

While working to understand how senescence is maintained in the presence of oncogenic Ras, Deschênes-Simard et al. find that attenuating ERK signaling prevents Ras-induced senescence. With ERK signaling inhibited, oncogenic Ras is then able to transform cells. ERK signaling promotes the proteasome-dependent degradation of proteins required for critical processes, such as cell cycle progression. In prostate neoplasms, high levels of phosphorylated ERK correlated with a lower Gleason score and delayed disease relapse after treatment.
Keywords: ERK, benign prostatic hyperplasia, oncogenic ras, proteasome, senescence
Abstract
Constitutive activation of growth factor signaling pathways paradoxically triggers a cell cycle arrest known as cellular senescence. In primary cells expressing oncogenic ras, this mechanism effectively prevents cell transformation. Surprisingly, attenuation of ERK/MAP kinase signaling by genetic inactivation of Erk2, RNAi-mediated knockdown of ERK1 or ERK2, or MEK inhibitors prevented the activation of the senescence mechanism, allowing oncogenic ras to transform primary cells. Mechanistically, ERK-mediated senescence involved the proteasome-dependent degradation of proteins required for cell cycle progression, mitochondrial functions, cell migration, RNA metabolism, and cell signaling. This senescence-associated protein degradation (SAPD) was observed not only in cells expressing ectopic ras, but also in cells that senesced due to short telomeres. Individual RNAi-mediated inactivation of SAPD targets was sufficient to restore senescence in cells transformed by oncogenic ras or trigger senescence in normal cells. Conversely, the anti-senescence viral oncoproteins E1A, E6, and E7 prevented SAPD. In human prostate neoplasms, high levels of phosphorylated ERK were found in benign lesions, correlating with other senescence markers and low levels of STAT3, one of the SAPD targets. We thus identified a mechanism that links aberrant activation of growth signaling pathways and short telomeres to protein degradation and cellular senescence.
Normal mammalian cells respond to oncogenic threats by triggering intrinsic tumor suppression mechanisms that curtail cell cycle progression and execute cell death or permanent cell cycle arrest (Lowe et al. 2004). These outcomes were originally discovered by studying the response of normal cells to Myc overexpression, which induces apoptosis (Evan et al. 1992), or Ras overexpression, which induces a terminal cell cycle arrest known as cellular senescence (Serrano et al. 1997). Subsequently, these mechanisms have been demonstrated in animal models expressing these oncogenes (Braig et al. 2005; Chen et al. 2005; Murphy et al. 2008; DeNicola et al. 2011). Apoptosis is a programmed response where proteolytic enzymes digest selective cellular targets, leading to a noninflammatory cell death process and eventually the engulfing of apoptotic cells by the immune system (Leist and Jaattela 2001). The effector mechanism of senescence remains unknown, although these cells seem to be also programmed to interact with the immune system, secreting large amounts of cytokines (Coppe et al. 2010) and being the target of immune-mediated clearance (Xue et al. 2007; Kang et al. 2011).
Ras-induced senescence depends on the concerted action of the p53, p16INK4a/RB, and PML tumor suppressor pathways (Serrano et al. 1997; Ferbeyre et al. 2000). The activation of p53 by ras and other oncogenes involves the DNA damage response (DDR) (Bartkova et al. 2006; Di Micco et al. 2006; Mallette et al. 2007), a consequence of DNA damage triggered by oncogenic activity. This DNA damage could be the result of a replication stress induced by aberrant activation of replication forks or the increased production of mitochondrial reactive oxygen species (ROS) (Mallette and Ferbeyre 2007). However there is still a gap between our current view of oncogene signaling and the molecular events leading to DNA damage. In addition, senescence can occur in the absence of DNA damage. For example, in normal fibroblasts expressing oncogenic ras, the inactivation of the DDR was not sufficient to bypass senescence (Mallette et al. 2007), and in mice, there is no evidence linking DNA damage to Ras-induced senescence (Efeyan et al. 2009). Other effects of oncogenic activity have been proposed as mediators of senescence and may compensate for or cooperate with the DDR. These include p19ARF expression (Ferbeyre et al. 2002), autophagy (Young et al. 2009), mitochondrial dysfunction (Moiseeva et al. 2009), cytokines (Coppe et al. 2010), PML bodies (Vernier et al. 2011), and heterochromatin formation (Narita et al. 2003). However, it remains puzzling why constitutive growth factor signaling pathways and the Ras/ERK pathway trigger these proliferation barriers in normal cells.
Here we performed an unbiased screen using shRNA libraries to discover genes required for oncogenic ras to regulate senescence in human normal fibroblasts. We identified the ERK/MAPK as essential mediators of senescence and surprisingly found that attenuating ERK expression in human or mouse primary fibroblasts allowed their transformation by oncogenic ras. We also found that aberrant Ras/ERK signaling led to a proteasome-dependent protein degradation process targeting proteins required for cell cycle progression, cell migration, mitochondrial functions, RNA metabolism, and cell signaling. These findings were validated in cells with short telomeres and in human prostate benign neoplasms where we found expression of senescence markers, high levels of phospho-ERK, and low levels of STAT3, one of the targets of the senescence-associated protein degradation (SAPD) process.
Results
ERK/MAPK are required for Ras-induced senescence in human fibroblasts
In order to identify genes that contribute to oncogene-induced senescence (OIS), we performed RNAi screening looking for shRNAs able to bypass Ras-induced senescence in human fibroblasts (Supplemental Fig. S1A). shRNAs targeting ERK2 (MAPK1) and HMGB1 were recovered from the screening. We confirmed the senescence bypass using several shRNAs against ERK1 and ERK2 that were all capable of inhibiting RasV12-induced senescence (Supplemental Fig. S1B). We found a good correlation between the degree of total ERK inhibition and the bypass of senescence (Supplemental Fig. S1B). More important, since several shRNAs against ERK1 or ERK2 bypassed Ras-induced senescence, it is very unlikely that off-target effects of shRNAs were responsible for the bypass. To further characterize the consequences of ERK inhibition for RAS signaling, we then used one shRNA that efficiently inhibited ERK2, the more abundant ERK isoform in fibroblasts. This shRNA did not inhibit the members of the pathway upstream of ERK (RAS, RAF, and MEK) (Fig. 1A) but efficiently inhibited the expression of several transcriptional targets of the ERK pathway (Fig. 1B; Pratilas et al. 2009). Interestingly, the phosphorylation of p90RSK at Ser380 did not decrease upon inhibition of ERK2 (Fig. 1A), suggesting that another kinase activated by RasV12 can catalyze this event or that the remaining levels of phospho-ERK in cells expressing shERK are sufficient to do it. ERK2 knockdown in cells expressing RasV12 inhibited the induction of senescence-associated β-galactosidase (SA-β-Gal) (Fig. 1C), PML bodies, and DNA damage foci (Supplemental Fig. S1C–G). Oncogenic ras engaged the p53/p21, p16INK4a/RB, and p38MAPK pathways in primary cells, and this was efficiently prevented by knockdown of ERK2 (Fig. 1D; Supplemental Fig. S1H,I). The induction of several senescence-associated cytokine genes by RasV12 was also efficiently blocked by ERK2 knockdown (Supplemental Fig. S1J–L). The inhibition of Ras-induced senescence by several shERKs was accompanied by the adoption of the distinctive cell morphology of small cells growing sometimes on top of each other (Fig. 1C), a complete rescue of the proliferation arrest (Fig. 1E), a stimulation of DNA synthesis as measured by BrdU incorporation and KI-67 staining (Fig. 1F), and the expression of mitotic markers such as phospho-H3S10 or phospho-H3S28 (Fig. 1D). Moreover, the high levels of ROS known to contribute to DNA damage during Ras-induced senescence (Moiseeva et al. 2009) were decreased in cells depleted of ERK2 (Fig. 1G). Taken together, the results indicate that reducing ERK levels shuts down the senescence tumor suppression response to oncogenic ras in normal human fibroblasts.
Figure 1.

