

Abstract
Hybrid structural methods have been used in recent years to understand protein-protein or protein-ligand interactions where high resolution crystallography or NMR data on the protein of interest has been limited. For G protein-coupled receptors (GPCRs), high resolution structures of native structural forms other than rhodopsin have not yet been achieved; gaps in our knowledge have been filled by creative crystallography studies that have developed stable forms of receptors by multiple means. The neurotransmitter serotonin (5-hydroxytryptamine) is a key GPCR-based signaling molecule affecting many physiological manifestations in humans ranging from mood and anxiety to bowel function. However, a high resolution structure of any of the serotonin receptors has not yet been solved. Here, we used structural mass spectrometry along with theoretical computations, modeling, and other biochemical methods to develop a structured model for human serotonin receptor subtype 4(b) in the presence and absence of its antagonist GR125487. Our data confirmed the overall structure predicted by the model and revealed a highly conserved motif in the ligand-binding pocket of serotonin receptors as an important participant in ligand binding. In addition, identification of waters in the transmembrane region provided clues as to likely paths mediating intramolecular signaling. Overall, this study reveals the potential of hybrid structural methods, including mass spectrometry, to probe physiological and functional GPCR-ligand interactions with purified native protein.
The neurotransmitter serotonin (5-hydroxytryptamine, 5-HT)1 is a key signaling molecule affecting many physiological manifestations in humans ranging from mood and anxiety to bowel function (2, 3). About 95% of total body 5-HT is released into the gut by intestinal enterochromaffin cells. Responses to 5-HT are elicited through a large family of G protein-coupled receptors (GPCRs) (4) called serotonergic receptors. Several subtypes of these receptors with specific functions are widely expressed (5). With the exception of 5-HT3, all other 5-HT receptors are members of the class A family of GPCRs. 5-HT4 receptors (5-HT4Rs), like some of the other family members (5-HT6R and 5-HT7R), are associated with an excitatory Gs protein that evokes a cAMP increase upon activation (6). There are four major isoforms of 5-HT4R translated from the same gene by alternate splicing of the C terminus (7). Human 5-HT4R isoform b, the longest transcript (388 residues), is the subject of this study.
The pharmacology of 5-HT4R is well understood for inverse agonists such as GR125487, GR113808, and SB204070, partial agonists like RS39604, and potent agonists like tegaserod (Zelnorm), cisapride (Propulsid), prucalopride, and renzapride (8, 9). Many of the aforementioned drugs used to treat constipation-predominant irritable bowel syndrome were shown to act through 5-HT4Rs (10). Restriction in the use of some of these drugs by the Food and Drug Administration for the treatment of constipation-predominant irritable bowel syndrome has prompted intense research to identify alternative compounds with greater 5-HT4R selectivity (11). A lack of understanding the mechanisms of binding and the action of the relevant drugs is in part due to the absence of 5-HT4R structural data.
Previously, we used radiolytic protein footprinting coupled to mass spectrometry (MS) to probe the conformational dynamics of the GPCR, bovine rhodopsin (12–15). In the context of high resolution crystallographic data on this receptor, we identified sites of conformational change as a function of light-induced activation and discovered that radiolysis could identify oxidized sites adjacent to internal water molecules identified by crystallographic data. Here, we extend these structural MS approaches to an examination of the 5-HT4R subtype b GPCR and its ligand interactions. Unlike rhodopsin, no high resolution crystallographic structure is available for any member of the 5-HT4R class subtype. Thus, we developed a homology model of this receptor based on available structural data, and by using limited proteolysis and MS, we tested the validity of the model and its predicted domain structure with native protein samples in the presence and absence of the antagonist GR125487. We also conducted radiolytic footprinting of this receptor in the presence and absence of ligand to locate internal water molecules and monitor the conformational dynamics of ligand binding. As our homology model explicitly included structural waters, we also compared the footprinting results to these proposed sites of water binding.
The biophysical study of membrane proteins is extremely challenging, due to the very nature of samples used, which are sensitive and tend to aggregate when solubilized outside of their membrane environment. The difficulty in sample handling limits the study of these classes of proteins despite their key role played in many cellular mechanisms. Recent developments led to the design of stabilized forms of many GPCRs produced as T4 lysozyme-modified versions, with T4 inserted in place of a third intracellular loop of GPCR. This smart engineering led to successful crystallization of many GPCRs as chimeric forms. Although these chimeras conserved binding properties, they are unable to couple to their secondary messenger partner, G protein. In this study, we were able to purify and study a full-length and active serotonin receptor subclass for our studies. This in itself is a rarity as the entire class of serotonergic receptors is notorious for their unstable nature outside the membrane environment. To add to the complications, the presence of detergents in the membrane protein samples requires additional sample preparation modifications for structural mass spectrometric analysis. These challenges must be overcome if GPCR research is to progress rapidly. High resolution structural data and models need to be tested by a range of hybrid structural methods applied to native protein samples captured in states of physiological interest. The present art of mass spectrometric structural techniques is emerging as a new tool to investigate the highly dynamic process of ligand binding and signal transduction. Yet, the task of pinpointing the residues involved in the signal transduction process is highly complicated. Here, we provide an overall structural model of 5-HT4R and probe its modes of binding with a key antagonist relevant to developing new therapeutic agents. These results allow the testing of a number of key hypotheses as to the general features of ligand binding by class A GPCRs. The study successfully applies mass spectrometric methods to a complex membrane protein to expand the state-of-the-art.
MATERIALS AND METHODS
Biochemical Methods
GPCR Expression and Purification
5-HT4R was expressed in an insect cell-baculovirus system (Sf9 cells). To assist purification, two affinity tags, T7 (MASMTGGQQMGSATASK) and 1D4 (TETSQVAPA), were added to the C-terminal sequence of the native full-length 5-HT4b receptor containing 388 residues (NCBI reference NP_000861.1). The receptor sequence then was subcloned into baculovirus expression vector pFastBac (Invitrogen). The multiplicity of infection ratio was 10:1, and cells were cultured in a baffled flask at 27°C in a shaker incubator and harvested 48 h after infection. Cell pellets obtained after centrifugation were frozen at −80°C until further purification.
Expression of the 5-HT4R in the crude pellet was verified with anti-rhodopsin 1D4 antibody (16). The band immunospecific for the 1D4 tag ran just above the 39-kDa molecular mass marker on SDS-PAGE (4–12% NuPAGE/MOPS, Invitrogen) under reducing conditions without sample heating. The calculated molecular mass of the full-length construct used for expression was 46,626.6 daltons with a theoretical pI around 8.05 and a calculated molar extinction coefficient of 67,475 m−1cm−1 with absorption at 280 nm of a 0.1% (1 mg/ml) solution in water of 1.447 OD, assuming all Cys residues were reduced.
Membrane Preparation and Solubilization
The purification protocol described here was adapted from those reported for β2-AR expressed in Sf9 insect cells (17) and 5-HT4R purified from mouse retinas (18). All procedures were carried out at 4°C. A Dounce tissue grinder was used to homogenize the Sf9 cell pellet into minimal buffer consisting of 10 mm Hepes, pH 7.5, 10 mm MgCl2, 10 μm GR125487, 20 mm KCl, and protease inhibitor tablets (Roche Applied Science, 1 tablet per 50 ml of suspension). After over 20 up-and-down strokes, the homogenate was centrifuged at 48,000 × g for 30 min. The resulting pellet was suspended in the above minimal buffer and subjected to the same homogenization and centrifugation process twice more. The thoroughly washed pellet was washed again, with 1 m NaCl and 10 μl of added benzonase (Novagen/EMD Millipore, Billerica, MA) to digest released nuclear material. The resulting pellet from an additional centrifugation (at 48,341 × g for 45 min) was further homogenized into 2× solubilization buffer (100 mm Hepes, pH 7.5, 300 mm NaCl, 10% glycerol, 4 mm EDTA, 16 mm MgCl2, and protease inhibitor tablets). After this homogenization, stock solutions of n-dodecyl-β-d-maltopyranoside (DDM) and cholesteryl hemisuccinate (CHS) were added. The resulting homogenate was diluted to final buffer concentrations of 50 mm Hepes, pH 7.5, with 150 mm NaCl, 5% glycerol, 2 mm EDTA, 8 mm MgCl2, 10 μm GR125487, 0.5% DDM, and 0.1% CHS and rotated for at least 6 h. Solubilized membrane proteins were separated by centrifugation at 45,000 × g for 45 min or until a tight pellet was obtained. The supernatant was used as the crude solubilized membrane preparation for further purification.
