lipton SA, Evans SR, Schifitto G, ACTG AACTG. HIV Clin. Trials. 2010;11:59. doi: 10.1310/hct1101-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (294).Tsuzuki N, Hama T, Kawada M, Hasui A, Konishi R, Shiwa S, Ochi Y, Futaki S, Kitagawa K. J. Pharm. Sci. 1994;83:481. doi: 10.1002/jps.2600830407. [DOI] [PubMed] [Google Scholar]
- (295).Balzarini J, De Clercq E, Kaminska B, Orzeszko A. Antivir. Chem. Chemoth. 2003;14:139. doi: 10.1177/095632020301400303. [DOI] [PubMed] [Google Scholar]
- (296).Cushman M, Golebiewski WM, Pommier Y, Mazunder A, Reymen D, De Clercq E, Grahm L, Rice WG. J. Med. Chem. 1995;38:443. doi: 10.1021/jm00003a007. [DOI] [PubMed] [Google Scholar]
- (297).Uckun FM, Mao C, Pendergrass S, Maher D, Zhu D, Tuel-Ahlgren L, Venkatachalam TK. Bioorg. Med. Chem. Lett. 1999;9:2721. doi: 10.1016/s0960-894x(99)00460-6. [DOI] [PubMed] [Google Scholar]
- (298).Balzarini J, Orzeszko B, Maurin JK, Orzeszko A. Eur. J. Med. Chem. 2007;42:993. doi: 10.1016/j.ejmech.2007.01.003. [DOI] [PubMed] [Google Scholar]
- (299).Balzarini J, Orzeszko-Krzesinska B, Maurin JK, Orzeszko A. Eur. J. Med. Chem. 2009;44:303. doi: 10.1016/j.ejmech.2008.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (300).Gonzalez ME, Carrasco L. FEBS Lett. 2003;552:28. doi: 10.1016/s0014-5793(03)00780-4. [DOI] [PubMed] [Google Scholar]
- (301).Fischer WB, Sansom MSP. BBA-Biomembranes. 2002;1561:27. doi: 10.1016/s0304-4157(01)00009-0. [DOI] [PubMed] [Google Scholar]
- (302).Sansom MS, Forrest LR, Bull R. Bioessays. 1998;20:992. doi: 10.1002/(SICI)1521-1878(199812)20:12<992::AID-BIES5>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- (303).Hout DR, Gomez ML, Pacyniak E, Gomez LM, Fegley B, Mulcahy ER, Hill MS, Culley N, Pinson DM, Nothnick W, Powers MF, Wong SW, Stephens EB. Virology. 2006;344:541. doi: 10.1016/j.virol.2005.08.022. [DOI] [PubMed] [Google Scholar]
- (304).Hout DR, Gomez LM, Pacyniak E, Miller JM, Hill MS, Stephens EB. Virology. 2006;348:449. doi: 10.1016/j.virol.2005.12.025. [DOI] [PubMed] [Google Scholar]
- (305).Park SH, Opella SJ. Protein Sci. 2007;16:2205. doi: 10.1110/ps.073041107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (306).Fredericksen BL, Wei BL, Yao J, Luo T, Garcia JV. J. Virol. 2002;76:11440. doi: 10.1128/JVI.76.22.11440-11446.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (307).Mahfoud R, Mylvaganam M, Lingwood CA, Fantini J. J. Lipid Res. 2002;43:1670. doi: 10.1194/jlr.m200165-jlr200. [DOI] [PubMed] [Google Scholar]
- (308).Fantini J, Hammache D, Delezay O, Yahi N, Andre-Barres C, Rico-Lattes I, Lattes A. J. Biol. Chem. 1997;272:7245. doi: 10.1074/jbc.272.11.7245. [DOI] [PubMed] [Google Scholar]
- (309).Lund N, Branch DR, Mylvaganam M, Chark D, Ma XZ, Sakac D, Binnington B, Fantini J, Puri A, Blumenthal R, Lingwood CA. AIDS. 2006;20:333. doi: 10.1097/01.aids.0000206499.78664.58. [DOI] [PubMed] [Google Scholar]
- (310).De Rosa MF, Ackerley C, Wang B, Ito S, Clarke DM, Lingwood C. J. Biol. Chem. 2008;283:4501. doi: 10.1074/jbc.M705473200. [DOI] [PubMed] [Google Scholar]
- (311).Balzarini J, Pannecouque C, De Clercq E, Pavlov AY, Printsevskaya SS, Miroshnikova OV, Reznikova MI, Preobrazhenskaya MN. J. Med. Chem. 2003;46:2755. doi: 10.1021/jm0300882. [DOI] [PubMed] [Google Scholar]
- (312).Solaja BA, Opsenica D, Smith KS, Milhous WK, Terzic N, Opsenica I, Burnett JC, Nuss J, Gussio R, Bavari S. J. Med. Chem. 2008;51:4388. doi: 10.1021/jm800737y. [DOI] [PubMed] [Google Scholar]
- (313).Singh C, Chaudhary S, Puri SK. J. Med. Chem. 2006;49:7227. doi: 10.1021/jm060826x. [DOI] [PubMed] [Google Scholar]
- (314).Singh C, Chaudhary S, Puri SK. Bioorg. Med. Chem. Lett. 2008;18:1436. doi: 10.1016/j.bmcl.2007.12.074. [DOI] [PubMed] [Google Scholar]
- (315).Wells TN, Alonso PL, Gutteridge WE. Nat. Rev. Drug Discov. 2009;8:879. doi: 10.1038/nrd2972. [DOI] [PubMed] [Google Scholar]
- (316).Kappe SH, Vaughan AM, Boddey JA, Cowman AF. Science. 2010;328:862. doi: 10.1126/science.1184785. [DOI] [PubMed] [Google Scholar]
- (317).McColm AA, Hommel M, Trigg PI. Mol. Biochem. Parasit. 1980;1:119. doi: 10.1016/0166-6851(80)90006-7. [DOI] [PubMed] [Google Scholar]
- (318).Evans SG, Havlik I. Biochem. Pharmacol. 1993;45:1168. doi: 10.1016/0006-2952(93)90264-w. [DOI] [PubMed] [Google Scholar]
- (319).Ginsburg H. Parasitol .Today. 1990;6:334. doi: 10.1016/0169-4758(90)90178-7. [DOI] [PubMed] [Google Scholar]
- (320).Evans SG, Havlik I. T. Roy. Soc. Trop. Med. H. 1994;88:683. doi: 10.1016/0035-9203(94)90229-1. [DOI] [PubMed] [Google Scholar]
- (321).Evans SG, Havlik I. Am. J. Trop. Med. Hyg. 1996;54:232. doi: 10.4269/ajtmh.1996.54.232. [DOI] [PubMed] [Google Scholar]
- (322).Johnson DJ, Fidock DA, Mungthin M, Lakshmanan V, Sidhu ABS, Bray PG, Ward SA. Mol. Cell. 2004;15:867. doi: 10.1016/j.molcel.2004.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (323).Wellems TE. Nat. Med. 2004;10:1169. doi: 10.1038/nm1104-1169. [DOI] [PubMed] [Google Scholar]
- (324).Singh C, Misra D, Saxena G, Chandra S. Bioorg. Med. Chem. Lett. 1992;2:497. [Google Scholar]
- (325).Vennerstrom JL, Arbe-Barnes S, Brun R, Charman SA, Chiu FCK, Chollet J, Dong Y, Dorn A, Hunziker D, Matile H, McIntosh K, Padmanilayam M, Santo Tomas J, Scheurer C, Scorneaux B, Tang Y, Urwyler H, Wittlin S, Charman WN. Nature. 2004;430:900. doi: 10.1038/nature02779. [DOI] [PubMed] [Google Scholar]
- (326).O'Neill PM, Posner GH. J. Med. Chem. 2004;47:2945. doi: 10.1021/jm030571c. [DOI] [PubMed] [Google Scholar]
- (327).Posner GH, O'Neill PM. Accounts Chem. Res. 2004;37:397. doi: 10.1021/ar020227u. [DOI] [PubMed] [Google Scholar]
- (328).Griesbaum K, Liu XJ, Dong YX. Tetrahedron. 1997;53:5463. [Google Scholar]
- (329).Griesbeck AG, El-Idreesy TT, Hoinck LO, Lex J, Brun R. Bioorg. Med. Chem. Lett. 2005;15:595. doi: 10.1016/j.bmcl.2004.11.043. [DOI] [PubMed] [Google Scholar]
- (330).Tang YQ, Dong YX, Karle JM, DiTusa CA, Vennerstrom JL. J. Org. Chem. 2004;69:6470. doi: 10.1021/jo040171c. [DOI] [PubMed] [Google Scholar]
- (331).Dong YX, Tang YQ, Chollet J, Matile H, Wittlin S, Charman SA, Charman WN, Tomas JS, Scheurer C, Snyder C, Scorneaux B, Bajpai S, Alexander SA, Wang XF, Padmanilayam M, Cheruku SR, Brun R, Vennerstrom JL. Bioorgan. Med. Chem. 2006;14:6368. doi: 10.1016/j.bmc.2006.05.041. [DOI] [PubMed] [Google Scholar]
- (332).Tang Y, Dong Y, Wang X, Sriraghavan K, Wood JK, Vennerstrom JL. J. Org. Chem. 2005;70:5103. doi: 10.1021/jo050385+. [DOI] [PubMed] [Google Scholar]
- (333).Perry CS, Charman SA, Prankerd RJ, Chiu FC, Scanlon MJ, Chalmers D, Charman WN. J. Pharm. Sci. 2006;95:146. doi: 10.1002/jps.20525. [DOI] [PubMed] [Google Scholar]
- (334).Creek DJ, Charman WN, Chiu FC, Prankerd RJ, McCullough KJ, Dong Y, Vennerstrom JL, Charman SA. J. Pharm. Sci. 2007;96:2945. doi: 10.1002/jps.20958. [DOI] [PubMed] [Google Scholar]
- (335).Zhou L, Alker A, Ruf A, Wang X, Chiu FC, Morizzi J, Charman SA, Charman WN, Scheurer C, Wittlin S, Dong Y, Hunziker D, Vennerstrom JL. Bioorg. Med. Chem. Lett. 2008;18:1555. doi: 10.1016/j.bmcl.2008.01.087. [DOI] [PubMed] [Google Scholar]
- (336).Dong Y, Wittlin S, Sriraghavan K, Chollet J, Charman SA, Charman WN, Scheurer C, Urwyler H, Santo Tomas J, Snyder C, Creek DJ, Morizzi J, Koltun M, Matile H, Wang X, Padmanilayam M, Tang Y, Dorn A, Brun R, Vennerstrom JL. J. Med. Chem. 2010;53:481. doi: 10.1021/jm901473s. [DOI] [PubMed] [Google Scholar]
- (337).Olliaro P, Wells TN. Clin. Pharmacol. Ther. 2009;85:584. doi: 10.1038/clpt.2009.51. [DOI] [PubMed] [Google Scholar]
- (338).Maerki S, Brun R, Charman SA, Dorn A, Matile H, Wittlin S. J. Antimicrob. Chemoth. 2006;58:52. doi: 10.1093/jac/dkl209. [DOI] [PubMed] [Google Scholar]
- (339).Kocken CH, van der Wel A, Arbe-Barnes S, Brun R, Matile H, Scheurer C, Wittlin S, Thomas AW. Exp. Parasitol. 2006;113:197. doi: 10.1016/j.exppara.2005.12.016. [DOI] [PubMed] [Google Scholar]
- (340).Osorio L, Murillo C, Aponte S, Mayagaya V, Scheurer C, Brun R, Matile H, Wittlin S. Am. J. Trop. Med. Hyg. 2007;76:1024. [PubMed] [Google Scholar]
- (341).Kaiser M, Wittlin S, Nehrbass-Stuedli A, Dong Y, Wang X, Hemphill A, Matile H, Brun R, Vennerstrom JL. Antimicrob. Agents Ch. 2007;51:2991. doi: 10.1128/AAC.00225-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (342).Uhlemann AC, Wittlin S, Matile H, Bustamante LY, Krishna S. Antimicrob. Agents Ch. 2007;51:667. doi: 10.1128/AAC.01064-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (343).Snyder C, Chollet J, Santo-Tomas J, Scheurer C, Wittlin S. Exp. Parasitol. 2007;115:296. doi: 10.1016/j.exppara.2006.09.016. [DOI] [PubMed] [Google Scholar]
- (344).Hofer S, Brun R, Maerki S, Matile H, Scheurer C, Wittlin S. J. Antimicrob. Chemoth. 2008;62:1061. doi: 10.1093/jac/dkn315. [DOI] [PubMed] [Google Scholar]
- (345).Fugi MA, Wittlin S, Dong Y, Vennerstrom JL. Antimicrob. Agents Ch. 2010;54:1042. doi: 10.1128/AAC.01305-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (346).Singh C, Kanchan R, Sharma U, Puri SK. J. Med. Chem. 2007;50:521. doi: 10.1021/jm0610043. [DOI] [PubMed] [Google Scholar]
- (347).Tripathi R, Mishra D, Rizvi A, Singh C. Life Sci. 2007;81:1544. doi: 10.1016/j.lfs.2007.09.023. [DOI] [PubMed] [Google Scholar]
- (348).Charman SA, Arbe-Barnes S, Bathurst IC, Brun R, Campbell M, Charman WN, Chiu FC, Chollet J, Craft JC, Creek DJ, Dong Y, Matile H, Maurer M, Morizzi J, Nguyen T, Papastogiannidis P, Scheurer C, Shackleford DM, Sriraghavan K, Stingelin L, Tang Y, Urwyler H, Wang X, White KL, Wittlin S, Zhou L, Vennerstrom JL. Proc. Natl. Acad. Sci. U.S.A. 2011;108:4400. doi: 10.1073/pnas.1015762108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (349).Griesbeck AG, Schlundt V. Synlett. 2011:2430. [Google Scholar]
- (350).Barton V, Ward SA, Chadwick J, Hill A, O'Neill PM. J. Med. Chem. 2010;53:4555. doi: 10.1021/jm100201j. [DOI] [PubMed] [Google Scholar]
- (351).Hartwig CL, Lauterwasser EMW, Mahajan SS, Hoke JM, Cooper RA, Renslo AR. J. Med. Chem. 2011;54:8207. doi: 10.1021/jm2012003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (352).Park BK, O'Neill PM, Maggs JL, Pirmohamed M. Brit. J. Clin. Pharmaco. 1998;46:521. doi: 10.1046/j.1365-2125.1998.00838.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (353).Barrett MP, Boykin DW, Brun R, Tidwell RR. Brit. J. Pharmacol. 2007;152:1155. doi: 10.1038/sj.bjp.0707354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (354).Rodgers J. J. Neuroimmunol. 2009;211:16. doi: 10.1016/j.jneuroim.2009.02.007. [DOI] [PubMed] [Google Scholar]
- (355).Kelly JM, Miles MA, Skinner AC. Antimicrob. Agents Ch. 1999;43:985. doi: 10.1128/aac.43.4.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (356).Kelly JM, Quack G, Miles MM. Antimicrob. Agents Ch. 2001;45:1360. doi: 10.1128/AAC.45.5.1360-1366.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (357).Papanastasiou I, Foscolos GB, Tsotinis A, Olah J, Ovadi J, Prathalingam SR, Kelly JM. Heterocycles. 2008;75:2043. [Google Scholar]
- (358).Papanastasiou I, Tsotinis A, Foscolos GB, Prathalingam SR, Kelly JM. J. Heterocyclic Chem. 2008;45:1401. [Google Scholar]
- (359).Papanastasiou I, Tsotinis A, Kolocouris N, Prathalingam SR, Kelly JM. J. Med. Chem. 2008;51:1496. doi: 10.1021/jm7014292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (360).Papanastasiou I, Tsotinis A, Zoidis G, Kolocouris N, Prathalingam SR, Kelly JM. ChemMedChem. 2009;4:1059. doi: 10.1002/cmdc.200900019. [DOI] [PubMed] [Google Scholar]
- (361).Duque MD, Camps P, Profire L, Montaner S, Vazquez S, Sureda FX, Mallol J, Lopez-Querol M, Naesens L, De Clercq E, Prathalingam SR, Kelly JM. Bioorgan. Med. Chem. 2009;17:3198. doi: 10.1016/j.bmc.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (362).Duque MD, Camps P, Torres E, Valverde E, Sureda FX, Lopez-Querol M, Camins A, Prathalingam SR, Kelly JM, Vazquez S. Bioorgan. Med. Chem. 2010;18:46. doi: 10.1016/j.bmc.2009.11.017. [DOI] [PubMed] [Google Scholar]
- (363).Zoidis G, Kolocouris N, Kelly JM, Prathalingam SR, Naesens L, De Clercq E. Eur. J. Med. Chem. 2010;45:5022. doi: 10.1016/j.ejmech.2010.08.009. [DOI] [PubMed] [Google Scholar]
- (364).Fytas C, Zoidis G, Tzoutzas N, Taylor MC, Fytas G, Kelly JM. J. Med. Chem. 2011;54:5250. doi: 10.1021/jm200217m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (365).Parkes JD, Calver DM, Zilkha KJ, Knill-Jones RP. Lancet. 1970;1:259. doi: 10.1016/s0140-6736(70)90634-3. [DOI] [PubMed] [Google Scholar]
