

Background: A novel chaperone, cpSRP43, recognizes and disassembles the aggregates formed by its client proteins.
Results: The client proteins of cpSRP43 form stable disc-shaped aggregates with the chaperone recognition motif displayed on the surface.
Conclusion: The surface-exposed motif on the aggregate allows it to be recognized by its chaperone.
Significance: Understanding the structure and energetics of protein aggregates provides insights into the mechanism of their disassembly.
Keywords: Aggregation, Enzyme Mechanisms, Membrane Biophysics, Molecular Chaperone, Protein Aggregation, Light-harvesting Complex, Membrane Protein Biogenesis, Signal Recognition Particle
Abstract
Protein aggregation is detrimental to the maintenance of proper protein homeostasis in all cells. To overcome this problem, cells have evolved a network of molecular chaperones to prevent protein aggregation and even reverse existing protein aggregates. The most extensively studied disaggregase systems are ATP-driven macromolecular machines. Recently, we reported an alternative disaggregase system in which the 38-kDa subunit of chloroplast signal recognition particle (cpSRP43) efficiently reverses the aggregation of its substrates, the light-harvesting chlorophyll a/b-binding (LHC) proteins, in the absence of external energy input. To understand the molecular mechanism of this novel activity, here we used biophysical and biochemical methods to characterize the structure and nature of LHC protein aggregates. We show that LHC proteins form micellar, disc-shaped aggregates that are kinetically stable and detergent-resistant. Despite the nonamyloidal nature, the LHC aggregates have a defined global organization, displaying the chaperone recognition motif on its solvent-accessible surface. These findings suggest an attractive mechanism for recognition of the LHC aggregate by cpSRP43 and provide important constraints to define the capability of this chaperone.
Introduction
The proper folding of proteins into their native structures is essential for the function and survival of cells. However, environmental stress, molecular crowding, and potential exposure of hydrophobic regions of proteins during their biogenesis (1–3) pose challenges to protein folding in vivo. In this setting, improper intra- or intermolecular interactions can lead to the aggregation of proteins. Aggregate formation is detrimental to cells as it removes functional proteins (4). Moreover, some aggregates, both amorphous ones and those that lead to highly ordered amyloid fibrils, are toxic to cells and have been implicated in a variety of protein folding diseases (5–7).
Cells have evolved elaborate mechanisms to overcome the problems associated with protein aggregation. A specialized class of molecular chaperones, the “disaggregases,” can perform the energetically uphill process of reversing protein aggregation. Thus far, studies of disaggregases have been dominated by the Clp/Hsp100 family of AAA+ 3 ATPases (ATPases associated with various cellular activities), such as ClpB in prokaryotes and Hsp104 in yeasts (3). Both are large hexameric rings (>500 kDa) powered by mechanical forces from ATP hydrolysis and require additional co-chaperones to efficiently disassemble a variety of protein aggregates (8, 9). The complexity of these disaggregase systems and the promiscuity in their substrate selection have made it difficult to pinpoint their molecular mechanisms of action. Further, AAA+ disaggregase machines were only found in prokaryote and yeast, and no homologues have been identified in higher eukaryotes outside of plastids and mitochondria. It is conceivable that alternative mechanisms of disaggregation, such as the recently described Hsp110–70–40 system (10, 11), could be used in higher eukaryotes. An understanding of alternative disaggregase systems can shed light on novel principles and mechanisms by which cellular chaperones overcome protein aggregates.
Previously, we identified an efficient disaggregase activity in the chloroplast signal recognition particle 43 subunit (cpSRP43) (12). This provides an example in which a relatively small protein scaffold (38 kDa) can recognize and disrupt large protein aggregates in an ATP-independent mechanism (12), in contrast to the Clp/Hsp100 family of disaggregases. cpSRP43 is part of the protein targeting machinery, the cpSRP, that mediates the delivery of the light-harvesting chlorophyll a/b-binding (LHC) family of proteins to the thylakoid membrane (13–15). The most abundant member of the LHC family, LHCP, comprises ∼30% of the proteins on the thylakoid membrane and is arguably the most abundant membrane protein on earth. The sheer abundance of these proteins and their highly hydrophobic nature demands highly effective chaperones that protect them from aggregation before arrival at the membrane. In the chloroplast stroma, this chaperone function is provided by cpSRP43, which effectively protects LHC proteins from aggregation and can even reverse preformed large LHC protein aggregates (12, 16). cpSRP43 recognizes a highly conserved 18-amino acid loop between the second and the third transmembrane (TM) domains of LHC proteins, termed L18 (17, 18). In previous work, we showed that the specific interaction of cpSRP43 with the L18 motif is crucial for the chaperone and disaggregase activity of cpSRP43 (12). This and other observations led us to propose that, in the absence of external energy input, cpSRP43 uses specific binding energy with its substrate proteins to remodel and rescue LHC protein aggregation (12).
To gain insights into the molecular mechanism that underlies the novel disaggregase activity of cpSRP43, we need to first understand the nature of the LHC aggregate and identify the structural features that facilitate its disassembly by cpSRP43. To this end, we examined the nature and structure of the LHC aggregate using biophysical and biochemical techniques. We show that LHC proteins form disc-like particles with a relatively amorphous hydrophobic core, but exhibit a defined interior/exterior organization in which the L18 recognition motif is displayed on the solvent-exposed surface. This suggests an attractive mechanism for cpSRP43 to recognize the LHC aggregates and thus initiate their disassembly.
EXPERIMENTAL PROCEDURES
Materials
LHCP, Lhcb5, and their mutants were purified under denaturing conditions as described (12), except that 6 m GdmHCl was used instead of 8 m urea for Lhcb5. Aβ1–40 and recrystallized thioflavin T (ThT) were generous gifts from Dr. J. W. Kelly. 1-Anilino-8-naphthalene sulfonate (ANS) and bis-ANS were from Sigma and Invitrogen, respectively. n-Dodecyl-N,N,-dimethylamine-N-oxide (LDAO), n-dodecyl-β-d-maltopyranoside (DDM), n-octyl-β-d-glucopyranoside (β-OG), and n-nonyl-β-d-glucopyranoside (BNG) were from Anatrace. Triton X-100 was from Sigma, and SDS was from Bio-Rad. Urea and GdmHCl were molecular biology grade from MP and Sigma, respectively. 1-Oxyl-2,2,5,5-tetramethylpyrroline-3-methyl methanethiosulfonate (MTSSL) was from Toronto Research Chemicals, N-ethyl-maleimide was from Sigma, and N-(1-pyrene)-maleimide was from Invitrogen.