Affinity Chromatography and Size Exclusion Chromatography
A T7 antibody resin (Novagen/EMD Millipore, Billerica, MA) was added at 1.5 ml per 50 ml of supernatant and rotated overnight at 4°C in a roller bottle to bind the 5-HT4R protein. The resin then was poured into an empty wide column and washed extensively (10 column volumes) with a buffer identical to the solubilization buffer except that the DDM concentration was reduced to 0.05% and CHS to 0.01%. Protein bound to the resin was eluted with a buffer containing excess T7 peptide (0.3–0.5 mm). The eluate was bound directly onto 1D4 affinity resin (0.5 ml/50 ml of solubilized crude membranes) connected in tandem to the T7 affinity column. At the end of elution, the 1D4 column was washed with 10 column volumes of washing buffer. Elution of 5-HT4R was achieved by competition with buffer containing 1D4 peptide. Fractions (1 ml) were collected and analyzed for sample purity (∼ 90%). Peak fractions were pooled and concentrated to a volume of 0.5 ml, deglycosylated with 25 μl of peptide-N-glycosidase F (New England Biolabs, Ipswich, MA) on ice for 36 h, and subjected to gel filtration chromatography on a Superose 6 10/300 GL column (GE Healthcare) with 20 mm Hepes, pH 7.5, 100 mm NaCl, 0.025% DDM, 10 μm GR125487, and 0.005% CHS as a running buffer. A typical gel filtration profile of a purified sample and SDS-PAGE of a final sample is shown in supplemental Fig. S1. Peak fractions were pooled and concentrated to ∼30 mg/ml. A typical yield from 15 liters of insect culture was ∼1.5 mg of purified sample. Ligand-free receptor was prepared by extensive washing of the antagonist-bound receptor at the 1D4 affinity chromatography step with a buffer that contained no GR125487 (20 mm Hepes, pH 7.5, 100 mm NaCl, 0.025% DDM, and 0.005% CHS). Eluted ligand-free receptor was further purified using a Superose 6 10/300 GL column equilibrated with 20 mm Hepes, pH 7.5, 200 mm NaCl, 0.025% DDM, and 0,005% CHS. The purified sample was diluted into buffers used for footprinting analysis.
Radioligand Binding and cAMP Assays
Radioligand binding of purified 5-HT4R was performed as described by Salom et al. (18) with the 5-HT4R antagonist [3H]GR113808 (GE Healthcare). For cAMP assays, insect cells expressing 5-HT4R were plated at 106 cells/well into a 6-well plate and allowed to attach by overnight growth. Attached cells were gently washed with PBS (10 mm sodium/potassium phosphate, pH 7.4, 2.7 mm KCl, 137 mm NaCl) before exposure to ligands. Exposure to excess concentrations of ligands (1 mm final concentration) was carried out at room temperature in a volume of medium just sufficient to submerge the monolayer. After 15–20 min of incubation, the supernatant was aspirated, and cells were killed by direct addition of 70% ethanol. Aspirated well contents were centrifuged, and supernatants containing cAMP were evaporated to dryness under vacuum. Dried pellets were dissolved in ELISA incubation buffer (100 μl of 50 mm phosphate buffer, pH 8.0), and cAMP was estimated by using an ELISA kit from Cayman Chemicals (Ann Arbor, MI). These assays were sensitive up to 10–20 fm cAMP/ml. All biological assays were performed in triplicate, and values represented are average values from three experiments.
Footprinting and MS Methods
Synchrotron X-ray Radiolysis
Purified free 5-HT4R and antagonist-bound 5-HT4R were diluted to a final concentration of 2.5 μm in saline phosphate buffer (10 mm sodium phosphate, pH 7.0, 0.05% DDM, and 150 mm NaCl) with and without 10 μm of agonist GR125487, respectively. Samples from three different preparations were exposed to broadband focused x-rays at Beamline X28C of the National Synchrotron Light Source, Brookhaven National Laboratory, Upton, NY (19, 20). X-ray beam parameters were optimized by using the standard fluorophore assay that monitors the loss of intensity of an Alexa 488 fluorophore (Invitrogen) to reveal the effective hydroxyl radical concentration under selected experimental conditions (19). No difference of effective hydroxyl radical concentration was observed between the free and antagonist-bound 5-HT4R samples; thus, no corrections to the data were deemed necessary. Protein samples were irradiated by a two-step flow method in a modified KinTek (KinTek Corp.) apparatus. In the first step, 200 μl of the sample was passed through a flow cell with an irradiation cell volume of 3.5 μl. The flow speed was varied to generate four to five x-ray irradiation time intervals ranging from 0 to 10 ms. In the second step, the flow speed was adjusted such that the samples were collected in methionine/amide solution (final concentration of 10 mm) within 40 ms to quench any peroxide-induced or free radical-induced secondary oxidation during the post-exposure period. All exposures were carried out at 10°C. Samples were frozen in dry ice and stored at −80°C before proteolytic cleavage and liquid chromatography/mass spectrometric analysis (LC-MS). Following x-ray exposure, protein samples were digested with either pepsin or elastase, and the resulting digests were subjected to liquid chromatography (LC) separation coupled to MS detection as described below.
Proteolysis
Prior to proteolysis, 10 mm dithiothreitol (DTT) was added to protein samples to reduce disulfide bonds, and then free Cys residues were alkylated with 55 mm iodoacetamide. Reduced protein samples were purified by acetone precipitation to remove detergents and small molecules. Cold acetone (320 μl at −20°C) was added to 80-μl protein samples, which then were maintained at −20°C overnight followed by two washes with 200 μl of ice-cold acetone.
When pepsin was used for protein digestion, protein pellets were solubilized by addition of 5 μl of formic acid, followed by 5 μl of acetonitrile and adjusted to a final volume of 50 μl with HPLC-grade water. Sequencing grade pepsin (1 μl of 0.5 μg/μl; Roche Applied Science) was added to each sample, which then was incubated at room temperature for 4 h to overnight. Protein digests were tested with matrix-assisted laser desorption/ionization (MALDI/MS) for digestion efficiency. If proteins were not completely digested, the samples were vacuum-dried and reconstituted in 10 μl of 0.1% formic acid, pH 2.5, followed by another overnight digestion with pepsin.
When elastase was used for complete protein digestion, protein pellets were solubilized by adding 10 μl of 8 m urea followed by sonication and then incubation for 30 min. Samples were reduced in the presence of 5 mm DTT for 30 min at 37°C and then alkylated with 25 mm iodoacetamide for another 30 min in the dark. Next, the samples were diluted with 100 mm Tris-HCl buffer, pH 7.0, to reduce the urea concentration to 2 m. Elastase (1 μl of 0.5 μg/μl; Sigma-Aldrich) was added, and samples were incubated at room temperature overnight. The resulting reaction mixtures were desalted by passage through a C18 column (PepClean C18 spin column, Thermo Scientific, Waltham, MA) before MS analysis.
For limited proteolysis, native protein in sample buffer was digested by trypsin for periods of 1, 4, and 24 h at 37°C. DTT (10 mm) was then added to the partially digested protein samples to reduce disulfide bonds, and free Cys residues were alkylated with 55 mm iodoacetamide. CNBr was used to digest the protein samples again in 70% formic acid overnight. Mixtures were spun in a vacuum and freed of detergent with Pierce detergent removal spin columns (Thermo Scientific Waltham, MA). All samples were spin-dried and reconstituted with HPLC-grade water prior to MS analyses.
Protein Sequence Coverage Revealed by MS
Total proteolysis and mass spectrometry provided high sequence coverage of 5-HT4R in both antagonist-bound and ligand-free states after either elastase or pepsin digestion. More than 200 proteolytic digests covering 95.4% of the 5-HT4R sequence for the antagonist-bound state and over 150 digests covering 94.5% of the ligand-free sequence were examined as shown in supplemental Fig. S2, A and B. This high sequence coverage included all the structurally important regions of the seven transmembrane helices (TM1–TM7), the three intracellular loops (ICL1–ICL3), the three extracellular loops (ECL1–ECL3), and the C-terminal helix (H8).
