

Abstract
In pursuit of a more detailed understanding of the structural requirements for the key side chain cannabinoid pharmacophore we have extended our SAR to cover a variety of conformationally modified side chains within the 9-keto and 9-hydroxyl tricyclic structures. Of the compounds described here, those with a seven-atom long side chain substituted with a cyclopentyl ring at C1′ position have very high affinities for both CB1 and CB2 (0.97 nM<Ki<5.25 nM), with no preference for either of the two receptors. However, presence of the smaller cyclobutyl group at C1′ position leads to an optimal affinity and selectivity interaction with CB1. Thus, two of the C1′-cyclobutyl analogs, namely, (6aR, 10aR)-3-(1-hexyl-cyclobut-1-yl)-6,6a,7,8,10,10a-hexahydro-1-hydroxy-6,6-dimethyl-9H-dibenzo[b,d]pyran-9-one and (6aR, 9R, 10aR)-3-(1-hexyl-cyclobut-1-yl)-6a,7,8,9,10,10a-hexahydro-6,6-dimethyl-6H-dibenzo[b,d]pyran-1,9 diol (7e-β, AM2389), exhibited remarkably high affinities (0.84 nM and 0.16 nM respectively) and significant selectivities (16- and 26-fold respectively) for CB1. Compound 7e-β was found to exhibit exceptionally high in vitro and in vivo potency with a relatively long duration of action.
Introduction
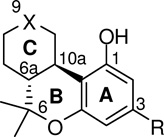
The recognition of two distinct Gi/o-protein-coupled cannabinoid receptors (CB1α and CB2)1–11 and the discovery of two endogenous ligands, arachidonoylethanolamine (anandamide)12 and 2-arachidonoylglycerol,13,14 has led to efforts to define the structural requirements for receptor specificity. A review of structure-activity relationship (SAR) studies15–19 recognizes two pharmacophores within the classical (−)-Δ8-tetrahydrocannabinol ((−)-Δ8-THC, 1a, Figure 1) template: a phenolic hydroxyl (PH), and a lipophilic side chain (SC). Of these, the aliphatic side chain was shown to play a pivotal role in determining cannabinergic potency. Optimal activity is obtained with a seven carbon length substituted with 1′,1′-dimethyl groups (1b) as was first demonstrated by Adams.20 It has also been shown that in hexahydrocannabinols (HHC, Figure 1) where the C-ring is fully saturated, presence of a keto21 or a hydroxyl22 group at the C9 position (e.g., nabilone, 1d) can have significant effects on the compound’s ability to interact with CB receptors.15–17 Based on the relative configuration at the C9 position of the 9-hydroxyhexahydrocannabinols (1e, 1f) we can obtain 9β- or 9α-OH isomers. Interestingly, a study involving the axial (9α) and the equatorial (9β) alcohols 1f and 1e respectively, indicates that only the 9β-hydroxyl isomer (1e) was an analgesic in mice.22
Figure 1.