- (366).Dallos V, Heathfield K, Stone P, Allen FA. Brit. Med. J. 1970;4:24. doi: 10.1136/bmj.4.5726.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (367).Fieschi C, Nardini M, Casacchia M, Tedone ME. Lancet. 1970;1:945. doi: 10.1016/s0140-6736(70)91065-2. [DOI] [PubMed] [Google Scholar]
- (368).Schwab RS, Poskanzer DC, England AC, Jr., Young RR. J. Am. Med. Assoc. 1972;222:792. doi: 10.1001/jama.222.7.792. [DOI] [PubMed] [Google Scholar]
- (369).Evidente VGH, Adler CH, Caviness JN, Gwinn-Hardy K. Clinical Neuropharmacology. 1999;22:30. doi: 10.1097/00002826-199901000-00006. [DOI] [PubMed] [Google Scholar]
- (370).Shannon KM, Goetz CG, Carroll VS, Tanner CM, Klawans HL. Clin. Neuropharmacol. 1987;10:522. doi: 10.1097/00002826-198712000-00003. [DOI] [PubMed] [Google Scholar]
- (371).Jorg J, Profrock A. Nervenheilkunde. 1995;14:76. [Google Scholar]
- (372).Cates LA, Gallio RL, Cramer MB. J. Pharm. Sci. 1973;62:1719. doi: 10.1002/jps.2600621034. [DOI] [PubMed] [Google Scholar]
- (373).Carlsson A, Lindqvist M, Magnusson TOR. Nature. 1957;180:1200. doi: 10.1038/1801200a0. [DOI] [PubMed] [Google Scholar]
- (374).Chakrabarti JK, Foulis MJ, Hotten TM, Szinai SS, Todd A. J. Med. Chem. 1974;17:602. doi: 10.1021/jm00252a007. [DOI] [PubMed] [Google Scholar]
- (375).Scherm A, Peteri D. 3,5-Dialkyl-1-aminoadamantanes for the treatment of parkinsonism. Ger. Offen. 1974 Oct 31; DE 2318461 A1. [Google Scholar]
- (376).Chakrabarti JK, Hotten TM, Sutton S, Tupper DE. J. Med. Chem. 1976;19:967. doi: 10.1021/jm00229a022. [DOI] [PubMed] [Google Scholar]
- (377).Henkel JG, Hane JT, Gianutsos G. J. Med. Chem. 1982;25:51. doi: 10.1021/jm00343a010. [DOI] [PubMed] [Google Scholar]
- (378).Dunn JP, Henkel JG, Gianutsos G. J. Pharm. Pharmacol. 1986;38:353. doi: 10.1111/j.2042-7158.1986.tb04586.x. [DOI] [PubMed] [Google Scholar]
- (379).Bailey EV, Stone TW. Arch. Int. Pharmacod. T. 1975;216:246. [PubMed] [Google Scholar]
- (380).Kornhuber J, Bormann J, Hubers M, Rusche K, Riederer P. Eur. J. Pharmacol. 1991;206:297. doi: 10.1016/0922-4106(91)90113-v. [DOI] [PubMed] [Google Scholar]
- (381).Nerobkova LN, Val'dman EA, Voronina TA, Markina NV, Sharkova LM. Eksp. Klin. Farm. 2000;63:3. [PubMed] [Google Scholar]
- (382).Skolimowski J, Kochman A, Gebicka L, Metodiewa D. Bioorgan. Med. Chem. 2003;11:3529. doi: 10.1016/s0968-0896(03)00299-2. [DOI] [PubMed] [Google Scholar]
- (383).Nakazono M, Hasegawa S, Yamamoto T, Zaitsu K. Bioorg. Med. Chem. Lett. 2004;14:5619. doi: 10.1016/j.bmcl.2004.08.053. [DOI] [PubMed] [Google Scholar]
- (384).Sarre S, Lanza M, Makovec F, Artusi R, Caselli G, Michotte Y. Eur. J. Pharmacol. 2008;584:297. doi: 10.1016/j.ejphar.2008.02.027. [DOI] [PubMed] [Google Scholar]
- (385).Losi G, Lanza M, Makovec F, Artusi R, Caselli G, Puia G. Neuropharmacology. 2006;50:277. doi: 10.1016/j.neuropharm.2005.09.002. [DOI] [PubMed] [Google Scholar]
- (386).Ehringer H, Hornykiewicz O. Klin. Wochenschr. 1960;38:1236. doi: 10.1007/BF01485901. [DOI] [PubMed] [Google Scholar]
- (387).Cox B, Tha SJ. Eur. J. Pharmacol. 1975;30:344. doi: 10.1016/0014-2999(75)90119-3. [DOI] [PubMed] [Google Scholar]
- (388).Grelak RP, Clark R, Stump JM, Vernier VG. Science. 1970;169:203. doi: 10.1126/science.169.3941.203. [DOI] [PubMed] [Google Scholar]
- (389).Scatton B, Cheramy A, Besson MJ, Glowinski J. Eur. J. Pharmacol. 1970;13:131. doi: 10.1016/0014-2999(70)90194-9. [DOI] [PubMed] [Google Scholar]
- (390).Farnebo LO, Fuxe K, Goldstein M, Hamberger B, Ungerstedt U. Eur. J. Pharmacol. 1971;16:27. doi: 10.1016/0014-2999(71)90053-7. [DOI] [PubMed] [Google Scholar]
- (391).Heimans RL, Rand MJ, Fennessy MR. J. Pharm. Pharmacol. 1972;24:875. doi: 10.1111/j.2042-7158.1972.tb08906.x. [DOI] [PubMed] [Google Scholar]
- (392).Papeschi R. Neuropharmacology. 1974;13:77. doi: 10.1016/0028-3908(74)90009-4. [DOI] [PubMed] [Google Scholar]
- (393).Baldessarini RJ, Lipinski JF, Chace KV. Biochem. Pharmacol. 1972;21:77. doi: 10.1016/0006-2952(72)90252-3. [DOI] [PubMed] [Google Scholar]
- (394).Fletcher EA, Redfern PH. J. Pharm. Pharmacol. 1970;22:957. doi: 10.1111/j.2042-7158.1970.tb08486.x. [DOI] [PubMed] [Google Scholar]
- (395).Symchowicz S, Korduba CA, Veals J. Eur. J. Pharmacol. 1973;21:155. doi: 10.1016/0014-2999(73)90220-3. [DOI] [PubMed] [Google Scholar]
- (396).Danysz W, Parsons CG, Kornhuber J, Schmidt WJ, Quack G. Neurosci. Biobehav. R. 1997;21:455. doi: 10.1016/s0149-7634(96)00037-1. [DOI] [PubMed] [Google Scholar]
- (397).Vaatstra WJ, Eigeman L. Eur. J. Pharmacol. 1974;25:185. doi: 10.1016/0014-2999(74)90048-x. [DOI] [PubMed] [Google Scholar]
- (398).Wesemann W, Dette-Wildenhahn G, Fellehner HJ. Neural Transm. 1979;44:263. doi: 10.1007/BF01250322. [DOI] [PubMed] [Google Scholar]
- (399).Grossmann A, Grossmann W, Jurna I. Eur. J. Pharmacol. 1976;35:379. doi: 10.1016/0014-2999(76)90241-7. [DOI] [PubMed] [Google Scholar]
- (400).Reiser G, Binmoller FJ, Koch R. Brain Res. 1988;443:338. doi: 10.1016/0006-8993(88)91630-7. [DOI] [PubMed] [Google Scholar]
- (401).Turski L, Bressler K, Rettig KJ, Loschmann PA, Wachtel H. Nature. 1991;349:414. doi: 10.1038/349414a0. [DOI] [PubMed] [Google Scholar]
- (402).Greenamyre JT, O'Brien CF. Arch. Neurol. 1991;48:977. doi: 10.1001/archneur.1991.00530210109030. [DOI] [PubMed] [Google Scholar]
- (403).Kornhuber J, Bormann J, Retz W, Hubers M, Riederer P. Eur. J. Pharmacol. 1989;166:589. doi: 10.1016/0014-2999(89)90384-1. [DOI] [PubMed] [Google Scholar]
- (404).Bormann J. Eur. J. Pharmacol. 1989;166:591. doi: 10.1016/0014-2999(89)90385-3. [DOI] [PubMed] [Google Scholar]
- (405).Kornhuber J, Quack G. Neurosci. Lett. 1995;195:137. doi: 10.1016/0304-3940(95)11785-u. [DOI] [PubMed] [Google Scholar]
- (406).Kornhuber J, Quack G, Danysz W, Jellinger K, Danielczyk W, Gsell W, Riederer P. Neuropharmacology. 1995;34:713. doi: 10.1016/0028-3908(95)00056-c. [DOI] [PubMed] [Google Scholar]
- (407).Gianutsos G, Chute S, Dunn JP. Eur. J. Pharmacol. 1985;110:357. doi: 10.1016/0014-2999(85)90564-3. [DOI] [PubMed] [Google Scholar]
- (408).Rojas P, Altagracia M, Kravzov J, Rios C. Drug Develop. Res. 1993;29:222. [Google Scholar]
- (409).Aronstam RS, Eldefrawi AT, Eldefrawi ME. Biochem. Pharmacol. 1980;29:1311. doi: 10.1016/0006-2952(80)90292-0. [DOI] [PubMed] [Google Scholar]
- (410).Masuo K, Enomoto K, Maeno T. Eur. J. Pharmacol. 1986;130:187. doi: 10.1016/0014-2999(86)90267-0. [DOI] [PubMed] [Google Scholar]
- (411).Matsubayashi H, Swanson KL, Albuquerque EX. J. Pharmacol. Exp. Ther. 1997;281:834. [PubMed] [Google Scholar]
- (412).Tsai MC, Chen ML, Lo SC, Tsai GC. Eur. J. Pharmacol. 1989;160:133. doi: 10.1016/0014-2999(89)90662-6. [DOI] [PubMed] [Google Scholar]
- (413).Maj J. Arznei-Forschung. 1982;32:1256. [PubMed] [Google Scholar]
- (414).Moryl E, Danysz W, Quack G. Pharmacol. Toxicol. 1993;72:394. doi: 10.1111/j.1600-0773.1993.tb01351.x. [DOI] [PubMed] [Google Scholar]
- (415).Stromberg U, Svensson TH. Acta Pharm. Toxicol. 1971;30:161. doi: 10.1111/j.1600-0773.1971.tb00646.x. [DOI] [PubMed] [Google Scholar]
- (416).Brown F, Redfern PH. Brit. J. Pharmacol. 1976;58:561. doi: 10.1111/j.1476-5381.1976.tb08624.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (417).Bubser M, Keseberg U, Notz PK, Schmidt WJ. Eur. J. Pharmacol. 1992;229:75. doi: 10.1016/0014-2999(92)90288-f. [DOI] [PubMed] [Google Scholar]
- (418).Von Voigtlander PF, Moore KE. J. Pharmacol. Exp. Ther. 1973;184:542. [PubMed] [Google Scholar]
- (419).Mistry R, Wilke R, Challiss RA. J. Brit. J. Pharmacol. 1995;114:797. doi: 10.1111/j.1476-5381.1995.tb13275.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (420).Quack G, Hesselink M, Danysz W, Spanagel RJ. Neural Transm.-Supp. 1995;46:97. [PubMed] [Google Scholar]
- (421).Spanagel R, Eilbacher B, Wilke R. Eur. J. Pharmacol. 1994;262:21. doi: 10.1016/0014-2999(94)90023-x. [DOI] [PubMed] [Google Scholar]
- (422).Parsons CG, Gruner R, Rozental J, Millar J, Lodge D. Neuropharmacology. 1993;32:1337. doi: 10.1016/0028-3908(93)90029-3. [DOI] [PubMed] [Google Scholar]
- (423).Parsons CG, Panchenko VA, Pinchenko VO, Tsyndrenko AY, Krishtal OA. Eur. J. Neurosci. 1996;8:446. doi: 10.1111/j.1460-9568.1996.tb01228.x. [DOI] [PubMed] [Google Scholar]
- (424).Lupp A, Lucking CH, Koch R, Jackisch R, Feuerstein TJ. J. Pharmacol. Exp. Ther. 1992;263:717. [PubMed] [Google Scholar]
- (425).Rohrbacher J, Bijak M, Misgeld U. Neurosci. Lett. 1994;182:95. doi: 10.1016/0304-3940(94)90215-1. [DOI] [PubMed] [Google Scholar]
- (426).Stoof JC, Booij J, Drukarch B, Wolters EC. Eur. J. Pharmacol. 1992;213:439. doi: 10.1016/0014-2999(92)90634-g. [DOI] [PubMed] [Google Scholar]
- (427).Danysz W, Gossel M, Zajaczkowski W, Dill D, Quack GJ. Neural Transm.-Park. 1994;7:155. doi: 10.1007/BF02253435. [DOI] [PubMed] [Google Scholar]
- (428).Kornhuber J, Weller M, Schoppmeyer K, Riederer PJ. Neural Transm.-Supp. 1994;43:91. [PubMed] [Google Scholar]
- (429).Porter RH, Greenamyre JT. J. Neurochem. 1995;64:614. doi: 10.1046/j.1471-4159.1995.64020614.x. [DOI] [PubMed] [Google Scholar]
- (430).Albuquerque EX, Pereira EFR, Braga MFM, Matsubayashi H, Alkondon M. Toxicol. Lett. 1998;103:211. doi: 10.1016/s0378-4274(98)00309-9. [DOI] [PubMed] [Google Scholar]
- (431).Uitti RJ, Rajput AH, Ahlskog JE, Offord KP, Schroeder DR, Ho MM, Prasad M, Rajput A, Basran P. Neurology. 1996;46:1551. doi: 10.1212/wnl.46.6.1551. [DOI] [PubMed] [Google Scholar]
- (432).Edby K, Larsson J, Eek M, Vonwendt L, Ostergard B. Child. Nerv. Syst. 1995;11:607. doi: 10.1007/BF00301001. [DOI] [PubMed] [Google Scholar]
- (433).Albrecht-Goepfert E, Schempp H, Elstner EF. Biochem. Pharmacol. 1998;56:141. doi: 10.1016/s0006-2952(98)00024-0. [DOI] [PubMed] [Google Scholar]
- (434).Drever BD, Anderson WG, Johnson H, O'Callaghan M, Seo S, Choi DY, Riedel G, Platt BJ. Alzheimers Dis. 2007;12:319. doi: 10.3233/jad-2007-12405. [DOI] [PubMed] [Google Scholar]
- (435).Busch AE, Karbach U, Miska D, Gorboulev V, Akhoundova A, Volk C, Arndt P, Ulzheimer JC, Sonders MS, Baumann C, Waldegger S, Lang F, Koepsell H. Mol. Pharmacol. 1998;54:342. doi: 10.1124/mol.54.2.342. [DOI] [PubMed] [Google Scholar]
- (436).Fisher A, Biggs CS, Starr MS. Amino Acids. 1998;14:43. doi: 10.1007/BF01345241. [DOI] [PubMed] [Google Scholar]
- (437).Fisher A, Starr MS. Brain Res. 2000;868:268. doi: 10.1016/s0006-8993(00)02339-8. [DOI] [PubMed] [Google Scholar]
- (438).Li XM, Juorio AV, Qi J, Boulton AA. J. Neurosci. Res. 1998:53–490. doi: 10.1002/(SICI)1097-4547(19980815)53:4<490::AID-JNR11>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- (439).Page G, Peeters M, Maloteaux JM, Hermans E. Eur. J. Pharmacol. 2000;403:75. doi: 10.1016/s0014-2999(00)00573-2. [DOI] [PubMed] [Google Scholar]
- (440).Giustizieri M, Cucchiaroni ML, Guatteo E, Bernardi G, Mercuri NB, Berretta N. J. Pharmacol. Exp. Ther. 2007:322–721. doi: 10.1124/jpet.107.122036. [DOI] [PubMed] [Google Scholar]
- (441).Iravani MM, Jenner P. J. Neural Transm. 2011:118–1661. doi: 10.1007/s00702-011-0698-2. [DOI] [PubMed] [Google Scholar]
- (442).Bido S, Marti M, Morari M. J. Neurochem. 2011;118:1043. doi: 10.1111/j.1471-4159.2011.07376.x. [DOI] [PubMed] [Google Scholar]
- (443).Kim JH, Lee HW, Hwang J, Kim J, Lee MJ, Han HS, Lee WH, Suk K. Neurobiol. Aging. 2011 doi: 10.1016/j.neurobiolaging.2011.08.011. [DOI] [PubMed] [Google Scholar]
- (444).Ossola B, Schendzielorz N, Chen SH, Bird GS, Tuominen RK, Mannisto PT, Hong JS. Neuropharmacology. 2011;61:574. doi: 10.1016/j.neuropharm.2011.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (445).Sommerauer C, Rebernik P, Reither H, Nanoff C, Pifl C. Neuropharmacology. 2012;62:1708. doi: 10.1016/j.neuropharm.2011.11.017. [DOI] [PubMed] [Google Scholar]
- (446).Frantz S. Nature. 2005;437:942. doi: 10.1038/437942a. [DOI] [PubMed] [Google Scholar]
- (447).Zambrowicz BP, Sands AT. Nat. Rev. Drug Discov. 2003;2:38. doi: 10.1038/nrd987. [DOI] [PubMed] [Google Scholar]
- (448).Kebabian JW, Britton DR, DeNinno MP, Perner R, Smith L, Jenner P, Schoenleber R, Williams M. Eur. J. Pharmacol. 1992;229:203. doi: 10.1016/0014-2999(92)90556-j. [DOI] [PubMed] [Google Scholar]
- (449).Blanchet PJ, Grondin R, Bedard PJ, Shiosaki K, Britton DR. Eur. J. Pharmacol. 1996;309:13. doi: 10.1016/0014-2999(96)00309-3. [DOI] [PubMed] [Google Scholar]