ERK/MAPK inhibition bypasses Ras-induced senescence. (A) Immunoblots for proteins in the ERK pathway using extracts from fibroblasts expressing H-RasV12 (R) or an empty vector (V) and shRNA against ERK2 (shERK) or a nontargeting shRNA (shCTR) obtained from cells 14 d after infection. (B) Quantitative PCR (qPCR) for ERK2 mRNA and mRNAs encoded by ERK-stimulated genes in cells as in A. (C) SA-β-Gal of cells as in A. Data were quantified from 100 cell counts in triplicate and are presented as the mean percentage of positive cells ± standard deviation (SD). (D) Immunoblots for cell cycle-regulated proteins in cells as in A. (E) Growth curves started with cells as in A. Data are presented as mean ± SD of triplicates. (F) Quantitation of BrdU incorporation (2 h of incubation with 10 μM BrdU) and KI-67 staining in cells as in A. Data were quantified from 100 cell counts in triplicate and are presented as the mean of positive cells ± SD. (***) P < 0.0005, two-sample t-test. (G) Superoxide levels in cells as before, measured by flow cytometry (FACS) after staining with 1 μM fluorescent probe dihydroethidium (DHE) during 1 h. The results are expressed as the percentage of maximum cell number. The maximum cell number is the number of cells for the most-represented fluorescence intensity in the cell population of a condition and is expressed as 100%. All experiments were performed a minimum of three times.
To assess the generality of these findings, we next studied the induction of senescence by oncogenic ras in primary human mammary epithelial cells (HMECs). Introduction of oncogenic ras by retroviral gene transfer in these cells induced a senescent phenotype, characterized by induction of PML bodies, DNA damage foci, and cell cycle arrest (Supplemental Fig. S2A–D). We also noticed that in cultures of HMECs expressing RasV12, some cells spontaneously escaped from senescence and started proliferating as small cells. All of these cells turned out to express very low levels of RasGTP and phospho-ERK (Supplemental Fig. S2E), consistent with the requirement for strong ERK/MAPK kinase signaling to sustain Ras-induced senescence. Then, we studied the effect of ERK2 knockdown on Ras-induced senescence in HMECs. As described for human fibroblasts, shERK2 (Fig. 2A) reduced the expression of ERK-dependent targets (Fig. 2B) and restored cell proliferation (Fig. 2C) and the expression of the mitotic markers phospho-H3S10 and phospho-H3S28. shERK2 allowed RB phosphorylation in HMECs expressing oncogenic ras, and the expression of E2F target genes such as MCM6 (Fig. 2D). shERK2 also prevented the induction of p53 target genes and cytokines by oncogenic ras in HMECs (Fig. 2E). The senescent phenotype was bypassed by shERK2 with a frequency several times higher than the spontaneous escape from senescence mentioned above (Fig. 2F,G).
Figure 2.

ERK/MAP kinases play a general role in cellular senescence. (A–G) Role of ERK/MAPK in H-RasV12-induced senescence in primary HMECs. (A) Immunoblots to confirm ERK knockdown and expression levels of H-RasV12 in extracts obtained 14 d after infection. (B) qPCR for ERK target genes to confirm the biological effect of ERK knockdown in cells expressing the indicated vectors. (C) qPCR for KI-67, a proliferation marker, in cells as in A. (D) Immunoblots for proteins in the RB pathway and mitosis markers in cells as in A. (E) qPCR for senescence markers in cells as in A. (F) Morphology of HMECs expressing H-RasV12-ER and shRNA against ERK2 (shERK) or a nontargeting shRNA (shCTR). Note the large size and vacuolated cytoplasm of senescent cells in contrast with the small growing cells that escape from senescence (arrows) due to low phospho-ERK levels. (G) Quantification of the bypass from senescence between cells with shControl (shCTR) and shERK2. Error bars represent SD. (**) P < 0.005, two-sample t-test. (H–P) Genetic inactivation of Erk2 bypasses Ras-induced senescence in MEFs. (H) Immunoblots for the indicated proteins in wild-type and Erk2−/− MEFs expressing H-RasV12, a vector control, or H-RasV12 + E1A 14 d after infection. (I) qPCR for Erk target genes in cells as in H. (J) SA-β-Gal markers in cells as in H. Data were quantified from 100 cell counts in triplicate and are presented as the mean percentage of positive cells ± SD. (K) Growth curves started with MEFs from wild-type and Erk2−/− animals expressing H-RasV12 or a vector control 14 d after infection. Data are presented as the mean ± SD of triplicates. (L–N) qPCR for the indicated genes in cells as in H. (O) Immunoblots for the indicated senescence markers in cells as in H. (P) Immunoblots against p53 and p53S15 from Erk2−/− MEFs treated for 24 h with doxorubicin (300 ng/mL) or vehicle. Experiments were performed n ≥ 3.
To demonstrate that reducing ERK levels in a genetic model also prevents Ras-induced senescence, we used mouse embryonic fibroblasts (MEFs) from Erk2 knockout mice (Voisin et al. 2010). In wild-type MEFs, oncogenic Ras induced Erk1/2 phosphorylation (Fig. 2H) and the expression of Erk target genes (Fig. 2I). The oncoprotein E1A, able to bypass Ras-induced senescence (Serrano et al. 1997), reduced Erk1/2 activation and also the expression of Erk target genes (Fig. 2H,I). In Erk2−/− MEFs, oncogenic Ras induced Erk1 but not Erk2 phosphorylation (Fig. 2H). This reduction in overall Erk activity was translated in a reduced induction of Erk target genes (Fig. 2I). More important, oncogenic Ras failed to induce growth arrest, p53, and senescence in Erk2−/− MEFs, in clear contrast to its effects in wild-type MEFs (Fig. 2J–O). In fact, Erk2−/− MEFs had a response to oncogenic ras similar to that of wild-type MEFs expressing E1A (Fig. 2J). We also looked at the expression of the tumor suppressor p19ARF in Erk2-null cells expressing RasV12. We found that RasV12 still induced p19ARF in these cells (Fig. 2O), but obviously, this was not sufficient to trigger senescence. We also found that p53 was stabilized and phosphorylated at Ser15 after treatment of Erk2−/− MEFs with doxorubicin, indicating that their resistance to Ras-induced senescence was not the result of an accidental loss of p53 (Fig. 2P).
Finally, we studied whether inhibition of ERK activity, not levels, was also sufficient to bypass RasV12-induced senescence. We used the MEK inhibitors U0126 and AZD6244, which inhibited ERK phosphorylation induced by RasV12 in human fibroblasts in a dose-dependent manner (Supplemental Fig. S2F) without altering ERK mRNA levels (Supplemental Fig. S2G). MEK inhibitors also prevented the induction of ERK target genes (Supplemental Fig. S2H,I) and restored RB phosphorylation and the expression of E2F targets in cells expressing RasV12 (Supplemental Fig. S2F). In agreement with previous results (Lin et al. 1998), MEK inhibitors blocked Ras-induced senescence (Supplemental Fig. S2J), growth arrest (Supplemental Fig. S2K–M), and the induction of the senescence genes CDKN1A and CDKN2A (Supplemental Fig. S2N). We thus conclude that reducing ERK activity by different manipulations in both human and mouse primary cells compromises Ras-induced senescence, preventing the induction of several tumor suppressor pathways. Next, we asked whether oncogenic ras was capable of transforming cells where reduced ERK levels prevented the activation of tumor suppressors.