Light Scattering Assay
Light scattering experiments were performed as described previously (12). For formation of aggregates (see Fig. 3, black), unfolded LHCP in 8 m urea was directly diluted into Buffer D (50 mm KHEPES, pH 7.5, 200 mm NaCl) to the desired final concentration; the final concentration of urea was equalized among different samples. The critical micelle concentration is obtained as the x-intercept from the linear fit of the data (19). For serial dilution experiments (see Fig. 3, red), the sample at 1 μm LHCP was serially diluted (by 2-fold) into fresh Buffer D and allowed 10–30 min to equilibrate before the measurement.
FIGURE 3.

LHCP forms aggregates after a critical concentration. Light scattering intensities during formation of the aggregate (black) are compared with those from serial dilution of preformed aggregates (red). The inset highlights the lag phase at low concentrations during formation of the aggregate. AU, arbitrary units.
TEM
LHCP aggregates were formed by diluting unfolded LHCP in 8 m urea into Buffer D to a final concentration of 2 μm. After incubation at 25 °C for 5 min, the sample was diluted 5-fold and immediately deposited onto a glow-discharged 200-mesh Formvar grid (Ted Pella Inc.). After a 45-s adsorption time, the grid was washed in water and then stained with 1% uranyl acetate for 45 s. Transmission electron microscopy (TEM) images were obtained on a 120-kV Tecnai T12 electron microscope coupled with a CCD camera. The diameters of the particles were measured using ImageJ (20).
AFM
1 μm LHCP aggregate in Buffer D was deposited onto a freshly cleaved mica and incubated for 5 min at 25 °C to allow equilibration. The wafer was rinsed with Millipore water and dried under the weak flux of nitrogen. Atomic force microscopy (AFM) images were taken immediately after the sample was prepared. A Digital Instrument Nanoscope IIIA AFM system in tapping mode was used throughout at ambient conditions. A sharp TESP tip (Veeco) was used in the experiment. Typical values for the force constant, resonance frequency, and tip radius were 42 newtons/m, 320 kHz, and 8 nm, respectively. Particle sizes were obtained by calculating the projected area of each particle at half-maximal height onto the surface. This is because the apparent lateral size of surface features is usually overestimated due to the broadening effect of the AFM tip. The cross-sectional area at half the maximum height provides a more realistic distribution of sizes of the particles.
Fluorescence
All fluorescence experiments were carried out in Buffer D using a FluoroLog 3-22 spectrofluorometer (HORIBA Jobin Yvon). For bis-ANS experiments, 1 mm bis-ANS was added to Buffer D with or without 1 μm LHCP aggregate. The samples were excited at 395 nm and then scanned from 410 to 620 nm, with excitation and emission band passes of 2 and 5 mm, respectively. For ThT experiments, 20 mm recrystallized ThT was added to Buffer D containing no aggregate, aggregates from 1 or 5 μm LHCP, or 15 μm freshly sonicated Aβ1–40 amyloid. The samples were excited at 440 nm and then scanned from 470 to 570 nm, with the excitation and emission band passes of 3 and 7 mm, respectively. For comparison, ThT fluorescence from 1 and 5 μm unfolded LHCP in 8 m urea was measured.
For pyrene excimer experiments, DTT-reduced single cysteine mutants of Lhcb5 in 6 m GdmHCl were labeled with a 30-fold molar excess of pyrene maleimide at room temperature in the dark for 2 h. Excess pyrene was removed by gel filtration, and the efficiency of spin labeling (90–100%) was determined by LC-MSDSL 1100 series (Agilent Technologies, Santa Clara, CA). The samples were prepared by diluting pyrene-labeled Lhcb5 pairs into Buffer D for a final concentration of 1.5 μm for each variant. Spectra were obtained from excitation at 317 nm and then scanned from 360 to 560 nm, with excitation and emission band passes of 3 and 6 mm, respectively. The amount of excimer fluorescence, indicated by a red shift to 445 nm, is normalized against the nonexcited fluorescence signal at 376 nm. Statistically, when two variants A and B are mixed, there is a population distribution of homo-pairs (e.g. 25% A-A and 25% B-B) and hetero-pairs (50% A-B). The equation below corrects for the real hetero-pair excimer (FAB)
where FAB,app is the apparent ratio of excimer fluorescence (I445/I376) between two pyrene-labeled variants, and FA and FB is the ratio of excimer fluorescence of each individual variants measured separately.
Sedimentation
Unfolded LHCP was diluted to 10 μm in Buffer D and incubated at 25 °C for 5 min. Aggregation was complete, judged by the absence of LHCP in the supernatant after centrifugation at 13,000 rpm in a microcentrifuge for 30 min. The pellet was dissolved with 50 μl of detergent or chemical denaturants at different concentrations for 30 min at 25 °C. The mixtures were then spun at 13,000 rpm in a microcentrifuge for 30 min, and soluble and pellet fractions were boiled and visualized by SDS-PAGE.
SDS Solubility
For Fig. 2B, the assay was performed as described for amyloid fibrils (21). Briefly, aggregation of 10 μm LHCP in Buffer D preceded for 5 min at 25 °C. The mixture was then mixed with 2% SDS-PAGE loading buffer and incubated at either 25 °C or 100 °C for 10 min prior to SDS-PAGE. Only the proteins that migrated into the resolving gel (e.g. solubilized portion) were visualized.
FIGURE 2.