Nano-ESI-MS/MS
MS experiments were carried out with an LTQ-FT mass spectrometer (Thermo Finnigan, San Jose, CA). Nano-reverse phase liquid chromatography separations were performed on a Dionex Ultimate U3000 HPLC (Dionex, Sunnyvale, CA) with a 5-cm × 75-μm Pico Frit C18 column (New Objective, Woburn, MA) directly connected to a nanospray emitter (10 μm, New Objectives). Chromatography was performed by using mobile phases A (0.1% formic acid in water) and B (80% acetonitrile, 0.04% formic acid in water) with a linear gradient of 1%/min, starting with 100% mobile phase A and finishing with 100% mobile phase B at a flow rate of 0.3 μl/min. All data were acquired in a positive ion mode. For these experiments, full MS scans (m/z 300–2000) were followed by MS2 scans of the eight most abundant peptide ions at normalized collision energy of 35%. High mass accuracy FT/MS was performed to detect precursor ions (resolution, 60,000; mass accuracy, 5 ppm); product ions were detected in an ion trap with relatively low mass accuracy (1 Da).
Data Analyses
Tandem MS data were analyzed with MassMatrix (2.4.2, Feb 22, 2012) software (21). Tandem MS data were searched against a protein database containing 5-HT4R sequences and decoy sequences with reversed sequences of this protein. Carbamidomethylated and all possible oxidized modifications were included as variable modifications for the Cys residue (22, 23). The enzyme was defined to cleave the protein nonspecifically. Mass tolerance was set to 10 ppm for the precursor ion and 1 Da for the product ion searches. Threshold limit of identification of the target protein was set to be greater than 40. Individual peptide mass spectra were identified with PP score greater than 5.0 and PPtag score greater than 2.0. All of the detected modified peptide mass spectra were confirmed manually.
Calculation of Oxidation Rates
The fraction of unoxidized peptides was calculated from the ratio of the chromatographic area under the ion signals for the unoxidized peptides to the sum of the unoxidized peptides and their radiolytic products (22, 24). Dose-response curves are presented as unmodified fractions versus exposure periods. The equation Y = Y0·e−kt (where Y and Y0 are the fractions of unmodified peptide at times t and 0 ms, and k is the first-order rate constant) was fit to the fraction of unmodified peptide. Datasets for the three labeling replicates were aligned in Origin 8.5.1 and fitted with the above first-order equation curve, and standard deviations were calculated by fitting all the data simultaneously. We report rate data for seven peptides (Table I). The S.D. associated with the rate determination was typically 30% or less. For other oxidized residues, the errors were higher, and the rates are not reported. We were not able to judge changes in oxidation rate as a function of ligand binding for these peptides. We do report a rate for peptide 302–308, even though the error for the free state was relatively high, because no oxidation was observed for the bound state, and we were able to confirm a significant change in conformation as a function of ligand binding.
Table I. Rates of 5-HT4R peptide oxidation calculated from radiolytic footprinting experiments.
Rates include the seven 5-HT4R peptides for which comparative rates of oxidation were calculated (see supplemental Fig. S6).The fraction of unoxidized peptides was calculated from the ratio of the chromatographic area under the ion signals for the unoxidized peptides to the sum of the unoxidized peptides and their radiolytic products (22, 24). Dose-response curves are presented as unmodified fractions versus exposure periods. The equation Y = Y0e−kt (where Y and Y0 are the fractions of unmodified peptide at times t and 0 ms, and k is the first-order rate constant) was fit to the fraction of unmodified peptide. The values are obtained from three replicates with four to five time points per experiment.
| Peptide | Range | Domain | Oxidized amino acids | Mass shift | Oxidation rate |
|
|---|---|---|---|---|---|---|
| Bound | Free | |||||
| Da | s−1 | |||||
| VMPFGA | 73–78 | TM2 helix | M742.58 | 16 | 3.2 ± 0.5 | 8.5 ± 2.3 |
| RNKMTPLRIAL | 130–140 | ICL2 loop and TM4 helix | M133 | 16 | 0.98 ± 0.0011 | 7.9 ± 1.9 |
| MVNKPYAIT | 187–195 | ECL2 loop | M187 and or Y192 | 16 or 32 | 6.7 ± 0.86 | 8.7 ± 2.2 |
| QMLQRAGASSESRPQSA | 227–243 | ICL3 loop | M228 | 16 | 23 ± 9.1 | 9.0 ± 2.7 |
| LQRAGASSESRPQSADQHSTHRMRTETKA | 236–257 | ICL3 loop | M251 | 16 | 47 ± 10 | 43 ± 13 |
| YINSGLN | 302–308 | TM7 helix | Y3027.43 | 16 | 0 | 1.1 ± 0.9 |
| AQPSDTLLQMA | 383–393 | C terminus (T7 Tag) | M392 | 16 | 108 ± 17 | 54 ± 11 |
Computational Methods
5-HT4R Model Generation
Comparative or homology modeling was performed with MODELLER Version 9.10 (Andrej Sali, University of California) (25). Six structures in the Protein Data Base (PDB) with a sequence similarity of about 40% were identified by a BLAST search (NCBI tool) that employed 5-HT4Rb (Homo sapiens) as the search sequence. PDB codes 2RH1 (β2-AR with partial inverse agonist carazolol), 3D4S (β2-AR with partial inverse agonist timolol), 3PDS (β2-AR with irreversible agonist FAUC50), 3PBL (dopamine D3 receptor with antagonist eticlopride), 2VT4 (mutant β1-AR antagonist cyanopindolol), and 2Y00 (mutant β1-AR with partial agonist, dobutamine) were selected. In a routine step of “compare” from MODELLER, all six of the above PDB structures were used as templates to identify the most appropriate structure for the queried sequence. The results showed that 2RH1, 3PDS, and 3D4S belong to one structural cluster, whereas 2VT4 and 2Y00 are close relatives. Finally, 2Y00 was selected over the others because of its sequence identity (48%, see supplemental Fig. S3) and superior data resolution (2.5 Å). An alignment of the sequences of 2Y00 with 5-HT4R displayed only some gaps and manifested considerable sequence similarity (complete sequence identity ∼30 and ∼48% partial to complete identity of 187 residues out of 388, see supplemental Fig. S3). A further modeling utility script was set up to build five models based on this sequence alignment. Models thus generated were further interrogated with the evaluation utilities in MODELLER, WHATCHECK, and PROCHECK to assess their quality.
Docking of Antagonist Ligand
To predict the binding of GR125487 to the 5-HT4R, an in silico docking calculation was performed with Autodock4.0 (Molecular Design Institute, University of California, San Francisco) (26). Positions of the various docked ligands were clustered based on their energy values, and the largest cluster with the lowest energy value was further analyzed for possible molecular interactions and biological relevance. The lowest energy clusters were assessed, and the one with the most biological relevance in light of our MS experimental data and previous biochemical results reported for the 5-HT4R was accepted. Details of energy and predicted affinities from docking analyses compared with experimental values are provided in supplemental Table S1. Because all five models were identical except for minor variations in the positions of loops, all of them yielded similar docking results.
RESULTS
Functional Characterization of Purified Receptors
Purified 5-HT4R samples of both ligand-free and antagonist-bound forms used in this study as analyzed by SDS-PAGE are shown in supplemental Fig. S1 (panel A before deglycosylation; panel B after deglycosylation, lanes 1 and 2). Size exclusion profiles of these samples indicate the presence of large aggregates and a homogeneous monomer population (supplemental Fig. S1C); fractions used for this study are indicated in the region defined by the arrow in supplemental Fig. S1C. Further characterization was accomplished by performing functional assays of 5-HTR receptors expressed in Sf9 cells (grown in a monolayer culture) (27). The second messenger produced upon activation of 5-HT4R is cAMP, which was assayed after exposing the 5-HT4R-expressing live cells to saturating concentrations (1 mm) of the synthetic agonist cisapride (Tocris Biosciences, Minneapolis, MN) for 15–20 min at room temperature. Although these Sf9 cells displayed some basal production of cAMP, exposure to cisapride significantly elevated cAMP levels above this level (supplemental Fig. S4A). Moreover, addition of GR125487 inhibited agonist-induced cAMP production. The purified receptor solubilized in DDM was also characterized for its ligand binding ability. When the synthetic antagonist [3H]GR113808 was used to determine the binding constant in a saturation binding assay, we estimated the purified receptors had a Kd of 4.7 nm (supplemental Fig. S4B). A subnanomolar (0.15 nm) Kd value was reported for the same compound in membranes from mammalian cells transiently expressing 5-HT4R (28), and a Kd of 1.1 nm was obtained for purified 5-HT4R in digitonin (18). Similarly, an assay we performed with competing concentrations of GR125487 against the [3H]GR113808 produced a curve with a Ki = 0.19 nm (data not shown). Overall, these data indicate that functional protein in a native-like state was used for these studies.