ERK/MAPK knockdown facilitates Ras-dependent transformation of primary human cells
To investigate the effect of ERK inhibition in ras-dependent transformation, we first used the experimental system where RasV12 cooperates with hTERT and the SV40 early region to transform normal human fibroblasts (Hahn et al. 2002). We reasoned that ERK2 inhibition could be genetically equivalent to the SV40 early region, which also inhibits the activation of p53 and RB by RasV12 (Hahn et al. 2002). We first prepared IMR90 cells expressing hTERT using a lentiviral vector. Then, we introduced vectors expressing shERK2 and RasV12 or the corresponding control vectors and confirmed the knockdown of ERK in these cells and the expression of oncogenic ras (Supplemental Fig. S3A). We found that ERK inhibition by shERK2 in RasV12-expressing cells enabled colony formation in soft agar (Fig. 3A, top) and inhibited the process of contact inhibition, as seen in a focus assay (Fig. 3A, middle). These two assays are strong indications of the transformation of normal cells into cancer cells. Consistent with these ex vivo models, primary human fibroblasts expressing hTERT, RasV12, and shERK2 formed tumors in nude mice (Fig. 3A [bottom], E). Upon histological examination, these tumors were characterized by large nuclei, abundant mitotic images, many blood vessels, and very low levels of phospho-ERK in the tumor cells (Supplemental Fig. S3B). These low levels of ERK were confirmed by immunoblots from tumor cells extracts (Supplemental Fig. S3C) or in cell lines established from the tumors (Supplemental Fig. S3D). We also found that adding c-MYC to the cells expressing hTERT, RasV12, and shERK2 further enhanced their transformation (Fig. 3A,E). When tested for their ability to grow on a monolayer of normal fibroblasts, cells recovered from these tumors formed as many colonies in focus assays as the parent populations (Supplemental Fig. S3E). In conjunction, these results suggest that cells expressing oncogenic ras and shERK2 did not undergo additional genetic changes in vivo that would have further enhanced their transformed phenotype. As shown before for the senescence bypass, a different anti-ERK shRNA cooperated with oncogenic ras to transform primary human fibroblasts or HMECs (Supplemental Fig. S3F–I).
Figure 3.

ERK/MAPK inhibition promotes Ras-induced transformation. (A, top) Soft agar assay with HeLa cells or IMR90 fibroblasts expressing the indicated vectors. Representative GFP-positive colonies are shown. A focus-forming assay (middle) and tumor formation in nude mice (bottom) were performed with cells as above. Numbers of colonies in soft agar and focus-forming assays are expressed as the mean ± SD of triplicates. (B) Soft agar assay with wild-type and Erk2−/− MEFs expressing the indicated vectors. (C) Quantification of B using the CyQuant GR dye. (RFU) Relative fluorescence units (a measure of growth in soft agar). Data are presented as mean ± SD of triplicates. (D) Tumor formation in nude mice of wild-type (WT) and Erk2−/− MEFs expressing the indicated vectors. (E) Quantification of tumor formation in nude mice. The number of injections that generated tumors and the time taken by the tumors to reach the threshold of significance (0.2 cm3) are shown.
Finally, we observed that oncogenic ras was able to transform primary MEFs from Erk2 knockout animals without the need to express any other cooperating oncogene (Fig. 3B–E). As shown for human fibroblasts, the ability of cells transformed by oncogenic Ras in Erk2 knockout MEFs to form colonies in soft agar was the same before and after forming tumors in mice, suggesting that no other genetic modifications occurred in vivo to further transform these cells (Supplemental Fig. S3J,K). As shown before (Serrano et al. 1997), oncogenic ras cooperated with E1A to transform primary rodent cells (Fig. 3B–E). In E1A-expressing MEFs, both phospho-Erk1 and phospho-Erk2 were reduced (Fig. 2H). The same was noticed in human cells (Supplemental Fig. S3A,C,D). Since decreasing ERK activity is sufficient to bypass Ras-induced senescence and promote transformation, these results suggest a novel mechanism by which E1A cooperates with oncogenic ras to transform primary cells. Taken together, these results reveal a tumor suppressor function of the ERK kinases in normal cells and dissociation between the transforming functions of ras, which do not require high ERK activity, and its ability to induce senescence, which requires high ERK activity.
Selective ERK-dependent protein degradation characterizes cellular senescence
Next, we addressed the mechanism by which the strength of ERK activation induces the stress signaling pathways leading to senescence. We used vectors able to drive either high or low expression levels of RasV12. As expected, high levels of RasV12 led to higher phosphorylation of ERK1/2 and some ERK targets such as ELKS383 and FAKS910 (Supplemental Fig. S4A). Low levels of RasV12 slightly stimulated ERK phosphorylation and p53 target gene expression (Supplemental Fig. S4A,B) but did not engage the RB tumor suppressor pathway (Supplemental Fig. S4B) and also failed to induce growth arrest, the DDR, and senescence (Supplemental Fig. S4C–H). We used these vectors and a battery of phospho-specific antibodies (Kinexus) to profile the state of ERK phosphorylation targets depending on the strength of Ras signaling. We found, as anticipated, that some ERK targets were highly phosphorylated in cells expressing high levels of RasV12. However, we also found unexpectedly that some phospho-ERK targets were less phosphorylated (Fig. 4A). Intriguingly, the reduction in phosphorylation of STAT3S727 and CaldesmonS789 in cells expressing high levels of Ras was also observed at the level of total protein (Supplemental Fig. S4I). As protein phosphorylation and protein degradation are often linked (Hunter 2007), we thought that one mechanism connecting high ERK signaling to senescence might involve the degradation of phosphorylated proteins due to aberrant ERK signaling.
Figure 4.

Selective and proteasome-dependent protein degradation characterizes Ras-induced senescence. (A) Summary of proteomic data obtained from cell lysates of IMR90 fibroblasts expressing a low level (LR) or high level (HR) of oncogenic ras. Cells were harvested 14 d after infection. Data show phosphorylation levels of 14 ERK targets measured by Western blot with phospho-specific antibodies by Kinexus (n = 1). The difference in the relative protein amount in cells that express HR is presented as a percentage of the relative protein amount in LR-expressing cells set as a reference (control). (B) Frequencies of phosphorylation motifs in phosphopeptides stabilized by MG132. Ras senescent cells (10 d after infection with H-RasV12) were treated for 18 h with DMSO (control) or 20 μM MG132. Then, cells were harvested, and protein extracts were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) for phosphoproteomics (n = 2, in triplicate each time). Phosphopeptides enriched in cells treated with MG132 were analyzed with the Motif-X software tool. Three motif families were identified (acidic, basic, and proline-directed), and almost half the phosphopeptides with an enriched motif have a proline-directed motif. (C) FatiGO single-enrichment analysis of phosphopeptides enriched in Ras-senescent cells treated MG132 with the Babelomics 4.3 platform. This platform was used to identify gene ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Reactome terms that were significantly enriched (Supplemental Fig. S5B). Then, these terms and their associated peptides were grouped in the indicated general categories. These categories were also classified according to their predicted ability to induce senescence or limit transformation when they are decreased. (D) Immunoblots for the indicated proteins and total ubiquitinylated proteins from fibroblasts expressing oncogenic ras or an empty vector (10 d after infection) and treated for 18 h with 20 μM MG132 or DMSO as control (n = 3). (E) Protein stability assays for the indicated proteins in cells as in D. Cells were treated with DMSO, 10 μg/mL cycloheximide, or 20 μM MG132 for the indicated times. The relative protein quantity was evaluated by immunoblotting and quantification with Adobe Photoshop CS4 or Image Lab 4.0 (n ≥ 2). (F,G) Immunoprecipitation of HSP70 or STAT3 in extracts from IMR90 cells expressing H-RasV12 or an empty vector 10 d after infection and treated for 18 h with 20 μM MG132 and immunoblotted against mono- and polyubiquitinylated conjugates (n ≥ 2). (H) Immunoprecipitation of c-MYC in extracts from cells expressing H-RasV12 or an empty vector (10 d after infection) and treated for 18 h with 20 μM MG132 and/or GSK3 inhibitor CHIR99021 (3 μM) and immunoblotted against mono- and polyubiquitinylated conjugates.
To support the model that increased ERK signaling leads to proteasome-dependent protein degradation in senescent cells, we used large-scale proteomics to look for phosphoproteins stabilized by the proteasome inhibitor MG132 in senescent cells. We identified an enrichment of nearly 3000 phosphopeptides from 1018 proteins. Most of the phosphorylation sites identified consisted of serine/threonine adjacent to a proline, consistent with ERK/MAPK phosphorylation sites (Fig. 4B; Supplemental Table SI). We also identified stabilized phosphopeptides where the phosphorylated residues were not part of the ERK consensus site that may be targets of ERK-regulated kinases. Motif analysis of the phosphopeptides revealed phosphorylation motifs for proline-directed kinases but also basophilic and acidophilic kinases (Supplemental Fig. S5). We analyzed the proteomics data using a FatiGO single enrichment from the bioinformatics platform Babelomics 4.3. We found that in senescent cells, phosphoproteins that control the response to growth factor stimulation and tumor progression are unstable. Hence, the degradation pattern may favor senescence and inhibit transformation (Fig. 4C). In Supplemental Table SI, we present a summary of the proteomics data with references to the implication of the proteins in cell senescence and cancer pathways, and in Supplemental Figure S6, we present the most significant functional categories affected by the degradation process. For example, the proteins FBXL11 (He et al. 2008), HSP70 (Gabai et al. 2009), RSL1D1 (Ma et al. 2008), and TBX2 (Martin et al. 2012) have been previously linked to the control of senescence, validating our results. In addition, the presence of multiple proteins linked to pseudopods, cell migration, and RNA metabolism (Supplemental Table SI) suggests that these pathways are targeted by a SAPD process.