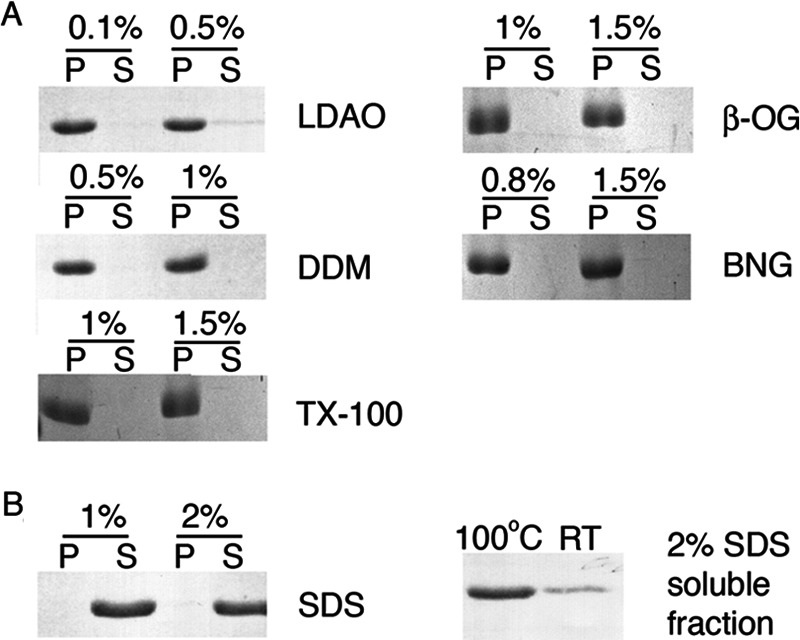
LHCP aggregates are resistant to many detergents. A, sedimentation analysis of the ability of various detergents to resolubilize LHCP aggregates. Critical micelle concentrations of LDAO, DDM, β-OG, BNG, and Triton X-100 (TX-100) are 0.023, 0.009, 0.53, 0.2, and 0.02%, respectively. P and S denote the pellet and soluble fractions, respectively. B, SDS solubility assay as described for amyloids (21) shows partial solubility of LHCP aggregates in 2% SDS (right panel). The samples were directly loaded onto the gel, and solubility was judged by the mobility of the protein into the resolving gel. Quantification using ImageJ revealed that 24% of the LHCP aggregates is soluble when the sample was not boiled (RT), when compared with 87% for the boiled sample (100 °C). The left panel shows complete solubilization of LHCP by SDS (critical micelle concentration 0.23%) after treatment as in A.
Spin Labeling and Electron Paramagnetic Resonance Measurements
Spin labeling reactions were performed in 6 m GdmHCl, 50 mm KHEPES, pH 7.5, and 2 mm EDTA. Reduced and degassed single cysteine mutants of Lhcb5 were labeled with a 3–5-fold molar excess of MTSSL at room temperature in the dark for 2–3 h. Excess MTSSL was removed by gel filtration, and the efficiency of spin labeling (80–100%) was determined by electron paramagnetic resonance (EPR) using a 2,2,6,6-tetramethyl peperidine-N-oxyl calibration curve according to manufacturer's instructions (Bruker). EPR spectra were acquired using a 9.4-GHz (X-band) EMX EPR spectrometer (Bruker) equipped with an ER 4119HS cavity at 20–23 °C. To form the aggregate, the individual spin-labeled proteins in GdmHCl were diluted into Buffer D. The concentrations of the aggregate samples were 30–100 μm. Data acquisition was as previously described (22).
NEM Alkylation And MS Analysis
Cysteine mutants of Lhcb5 in 6 m GdmHCl were reduced with 2.5 mm tris(2-carboxyethyl)phosphine at room temperature for 2 h. Each mutant was diluted into Buffer D to a final concentration of 3.3 μm and incubated on ice for 10 min to form the aggregate followed by the addition of 100 μm N-ethyl-maleimide (NEM). The reaction was quenched with 50 mm DTT at various time points, concentrated under vacuum, and redissolved in 0.2% formic acid, and ∼25 pmol of protein was analyzed on an LC-MSD SL 1100 series (Agilent). The samples were chromatographed on a 2.1 × 150-mm ZORBAX 300SB-C3 column (Agilent) using a gradient consisting of 0.2% formic acid and 0.2% formic acid in acetonitrile (89.8%) and methanol (10%). Intact masses were measured in the single quadrupole and quantified using the software ChemStation software (Agilent). Control experiments where different ratios of unalkylated and alkylated proteins were mixed and subjected to MS analysis show the quantification of ratios of alkylated species to be reliable (see Fig. 6E). The reported accessibilities were calculated as a ratio between the alkylation of each cysteine mutant under aggregation Buffer D versus denaturing 6 m GdmHCl.
FIGURE 6.

Mapping the LHC aggregates reveals exposed motif. A, Lhcb5 sequence, with residues mutated to cysteine in blue, the TMs underlined in green, and the L18 peptide underlined in red. B, light scattering from aggregates (A360) 5 min after dilution of each single-cysteine mutant into Buffer D, final concentration, 1 μm Lhcb5 proteins. Values are relative to that of wild-type protein. rel A360, relative A360. C, representative EPR spectra of the spin probes placed at buried site L130C of TM2 (green), at exposed site L170C of L18 (red) upon Lhcb5 aggregation, and at L170C when Lhcb5 was solubilized in 6 m GdmHCl (black). a. u., arbitrary units. D, summary of the mobility of different residues in the Lhcb5 aggregate, reported in values of ΔH−1. Residues in the TMs are in green, L18 is in red, and the remainder of Lhcb5 is in black. E, control experiment shows that intact mass spectrometry can be used for the quantification of the efficiency of NEM alkylation. Wild-type Lhcb5, which contains one native cysteine (Cys-100), was reacted with NEM to completion in 6 m GdmHCl. Different known ratios of the NEM-modified Lhcb5 were mixed with unreacted protein and submitted for MS analysis. F, time courses for the alkylation reactions of representative cysteines at residues L130C and L170C in Lhcb5 aggregates and at residue L170C when Lhcb5 was dissolved in 6 m GdmHCl. G, summary of NEM accessibility of the single-site cysteines in the Lhcb5 aggregate. The color scheme is the same as in D.
RESULTS
LHCP Aggregates Contain Exposed Hydrophobic Grooves
To characterize the surface features of LHC protein aggregates, we used an established collection of small molecule dyes. Exposure of hydrophobic patches or crevices within aggregates can be probed by extrinsic fluorescent molecular dyes such as ANS and bis-ANS (23, 24). We tested whether the aggregates of LHCP, the most abundant member of the LHC protein family, share this feature. Indeed, the fluorescence of both ANS (data not shown) and bis-ANS (Fig. 1A) increased significantly in the presence of 1 μm LHCP aggregate, accompanied by a blue shift of the fluorescence emission spectra. These results strongly suggest that LHCP aggregates contain exposed hydrophobic microdomains that allow the binding of these dyes, consistent with the highly hydrophobic nature of this protein.
FIGURE 1.

LHCP aggregates contain exposed hydrophobic surfaces as detected by small molecule dyes. A, fluorescence emission spectra of 1 mm bis-ANS with (blue) or without (black) 1 μm LHCP aggregate. AU, arbitrary units. B, fluorescence spectra of 20 mm ThT in the absence (black) and presence of 1 (light blue) or 5 (dark blue) μm LHCP aggregate or 15 μm Aβ1–40 (red). C, quantification of the ThT fluorescence change at 484 nm per μm of protein (aggregate).
