

Abstract
Serine-rich repeat glycoproteins (SRRPs) are important bacterial adhesins conserved in streptococci and staphylococci. Fap1, a SRRP identified in Streptococcus parasanguinis, is the major constituent of bacterial fimbriae and is required for adhesion and biofilm formation. An 11-gene cluster is required for Fap1 glycosylation and secretion; however, the exact mechanism of Fap1 biogenesis remains a mystery. Two glycosylation-associated proteins within this cluster—Gap1 and Gap3—function together in Fap1 biogenesis. Here we report the role of the third glycosylation-associated protein, Gap2. A gap2 mutant exhibited the same phenotype as the gap1 and gap3 mutants in terms of Fap1 biogenesis, fimbrial assembly, and bacterial adhesion, suggesting that the three proteins interact. Indeed, all three proteins interacted with each other independently and together to form a stable protein complex. Mechanistically, Gap2 protected Gap3 from degradation by ClpP protease, and Gap2 required the presence of Gap1 for expression at the wild-type level. Gap2 augmented the function of Gap1 in stabilizing Gap3; this function was conserved in Gap homologs from Streptococcus agalactiae. Our studies demonstrate that the three Gap proteins work in concert in Fap1 biogenesis and reveal a new function of Gap2. This insight will help us elucidate the molecular mechanism of SRRP biogenesis in this bacterium and in pathogenic species.
INTRODUCTION
Two of the most prevalent infectious diseases of humans are dental caries and inflammatory periodontal disease. Oral streptococci constitute a large proportion of oral bacterial species in dental plaque and are among the first colonizers of the tooth surface (1–3). As such, oral streptococci will encounter not only host oral epithelial cells but also other microbial cells, of which there are >500 species in the oral cavity, including the major periodontal pathogens—which often cannot colonize unless a layer of initial colonizers, such as oral streptococci, has developed first (4–8). Like other oral streptococci, Streptococcus parasanguinis has several colonization and adhesion factors; one of its adhesion factors is long peritrichous fimbriae (9). S. parasanguinis fimbriae are made of Fap1 (fimbria-associated protein 1), a 200-kDa cell wall-anchored serine-rich repeat glycoprotein (SRRP) (10). Fap1 is required for fimbrial formation, bacterial adhesion (1, 11), and biofilm formation (10, 12). Since the discovery of Fap1 (13, 14), Fap1-like SRRPs have been identified in many streptococci, staphylococci, and other Gram-positive bacteria and have been implicated in bacterial interactions with hosts, adhesion, biofilm formation, and pathogenesis (10, 11, 15–20). They include GspB and Hsa of Streptococcus gordonii (21, 22), SraP of Streptococcus sanguinis (23), PsrP of Streptococcus pneumoniae (18), Srr-1 and Srr-2 of Streptococcus agalactiae (16, 17), SrpA of Streptococcus cristatus (24), SraP of Staphylococcus aureus (10, 19), and FimS of Streptococcus salivarius (25).
The exact mechanism of SRRP biogenesis is not well understood. The chromosomal region dedicated to SRRP glycosylation and secretion is quite large and highly conserved. For Fap1, the cluster is separated into two regions: a core region that is conserved in every genome (secY2, gap1 to gap3, secA2, and gtf1 and gtf2) and a variable region that includes genes encoding several putative glycosyltransferases (gly, nss, galT1, and galT2) (10). gtf1 and gtf2 and the genes from the gly-gtf3-galT1-galT2 locus mediate Fap1 glycosylation (13, 26–30); Fap1 is glycosylated in the cytoplasm with several monosaccharides, including glucose, N-acetylglucosamine, N-acetylgalactosamine, and rhamnose (11, 29). The secY2-gap1-gap2-gap3-secA2 locus is responsible for the secretion of Fap1 (28, 29, 31). SecA2 and SecY2 have homology to their counterparts in the canonical Sec pathway and are required for the export of mature Fap1 to the cell wall surface (28, 29). There is no known homology for the remainder of the locus—gap1-gap2-gap3—outside of the SRRP family. We have shown previously that both gap1 and gap3 mutants produce similar immature forms of Fap1 and that the interaction between Gap1 and Gap3 is required for Fap1 biogenesis, indicating that Gap1 and Gap3 are involved in the biogenesis of mature Fap1 (32–34). However, to date, the function of Gap2 is unknown.
In this study, we determined the role of Gap2 and found that it is involved in Fap1 biogenesis by stabilizing Gap3 through interactions with Gap1 and Gap3. Thus, this study reveals an activity of Gap2 and its homolog that was previously unknown.
MATERIALS AND METHODS
Bacterial strains, plasmids, and primers and DNA manipulation.
The bacterial strains and plasmids used in this study are listed in Table 1. Escherichia coli and S. parasanguinis strains were cultured as described previously (15). S. parasanguinis cell concentrations were determined by the absorbance at 470 nm. Antibiotics were used at the following concentrations: 10 μg/ml erythromycin,125 μg/ml kanamycin, and 250 μg/ml spectinomycin in Todd-Hewitt (TH) broth or agar plates for S. parasanguinis; 300 μg/ml erythromycin, 50 μg/ml kanamycin, 50 μg/ml ampicillin, and 50 μg/ml spectinomycin in Luria-Bertani (LB) broth or agar plates for E. coli. Standard recombinant DNA techniques were used for DNA preparation and analyses (35). Plasmid DNA preparations were isolated with a QIAprep Miniprep kit (Qiagen). The primers used in this study are listed in Table 2. PCR was carried out with Taq DNA polymerase (Promega) or KOD DNA polymerase (Novagen). PCR products were purified with a QIAquick PCR purification kit (Qiagen). DNA digestion, ligation, and transformation were performed using standard methods. Competent cells for S. parasanguinis electroporation were prepared as described previously (36).
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristic(s) | Reference or source |
|---|---|---|
| Strains | ||
| S. parasanguinis | ||
| FW213 | Wild type | 9 |
| FW213 fap1 | fap1 insertion mutant; Kanr | 1 |
| FW213 secY2 | secY2 insertion mutant; Kanr | 28 |
| FW213 gap1 | gap1 insertion mutant; Kanr | 34 |
| FW213 gap2 | gap2 insertion mutant; Kanr | This study |
| FW213 gap3 | gap3 insertion mutant; Kanr | 32 |
| FW213 clpP | clpP insertion mutant; Kanr | 38 |
| FW213 gap2 clpP | FW213 gap2 with clpP insertion mutant; Kanr Specr | This study |
| FW213 gap1/pVPT | FW213 gap1 containing pVPT-gfp; vector control strain; Ermr Kanr | 38 |
| FW213 gap2/pVPT | FW213 gap2 containing pVPT-gfp; vector control strain; Ermr Kanr | This study |
| FW213 gap3/pVPT | FW213 gap3 containing pVPT-gfp; vector control strain; Ermr Kanr | This study |
| FW213 gap1/pVPT-gap1 | FW213 gap1 containing plasmid pVPT-gap1-gfp; Ermr Kanr | 38 |
| FW213 gap2/pVPT-gap2 | FW213 gap2 containing plasmid pVPT-gap2-gfp; Ermr Kanr | This study |
| FW213 gap3/pVPT-gap3 | FW213 gap3 containing plasmid pVPT-gap3; Ermr Kanr | This study |
| FW213/pIB184-gap3 | FW213 containing plasmid pIB184-gap3-gfp; Ermr | This study |
| FW213 gap1/pIB184-gap3 | FW213 gap1 containing plasmid pIB184-gap3-gfp; Ermr Kanr | This study |