We characterized in detail the degradation of c-MYC, HSP27, HSP70, KAP1, RSL1D1, and STAT3. The total level of these proteins was down-regulated in senescent cells, and all but HSP27 were restored by treatment with the proteasome inhibitor MG132, consistent with the anticipated role of the proteasome in SAPD (Fig. 4D). This proteasome-dependent degradation seems to be selective because total levels of ubiquitinated proteins did not change significantly in cells expressing RasV12 (Fig. 4D). We also evaluated the half-life of several of these proteins after inhibition of protein synthesis with cycloheximide. As expected, c-MYC, HSP70, KAP1, RSL1D1, and STAT3 had a reduced half-life that was restored by the proteasome inhibitor MG132 in primary fibroblasts expressing RasV12 (Fig. 4E). In addition, in RasV12-expressing cells treated with MG132, the levels of ubiquitinated HSP70 and STAT3 were increased (Fig. 4F-G). Together, the results are consistent with a model of selective protein degradation triggered by aberrant ERK signaling during Ras-induced senescence. However, not all phosphoproteins found unstable in our proteomic analysis are expected to be depleted in senescent cells, since their reduction will depend on the extent of phosphorylation of each protein pool and compensatory biosynthesis and the degradation rate for each protein in normal conditions. In addition, although most phosphoproteins contain candidate ERK phosphorylation sites, others do not, suggesting that other kinases may be engaged by ERK in the process. The case of c-MYC illustrates this point. c-MYC is phosphorylated by ERK at Ser62, and this phosphorylation facilitates phosphorylation at Thr58 by GSK3; it is this latter event that targets c-MYC to the proteasome (Yeh et al. 2004). In agreement with this model, the GSK3 inhibitor CHIR99021 inhibited the ubiquitination of c-MYC in Ras-expressing cells (Fig. 4H). Nevertheless, the levels of c-MYC, HSP27, HSP70, KAP1, RSL1D1, and STAT3 were restored by shERK2 in Ras-expressing IMR90 cells (Fig. 5A) or HMECs (Fig. 5B), indicating that ERK signaling, directly or indirectly, triggers their degradation. In MEFs, we also observed a decrease in levels of Hsp70, Kap1, and Stat3, but not c-Myc, in response to RasV12, and this down-regulation did not occur in Erk2−/− MEFs (Fig. 5C). In all cell types, the ERK-dependent decrease in c-MYC, HSP70, KAP1, RSL1D1, and STAT3 could not be explained by a reduction in their mRNA levels (Supplemental Fig. S7A–C). However, HSP27 levels were reduced at the mRNA level by an ERK-independent mechanism, explaining why its levels could not be restored by MG132 (Supplemental Fig. S7A,B). Of note, RasV12 induced RSL1D1 mRNA levels in human cells, perhaps as a feedback response to the reduction in protein levels (Supplemental Fig. S7A,B). To investigate whether protein degradation is dependent on ERK levels only or ERK signaling, we used MEK inhibitors. Both AZD6244 and U0126 restored the levels of selected proteins in RasV12-expressing cells (Fig. 5D) without corresponding changes in mRNA levels (Supplemental Fig. S7D–I), consistent with their ability to bypass RasV12-induced senescence (Supplemental Fig. S2F–N).
Figure 5.
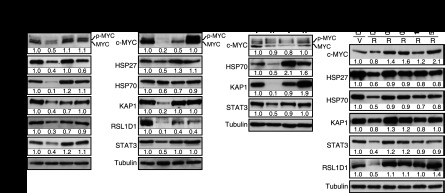
Role of ERK kinases in SAPD. (A) Immunoblots showing the levels of indicated proteins in IMR90 cells expressing H-RasV12 (R) or a vector control (V) and shERK2 or a control shRNA (shCTR) 14 d after infection (n ≥ 3). (B) Immunoblots for proteins and conditions as in A but for HMECs (n = 3). (C) Immunoblots showing the levels of the indicated proteins in wild-type or Erk2−/− MEFs expressing oncogenic ras or a vector control 14 d after infection (n = 3). (D) Immunoblots for the indicated proteins in IMR90 cells with a vector control (V) or H-RasV12 (R) and treated with the indicated chemicals. The treatments started immediately after infection, and the medium was changed every 2 d. Cells were harvested 10 d after infection (n = 2).
Cell senescence can also be induced by other stresses, such as radiation and short telomeres (replicative senescence). These situations commonly trigger a persistent DDR (d'Adda di Fagagna et al. 2003), mitochondrial dysfunction, accumulation of ROS (Passos et al. 2007), and a constitutive ERK activation (Satyanarayana et al. 2004). We thus reasoned that sustained ERK activation may accelerate protein degradation during replicative senescence and contribute to the establishment and maintenance of this phenotype. We cultured young normal human fibroblasts until their replicative senescence in the presence of two concentrations of the MEK inhibitors AZD6244 and U0126. We confirmed that senescent (old) fibroblasts displayed a constitutive high ERK activity, but MEK inhibitors reduced this trait almost to young levels (Fig. 6A). This inhibition of phospho-ERK levels translated in a decrease of p53 phosphorylation at Ser15 and an increase in RB phosphorylation, indicating a cell cycle pattern of young healthy cells (Fig. 6A). In addition, the levels of ERK target genes LIF and DUSP6 increased with replicative senescence and were decreased by MEK inhibitors (Fig. 6B,C). MEK inhibitors improved cell proliferation and the life span of normal human fibroblasts according to several criteria. First, they increased the expression of the proliferation markers KI-67 and MCM6 (Fig. 6D). Second, they increased the number of passages upon serial culture of normal human fibroblasts (Fig. 6E). Third, in a proliferation assay performed over 14 d, cells from late-passage cultures proliferated faster if they were previously treated with MEK inhibitors (Fig. 6F). Finally, since the loss of proliferation potential of normal human fibroblasts upon serial culturing is due to cellular senescence, we measured the senescence markers IL6 (part of the senescence-associated secretory phenotype) and SA-β-Gal. MEK inhibitors reduced significantly the accumulation of both markers in late-passage cultures (Fig. 6G,H). Taken together, our data indicate that ERK signaling is required for both replicative senescence and OIS and raise the question of whether the SAPD phenotype is also relevant for replicative senescence.
Figure 6.

ERK and the SAPD during replicative senescence. (A) Immunoblots for the indicated proteins in young (population doublings [PDL] = 21.5) and old (PDL ≥ 40) IMR90 cells treated with the indicated concentrations of MEK inhibitors or DMSO as control for 27 d. Fresh medium and inhibitors were added every 2 d. (B–D) qPCR for mRNAs encoded by ERK-stimulated genes and proliferation markers in cells as in A. (E) PDL of normal human fibroblasts in the presence of the indicated concentrations of MEK inhibitors or vehicle during 80 d. The experiment was started with 1 × 106 middle-age (PDL = 34) IMR90 cells for each condition. (F) Relative growth of late-passage human fibroblasts (PLD ≥ 41) after 60 d of treatments with MEK inhibitors or vehicle evaluated by a crystal violet assay. (G) qPCR for IL6 in cells as in A. (H) SA-β-Gal of cells as before after 40 d of treatments. Data were quantified from 100 cell counts in triplicate and are presented as the mean percentage of positive cells ± SD (shown in the bottom right of every panel). (I) Immunoblots for the indicated proteins in young (Y) (PDL = 21.5) and old (O) (PDL = 40) IMR90 cells treated with 20 μM MG132 or vehicle (n = 2). (J) Relative decrease of half-lives for the indicated proteins calculated from cycloheximide stability assays as presented in Figure 4E. (K) Immunoblots for the indicated proteins in cells as in A. (L) High-strength ERK signals or short telomeres lead to protein degradation, which in turn activates multiple stress responses that characterize cellular senescence. Note that the process could be self-sustained by multiple positive feedback loops. Lower levels of ERK signaling are permissive for Ras-dependent transformation in cooperation with other signals stimulated by oncogenic ras.