- (450).Lin CW, Bianchi BR, Miller TR, Stashko MA, Wang SS, Curzon P, Bednarz L, Asin KE, Britton DR. J. Pharmacol. Exp. Ther. 1996;276:1022. [PubMed] [Google Scholar]
- (451).Pearce RK, Jackson M, Britton DR, Shiosaki K, Jenner P, Marsden CD. Psychopharmacology. 1999;142:51. doi: 10.1007/s002130050861. [DOI] [PubMed] [Google Scholar]
- (452).Smith LA, Jackson MJ, Al-Barghouthy G, Jenner PJ. Neural Transm. 2002;109:123. doi: 10.1007/s007020200009. [DOI] [PubMed] [Google Scholar]
- (453).Saklayen SS, Mabrouk OS, Pehek EA. J. Pharmacol. Exp. Ther. 2004;311:342. doi: 10.1124/jpet.104.067991. [DOI] [PubMed] [Google Scholar]
- (454).Ryman-Rasmussen JP, Griffith A, Oloff S, Vaidehi N, Brown JT, Goddard WA, 3rd, Mailman RB. Neuropharmacology. 2007;52:562. doi: 10.1016/j.neuropharm.2006.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (455).Crosby N, Deane KH, Clarke CE. Cochrane DB Syst. Rev. 2009;(Issue 1):CD003468. doi: 10.1002/14651858.CD003468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (456).Crosby NJ, Deane KH, Clarke CE. Cochrane DB Syst. Rev. 2009;(Issue 1):CD003467. doi: 10.1002/14651858.CD003467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (457).Stark H, Reichert U, Grassmann S. Pharm. Unserer Zeit. 2000;29:228. doi: 10.1002/1615-1003(200007)29:4<228::AID-PAUZ228>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- (458).Palmer GC. Curr. Drug Targets. 2001;2:241. doi: 10.2174/1389450013348335. [DOI] [PubMed] [Google Scholar]
- (459).Chen HSV, Lipton SA. J. Neurochem. 2006;97:1611. doi: 10.1111/j.1471-4159.2006.03991.x. [DOI] [PubMed] [Google Scholar]
- (460).Inzelberg R, Bonuccelli U, Schechtman E, Miniowich A, Strugatsky R, Ceravolo R, Logi C, Rossi C, Klein C, Rabey JM. Movement Disord. 2006;21:1375. doi: 10.1002/mds.20968. [DOI] [PubMed] [Google Scholar]
- (461).Nichols CG. Nature. 2006;440:470. doi: 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- (462).Smith AJ, Taneja TK, Mankouri J, Sivaprasadarao A. Expert Rev. Mol. Med. 2007;9:1. doi: 10.1017/S1462399407000403. [DOI] [PubMed] [Google Scholar]
- (463).Garrino MG, Henquin JC. Brit. J. Pharmacol. 1987;90:583. doi: 10.1111/j.1476-5381.1987.tb11209.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (464).Meszaros J, Kovacs T, Dinya A, Szegi J. Acta Physiol. Acad. Sci. Hung. 1981;58:79. [PubMed] [Google Scholar]
- (465).Meszaros J, Kelemen K, Marko R, Kecskemeti V, Szegi J. Eur. J. Pharmacol. 1982;84:151. doi: 10.1016/0014-2999(82)90197-2. [DOI] [PubMed] [Google Scholar]
- (466).Meisheri KD, Humphrey SJ, Khan SA, Cipkus-Dubray LA, Smith MP, Jones AW. J. Pharmacol. Exp. Ther. 1993;266:655. [PubMed] [Google Scholar]
- (467).Ohrnberger CE, Khan SA, Meisheri KD. J. Pharmacol. Exp. Ther. 1993;267:25. [PubMed] [Google Scholar]
- (468).Ploug KB, Boni LJ, Baun M, Hay-Schmidt A, Olesen J, Jansen-Olesen I. Brit. J. Pharmacol. 2008;154:72. doi: 10.1038/bjp.2008.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (469).Clayton A, Siebold C, Gilbert RJC, Sutton GC, Harlos K, McIlhinney RAJ, Jones EY, Aricescu AR. J. Mol. Biol. 2009;392:1125. doi: 10.1016/j.jmb.2009.07.082. [DOI] [PubMed] [Google Scholar]
- (470).Fleming JJ, England PM. Bioorgan. Med. Chem. 2010;18:1381. doi: 10.1016/j.bmc.2009.12.072. [DOI] [PubMed] [Google Scholar]
- (471).Antonov SM, Johnson JW, Lukomskaya NY, Potapyeva NN, Gmiro VE, Magazanik LG. Mol. Pharmacol. 1995;47:558. [PubMed] [Google Scholar]
- (472).Magazanik LG, Buldakova SL, Samoilova MV, Gmiro VE, Mellor IR, Usherwood PNR. J. Physiol.-London. 1997;505:655. doi: 10.1111/j.1469-7793.1997.655ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (473).Andersen TF, Tikhonov DB, Bolcho U, Bolshakov K, Nelson JK, Pluteanu F, Mellor IR, Egebjerg J, Stromgaard K. J. Med. Chem. 2006;49:5414. doi: 10.1021/jm060606j. [DOI] [PubMed] [Google Scholar]
- (474).Tikhonov DB, Zhorov BS, Magazanik LG. Biophys. J. 1999;77:1914. doi: 10.1016/S0006-3495(99)77033-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (475).Tikhonov DB, Mellor JR, Usherwood PN, Magazanik LG. Biophys. J. 2002;82:1884. doi: 10.1016/S0006-3495(02)75538-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (476).Jensen LS, Bolcho U, Egebjerg J, Stromgaard K. ChemMedChem. 2006;1:419. doi: 10.1002/cmdc.200500093. [DOI] [PubMed] [Google Scholar]
- (477).Bolshakov KV, Tikhonov DB, Gmiro VE, Magazanik LG. Neurosci. Lett. 2000;291:101. doi: 10.1016/s0304-3940(00)01386-0. [DOI] [PubMed] [Google Scholar]
- (478).Tikhonov DB, Samoilova MV, Buldakova SL, Gmiro VE, Magazanik LG. Brit. J. Pharmacol. 2000;129:265. doi: 10.1038/sj.bjp.0703043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (479).Tikhonova TB, Barygin OI, Gmiro VE, Tikhonov DB, Magazanik LG. Neuropharmacology. 2008;54:653. doi: 10.1016/j.neuropharm.2007.11.014. [DOI] [PubMed] [Google Scholar]
- (480).Tikhonova TB, Tikhonov DB, Magazanik LG. J. Mol. Neurosci. 2009 doi: 10.1007/s12031-008-9172-5. [DOI] [PubMed] [Google Scholar]
- (481).Buldakova SL, Vorobjev VS, Sharonova IN, Samoilova MV, Magazanik LG. Brain Res. 1999;846:52. doi: 10.1016/s0006-8993(99)01970-8. [DOI] [PubMed] [Google Scholar]
- (482).Buldakova SL, Kim KK, Tikhonov DB, Magazanik LG. Neuroscience. 2007;144:88. doi: 10.1016/j.neuroscience.2006.09.005. [DOI] [PubMed] [Google Scholar]
- (483).Lukomskaya NY, Lavrent'eva VV, Starshinova LA, Zhabko EP, Gorbunova LV, Tikhonova TB, Gmiro VE, Magazanik LG. Neurosci. Behav. Physiol. 2007;37:75. doi: 10.1007/s11055-007-0152-y. [DOI] [PubMed] [Google Scholar]
- (484).Majdi M, Chen H-SV. J. Recept. Ligand Channel Res. 2009;2:59. [Google Scholar]
- (485).Scherm M, Peter D, Jamiak B. Arzneimittel, insbesondere zur Bekaempfung der Parkinson'schen Krankheit. Ger. Offen. DE2219256. 1973 Nov 8; [Google Scholar]
- (486).Scherm A, Peteri D, Janiak B. Aminoadamantan-Verbindungen und Verfahren zu ihrer Herstellung. Ger. Offen. DE2232735. 1974 Jan 24; [Google Scholar]
- (487).Svensson TH. Eur. J. Pharmacol. 1973;23:232. doi: 10.1016/0014-2999(73)90088-5. [DOI] [PubMed] [Google Scholar]
- (488).Wesemann W, Sontag KH, Maj J. Arznei-Forschung. 1983;33:1122. [PubMed] [Google Scholar]
- (489).Fleischhacker WW, Buchgeher A, Schubert H. Prog. Neuropsychopharmacol. Biol. Psych. 1986;10:87. doi: 10.1016/0278-5846(86)90047-3. [DOI] [PubMed] [Google Scholar]
- (490).Bormann J, Gold MR, Schatton W. Verwendung von Adamantan-Derivaten zur Praevention und Behandlung der cerebralen Ischaemie Eur. Pat. Appl. EP0392059. 1989 Apr 14; [Google Scholar]
- (492).Wesemann W, Sturm G, Funfgeld EW. J. Neural Transm.-Supp. 1980;143 doi: 10.1007/978-3-7091-8582-7_15. [DOI] [PubMed] [Google Scholar]
- (493).Marx J. Science. 2000;289:375. doi: 10.1126/science.289.5478.375b. [DOI] [PubMed] [Google Scholar]
- (494).Winblad B, Poritis N. Int. J. Geriatr. Psych. 1999;14:135. doi: 10.1002/(sici)1099-1166(199902)14:2<135::aid-gps906>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- (495).Reisberg B, Doody R, Stoffler A, Schmitt F, Ferris S, Mobius HJ. New Engl. J. Med. 2003;348:1333. doi: 10.1056/NEJMoa013128. [DOI] [PubMed] [Google Scholar]
- (496).McShane R, Areosa Sastre A, Minakaran N. Cochrane DB Syst. Rev. 2009;(Issue 1) doi: 10.1002/14651858.CD003154.pub5. Art. No.: CD003154. [DOI] [PubMed] [Google Scholar]
- (497).Winblad B, Jones RW, Wirth Y, Stoffler A, Mobius HJ. Dement. Geriatr. Cogn. 2007;24:20. doi: 10.1159/000102568. [DOI] [PubMed] [Google Scholar]
- (498).Newsbriefs Am. J. Alzheimers. Dis. Other Demen. 2003;18:329. [Google Scholar]
- (499).Schneider LS, Dagerman KS, Higgins JPT, McShane R. Arch. Neurol.-Chicago. 2011;68:991. doi: 10.1001/archneurol.2011.69. [DOI] [PubMed] [Google Scholar]
- (500).Saxton J, Hofbauer RK, Woodward M, Gilchrist NL, Potocnik F, Hsu HA, Miller ML, Pejovic V, Graham SM, Perhach JL. J. Alzheimers Dis. 2012;28:109. doi: 10.3233/JAD-2011-110947. [DOI] [PubMed] [Google Scholar]
- (501).Zucker RS, Lando L. Science. 1986;231:574. doi: 10.1126/science.2868525. [DOI] [PubMed] [Google Scholar]
- (502).Khachaturian ZS. Ann. N Y Acad. Sci. 1994;747:1. doi: 10.1111/j.1749-6632.1994.tb44398.x. [DOI] [PubMed] [Google Scholar]
- (503).Berridge MJ. Pflug. Aech. Eur. J. Phy. 2009;459:441. doi: 10.1007/s00424-009-0736-1. [DOI] [PubMed] [Google Scholar]
- (504).Pellegrini JW, Lipton SA. Ann. Neurol. 1993;33:403. doi: 10.1002/ana.410330414. [DOI] [PubMed] [Google Scholar]
- (505).Bresink I, Danysz W, Parsons CG, Tiedtke P, Mutschler E. J. Neural Transm.-Park. 1995;10(11) doi: 10.1007/BF02256626. [DOI] [PubMed] [Google Scholar]
- (506).Parsons CG, Danysz W, Quack G. Neuropharmacology. 1999;38:735. doi: 10.1016/s0028-3908(99)00019-2. [DOI] [PubMed] [Google Scholar]
- (507).Danysz W, Parsons CG. Int. J. Geriatr. Psych. 2003(18):S23. doi: 10.1002/gps.938. [DOI] [PubMed] [Google Scholar]
- (508).Wenk GL, Quack G, Moebius HJ, Danysz A. Life Sci. 2000;66:1079. doi: 10.1016/s0024-3205(00)00411-2. [DOI] [PubMed] [Google Scholar]
- (509).Helmuth L. Science. 2002;297:1260. doi: 10.1126/science.297.5585.1260. [DOI] [PubMed] [Google Scholar]
- (510).Bolshakov KV, Gmiro VE, Tikhonov DB, Magazanik LG. J. Neurochem. 2003;87:56. doi: 10.1046/j.1471-4159.2003.01956.x. [DOI] [PubMed] [Google Scholar]
- (511).Chen HS, Lipton SA. J. Pharmacol. Exp. Ther. 2005;314:961. doi: 10.1124/jpet.105.085142. [DOI] [PubMed] [Google Scholar]
- (512).Joubert J, van Dyk S, Green IR, Malan SF. Eur. J. Med. Chem. 2011;46:5010. doi: 10.1016/j.ejmech.2011.08.008. [DOI] [PubMed] [Google Scholar]
- (513).Motawaj M, Burban A, Davenas E, Arrang JM. J. Pharmacol. Exp. Ther. 2011;336:479. doi: 10.1124/jpet.110.174458. [DOI] [PubMed] [Google Scholar]
- (514).Rogawski MA, Wenk GL. CNS Drug Rev. 2003;9:275. doi: 10.1111/j.1527-3458.2003.tb00254.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (515).Lipton SA. Curr. Alzheimer Res. 2005;2:155. doi: 10.2174/1567205053585846. [DOI] [PubMed] [Google Scholar]
- (516).Parsons CG, Stoffler A, Danysz W. Neuropharmacology. 2007;53:699. doi: 10.1016/j.neuropharm.2007.07.013. [DOI] [PubMed] [Google Scholar]
- (517).Rammes G, Danysz W, Parsons CG. Curr. Neuropharmacol. 2008;6:55. doi: 10.2174/157015908783769671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (518).McKeage K. CNS Drugs. 2009;23:881. doi: 10.2165/11201020-000000000-00000. [DOI] [PubMed] [Google Scholar]
- (519).Kmietowicz Z. Brit. Med. J. 2005;330:495. doi: 10.1136/bmj.330.7490.495-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (520).Witt A, Macdonald N, Kirkpatrick P. Nat. Rev. Drug Discov. 2004;3:109. doi: 10.1038/nrd1311. [DOI] [PubMed] [Google Scholar]
- (521).Heidbreder CA, Hagan J. J. Curr. Opin. Pharmacol. 2005;5:107. doi: 10.1016/j.coph.2004.08.013. [DOI] [PubMed] [Google Scholar]
- (522).Lieberman JA, Papadakis K, Csernansky J, Litman R, Volavka J, Jia XD, Gage A. Neuropsychopharmacol. 2008:1322. doi: 10.1038/npp.2008.200. [DOI] [PubMed] [Google Scholar]
- (523).Zdanys K, Tampi RR. Prog. Neuro-Psychopha. 2008;32:1362. doi: 10.1016/j.pnpbp.2008.01.008. [DOI] [PubMed] [Google Scholar]
- (524).Kotermanski SE, Johnson JW. J. Neurosci. 2009;29:2774. doi: 10.1523/JNEUROSCI.3703-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (525).Ahmed MM, Hoshino H, Chikuma T, Yamada M, Kato T. Neuroscience. 2004;126:639. doi: 10.1016/j.neuroscience.2004.04.024. [DOI] [PubMed] [Google Scholar]
- (526).Caumont AS, Octave JN, Hermans E. Neurosci. Lett. 2006;394:196. doi: 10.1016/j.neulet.2005.10.027. [DOI] [PubMed] [Google Scholar]
- (527).Wu HM, Tzeng NS, Qian L, Wei SJ, Hu XM, Chen SH, Rawls SM, Flood P, Hong JS, Lu RB. Neuropsychopharmacol. 2009;34:2344. doi: 10.1038/npp.2009.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (528).Maekawa M, Namba T, Suzuki E, Yuasa S, Kohsaka S, Uchino S. Neurosci. Res. 2009;63:259. doi: 10.1016/j.neures.2008.12.006. [DOI] [PubMed] [Google Scholar]
- (529).Jin K, Xie L, Mao XO, Greenberg DA. Brain Res. 2006;1085:183. doi: 10.1016/j.brainres.2006.02.081. [DOI] [PubMed] [Google Scholar]
- (530).Meisner F, Scheller C, Kneitz S, Sopper S, Neuen-Jacob E, Riederer P, ter Meulen V, Koutsilieri E. Neuropsychopharmacol. 2008;33:2228. doi: 10.1038/sj.npp.1301615. [DOI] [PubMed] [Google Scholar]
- (531).Li B, Wanka L, Blanchard J, Liu F, Chohan MO, Iqbal K, Grundke-Iqbal I. FEBS Lett. 2010;584:3359. doi: 10.1016/j.febslet.2010.06.025. [DOI] [PubMed] [Google Scholar]