| FW213 gap2/pIB184-gap3 | FW213 gap2 containing plasmid pIB184-gap3-gfp; Ermr Kanr | This study |
| FW213/pIB184-gap2-3 | FW213 containing plasmid pIB184-gap2-gap3-gfp; Ermr | This study |
| FW213 gap1/pIB184-gap2-3 | FW213 gap1 containing plasmid pIB184-gap2-gap3-gfp; Ermr Kanr | This study |
| FW213 gap2/pIB184-gap2-3 | FW213 gap2 containing plasmid pIB184-gap2-gap3-gfp; Ermr Kanr | This study |
| FW213/pIB184-gap1-2-3 | FW213 containing plasmid pIB184-gap1-gap2-gap3-gfp; Ermr | This study |
| FW213 gap1/pIB184-gap1-2-3 | FW213 gap1 containing plasmid pIB184-gap1-gap2-gap3-gfp; Ermr Kanr | This study |
| FW213 gap2/pIB184-gap1-2-3 | FW213 gap2 containing plasmid pIB184-gap1-gap2-gap3-gfp; Ermr Kanr | This study |
| FW213/pIB184-gap2 | FW213 containing plasmid pIB184-gap2-hsv-his; Ermr | This study |
| FW213 gap1/pIB184-gap2 | FW213 gap1 containing plasmid pIB184-gap2-hsv-his; Ermr Kanr | This study |
| FW213 gap2/pIB184-gap2 | FW213 gap2 containing plasmid pIB184-gap2-hsv-his; Ermr Kanr | This study |
| FW213/pIB184-gap1-2 | FW213 containing plasmid pIB184-gap1-gap2-hsv-his; Ermr | This study |
| FW213 gap1/pIB184-gap1-2 | FW213 gap1 containing plasmid pIB184-gap1-gap2-hsv-his; Ermr Kanr | This study |
| FW213 gap2/pIB184-gap1-2 | FW213 gap2 containing plasmid pIB184-gap1-gap2-hsv-his; Ermr Kanr | This study |
| FW213 gap2/pIB184-asp3 | FW213 gap2 containing plasmid pIB184-asp3-gfp; Ermr Kanr | This study |
| FW213 gap2/pIB184-asp2-3 | FW213 gap2 containing plasmid pIB184-asp2-asp3-gfp; Ermr Kanr | This study |
| FW213 gap2/pIB184-asp1-2-3 | FW213 gap2 containing plasmid pIB184-asp1-asp2-asp3-gfp; Ermr Kanr | This study |
| FW213/pIB184-asp2 | FW213 containing plasmid pIB184-asp2-hsv-his; Ermr | This study |
| FW213 gap1/pIB184-asp2 | FW213 gap1 containing plasmid pIB184-asp2-hsv-his; Ermr Kanr | This study |
| FW213/pIB184-asp1-2 | FW213 containing plasmid pIB184-asp1-asp2-hsv-his; Ermr | This study |
| FW213 gap1/pIB184-asp1-2 | FW213 gap1 containing plasmid pIB184-asp1-asp2-hsv-his; Ermr Kanr | This study |
| S. agalactiae J48 | Wild type | 17 |
| E. coli | ||
| Top10 | Host strain for cloning | Invitrogen |
| BL21 | Host strain for protein expression | Invitrogen |
| Plasmids | ||
| pVPT-gfp | E. coli-S. parasanguinis shuttle vector; Ermr | 40 |
| pVPT-Gap1-gfp | gap1 from FW213 cloned into pVPT-gfp; Ermr | This study |
| pVPT-Gap2-gfp | gap2 from FW213 cloned into pVPT-gfp; Ermr | This study |
| pVPT-Gap3-gfp | gap3 from FW213 cloned into pVPT-gfp; Ermr | This study |
| pIB184 | E. coli-S. parasanguinis shuttle vector; Ermr | 41 |
| pIB184-gfp | E. coli-S. parasanguinis shuttle vector with gfp tag; Ermr | This study |
| pIB184-hsv-his | E. coli-S. parasanguinis shuttle vector with hsv-his tag; Ermr | This study |
| pIB184-Gap3-gfp | gap3 from FW213 cloned into pIB184-gfp; Ermr | This study |
| pIB184-Gap2-3-gfp | gap2 and gap3 from FW213 cloned into pIB184-gfp; Ermr | This study |
| pIB184-Gap1-2-3-gfp | gap1, gap2, and gap3 from FW213 cloned into pIB184 gfp; Ermr | This study |
| pIB184-Gap2-hsv-his | gap2 from FW213 cloned into pIB184-hsv-his; Ermr | This study |
| pIB184-Gap1-2-hsv-his | gap1 and gap2 from FW213 cloned into pIB184-hsv-his; Ermr | This study |
| pIB184-Asp3-gfp | asp3 from J48 cloned into pIB184-gfp; Ermr | This study |
| pIB184-Asp2-3-gfp | asp2 and asp3 from J48 cloned into pIB184-gfp; Ermr | This study |
| pIB184-Asp1-2-3-gfp | asp1, asp2, and asp3 from J48 cloned into pIB184-gfp; Ermr | This study |
| pIB184-Asp2-hsv-his | asp2 from J48 cloned into pIB184-hsv-his; Ermr | This study |
| pIB184-Asp1-2-hsv-his | asp1 and asp2 from J48 cloned into pIB184-hsv-his; Ermr | This study |
| pGEX-GST-Gap1 | pGEX-GST vector containing gap1 gene from FW213; Ampr | 34 |
| pGEX-GST-Gap2 | pGEX-GST vector containing gap2 gene from FW213; Ampr | 34 |
| pGEX-GST-Gap3 | pGEX-GST vector containing gap3 gene from FW213; Ampr | This study |
| pET-His-SUMO-Gap1-3 | pET-His-SUMO vector containing gap1 and gap3 genes; Kanr | 38 |
| pET-His-SUMO-Gap1-2-3 | pET-His-SUMO vector containing gap1, gap2, and gap3 genes; Kanr | This study |
| pGEM::Δgap2-aphA3 | pGEM vector containing gap2 with aphA3 insertion; Kanr | This study |
| pGEM::ΔclpP-aphA3 | pGEM vector containing clpP with aphA3 insertion; Kanr | 38 |
| pGEM::ΔclpP-spec | pGEM vector containing clpP with spec insertion; Specr | This study |
Table 2.
Primers used in this study
| Primer | Sequence |
|---|---|
| Gap1-SalI-F | ATACGCGTCGACATGTTTTATTTTGTACCTTC |
| Gap1-KpnI-R | CGGGGTACCTTTCTTTTTTAGCATACCTTTCC |
| Gap2-SalI-F | ATACGCGTCGACATGAAGATTTTACAATTGGC |
| Gap2-KpnI-R | CGCGGTACCTCTTCCAAACTGATCTTCTAG |
| Gap3-SalI-F | ACTCGCGTCGACATGACTAAACAGTTAATTTCTG |
| Gap3-KpnI-R | CGCGGTACCAATATATTCTATTAAATTTTTCACC |
| Gap2+Flank-F | ATACGCGTCGACATGAAG ATTTTACAAATTGGCCG |
| Gap2+Flank-R | CGGGGTACCTCTTCCAAACTGATCTTC TAG |
| Gap2-StuI-F | GCAGAGGCCTACAAGTGCTGATATGCTACTG |
| Gap2-StuI-R | GCAGAGGCCTCTTTGCTCCGTATTGACTAC |
| Spec-HindIII-F | CGGCCGCAAGCTTGTGAGGAGGATATATTTGAA |
| Spec-HindIII-R | CGGGCGCCGCAAGCTTTTATAATTTTTTTAATCTG |
| Gap1-BamHI-F | CCGGCGCCGGATCCGGATGTTTTATTTTGTACCTTCTTGG |
| Gap2-BamHI-F | GAGCGGATCCGGATGAAGATTTTACAAATTGGCCG |
| Gap2-XmaI-R | CCGCTGCCCGGGTCTTCCAAACTGATCTTCTA |
| Gap3-BamHI-F | GCGGCCTCGCGGATCCGAATGACTAAACAGTTAATTTCTG |
| Gap3-XmaI-R | GGCTCGCCGCGGTCCCGGGAATATATTCTATTAAATTTTTCACCAAATC |
| GFP-XmaI-F | GACGCCCGGGATGAGTAAAGGAGAAGAACTTTTCACTG |
| GFP-SacI-R | GCCGCGAGCTCCTATTTGTATAGTTCATCCATGCC |
| HsvHis-XmaI-F | ATATAACCCGGGAGCCAGCCAGAACTCGC |
| HsvHis-SacI-R | TATTGAGCTCTCAGTGGTGGTGGTGGTGGTGC |
| Asp1-BamHI-F | GGCGCGCGGATCCGGATGTTTTATTTTATTCCTTCGTGG |
| Asp2-BamHI-F | CGCCGCCGGCGGATCCGGATGGAAAAATTAAAAATTTTGCAG |
| Asp2-XmaI-R | GATCCCCGGGACCACTAAACACTCTCCCAAAAT |
| Asp3-BamHI-F | GCCGATCGGATCCGGATGATTTTGGGAGAGTGTTTAG |
| Asp3-XmaI-R | GCGGCCGGATGCCCGGGCGATTTTTTATCCTTAGAAAATGCTATCAACG |
| Gap2-EcoRI-F | GACGAATTCATGAAGATTTTACAATTGGC |
| Gap2-BamHI-R | TGTGGATCCTCTTCCAAACTGATCTTCTAG |
| Gap1-NotI-1F | AAGGAAAAAAGCGGCCGCATGTTTTATTTTGTACCTTCTTGG |
| Gap3-XhoI-R | ACCGCTCGAGTTAAATATATTCTATTAAATTTTTC |
Western blot analysis.
All S. parasanguinis strains were grown to an optical density at 470 nm (OD470) of 0.5 to 0.6 and were centrifuged. The cell pellets were treated with amidase to lyse the cells (28). Cell lysates were boiled in sample buffer (0.0625 M Tris [pH 6.8], 2% sodium dodecyl sulfate [SDS], 10% glycerol, 0.01% bromophenol blue) for 10 min before being loaded onto 10% SDS-polyacrylamide gel electrophoresis (PAGE) gels and subjected to Western blotting. Two monoclonal antibodies (MAbs) were used to detect Fap1: MAb E42, which is specific to the peptide backbone of Fap1, and MAb F51, which is specific to mature Fap1 (11). MAb F51 recognizes only the 200-kDa mature Fap1, whereas MAb E42 recognizes both the 200-kDa mature Fap1 and the 470-kDa Fap1 precursor. Rabbit polyclonal antibodies against Gap1, Gap2, and Gap3 were custom-produced using recombinant Gap1, Gap2, or Gap3, or the Gap1/2/3 complex, as an antigen. A monoclonal antibody against Hsv (Novagen) was used to detect tagged proteins. A polyclonal antibody against FimA was used to standardize the protein loading of S. parasanguinis proteins.