To investigate whether the proteins that we found down-regulated in Ras-induced senescence were also reduced during replicative senescence, we measured their levels in old and young cultures by immunoblotting. Again, we characterized these cells for a variety of cell cycle and senescence markers (Supplemental Fig. S8A–H). We found that c-MYC, HSP27, HSP70, KAP1, RSL1D1, and STAT3 were all found down-regulated in replicative senescence, and their levels were restored by the proteasome inhibitor MG132 (Fig. 6I). The mRNAs coding for these proteins were not down-regulated in senescent cells, and some of them were found slightly increased (Supplemental Fig. S8I). The half-life of all of these proteins was decreased in senescent cells (Fig. 6J) and restored by MG132 (Supplemental Fig. S8J). Culturing cells with MEK inhibitors prevented the proteasome-dependent down-regulation of these proteins (Fig. 6K), and the mRNAs for all of them but HSP27 were not changed (Supplemental Fig. S8K). Altogether, the results indicate that replicative senescence and OIS involve a proteasome-dependent degradation of multiple proteins (SAPD), which can potentially explain the activation of many stress signaling pathways in senescent cells and their resistance to proliferating stimuli (Fig. 6L). The mechanistic connections between the SAPD and the stresses that characterize senescence suggest multiple positive feedback loops in the process that may contribute to the robustness of senescence as a tumor suppressor mechanism.
If SAPD is important for senescence, it should be possible to induce senescence by mimicking the process and bypass senescence by inhibiting it. We tested this idea first by using shRNAs against HSP27, HSP70, KAP1, RSL1D1, and STAT3 (Supplemental Fig. S9A,B), and, as expected, the individual knockdown of these proteins triggers cellular senescence in both primary fibroblasts and IMR90 cells transformed by hTERT, RasV12, and shERK2 (Fig. 7A; Supplemental Fig. S9A,B). Unfortunately, we could not rescue senescence with proteasome inhibitors because they stabilize multiple tumor suppressors upon long-term incubation, leading to cell death. On the other hand, ectopic expression of the viral oncoproteins E1A and E6/E7 (Fig. 7B-D) restored cell proliferation and inhibited senescence (Fig. 7E; Supplemental Fig. S9C–E). The expression of all tested SAPD targets—c-MYC, HSP27, HSP70, KAP1, RSL1D1, and STAT3—was restored by these viral oncoproteins (Fig. 7F), while the expression of their mRNAs was not generally increased (Supplemental Fig. S9F–H), indicating a close correlation between SAPD inhibition and bypass of senescence.
Figure 7.

Inactivation and stabilization of targets of SAPD (A) SA-β-Gal of wild-type IMR90 cells or transformed IMR90 cells (hTERT, H-RasV12, and shERK2) expressing the indicated shRNA expression vectors fixed 10 d after infection. Data were quantified from 100 cell counts in triplicate and are presented as the mean percentage of positive cells ± SD. These experiments were done in triplicate and at least two times. (B–D) qPCR for the viral oncoproteins E6, E7, and E1A expressed from retroviral vectors in IMR90 cells. (E) SA-β-Gal of IMR90 fibroblasts expressing an empty vector, the human papillomavirus E6/E7 oncoproteins, or the adenovirus E1A oncoproteins together with H-RasV12 or an empty vector. SA-β-Gal activity was measured 14 d after infection. Data were quantified from 100 cell counts in triplicate and are presented as the mean percentage of positive cells ± SD. (F) Immunoblots for cell cycle-regulated proteins and SAPD targets using extracts from cells as in E.
High levels of ERK/MAPK activation characterize benign prostate tumors and predict a better outcome in malignant tumors
To confirm the biological and clinical importance of senescence stimulated by hyperactivation of ERK and the SAPD process, we next investigated several prostate tumors. In cases of benign prostatic hyperplasia (BPH), which can be considered senescent lesions (Choi et al. 2000; Vernier et al. 2011), we found very high levels of phospho-ERK as well as the senescence markers p16INK4a and PML in epithelial cells of prostate acini (Fig. 8A,B; Supplemental Fig. S10A,B). In prostate carcinomas, ERK activation was never seen as high as in BPH (Fig. 8A,B vs. C). Moreover, in epithelial cells, nuclear phospho-ERK levels had an inverse correlation with the Gleason pattern (P < 0.05) (Supplemental Fig. S10C) and a positive correlation with the presence of regions of BPH in the same prostate (P < 0.05) (Supplemental Fig. S10D). We also found a negative correlation between phospho-ERK staining in the nucleus of the tumor cells or in the tissue adjacent to the tumor and patients' biochemical relapse (measured by their prostate-specific antigen [PSA] levels; P < 0.06) (Fig. 8C,D). Consistent with our results, ERK activation is rare in human pancreatic cancers, where ras mutations occur in ∼90% of the cases (Yip-Schneider et al. 2001), and correlations between ERK activation and good prognosis were previously reported in prostate (Malik et al. 2002) and breast cancer patients (Svensson et al. 2005).
Figure 8.

Phospho-ERK and STAT3 in the normal prostate and BPH. (A) Phospho-ERK staining in samples from patients with BPH or normal controls. White arrows point to the stroma, and black arrows point to epithelial cells. (B) Quantification of phospho-ERK data in normal and BPH patients according to three degrees of staining intensity (none, 0; moderate, 1; and high, 2). The differences between normal tissues and BPH were evaluated with the nonparametric Mann-Whitney U-test. (C) Degrees of phospho-ERK staining (0, 1, and 2) in prostate cancer. The survival Kaplan-Meier curves describe the time for biochemical relapse (BCR) in patients with different levels of staining. (D) Degrees of phospho-ERK staining (0, 1, and 2) in normal tissue adjacent to prostate cancer and survival Kaplan-Meier curves as in C. (E) STAT3 staining in samples from patients with BPH (n = 43) or normal controls (n = 3) or cancer patients (n = 20). The percentage of patients with a STAT3 staining of four different degrees of intensity (none, 0; low, 1; moderate, 2; and high, 3) is shown in the right panel. The differences between normal and tumor tissues versus BPH were evaluated with the nonparametric Mann-Whitney U-test, P = 0.0003.
We also stained sections of normal prostate, BPH, and prostate cancer for one of the targets of SAPD, STAT3. Consistent with the results shown above characterizing BPH as senescent benign lesions with high levels of phospho-ERK, STAT3 was found down-regulated in BPH when compared with normal tissues, while most malignant tumors displayed high levels of STAT3 (Fig. 8E).
Discussion
We show here that the outcome of the stimulation of the ERK/MAP kinases depends on the expression levels and activity of ERK1/2. In normal cells, high levels of stimulation led to cellular senescence. In contrast, reducing ERK levels and, as a consequence, its activity rescued cells from senescence and facilitated cell transformation by oncogenic ras. Our results are consistent with previous data showing that MEK inhibitors can bypass Ras-induced senescence (Lin et al. 1998) and that shRNAs against MEK increased tumor formation in Myc-expressing cells (Bric et al. 2009). Collectively, these studies reveal a tumor suppressor role for the ERK/MAP kinase pathway, which depends on the strength of its activation, and anticipate that molecular mechanisms controlling ERK kinase levels and activity are critical for tumor suppression and are likely targets of the transformation process.