We next used ThT to probe the structural organization of the LHCP aggregate. ThT is often used as a diagnostic for the formation of amyloid fibrils generated by amyloid-β (Aβ), α-synuclein, and other amyloidogenic proteins (25). Similar to bis-ANS, the fluorescence of ThT exhibited a significant increase in intensity and a blue shift in spectrum in the presence of the LHCP aggregate (Fig. 1B, blue lines). The extent of these fluorescence changes is comparable with that induced by mature amyloid fibrils generated by the Aβ1–40 peptide (Fig. 1B, red versus blue, and Fig. 1C). As microscopy analyses did not indicate fibril formation in the LHCP aggregate (see below), these results suggest that ThT is not highly specific for amyloid fibrils, consistent with recent work observing ThT fluorescence of nonfibrillar aggregates of β-lactoglobulin and transthyretin (19, 26). Instead, this dye possibly binds to hydrophobic grooves that are often present in amyloid fibrils but can also be generated by other types of aggregates (27).
LHCP Forms Stable Aggregates
To probe the stability of the LHCP aggregate, we tested its solubility in various detergents, including LDAO, DDM, β-OG, BNG, and Triton X-100. By analyzing the amount of proteins in the soluble and insoluble fractions after medium speed sedimentation (see “Experimental Procedures”), we showed that none of these detergents were able to solubilize the LHCP aggregate at or above their respective concentrations typically used for membrane protein solubilization (Fig. 2A).
In addition, we tested the solubility of the LHCP aggregate in SDS using an established protocol for amyloid fibrils (21). This assay evaluated solubility of the aggregate based on the mobility of the protein in SDS-PAGE after incubation with SDS-containing buffer at room temperature (see “Experimental Procedures”). “SDS-insoluble” amyloid fibrils or oligomeric protein aggregates cannot enter the resolving gel unless boiled (21). LHCP aggregate showed significant resistance to 2% SDS in this procedure as only 24% of the aggregates could be solubilized and migrated into the gel without boiling (Fig. 2B, right panel). SDS could solubilize large LHCP aggregates only after extensive incubation and boiling of the sample (Fig. 2B, left panel). Taken together, the detergent resistance of the LHC protein aggregate suggests the presence of highly stable packing interactions within the aggregate that must be overcome by cpSRP43.
LHCP Forms Micellar, Disc-shaped Aggregates
Formation of large LHC aggregates can be monitored based on light scattering at 360 nm (12). The scattering intensity increases linearly with LHCP concentration above ∼100 nm (Fig. 3, black), suggesting that aggregate formation was complete under these conditions. However, the linearity broke down at lower LHCP concentrations (Fig. 3 and inset, black). This was not due to limitations in instrument sensitivity; when preformed LHCP aggregates were diluted, linearity in light scattering intensity was observed at all concentrations and extrapolated through zero (Fig. 3 and inset, red). These observations show that: (i) the LHCP aggregate is kinetically stable and virtually irreversible once it has formed; and (ii) formation of the LHCP aggregate requires a critical protein concentration, reminiscent of the critical micelle concentration during micelle formation. An analogous, “critical aggregate concentration” of 125 nm was obtained for the LHCP aggregates from these data (see “Experimental Procedures”). This micelle-like characteristic begins to suggest a globular morphology of the LHC aggregates.
To directly observe the global structure of LHC aggregates, we examined them using TEM and AFM. Negatively stained TEM images revealed LHCP aggregates to be circular particles (Fig. 4, A and B). Analysis of the size of these particles resulted in a distribution that fits well to a Gaussian function, with diameters of 12 ± 2 nm (Fig. 4C). Consistent with the EM images, AFM analysis also showed LHCP aggregates to be disc-shaped particles (Fig. 5, A and B) with mean area of 214 ± 94 nm2 (Fig. 5C) or mean diameter of 16 ± 5 nm, in good agreement with the EM measurements. Strikingly, the heights of the aggregates measured by AFM are “quantized” and peaked at integrals of 0.7–0.8 nm (Fig. 5D and inset). These results suggest that LHC proteins form disc-shaped aggregates with a height of 0.7–0.8 nm, and these discs can further stack upon one another.
FIGURE 4.

TEM analysis of LHCP aggregates. A, large field view of a negatively stained TEM image of LHCP aggregates. B, a zoomed-in image shows that LHCP aggregates are round particles. C, size distribution of the LHCP aggregates, measured from several independent experiments. The mean diameter is 12 ± 2 nm.
FIGURE 5.

AFM analysis of LHCP aggregates. A, large field view of AFM topographic image showing well separated LHCP aggregates. Large clusters are occasionally observed. The scale bar is 500 nm. B, a zoomed-in region of the image reveals disc-shaped particles. The lines indicate particles whose heights were measured (red, blue, and green). The scale bar is 100 nm. C, size distribution of LHCP aggregates, measured from several regions on the surface. The red line is a Gaussian fit to the data, which gave a mean area of the particle of 214 nm2. D, height distribution of LHCP aggregates shows three populations of 0.8, 1.4, and 2.1 nm. The inset shows the height profiles for the representative particles indicated in B. Curves are vertically displaced for clarity.
The L18 Recognition Motif Is Displayed on the Aggregate Surface
To probe the global structure of LHC aggregates at higher resolution, we engineered 30 single-cysteine mutations, which span every 5–10 residues throughout the entire protein sequence of Lhcb5 (Fig. 6A, blue). Lhcb5 is a close homologue of LHCP (supplemental Fig. S1) that strongly depends on the cpSRP pathway for its biogenesis. Its aggregation is efficiently prevented and disassembled by cpSRP43 analogously to LHCP (12, 28). All single-cysteine mutants were able to form light scattering aggregates with the same extent and kinetics as wild-type Lhcb5, and thus can be used to probe the assembly of the wild-type aggregate (Fig. 6B).
With each single-cysteine mutant, we used two independent methods to measure their relative positions on the LHC aggregate. In the first approach, we labeled each cysteine with the nitroxide spin probe MTSSL in 6 m GdmHCl, allowed for aggregation in aqueous buffer, and used EPR spectroscopy to investigate the local backbone mobility of each specific site in the aggregate. A probe buried inside the aggregate will engage in strong interactions and have more restricted motions than those on the solvent-exposed surface and hence exhibit broader central line width (ΔH) and hyperfine splitting in the overall spectral width (Fig. 6C, green versus red spectra). As a control, EPR measurements were carried out for each protein variant solubilized in 6 m GdmHCl; all spin labels displayed similar, low values of ΔH under these conditions, indicating the high mobility of the residues in the unfolded protein (Fig. 6C, black).