Construction of the insertional gap2 mutant and the gap2 clpP double mutant.
A gap2 mutant was constructed by allelic replacement of gap2 with a kanamycin resistance cassette, aphA3 (encoding aminoglycoside phosphotransferase). A fragment containing the gap2 gene and its flanking regions was amplified from S. parasanguinis chromosomal DNA using Gap2+Flank-F/Gap2+Flank-R. The PCR fragment was ligated into pGEM-T Easy (Promega). An 850-bp region of gap2 was deleted by inverse PCR using Gap2-StuI-F/Gap2-StuI-R. The inverse PCR product was digested with StuI and was ligated with a promoterless aphA3 kanamycin resistance cassette from pALH124 (37) to generate pGEM::Δgap2-aphA3. Finally, the gap2 insertion mutant was constructed by transformation of FW213 with pGEM::Δgap2-aphA3, followed by the selection of kanamycin-resistant colonies. The in-frame insertion was further examined by DNA sequencing analyses. Western blot analysis with an antiserum against SecA2, a protein encoded by a gene downstream of gap2, was performed to confirm that the mutation was nonpolar (data not shown). The fap1 (1), secY2 (28), gap1 (34), gap3 (32), and clpP (38) mutants were constructed by a similar method. For the gap2 clpP double mutant, a spectinomycin resistance cassette (Spec) was inserted into clpP in the gap2 mutant. The pGEM::ΔclpP-aphA3 construct (38) was digested with HindIII to remove the kanamycin resistance cassette and was then ligated in-frame with the spectinomycin resistance cassette amplified from pCG1 (39) to construct pGEM::ΔclpP-spec. The gap2 clpP double mutant was constructed by transformation of the gap2 mutant with pGEM::ΔclpP-spec, followed by the selection of kanamycin- and spectinomycin-resistant colonies. The in-frame insertion was further examined by DNA sequencing analyses.
Complementation of the gap1, gap2, and gap3 mutants.
The full-length gap1, gap2, and gap3 genes were amplified from FW213 genomic DNA by PCR using primers Gap1-SalI-F/Gap1-KpnI-R, Gap2-SalI-F/Gap2-KpnI-R, and Gap3-SalI-F/Gap3-KpnI-R, respectively (Table 2). The purified gap1, gap2, and gap3 PCR products were digested with SalI and KpnI and were then cloned into the E. coli-Streptococcus shuttle vector pVPT-gfp (40) to generate the corresponding complementation plasmids pVPT-gap1-gfp, pVPT-gap2-gfp, and pVPT-gap3 (no gfp). The plasmid and its control vector pVPT-gfp were then transformed into the gap1, gap2, and gap3 mutants via electroporation. The transformants were selected on TH agar plates containing kanamycin and erythromycin.
Modification of the E. coli-Streptococcus shuttle vector pIB184.
A second E. coli-Streptococcus shuttle vector, pIB184 (41), was used in this study for better expression and genetic manipulation. To enhance the utility of this vector, pIB184 was modified by cloning in gfp and hsv-his tags within the multiple cloning site. Full-length gfp and hsv-his were amplified from pVPT-gfp and pET27b (Novagen) using primers GFP-XmaI-F/GFP-SacI-R and HsvHis-Xmal-F/HsvHis-SacI-R, respectively. The purified gfp and hsv-his PCR products were digested with XmaI and SacI and were then ligated with the pIBI84 vector to create pIB184-gfp and pIB184-hsv-his.
Construction of overexpression strains in S. parasanguinis FW213.
Full-length gap3, gap2-gap3, and gap1-gap2-gap3 were amplified from FW213 genomic DNA by PCR using primers Gap3-BamHI-F/Gap3-XmaI-R, Gap2-BamHI-F/Gap3-XmaI-R, and Gap1-BamHI-F/Gap3-XmaI-R, respectively (Table 2). The purified gap3, gap2-gap3, and gap1-gap2-gap3 PCR products were digested with BamHI and XmaI and were then cloned into the E. coli-Streptococcus shuttle vector pIB184-gfp to generate pIB184-gap3-gfp, pIB184-gap2-gap3-gfp, and pIB184-gap1-gap2-gap3-gfp, where Gap3 is tagged with green fluorescent protein (GFP) in all vectors. The plasmids were then transformed into the wild type and the gap1 and gap2 mutants via electroporation. pIB184-gap2-hsv-his and pIB184-gap1-gap2-hsv-his were created in the same fashion, using pIB184-hsv-his and primer pairs Gap2-BamHI-F/Gap2-XmaI-R and Gap1-BamHI-F/Gap2-XmaI-R, respectively. The Gap homologs Asp1, Asp2, and Asp3, from wild-type S. agalactiae J48, were used to check for conservation of function. pIB184-asp3-gfp, pIB184-asp2-gap3-gfp, pIB184-asp1-asp2-asp3-gfp, pIB184-asp2-hsv-his, and pIB184-asp1-asp2-hsv-his were created in the manner described above, using primers Asp3-BamHI-F/Asp3-XmaI-R, Asp2-BamHI-F/Asp3-XmaI-R, Asp1-BamHI-F/Asp3-XmaI-R, Asp2-BamHI-F/Asp2-XmaI-R, and Asp1-BamHI-F/Asp2-XmaI-R, respectively (Table 2). The resulting plasmids were then transformed into FW213 and gap1 and gap2 mutants via electroporation. The transformants were selected on TH agar plates containing erythromycin (wild type) or kanamycin and erythromycin (mutants).
Bacterial adhesion assay.
Saliva-coated hydroxyapatite (SHA) was used as an in vitro tooth model to test the binding abilities of S. parasanguinis and the relevant derivatives as described previously (42). Briefly, [3H]thymidine-labeled bacteria at an OD470 of 1.0 in adhesion buffer (67 mM phosphate buffer, pH 6.0) were sonicated for 15 s at 85 W using an ultrasonic cup horn system (Heat Systems-Ultrasonics). One milliliter of sonicated bacteria (in triplicate) was added to 7-ml scintillation vials containing SHA and was incubated for 1 h at 37°C with gentle shaking. The supernatant fluids were removed, and the beads were washed 3 times with adhesion buffer. The amounts of unbound bacteria in the supernatant fluids and bacteria bound to SHA were determined in an LS6500 scintillation counter (Beckman Coulter) (1). Differences in SHA adhesion were analyzed via a 2-tailed Student t test for two samples with equal variances.
Transmission electron microscopy.
S. parasanguinis cell cultures (5 ml) grown to an OD470 of 0.4 were harvested by centrifugation. Cell pellets were washed twice with ice-cold phosphate-buffered saline (PBS) and were resuspended in 100 μl PBS. Five microliters of the bacterial suspension was diluted in PBS and was applied to 400-mesh copper grids coated with a thin carbon film. The grids were first washed with several drops of PBS buffer. The samples were stained with a few drops of 2% phosphotungstic acid (PTA), pH 7.0, over the grid surfaces. The excess liquid was wicked off, and the grids were quickly air dried. The grids were observed on a Tecnai 12 Philips electron microscope (FEI, The Netherlands) equipped with a LaB6 cathode operated in point mode (Kimball) and a 2,048- by 2,048-pixel charge-coupled device (CCD) camera (TVIPS, Germany). The microscope was run to obtain images that show Thon rings beyond a 0.9-nm resolution in vitreous ice preparations (43). Images were recorded at an accelerating voltage of 100 kV and at nominal magnifications in the range of ×40,000 to ×70,000 under low-dose conditions on either film (SO-163; Kodak) or the CCD camera. Images were converted to SPIDER format (44), and a high-pass filter was used to remove the background.
In vitro GST pulldown assays.
The glutathione S-transferase (GST) pulldown protocol was developed to determine protein-protein interactions in solutions (45). Gap1-pGADT7 and Gap3-pGADT7 were constructed as described previously (34). Gap2-pGADT7 was constructed by PCR amplification of gap2 from FW213 chromosomal DNA using primers Gap2-EcoRI-F/Gap2-BamHI-R (Table 2), digestion with EcoRI and BamHI, and ligation into pGADT7. The GST-Gap1, GST-Gap2, and GST-Gap3 fusion proteins were created by cloning of EcoRI- and XhoI-digested fragments from Gap1-pGADT7, Gap2-pGADT7, and Gap3-pGADT7, respectively, into pGEX-5X-2. The GST fusion proteins were expressed and purified using glutathione Sepharose 4B beads. The same amounts of GST or GST fusion proteins (5 μg) immobilized on beads and estimated by SDS-PAGE analysis were resuspended in NETN washing buffer (20 mM Tris-HCl [pH 7.0], 150 mM NaCl, 1 mM EDTA, 0.5% NP-40) and were mixed with 5 μl of an in vitro-translated c-Myc-Gap1, c-Myc-Gap2, or c-Myc-Gap3 fusion protein product (34). The mixtures were reconstituted in a final volume of 200 μl with NETN binding buffer and were incubated at 4°C overnight on a rotary shaker. The beads were washed three times with 600 μl of NETN washing buffer. The proteins bound to the beads were eluted by boiling in SDS loading buffer and were subjected to Western blot analyses using an anti-c-Myc antibody (Invitrogen). The interaction between Gap1 and Gap3 has been confirmed previously (34) and was used here as a control.