- (532).Aracava Y, Pereira EFR, Maelicke A, Albuquerque EX. J. Pharmacol. Exp. Ther. 2005;312:1195. doi: 10.1124/jpet.104.077172. [DOI] [PubMed] [Google Scholar]
- (533).Aracava Y, Pereira EFR, Maelicke A, Albuquerque EX. J. Pharmacol. Exp. Ther. 2005;313:930. doi: 10.1124/jpet.104.077172. [DOI] [PubMed] [Google Scholar]
- (534).De Sarno P, Bijur GN, Zmijewska AA, Li X, Jope RS. Neurobiol. Aging. 2006;27:413. doi: 10.1016/j.neurobiolaging.2005.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (535).Wang Y, Eu J, Washburn M, Gong T, Chen HS, James WL, Lipton SA, Stamler JS, Went GT, Porter S. Curr. Alzheimer Res. 2006;3:201. doi: 10.2174/156720506777632808. [DOI] [PubMed] [Google Scholar]
- (536).Dias CP, De Lima MNM, Presti-Torres J, Dornelles A, Garcia VA, Scalco FS, Guimaraes MR, Constantino L, Budni P, Dal-Pizzol F, Schroder N. Neuroscience. 2007;146:1719. doi: 10.1016/j.neuroscience.2007.03.018. [DOI] [PubMed] [Google Scholar]
- (537).Annweiler C, Beauchet O. Drug. Aging. 2012;29:81. doi: 10.2165/11597550-000000000-00000. [DOI] [PubMed] [Google Scholar]
- (538).Bores GM, Huger FP, Petko W, Mutlib AE, Camacho F, Rush DK, Selk DE, Wolf V, Kosley RW, Jr., Davis L, Vargas HM. J. Pharmacol. Exp. Ther. 1996;277:728. [PubMed] [Google Scholar]
- (539).Seeman P, Caruso C, Lasaga M. Synapse. 2008;62:149. doi: 10.1002/syn.20472. [DOI] [PubMed] [Google Scholar]
- (540).Roberson ED, Mucke L. Science. 2006;314:781. doi: 10.1126/science.1132813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (541).Li L, Sengupta A, Haque N, Grundke-Iqbal I, Iqbal K. FEBS Lett. 2004;566:261. doi: 10.1016/j.febslet.2004.04.047. [DOI] [PubMed] [Google Scholar]
- (542).Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Proc. Natl. Acad. Sci. U.S.A. 1986;83:4913. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (543).Tanimukai H, Grundke-Iqbal I, Iqbal K. Am. J. Pathol. 2005;166:1761. doi: 10.1016/S0002-9440(10)62486-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (544).Iqbal K, Del C, Alonso A, Chen S, Chohan MO, El-Akkad E, Gong C-X, Khatoon S, Li B, Liu F, Rahman A, Tanimukai H, Grundke-Iqbal I. BBA-Mol. Basis. Dis. 2005;1739:198. doi: 10.1016/j.bbadis.2004.09.008. [DOI] [PubMed] [Google Scholar]
- (545).Degerman Gunnarsson M, Kilander L, Basun H, Lannfelt L. Dement. Geriatr. Cogn. 2007;24:247. doi: 10.1159/000107099. [DOI] [PubMed] [Google Scholar]
- (546).Chebib M, Johnston GAR. J. Med. Chem. 2000;43:1427. doi: 10.1021/jm9904349. [DOI] [PubMed] [Google Scholar]
- (547).Johnston GAR. Curr. Pharm. Design. 2005;11:1867. doi: 10.2174/1381612054021024. [DOI] [PubMed] [Google Scholar]
- (548).Krogsgaard-Larsen P, Frolund B, Frydenvang K. Curr. Pharm. Design. 2000;6:1193. doi: 10.2174/1381612003399608. [DOI] [PubMed] [Google Scholar]
- (549).Taylor CP. Neurology. 1994;44:S10. [Google Scholar]
- (550).Field MJ, Li Z, Schwarz JB. J. Med. Chem. 2007;50:2569. doi: 10.1021/jm060650z. [DOI] [PubMed] [Google Scholar]
- (551).Bryans JS, Davies N, Gee NS, Dissanayake VUK, Ratcliffe GS, Horwell DC, Kneen CO, Morrell AI, Oles RJ, O'Toole JC, Perkins GM, Singh L. J. Med. Chem. 1998;41:1838. doi: 10.1021/jm970649n. [DOI] [PubMed] [Google Scholar]
- (552).Andrade A, Sandoval A, Gonzalez-Ramirez R, Lipscombe D, Campbell KP, Felix R. Cell Calcium. 2009;46:282. doi: 10.1016/j.ceca.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (553).Verspohl EJ, Koch R, Schatton WJ. Pharm. Pharmacol. 1988;40:111. doi: 10.1111/j.2042-7158.1988.tb05192.x. [DOI] [PubMed] [Google Scholar]
- (554).Davey AE, Horwell DC. Bioorgan. Med. Chem. 1993;1:45. doi: 10.1016/s0968-0896(00)82102-1. [DOI] [PubMed] [Google Scholar]
- (555).Dezube M, Sugg EE, Birkemo LS, Croom DK, Dougherty RW, Jr., Ervin GN, Grizzle MK, James MK, Johnson MF, Mosher JT, Queen KL, Rimele TJ, Sauls HR, Jr., Triantafillou JA. J. Med. Chem. 1995;38:3384. doi: 10.1021/jm00017a022. [DOI] [PubMed] [Google Scholar]
- (556).Hughes J, Dockray GJ, Hill D, Garcia L, Pritchard MC, Forster E, Toescu E, Woodruff G, Horwell DC. Regul. Peptides. 1996;65:15. doi: 10.1016/0167-0115(96)00067-5. [DOI] [PubMed] [Google Scholar]
- (557).Trivedi BK, Padia JK, Holmes A, Rose S, Wright DS, Hinton JP, Pritchard MC, Eden JM, Kneen C, Webdale L, Suman-Chauhan N, Boden P, Singh L, Field MJ, Hill DJ. Med. Chem. 1998;41:38. doi: 10.1021/jm970065l. [DOI] [PubMed] [Google Scholar]
- (558).Ekhato IV, Huang Y. J. Labelled Compd. Rad. 1997;39:1019. [Google Scholar]
- (559).Harper EA, Shankley NP, Black JW. Brit. J. Pharmacol. 1999;126:1504. doi: 10.1038/sj.bjp.0702447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (560).Bernad N, Burgaud BG, Horwell DC, Lewthwaite RA, Martinez J, Pritchard MC. Bioorg. Med. Chem. Lett. 2000;10:1245. doi: 10.1016/s0960-894x(00)00198-0. [DOI] [PubMed] [Google Scholar]
- (561).Martin-Martinez M, Latorre M, Garcia-Lopez MT, Cenarruzabeitia E, Del Rio J, Gonzalez-Muniz R. Bioorg. Med. Chem. Lett. 2002;12:109. doi: 10.1016/s0960-894x(01)00630-8. [DOI] [PubMed] [Google Scholar]
- (562).Munoz-Ruiz P, Garcia-Lopez MT, Cenarruzabeitia E, Del Rio J, Dufresne M, Foucaud M, Fourmy D, Herranz R. J. Med. Chem. 2004;47:5318. doi: 10.1021/jm0498755. [DOI] [PubMed] [Google Scholar]
- (563).Buck IM, Black JW, Cooke T, Dunstone DJ, Gaffen JD, Griffin EP, Harper EA, Hull RAD, Kalindjian SB, Lilley EJ, Linney ID, Low CMR, McDonald IM, Pether MJ, Roberts SP, Shankley NP, Shaxted ME, Steel KIM, Sykes DA, Tozer MJ, Watt GF, Walker MK, Wright L, Wright PT. J. Med. Chem. 2005;48:6803. doi: 10.1021/jm0490686. [DOI] [PubMed] [Google Scholar]
- (564).Low CMR, Buck IM, Cooke T, Cushnir JR, Kalindjian SB, Kotecha A, Pether MJ, Shankley NP, Vinter JG, Wright L. J. Med. Chem. 2005;48:6790. doi: 10.1021/jm049069y. [DOI] [PubMed] [Google Scholar]
- (565).Low CMR, Vinter JG. J. Med. Chem. 2008;51:565. doi: 10.1021/jm070880t. [DOI] [PubMed] [Google Scholar]
- (566).Fornai M, Colucci R, Antonioli L, Crema F, Buccianti P, Chiarugi M, Baschiera F, Ghisu N, Tuccori M, Blandizzi C, Del Tacca M. Brit. J. Pharmacol. 2007;151:1246. doi: 10.1038/sj.bjp.0707339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (567).Christos TE, Arvanitis A, Cain GA, Johnson AL, Potorf RS, Tam SW, Schmidt WK. Bioorg. Med. Chem. Lett. 1993;3:1035. [Google Scholar]
- (568).Gully D, Labeeuw B, Biogegrain R, Oury-Donat F, Bachy A, Poncelet M, Steinberg R, Suaud-Chagny MF, Santucci V, Vita N, Pecceu F, Labbe-Jullie C, Kitabgi P, Soubrie P, Le Fur G, Maffrand JP. J. Pharmacol. Exp. Ther. 1997;280:802. [PubMed] [Google Scholar]
- (569).Betancur C, Canton M, Burgos A, Labeeuw B, Gully D, Rostene W, Pelaprat D. Eur. J. Pharmacol. 1998;343:67. doi: 10.1016/s0014-2999(97)01510-0. [DOI] [PubMed] [Google Scholar]
- (570).Binder EB, Kinkead B, Owens MJ, Kilts CD, Nemeroff CB. J. Neurosci. 2001;21:601. doi: 10.1523/JNEUROSCI.21-02-00601.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (571).Barroso S, Richard F, Nicolas-Etheve D, Reversat J-L, Bernassau J-M, Kitabgi P, Labbe-Jullie C. J. Biol. Chem. 2000;275:328. doi: 10.1074/jbc.275.1.328. [DOI] [PubMed] [Google Scholar]
- (572).Costa FG, Frussa-Filho R, Felicio LF. Eur. J. Pharmacol. 2001;428:97. doi: 10.1016/s0014-2999(01)01271-7. [DOI] [PubMed] [Google Scholar]
- (573).Mitchell VA, Kawahara H, Vaughan CW. J. Physiol. 2009;587:2511. doi: 10.1113/jphysiol.2008.167429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (574).Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG. Nature. 1994;369:744. doi: 10.1038/369744a0. [DOI] [PubMed] [Google Scholar]
- (575).Lammek B, Wang Y-X, Gavras I, Gavras H. Peptides. 1990;11:1041. doi: 10.1016/0196-9781(90)90031-y. [DOI] [PubMed] [Google Scholar]
- (576).Lammek B, Wang YX, Gavras I, Gavras H. J. Pharm. Pharmacol. 1991;43:887. doi: 10.1111/j.2042-7158.1991.tb03205.x. [DOI] [PubMed] [Google Scholar]
- (577).Ehringer WD, Edwards MJ, Gray RD, Miller FN. Inflammation. 1997;21:279. doi: 10.1023/a:1027345832138. [DOI] [PubMed] [Google Scholar]
- (578).Matsuda H, Hayashi K, Wakino S, Kubota E, Honda M, Tokuyama H, Takamatsu I, Tatematsu S, Saruta T. Hypertension. 2004;43:603. doi: 10.1161/01.HYP.0000118053.42262.71. [DOI] [PubMed] [Google Scholar]
- (579).Dear JW, Wirth K, Scadding GK, Foreman JC. Brit. J. Pharmacol. 1996;119:1054. doi: 10.1111/j.1476-5381.1996.tb15777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (580).Zenteno-Savin T, Sada-Ovalle I, Ceballos G, Rubio R. Eur. J. Pharmacol. 2000;410:15. doi: 10.1016/s0014-2999(00)00853-0. [DOI] [PubMed] [Google Scholar]
- (581).Sleszynska M, Wierzba TH, Malinowski K, Borovickova L, Maluch I, Sobolewski D, Lammek B, Slaninova J, Prahl A. J. Pept. Sci. 2011;17:366. doi: 10.1002/psc.1351. [DOI] [PubMed] [Google Scholar]
- (582).Tanabe S, Shishido Y, Nakayama Y, Furushiro M, Hashimoto S, Terasaki T, Tsujimoto G, Yokokura T. Pharmacol. Biochem. Be. 1999;63:549. doi: 10.1016/s0091-3057(99)00034-9. [DOI] [PubMed] [Google Scholar]
- (583).Salvador AM. Eur. J. Pharmacol. 1975;31:38. doi: 10.1016/0014-2999(75)90076-x. [DOI] [PubMed] [Google Scholar]
- (584).Flouret G, Majewski T, Balaspiri L, Brieher W, Mahan K, Chaloin O, Wilson L, Jr., Slaninova J. J. Pept. Sci. 2002;8:314. doi: 10.1002/psc.390. [DOI] [PubMed] [Google Scholar]
- (585).Hughes J, Smith TW, Kosterlitz HW, Fothergill LA, Morgan BA, Morris HR. Nature. 1975;258:577. doi: 10.1038/258577a0. [DOI] [PubMed] [Google Scholar]
- (586).Comb M, Seeburg PH, Adelman J, Eiden L, Herbert E. Nature. 1982;295:663. doi: 10.1038/295663a0. [DOI] [PubMed] [Google Scholar]
- (587).Mazur RH. N-Adamantane-substituted Tetrapeptidamide and its Pharmacologically Compatible Salts. Ger. Offen. DE3044793. 1981 Jun 19; [Google Scholar]
- (588).Kim Quang D, Schwyzer R. Helv. Chim. Acta. 1981;64:2084. [Google Scholar]
- (589).Roscic M, Sabljic V, Mlinaric-Majerski K, Horvat S. Croat. Chem. Acta. 2008;81:637. [Google Scholar]
- (590).Tsuzuki N, Hama T, Hibi T, Konishi R, Futaki S, Kitagawa K. Biochem. Pharmacol. 1991;41:R5. doi: 10.1016/0006-2952(91)90616-d. [DOI] [PubMed] [Google Scholar]
- (591).Simonin F, Schmitt M, Laulin JP, Laboureyras E, Jhamandas JH, MacTavish D, Matifas A, Mollereau C, Laurent P, Parmentier M, Kieffer BL, Bourguignon JJ, Simonnet G. Proc. Natl. Acad. Sci. U.S.A. 2006;103:466. doi: 10.1073/pnas.0502090103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (592).Fang Q, Guo J, Peng Y.-l., Chang M, He F, Chen Q, Wang R. Peptides. 2006;27:1297. doi: 10.1016/j.peptides.2005.10.021. [DOI] [PubMed] [Google Scholar]
- (593).Fang Q, Wang YQ, He F, Guo J, Guo J, Chen Q, Wang R. Regul. Peptides. 2008;147:45. doi: 10.1016/j.regpep.2007.12.007. [DOI] [PubMed] [Google Scholar]
- (594).Wang YQ, Wang SB, Ma JL, Guo J, Fang Q, Sun T, Zhuang Y, Wang R. Peptides. 2011;32:702. doi: 10.1016/j.peptides.2010.12.001. [DOI] [PubMed] [Google Scholar]
- (595).Wang Y-Q, Guo J, Wang S-B, Fang Q, He F, Wang R. Peptides. 2008;29:1183. doi: 10.1016/j.peptides.2008.02.016. [DOI] [PubMed] [Google Scholar]
- (596).Ma L, MacTavish D, Simonin F, Bourguignon J-J, Watanabe T, Jhamandas Jack H. The European journal of neuroscience. 2009;30:1585. doi: 10.1111/j.1460-9568.2009.06956.x. [DOI] [PubMed] [Google Scholar]
- (597).Talmont F, Mouledous L, Piedra-Garcia L, Schmitt M, Bihel F, Bourguignon J-J, Zajac J-M, Mollereau C. Peptides. 2010;31:215. doi: 10.1016/j.peptides.2009.11.020. [DOI] [PubMed] [Google Scholar]
- (598).Pineda R, Garcia-Galiano D, Sanchez-Garrido MA, Romero M, Ruiz-Pino F, Aguilar E, Dijcks FA, Blomenrohr M, Pinilla L, van Noort PI, Tena-Sempere M. Endocrinology. 2010;151:1902. doi: 10.1210/en.2009-1259. [DOI] [PubMed] [Google Scholar]
- (599).Raghupathi RK, Rydelek-Fitzgerald L, Teitler M, Glennon RA. J. Med. Chem. 1991;34:2633. doi: 10.1021/jm00112a043. [DOI] [PubMed] [Google Scholar]
- (600).Uchida H, Inagawa K, Tameda C, Miyauchi T. Prog. Neuropsychopharmacol. Biol. Psych. 1995;19:1201. doi: 10.1016/0278-5846(95)00237-5. [DOI] [PubMed] [Google Scholar]
- (601).Inagawa K, Tameda C, Uchida H, Miyauchi T. Prog. Neuropsychopharmacol. Biol. Psych. 1996;20:129. doi: 10.1016/0278-5846(95)00300-2. [DOI] [PubMed] [Google Scholar]
- (602).Cohen ML, Bloomquist W, Schaus JM, Thompson DC, Susemichel AD, Calligaro DA, Cohen I. J. Pharmacol. Exp. Ther. 1996;277:97. [PubMed] [Google Scholar]