Upon formation of the aggregate, the spin probes in all the TMs, loop 1 (between TM1 and TM2), and the C terminus of Lhcb5 displayed high ΔH values and broad EPR spectra, suggesting that they are highly immobile and likely engaged in strong inter- or intramolecular interactions (Fig. 6D, supplemental Table 1, ΔH−1 values are plotted). In contrast, spin probes placed in the L18 motif and the N terminus of TM3 are highly mobile, indicating that these regions are free from any extensive interactions in the aggregate and are likely solvent-exposed (Fig. 6D). In addition, spin labels at the N terminus of Lhcb5 also displayed highly mobile spectra.
To independently probe the global architecture of LHC protein aggregates, we examined the susceptibility of the individual cysteine residues to alkylation with NEM. The cysteine residues on the solvent-accessible surface of the aggregate will react rapidly and efficiently with NEM, whereas those buried within the aggregate will be alkylated much less efficiently (Fig. 6F, green versus red curves). The efficiency of alkylation can be quantified by intact mass spectrometry and provides a direct measure for the solvent accessibility of individual residues in the LHC aggregate (Fig. 6E). As a control for the intrinsic bias in the reactivity of cysteines at different positions, each single-cysteine mutant was solubilized in 6 m GdmHCl and tested in parallel experiments (Fig. 6F, black).
In agreement with the results of EPR measurements, the residues within the TMs exhibit low efficiency of alkylation, ranging from 20 to 40%, in contrast to the almost complete alkylation of the respective cysteines under denaturing conditions (Fig. 6G and supplemental Table 1). Residues in loop 1 and the C terminus of the LHC protein exhibit slightly higher alkylation efficiency, 40–60%, indicating that these regions are partially buried in the aggregate but to a lesser extent than the TMs. In contrast, residues on the L18 loop and the N-terminal end of TM3 proximal to L18 are almost completely alkylated (90–100%), suggesting that these sites are highly solvent-accessible and presented on the exterior of the aggregate. Finally, residues in the N terminus of Lhcb5 showed almost 100% reactivity, again demonstrating the exposure of this region on the aggregate surface.
Although the burial of TMs in the Lhcb5 aggregate is expected due to their hydrophobic nature, the low mobility and inaccessibility of loop 1 and the C terminus of Lhcb5 were surprising. We therefore asked whether the burial of these loop regions results from topological constraints imposed by the neighboring TMs or from the inherent physicochemical property of the looping sequence. To address this question, we altered the location of L18 in Lhcb5 by either swapping it with loop 1 (TM1-L18-TM2-TM3) to construct the LoopSwap mutant or swapping it with the protein sequence C-terminal to TM3 (TM1-TM2-TM3-L18) to construct the C-terminal mutant. The aggregate formed by both constructs can be rescued by cpSRP43 (Fig. 7A), suggesting that cpSRP43, despite its specific interaction with L18, can tolerate variations in the remainder of the sequence of its substrate.
FIGURE 7.

L18 has a strong tendency to be exposed on the surface of the aggregate. A, the extent of wild-type and mutant Lhcb5 aggregates (1 μm) resolubilized by 10 (white), 20 (gray), and 40 (black) μm cpSRP43. The aggregates formed by L18-swapped mutants TM1-L18-TM2-TM3 (LoopSwap (LS)) and TM1-TM2-TM3-L18 (C-terminal (Cterm)) can be rescued by cpSRP43, although the LoopSwap mutant required a higher concentration of cpSRP43. B, NEM accessibility analysis of residues in the wild-type aggregates. Regions probed include the N terminus (G50C), TM1 (A80C), loop 1 (N120C), L18 (G162C), and C terminus (A230C). C and D, NEM accessibility analysis of the same residues in the LoopSwap construct (C) and in the C-terminal construct (D). Error bars in all panels indicate S.D.
To probe the accessibility of individual motifs in the L18-swapped mutants, we probed the accessibility of engineered single cysteines in each domain by NEM alkylation. The alkylation efficiency of each motif in both L18-swapped mutants is similar to that of wild-type Lhcb5; the L18 motif is highly accessible and almost completely alkylated, whereas loop 1 and the C terminus regions exhibit medium levels of alkylation (Fig. 7, B–D). These results indicate that the intrinsic properties of these sequences/domains determine their accessibility in the aggregate and demonstrate that the L18 motif has a strong tendency to be displayed on the surface of protein aggregates.
LHC Aggregates Contain an Amorphous Hydrophobic Core
To determine whether the TMs of the LHC protein make specific intermolecular contacts in the buried core of the aggregate, we exploited the ability of pyrene labels to form excited state dimers (excimers) when they are within 4–10 Å of each other. High pyrene excimer fluorescence reports on close proximity between specific sites within the aggregate. To this end, we mixed two proteins, each labeled with pyrene at a single cysteine residue, in all pair-wise combinations, allowed them to form the aggregates, and monitored for pyrene excimer fluorescence at 455 nm relative to the monomer fluorescence at 375 nm (see “Experimental Procedures”). As a positive control, we used a double-cysteine mutant V139C/L140C and labeled both positions with pyrene probes. The excimer ratio of this construct was high, ∼0.8 (Fig. 8A). As the negative control, pyrene-labeled L180C was used (excimer ratio = 0.08; Fig. 8A).
FIGURE 8.

Analysis of pyrene excimer fluorescence reveals an amorphous aggregate core. A, representative pyrene spectra for the homo-pair L180C were recorded when it was solubilized in 6 m GdmHCl (black trace). Pyrene-labeled homo-pair A67C and L180C after aggregate formation in Buffer D show excimer fluorescence (red and blue traces, respectively). Doubly pyrene-labeled Lhcb5 mutant V139C/L140C in Buffer D served as positive control for pyrene excimer fluorescence between residues in close proximity (orange trace). a.u., arbitrary units. B, the chart lists pyrene excimer fluorescence (I445/I376) for all pairwise combinations (see “Experimental Procedures”); excimer fluorescence values between 0.21 and 0.29 are highlighted in yellow, and values ≥0.3 are in orange.
Two important lessons were learned from the results of these measurements. First, many pyrene pairs exhibit excimer fluorescence intensities substantially above the background and above the other pyrene pairs, with excimer ratios of ≥0.30 for homo-pyrene pairs at multiple residues in TM1 (67, 72, 88, and 92) and for multiple hetero-pyrene pairs in all three TMs (Fig. 8B). Second, these excimer fluorescence intensities were still modest, up to 0.36 (Fig. 8B). These values are far below the values of 0.6–0.8 expected for specifically interacting pairs that are always in close proximity. These data indicate that in the LHC aggregate, the TMs form extensive intermolecular contacts in its hydrophobic core, but these interactions are much less specific than those observed in amyloid fibrils (29).