Analytical ultracentrifugation. (i) Sample preparation.
A fusion plasmid was constructed to express His-SUMO-tagged Gap1/2/3 by the same method used in the construction of His-SUMO-tagged Gap1/3 (38). Briefly, full-length gap1-gap2-gap3 was amplified from S. parasanguinis FW213 genomic DNA using Gap1-NotI-1F/Gap3-XhoI-R, digested by NotI and XhoI, and ligated into pET-SUMO to construct the His-SUMO-Gap1/2/3 fusion protein. The constructed plasmid was verified by DNA sequence analysis and was then transformed into E. coli BL21(DE3). Gap1/3 and Gap1/2/3 were expressed and purified as described previously (26). Peak fractions from gel filtration were collected and used for ultracentrifugation. Concentrations of the proteins were determined by measuring the sample absorbance at 280 nm using a Beckman DU-640 spectrophotometer (International Mi-Ss, Inc., Corona, CA). The sample proteins were diluted to the desired concentrations with buffer G (26).
(ii) SE.
Sedimentation equilibrium (SE) experiments were performed at 20°C using six-channel centerpieces in a Beckman Optima XL-A analytical centrifuge with absorption optics. Three concentrations (0.2 mg/ml, 0.4 mg/ml, and 0.9 mg/ml) were analyzed at two rotor speeds (17,000 rpm and 20,000 rpm) with detection by absorbance at 280 nm. All data sets from different protein concentrations and rotor speeds were fit to a single global model (global fits) to determine the stoichiometry and equilibrium constants. Model fittings of the SE data were performed by HETEROANALYSIS software (Biotechnology/Bioservices Center, University of Connecticut, Storrs, CT).
RESULTS
The Gap2 mutant exhibits same phenotype as the Gap1 and Gap3 mutants.
Gap1 and Gap3 have been shown to be involved in Fap1 biogenesis (32, 34). However, there have been no reports on the function of the third glycosylation-associated protein, Gap2. In this study, we generated a Gap2-deficient mutant and examined its phenotype. Fap1 production in the Gap2-deficient strain was similar to that in the strains deficient in Gap1, Gap3, and SecY2, where mature Fap1 (Fig. 1, lane 1), recognized by F51, was undetectable and a larger band, corresponding to immature Fap1 (lanes 3 to 6), was observed upon probing with E42, a peptide-specific antibody. The wild-type phenotype was restored upon complementation (Fig. 1, lanes 7 to 9); the empty vectors could not restore the wild-type phenotype (lanes 10 to 12). This result demonstrates that Gap2, like Gap1 and Gap3, is required for the production of mature Fap1 (32, 34).
Fig 1.
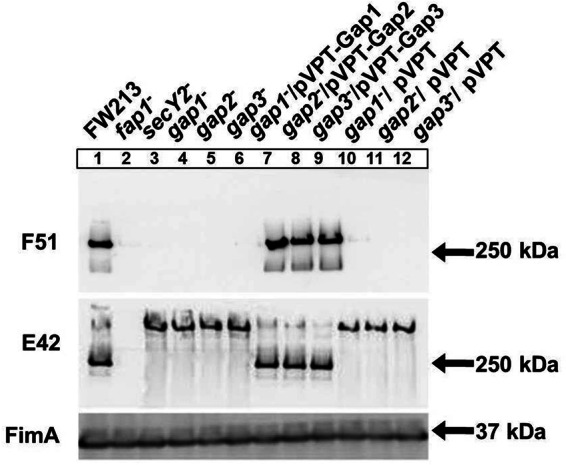
Biogenesis of mature Fap1 requires Gap2, as well as Gap1 and Gap3. Western blot analysis for Fap1 was performed on S. parasanguinis cell lysates. The strains used include wild-type FW213, insertional mutants of fap1, secY2, gap1, gap2, and gap3, complemented strains of the gap1, gap2, and gap3 mutants with the full gene in pVPT, and control strains with the empty vector. The antibodies used include F51 (specific to mature Fap1), E42 (specific to the Fap1 polypeptide), and FimA (a loading control).
Since Fap1 is required for the assembly of S. parasanguinis fimbriae (1), the cell surface structures of S. parasanguinis variants were examined using transmission electron microscopy. In the gap2 mutant, fimbriae were no longer detected (Fig. 2C), while they were detected in the wild-type strain FW213 (Fig. 2A). However, the Gap2 deficiency had no effect on a smaller fibril (indicated by the white arrows in Fig. 2B to D), which has been identified previously as BapA1 (46). This fimbrial phenotype is comparable to that of the gap1 (Fig. 2B) and gap3 (Fig. 2D) mutants. Furthermore, the Gap2 deficiency decreased bacterial adherence to SHA (Fig. 3). This phenotype was similar to that observed in strains deficient in Gap1 or Gap3. For all three strains, complementation restored adhesion levels nearly to that of the wild type (Fig. 3). These results indicate that Gap2 functions in concert with Gap1 and Gap3 in Fap1 biogenesis, with a subsequent effect on fimbrial biogenesis and adhesion levels.
Fig 2.

Gap2 is necessary for the production of wild-type fimbriae. Shown are transmission electron micrographs of wild-type S. parasanguinis and mutants. (A) FW213 (wild type); (B) gap1 mutant; (C) gap2 mutant; (D) gap3 mutant. The black arrow points to long fimbriae. White arrows point to short fibrils. Bar, 100 nm.
Fig 3.
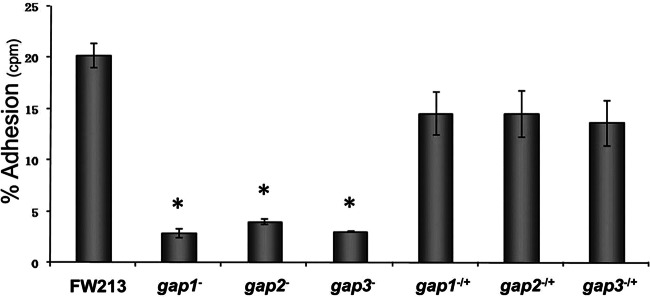
Gap2, like Gap1 and Gap3, is required for the adhesion of S. parasanguinis to SHA. Levels of in vitro adhesion to SHA are shown for S. parasanguinis FW213, its gap1, gap2, and gap3 insertional mutants, and the corresponding complemented strains. The data were obtained from two independent experiments in three replicates and are presented as means ± standard deviations. An asterisk indicates that the level of adhesion was significantly lower than that observed for FW213 (P < 0.003).
Gap1, Gap2, and Gap3 interact with each other to form a complex.
Not only do Gap1-, Gap2-, and Gap3-deficient strains have similar phenotypes (Fig. 1 to 3), but also the interaction between Gap1 and Gap3 is required for the biogenesis of Fap1 (34, 38). Therefore, it is likely that Gap2 interacts with Gap1 and Gap3 as well. To determine this, we coexpressed all three proteins in E. coli, with Gap1 tagged with GST, and performed GST pulldown assays. Gap2 and Gap3 were invariably pulled down by GST-Gap1 (Fig. 4A). GST itself did not pull down Gap2 and Gap3 (data not shown). To address whether Gap2 could interact with Gap1 and Gap3 independently, we expressed each protein tagged with GST individually and incubated them with in vitro-translated c-Myc fusion proteins. In GST pulldown assays, GST-tagged Gap2 pulled down Gap1 and Gap3, and Gap2 was pulled down by GST-tagged Gap1 and Gap3 (Fig. 4B). These results indicate that Gap2 can interact with both Gap1 and Gap3 directly. The interaction between Gap1 and Gap3 was used as a positive assay control. In negative controls, neither Gap1, Gap2, nor Gap3 interacted with GST alone, indicating that the interactions between Gap2 and Gap1 and between Gap2 and Gap3 were specific.
Fig 4.