- (603).Schaus JM, Thompson DC, Bloomquist WE, Susemichel AD, Calligaro DO, Cohen ML. J. Med. Chem. 1998;41:1943. doi: 10.1021/jm970857f. [DOI] [PubMed] [Google Scholar]
- (604).Abou-Gharbia MA, Childers WE, Jr., Fletcher H, McGaughey G, Patel U, Webb MB, Yardley J, Andree T, Boast C, Kucharik RJ, Marquis K, Morris H, Scerni R, Moyer JA. J. Med. Chem. 1999;42:5077. doi: 10.1021/jm9806704. [DOI] [PubMed] [Google Scholar]
- (605).Abou-Gharbia M. J. Med. Chem. 2008;52:2. doi: 10.1021/jm8012823. [DOI] [PubMed] [Google Scholar]
- (606).Fujio M, Kuroita T, Sakai Y, Nakagawa H, Matsumoto Y. Bioorg. Med. Chem. Lett. 2000;10:2457. doi: 10.1016/s0960-894x(00)00492-3. [DOI] [PubMed] [Google Scholar]
- (607).Bojarski AJ, Mokrosz MJ, Minol SC, Koziol A, Wesolowska A, Tatarczynska E, Klodzinska A, Chojnacka-Wojcik E. Bioorgan. Med. Chem. 2002;10:87. doi: 10.1016/s0968-0896(01)00236-x. [DOI] [PubMed] [Google Scholar]
- (608).Zajdel P, Subra G, Bojarski AJ, Duszynska B, Pawlowski M, Martinez J. J. Comb. Chem. 2004;6:761. doi: 10.1021/cc049970z. [DOI] [PubMed] [Google Scholar]
- (609).Becker DP, Flynn DL, Shone RL, Gullikson G. Bioorg. Med. Chem. Lett. 2004;14:5509. doi: 10.1016/j.bmcl.2004.09.005. [DOI] [PubMed] [Google Scholar]
- (610).Maeda K, Kawata E, Sakai K, Chihara K. Eur. J. Pharmacol. 1993;233:227. doi: 10.1016/0014-2999(93)90054-l. [DOI] [PubMed] [Google Scholar]
- (611).Davies P, Katzman R, Terry RD. Nature. 1980;288:279. doi: 10.1038/288279a0. [DOI] [PubMed] [Google Scholar]
- (612).Patel YC, Wheatley T. Endocrinology. 1983;112:220. doi: 10.1210/endo-112-1-220. [DOI] [PubMed] [Google Scholar]
- (613).Weckbecker G, Lewis I, Albert R, Schmid HA, Hoyer D, Bruns C. Nat. Rev. Drug Discov. 2003;2:999. doi: 10.1038/nrd1255. [DOI] [PubMed] [Google Scholar]
- (614).Blakeney JS, Reid RC, Le GT, Fairlie DP. Chem. Rev. 2007;107:2960. doi: 10.1021/cr050984g. [DOI] [PubMed] [Google Scholar]
- (615).Miyazaki A, Tsuda Y, Fukushima S, Yokoi T, Vantus T, Bokonyi G, Szabo E, Horvath A, Keri G, Okada Y. J. Med. Chem. 2008;51:5121. doi: 10.1021/jm701599w. [DOI] [PubMed] [Google Scholar]
- (616).Salvadori S, Tomatis R, Sarto G. Farmaco-Ed. Sci. 1982;37:669. [PubMed] [Google Scholar]
- (617).Erspamer V, Melchiorri P, Falconieri-Erspamer G, Negri L, Corsi R, Severini C, Barra D, Simmaco M, Kreil G. Proc. Natl. Acad. Sci. U.S.A. 1989;86:5188. doi: 10.1073/pnas.86.13.5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (618).Papaioannou S, Hansen D, Jr., Babler M, Yang PC, Bittner S, Miller A, Clare M. Clin. Exp. Hypertens. A. 1985;7:1243. doi: 10.3109/10641968509073588. [DOI] [PubMed] [Google Scholar]
- (619).Lazarus LH, Salvadori S, Tomatis R, Wilson WE. Biochem. Bioph. Res. Co. 1991;178:110. doi: 10.1016/0006-291x(91)91786-c. [DOI] [PubMed] [Google Scholar]
- (620).Horvat S, Varga-Defterdarovic L, Horvat J, Jukic R, Kantoci D, Chung NN, Schiller PW, Biesert L, Pfutzner A, Suhartono H, Rubsamen-Waigmann H. J. Pept. Sci. 1995;1:303. doi: 10.1002/psc.310010505. [DOI] [PubMed] [Google Scholar]
- (621).Salvadori S, Guerrini R, Balboni G, Bianchi C, Bryant SD, Cooper PS, Lazarus LH. J. Med. Chem. 1999;42:5010. doi: 10.1021/jm990165m. [DOI] [PubMed] [Google Scholar]
- (622).Lovekamp T, Cooper PS, Hardison J, Bryant SD, Guerrini R, Balboni G, Salvadori S, Lazarus LH. Brain Res. 2001;902:131. doi: 10.1016/s0006-8993(01)02363-0. [DOI] [PubMed] [Google Scholar]
- (623).Bryant SD, George C, Flippen-Anderson JL, Deschamps JR, Salvadori S, Balboni G, Guerrini R, Lazarus LH. J. Med. Chem. 2002;45:5506. doi: 10.1021/jm020330p. [DOI] [PubMed] [Google Scholar]
- (624).Fujita Y, Tsuda Y, Li T, Motoyama T, Takahashi M, Shimizu Y, Yokoi T, Sasaki Y, Ambo A, Kita A, Jinsmaa Y, Bryant SD, Lazarus LH, Okada Y. J. Med. Chem. 2004;47:3591. doi: 10.1021/jm030649p. [DOI] [PubMed] [Google Scholar]
- (625).Leonard BE. Pharmacopsychiatry. 2004;37(Suppl 3):S166. doi: 10.1055/s-2004-832674. [DOI] [PubMed] [Google Scholar]
- (626).Maurice T, Su TP. Pharmacol. Therapeut. 2009;124:195. doi: 10.1016/j.pharmthera.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (627).Guitart X, Codony X, Monroy X. Psychopharmacology. 2004;174:301. doi: 10.1007/s00213-004-1920-9. [DOI] [PubMed] [Google Scholar]
- (628).Peeters M, Romieu P, Maurice T, Su TP, Maloteaux JM, Hermans E. Eur. J. Neurosci. 2004;19:2212. doi: 10.1111/j.0953-816X.2004.03297.x. [DOI] [PubMed] [Google Scholar]
- (629).Scherz MW, Fialeix M, Fischer JB, Reddy NL, Server AC, Sonders MS, Tester BC, Weber E, Wong ST, Keana JFW. J. Med. Chem. 1990;33:2421. doi: 10.1021/jm00171a016. [DOI] [PubMed] [Google Scholar]
- (630).Ronsisvalle G, Marrazzo A, Prezzavento O, Pasquinucci L, Falcucci B, Di Toro R, Spampinato S. Bioorgan. Med. Chem. 2000;8:1503. doi: 10.1016/s0968-0896(00)00072-9. [DOI] [PubMed] [Google Scholar]
- (631).Bucolo C, Marrazzo A, Ronsisvalle S, Ronsisvalle G, Cuzzocrea S, Mazzon E, Caputi A, Drago F. Eur. J. Pharmacol. 2006;536:200. doi: 10.1016/j.ejphar.2006.02.026. [DOI] [PubMed] [Google Scholar]
- (632).Bourrie B, Bribes E, De Nys N, Esclangon M, Garcia L, Galiegue S, Lair P, Paul R, Thomas C, Vernieres J-C, Casellas P. Eur. J. Pharmacol. 2002;456:123. doi: 10.1016/s0014-2999(02)02646-8. [DOI] [PubMed] [Google Scholar]
- (633).Murdoch FE, Gorski J. Mol. Cell. Endocrinol. 1991;78:C103. doi: 10.1016/0303-7207(91)90114-8. [DOI] [PubMed] [Google Scholar]
- (634).Yamakoshi Y, Otani Y, Fujii S, Endo Y. Biol. Pharm. Bull. 2000;23:259. doi: 10.1248/bpb.23.259. [DOI] [PubMed] [Google Scholar]
- (635).Nikov GN, Eshete M, Rajnarayanan RV, Alworth WL. J. Endocrinol. 2001;170:137. doi: 10.1677/joe.0.1700137. [DOI] [PubMed] [Google Scholar]
- (636).Liu R, Yang SH, Perez E, Yi KD, Wu SS, Eberst K, Prokai L, Prokai-Tatrai K, Cai ZY, Covey DF, Day AL, Simpkins JW. Stroke. 2002;33:2485. doi: 10.1161/01.str.0000030317.43597.c8. [DOI] [PubMed] [Google Scholar]
- (637).Perez E, Liu R, Yang S-H, Cai ZY, Covey DF, Simpkins JW. Brain Res. 2005;1038:216. doi: 10.1016/j.brainres.2005.01.026. [DOI] [PubMed] [Google Scholar]
- (638).Kumar DM, Perez E, Yun Cai Z, Aoun P, Brun-Zinkernagel A-M, Covey DF, Simpkins JW, Agarwal N. Free Radical Bio. Med. 2005;38:1152. doi: 10.1016/j.freeradbiomed.2004.12.007. [DOI] [PubMed] [Google Scholar]
- (639).Yi KD, Cai ZY, Covey DF, Simpkins JW. J. Pharmacol. Exp. Ther. 2008;324:1188. doi: 10.1124/jpet.107.132308. [DOI] [PubMed] [Google Scholar]
- (640).Endo Y, Yoshimi T, Ohta K, Suzuki T, Ohta S. J. Med. Chem. 2005;48:3941. doi: 10.1021/jm050195r. [DOI] [PubMed] [Google Scholar]
- (641).Muthyala RS, Sheng S, Carlson KE, Katzenellenbogen BS, Katzenellenbogen JA. J. Med. Chem. 2003;46:1589. doi: 10.1021/jm0204800. [DOI] [PubMed] [Google Scholar]
- (642).Gever J, Cockayne D, Dillon M, Burnstock G, Ford A. Pflug. Aech. Eur. J. Phy. 2006;452:513. doi: 10.1007/s00424-006-0070-9. [DOI] [PubMed] [Google Scholar]
- (643).Baxter A, Bent J, Bowers K, Braddock M, Brough S, Fagura M, Lawson M, McInally T, Mortimore M, Robertson M, Weaver R, Webborn P. Bioorg. Med. Chem. Lett. 2003;13:4047. doi: 10.1016/j.bmcl.2003.08.034. [DOI] [PubMed] [Google Scholar]
- (644).Baraldi PG, Virgilio F. D. i., Romagnoli R. Curr. Top. Med. Chem. 2004;4:1707. doi: 10.2174/1568026043387223. [DOI] [PubMed] [Google Scholar]
- (645).Romagnoli R, Baraldi PG, Di Virgilio F. Expert Opin. Ther. Pat. 2005;15:271. [Google Scholar]
- (646).Nelson DW, Gregg RJ, Kort ME, Perez-Medrano A, Voight EA, Wang Y, Grayson G, Namovic MT, Donnelly-Roberts DL, Niforatos W, Honore P, Jarvis MF, Faltynek CR, Carroll WA. J. Med. Chem. 2006;49:3659. doi: 10.1021/jm051202e. [DOI] [PubMed] [Google Scholar]
- (647).Furber M, Alcaraz L, Bent JE, Beyerbach A, Bowers K, Braddock M, Caffrey MV, Cladingboel D, Collington J, Donald DK, Fagura M, Ince F, Kinchin EC, Laurent C, Lawson M, Luker TJ, Mortimore MM, Pimm AD, Riley RJ, Roberts N, Robertson M, Theaker J, Thorne PV, Weaver R, Webborn P, Willis P. J. Med. Chem. 2007;50:5882. doi: 10.1021/jm700949w. [DOI] [PubMed] [Google Scholar]
- (648).Guile SD, Alcaraz L, Birkinshaw TN, Bowers KC, Ebden MR, Furber M, Stocks MJ. J. Med. Chem. 2009;52:3123. doi: 10.1021/jm801528x. [DOI] [PubMed] [Google Scholar]
- (649).Gomez GA, Morisseau C, Hammock BD, Christianson DW. Biochemistry. 2004;43:4716. doi: 10.1021/bi036189j. [DOI] [PubMed] [Google Scholar]
- (650).Morisseau C, Goodrow MH, Dowdy D, Zheng J, Greene JF, Sanborn JR, Hammock BD. Proc. Natl. Acad. Sci. U.S.A. 1999;96:8849. doi: 10.1073/pnas.96.16.8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (651).Imig JD, Zhao X, Zaharis CZ, Olearczyk JJ, Pollock DM, Newman JW, Kim IH, Watanabe T, Hammock BD. Hypertension. 2005;46:975. doi: 10.1161/01.HYP.0000176237.74820.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (652).Jung O, Brandes RP, Kim IH, Schweda F, Schmidt R, Hammock BD, Busse R, Fleming I. Hypertension. 2005;45:759. doi: 10.1161/01.HYP.0000153792.29478.1d. [DOI] [PubMed] [Google Scholar]
- (653).Kim I-H, Heirtzler FR, Morisseau C, Nishi K, Tsai H-J, Hammock BD. J. Med. Chem. 2005;48:3621. doi: 10.1021/jm0500929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (654).Hwang SH, Morisseau C, Do Z, Hammock BD. Bioorg. Med. Chem. Lett. 2006;16:5773. doi: 10.1016/j.bmcl.2006.08.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (655).Kim IH, Nishi K, Tsai HJ, Bradford T, Koda Y, Watanabe T, Morisseau C, Blanchfield J, Toth I, Hammock BD. Bioorgan. Med. Chem. 2007;15:312. doi: 10.1016/j.bmc.2006.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (656).Anandan SK, Webb HK, Chen D, Wang YX, Aavula BR, Cases S, Cheng Y, Do ZN, Mehra U, Tran V, Vincelette J, Waszczuk J, White K, Wong KR, Zhang LN, Jones PD, Hammock BD, Patel DV, Whitcomb R, MacIntyre DE, Sabry J, Gless R. Bioorg. Med. Chem. Lett. 2011;21:983. doi: 10.1016/j.bmcl.2010.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (657).Schmelzer KR, Kubala L, Newman JW, Kim IH, Eiserich JP, Hammock BD. Proc. Natl. Acad. Sci. U.S.A. 2005;102:9772. doi: 10.1073/pnas.0503279102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (658).Parrish AR, Chen G, Burghardt RC, Watanabe T, Morisseau C, Hammock BD. Cell Biol. Toxicol. 2009;25:217. doi: 10.1007/s10565-008-9071-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (659).Simpkins AN, Rudic RD, Schreihofer DA, Roy S, Manhiani M, Tsai HJ, Hammock BD, Imig JD. Am J. Pathol. 2009;174:2086. doi: 10.2353/ajpath.2009.080544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (660).Koeners MP, Wesseling S, Ulu A, Sepulveda RL, Morisseau C, Braam B, Hammock BD, Joles JA. Am. J. Physiol-Endoc. M. 2011;300:E691. doi: 10.1152/ajpendo.00710.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (661).Zhang LN, Vincelette J, Chen D, Gless RD, Anandan SK, Rubanyi GM, Webb HK, MacIntyre DE, Wang YX. Eur. J. Pharmacol. 2011;654:68. doi: 10.1016/j.ejphar.2010.12.016. [DOI] [PubMed] [Google Scholar]
- (662).Mirmira RG, Tager HS. Biochemistry. 1991;30:8222. doi: 10.1021/bi00247a019. [DOI] [PubMed] [Google Scholar]
- (663).Drucker DJ, Nauck MA. Lancet. 2006;368:1696. doi: 10.1016/S0140-6736(06)69705-5. [DOI] [PubMed] [Google Scholar]
- (664).Ahren B, Gomis R, Standl E, Mills D, Schweizer A. Diabetes Care. 2004;27:2874. doi: 10.2337/diacare.27.12.2874. [DOI] [PubMed] [Google Scholar]
- (665).Ahren B, Landin-Olsson M, Jansson PA, Svensson M, Holmes D, Schweizer A. J. Clin. Endocr. Metab. 2004;89:2078. doi: 10.1210/jc.2003-031907. [DOI] [PubMed] [Google Scholar]
- (666).Ahren B, Pacini G, Foley JE, Schweizer A. Diabetes Care. 2005;28:1936. doi: 10.2337/diacare.28.8.1936. [DOI] [PubMed] [Google Scholar]
- (667).Ahren B. Expert Opin. Investig. Drugs. 2006;15:431. doi: 10.1517/13543784.15.4.431. [DOI] [PubMed] [Google Scholar]
- (668).Brandt I, Joossens J, Chen X, Maes MB, Scharpe S, De Meester I, Lambeir AM. Biochem. Pharmacol. 2005;70:134. doi: 10.1016/j.bcp.2005.04.009. [DOI] [PubMed] [Google Scholar]
- (669).Burkey BF, Li X, Bolognese L, Balkan B, Mone M, Russell M, Hughes TE, Wang PR. J. Pharmacol. Exp. Ther. 2005;315:688. doi: 10.1124/jpet.105.087064. [DOI] [PubMed] [Google Scholar]
- (670).Gill I, Patel R. Bioorg. Med. Chem. Lett. 2006;16:705. doi: 10.1016/j.bmcl.2005.10.021. [DOI] [PubMed] [Google Scholar]
- (671).Hanson RL, Goldberg SL, Brzozowski DB, Tully TP, Cazzulino D, Parker WL, Lyngberg OK, Vu TC, Wong MK, Patel RN. Adv. Synth. Catal. 2007;349:1369. [Google Scholar]