Taken together, the results demonstrate that: (i) LHC proteins form highly stable, disc-shaped aggregates; (ii) despite the possibly amorphous nature of the LHC aggregate core, it contains a defined global organization that can be reliably probed; and (iii) the L18 motif, among other regions of the LHC protein, is displayed on the surface of the aggregate and thus poised for interactions with cpSRP43 (Fig. 9). These results suggest an attractive model in which cpSRP43 could recognize the L18 motif presented on the surface of the aggregate, providing a starting point for its action as a disaggregase. Further, the exposure of the N terminus and the N-terminal end of TM3 suggests additional potential interaction sites with cpSRP43 during aggregate recognition.
FIGURE 9.

Model for the global organization of LHC proteins in the aggregate. L18 is in red, the TMs are in different shades of green, and the other looping sequences are in black. The shaded region depicts the buried core of the aggregate. For clarity, only one LHCP molecule in the aggregate is highlighted.
DISCUSSION
The ability of cpSRP43 to prevent and reverse LHC protein aggregation demonstrates the diversity and capability of cellular chaperones and highlights a disaggregation mechanism that relies on binding interactions instead of external energy input. The robustness and simplicity of the cpSRP43-LHC disaggregase system provide an opportunity to unravel the mechanism by which a relatively small, ATP-independent chaperone can rescue insoluble protein aggregates. In this work, biophysical and biochemical analyses of the structure and energetics of the LHCP aggregate help define the capability of cpSRP43 as a protein disaggregase and suggest an attractive mechanism for how this chaperone recognizes the LHC protein aggregates to initiate the disaggregation reaction.
Using kinetic analyses, we previously showed that cpSRP43 can actively remodel and disassemble LHC aggregates (12). To gauge the amount of energy cpSRP43 must overcome during disaggregation, here we examined the stability of the LHC aggregates. The results indicate that LHC aggregates are stable both kinetically and thermodynamically. First, extensive dilution of the aggregate did not lead to resolubilization, suggesting that LHC aggregates, once formed, are kinetically stable. This is in contrast to the “salting out” effect, in which protein precipitates are reversibly produced when the protein concentration exceeds the solubility limit (30). Second, LHC aggregates are resistant to a variety of detergents, even up to 2% SDS, akin to highly stable fibrils and insoluble amyloid oligomers (31). The stability of the LHC aggregate further supports the notion that its reversal requires the active participation of cpSRP43 to engage and disrupt the aggregate and showcases the capacity of this chaperone as a disaggregase.
The morphology of the LHC aggregates bears resemblance to those of the soluble oligomeric intermediates that often precede amyloid fibril formation, which are disc-shaped, 9–25 nm in diameter, and 2–3 nm in height (32, 33). Although earlier work tends to categorically describe these protein aggregates as amorphous, accumulating data suggest that there are nonetheless degrees of organization in some of these aggregates (34, 35). For instance, the folding intermediates of bovine growth hormone, phosphoglycerate kinase, P22 tailspike, and coat proteins participate in specific intermolecular interactions in their aggregation pathways (36–38). Likewise, although highly specific intermolecular interactions have not been detected in the LHC aggregates, more detailed analyses at the individual residue level provide convincing evidence that LHC aggregates have a defined interior and exterior that can be reliably probed, arguing against complete disorder in these aggregates.
What features of the LHC aggregate allow cpSRP43 to recognize it and initiate the disaggregation process? Answers to this question are central to understanding the mechanism of the disaggregase activity of cpSRP43. The results here strongly suggest that the formation of LHC aggregates is driven largely by hydrophobic collapse to bury its three TM domains. Importantly, we showed that the N terminus of the LHC protein, the L18 motif, and the N-terminal segment of TM3 are displayed on the solvent-accessible surface when LHC proteins form aggregates. As the L18 motif is the primary recognition element for cpSRP43, its presentation on the exterior of the aggregate provides a very attractive mechanism by which cpSRP43 could recognize and anchor onto the aggregate to initiate the disassembly process (Fig. 9). Conceivably, the N-terminal fragment of TM3 proximal to the L18 motif could also contribute to this initial recognition as previous work has detected cross-links between cpSRP43 and residues at the N terminus of TM3 (39). This and additional mutational studies suggest that TM3 is a likely candidate for cpSRP43 to initiate disruptions of the internal packing within the LHC aggregate (see accompanying manuscript (45)).
Further, the L18 swap experiments show that the L18 motif is a dominant sequence element that has a strong tendency to be displayed on the surface of a protein aggregate, likely due to its relatively polar amino acid composition and high propensity for disorder (Fig. 7). Considering that cpSRP43 has co-evolved with and is dedicated to the chaperoning of the LHC family of proteins, it is intriguing that the latter evolved a polar L18 recognition motif and made it accessible even when they form aggregates, which would enable cpSRP43 to readily recognize the aggregated LHC proteins. Although the physiological significance of the disaggregase activity of cpSRP43 remains to be directly established, this observation is consistent with the possibility that this activity is beneficial as it would enable cpSRP43 to rescue aggregated, off-pathway intermediates during the targeting or insertion of its substrate proteins (40).
The mechanism of aggregate recognition proposed here for cpSRP43 is distinct from those proposed for ClpB/Hsp104, where exposed patches enriched in charged and hydrophobic amino acids are recognized by the disaggregases (41, 42). It can be speculated that a generalized mode of substrate recognition is optimal for Hsp70 and/or the AAA+ disaggregases, which must handle a broad range of substrates. In contrast, dedication of cpSRP43 to the LHC family of proteins allows them to adopt a more specific and effective mechanism, in which an exposed polar motif is used for recognition and ultimately enables the chaperone to gain access to the hydrophobic core. This mechanism of aggregate recognition could explain analogous disaggregase systems reported previously, such as the mitochondria import stimulation factor (MSF), whose ability to rescue aggregated mitochondrial precursor proteins also depends on the basic mitochondrial signal sequence that is likely displayed on the aggregate surface (43, 44). On the other hand, the L18 swap experiments here and additional mutagenesis studies (see accompanying manuscript (45)) strongly suggest that the interaction of cpSRP43 with the remainder of the LHC protein, apart from L18, is highly adaptable as a wide range of unnatural substrates can be effectively bound, chaperoned, and rescued by cpSRP43.