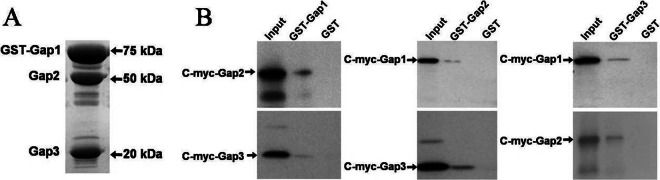
Gap2 interacts with Gap1 and Gap3 individually and together to form a complex. Shown are results of in vitro GST pulldown assays to detect interactions among Gap1, Gap2, and Gap3. (A) SDS-PAGE analysis of E. coli cell lysates expressing GST-Gap1, Gap2, and Gap3. Gap2 and Gap3 are invariably pulled down by GST-Gap1. (B) Western blot analysis of GST pulldown assays between Gap1, Gap2, and Gap3. An antibody against c-Myc was used.
Analytical ultracentrifugation sedimentation equilibrium (SE) experiments were performed to further characterize the interactions among Gap1, Gap2, and Gap3. The SE data showed that the Gap1/3 complex fit a single-species model well (Fig. 5A), suggesting that the binding between Gap1 and Gap3 was tight. The binding of Gap2 to the already formed Gap1/3 complex fit a heterodimer model (“A + B ⇌ AB,” where A represents Gap1/3 and B represents Gap2; dissociation constant [Kd], 4.4E−07 M) (Fig. 5B), suggesting that Gap2 binds to Gap1/3 to form a Gap1/2/3 complex in a reversible manner. The experimental data fit the models regardless of the concentration (0.2 mg/ml, 0.4 mg/ml, or 0.9 mg/ml) (Fig. 5) or speed (17,000 rpm [Fig. 5] or 20,000 rpm [data not shown]) used.
Fig 5.
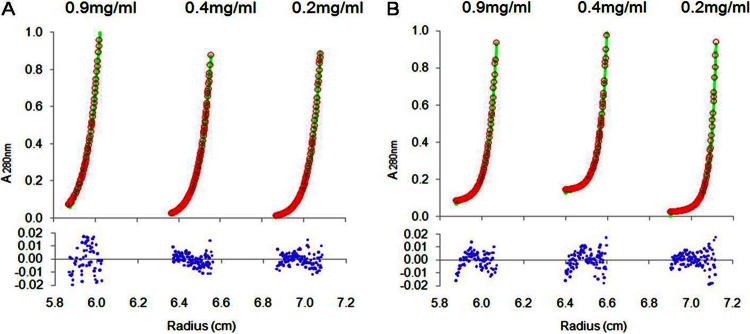
Gap2 binds to the Gap1/3 complex in a reversible manner. Sedimentation equilibrium analyses of the protein complexes Gap1/3 (A) and Gap1/2/3 (B) were performed. Three concentrations (0.2 mg/ml, 0.4 mg/ml, and 0.9 mg/ml) were analyzed at 17,000 rpm with detection by absorbance at 280 nm. (A) The sedimentation equilibrium data from Gap1/3 fit a single-species model well. Root mean square deviation, 0.00736. (B) The sedimentation equilibrium data from Gap1/2/3 fit a heterodimer (consisting of monomers Gap2 and Gap1/3) model well, with a Kd of 4.4E−07 M. Root mean square deviation, 0.00643. Green curves, calculations for the sample based on the model fitting; red circles, experimental data points of the distribution of Gap1/3 or Gap1/2/3 concentrations along the radius. Blue dots stand for residuals, which represent the difference between the sample and model values. All residuals were randomly distributed.
Gap2 levels are increased with Gap1 overexpression.
Previously, we have shown that Gap1 is required for the stability of Gap3 (38). In this study, we demonstrated that Gap2 interacted with both Gap1 and Gap3. In order to determine how Gap2 affects or is affected by Gap1, Gap1 and Gap2 protein levels in wild-type and gap mutant variants were determined (Fig. 6A). A gap2 mutant had no effect on the amount of Gap1 (Fig. 6A, lane 3). On the other hand, in the absence of Gap1 (Fig. 6A, lane 2), the level of Gap2 was decreased from that in the wild type (lane 1). The gap1 complemented strain restored the wild-type phenotype (Fig. 6A, lane 5); expression of Gap1-GFP was observed as a band slightly above 75 kDa upon probing with the Gap1-specific antibody. The negative-control vector had no effect on the decreased amount of Gap2 (Fig. 6A, lane 8). These results suggest that Gap1 expression increases the amount of Gap2. To confirm this, we compared the expression of Gap2 in a strain overexpressing Gap2 alone to that in a strain overexpressing both Gap1 and Gap2 (Fig. 6B). Gap2 expression was greatly increased when both Gap1 and Gap2 (Fig. 6B, lanes 2, 4, and 6) were overexpressed over that observed when Gap2 alone was overexpressed (lanes 1, 3, and 5); expression of Gap2-HH was observed as a band of about 65 kDa upon probing with the Gap2-specific antibody. This phenotype was observed for the wild-type strain, as well as for the gap1 and gap2 mutants. Reverse transcription-PCR (RT-PCR) analysis of gap2 transcription demonstrated no difference between the wild type and the gap1 mutant, indicating that the effect of Gap1 on Gap2 occurs at the posttranscriptional level (see Fig. S1A in the supplemental material). Together, these data demonstrate that the amount of Gap2 is modulated by Gap1.
Fig 6.
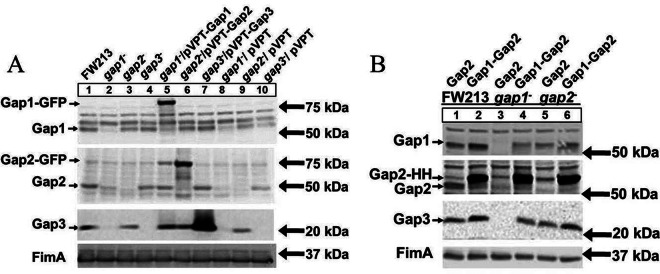
The amount of Gap2 is increased with overexpression of Gap1. Western blot analysis for Gap1, Gap2, and Gap3 was performed on S. parasanguinis cell lysates. (A) The strains used include wild-type FW213, insertional mutants of gap1, gap2, and gap3, complemented strains of the gap1, gap2, and gap3 mutants with the full gene in pVPT, and control strains with the empty vector. In the gap1 and gap2 complemented strains, Gap1 and Gap2 are tagged with GFP. (B) The strains used include wild-type FW213, the gap1 mutant, and the gap2 mutant, all overexpressing either Gap2 alone or both Gap1 and Gap2 in the pIB184-hsv-his vector, where Gap2 is tagged with Hsv-His (abbreviated as HH) in all strains. Polyclonal antibodies against Gap1, Gap2, Gap3, and FimA (a loading control) were used.
Gap2 expression results in increased Gap3 levels.
To determine the association between Gap2 and Gap3, we examined the effect of Gap2 deficiency on Gap3 (Fig. 6A). In the absence of Gap2 (Fig. 6A, lane 3), the amount of Gap3 was decreased from that in the wild type. Further, in the gap2 complemented strain (Fig. 6A, lane 6), the amount of Gap3 was restored to the wild-type level; expression of Gap2-GFP was observed as a stronger band compared to a nonspecific band present at 75 kDa upon probing with the anti-Gap2 antibody. The negative-control vector had no effect on the decreased amount of Gap3 (Fig. 6A, lane 9). However, the gap3 mutant had no effect on the amount of Gap2 (Fig. 6A, lane 4). This result suggested that Gap2 expression increases the amount of Gap3. To confirm this, we overexpressed Gap2 and determined its impact on Gap3 (Fig. 7A). Overexpression of Gap2 in the wild-type strain (Fig. 7A, lane 4) did indeed increase the amount of Gap3. In the gap2 mutant (Fig. 7A, lane 6) background, overexpression of Gap2 did not quite restore the amount of Gap3 to the wild-type level. However, this could be due to the smaller amount of Gap2 in the mutant strain than in the wild-type strain. RT-PCR analysis of gap3 transcription demonstrates no difference between the wild type and the gap2 mutant, indicating that the effect of Gap2 on Gap3 occurs at the posttranscriptional level (see Fig. S1A in the supplemental material). These data demonstrate that Gap2 modulates the amount of Gap3.
Fig 7.

Overexpression of Gap2 increases the amount of Gap3, and the addition of Gap1 to overexpressed Gap2 results in an even greater amount of Gap3. Western blot analysis for Gap1, Gap2, and Gap3 was performed on S. parasanguinis cell lysates. (A) The strains used include wild-type FW213, the gap1 mutant, and the gap2 mutant, as well as FW213, the gap1 mutant, and the gap2 mutant overexpressing Gap2 in the pIB184-hsv-his vector (HH stands for the Hsv-His tag). (B) The strains used include wild-type FW213, the gap1 mutant, and the gap2 mutant overexpressing either Gap3 alone, both Gap2 and Gap3, or Gap1, Gap2, and Gap3 in the pIB184-gfp vector, where Gap3 is tagged with GFP in all strains. Polyclonal antibodies against Gap1, Gap2, Gap3, and FimA (a loading control) were used.
Gap2 modulates the amount of Gap3 independently of Gap1.