Partial Proteolysis Data Reveal Accessible Domains of the 5-HT4R
To probe accessible regions of purified solubilized 5-HT4R in 0.05% DDM and Hepes buffer, pH 7.0, we performed tryptic proteolysis for periods of 1, 4, and 24 h. Following partial proteolysis, protein samples were denatured and digested with CNBr. As the lack of cleavage of inaccessible sites could produce fragments too long to sequence by MS, this second cleavage step after denaturation was used to generate sets of moderate length fragments, thereby ensuring that missed tryptic cleavages were scored. The peptide cleavage maps observed for ligand-free 5-HT4R (A) and GR125487-bound samples (B) are shown in supplemental Fig. S5. The data revealed sets of trypsin cleavage sites (Arg and Lys) both easily cleaved and refractory to cleavage, providing guidance as to the local topology of the protein. However, we did not have extensive coverage for some domains shown in black in supplemental Fig. S5 (in TM2, TM6, ECL3, and TM7), so these could not be probed.
A critical finding is that the patterns of cleavage in panels A and B of supplemental Fig. S5 (Fig. 1) are virtually identical; thus, this approach revealed no large scale changes in topology upon ligand binding. The trypsin-sensitive sites are expected to correspond to accessible domains of the native protein as the cleavage was carried out on a completely folded and active form of 5-HT4R in DDM/CHS micelles. Almost all trypsin-accessible sites were distributed in domains predicted by the model to be exposed to the extracellular or intracellular side of this receptor as follows: K3 and K19 belong to the extreme ends of the N terminus; K52 and K54 belong to intracellular loop 1 (ICL1); K132 is part of ICL2; K172 and K190 are in extracellular loop 2 (ECL2); R231, R239, R250, and R252 are in ICL3; and R335, R336, R359, and K408 are in the unstructured C terminus. There are several residues cleaved at regions that are at the junction of the membrane, including R3218.55 in helix 8 (H8), K2566.33 in TM6, and K2205.66 in TM5, all in reasonable agreement with the model. The model also predicts that R963.28 and R1183.50 are in TM3, but their behavior as cleavage sites could indicate that the TM3 helix extends beyond the membrane. The predicted accessibility of R963.28 in TM3 is supported by its suggested role in ligand binding of 5-HT4R (29). Accessibility of R1183.50 is also notable due to its location in the “DRY” motif, which was suggested to interact directly with the G protein (30). Residues R1374.41, R2145.60, and K2596.36 are predicted to be deeply buried within the membrane; none of these were cleaved by trypsin in any sample analyzed.
Fig. 1.
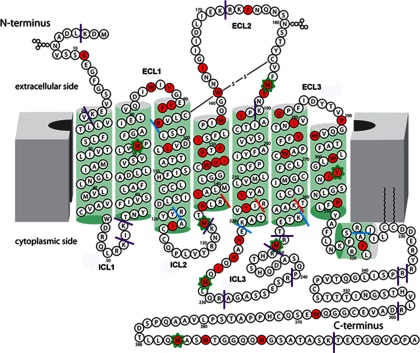
Two-dimensional representation of the 5-HT4R sequence. Residues colored red were identified to undergo oxidation in footprinting experiments. The approximate membrane lipid bilayer is shown as solid gray blocks at both sides, and helices are represented as green cylinders. The sites of trypsin cleavages experimentally observed are shown as solid lines. Purple solid lines indicate exposed and expected cleavage sites; blue solid lines indicate juxtamembrane cleavage sites, and red solid lines indicate sites not accessible to trypsin. Seven residues with calculated oxidation rates by mass spectrometry are indicated by an asterisk around that residue.
Based on our understanding of domain boundaries from the limited proteolysis observations and the alignment of the protein sequence with the crystal structure PDB code 2Y00, we built a two-dimensional diagram of 5-HT4R as shown in Fig. 1. 5-HT4R has a predicted long C terminus with ICL3 and ECL2 forming the largest loops (31). The membrane and helix boundaries are deduced from the proteolysis analysis. Overall, the translation of the model to the two-dimensional map required minimal topological rearrangement based on the limited proteolysis data, with the exception that the length of TM3 may be overestimated based on the limited proteolysis results above.
Radiolytic Protein Footprinting Analyses
The rate of oxidation of a residue side chain depends on both its reactivity with hydroxyl radicals and its accessibility to water from which hydroxyl radicals are formed during radiolysis (19, 22, 23, 32). Forty four residues within the 5HT4R sequence were found modified by oxidation, plus three residues were modified in the C-terminal tag in the 41 unique peptides detected by MS. These oxidized residues are colored red in Fig. 1.
The extent of side chain oxidation was calculated from total ion currents for the unmodified and modified peptides of interest. For seven peptides (Table I) we were able to construct dose-response curves (with S.D. <30%) illustrating the explicit rates of oxidation for both free and bound states. These overall oxidation rates (33) ranged from zero to 108 s−1 based on data from three replicates based on three different batches of sample (22). The explicit sites of oxidation for these peptides are presented in Table I, and the tandem MS mapping of all the modification sites is shown in supplemental Fig. S6. Although the rates of oxidation were calculated from the quantification of MS1 intensity data at the peptide level, the oxidation for each one of these seven peptides is dominated by one or two residues that represent explicit structural probes. Thus, our experiments show that M742.58 in TM2, M133, in ICL2 directly adjacent to TM4, and Y3027.43 in TM7, showed decreased oxidation rates upon ligand binding. Particularly, we detected tandem MS of peptides containing oxidized Y3027.43 only in the free state and not in the bound state. However, M228 in ICL3 and M392 in the C-terminal T7 tag showed increased oxidation rates upon ligand binding. M187 and Y192 mapped onto ECL2 and M251 in ICL3 showed minimal differences in oxidation between the ligand-free and antagonist-bound protein forms. Notably, of the eight modified residues, several are in ICL3, ECL3, and at the N or C terminus and are predicted to be accessible to bulk solvent. Moreover, M742.58 and Y3027.43 are predicted to be in buried TM regions. As the model predicts, the latter have no access to bulk water, and we expect they are located near internal waters that drive their oxidation. Changes in the structural relationship of side chains to their water network are likely responsible for the changes in the observed oxidation rates (14).
Apart from the above mentioned eight oxidized residues from seven peptides, we identified 39 additional modified residues in 34 additional peptides (supplemental Table S2). The dose-response curves for these peptides had standard deviations of the fitted rates above 50%, and therefore we did not report these rates. However, the oxidized residues were reproducibly identified. These modifications spanned an extensive stretch of various TM helices, ECLs, and ICLs that are critical for ligand recognition and signal transduction. E11 was the only residue modified in the presumably unstructured N-terminal peptide stretch (VSSEEGF) for which there is very little explicit structural information available. In crystal structures of GPCRs solved to date, the N terminus was either removed to facilitate crystallization or found to be unstructured (17, 34). Residues W86, Y88, V913.23, F923.24, and R963.28 (mapping onto ECL1 and the N terminus of TM3) and M1584.62, W161, I164, F175, M187, and V188 (all belonging to the ECL2 domain, except M1584.62, which is located on C terminus of TM4) were found to be modified and spatially distributed on the extracellular side of the receptor in addition to E11 of the N terminus (Fig. 1). These residues are oxidized by bulk water based on our model, and they appear moderately accessible to solvent.