- (672).Madar DJ, Kopecka H, Pireh D, Yong H, Pei Z, Li X, Wiedeman PE, Djuric SW, VonGeldern TW, Fickes MG, Bhagavatula L, McDermott T, Wittenberger S, Richards SJ, Longenecker KL, Stewart KD, Lubben TH, Ballaron SJ, Stashko MA, Long MA, Wells H, Zinker BA, Mika AK, Beno DWA, Kempf-Grote AJ, Polakowski J, Segreti J, Reinhart GA, Fryer RM, Sham HL, Trevillyan JM. J. Med. Chem. 2006;49:6416. doi: 10.1021/jm060777o. [DOI] [PubMed] [Google Scholar]
- (673).Wright SW, Ammirati MJ, Andrews KM, Brodeur AM, Danley DE, Doran SD, Lillquist JS, McClure LD, McPherson RK, Orena SJ, Parker JC, Polivkova J, Qiu X, Soeller WC, Soglia CB, Treadway JL, VanVolkenburg MA, Wang H, Wilder DC, Olson TV. J. Med. Chem. 2006;49:3068. doi: 10.1021/jm0600085. [DOI] [PubMed] [Google Scholar]
- (674).Metzler WJ, Yanchunas J, Weigelt C, Kish K, Klei HE, Xie DL, Zhang YQ, Corbett M, Tamura JK, He B, Hamann LG, Kirby MS, Marcinkeviciene J. Protein Sci. 2008;17:240. doi: 10.1110/ps.073253208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (675).Cao K, Bonacorsi SJ, Balasubramanian B, Hanson RL, Manchand P, Godfrey JD, Fox R, Christopher LJ, Su H, Iyer R. J. Labelled Compd. Rad. 2007;50:1224. [Google Scholar]
- (676).Ciszewska G, Allentoff A, Jones L, Wu A, Ray T. J. Labelled Compd. Rad. 2007;50:593. [Google Scholar]
- (677).Simpkins LM, Bolton S, Pi Z, Sutton JC, Kwon C, Zhao G, Magnin DR, Augeri DJ, Gungor T, Rotella DP, Sun Z, Liu Y, Slusarchyk WS, Marcinkeviciene J, Robertson JG, Wang A, Robl JA, Atwal KS, Zahler RL, Parker RA, Kirby MS, Hamann LG. Bioorg. Med. Chem. Lett. 2007;17:6476. doi: 10.1016/j.bmcl.2007.09.090. [DOI] [PubMed] [Google Scholar]
- (678).Kim YB, Kopcho LM, Kirby MS, Hamann LG, Weigelt CA, Metzler WJ, Marcinkeviciene J. Arch. Biochem. Biophys. 2006;445:9. doi: 10.1016/j.abb.2005.11.010. [DOI] [PubMed] [Google Scholar]
- (679).Sauvanet JP. Diabetes Metab. 2007;33 [Google Scholar]
- (680).Bosi E, Lucotti P, Setola E, Monti L, Piatti PM. Diabetes Res. Clin. Pr. 2008;82(Suppl 2):S102. doi: 10.1016/j.diabres.2008.10.003. [DOI] [PubMed] [Google Scholar]
- (681).Gwaltney SL. Curr. Top. Med. Chem. 2008;8:1545. doi: 10.2174/156802608786413519. [DOI] [PubMed] [Google Scholar]
- (682).Maltais R, Luu-The V, Poirier D. J. Med. Chem. 2002;45:640. doi: 10.1021/jm010286y. [DOI] [PubMed] [Google Scholar]
- (683).Olson S, Aster SD, Brown K, Carbin L, Graham DW, Hermanowski-Vosatka A, LeGrand CB, Mundt SS, Robbins MA, Schaeffer JM, Slossberg LH, Szymonifka MJ, Thieringer R, Wright SD, Balkovec JM. Bioorg. Med. Chem. Lett. 2005;15:4359. doi: 10.1016/j.bmcl.2005.06.040. [DOI] [PubMed] [Google Scholar]
- (684).Hermanowski-Vosatka A, Balkovec JM, Cheng K, Chen HY, Hernandez M, Koo GC, Le Grand CB, Li Z, Metzger JM, Mundt SS, Noonan H, Nunes CN, Olson SH, Pikounis B, Ren N, Robertson N, Schaeffer JM, Shah K, Springer MS, Strack AM, Strowski M, Wu K, Wu T, Xiao J, Zhang BB, Wright SD, Thieringer R. J. Exp. Med. 2005;202:517. doi: 10.1084/jem.20050119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (685).Sorensen B, Rohde J, Wang J, Fung S, Monzon K, Chiou W, Pan L, Deng X, Stolarik D, Frevert EU, Jacobson P, Link JT. Bioorg. Med. Chem. Lett. 2006;16:5958. doi: 10.1016/j.bmcl.2006.08.129. [DOI] [PubMed] [Google Scholar]
- (686).Yeh VSC, Kurukulasuriya R, Kerdesky FA. Org. Lett. 2006;8:3963. doi: 10.1021/ol061429j. [DOI] [PubMed] [Google Scholar]
- (687).Yeh VSC, Patel JR, Yong H, Kurukulasuriya R, Fung S, Monzon K, Chiou W, Wang J, Stolarik D, Imade H, Beno D, Brune M, Jacobson P, Sham H, Link JT. Bioorg. Med. Chem. Lett. 2006;16:5414. doi: 10.1016/j.bmcl.2006.07.055. [DOI] [PubMed] [Google Scholar]
- (688).Patel JR, Shuai Q, Dinges J, Winn M, Pliushchev M, Fung S, Monzon K, Chiou W, Wang J, Pan L, Wagaw S, Engstrom K, Kerdesky FA, Longenecker K, Judge R, Qin W, Imade HM, Stolarik D, Beno DW, Brune M, Chovan LE, Sham HL, Jacobson P, Link JT. Bioorg. Med. Chem. Lett. 2007;17:750. doi: 10.1016/j.bmcl.2006.10.074. [DOI] [PubMed] [Google Scholar]
- (689).Rohde JJ, Pliushchev MA, Sorensen BK, Wodka D, Shuai Q, Wang J, Fung S, Monzon KM, Chiou WJ, Pan L, Deng X, Chovan LE, Ramaiya A, Mullally M, Henry RF, StolarikD.F.; Imade HM, Marsh KC, Beno DWA, Fey TA, Droz BA, Brune ME, Camp HS, Sham HL, Frevert EU, Jacobson PB, Link JT. J. Med. Chem. 2007;50:149. doi: 10.1021/jm0609364. [DOI] [PubMed] [Google Scholar]
- (690).Sorensen B, Winn M, Rohde J, Shuai Q, Wang J, Fung S, Monzon K, Chiou W, Stolarik D, Imade H, Pan L, Deng X, Chovan L, Longenecker K, Judge R, Qin W, Brune M, Camp H, Frevert EU, Jacobson P, Link JT. Bioorg. Med. Chem. Lett. 2007;17:527. doi: 10.1016/j.bmcl.2006.10.008. [DOI] [PubMed] [Google Scholar]
- (691).Webster SP, Ward P, Binnie M, Craigie E, McConnell KM, Sooy K, Vinter A, Seckl JR, Walker BR. Bioorg. Med. Chem. Lett. 2007;17:2838. doi: 10.1016/j.bmcl.2007.02.057. [DOI] [PubMed] [Google Scholar]
- (692).Johansson L, Fotsch C, Bartberger MD, Castro VM, Chen M, Emery M, Gustafsson S, Hale C, Hickman D, Homan E, Jordan SR, Komorowski R, Li A, McRae K, Moniz G, Matsumoto G, Orihuela C, Palm G, Veniant M, Wang M, Williams M, Zhang J. J. Med. Chem. 2008;51:2933. doi: 10.1021/jm701551j. [DOI] [PubMed] [Google Scholar]
- (693).Su XD, Vicker N, Trusselle M, Halem H, Culler MD, Potter BVL. Mol. Cell. Endocrinol. 2009;301:169. doi: 10.1016/j.mce.2008.08.006. [DOI] [PubMed] [Google Scholar]
- (694).Nicolaou KC, Chen JS, Dalby SM. Bioorgan. Med. Chem. 2009;17:2290. doi: 10.1016/j.bmc.2008.10.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (695).Habich D, von Nussbaum F. ChemMedChem. 2006;1:951. doi: 10.1002/cmdc.200600145. [DOI] [PubMed] [Google Scholar]
- (696).Herath KB, Attygalle AB, Singh SB. J. Am. Chem. Soc. 2007;129:15422. doi: 10.1021/ja0758943. [DOI] [PubMed] [Google Scholar]
- (697).Smanski MJ, Peterson RM, Rajski SR, Shen B. Antimicrob. Agents Ch. 2009;53:1299. doi: 10.1128/AAC.01358-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (698).Jinchao Z, Dandan L, Yaping L, Jing S, Liwei W, Aimin Z. Mini-Rev. Med. Chem. 2009;9:1357. [Google Scholar]
- (699).Kelland LR, Barnard FJ, Evans IG, Murrer BA, Theobald BRC, Wyer SB, Goddard PM, Jones M, Valenti M, Bryant A, Rogers PM, Harrap KR. J. Med. Chem. 1995;38:3016. doi: 10.1021/jm00016a004. [DOI] [PubMed] [Google Scholar]
- (700).Zak F, Turanek J, Kroutil A, Sova P, Mistr A, Poulova A, Mikolin P, Zak Z, Kasna A, Zaluska D, Neca J, Sindlerova L, Kozubik A. J. Med. Chem. 2004;47:761. doi: 10.1021/jm030858+. [DOI] [PubMed] [Google Scholar]
- (701).Turanek J, Kasna A, Zaluska D, Neca J, Kvardova V, Knotigova P, Horvath V, L SI, Kozubik A, Sova P, Kroutil A, Zak F, Mistr A. Colloq. Inse. 2004;15:537. doi: 10.1097/01.cad.0000127147.57796.e5. [DOI] [PubMed] [Google Scholar]
- (702).Kozubik A, Horvath V, Svihalkova-Sindlerova L, Soucek K, Hofmanova J, Sova P, Kroutil A, Zak F, Mistr A, Turanek J. Biochem. Pharmacol. 2005;69:373. doi: 10.1016/j.bcp.2004.09.005. [DOI] [PubMed] [Google Scholar]
- (703).Sova P, Mistr A, Kroutil A, Zak F, Pouckova P, Zadinova M. Colloq. Inse. 2005;16:653. doi: 10.1097/00001813-200507000-00010. [DOI] [PubMed] [Google Scholar]
- (704).Sova P, Mistr A, Kroutil A, Zak F, Pouckova P, Zadinova M. Colloq. Inse. 2006;17:201. doi: 10.1097/00001813-200602000-00012. [DOI] [PubMed] [Google Scholar]
- (705).Horvath V, Blanarova O, Svihalkova-Sindlerova L, Soucek K, Hofmanova J, Sova P, Kroutil A, Fedorocko P, Kozubik A. Gynecol. Oncol. 2006;102:32. doi: 10.1016/j.ygyno.2005.11.016. [DOI] [PubMed] [Google Scholar]
- (706).Horvath V, Soucek K, Svihalkova-Sindlerova L, Vondracek J, Blanarova O, Hofmanova J, Sova P, Kozubik A. Invest. New Drug. 2007;25:435. doi: 10.1007/s10637-007-9062-7. [DOI] [PubMed] [Google Scholar]
- (707).Lane DP. Nature. 1992;358:15. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- (708).Prochazka L, Turanek J, Tesarik R, Knotigova P, Polaskova P, Andrysik Z, Kozubik A, Zak F, Sova P, Neuzil J, Machala M. Arch. Biochem. Biophys. 2007;462:54. doi: 10.1016/j.abb.2007.03.021. [DOI] [PubMed] [Google Scholar]
- (709).Hrstka R, Powell DJ, Kvardova V, Roubalova E, Bourougaa K, Candeias MM, Sova P, Zak F, Fahraeus R, Vojtesek B. Colloq. Inse. 2008;19:369. doi: 10.1097/CAD.0b013e3282f7f500. [DOI] [PubMed] [Google Scholar]
- (710).Roubalova E, Kvardova V, Hrstka R, Borilova S, Michalova E, Dubska L, Muller P, Sova P, Vojtesek B. Invest. New Drug. 2009;28:445. doi: 10.1007/s10637-009-9270-4. [DOI] [PubMed] [Google Scholar]
- (711).Kvardova V, Hrstka R, Walerych D, Muller P, Matoulkova E, Hruskova V, Stelclova D, Sova P, Vojtesek B. Mol. Cancer. 2010;9:147. doi: 10.1186/1476-4598-9-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (712).Vondalova Blanarova O, Jelinkova I, Szoor A, Skender BA, Soucek K, Horvath V, Vaculova A, Andera L, Sova P, Szollosi J, Hofmanova J, Vereb G, Kozubik A. Carcinogenesis. 2011;32:42. doi: 10.1093/carcin/bgq220. [DOI] [PubMed] [Google Scholar]
- (713).Halamikova A, Heringova P, Kasparkova J, Intini FP, Natile G, Nemirovski A, Gibson D, Brabec V. J. Inorg. Biochem. 2008;102:1077. doi: 10.1016/j.jinorgbio.2007.12.015. [DOI] [PubMed] [Google Scholar]
- (714).Narayanan VL, Sausville EA, Kaur G, Varma RK. Preparation of disubstituted lavendustin a analogs and their pharmaceutical compositions WO9943636A2. 1999 [Google Scholar]
- (715).Svingen PA, Tefferi A, Kottke TJ, Kaur G, Narayanan VL, Sausville EA, Kaufmann SH. Clin. Cancer Res. 2000;6:237. [PubMed] [Google Scholar]
- (716).Kaur G, Narayanan VL, Risbood PA, Hollingshead MG, Stinson SF, Varma RK, Sausville EA. Bioorgan. Med. Chem. 2005;13:1749. doi: 10.1016/j.bmc.2004.12.003. [DOI] [PubMed] [Google Scholar]
- (717).Mow BM, Chandra J, Svingen PA, Hallgren CG, Weisberg E, Kottke TJ, Narayanan VL, Litzow MR, Griffin JD, Sausville EA, Tefferi A, Kaufmann SH. Blood. 2002;99:664. doi: 10.1182/blood.v99.2.664. [DOI] [PubMed] [Google Scholar]
- (718).Avramis IA, Christodoulopoulos G, Suzuki A, Laug WE, Gonzalez-Gomez I, McNamara G, Sausville EA, Avramis VI. Cancer Chemoth. Pharm. 2002;50:479. doi: 10.1007/s00280-002-0507-6. [DOI] [PubMed] [Google Scholar]
- (719).Long J, Manchandia T, Ban K, Gao S, Miller C, Chandra J. Cancer Chemoth. Pharm. 2007;59:527. doi: 10.1007/s00280-006-0295-5. [DOI] [PubMed] [Google Scholar]
- (720).Mukhopadhyay I, Sausville EA, Doroshow JH, Roy KK. J. Biol. Chem. 2006;281:37330. doi: 10.1074/jbc.M605569200. [DOI] [PubMed] [Google Scholar]
- (721).Fer ND, Shoemaker RH, Monks A. J. Exp. Clin. Canc. Res. 2010;29 doi: 10.1186/1756-9966-29-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (722).Orsolic N, Golemovic M, Quintas-Cardama A, Scappini B, Manshouri T, Chandra J, Basic I, Giles F, Kantarjian H, Verstovsek S. Cancer Sci. 2006;97:952. doi: 10.1111/j.1349-7006.2006.00269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (723).Chandra J, Hackbarth J, Le S, Loegering D, Bone N, Bruzek LM, Narayanan VL, Adjei AA, Kay NE, Tefferi A, Karp JE, Sausville EA, Kaufmann SH. Blood. 2003;102:4512. doi: 10.1182/blood-2003-02-0562. [DOI] [PubMed] [Google Scholar]
- (724).Yu C, Rahmani M, Almenara J, Sausville EA, Dent P, Grant S. Oncogene. 2004;23:1364. doi: 10.1038/sj.onc.1207248. [DOI] [PubMed] [Google Scholar]
- (725).Hose C, Kaur G, Sausville EA, Monks A. Clin. Cancer Res. 2005;11:6370. doi: 10.1158/1078-0432.CCR-05-0291. [DOI] [PubMed] [Google Scholar]
- (726).Dasmahapatra G, Nguyen TK, Dent P, Grant S. Leukemia Res. 2006;30:1263. doi: 10.1016/j.leukres.2006.01.005. [DOI] [PubMed] [Google Scholar]
- (727).Chandra J, Tracy J, Loegering D, Flatten K, Verstovsek S, Beran M, Gorre M, Estrov Z, Donato N, Talpaz M, Sawyers C, Bhalla K, Karp J, Sausville E, Kaufmann SH. Blood. 2006;107:2501. doi: 10.1182/blood-2005-07-2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (728).Barnes DJ, De S, van HP, Moravcsik E, Melo JV. Leukemia. 2007;21:421. doi: 10.1038/sj.leu.2404533. [DOI] [PubMed] [Google Scholar]
- (729).Le SB, Hailer MK, Buhrow S, Wang Q, Flatten K, Pediaditakis P, Bible KC, Lewis LD, Sausville EA, Pang YP, Ames MM, Lemasters JJ, Holmuhamedov EL, Kaufmann SH. J. Biol. Chem. 2007;282:8860. doi: 10.1074/jbc.M611777200. [DOI] [PubMed] [Google Scholar]
- (730).Podar K, Raab MS, Tonon G, Sattler M, Barila D, Zhang J, Tai Y-T, Yasui H, Raje N, DePinho RA, Hideshima T, Chauhan D, Anderson KC. Cancer Res. 2007;67:1680. doi: 10.1158/0008-5472.CAN-06-1863. [DOI] [PubMed] [Google Scholar]