Gap2 deficiency resulted in a diminished amount of Gap3, and overexpression of Gap2 led to a greater amount of Gap3. However, from these data, we cannot determine whether Gap2 functions independently of Gap1; in the absence of Gap1, native Gap3 was no longer detected, even when Gap2 was overexpressed (Fig. 7A, lanes 2 and 5). To determine if Gap2 can affect Gap3 independently of Gap1, strains were created that overexpressed Gap3 alone, Gap2 and Gap3, or Gap1, Gap2, and Gap3 in the wild-type strain and in gap1 and gap2 mutants (Fig. 7B); expression of Gap3-GFP was observed as a band slightly below 50 kDa upon probing with the anti-Gap3 antibody. Again, when Gap2 was overexpressed, both native and overexpressed Gap3 levels were increased (Fig. 7B, lane 2) over those in the strain overexpressing Gap3 alone (lane 1). Moreover, the amount of Gap3 was increased even further when both Gap1 and Gap2 were overexpressed along with Gap3 (Fig. 7B, lane 3). This phenomenon was not limited to the wild type: it also occurred in the gap1 (Fig. 7B, lanes 4 to 6) and gap2 (lanes 7 to 9) mutant strains, although the overall levels were lower than those in the wild type. RT-PCR analysis of gap3 transcription demonstrated no difference between overexpressing strains, indicating that the effects of Gap1 and Gap2 on Gap3 occur at the posttranscriptional level (see Fig. S1B in the supplemental material). These data demonstrate that increasing Gap2 expression can increase the amount of overexpressed Gap3 in the absence of Gap1, suggesting that Gap2 augments the function of Gap1 in stabilizing Gap3.
Gap homologs from S. agalactiae displayed the same conserved functions as Gap proteins.
Gap1, Gap2, and Gap3 are highly conserved in SRRP-containing Gram-positive bacteria. We have shown previously that the Gap1 homolog from S. agalactiae stabilizes the Gap3 homolog, much as Gap1 acts as a chaperone for Gap3 (38). To determine if the relationships between Gap2 and Gap1 and between Gap2 and Gap3 are conserved, we expressed Gap homologs from S. agalactiae (Asp1, Asp2, and Asp3) in S. parasanguinis (Fig. 8). In wild-type S. parasanguinis, Asp2 was detected when both Asp1 and Asp2 were expressed (Fig. 8A, lane 2) but was undetectable when expressed alone (lane 1). This result suggests that the amount of Asp2 is increased in the presence of Asp1, a finding much like that for the Gap proteins in S. parasanguinis (Fig. 6B). This phenomenon was also observed in the absence of Gap1 (Fig. 8A, lanes 3 and 4), further demonstrating that Asp1 can increase the amount of Asp2. To determine if the function of Gap2 is conserved, we expressed Gap homologs (Asp1, Asp2, and Asp3) from S. agalactiae in S. parasanguinis strains lacking Gap2 (gap2 mutant). In these strains, Asp3 was expressed either by itself, with Asp2, or with Asp1 and Asp2 (Fig. 8B). When Asp2 was expressed along with Asp3 (Fig. 8B, lane 2), the amount of Asp3 increased over that observed when Asp3 was expressed alone (lane 1); when Asp1 was expressed with Asp2 and Asp3 (lane 3), the amount of Asp3 was even greater. Because this trend is similar to that observed for the S. parasanguinis homologs (Fig. 7B), this result indicates that Asp2 can function similarly to Gap2. Together, these data suggest that the relationship among the Gap proteins is conserved.
Fig 8.
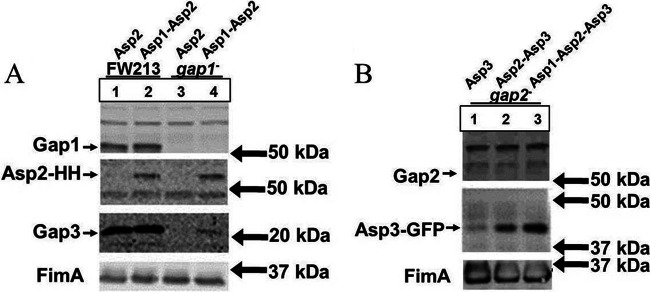
Gap homologs from S. agalactiae display the same conserved functions as Gap proteins. Western blot analysis for Gap1, Gap2, Gap3, and the Gap homologs was performed on S. parasanguinis cell lysates. (A) To check conservation of function, Asp1 and Asp2, the Gap homologs from S. agalactiae J48, were transformed into wild-type S. parasanguinis and the gap1 mutant. Strains used included wild-type S. parasanguinis and the gap1 mutant overexpressing either Asp2 alone or both Asp1 and Asp2 in the pIB184-hsv-his vector, where Asp2 is tagged with Hsv-His (abbreviated as HH) in all strains. (B) Gap homologs Asp1, Asp2, and Asp3 were transformed into the S. parasanguinis gap2 mutant. Strains used included the gap2 mutant overexpressing either Asp3 alone, both Asp2 and Asp3, or Asp1, Asp2, and Asp3 in the pIB184-gfp vector, where Asp3 is tagged with GFP in all strains. Polyclonal antibodies against Gap1, Gap2, and Gap3 were used. Monoclonal antibodies against Hsv (A) and GFP (B) were used to detect Asp2 and Asp3, respectively. An antibody against FimA was used as a loading control.
Gap2 prevents Gap3 degradation by ClpP protease.
Proteases are often involved in the degradation of misfolded proteins. Previously, the protease ClpP was shown to be responsible for the degradation of Gap3 in the absence of Gap1, a specific chaperone of Gap3 (38). Here, we wanted to determine if Gap2 protected Gap3 in a similar fashion. We constructed a clpP mutant and a gap2 clpP double mutant in order to examine the ability of Gap2 to shield Gap3 from degradation by ClpP (Fig. 9). No difference in the amount of Gap3 was observed between the wild type (Fig. 9, lane 1) and the clpP mutant (lane 2). In the absence of both ClpP and Gap2 (Fig. 9, lane 4), the amount of Gap3 was increased (nearly restored to the wild-type level) over that in the gap2 single mutant (lane 3). This result suggests that Gap2, similarly to Gap1, protects Gap3 from degradation by ClpP.
Fig 9.
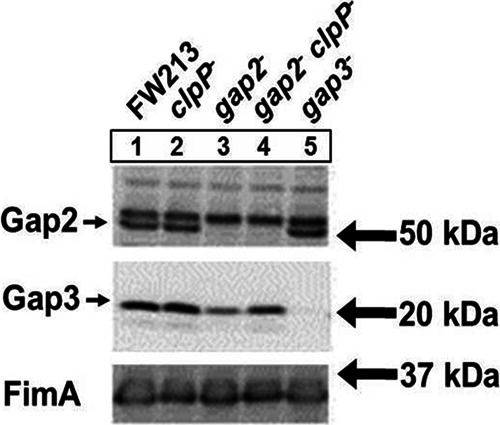
A ClpP deficiency in the gap2 mutant restores the amount of Gap3 nearly to the wild-type level. Western blot analysis for Gap2 and Gap3 was performed on S. parasanguinis cell lysates. The strains used include wild-type FW213, the clpP mutant, the gap2 mutant, the gap2 clpP double mutant, and the gap3 mutant. Polyclonal antibodies against Gap2, Gap3, and FimA (a loading control) were used.
DISCUSSION
Biogenesis of SRRPs is mediated by glycosylation and accessory secretory loci, which are highly conserved in many streptococci and staphylococci (10). In S. parasanguinis, an 11-gene cluster including glycosyltransferase genes and genes involved in protein secretion has been identified for Fap1 biosynthesis. Accessory secretion components—containing SecA2, SecY2, and glycosylation-associated proteins Gap1, Gap2, and Gap3 (10, 28, 29)—are implicated in Fap1 secretion and maturation. The exact roles of Gap1, Gap2, and Gap3 in Fap1 biogenesis remain unknown. We have shown previously that Gap1 and Gap3 are required for the production of mature Fap1, the formation of fimbriae, and adhesion to SHA (32, 34). In this study, we have determined the function of Gap2. Like Gap1 and Gap3, Gap2 was necessary for mature Fap1 biogenesis, with direct effects on the production of fimbriae and adhesion to an in vitro tooth surface model (Fig. 1 to 3). Because all three of the gap mutants had similar phenotypes, it is likely that they interact and work in concert to complete Fap1 biogenesis. Indeed, we show here that Gap1, Gap2, and Gap3 interact to form a complex (Fig. 4). The formation of a protein complex by Gap homologs has been demonstrated in S. gordonii as well (47); however, the details of the interactions were not characterized. Through ultracentrifugation, we determined that Gap2 could interact with an already formed Gap1/3 complex in a reversible manner. While Gap1 and Gap3 bind tightly to each other, Gap2 has a lower binding affinity for the Gap1/3 complex, suggesting that Gap2 may have regulatory activity toward the Gap1/3 complex (Fig. 5).