Most functions of the ERK/MAP kinase pathway depend on the ability of the ERK kinases to phosphorylate multiple target proteins. How, then, do these kinases stimulate or inhibit cell proliferation, depending on the strength of their activity? An unbiased proteomic study comparing the levels and phosphorylation of several ERK targets in conditions of high or moderate activity led to the identification of a selective and ERK-dependent protein degradation process in senescent cells that we named SAPD. SAPD provides a direct mechanism to explain the anti-proliferative prosenescence functions of the ERK/MAP kinase pathway because phosphorylation and protein degradation are tightly linked (Hunter 2007). During normal signaling, this process will only reduce a minor fraction of every ERK target protein because only a minor fraction of each protein is phosphorylated (Olsen et al. 2010). However, aberrant ERK signaling can effectively deplete some ERK targets because the fraction of phosphorylation will be dramatically increased (Supplemental Fig. S11). SAPD also explains the inability of cells with activated oncogenes to proliferate despite activation of growth signaling pathways. Analysis of individual SAPD targets likely explains many traits of senescence, including the cell cycle arrest, DDR, telomere dysfunction, and cell motility defects (Supplemental Table SI). For example, the down-regulation of c-MYC or HSP70 was reported to be sufficient to trigger senescence (Guney et al. 2006; Wu et al. 2007; Gabai et al. 2009), and the down-regulation of STAT3 and several mitochondrial import proteins could account for the mitochondrial dysfunction associated with Ras-induced senescence (Gough et al. 2009; Moiseeva et al. 2009). Interestingly, both c-Myc and STAT3 cooperate with oncogenic ras to transform primary cells (Land et al. 1983; Gough et al. 2009) and are targets of ERK-dependent protein degradation. Remarkably, the oncoprotein E1A cooperates with ras for transformation, but it is not clear how E1A may block p53 functions or stabilize c-MYC. We show now that by reducing overall ERK phosphorylation, perhaps via the induction of ERK-specific phosphatases (Callejas-Valera et al. 2008), E1A can cooperate with RasV12 as ERK knockdown or the SV40 early region.
The mechanistic links between ERK activity and protein degradation will require further studies. However, most phosphopeptides stabilized by proteasome inhibitors in Ras senescent cells contained serine or threonine residues adjacent to proline (Fig. 4B; Supplemental Table SI), a characteristic of ERK phosphorylation sites. However, not all phosphoproteins stabilized by proteasome inhibitors in senescent cells were recognized ERK targets or contained ERK phosphorylation sites. This suggests that other kinases downstream from ERK or the DDR may provide signals for phosphorylation-dependent protein degradation, leading to senescence. In addition, an overall reduction of phosphatase activity in senescent cells due to ROS may also contribute to phosphorylation-dependent protein degradation and likely explains why SAPD is also observed in cells with short telomeres, which are characterized by mitochondrial dysfunction and oxidative stress (Passos et al. 2007; Sahin and Depinho 2010).
The increase in phospho-ERK levels during replicative senescence and the increase in the life span of normal human fibroblasts after using inhibitors of the ERK pathway suggest a role for ERK signaling in the activation of the senescence program by short telomeres. We found that the phosphorylation of p53 at Ser15, a hallmark of the DDR, was prevented by MEK inhibitors. This observation is consistent with results showing that inhibition of the DDR can restore cell cycle progression despite telomere shortening (d'Adda di Fagagna et al. 2003). In some contexts, preventing telomere shortening by expressing telomerase can delay aging in mice (Tomas-Loba et al. 2008; Jaskelioff et al. 2011). Since MEK inhibitors were able to extend the replicative life span of normal human fibroblasts, which is normally limited by short telomeres, they could be tested to mitigate signals from short telomeres during aging or age-related diseases.
Clinical studies in a variety of cancers indicate that in some tumors, phospho-ERK levels are remarkably low. The most compelling example of phospho-ERK down-regulation was described in pancreatic tumors where ras mutations occur frequently (Yip-Schneider et al. 2001). Presumably, pancreatic carcinogenesis selects early for events that down-regulate ERK levels/activity to avoid senescence and other anti-proliferative consequences of high ERK activity. Interestingly, patients with pancreatic tumors with high ERK levels had better survival and responded better to treatment (Chadha et al. 2006), suggesting that some of the tumor suppressor functions of the ERK pathway could be reactivated in cancer patients. Likewise, in mammary carcinomas, phospho-ERK levels correlated with good prognosis and a less aggressive phenotype (Milde-Langosch et al. 2005; Svensson et al. 2005), and similar correlations were found in brain tumors as well (Mawrin et al. 2003, 2005). We extend these observations here by showing that phospho-ERK levels were remarkably low in the most aggressive prostate tumors. Also, patients with tumors having high phospho-ERK levels had a better prognosis. In agreement with our findings, it has been reported that advanced prostate cancer correlates with low phospho-ERK and high AKT levels (Malik et al. 2002). It has been reported as well that, in some tumors, phospho-ERK levels are very high and that MEK inhibitors may have therapeutic value (Sebolt-Leopold 2008). We anticipate that such tumors contain genetic or epigenetic lesions that inactivate the protein degradation mechanism acting downstream from ERK to mediate tumor suppression.
Astonishingly, we found a dramatic increase in phospho-ERK staining along with the senescence markers PML and p16INK4a in BPH. Senescent cells have been also observed in benign human nevi, which, like BPH, rarely progress into malignant melanomas (Michaloglou et al. 2005). On the other hand, in some mouse models of prostate cancer, premalignant lesions (prostatic intraepithelial neoplasias [PINs]) display some markers of senescence in association with less aggressive but detectable tumor progression (Chen et al. 2005). It will be very important to characterize which traits associated with senescence determine the reversibility of the cell cycle arrest and the potential for further tumor progression. The incidence of both prostate cancer and BPH increases with age, and both require androgens for growth, but unlike PIN, BPH is not a premalignant lesion (Bostwick et al. 1992). Comparison of lesions that do not progress, such as BPH, and lesions that eventually become malignant tumors, such as PIN (Bostwick 1996), can help us understand how senescence prevents tumor formation in vivo. One such factor is the presence of PML bodies, which accumulate in an ERK-dependent manner during OIS in cell culture and are highly expressed in BPH but absent in PIN lesions (Vernier et al. 2011). To circumvent ERK-dependent tumor suppression, some tumors may select for a reduction of ERK levels/activity, while others may disable the SAPD mechanism. The relationship between ERK levels, senescence, and transformation is also relevant for anti-cancer therapeutics, as drugs that inhibit the pathway may inhibit tumor suppression in some contexts, while drugs increasing ERK activity beyond the threshold required for senescence may have anti-tumor effects. In conclusion, we describe a plausible mechanism linking hyperactivation of signaling pathways with protein degradation and the cellular defects and malfunctions associated with cellular senescence, suggesting that attenuation of excessive or aberrant signaling can have anti-aging effects by preventing cellular senescence.
Materials and methods
Mice and cells
All mouse experiments were conducted in accordance with institutional and national guidelines and regulations. Conditional Erk2flox/flox female mice (Voisin et al. 2010) were crossed with male mice carrying a Cre-expressing transgene under the control of a Sox2 promoter (Hayashi et al. 2002) to generate heterozygous Erk2−/+ animals. Erk2 knockout embryos (Erk2−/−) were obtained in normal Mendelian frequencies when male Sox2-Cre; Erk2−/+ mice were crossed to Erk2flox/flox females. Sox2-mediated excision by the Cre recombinase permitted the rescue of the otherwise embryonic-lethal phenotype caused by the absence of Erk2 by specifically excising Erk2 floxed alleles in the epiblast and maintaining functional floxed alleles in the extraembryonic tissues of the embryo. MEFs were prepared at embryonic day 14.5 (E14.5) as previously reported (Voisin et al. 2010).
For xenografts, BALB/c 6-wk-old nude mice (Charles River) were injected subcutaneously in both flanks with 106 cells resuspended in 250 μL of PBS mixed with 250 μL of Matrigel (BD Biosciences) at 4°C. Tumor formation was evaluated over a period of 60 d. Tumors >0.2 cm3 were counted, and mice were euthanized before the end point of the experiment when tumor volume reached 2 cm3.
Cell lines, reagents, plasmids, quantitative PCR (qPCR), cell growth analysis, soft agar, focus-forming assay, and protein analysis (immunoblotting and immunofluorescence) are described in the Supplemental Material.