Interestingly, many modified residues were found in predicted transmembrane regions and near regions of functional interest. For example, modified residue W2726.48, seen in peptide CWAPFF of TM6, is a highly conserved residue among the rhodopsin class of GPCRs that flanks the ligand-binding pocket (35). V2776.53 found modified in peptide VTNIVDPFI of TM6 is close to F2756.51 and F2766.52, two other conserved residues involved in both ligand binding and signaling processes (36). Peptides YTVPGQVW (W2947.35), YINSGLN (where Y3027.43 showed ligand-dependent protection), and GLNPFL (P3097.50) all are located in the TM7 helix.
Another major stretch of modified residues was identified between residues 133 and 158 in TM4. These included M133, P1354.39, I1384.42 V1474.51, I1484.52, P1494.53, W1464.50, F1514.55, I1524.56, and M1584.62. This observation suggests significant water occupancy across the length of TM4. Finally, a stretch of five modified residues (Y2135.58, Y2165.62, M228, H222, H224), and I226 from the peptide representing residue numbers 212 to 226 span the C-terminal region of TM5 and extend into the ICL3 domain. Their oxidation is consistent with the solvent accessibility of this region (Fig. 1).
DISCUSSION
5-HT4R is a serotonin receptor subtype that can be expressed and purified to reasonable purity and homogeneity. In addition, the pharmacokinetics of its ligand interactions have been well studied (37) making it an excellent model system for exploring GPCR structure-function. In this study, we analyzed the ligand binding properties of a physiologically active form of 5-HT4R that was stable under various experimental conditions. In addition, using a hybrid approach and relying heavily on structural mass spectrometry, we assessed the overall membrane protein structure of 5-HT4R and evaluated its changes in conformation upon binding the antagonist GR125487.
An interesting feature of the data was the observation of many sites of radiolytic modification across various domains of the 5-HT4R, including those within the transmembrane region of this structure. Also, changes in modification as a function of ligand binding hinted at potential sites of ligand interaction as well as possible allosteric effects of ligand binding. Identification of radiolytic modifications of residues induced by synchrotron radiation in protein samples has helped identify side chains interacting with bulk water as well as side chains interacting with waters buried inside the protein structure, including those within the transmembrane domains of membrane proteins (14, 15).
Conserved Structural Motifs in Homology Model of 5HT4R
To put our experimental results into a structural context, we first built a three-dimensional model of the 5-HT4R based on a homologous crystal structure of the β1-AR (Fig. 2A). Then, to evaluate the effects of ligand binding, we docked the antagonist GR125487 into the generated model (Fig. 2B). The proposed configuration of the docked ligand along with the modification patterns observed for the receptor reveals that 5-HT4R shares several ligand binding characteristics described below that are typical of class A GPCRs. In addition, we interpreted the overall modification pattern in the context of conserved water molecules observed in class A GPCRs derived from a previous footprinting analysis of rhodopsin (14). Evidence that this model reflects the native structure in detergent (and that the biochemical preparation reflects the structure) is provided by the limited proteolysis data, which support an overall agreement between the observed domain structural modifications and the model. However, this mapping has some limitations. For example, limited proteolysis is only sensitive to local structural factors and cannot reveal a three-dimensional orientation.
Fig. 2.

Structural model of 5-HT4R with water molecules. A, three-dimensional model generated for 5-HT4R. Modified residues identified by MS footprinting analysis are represented as yellow sticks spatially distributed throughout this receptor. B, waters were placed into the 5-HT4R model by structural alignment of the main carbon skeleton to homologous class A GPCR crystal structures to analyze the general distribution of modified residues (see Fig. 5 for a detailed analysis of bound waters based on residue conservation); cyan balls from rhodopsin bound to 11-cis-retinal (PDB code 1F88); pink balls, β1-AR bound to dobutamine (PDB code 2Y00); dark blue balls, β1-AR bound to cyanopindolol (PDB code 2Y00); and golden brown balls, β2-AR bound to carazolol (PDB code 2RH1).
When structurally aligned, the 5-HT4R model and relevant homologous GPCR crystal structures (PDB codes 1F88, 2RH1, and 2Y00) showed remarkable conservation of several motifs, especially in their binding pockets (Fig. 3, A–C). For example, when 11-cis-retinal (the configuration seen in Fig. 3A) isomerizes into the all-trans form to activate rhodopsin (moving away from the position in Fig. 3A), it allows a fourth conserved Trp, W2656.48, to swing into the space between Y2686.51 and F2125.47 (38). A similar “toggle switch ” mechanism was proposed for β1-AR and β2-AR where the particular residues relevant for rhodopsin activation are conserved (F6.51, F6.52, F5.47, and W6.48) and form a similar overall architecture (Fig. 3, B and C). Our model of 5-HT4R includes these identical residues in positions similar to that of the other homologs suggesting that the toggle switch mechanism may also be a feature of 5-HT4R activation. When the 11-cis-retinal·opsin complex is compared with the model of the GR125487·5-HT4R complex, the heteroaromatic headgroup of GR125487 does not bind as deeply as the β-ionone ring of retinal in rhodopsin (flanked by Y2686.51 and F2125.47, Fig. 3A) (36, 39, 40). This deeper positioning of the β-ionone ring in rhodopsin is possible due to the presence of an Ala residue at position 6.52, whereas in β1-AR, β2-AR, and 5-HT4R, this position is occupied by a more bulky conserved residue F6.52 (Fig. 3, B and C) that tends to push their ligands' heteroaromatic headgroup away.
Fig. 3.

Comparison of the ligand-binding sites in GPCRs. A comparative analysis of docked antagonist GR125487 within the 5-HT4R model to crystal structures of 11-cis-retinal bound to rhodopsin (1F88) (A), carazolol bound to β2-AR (2RH1) (B), and cyanopindolol bound to β1-AR (C). The proposed toggle switch mechanism of rhodopsin activation can be seen in A. The β-ionone ring of retinal in the 11-cis-conformation (light pink) is sandwiched between Y2686.51 and F2125.47 (purple residues). The flip of the β-ionone ring forming all-trans-retinal allows swinging-in of W2656.48 to permit signaling in rhodopsin. B, comparison of the 5-HT4R-GR125487 model with the β2-AR·carazolol complex. The toggle switch mechanism of β2-AR is represented by four residues as follows: F2906.52 sandwiched between F2896.51, F2085.47, and W2866.48. Activation is proposed to trigger a rotation of W2866.48 to displace F2906.52. In 5-HT4R, residues F2756.51, F2766.52, F2015.47, and W2726.48 are conserved and positioned similarly around GR125487. C, comparison of binding of cyanopindolol to β1-AR with docked superposed GR125487. The four residues corresponding to the toggle switch are conserved in β1-AR, including F3066.51, F3076.52, F2165.47, and W3036.48. The length and branching of the tail group of GR125487 make its orientation different from those of the tail groups of carazolol and cyanopindolol. The green cartoon trace represents rhodopsin bound to 11-cis-retinal; the yellow trace is the β2-AR-T4L chimera bound to carazolol; the sea blue trace is the 5-HT4R model bound to GR125487, and the silver gray trace is the β1-AR bound to cyanopindolol.
In β2-AR, the toggle switch mechanism is facilitated by movement of F2906.52, which is sandwiched between F2896.51 and F2085.47 (Fig. 3B, yellow trace). Upon activation, W2866.48 is proposed to rotate into the position occupied by F2906.52 in the inactive form of the β2-AR. Thus, steric constraints imposed by F2906.52 mimic the interaction of retinal's β-ionone ring with W2656.48 and F2125.47 of rhodopsin. In the β1-AR, all four residues proposed to mediate the toggle switch mechanism also are conserved and flank the sides of the binding pocket (Fig. 3C, silver trace). Our model proposes a docked position of GR125487 in 5-HT4R similar to that of the antagonists in the β2-AR·carazolol and β1-AR·cyanopindolol complexes suggesting that these proposed antagonist configurations are similarly inhibitory to toggle switch signaling (17, 41).