Based on the data obtained from the current study (summarized in Fig. 10), we can expand our previous model of Fap1 biogenesis. In this model, Gap1 binds to Gap3 (38) (Fig. 5A). This is then followed by the binding of Gap2, which can further stabilize Gap3 and is itself stabilized by Gap1 (Fig. 5B, 6, and 7). Such binding and stabilization were also observed for Gap homologs from S. agalactiae (Fig. 8), suggesting that this new function of Gap2 is conserved among SRRP-containing Gram-positive bacteria. Further, the current study indicates that Gap2 protects Gap3 from degradation by ClpP (Fig. 9). Similarly, we have shown previously that the protease ClpP is responsible for the degradation of Gap3 in the absence of Gap1, which acts as a specific chaperone of Gap3 (38). How ClpP gains access to the Gap3 protein remains to be determined.
Fig 10.
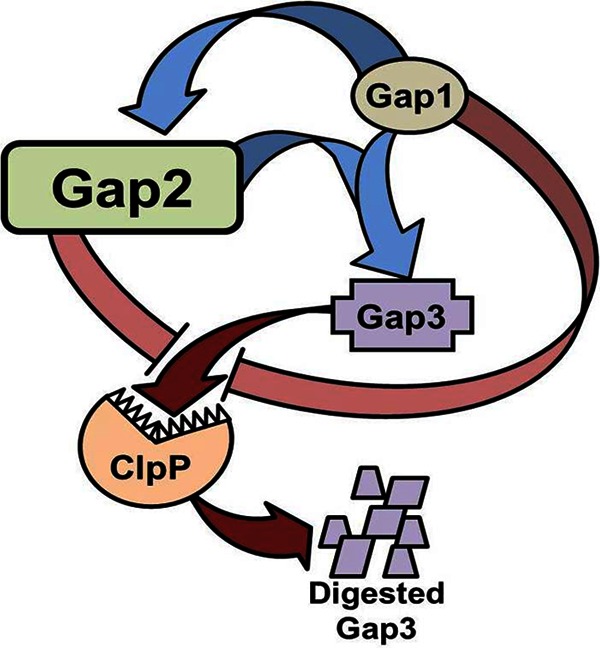
Model representation of Gap interactions. Gap2 is stabilized by Gap1 and augments the ability of Gap1 to stabilize Gap3 (indicated by blue arrows). Gap2, like Gap1, inhibits (pink bands) the degradation of Gap3 by ClpP (red arrows).
Since Gap2 works in concert with Gap1 to stabilize Gap3—the putative key scaffolding protein required for the formation of the Fap1 biosynthetic protein complex—we believe the function of Gap2 is to ensure Gap3 activity, which promotes Fap1 biogenesis. A similar proposition has been made for S. gordonii, in which Asp2 interacts with Asp3, which interacts Asp1 and SecA2, for optimal export of GspB (47). Gap2 can interact with the Gap1/3 complex, which then interacts with SecA2 and SecY2 to aid in Fap1 secretion (31). However, the precise biochemical function of this Gap complex in the conversion of an immature form of Fap1 to the mature form remains to be elucidated. Recent work with S. gordonii indicates that Asp2 is required for the export of GspB as well as for the conversion to the final glycoform of GspB, since Asp2 mutants resulted in altered GspB glycoforms with increased GlcNAc contents (48). Our previous study also suggested that the Gap1 deficiency altered the glycosyl composition of Fap1 (34).
Although these data provide insights into the function of the accessory secretion components, the details of the biochemical activity of the complex still remain unresolved. It is possible that by binding to the Gap1/3 complex, Gap2 is brought within a distance appropriate for monitoring the glycosylation status of Fap1 so as to ensure the export of correctly folded Fap1—possibly suggesting a role for Gap2 as a glycoside hydrolase, an important activity in the quality control of glycoproteins in eukaryotes (49, 50). This activity is often associated with the removal of sugar residues and typically functions through the Ser-Asp-His catalytic triads identified in the Gap2 homolog (48). Indeed, analysis of the Gap2 sequence with the Phyre fold prediction program predicted that Gap2 is a hydrolase (51). In S. gordonii, Asp2 alone does not exhibit detectable enzymatic activity against a panel of hydrolase substrates, suggesting that the catalytic activity requires additional cofactors (48). Alternatively, Gap2 may also bind to Fap1, bringing Gap3 into proximity with Fap1, thereby modulating Fap1 maturation. Indeed, in S. gordonii, Asp2, along with Asp3, is capable of binding the unglycosylated serine-rich repeat domains of GspB, and these interactions are required for optimal GspB export (52). Along the lines of this alternative, Gap2 may possess some sort of regulatory function, which may then become a means of controlling Fap1 fimbrial assembly and fine tuning bacterial adhesion levels.
In this study, we identify the necessity of Gap2 for mature Fap1 biogenesis, production of fimbriae, and adhesion to the in vitro tooth surface model, and we demonstrate that Gap2 forms a complex with Gap1/3 and is required for the production of wild-type levels of Gap3. However, whether and how Gap2 acts as a regulatory protein for Fap1 biogenesis remains to be determined.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH grants R01 DE017954, T32 DE017607, and F31 DE022995 from the National Institutes of Dental and Craniofacial Research and by the National Natural Science Foundation of China (30970060).
Footnotes
Published ahead of print 8 March 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02255-12.
REFERENCES
- 1. Wu H, Mintz KP, Ladha M, Fives-Taylor PM. 1998. Isolation and characterization of Fap1, a fimbriae-associated adhesin of Streptococcus parasanguis FW213. Mol. Microbiol. 28:487–500 [DOI] [PubMed] [Google Scholar]
- 2. Palmer RJ, Jr, Gordon SM, Cisar JO, Kolenbrander PE. 2003. Coaggregation-mediated interactions of streptococci and actinomyces detected in initial human dental plaque. J. Bacteriol. 185:3400–3409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Morris EJ, McBride BC. 1984. Adherence of Streptococcus sanguis to saliva-coated hydroxyapatite: evidence for two binding sites. Infect. Immun. 43:656–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jenkinson HF, Lamont RJ. 1997. Streptococcal adhesion and colonization. Crit. Rev. Oral Biol. Med. 8:175–200 [DOI] [PubMed] [Google Scholar]
- 5. Whittaker CJ, Klier CM, Kolenbrander PE. 1996. Mechanisms of adhesion by oral bacteria. Annu. Rev. Microbiol. 50:513–552 [DOI] [PubMed] [Google Scholar]
- 6. Lamont RJ, Hersey SG, Rosan B. 1992. Characterization of the adherence of Porphyromonas gingivalis to oral streptococci. Oral Microbiol. Immunol. 7:193–197 [DOI] [PubMed] [Google Scholar]
- 7. Yao ES, Lamont RJ, Leu SP, Weinberg A. 1996. Interbacterial binding among strains of pathogenic and commensal oral bacterial species. Oral Microbiol. Immunol. 11:35–41 [DOI] [PubMed] [Google Scholar]
- 8. Slots J, Gibbons RJ. 1978. Attachment of Bacteroides melaninogenicus subsp. asaccharolyticus to oral surfaces and its possible role in colonization of the mouth and of periodontal pockets. Infect. Immun. 19:254–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fives-Taylor PM, Thompson DW. 1985. Surface properties of Streptococcus sanguis FW213 mutants nonadherent to saliva-coated hydroxyapatite. Infect. Immun. 47:752–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhou M, Wu H. 2009. Glycosylation and biogenesis of a family of serine-rich bacterial adhesins. Microbiology 155:317–327 [DOI] [PubMed] [Google Scholar]
- 11. Stephenson AE, Wu H, Novak J, Tomana M, Mintz K, Fives-Taylor P. 2002. The Fap1 fimbrial adhesin is a glycoprotein: antibodies specific for the glycan moiety block the adhesion of Streptococcus parasanguis in an in vitro tooth model. Mol. Microbiol. 43:147–157 [DOI] [PubMed] [Google Scholar]
- 12. Froeliger EH, Fives-Taylor P. 2001. Streptococcus parasanguis fimbria-associated adhesin Fap1 is required for biofilm formation. Infect. Immun. 69:2512–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wu H, Zeng M, Fives-Taylor P. 2007. The glycan moieties and the N-terminal polypeptide backbone of a fimbria-associated adhesin, Fap1, play distinct roles in the biofilm development of Streptococcus parasanguinis. Infect. Immun. 75:2181–2188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wu H, Fives-Taylor PM. 1999. Identification of dipeptide repeats and a cell wall sorting signal in the fimbriae-associated adhesin, Fap1, of Streptococcus parasanguis. Mol. Microbiol. 34:1070–1081 [DOI] [PubMed] [Google Scholar]
- 15. Zhou M, Peng Z, Fives-Taylor P, Wu H. 2008. A conserved C-terminal 13-amino-acid motif of Gap1 is required for Gap1 function and necessary for the biogenesis of a serine-rich glycoprotein of Streptococcus parasanguinis. Infect. Immun. 76:5624–5631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Samen U, Eikmanns BJ, Reinscheid DJ, Borges F. 2007. The surface protein Srr-1 of Streptococcus agalactiae binds human keratin 4 and promotes adherence to epithelial HEp-2 cells. Infect. Immun. 75:5405–5414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Seifert KN, Adderson EE, Whiting AA, Bohnsack JF, Crowley PJ, Brady LJ. 2006. A unique serine-rich repeat protein (Srr-2) and novel surface antigen (epsilon) associated with a virulent lineage of serotype III Streptococcus agalactiae. Microbiology 152:1029–1040 [DOI] [PubMed] [Google Scholar]