- (731).Stockwin LH, Bumke MA, Yu SX, Webb SP, Collins JR, Hollingshead MG, Newton DL. Clin. Cancer Res. 2007;13:3667. doi: 10.1158/1078-0432.CCR-07-0025. [DOI] [PubMed] [Google Scholar]
- (732).Zhang J, Sattler M, Tonon G, Grabher C, Lababidi S, Zimmerhackl A, Raab MS, Vallet S, Zhou Y, Cartron MA, Hideshima T, Tai YT, Chauhan D, Anderson KC, Podar K. Cancer Res. 2009;69:5082. doi: 10.1158/0008-5472.CAN-08-4603. [DOI] [PubMed] [Google Scholar]
- (733).Avramis IA, Laug WE, Sausville EA, Avramis VI. Cancer Chemoth. Pharm. 2003;52:307. doi: 10.1007/s00280-003-0668-y. [DOI] [PubMed] [Google Scholar]
- (734).Shanafelt TD, Lee YK, Bone ND, Strege AK, Narayanan VL, Sausville EA, Geyer SM, Kaufmann SH, Kay NE. Blood. 2005;105:2099. doi: 10.1182/blood-2004-06-2205. [DOI] [PubMed] [Google Scholar]
- (735).Yu C, Bruzek LM, Meng XW, Gores GJ, Carter CA, Kaufmann SH, Adjei AA. Oncogene. 2005;24:6861. doi: 10.1038/sj.onc.1208841. [DOI] [PubMed] [Google Scholar]
- (736).Dasmahapatra G, Rahmani M, Dent P, Grant S. Blood. 2006;107:232. doi: 10.1182/blood-2005-06-2302. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- (737).Liu WH, Chang LS. Biochem. J. 2011;439:453. doi: 10.1042/BJ20110725. [DOI] [PubMed] [Google Scholar]
- (738).Huang RL, Wallqvist A, Covell DG. J. Med. Chem. 2006;49:1964. doi: 10.1021/jm051029m. [DOI] [PubMed] [Google Scholar]
- (739).Li M, Wang H, Hill DL, Stinson S, Veley K, Grossi I, Peggins J, Covey JM, Zhang R. Cancer Chemoth. Pharm. 2006;57:607. doi: 10.1007/s00280-005-0094-4. [DOI] [PubMed] [Google Scholar]
- (740).Altucci L, Gronemeyer H. Nat. Rev. Cancer. 2001;1:181. doi: 10.1038/35106036. [DOI] [PubMed] [Google Scholar]
- (741).Charpentier B, Bernardon J-M, Eustache J, Millois C, Martin B, Michel S, Shroot B. J. Med. Chem. 1995;38:4993. doi: 10.1021/jm00026a006. [DOI] [PubMed] [Google Scholar]
- (742).Shroot B, Michel S. J. Am. Acad. Dermatol. 1997;36:S96. doi: 10.1016/s0190-9622(97)70050-1. [DOI] [PubMed] [Google Scholar]
- (743).Verschoore M, Langner A, Wolska H, Jablonska S, Czernielewski J, Schaefer H. Brit. J. Dermatol. 1991;124:368. doi: 10.1111/j.1365-2133.1991.tb00600.x. [DOI] [PubMed] [Google Scholar]
- (744).Griffiths CEM, Elder JT, Bernard BA, Rossio P, Cromie MA, Finkel LJ, Shroot B, Voorhees JJ. J. Invest. Dermatol. 1993;101:325. doi: 10.1111/1523-1747.ep12365480. [DOI] [PubMed] [Google Scholar]
- (745).Thiboutot DM, Weiss J, Bucko A, Eichenfield L, Jones T, Clark S, Liu Y, Graeber M, Kang S. J. Am. Acad. Dermatol. 2007;57:791. doi: 10.1016/j.jaad.2007.06.006. [DOI] [PubMed] [Google Scholar]
- (746).Milanese A, Gorincioi E, Rajabi M, Vistoli G, Santaniello E. Bioorg. Chem. 2011;39:151. doi: 10.1016/j.bioorg.2011.07.003. [DOI] [PubMed] [Google Scholar]
- (747).Bernard BA, Bernardon JM, Delescluse C, Martin B, Lenoir MC, Maignan J, Charpentier B, Pilgrim WR, Reichert U, Shroot B. Biochem. Bioph. Res. Co. 1992;186:977. doi: 10.1016/0006-291x(92)90842-9. [DOI] [PubMed] [Google Scholar]
- (748).Sun SY, Yue P, Dawson MI, Shroot B, Michel S, Lamph WW, Heyman RA, Teng M, Chandraratna RA, Shudo K, Hong WK, Lotan R. Cancer Res. 1997;57:4931. [PubMed] [Google Scholar]
- (749).Dawson MI, Hobbs PD, Peterson VJ, Leid M, Lange CW, Feng KC, Chen G, Gu J, Li H, Kolluri SK, Zhang X, Zhang Y, Fontana JA. Cancer Res. 2001;61:4723. [PubMed] [Google Scholar]
- (750).Ortiz MA, Bayon Y, Lopez-Hernandez FJ, Piedrafita FJ. Drug. Resist. Update. 2002;5:162. doi: 10.1016/s1368-7646(02)00050-x. [DOI] [PubMed] [Google Scholar]
- (751).Lorenzo P, Alvarez R, Ortiz MA, Alvarez S, Piedrafita FJ, de Lera AR. J. Med. Chem. 2008;51:5431. doi: 10.1021/jm800285f. [DOI] [PubMed] [Google Scholar]
- (752).Shroot B, Eustache J, Bernardon JM. Benzonaphthalenic derivatives and their use in pharmacy and cosmetics EP199636A1. 1986 [Google Scholar]
- (753).Gianni M, Zanotta S, Terao M, Garattini S, Garattini E. Biochem. Bioph. Res. Co. 1993;196:252. doi: 10.1006/bbrc.1993.2242. [DOI] [PubMed] [Google Scholar]
- (754).Shao ZM, Dawson MI, Li XS, Rishi AK, Sheikh MS, Han QX, Ordonez JV, Shroot B, Fontana JA. Oncogene. 1995;11:493. [PubMed] [Google Scholar]
- (755).Li XS, Rishi AK, Shao ZM, Dawson MI, Jong L, Shroot B, Reichert U, Ordonez J, Fontana JA. Cancer Res. 1996;56:5055. [PubMed] [Google Scholar]
- (756).Hsu CA, Rishi AK, SuLi X, Gerald TM, Dawson MI, Schiffer C, Reichert U, Shroot B, Poirer GC, Fontana JA. Blood. 1997;89:4470. [PubMed] [Google Scholar]
- (757).Sun S-Y, Yue P, Shroot B, Hong WK, Lotan R. J. Cell. Physiol. 1997;173:279. doi: 10.1002/(SICI)1097-4652(199711)173:2<279::AID-JCP36>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- (758).Schadendorf D, Kern MA, Artuc M, Pahl HL, Rosenbach T, Fichtner I, Nurnberg W, Stuting S, von Stebut E, Worm M, Makki A, Jurgovsky K, Kolde G, Henz BM. J. Cell Biol. 1996;135:1889. doi: 10.1083/jcb.135.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (759).Adachi H, Preston G, Harvat B, Dawson MI, Jetten AM. Am. J. Resp. Cell Mol. 1998;18:323. doi: 10.1165/ajrcmb.18.3.2974. [DOI] [PubMed] [Google Scholar]
- (760).Langdon SP, Rabiasz GJ, Ritchie AA, Reichert U, Buchan P, Miller WR, Smyth JF. Cancer Chemoth. Pharm. 1998;42:429. doi: 10.1007/s002800050841. [DOI] [PubMed] [Google Scholar]
- (761).Oridate N, Higuchi M, Suzuki S, Shroot B, Hong WK, Lotan R. Int. J. Cancer. 1997;70:484. doi: 10.1002/(sici)1097-0215(19970207)70:4<484::aid-ijc21>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- (762).Li Y, Lin BZ, Agadir A, Liu R, Dawson MI, Reed JC, Fontana JA, Bost F, Hobbs PD, Zheng Y, Chen GQ, Shroot B, Mercola D, Zhang XK. Mol. Cell. Biol. 1998;18:4719. doi: 10.1128/mcb.18.8.4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (763).Sun SY, Yue P, Shroot B, Hong WK, Lotan R. Oncogene. 1999;18:3894. doi: 10.1038/sj.onc.1202771. [DOI] [PubMed] [Google Scholar]
- (764).Sun SY, Yue P, Wu GS, El-Deiry WS, Shroot B, Hong WK, Lotan R. Cancer Res. 1999;59:2829. [PubMed] [Google Scholar]
- (765).Sun SY, Yue P, Wu GS, El-Deiry WS, Shroot B, Hong WK, Lotan R. Oncogene. 1999;18:2357. doi: 10.1038/sj.onc.1202543. [DOI] [PubMed] [Google Scholar]
- (766).Sun SY, Yue P, Hong WK, Lotan R. Cancer Res. 2000;60:7149. [PubMed] [Google Scholar]
- (767).Sun SY, Yue P, Hong WK, Lotan R. Cancer Res. 2000;60:6537. [PubMed] [Google Scholar]
- (768).Sun SY, Yue P, Chen X, Hong WK, Lotan R. Cancer Res. 2002;62:2430. [PubMed] [Google Scholar]
- (769).Gianni M, de The H. Leukemia. 1999;13:739. doi: 10.1038/sj.leu.2401419. [DOI] [PubMed] [Google Scholar]
- (770).Mologni L, Ponzanelli I, Bresciani F, Sardiello G, Bergamaschi D, Gianni M, Reichert U, Rambaldi A, Terao M, Garattini E. Blood. 1999;93:1045. [PubMed] [Google Scholar]
- (771).Zhang YX, Huang Y, Rishi AK, Sheikh MS, Shroot B, Reichert U, Dawson M, Poirier G, Fontana JA. Exp. Cell. Res. 1999;247:233. doi: 10.1006/excr.1998.4350. [DOI] [PubMed] [Google Scholar]
- (772).Ponzanelli I, Gianni M, Giavazzi R, Garofalo A, Nicoletti I, Reichert U, Erba E, Rambaldi A, Terao M, Garattini E. Blood. 2000;95:2672. [PubMed] [Google Scholar]
- (773).Zang Y, Beard RL, Chandraratna RA, Kang JX. Cell Death Differ. 2001;8:477. doi: 10.1038/sj.cdd.4400843. [DOI] [PubMed] [Google Scholar]
- (774).Lopez-Hernandez FJ, Ortiz MA, Bayon Y, Piedrafita FJ. Mol. Cancer Ther. 2003;2:255. [PubMed] [Google Scholar]
- (775).Hsu SL, Yin SC, Liu MC, Reichert U, Ho WL. Exp. Cell. Res. 1999;252:332. doi: 10.1006/excr.1999.4625. [DOI] [PubMed] [Google Scholar]
- (776).Gonda K, Tsuchiya H, Sakabe T, Akechi Y, Ikeda R, Nishio R, Terabayashi K, Ishii K, Matsumi Y, Ashla AA, Okamoto H, Takubo K, Matsuoka S, Watanabe Y, Hoshikawa Y, Kurimasa A, Shiota G. Biochem. Bioph. Res. Co. 2008;370:629. doi: 10.1016/j.bbrc.2008.04.008. [DOI] [PubMed] [Google Scholar]
- (777).Liang JY, Fontana JA, Rao JN, Ordonez JV, Dawson MI, Shroot B, Wilber JF, Feng P. Prostate. 1999;38:228. doi: 10.1002/(sici)1097-0045(19990215)38:3<228::aid-pros7>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- (778).Sun SY, Yue P, Lotan R. Oncogene. 2000;19:4513. doi: 10.1038/sj.onc.1203810. [DOI] [PubMed] [Google Scholar]
- (779).Farhana L, Dawson M, Rishi AK, Zhang YX, Van Buren E, Trivedi C, Reichert U, Fang GW, Kirschner MW, Fontana JA. Cancer Res. 2002;62:3842. [PubMed] [Google Scholar]
- (780).Jin FS, Liu XG, Zhou ZM, Yue P, Lotan R, Khuri FR, Chung LWK, Sun SY. Cancer Res. 2005;65:6354. doi: 10.1158/0008-5472.CAN-04-4061. [DOI] [PubMed] [Google Scholar]
- (781).Widschwendter M, Widschwendter A, Welte T, Daxenbichler G, Zeimet AG, Bergant A, Berger J, Peyrat JP, Michel S, Doppler W, Marth C. Brit. J. Cancer. 1999;79:204. doi: 10.1038/sj.bjc.6690034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (782).Fontana JA, Dawson MI, Leid M, Rishi AK, Zhang YX, Hsu CA, Lu JS, Peterson VJ, Jong L, Hobbs P, Chao WR, Shroot B, Reichert U. Int. J. Cancer. 2000;86:474. doi: 10.1002/(sici)1097-0215(20000515)86:4<474::aid-ijc5>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- (783).Belzacq AS, El Hamel C, Vieira HLA, Cohen I, Haouzi D, Metivier D, Marchetti P, Brenner C, Kroemer G. Oncogene. 2001;20:7579. doi: 10.1038/sj.onc.1204953. [DOI] [PubMed] [Google Scholar]
- (784).Rishi AK, Zhang LY, Boyanapalli M, Wali A, Mohammad RM, Yu YJ, Fontana JA, Hatfield JS, Dawson MI, Majumdar APN, Reichert U. J. Biol. Chem. 2003;278:33422. doi: 10.1074/jbc.M303173200. [DOI] [PubMed] [Google Scholar]
- (785).Holmes WF, Dawson MI, Soprano DR, Soprano KJ. J. Cell. Physiol. 2000;185:61. doi: 10.1002/1097-4652(200010)185:1<61::AID-JCP5>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- (786).Kumar A, Soprano DR, Parekh HK. Cancer Res. 2001;61:7552. [PubMed] [Google Scholar]
- (787).Holmes WF, Soprano DR, Soprano KJ. J. Biol. Chem. 2002;277:45408. doi: 10.1074/jbc.M204600200. [DOI] [PubMed] [Google Scholar]
- (788).Holmes WF, Soprano DR, Soprano KJ. Oncogene. 2003;22:6377. doi: 10.1038/sj.onc.1206694. [DOI] [PubMed] [Google Scholar]
- (789).Holmes WF, Soprano DR, Soprano KJ. J. Cell Biol. 2003;89:262. doi: 10.1002/jcb.10505. [DOI] [PubMed] [Google Scholar]
- (790).Jiang T, Soprano DR, Soprano KJ. J. Cell. Physiol. 2007;212:771. doi: 10.1002/jcp.21073. [DOI] [PubMed] [Google Scholar]
- (791).Matsuoka S, Tsuchiya H, Sakabe T, Watanabe Y, Hoshikawa Y, Kurimasa A, Itamochi H, Harada T, Terakawa N, Masutani H, Yodoi J, Shiota G. Cancer Sci. 2008;99:2485. doi: 10.1111/j.1349-7006.2008.00979.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (792).Watanabe Y, Tsuchiya H, Sakabe T, Matsuoka S, Akeehi Y, Fujimoto Y, Yamane K, Ikeda R, Nishio R, Terabayashi K, Ishii K, Gonda K, Matsumi Y, Ashla AA, Okamoto H, Takubo K, Tomita A, Hoshikawa Y, Kurimasa A, Itamochi H, Harada T, Terakawa N, Shiota G. Biochem. Bioph. Res. Co. 2008;366:840. doi: 10.1016/j.bbrc.2007.12.028. [DOI] [PubMed] [Google Scholar]
- (793).Marchetti P, Zamzami N, Joseph B, Schraen-Maschke S, Mereau-Richard C, Costantini P, Metivier D, Susin SA, Kroemer G, Formstecher P. Cancer Res. 1999;59:6257. [PubMed] [Google Scholar]
- (794).Joseph B, Marchetti P, Lefebvre O, Schraen-Maschke S, Mereau-Richard C, Formstecher P. BBA-Mol. Cell Res. 2003;1593:277. doi: 10.1016/s0167-4889(02)00399-3. [DOI] [PubMed] [Google Scholar]
- (795).Sun SY, Kurie JM, Yue P, Dawson MI, Shroot B, Chandraratna RA, Hong WK, Lotan R. Clin. Cancer Res. 1999;5:431. [PubMed] [Google Scholar]
- (796).Boisvieux-Ulrich E, Sourdeval M, Marano F. Exp. Cell. Res. 2005;307:76. doi: 10.1016/j.yexcr.2005.02.005. [DOI] [PubMed] [Google Scholar]
- (797).Lovat PE, Ranalli M, Bernassola F, Tilby M, Malcolm AJ, Pearson ADJ, Piacentini M, Melino G, Redfern CPF. Int. J. Cancer. 2000;88:977. doi: 10.1002/1097-0215(20001215)88:6<977::aid-ijc22>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- (798).Costa SL, Paillaud E, Fages C, Rochette-Egly C, Plassat JL, Jouault H, Perzelova A, Tardy M. Eur. J. Cancer. 2001;37:520. doi: 10.1016/s0959-8049(00)00430-5. [DOI] [PubMed] [Google Scholar]
- (799).Lena A, Rechichi M, Salvetti A, Bartoli B, Vecchio D, Scarcelli V, Amoroso R, Benvenuti L, Gagliardi R, Gremigni V, Rossi L. J. Transl. Med. 2009;7:13. doi: 10.1186/1479-5876-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (800).Sun SY, Yue P, Chandraratna RA, Tesfaigzi Y, Hong WK, Lotan R. Mol. Pharmacol. 2000;58:508. doi: 10.1124/mol.58.3.508. [DOI] [PubMed] [Google Scholar]