Mechanisms of GPCR, Ligand Interactions
Studies aimed at understanding the interaction of the natural ligand 5-HT with its receptors have revealed a key role for the highly conserved residue aspartate at position 3.32. Mutation of residue D1163.32 to N reduced the ability of the 5-HT1aR to bind 5-HT (42), whereas it had no effect on binding the inverse agonist pindolol. Likewise, mutation of residue D1043.32 to L in the β1-AR resulted in a similar reduction of natural ligand binding (43). In yet another aminergic GPCR, the α2-AR, mutation of residue D1133.32 to N, also reduced agonist binding efficiency significantly (44). Binding of 5-HT to 5-HTRs thus was predicted to involve a strong ionic interaction between its amine group and the carboxylate group of the conserved D3.32 residue. In addition, positioning of the 5-HT amine to form a polar contact with D3.32 could promote the re-arrangement of other transmembrane helices and the toggle switch. Based on their observations of an antagonist-bound crystal structure for the β1-AR, Warne et al. (41) proposed that a large 2–3 Å tightening of the TM helices around the binding pocket occurs upon epinephrine binding.
For 5HT4R, Mialet et al. (45) have found that mutation of D3.32 to N completely abolished the binding of 5-HT, although tight binding of [3H]GR113808 (an antagonist very similar to GR125487) was maintained. Additionally, mutations of Y302A7.43, F275A6.51, or F276V6.52 had no effect on 5-HT binding, although they markedly decreased [3H]GR113808 binding. In our model, we suggest a set of hydrogen bonding and aromatic interactions between Y3027.43, F2756.51, or F2766.52 and the antagonist, which is consistent with the mutational data. These mutational results also suggest that critical ligand binding interactions for the agonist 5-HT within the ligand binding pocket conformation is quite different for agonists compared with antagonists. The mode of binding for smaller ligands like 5-HT within the 5-HT receptors could be relatively simple, involving ionic interactions with residue D1003.32. In this mechanism, and based on our model of the free 5-HT4R, binding of ligand requires breakage of an ionic bond between D1003.32 and Y3027.43 present in the unliganded structure. This conformational change contributes to toggle switch activation. During antagonist binding, the ionic bond between Y3027.43 and D1003.32 also breaks, but in this case Y3027.43 rotates to a position where it may have an interaction with the antagonist ligand, as illustrated in Fig. 4. Although the 3.9 Å distance illustrated in Fig. 4 is relatively long, it is interesting that the distances from the conserved Y7.43 to the similarly placed nitrogen atoms for the antagonists' carazolol and cyanopindolol are quite similar at 3.4 and 3.8 Å, respectively (supplemental Fig. S7). In addition to these potential hydrogen-binding interactions, Y3027.43 and the heteroaromatic ring of the antagonist are predicted to be in positions to facilitate a series of aromatic ring face interactions with multiple residues of the toggle switch (Fig. 4B). Through the formation of this rigid network (as suggested by Schertler and co-workers (46)), antagonists may stabilize an inactive conformation of the type A GPCRs.
Fig. 4.

GR125487 interactions with hydrophobic residues in the binding site of 5-HT4R. A and B are views of binding pocket with a rotation of 90° along the vertical axis. For 5-HT4R, our model posits ring-face aromatic interactions of binding site residues that translate from the back face of the hetero-aromatic ring structure of GR125487 through a series of aligned aromatic residues. Apart from the highly conserved alignment of the toggle switch residues, a key difference in the binding site interactions for GR125487 in 5-HT4R involves W2947.35 on the back face of the heteroaromatic ring. The bulky aromatic ring of W294 interacts strongly with the heteroaromatic moiety of GR125487 through F2756.51 (purple), which continues down to F2766.52 (purple) and F2015.47 (purple), which in turn interact with W2726.48 (purple) and continues to Y3027.43 (green) on the front face. These strong ring-face aromatic hydrophobic interactions suppress toggle switch activation as long as the heteroaromatic headgroup of GR125487 remains bound in the 5-HT4R pocket to Y3027.43. Residues M742.58 and Y3027.43 are highlighted (forest green). Both of these residues are significantly protected upon antagonist binding, supporting their proximity to the binding site. This change is in contrast to M184, which shows no change in oxidation upon ligand binding. D1003.32 a key binding site residue, binds to 5-HT (serotonin) but not the antagonist (cyan). In our model of the ligand free state (not shown) D1003.32 is hydrogen bonded to Y3027.43.
Locating Transmembrane Waters in the 5-HT4R Structure Using Modeling and Footprinting
As outlined above, 5-HT4R aligns well with many structural features seen in homologous high resolution structures of multiple class A GPCRs (48). We have previously proposed that the positions of waters observed in these structures are conserved (15, 49). Through alignment of the model generated for 5-HT4R with selected GPCR crystal structures (PDB codes 2Y00, 2RH1, and 1F88), we positioned potential conserved waters in the 5-HT4R model. Fig. 5 shows the various sequences, the relevant domains, and the adjacent water molecules across the four proteins, including the waters proposed for 5-HT4R. In previous studies of bovine rhodopsin using radiolytic footprinting and MS, internal waters near residue side chains buried within the bilayers were shown to mediate side-chain oxidation (13, 15). This correlation of water positions and side-chain oxidation was analyzed in the context of footprinting data obtained from experiments with 5-HT4R.
Fig. 5.

Sequence alignments (ClustalW) of GPCRs used in this study are shown together with the 5-HT4R sequence. These GPCRs include PDB code 2Y00-β1-AR bound to dobutamine; PDB code 2RH1-β2-AR bound to carazolol, and PDB code 1F88-rhodopsin bound to 11-cis-retinal. Crystallographic waters from these structures are shown as numbers above and below the sequences with lines drawn to residues with which they make polar contacts. Residues conserved across the four sequences are represented as follows: completely conserved residues are indicated by filled red boxes, and partially conserved residues are identified by empty blue boxes with red letters. Brick-red boxed residues shown in the 5-HT4R sequence and green boxed residues in the rhodopsin represent oxidized residues observed by MS footprinting analysis. Thick brick-red lines drawn above the sequences represent helices (TM1 to H8). The green lines drawn below the sequences indicate four stretches of peptides representing regions of the serotonin receptor susceptible to radiolytic oxidation upon antagonist binding. Numbers in the 2000 series are waters from PDB code 2Y00 (β1-AR bound to dobutamine). Numbers in the 500 series are waters from PDB code 2RH1. Numbers in the 900 to 2000 series in blue are waters from PDB code 1F88. The two residues critical for conveying the inhibitory signal to the cytoplasmic side of the serotonin receptor are indicated inside black boxes; these are connected by a chain of conserved buried water molecules that extend from TM6 (W2726.48) to TM7 (P3097.50). Residue numbers on the bottom line of aligned sequences correspond to those of PDB code 1F88.
A group of conserved waters (numbers 2001, 2003, 2006, 2007, 2014, 2015, 2017, and 2018 from PDB code 2Y00 and 506, 519, 529, 528, 532, 534, 546, and 548 from PDB code 2RH1; Figs. 5 and 6 and supplemental Figs. S8 and S9) surround modified residue W2726.48, part of the toggle switch relay in 5-HT4R, and extend down the length of the respective transmembrane segments between TM6 and TM7 to the cytoplasmic face of the receptor. These conserved waters near W2726.48 appear perfectly positioned to form interacting polar contacts leading down to P3097.50 at the C terminus of TM7 (Fig. 6). Not only are these waters seen in the crystallographic data for multiple GPCRs, but the footprinting results of 5-HT4R reveal multiple modified residues lining TM7, including P2907.31, W2947.35, W2997.40, Y3027.43, I3037.44, and P3097.50, many of which are opposite W2726.48 on TM6 and lead down to P3097.50 (supplemental Table S2). This trail of modified residues provides direct evidence of water in a channel between TM6 and TM7 in the native 5-HT4R (14, 15, 46).
Fig. 6.
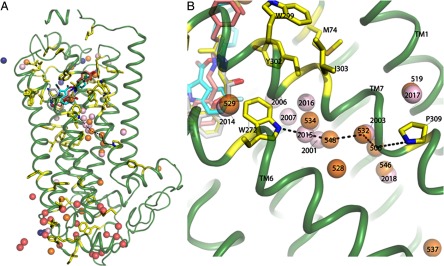
Radiolytic oxidation guided placement of waters near the conserved NPXXY motif of 5-HT4R. A, distribution of waters placed into the 5-HT4R model based on residue conservation. B, possible pathway of signal communication from critical residue W2726.48 through a water channel to P3097.50 for inhibition of signaling mediated through H8 and the C terminus of the 5-HT4R.