In summary, in-depth characterization of the nature and structure of the LHC protein aggregate suggests an attractive mechanism for its recognition by cpSRP43 and provides important constraints for the capability and limitation of the disaggregase activity of cpSRP43. In the accompanying manuscript (Jaru-Ampornpan et al. (45)), the lessons learned from this work are leveraged against structure-function analyses to propose a multistep mechanism for the disaggregase reaction mediated by cpSRP43. These results provide a foundation for understanding the molecular basis of ATP-independent disaggregase systems and guide the engineering of specific chaperone-substrate interactions for aggregates of similar nature.
Acknowledgments
We thank A. N. Murray and Dr. J. W. Kelly for recrystallized ThT and fibrilized Aβ1–40, A. McDowall for EM analyses, members of the Proteome Exploration Laboratory for mass spectrometry assistance, and members of the Shan group for helpful comments on the manuscript. The Proteome Exploration Laboratory is supported by the Betty and Gordon Moore Foundation and the Beckman Institute. AFM studies were supported by United States Department of Energy Grant DE-FG03-01ER46175 (to J. Heath).

This article contains supplemental Fig. S1 and Table 1.
- AAA+
- ATPases associated with various cellular activities
- cpSRP
- chloroplast signal recognition particle
- LHC
- light-harvesting chlorophyll a/b-binding
- LHCP
- LHC protein
- TM
- transmembrane
- Aβ
- amyloid β
- TEM
- transmission electron microscopy
- AFM
- atomic force microscopy
- ThT
- thioflavin T
- ANS
- 1-anilino-8-naphthalene sulfonate
- LDAO
- n-dodecyl-N,N,-dimethylamine-N-oxide
- DDM
- n-dodecyl-β-d-maltopyranoside
- β-OG
- n-octyl-β-d-glucopyranoside
- BNG
- n-nonyl-β-d-glucopyranoside
- MTSSL
- 1-oxyl-2,2,5,5-tetramethylpyrroline-3-methyl methanethiosulfonate
- NEM
- N-ethyl-maleimide
- GdmHCl
- guanidinium hydrochloride.
REFERENCES
- 1. Hartl F. U., Hayer-Hartl M. (2002) Molecular chaperones in the cytosol: from nascent chain to folded protein. Science 295, 1852–1858 [DOI] [PubMed] [Google Scholar]
- 2. Balch W. E., Morimoto R. I., Dillin A., Kelly J. W. (2008) Adapting proteostasis for disease intervention. Science 319, 916–919 [DOI] [PubMed] [Google Scholar]
- 3. Doyle S. M., Wickner S. (2009) Hsp104 and ClpB: protein disaggregating machines. Trends Biochem. Sci. 34, 40–48 [DOI] [PubMed] [Google Scholar]
- 4. Weibezahn J., Tessarz P., Schlieker C., Zahn R., Maglica Z., Lee S., Zentgraf H., Weber-Ban E. U., Dougan D. A., Tsai F. T., Mogk A., Bukau B. (2004) Thermotolerance requires refolding of aggregated proteins by substrate translocation through the central pore of ClpB. Cell 119, 653–665 [DOI] [PubMed] [Google Scholar]
- 5. Luheshi L. M., Dobson C. M. (2009) Bridging the gap: from protein misfolding to protein misfolding diseases. FEBS Lett. 583, 2581–2586 [DOI] [PubMed] [Google Scholar]
- 6. Broadley S. A., Hartl F. U. (2009) The role of molecular chaperones in human misfolding diseases. FEBS Lett. 583, 2647–2653 [DOI] [PubMed] [Google Scholar]
- 7. Powers E. T., Morimoto R. I., Dillin A., Kelly J. W., Balch W. E. (2009) Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 78, 959–991 [DOI] [PubMed] [Google Scholar]
- 8. Doyle S. M., Shorter J., Zolkiewski M., Hoskins J. R., Lindquist S., Wickner S. (2007) Asymmetric deceleration of ClpB or Hsp104 ATPase activity unleashes protein-remodeling activity. Nat. Struct. Mol. Biol. 14, 114–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Glover J. R., Lindquist S. (1998) Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell 94, 73–82 [DOI] [PubMed] [Google Scholar]
- 10. Duennwald M. L., Echeverria A., Shorter J. (2012) Small heat shock proteins potentiate amyloid dissolution by protein disaggregases from yeast and humans. PLoS Biol. 10, e1001346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rampelt H., Kirstein-Miles J., Nillegoda N. B., Chi K., Scholz S. R., Morimoto R. I., Bukau B. (2012) Metazoan Hsp70 machines use Hsp110 to power protein disaggregation. EMBO J. 31, 4221–4235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jaru-Ampornpan P., Shen K., Lam V. Q., Ali M., Doniach S., Jia T. Z., Shan S. (2010) ATP-independent reversal of a membrane protein aggregate by a chloroplast SRP subunit. Nat. Struct. Mol. Biol. 17, 696–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schuenemann D., Gupta S., Persello-Cartieaux F., Klimyuk V. I., Jones J. D., Nussaume L., Hoffman N. E. (1998) A novel signal recognition particle targets light-harvesting proteins to the thylakoid membranes. Proc. Natl. Acad. Sci. U.S.A. 95, 10312–10316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Groves M. R., Mant A., Kuhn A., Koch J., Dübel S., Robinson C., Sinning I. (2001) Functional characterization of recombinant chloroplast signal recognition particle. J. Biol. Chem. 276, 27778–27786 [DOI] [PubMed] [Google Scholar]
- 15. Tu C.-J., Schuenemann D., Hoffman N. E. (1999) Chloroplast FtsY, chloroplast signal recognition particle, and GTP are required to reconstitute the soluble phase of light-harvesting chlorophyll protein transport into thylakoid membranes. J. Biol. Chem. 274, 27219–27224 [DOI] [PubMed] [Google Scholar]
- 16. Falk S., Sinning I. (2010) cpSRP43 is a novel chaperone specific for light-harvesting chlorophyll a,b-binding proteins. J. Biol. Chem. 285, 21655–21661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. DeLille J., Peterson E. C., Johnson T., Moore M., Kight A., Henry R. (2000) A novel precursor recognition element facilitates posttranslational binding to the signal recognition particle in chloroplasts. Proc. Natl. Acad. Sci. U.S.A. 97, 1926–1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tu C. J., Peterson E. C., Henry R., Hoffman N. E. (2000) The L18 domain of light-harvesting chlorophyll proteins binds to chloroplast signal recognition particle 43. J. Biol. Chem. 275, 13187–13190 [DOI] [PubMed] [Google Scholar]