- (801).Zhang Y, Rishi AK, Dawson MI, Tschang R, Farhana L, Boyanapalli M, Reichert U, Shroot B, Van Buren EC, Fontana JA. Cancer Res. 2000;60:2025. [PubMed] [Google Scholar]
- (802).Hail N, Jr., Lotan R. J. Cell. Physiol. 2001;186:24. doi: 10.1002/1097-4652(200101)186:1<24::AID-JCP1006>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- (803).Wanner RW, Henseleit-Walter UH-W, Wittig BW, Kolde GK. J. Mol. Med. 2002;80:61. doi: 10.1007/s00109-001-0288-0. [DOI] [PubMed] [Google Scholar]
- (804).Hail N, Jr., Youssef EM, Lotan R. Cancer Res. 2001;61:6698. [PubMed] [Google Scholar]
- (805).Danielsson C, Torma H, Vahlquist A, Carlberg C. Int. J. Cancer. 1999;81:467. doi: 10.1002/(sici)1097-0215(19990505)81:3<467::aid-ijc22>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- (806).Zhao X, Demary K, Wong L, Vaziri C, McKenzie AB, Eberlein TJ, Spanjaard RA. Cell Death Differ. 2001;8:878. doi: 10.1038/sj.cdd.4400894. [DOI] [PubMed] [Google Scholar]
- (807).Pan M, Geng SM, Xiao SX, Ren JW, Liu Y, Li XL, Li ZX, Peng ZH. Arch. Dermatol. Res. 2009;301:15. doi: 10.1007/s00403-008-0902-x. [DOI] [PubMed] [Google Scholar]
- (808).Farso MC, Krantic S, Rubio M, Sarfati M, Quirion R. Neuroscience. 2011;192:172. doi: 10.1016/j.neuroscience.2011.06.053. [DOI] [PubMed] [Google Scholar]
- (809).Fontana JA, Rishi AK. Leukemia. 2002;16:463. doi: 10.1038/sj.leu.2402414. [DOI] [PubMed] [Google Scholar]
- (810).Debatin KM, Poncet D, Kroemer G. Oncogene. 2002;21:8786. doi: 10.1038/sj.onc.1206039. [DOI] [PubMed] [Google Scholar]
- (811).Zhao XS, Spanjaard RA. J. Biomed. Sci. 2003;10:44. doi: 10.1007/BF02255996. [DOI] [PubMed] [Google Scholar]
- (812).Holmes WF, Soprano DR, Soprano KJ. J. Cell. Physiol. 2004;199:317. doi: 10.1002/jcp.10338. [DOI] [PubMed] [Google Scholar]
- (813).Piedrafita FJ, Ortiz MA. Curr. Cancer Ther. Rev. 2006;2:185. [Google Scholar]
- (814).Sun SY, Yue P, Mao L, Dawson MI, Shroot B, Lamph WW, Heyman RA, Chandraratna RA, Shudo K, Hong WK, Lotan R. Clin. Cancer Res. 2000;6:1563. [PubMed] [Google Scholar]
- (815).Pfahl M, Piedrafita FJ. Oncogene. 2003;22:9058. doi: 10.1038/sj.onc.1207109. [DOI] [PubMed] [Google Scholar]
- (816).Zhang Y, Dawson MI, Mohammad R, Rishi AK, Farhana L, Feng KC, Leid M, Peterson V, Zhang XK, Edelstein M, Eilander D, Biggar S, Wall N, Reichert U, Fontana JA. Blood. 2002;100:2917. doi: 10.1182/blood.V100.8.2917. [DOI] [PubMed] [Google Scholar]
- (817).Cincinelli R, Dallavalle S, Merlini L, Penco S, Pisano C, Carminati P, Giannini G, Vesci L, Gaetano C, Illy B, Zuco V, Supino R, Zunino F. J. Med. Chem. 2003;46:909. doi: 10.1021/jm025593y. [DOI] [PubMed] [Google Scholar]
- (818).Dawson MI, Harris DL, Liu G, Hobbs PD, Lange CW, Jong L, Bruey-Sedano N, James SY, Zhang XK, Peterson VJ, Leid M, Farhana L, Rishi AK, Fontana JA. J. Med. Chem. 2004;47:3518. doi: 10.1021/jm030524k. [DOI] [PubMed] [Google Scholar]
- (819).Farhana L, Dawson MI, Huang Y, Zhang YX, Rishi AK, Reddy KB, Freeman RS, Fontana JA. Oncogene. 2004;23:1874. doi: 10.1038/sj.onc.1207311. [DOI] [PubMed] [Google Scholar]
- (820).Cincinelli R, Dallavalle S, Nannei R, Carella S, DeZani D, Merlini L, Penco S, Garattini E, Giannini G, Pisano C, Vesci L, Carminati P, Zuco V, Zanchi C, Zunino F. J. Med. Chem. 2005;48:4931. doi: 10.1021/jm049440h. [DOI] [PubMed] [Google Scholar]
- (821).Garattini E, Parrella E, Diomede L, Gianni M, Kalac Y, Merlini L, Simoni D, Zanier R, Ferrara FF, Chiarucci I, Carminati P, Terao M, Pisano C. Blood. 2004;103:194. doi: 10.1182/blood-2003-05-1577. [DOI] [PubMed] [Google Scholar]
- (822).Garattini E, Gianni M, Terao M. Curr. Pharm. Design. 2004;10:433. doi: 10.2174/1381612043453351. [DOI] [PubMed] [Google Scholar]
- (823).Dallavalle S, Zunino F. Expert Opin. Ther. Pat. 2005;15:1625. [Google Scholar]
- (824).Zuco V, Zanchi C, Cassinelli G, Lanzi C, Supino R, Pisano C, Zanier R, Giordano V, Garattini E, Zunino F. Cell Death Differ. 2004;11:280. doi: 10.1038/sj.cdd.4401304. [DOI] [PubMed] [Google Scholar]
- (825).Zanchi C, Zuco V, Lanzi C, Supino R, Zunino F. Cancer Res. 2005;65:2364. doi: 10.1158/0008-5472.CAN-04-2495. [DOI] [PubMed] [Google Scholar]
- (826).Pisano C, Vesci L, Fodera R, Ferrara FF, Rossi C, De Cesare M, Zuco V, Pratesi G, Supino R, Zunino F. Ann. Oncol. 2007;18:1500. doi: 10.1093/annonc/mdm195. [DOI] [PubMed] [Google Scholar]
- (827).Farhana L, Dawson MI, Fontana JA. Cancer Res. 2005;65:4909. doi: 10.1158/0008-5472.CAN-04-4124. [DOI] [PubMed] [Google Scholar]
- (828).Zuco V, Zanchi C, Lanzi C, Beretta GL, Supino R, Pisano C, Barbarino M, Zanier R, Bucci F, Aulicino C, Carminati P, Zunino F. Neoplasia. 2005;7:667. doi: 10.1593/neo.05127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (829).Parrella E, Gianni M, Fratelli M, Barzago MM, Raska I, Diomede L, Kurosaki M, Pisano C, Carminati P, Merlini L, Dallavalle S, Tavecchio M, Rochette-Egly C, Terao M, Garattini E. Mol. Pharmacol. 2006;70:909. doi: 10.1124/mol.106.023614. [DOI] [PubMed] [Google Scholar]
- (830).Cincinelli R, Dallavalle S, Nannei R, Merlini L, Penco S, Giannini G, Pisano C, Vesci L, Ferrara FF, Zuco V, Zanchi C, Zunino F. Bioorgan. Med. Chem. 2007;15:4863. doi: 10.1016/j.bmc.2007.04.057. [DOI] [PubMed] [Google Scholar]
- (831).Di Francesco AM, Meco D, Torella AR, Barone G, D'Incalci M, Pisano C, Carminati P, Riccardi R. Biochem. Pharmacol. 2007;73:643. doi: 10.1016/j.bcp.2006.10.033. [DOI] [PubMed] [Google Scholar]
- (832).Farhana L, Dawson MI, Leid M, Wang L, Moore DD, Liu G, Xia Z, Fontana JA. Cancer Res. 2007;67:318. doi: 10.1158/0008-5472.CAN-06-2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (833).Dawson MI, Xia Z, Liu G, Ye M, Fontana JA, Farhana L, Patel BB, Arumugarajah S, Bhuiyan M, Zhang XK, Han YH, Stallcup WB, Fukushi J, Mustelin T, Tautz L, Su Y, Harris DL, Waleh N, Hobbs PD, Jong L, Chao WR, Schiff LJ, Sani BP. J. Med. Chem. 2007;50:2622. doi: 10.1021/jm0613323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (834).Dawson MI, Xia Z, Jiang T, Ye M, Fontana JA, Farhana L, Patel B, Xue LP, Bhuiyan M, Pellicciari R, Macchiarulo A, Nuti R, Zhang XK, Han YH, Tautz L, Hobbs PD, Jong L, Waleh N, Chao WR, Feng GS, Pang Y, Su Y. J. Med. Chem. 2008;51:5650. doi: 10.1021/jm800456k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (835).Valli C, Paroni G, Di Francesco AM, Riccardi R, Tavecchio M, Erba E, Boldetti A, Gianni M, Fratelli M, Pisano C, Merlin L, Antoccia A, Cenciarelli C, Terao M, Garattini E. Mol. Cancer Ther. 2008;7:2941. doi: 10.1158/1535-7163.MCT-08-0419. [DOI] [PubMed] [Google Scholar]
- (836).Farhana L, Dawson MI, Dannenberg JH, Xu L, Fontana JA. Mol. Cancer Ther. 2009;8:1625. doi: 10.1158/1535-7163.MCT-08-0964. [DOI] [PubMed] [Google Scholar]
- (837).Dawson MI, Fontana JA. Mini-Rev. Med. Chem. 2010;10:455. doi: 10.2174/138955710791384045. [DOI] [PubMed] [Google Scholar]
- (838).Farhana L, Dawson MI, Xia Z, Aboukameel A, Xu L, Liu G, Das JK, Hatfield J, Levi E, Mohammad R, Fontana JA. Mol. Cancer Ther. 2010;9:2903. doi: 10.1158/1535-7163.MCT-10-0546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (839).Xia Z, Farhana L, Correa RG, Das JK, Castro DJ, Yu J, Oshima RG, Reed JC, Fontana JA, Dawson MI. J. Med. Chem. 2011;54:3793. doi: 10.1021/jm200051z. [DOI] [PubMed] [Google Scholar]
- (840).Zhang Y, Soto J, Park K, Viswanath G, Kuwada S, Abel ED, Wang L. Mol. Cell. Biol. 2010;30:1341. doi: 10.1128/MCB.01076-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (841).Zhang L, Wali A, Fontana JA, Dawson MI, Rishi AK. J. Mol. Signal. 2010;5:12. doi: 10.1186/1750-2187-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (842).Hashiguchi K, Tsuchiya H, Tomita A, Ueda C, Akechi Y, Sakabe T, Kurimasa A, Nozaki M, Yamada T, Tsuchida S, Shiota G. Biochem. Bioph. Res. Co. 2010;391:621. doi: 10.1016/j.bbrc.2009.11.109. [DOI] [PubMed] [Google Scholar]
- (843).Zuco V, Benedetti V, De Cesare M, Zunino F. Int. J. Cancer. 2010;126:1246. doi: 10.1002/ijc.24819. [DOI] [PubMed] [Google Scholar]
- (844).Farhana L, Dawson MI, Murshed F, Fontana JA. Cell Death Differ. 2011;18:164. doi: 10.1038/cdd.2010.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (845).Milli A, Perego P, Beretta GL, Corvo A, Righetti PG, Carenini N, Corna E, Zuco V, Zunino F, Cecconi D. J. Proteome Res. 2011 doi: 10.1021/pr100963n. in press. [DOI] [PubMed] [Google Scholar]
- (846).Miao J, Choi SE, Seok SM, Yang L, Zuercher WJ, Xu Y, Willson TM, Xu HE, Kemper JK. Mol. Endocrinol. 2011;25:1159. doi: 10.1210/me.2011-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (847).Mishur RJ, Griffin ME, Battle CH, Shan B, Jayawickramarajah J. Bioorg. Med. Chem. Lett. 2011;21:857. doi: 10.1016/j.bmcl.2010.11.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (848).Bleidner WE, Harmon JB, Hewes WE, Lynes TE, Hermann EC. J. Pharmacol. Exp. Ther. 1965;150:484. [PubMed] [Google Scholar]
- (849).Koppel C, Tenczer J. Biomed. Mass. Spectrom. 1985;12:499. doi: 10.1002/bms.1200120910. [DOI] [PubMed] [Google Scholar]
- (850).Wesemann W, Schollmeyer JD, Sturm G. Arznei-Forschung. 1982;32:1243. [PubMed] [Google Scholar]
- (851).Rubio FR, Fukuda EK, Garland WA. Drug Metab. Dispos. 1988;16:773. [PubMed] [Google Scholar]
- (852).Brown SY, Garland WA, Fukuda EK. Drug Metab. Dispos. 1990;18:546. [PubMed] [Google Scholar]
- (853).Loh AC, Szuna AJ, Williams TH, Sasso GJ, Leinweber FJ. Drug Metab. Dispos. 1991;19:381. [PubMed] [Google Scholar]
- (854).Kajbaf M, Marchioro C, Barnaby JR, Pellegatti M. Xenobiotica. 1998;28:785. doi: 10.1080/004982598239209. [DOI] [PubMed] [Google Scholar]
- (855).Kajbaf M, Rossato P, Barnaby JR, Pellegatti M. Xenobiotica. 1998;28:167. doi: 10.1080/004982598239669. [DOI] [PubMed] [Google Scholar]
- (856).He H, Tran P, Yin H, Smith H, Batard Y, Wang L, Einolf H, Gu H, Mangold JB, Fischer V, Howard D. Drug Metab. Dispos. 2009;37:536. doi: 10.1124/dmd.108.023010. [DOI] [PubMed] [Google Scholar]
- (857).He H, Tran P, Yin H, Smith H, Flood D, Kramp R, Filipeck R, Fischer V, Howard D. Drug Metab. Dispos. 2009;37:545. doi: 10.1124/dmd.108.023002. [DOI] [PubMed] [Google Scholar]
- (858).Fura A, Khanna A, Vyas V, Koplowitz B, Chang SY, Caporuscio C, Boulton DW, Christopher LJ, Chadwick KD, Hamann LG, Humphreys WG, Kirby M. Drug Metab. Dispos. 2009;37:1164. doi: 10.1124/dmd.108.026088. [DOI] [PubMed] [Google Scholar]
- (859).Bhamidipati RK, Morizzi J, Chiu FC, Shackleford DM, Charman SA. J. Chromatogr. B. 2009;877:2989. doi: 10.1016/j.jchromb.2009.07.015. [DOI] [PubMed] [Google Scholar]
- (860).Sala F, Zucchetti M, Bagnati R, D'Incalci M, Pace S, Capocasa F, Marangon E. J. Chromatogr. B. 2009;877:3118. doi: 10.1016/j.jchromb.2009.08.001. [DOI] [PubMed] [Google Scholar]
- (861).Raag R, Poulos TL. Biochemistry. 1991;30:2674. doi: 10.1021/bi00224a016. [DOI] [PubMed] [Google Scholar]
- (862).Fokin AA, Tkachenko BA, Gunchenko PA, Gusev DV, Schreiner PR. Chem.-Eur. J. 2005;11:7091. doi: 10.1002/chem.200500031. [DOI] [PubMed] [Google Scholar]
- (863).Tkachenko BA, Fokina NA, Chernish LV, Dahl JEP, Liu SG, Carlson RMK, Fokin AA, Schreiner PR. Org. Lett. 2006;8:1767. doi: 10.1021/ol053136g. [DOI] [PubMed] [Google Scholar]
- (864).Fokina NA, Tkachenko BA, Merz A, Serafin M, Dahl JEP, Carlson RMK, Fokin AA, Schreiner PR. Eur. J. Org. Chem. 2007:4738. [Google Scholar]
- (865).Schwertfeger H, Wuertele C, Serafin M, Hausmann H, Carlson RMK, Dahl JEP, Schreiner PR. J. Org. Chem. 2008;73:7789. doi: 10.1021/jo801321s. [DOI] [PubMed] [Google Scholar]
- (866).Fokin AA, Zhuk TS, Pashenko AE, Dral PO, Gunchenko PA, Dahl JEP, Carlson RMK, Koso TV, Serafin M, Schreiner PR. Org. Lett. 2009;11:3068. doi: 10.1021/ol901089h. [DOI] [PubMed] [Google Scholar]
- (867).Schwertfeger H, Wurtele C, Hausmann H, Dahl JEP, Carlson RMK, Fokin AA, Schreiner PR. Adv. Synth. Catal. 2009;351:1041. [Google Scholar]
- (868).Schwertfeger H, Wurtele C, Schreiner PR. Synlett. 2010;3:493. [Google Scholar]
- (869).Wanka L, Cabrele C, Vanejews M, Schreiner PR. Eur. J. Org. Chem. 2007;9:1474. [Google Scholar]
- (870).Black RM. J. Chem. Soc. Perk. T 1. 1982;1:73. [Google Scholar]
- (871).Lee GS, Bashara JN, Sabih G, Oganesyan A, Godjoian G, Duong HM, Marinez ER, Gutierrez CG. Org. Lett. 2004;6:1705. doi: 10.1021/ol036526g. [DOI] [PubMed] [Google Scholar]