Immunohistochemistry
We used samples from seven patients diagnosed with BPH and five different tissue microarrays (TMAs) (Diallo et al. 2007). The first is comprised of 49 normal prostate specimens from autopsies. The four others contain tissues obtained from 64 patients with primary prostate cancer. They are comprised of related nonneoplasic tissues adjacent to prostate cancer and cancerous tissues from 64 patients who underwent radical prostatectomy. Regions of normal, intraepithelial neoplasia or cancerous epithelial tissue were identified by two pathologists and subsequently spotted on TMAs (Diallo et al. 2007). Specimens were obtained from consenting patients, and the institutional ethics review committee approved the study. We also purchased TMAs from Biochain Institute (catalog no. Z5070001). They comprised 63 cores, with 43 cases of BPH and 16 cases of adenocarcinomas. Normal prostate samples were obtained from BioChain Institute.
Tissue sections and TMAs were stained with a mouse monoclonal anti-phospho-ERK1/2T202/Y204 (1:175; clone E10, no. 9106, Cell Signaling Technology), anti-PML antibody (1:300; clone PG-M3, Sc-966, Santa Cruz Biotechnology), and anti-p16INK4a (1:25; clone F-12, Sc-1661, Santa Cruz Biotechnology). Primary antibody detection was done using the LSAB 2 peroxidase system from DAKO. Briefly, tissue samples were deparaffinized, rehydrated, and treated with 0.3% H2O2 in methanol to eliminate endogenous peroxidase activity. Antigen epitope retrieval was performed by heating for 15 min at 95°C in Tris–EDTA buffer (10 mM Tris Base, 1 mM EDTA solution at pH 8.0) for phospho-ERK and PML or 10 mM citrate buffer (pH 6.0) for p16INK4a. All subsequent steps were done at room temperature. The sections were blocked with a protein-blocking serum-free reagent (DAKO) and incubated with primary antibody for 60 min followed by a 20-min treatment with the secondary biotinylated antibody (DAKO) and then incubated for 20 min with streptavidin–peroxidase label (DAKO). Reaction products were developed with diaminobenzidine (DAKO) containing 0.3% H2O2 as a substrate for peroxidase. Nuclei were counterstained with Harris hematoxylin (Sigma-Aldrich).
Statistics
Statistical analysis was performed using SPSS software 16.0 (SPSS, Inc.). The expression level of phosphorylated ERK1/2 was evaluated on a scale of 0 (for no expression) to 2 (strong expression) in both the epithelial cells (nucleus and cytoplasm) and the stroma of normal prostate samples, normal adjacent tissues, prostate tumor tissues, and BPH. The nonparametric Mann-Whitney U-test was used to show significant differences between the normal, normal adjacent, PIN, tumor, and BPH groups. Correlations in expression between cell types and/or subcellular expression levels were done using the nonparametric Spearman's rank correlation coefficient or the Pearson χ2 analysis.
Acknowledgments
We thank James R. Davie, Elliot Drobetsky, Philippe Roux, Patrick J. Padisson, Peiqing Sun, Stéphane Roy, Jacques Landry, and Jason C. Young for critical reading, reagents, and/or support. We thank Éric Bonneil and the Institut de Recherche en Immunologie et Cancérologie (IRIC) proteomic service for the phosphopeptide analysis and identification, and Louise Cournoyer, Catherine Ménard, and Frédérique Badeaux for technical assistance. This work was supported by grants from the Canadian Institute of Health and Research (CIHR) to G.F. and S.M. G.F. is a FRSQ senior fellow. S.M. holds the Canada Research Chair in Cellular Signaling. X.D.-S. is a fellow of the Vanier Canada Graduate Scholarships Program. M.F.G.-L. is a CIHR fellow.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.203984.112.
References
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, et al. 2006. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444: 633–637 [DOI] [PubMed] [Google Scholar]
- Bostwick DG 1996. Prospective origins of prostate carcinoma. Prostatic intraepithelial neoplasia and atypical adenomatous hyperplasia. Cancer 78: 330–336 [DOI] [PubMed] [Google Scholar]
- Bostwick DG, Cooner WH, Denis L, Jones GW, Scardino PT, Murphy GP 1992. The association of benign prostatic hyperplasia and cancer of the prostate. Cancer 70: 291–301 [DOI] [PubMed] [Google Scholar]
- Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, Stein H, Dorken B, Jenuwein T, Schmitt CA 2005. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 436: 660–665 [DOI] [PubMed] [Google Scholar]
- Bric A, Miething C, Bialucha CU, Scuoppo C, Zender L, Krasnitz A, Xuan Z, Zuber J, Wigler M, Hicks J, et al. 2009. Functional identification of tumor-suppressor genes through an in vivo RNA interference screen in a mouse lymphoma model. Cancer Cell 16: 324–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callejas-Valera JL, Guinea-Viniegra J, Ramirez-Castillejo C, Recio JA, Galan-Moya E, Martinez N, Rojas JM, Ramon y Cajal S, Sanchez-Prieto R 2008. E1a gene expression blocks the ERK1/2 signaling pathway by promoting nuclear localization and MKP up-regulation: Implication in v-H-Ras-induced senescence. J Biol Chem 283: 13450–13458 [DOI] [PubMed] [Google Scholar]
- Chadha KS, Khoury T, Yu J, Black JD, Gibbs JF, Kuvshinoff BW, Tan D, Brattain MG, Javle MM 2006. Activated Akt and Erk expression and survival after surgery in pancreatic carcinoma. Ann Surg Oncol 13: 933–939 [DOI] [PubMed] [Google Scholar]
- Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, et al. 2005. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 436: 725–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Shendrik I, Peacocke M, Peehl D, Buttyan R, Ikeguchi EF, Katz AE, Benson MC 2000. Expression of senescence-associated β-galactosidase in enlarged prostates from men with benign prostatic hyperplasia. Urology 56: 160–166 [DOI] [PubMed] [Google Scholar]
- Coppe JP, Desprez PY, Krtolica A, Campisi J 2010. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu Rev Pathol 5: 99–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP 2003. A DNA damage checkpoint response in telomere-initiated senescence. Nature 426: 194–198 [DOI] [PubMed] [Google Scholar]
- DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, et al. 2011. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475: 106–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diallo JS, Aldejmah A, Mouhim AF, Peant B, Fahmy MA, Koumakpayi IH, Sircar K, Begin LR, Mes-Masson AM, Saad F 2007. NOXA and PUMA expression add to clinical markers in predicting biochemical recurrence of prostate cancer patients in a survival tree model. Clin Cancer Res 13: 7044–7052 [DOI] [PubMed] [Google Scholar]
- Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre M, Nuciforo PG, Bensimon A, et al. 2006. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444: 638–642 [DOI] [PubMed] [Google Scholar]
- Efeyan A, Murga M, Martinez-Pastor B, Ortega-Molina A, Soria R, Collado M, Fernandez-Capetillo O, Serrano M 2009. Limited role of murine ATM in oncogene-induced senescence and p53-dependent tumor suppression. PLoS ONE 4: e5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, Waters CM, Penn LZ, Hancock DC 1992. Induction of apoptosis in fibroblasts by c-myc protein. Cell 69: 119–128 [DOI] [PubMed] [Google Scholar]
- Ferbeyre G, de Stanchina E, Querido E, Baptiste N, Prives C, Lowe SW 2000. PML is induced by oncogenic ras and promotes premature senescence. Genes Dev 14: 2015–2027 [PMC free article] [PubMed] [Google Scholar]
- Ferbeyre G, de Stanchina E, Lin AW, Querido E, McCurrach ME, Hannon GJ, Lowe SW 2002. Oncogenic ras and p53 cooperate to induce cellular senescence. Mol Cell Biol 22: 3497–3508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabai VL, Yaglom JA, Waldman T, Sherman MY 2009. Heat shock protein Hsp72 controls oncogene-induced senescence pathways in cancer cells. Mol Cell Biol 29: 559–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE 2009. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 324: 1713–1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guney I, Wu S, Sedivy JM 2006. Reduced c-Myc signaling triggers telomere-independent senescence by regulating Bmi-1 and p16(INK4a). Proc Natl Acad Sci 103: 3645–3650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn WC, Dessain SK, Brooks MW, King JE, Elenbaas B, Sabatini DM, DeCaprio JA, Weinberg RA 2002. Enumeration of the simian virus 40 early region elements necessary for human cell transformation. Mol Cell Biol 22: 2111–2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S, Lewis P, Pevny L, McMahon AP 2002. Efficient gene modulation in mouse epiblast using a Sox2Cre transgenic mouse strain. Mech Dev 119: S97–S101 [DOI] [PubMed] [Google Scholar]