GPCR Signal Activation Down the TM6/TM7 Water Channel
M742.58, located near the ligand binding pocket (Fig. 6B), exhibits clear and consistent conformational changes upon ligand binding in the footprinting experiments seen here. Y3027.43, another residue located in close proximity of the ligand binding pocket of docked antagonist, also showed consistent oxidation in free samples (and none in bound samples). However, we did not observe changes in oxidation for other 5-HT4R residues within TM6 or TM7 upon ligand binding. For example, M251 in ICL3 near TM6 had no change in oxidation, and other residues in TM6/TM7 were always oxidized. Conversely, in our previous experiments on rhodopsin, we observed significant changes in oxidation (and thus changes in conformation) near the retinal-binding site upon activation of ground state rhodopsin to the Meta-II form, including changes in oxidation of M862.53 (TM2) and F1163.31 (TM3) near the retinal-binding site as well as changes in multiple residues in TM7, including M2887.35, Y3017.48, P3037.50 (the homologous residue to P3097.50 in Fig. 6B), and Y3067.53. These data provide supportive evidence for activation-based signaling from the retinal site down TM7 toward the intracellular surface in rhodopsin. In contrast, there were no changes in oxidation in TM6/TM7 for 5-HT4R upon antagonist binding, consistent with our hypothesis that upon antagonist binding Y3027.43 alters its conformation to productively interact with the antagonist, and the resultant rigid conformation in the ligand-binding pocket locks the toggle switch in place of blocking signaling. The conservation of the water channel in 5-HT4R and other type A GPCRs indicates that this communication path may be critically important for agonist-induced signaling. This hypothesis could be specifically tested by examining the structural properties of the proposed water channel for the 5-HT agonist·receptor complex, where we would predict activation-dependent oxidation changes in TM7 similar to those seen for rhodopsin.
CONCLUSION
Our studies clearly show the power of MS techniques to study native GPCR structure and ligand interactions. Unlike many other structural tools, MS methods have unique advantages in probing subtle local structural changes within native membrane protein samples. Disadvantages of this approach include its inability to provide three-dimensional structural information. This makes it essential to analyze the MS data in the context of three-dimensional structures or structural models. In this study, we found over 40 modification sites in 5-HT4R through radiolytic footprinting experiments. However, improvements in instrument sensitivity may be required to provide quantitative information about the relative reactivity of all the residues in their different states. Use of targeted MS methods, such as single reaction monitoring, may improve performance and permit detailed measures of signaling mechanisms for GPCRs poised in native-like states (50). Also, recent advances in footprinting technology, through examining the temperature dependence of oxidation to precisely define sites oxidized by bound waters and using O-18 to measure rates of exchange, can more precisely define water-mediated communication channels that could be critical for GPCR signaling (12).
Supplementary Material
Acknowledgments
We thank Rujitha Di Mello for technical assistance at the X28C beamline.
Footnotes
* This work was supported, in whole or in part, by National Institutes of Health Grants P30-EB-9998, R01-EB-9688, and U01-GM-94612 (to M.R.C.) and Grant R01-EY-8061 (to K.P.).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- 5-HT
- 5-hydroxytryptamine (serotonin)
- 5-HT4R
- serotonin type 4 receptor
- AR
- adrenergic receptor
- CHS
- cholesteryl hemisuccinate
- DDM
- n-dodecyl-β-d-maltopyranoside
- ECL
- extracellular loop
- GPCR
- G protein-coupled receptor
- ICL
- intracellular loop
- PDB
- Protein Data Bank
- TM
- transmembrane; .Protein residues are designated by single-letter amino acid codes followed by the residue number. The Ballesteros and Weinstein numbering system (1) is used to identify conserved residues of GPCRs.
REFERENCES
- 1. Ballesteros J. A., Jensen A. D., Liapakis G., Rasmussen S. G., Shi L., Gether U., Javitch J. A. (2001) Activation of the β2-adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J. Biol. Chem. 276, 29171–29177 [DOI] [PubMed] [Google Scholar]
- 2. Berger M., Gray J. A., Roth B. L. (2009) The expanded biology of serotonin. Annu. Rev. Med. 60, 355–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gershon M. D., Tack J. (2007) The serotonin signaling system: from basic understanding to drug development for functional GI disorders. Gastroenterology 132, 397–414 [DOI] [PubMed] [Google Scholar]
- 4. Salom D., Wang B., Dong Z., Sun W., Padayatti P., Jordan S., Salon J. A., Palczewski K. (2012) Post-translational modifications of the serotonin type 4 receptor heterologously expressed in mouse rod cells. Biochemistry 51, 214–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hegde S. S., Eglen R. M. (1996) Peripheral 5-HT4 receptors. FASEB J. 10, 1398–1407 [DOI] [PubMed] [Google Scholar]
- 6. Hamblin M. W., Guthrie C. R., Kohen R., Heidmann D. E. (1998) Gs protein-coupled serotonin receptors: receptor isoforms and functional differences. Ann. N.Y. Acad. Sci. 861, 31–37 [DOI] [PubMed] [Google Scholar]
- 7. Claeysen S., Sebben M., Becamel C., Bockaert J., Dumuis A. (1999) Novel brain-specific 5-HT4 receptor splice variants show marked constitutive activity: role of the C-terminal intracellular domain. Mol. Pharmacol. 55, 910–920 [PubMed] [Google Scholar]
- 8. Chang W. C., Ng J. K., Nguyen T., Pellissier L., Claeysen S., Hsiao E. C., Conklin B. R. (2007) Modifying ligand-induced and constitutive signaling of the human 5-HT4 receptor. PloS one 2, e1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gale J. D., Grossman C. J., Whitehead J. W., Oxford A. W., Bunce K. T., Humphrey P. P. (1994) GR113808: a novel, selective antagonist with high affinity at the 5-HT4 receptor. Br. J. Pharmacol. 111, 332–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Beattie D. T., Smith J. A., Marquess D., Vickery R. G., Armstrong S. R., Pulido-Rios T., McCullough J. L., Sandlund C., Richardson C., Mai N., Humphrey P. P. (2004) The 5-HT4 receptor agonist, tegaserod, is a potent 5-HT2B receptor antagonist in vitro and in vivo. Br. J. Pharmacol. 143, 549–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Long D. D., Armstrong S. R., Beattie D. T., Choi S. K., Fatheree P. R., Gendron R. A., Goldblum A. A., Humphrey P. P., Marquess D. G., Shaw J. P., Smith J. A., Derek Turner S., Vickery R. G. (2012) Discovery, oral pharmacokinetics, and in vivo efficacy of a highly selective 5-HT(4) receptor agonist: Clinical compound TD-2749. Bioorg. Med. Chem. Lett. 22, 4849–4853 [DOI] [PubMed] [Google Scholar]
- 12. Gupta S., D'Mello R., Chance M. R. (2012) Structure and dynamics of protein waters revealed by radiolysis and mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 109, 14882–14887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Orban T., Gupta S., Palczewski K., Chance M. R. (2010) Visualizing water molecules in transmembrane proteins using radiolytic labeling methods. Biochemistry 49, 827–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Angel T. E., Gupta S., Jastrzebska B., Palczewski K., Chance M. R. (2009) Structural waters define a functional channel mediating activation of the GPCR, rhodopsin. Proc. Natl. Acad. Sci. U.S.A. 106, 14367–14372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Angel T. E., Chance M. R., Palczewski K. (2009) Conserved waters mediate structural and functional activation of family A (rhodopsin-like) G protein-coupled receptors. Proc. Natl. Acad. Sci. U.S.A. 106, 8555–8560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. MacKenzie D., Arendt A., Hargrave P., McDowell J. H., Molday R. S. (1984) Localization of binding sites for carboxyl-terminal specific anti-rhodopsin monoclonal antibodies using synthetic peptides. Biochemistry 23, 6544–6549 [DOI] [PubMed] [Google Scholar]