- 19. Hurshman A. R., White J. T., Powers E. T., Kelly J. W. (2004) Transthyretin aggregation under partially denaturing conditions is a downhill polymerization. Biochemistry 43, 7365–7381 [DOI] [PubMed] [Google Scholar]
- 20. Abramoff M. D., Magelhães P. J., Ram S. J. (2004) Image processing with Image J. Biophotonics Int. 11, 36–42 [Google Scholar]
- 21. Chernoff Y. O., Uptain S. M., Lindquist S. L. (2002) Analysis of prion factors in yeast. Methods Enzymol. 351, 499–538 [DOI] [PubMed] [Google Scholar]
- 22. Lam V. Q., Akopian D., Rome M., Henningsen D., Shan S. (2010) Lipid activation of the signal recognition particle receptor provides spatial coordination of protein targeting. J. Cell Biol. 190, 623–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stryer L. (1965) The interaction of a naphthalene dye with apomyoglobin and apohemoglobin. A fluorescent probe of non-polar binding sites. J. Mol. Biol. 13, 482–495 [DOI] [PubMed] [Google Scholar]
- 24. Rosen C. G., Weber G. (1969) Dimer formation from 1-anilino-8-naphthalenesulfonate catalyzed by bovine serum albumin. A new fluorescent molecule with exceptional binding properties. Biochemistry 8, 3915–3920 [DOI] [PubMed] [Google Scholar]
- 25. LeVine H. (1999) Quantification of β-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 309, 274–284 [DOI] [PubMed] [Google Scholar]
- 26. Carrotta R., Bauer R., Waninge R., Rischel C. (2001) Conformational characterization of oligomeric intermediates and aggregates in β-lactoglobulin heat aggregation. Protein Sci. 10, 1312–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Groenning M., Olsen L., van de Weert M., Flink J. M., Frokjaer S., Jørgensen F. S. (2007) Study on the binding of thioflavin T to β-sheet-rich and non-β-sheet cavities. J. Struct. Biol. 158, 358–369 [DOI] [PubMed] [Google Scholar]
- 28. Jansson S. (1999) A guide to the Lhc genes and their relatives in Arabidopsis. Trends Plant Sci. 4, 236–240 [DOI] [PubMed] [Google Scholar]
- 29. Krishnan R., Lindquist S. (2005) Structural insights into a yeast prion illuminate nucleation and strain diversity. Nature 435, 765–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fink A. L. (1998) Protein aggregation: folding aggregates, inclusion bodies, and amyloid. Fold Des. 3, R9–R23 [DOI] [PubMed] [Google Scholar]
- 31. Serio T. R., Cashikar A. G., Kowal A. S., Sawicki G. J., Moslehi J. J., Serpell L., Arnsdorf M. F., Lindquist S. L. (2000) Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science 289, 1317–1321 [DOI] [PubMed] [Google Scholar]
- 32. Goldsbury C. S., Wirtz S., Müller S. A., Sunderji S., Wicki P., Aebi U., Frey P. (2000) Studies on the in vitro assembly of Aβ 1–40: Implications for the search for Aβ fibril formation inhibitors. J. Struct. Biol. 130, 217–231 [DOI] [PubMed] [Google Scholar]
- 33. Mastrangelo I. A., Ahmed M., Sato T., Liu W., Wang C., Hough P., Smith S. O. (2006) High-resolution atomic force microscopy of soluble Aβ42 oligomers. J. Mol. Biol. 358, 106–119 [DOI] [PubMed] [Google Scholar]
- 34. Zettlmeissl G., Rudolph R., Jaenicke R. (1979) Reconstitution of lactic dehydrogenase. Noncovalent aggregation vs. reactivation. 1. Physical properties and kinetics of aggregation. Biochemistry 18, 5567–5571 [DOI] [PubMed] [Google Scholar]
- 35. Kopito R. R., Sitia R. (2000) Aggresomes and Russell bodies. Symptoms of cellular indigestion? EMBO Rep. 1, 225–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Brems D. N., Plaisted S. M., Havel H. A., Tomich C. S. (1988) Stabilization of an associated folding intermediate of bovine growth hormone by site-directed mutagenesis. Proc. Natl. Acad. Sci. U.S.A. 85, 3367–3371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mitraki A., Betton J. M., Desmadril M., Yon J. M. (1987) Quasi-irreversibility in the unfolding-refolding transition of phosphoglycerate kinase induced by guanidine hydrochloride. Eur. J. Biochem. 163, 29–34 [DOI] [PubMed] [Google Scholar]
- 38. Speed M. A., Wang D. I., King J. (1996) Specific aggregation of partially folded polypeptide chains: the molecular basis of inclusion body composition. Nat. Biotechnol. 14, 1283–1287 [DOI] [PubMed] [Google Scholar]
- 39. Cain P., Holdermann I., Sinning I., Johnson A. E., Robinson C. (2011) Binding of chloroplast signal recognition particle to a thylakoid membrane protein substrate in aqueous solution and delineation of the cpSRP43-substrate interaction domain. Biochem. J. 437, 149–155 [DOI] [PubMed] [Google Scholar]
- 40. Henry R. (2010) SRP: adapting to life in the chloroplast. Nat. Struct. Mol. Biol. 17, 676–677 [DOI] [PubMed] [Google Scholar]
- 41. Lum R., Niggemann M., Glover J. R. (2008) Peptide and protein binding in the axial channel of Hsp104: insights into the mechanism of protein unfolding. J. Biol. Chem. 283, 30139–30150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schlieker C., Weibezahn J., Patzelt H., Tessarz P., Strub C., Zeth K., Erbse A., Schneider-Mergener J., Chin J. W., Schultz P. G., Bukau B., Mogk A. (2004) Substrate recognition by the AAA+ chaperone ClpB. Nat. Struct. Mol. Biol. 11, 607–615 [DOI] [PubMed] [Google Scholar]
- 43. Hachiya N., Alam R., Sakasegawa Y., Sakaguchi M., Mihara K., Omura T. (1993) A mitochondrial import factor purified from rat liver cytosol is an ATP-dependent conformational modulator for precursor proteins. EMBO J. 12, 1579–1586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hachiya N., Komiya T., Alam R., Iwahashi J., Sakaguchi M., Omura T., Mihara K. (1994) MSF, a novel cytoplasmic chaperone which functions in precursor targeting to mitochondria. EMBO J. 13, 5146–5154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jaru-Ampornpan P., Liang F.-C., Nisthal A., Nguyen T. X., Wang P., Shen K., Mayo S. L., Shan S. (2013) Mechanism of an ATP-independent protein disaggregase: II. Distinct molecular interactions drive multiple steps during aggregate disassembly. J. Biol. Chem. 288, 13431–13445 [DOI] [PMC free article] [PubMed] [Google Scholar]