- He J, Kallin EM, Tsukada Y, Zhang Y 2008. The H3K36 demethylase Jhdm1b/Kdm2b regulates cell proliferation and senescence through p15(Ink4b). Nat Struct Mol Biol 15: 1169–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter T 2007. The age of crosstalk: Phosphorylation, ubiquitination, and beyond. Mol Cell 28: 730–738 [DOI] [PubMed] [Google Scholar]
- Jaskelioff M, Muller FL, Paik JH, Thomas E, Jiang S, Adams AC, Sahin E, Kost-Alimova M, Protopopov A, Cadinanos J, et al. 2011. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature 469: 102–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A, et al. 2011. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 479: 547–551 [DOI] [PubMed] [Google Scholar]
- Land H, Parada LF, Weinberg RA 1983. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature 304: 596–602 [DOI] [PubMed] [Google Scholar]
- Leist M, Jaattela M 2001. Four deaths and a funeral: From caspases to alternative mechanisms. Nat Rev Mol Cell Biol 2: 589–598 [DOI] [PubMed] [Google Scholar]
- Lin AW, Barradas M, Stone JC, van Aelst L, Serrano M, Lowe SW 1998. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev 12: 2997–3007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe SW, Cepero E, Evan G 2004. Intrinsic tumour suppression. Nature 432: 307–315 [DOI] [PubMed] [Google Scholar]
- Ma L, Chang N, Guo S, Li Q, Zhang Z, Wang W, Tong T 2008. CSIG inhibits PTEN translation in replicative senescence. Mol Cell Biol 28: 6290–6301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik SN, Brattain M, Ghosh PM, Troyer DA, Prihoda T, Bedolla R, Kreisberg JI 2002. Immunohistochemical demonstration of phospho-Akt in high Gleason grade prostate cancer. Clin Cancer Res 8: 1168–1171 [PubMed] [Google Scholar]
- Mallette FA, Ferbeyre G 2007. The DNA damage signaling pathway connects oncogenic stress to cellular senescence. Cell Cycle 6: 1831–1836 [DOI] [PubMed] [Google Scholar]
- Mallette FA, Gaumont-Leclerc MF, Ferbeyre G 2007. The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev 21: 43–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin N, Benhamed M, Nacerddine K, Demarque MD, van Lohuizen M, Dejean A, Bischof O 2012. Physical and functional interaction between PML and TBX2 in the establishment of cellular senescence. EMBO J 31: 95–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mawrin C, Diete S, Treuheit T, Kropf S, Vorwerk CK, Boltze C, Kirches E, Firsching R, Dietzmann K 2003. Prognostic relevance of MAPK expression in glioblastoma multiforme. Int J Oncol 23: 641–648 [PubMed] [Google Scholar]
- Mawrin C, Sasse T, Kirches E, Kropf S, Schneider T, Grimm C, Pambor C, Vorwerk CK, Firsching R, Lendeckel U, et al. 2005. Different activation of mitogen-activated protein kinase and Akt signaling is associated with aggressive phenotype of human meningiomas. Clin Cancer Res 11: 4074–4082 [DOI] [PubMed] [Google Scholar]
- Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS 2005. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 436: 720–724 [DOI] [PubMed] [Google Scholar]
- Milde-Langosch K, Bamberger AM, Rieck G, Grund D, Hemminger G, Muller V, Loning T 2005. Expression and prognostic relevance of activated extracellular-regulated kinases (ERK1/2) in breast cancer. Br J Cancer 92: 2206–2215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moiseeva O, Bourdeau V, Roux A, Deschenes-Simard X, Ferbeyre G 2009. Mitochondrial dysfunction contributes to oncogene-induced senescence. Mol Cell Biol 29: 4495–4507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DJ, Junttila MR, Pouyet L, Karnezis A, Shchors K, Bui DA, Brown-Swigart L, Johnson L, Evan GI 2008. Distinct thresholds govern Myc's biological output in vivo. Cancer Cell 14: 447–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M, Nunez S, Heard E, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW 2003. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113: 703–716 [DOI] [PubMed] [Google Scholar]
- Olsen JV, Vermeulen M, Santamaria A, Kumar C, Miller ML, Jensen LJ, Gnad F, Cox J, Jensen TS, Nigg EA, et al. 2010. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal 3: ra3. [DOI] [PubMed] [Google Scholar]
- Passos JF, Saretzki G, Ahmed S, Nelson G, Richter T, Peters H, Wappler I, Birket MJ, Harold G, Schaeuble K, et al. 2007. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol 5: e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratilas CA, Taylor BS, Ye Q, Viale A, Sander C, Solit DB, Rosen N 2009. (V600E)BRAF is associated with disabled feedback inhibition of RAF–MEK signaling and elevated transcriptional output of the pathway. Proc Natl Acad Sci 106: 4519–4524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin E, Depinho RA 2010. Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature 464: 520–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satyanarayana A, Greenberg RA, Schaetzlein S, Buer J, Masutomi K, Hahn WC, Zimmermann S, Martens U, Manns MP, Rudolph KL 2004. Mitogen stimulation cooperates with telomere shortening to activate DNA damage responses and senescence signaling. Mol Cell Biol 24: 5459–5474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebolt-Leopold JS 2008. Advances in the development of cancer therapeutics directed against the RAS-mitogen-activated protein kinase pathway. Clin Cancer Res 14: 3651–3656 [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW 1997. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88: 593–602 [DOI] [PubMed] [Google Scholar]
- Svensson S, Jirstrom K, Ryden L, Roos G, Emdin S, Ostrowski MC, Landberg G 2005. ERK phosphorylation is linked to VEGFR2 expression and Ets-2 phosphorylation in breast cancer and is associated with tamoxifen treatment resistance and small tumours with good prognosis. Oncogene 24: 4370–4379 [DOI] [PubMed] [Google Scholar]
- Tomas-Loba A, Flores I, Fernandez-Marcos PJ, Cayuela ML, Maraver A, Tejera A, Borras C, Matheu A, Klatt P, Flores JM, et al. 2008. Telomerase reverse transcriptase delays aging in cancer-resistant mice. Cell 135: 609–622 [DOI] [PubMed] [Google Scholar]
- Vernier M, Bourdeau V, Gaumont-Leclerc MF, Moiseeva O, Begin V, Saad F, Mes-Masson AM, Ferbeyre G 2011. Regulation of E2Fs and senescence by PML nuclear bodies. Genes Dev 25: 41–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voisin L, Saba-El-Leil MK, Julien C, Fremin C, Meloche S 2010. Genetic demonstration of a redundant role of extracellular signal-regulated kinase 1 (ERK1) and ERK2 mitogen-activated protein kinases in promoting fibroblast proliferation. Mol Cell Biol 30: 2918–2932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CH, van Riggelen J, Yetil A, Fan AC, Bachireddy P, Felsher DW 2007. Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc Natl Acad Sci 104: 13028–13033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW 2007. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445: 656–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, Hahn WC, Stukenberg PT, Shenolikar S, Uchida T, et al. 2004. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol 6: 308–318 [DOI] [PubMed] [Google Scholar]
- Yip-Schneider MT, Lin A, Marshall MS 2001. Pancreatic tumor cells with mutant K-ras suppress ERK activity by MEK-dependent induction of MAP kinase phosphatase-2. Biochem Biophys Res Commun 280: 992–997 [DOI] [PubMed] [Google Scholar]
- Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, Tavare S, Arakawa S, Shimizu S, Watt FM, et al. 2009. Autophagy mediates the mitotic senescence transition. Genes Dev 23: 798–803 [DOI] [PMC free article] [PubMed] [Google Scholar]