- 17. Cherezov V., Rosenbaum D. M., Hanson M. A., Rasmussen S. G., Thian F. S., Kobilka T. S., Choi H. J., Kuhn P., Weis W. I., Kobilka B. K., Stevens R. C. (2007) High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science 318, 1258–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Salom D., Wu N., Sun W., Dong Z., Palczewski K., Jordan S., Salon J. A. (2008) Heterologous expression and purification of the serotonin type 4 receptor from transgenic mouse retina. Biochemistry 47, 13296–13307 [DOI] [PubMed] [Google Scholar]
- 19. Gupta S., Sullivan M., Toomey J., Kiselar J., Chance M. R. (2007) The Beamline X28C of the Center for Synchrotron Biosciences: a national resource for biomolecular structure and dynamics experiments using synchrotron footprinting. J. Synchrotron Radiat. 14, 233–243 [DOI] [PubMed] [Google Scholar]
- 20. Sullivan M. R., Rekhi S., Bohon J., Gupta S., Abel D., Toomey J., Chance M. R. (2008) Installation and testing of a focusing mirror at beamline X28C for high flux x-ray radiolysis of biological macromolecules. Rev. Sci. Instrum. 79, 025101 [DOI] [PubMed] [Google Scholar]
- 21. Xu H., Freitas M. A. (2007) A mass accuracy sensitive probability based scoring algorithm for database searching of tandem mass spectrometry data. BMC Bioinformatics 8, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Takamoto K., Chance M. R. (2006) Radiolytic protein footprinting with mass spectrometry to probe the structure of macromolecular complexes. Annu. Rev. Biophys. Biomol. Struct. 35, 251–276 [DOI] [PubMed] [Google Scholar]
- 23. Xu G., Chance M. R. (2005) Radiolytic modification and reactivity of amino acid residues serving as structural probes for protein footprinting. Anal. Chem. 77, 4549–4555 [DOI] [PubMed] [Google Scholar]
- 24. Kiselar J. G., Mahaffy R., Pollard T. D., Almo S. C., Chance M. R. (2007) Visualizing Arp2/3 complex activation mediated by binding of ATP and WASp using structural mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 104, 1552–1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sali A., Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 26. Morris G. M., Goodsell D. S., Halliday R. S., Huey R., Hart W. E., Belew R. K., Olson A. J. (1998) Automated docking using a Lamarckian gentic algorithm and epirical bining free energy function. J. Comput. Chem. 19, 1639–1662 [Google Scholar]
- 27. Schneider E. H., Seifert R. (2010) Sf9 cells: a versatile model system to investigate the pharmacological properties of G protein-coupled receptors. Pharmacol. Ther. 128, 387–418 [DOI] [PubMed] [Google Scholar]
- 28. Van den Wyngaert I., Gommeren W., Verhasselt P., Jurzak M., Leysen J., Luyten W., Bender E. (1997) Cloning and expression of a human serotonin 5-HT4 receptor cDNA. J. Neurochem. 69, 1810–1819 [DOI] [PubMed] [Google Scholar]
- 29. Rivail L., Giner M., Gastineau M., Berthouze M., Soulier J. L., Fischmeister R., Lezoualc'h F., Maigret B., Sicsic S., Berque-Bestel I. (2004) New insights into the human 5-HT4 receptor-binding site: exploration of a hydrophobic pocket. Br. J. Pharmacol. 143, 361–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rovati G. E., Capra V., Neubig R. R. (2007) The highly conserved DRY motif of class A G protein-coupled receptors: beyond the ground state. Mol. Pharmacol. 71, 959–964 [DOI] [PubMed] [Google Scholar]
- 31. Westkaemper R. B., Glennon R. A. (1991) Approaches to molecular modeling studies and specific application to serotonin ligands and receptors. Pharmacol. Biochem. Behav. 40, 1019–1031 [DOI] [PubMed] [Google Scholar]
- 32. Kiselar J. G., Janmey P. A., Almo S. C., Chance M. R. (2003) Structural analysis of gelsolin using synchrotron protein footprinting. Mol. Cell. Proteomics 2, 1120–1132 [DOI] [PubMed] [Google Scholar]
- 33. Kaur P., Kiselar J. G., Chance M. R. (2009) Integrated algorithms for high throughput examination of covalently labeled biomolecules by structural mass spectrometry. Anal. Chem. 81, 8141–8149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tebben A. J., Schnur D. M. (2011) Beyond rhodopsin: G protein-coupled receptor structure and modeling incorporating the β2-adrenergic and adenosine A(2A) crystal structures. Methods Mol. Biol. 672, 359–386 [DOI] [PubMed] [Google Scholar]
- 35. Mirzadegan T., Benkö G., Filipek S., Palczewski K. (2003) Sequence analyses of G-protein-coupled receptors: similarities to rhodopsin. Biochemistry 42, 2759–2767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Palczewski K. (2006) G protein-coupled receptor rhodopsin. Ann. Rev. Biochem. 75, 743–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bureau R., Boulouard M., Dauphin F., Lezoualc'h F., Rault S. (2010) Review of 5-HT4R ligands: state of art and clinical applications. Curr. Top. Med. Chem. 10, 527–553 [DOI] [PubMed] [Google Scholar]
- 38. Choe H. W., Kim Y. J., Park J. H., Morizumi T., Pai E. F., Krauss N., Hofmann K. P., Scheerer P., Ernst O. P. (2011) Crystal structure of metarhodopsin II. Nature 471, 651–655 [DOI] [PubMed] [Google Scholar]
- 39. Filipek S., Stenkamp R. E., Teller D. C., Palczewski K. (2003) G protein-coupled receptor rhodopsin: a prospectus. Annu. Rev. Physiol. 65, 851–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Filipek S., Teller D. C., Palczewski K., Stenkamp R. (2003) The crystallographic model of rhodopsin and its use in studies of other G protein-coupled receptors. Annu. Rev. Biophys. Biomol. Struct. 32, 375–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Warne T., Serrano-Vega M. J., Baker J. G., Moukhametzianov R., Edwards P. C., Henderson R., Leslie A. G., Tate C. G., Schertler G. F. (2008) Structure of a β1-adrenergic G-protein-coupled receptor. Nature 454, 486–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ho B. Y., Karschin A., Branchek T., Davidson N., Lester H. A. (1992) The role of conserved aspartate and serine residues in ligand binding and in function of the 5-HT1A receptor: a site-directed mutation study. FEBS Lett. 312, 259–262 [DOI] [PubMed] [Google Scholar]
- 43. Hossain M., Ahmed M., Bhuiyan M. A., Ishiguro M., Nakamura T., Ozaki M., Nagatomo T. (2008) Mutation of important amino acid residue of Asp104Lys in human β(1)-adrenergic receptor triggers functional and constitutive inactivation. Biol. Pharm. Bull. 31, 1517–1522 [DOI] [PubMed] [Google Scholar]
- 44. Wang C. D., Buck M. A., Fraser C. M. (1991) Site-directed mutagenesis of α2A-adrenergic receptors: identification of amino acids involved in ligand binding and receptor activation by agonists. Mol. Pharmacol. 40, 168–179 [PubMed] [Google Scholar]
- 45. Mialet J., Dahmoune Y., Lezoualc'h F., Berque-Bestel I., Eftekhari P., Hoebeke J., Sicsic S., Langlois M., Fischmeister R. (2000) Exploration of the ligand-binding site of the human 5-HT(4) receptor by site-directed mutagenesis and molecular modeling. Br. J. Pharmacol. 130, 527–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Deupi X., Standfuss J., Schertler G. (2012) Conserved activation pathways in G-protein-coupled receptors. Biochem. Soc. Trans. 40, 383–388 [DOI] [PubMed] [Google Scholar]
- 47. Xu G., Chance M. R. (2007) Hydroxyl radical-mediated modification of proteins as probes for structural proteomics. Chem. Rev. 107, 3514–3543 [DOI] [PubMed] [Google Scholar]
- 48. Katritch V., Cherezov V., Stevens R. C. (2012) Diversity and modularity of G protein-coupled receptor structures. Trends Pharmacol. Sci. 33, 17–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Renthal R. (2008) Buried water molecules in helical transmembrane proteins. Protein Sci. 17, 293–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kiselar J. G., Chance M. R. (2010) Future directions of structural mass spectrometry using hydroxyl radical footprinting. J. Mass Spectrom. 45, 1373–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.