- 18. Shivshankar P, Sanchez C, Rose LF, Orihuela CJ. 2009. The Streptococcus pneumoniae adhesin PsrP binds to keratin 10 on lung cells. Mol. Microbiol. 73:663–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Siboo IR, Chambers HF, Sullam PM. 2005. Role of SraP, a serine-rich surface protein of Staphylococcus aureus, in binding to human platelets. Infect. Immun. 73:2273–2280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Takamatsu D, Bensing BA, Cheng H, Jarvis GA, Siboo IR, Lopez JA, Griffiss JM, Sullam PM. 2005. Binding of the Streptococcus gordonii surface glycoproteins GspB and Hsa to specific carbohydrate structures on platelet membrane glycoprotein Ibα. Mol. Microbiol. 58:380–392 [DOI] [PubMed] [Google Scholar]
- 21. Takamatsu D, Bensing BA, Sullam PM. 2004. Four proteins encoded in the gspB-secY2A2 operon of Streptococcus gordonii mediate the intracellular glycosylation of the platelet-binding protein GspB. J. Bacteriol. 186:7100–7111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bensing BA, Sullam PM. 2002. An accessory sec locus of Streptococcus gordonii is required for export of the surface protein GspB and for normal levels of binding to human platelets. Mol. Microbiol. 44:1081–1094 [DOI] [PubMed] [Google Scholar]
- 23. Plummer C, Wu H, Kerrigan SW, Meade G, Cox D, Ian Douglas CW. 2005. A serine-rich glycoprotein of Streptococcus sanguis mediates adhesion to platelets via GPIb. Br. J. Haematol. 129:101–109 [DOI] [PubMed] [Google Scholar]
- 24. Handley PS, Correia FF, Russell K, Rosan B, DiRienzo JM. 2005. Association of a novel high molecular weight, serine-rich protein (SrpA) with fibril-mediated adhesion of the oral biofilm bacterium Streptococcus cristatus. Oral Microbiol. Immunol. 20:131–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Levesque C, Vadeboncoeur C, Chandad F, Frenette M. 2001. Streptococcus salivarius fimbriae are composed of a glycoprotein containing a repeated motif assembled into a filamentous nondissociable structure. J. Bacteriol. 183:2724–2732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhu F, Erlandsen H, Ding L, Li J, Huang Y, Zhou M, Liang X, Ma J, Wu H. 2011. Structural and functional analysis of a new subfamily of glycosyltransferases required for glycosylation of serine-rich streptococcal adhesins. J. Biol. Chem. 286:27048–27057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhou M, Zhu F, Dong S, Pritchard DG, Wu H. 2010. A novel glucosyltransferase is required for glycosylation of a serine-rich adhesin and biofilm formation by Streptococcus parasanguinis. J. Biol. Chem. 285:12140–12148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wu H, Bu S, Newell P, Chen Q, Fives-Taylor P. 2007. Two gene determinants are differentially involved in the biogenesis of Fap1 precursors in Streptococcus parasanguis. J. Bacteriol. 189:1390–1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen Q, Wu H, Fives-Taylor PM. 2004. Investigating the role of secA2 in secretion and glycosylation of a fimbrial adhesin in Streptococcus parasanguis FW213. Mol. Microbiol. 53:843–856 [DOI] [PubMed] [Google Scholar]
- 30. Bu S, Li Y, Zhou M, Azadin P, Zeng M, Fives-Taylor P, Wu H. 2008. Interaction between two putative glycosyltransferases is required for glycosylation of a serine-rich streptococcal adhesin. J. Bacteriol. 190:1256–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhou M, Zhang H, Zhu F, Wu H. 2011. Canonical SecA associates with an accessory secretory protein complex involved in biogenesis of a streptococcal serine-rich repeat glycoprotein. J. Bacteriol. 193:6560–6566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Peng Z, Wu H, Ruiz T, Chen Q, Zhou M, Sun B, Fives-Taylor P. 2008. Role of gap3 in Fap1 glycosylation, stability, in vitro adhesion, and fimbrial and biofilm formation of Streptococcus parasanguinis. Oral Microbiol. Immunol. 23:70–78 [DOI] [PubMed] [Google Scholar]
- 33. Peng Z, Fives-Taylor P, Ruiz T, Zhou M, Sun B, Chen Q, Wu H. 2008. Identification of critical residues in Gap3 of Streptococcus parasanguinis involved in Fap1 glycosylation, fimbrial formation and in vitro adhesion. BMC Microbiol. 8:52 doi:10.1186/1471-2180-8-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li Y, Chen Y, Huang X, Zhou M, Wu R, Dong S, Pritchard DG, Fives-Taylor P, Wu H. 2008. A conserved domain of previously unknown function in Gap1 mediates protein-protein interaction and is required for biogenesis of a serine-rich streptococcal adhesin. Mol. Microbiol. 70:1094–1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 36. Fenno JC, Shaikh A, Fives-Taylor P. 1993. Characterization of allelic replacement in Streptococcus parasanguis: transformation and homologous recombination in a ‘nontransformable’ streptococcus. Gene 130:81–90 [DOI] [PubMed] [Google Scholar]
- 37. Kremer BH, van der Kraan M, Crowley PJ, Hamilton IR, Brady LJ, Bleiweis AS. 2001. Characterization of the sat operon in Streptococcus mutans: evidence for a role of Ffh in acid tolerance. J. Bacteriol. 183:2543–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhou M, Zhu F, Li Y, Zhang H, Wu H. 2012. Gap1 functions as a molecular chaperone to stabilize its interactive partner Gap3 during biogenesis of serine-rich repeat bacterial adhesin. Mol. Microbiol. 83:866–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen YY, Shieh HR, Lin CT, Liang SY. 2011. Properties and construction of plasmid pFW213, a shuttle vector with the oral Streptococcus origin of replication. Appl. Environ. Microbiol. 77:3967–3974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhou M, Fives-Taylor P, Wu H. 2008. The utility of affinity-tags for detection of a streptococcal protein from a variety of streptococcal species. J. Microbiol. Methods 72:249–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Biswas I, Jha JK, Fromm N. 2008. Shuttle expression plasmids for genetic studies in Streptococcus mutans. Microbiology 154:2275–2282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fachon-Kalweit S, Elder BL, Fives-Taylor P. 1985. Antibodies that bind to fimbriae block adhesion of Streptococcus sanguis to saliva-coated hydroxyapatite. Infect. Immun. 48:617–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ruiz T, Lenox C, Radermacher M, Mintz KP. 2006. Novel surface structures are associated with the adhesion of Actinobacillus actinomycetemcomitans to collagen. Infect. Immun. 74:6163–6170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A. 1996. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J. Struct. Biol. 116:190–199 [DOI] [PubMed] [Google Scholar]
- 45. Kaelin WG, Jr, Pallas DC, DeCaprio JA, Kaye FJ, Livingston DM. 1991. Identification of cellular proteins that can interact specifically with the T/E1A-binding region of the retinoblastoma gene product. Cell 64:521–532 [DOI] [PubMed] [Google Scholar]
- 46. Liang X, Chen YY, Ruiz T, Wu H. 2011. New cell surface protein involved in biofilm formation by Streptococcus parasanguinis. Infect. Immun. 79:3239–3248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Seepersaud R, Bensing BA, Yen YT, Sullam PM. 2010. Asp3 mediates multiple protein-protein interactions within the accessory Sec system of Streptococcus gordonii. Mol. Microbiol. 78:490–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Seepersaud R, Bensing BA, Yen YT, Sullam PM. 2012. The accessory Sec protein Asp2 modulates GlcNAc deposition onto the serine rich-repeat glycoprotein GspB. J. Bacteriol. 194:5564–5575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hebert DN, Foellmer B, Helenius A. 1995. Glucose trimming and reglucosylation determine glycoprotein association with calnexin in the endoplasmic reticulum. Cell 81:425–433 [DOI] [PubMed] [Google Scholar]
- 50. Hurtley SM, Helenius A. 1989. Protein oligomerization in the endoplasmic reticulum. Annu. Rev. Cell Biol. 5:277–307 [DOI] [PubMed] [Google Scholar]
- 51. Bennett-Lovsey RM, Herbert AD, Sternberg MJ, Kelley LA. 2008. Exploring the extremes of sequence/structure space with ensemble fold recognition in the program Phyre. Proteins 70:611–625 [DOI] [PubMed] [Google Scholar]
- 52. Yen YT, Seepersaud R, Bensing BA, Sullam PM. 2011. Asp2 and Asp3 interact directly with GspB, the export substrate of the Streptococcus gordonii accessory Sec system. J. Bacteriol. 193:3165–3174 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.