In earlier work we have focused on novel (−)-Δ8-tetrahydrocannabinol analogs (1c) that bear cyclic moieties at the C1′-position of the side chain.23–28 This careful SAR led to (−)-Δ8-THC analogs with high affinities for both CB1 and CB2 cannabinoid receptors and provided a good starting point for the development of more subtype selective second generation ligands.
In the current study we chose to investigate the effect of modifying the C3 side chain in HHC’s with regard to affinity and selectivity for the CB1 and CB2 cannabinoid receptors. Toward this end a number of novel side chain hexahydrocannabinol analogs represented by the general structure 1g were synthesized. Our design retains the keto or hydroxyl groups at C9 and replaces the 1′,1′-gem-dimethyl group with the larger and sterically more confined cyclobutyl and cyclopentyl groups as well as with the less hydrophobic dithiolane 5-membered ring. As with earlier work on the THC template, the design of our HHC analogs has included a seven-atom-long side chain and methyl-, bromo- or cyano-substitution at the terminal carbon (analogs 6b, 6c, 6e, 15b, 16, 22, 7c-β, 7e-β, 23, and 24, Table 1). Additionally, we have explored the pharmacophoric limits of side chain length and the effects of conformational restriction at the C2′–C3′ bond by synthesizing the C1′-cyclopentyl analogs with six- and eight-atom-long side chains (compounds 15a and 15c, Table 1), as well as those incorporating the respective C2′–C3′-Z-heptenyl chains (compounds 6a and 6d, Table 1).
Table 1.
Affinities (Ki) of Hexahydrocannabinol Analogs for rCB1 and mCB2 Cannabinoid Receptors (95% Confidence Limits).
 | |||||
|---|---|---|---|---|---|
| compd | X | R | rCB1 (Ki, nM)a | mCB2 (Ki, nM)a | mCB2/rCB1 |
| 1h | 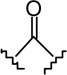 |
333 | 265 | 0.8 | |
| 1b | 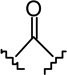 |
 |
2.2b | 1.8b | 0.8 |
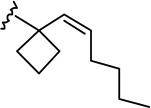
| 6a | 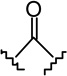 |
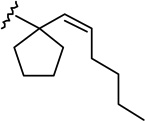 |
1.23 ± 0.22 | 5.25 ± 1.02 | 4.3 |
| 6b | 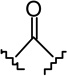 |
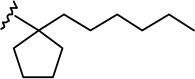 |
1.76 ± 0.41 | 0.97 ± 0.29 | 0.6 |
| 6c | 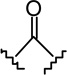 |
 |
6.57 ± 1.55 | 42.3 ± 11.0 | 6.4 |
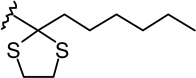
| 6d | 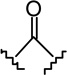 |
 |
1.13 ± 0.28 | 12.0 ± 3.5 | 10.6 |
| 6e | 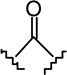 |
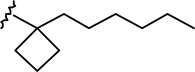 |
0.84 ± 0.18 | 13.7 ± 3.2 | 16.3 |
| 15a | 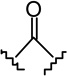 |
 |
13.1 ± 3.5 | 13.9 ± 3.8 | 1.1 |
| 15b | 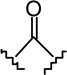 |
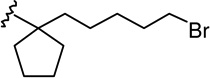 |
1.03 ± 0.21 | 2.59 ± 0.85 | 2.5 |
| 15c | 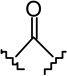 |
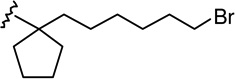 |
4.96 ± 1.24 | 1.60 ± 0.38 | 0.3 |
| 16 | 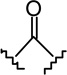 |
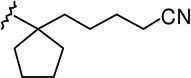 |
3.14 ± 0.51 | 2.78 ± 0.67 | 0.9 |
| 22 | 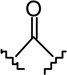 |
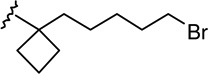 |
2.33 ± 0.55 | 7.56 ± 1.79 | 3.2 |
| 7c-β | 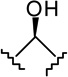 |
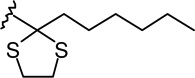 |
4.51 ± 0.72 | 13.9 ± 3.4 | 3.1 |
|
7e-β AM2389 |
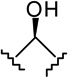 |
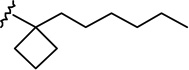 |
0.16 ± 0.05 | 4.21 ± 0.93 | 26.3 |
| 23 | 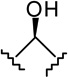 |
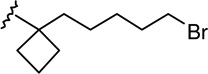 |
1.37 ± 0.35 | 2.76 ± 0.63 | 2 |
| 24 | 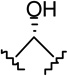 |
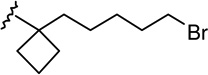 |
1.50 ± 0.33 | 1.67 ± 0.43 | 1.1 |
Affinities for CB1 and CB2 were determined using rat brain (CB1) or mouse spleen (CB2) membranes and [3H]CP-55,940 as the radioligand following previously described procedures.25,27,37 Data were analyzed using nonlinear regression analysis. Ki values were obtained from three independent experiments run in duplicate and are expressed as the mean of the three values.
Reported previously.15
All synthesized analogs were tested for their respective affinities for CB1 and CB2. Hexahydrocannabinols carrying seven-atom-long side chains substituted with cyclobutyl and cyclopentyl groups at the C1′-position exhibited the highest affinities for CB1 and CB2. More interestingly, within this series the cyclobutylhexenyl (6d) and the cyclobutylhexyl (6e and 7e-β) analogs (Table-1) were found to have remarkably high affinities and significant selectivities (11- to 26-fold) for CB1. The analog with the highest CB1 binding affinity and selectivity (7e-β, AM238929) was tested in rats for its ability to induce hypothermia and analgesia. The compound was found to exhibit exceptionally high in vivo potency with a relatively long duration of action. In addition, functional characterization found 7e-β to be a potent CB1 agonist with an EC50 = 1.5 ± 0.3 nM.
Chemistry
Resorcinols 4a–4e, (Scheme 1) bearing C1′-cyclopentyl, C1′-dithiolanyl, and C1′-cyclobutyl-ring substituents were synthesized in four to eight steps from 3,5-dimethoxybenzaldehyde (2) by following our previously reported procedures.23–27,30 We have recently described the efficient synthesis of the mixture of chiral terpene acetates 3 which was used in the synthesis of (−)-Δ9-tetrahydrocannabinol and (−)-Δ9-tetrahydrocannabivarin metabolites in their native 6aR, 10aR absolute configuration.31 This mixture served as the starting point for the synthesis of the bicyclic intermediates 5a–5e. Thus, coupling of resorcinol derivatives 4a–4e with 3 in the presence of p-toluenesulfonic acid (p-TSA), under our optimized reaction conditions31 led to norpinanones 5a–5e (24–58%). The structure of 5b was established using 1D NMR spectra as well as 13C DEPT spectrum, COSY, HSQC, HMBC, and NOESY correlations (available under supporting information). Dibenzo[b,d]pyran ring closure proceeded smoothly with catalytic trimethylsilyl triflate31 to give ketones 6a–6e in 51–67% yield. Sodium borohydride reduction of ketones 6c and 6e at −40°C gave the corresponding equatorial hydroxyl compounds 7c-β and 7e-β along with traces (3–4% by 1H NMR) of the respective axial isomers 7c-α and 7e-α. This was followed by flash column chromatography purification to give pure equatorial hydroxyl analogs 7c-β and 7e-β in 85–95% yield. The stereochemistry of the hydroxyl groups of 7c-β/7c-α and 7e-β/7e-α isomeric pairs was assigned on the basis of 1H NMR (500 MHz) spectral data. Thus, in compounds 7c-β and 7e-β the peak half-width for the C9 protons was found to be 21–22 Hz while in compounds 7c-α and 7e-α was 10.5 Hz (see experimental section). This correlates well with an axial C9 proton in the former two compounds and an equatorial C9 proton in the latter two.
Scheme 1.

Reagents and conditions: (a) 3, p-TSA, CHCl3, 0°C to room temperature, 3 days, 24–58%; (b) TMSOTf, CH2Cl2/CH3NO2 (3:1), 0°C to room temperature, 3 h, 51–67%. (c) NaBH4, MeOH, −40°C, 1.5 hours, 85–95%.
Synthesis of the C1′-cyclopentyl analogs 15a–15c and 16 was accomplished by a reaction sequence shown in Scheme 2. Commercially available phenoxyalkyl bromides 8a, 8b and 8c were reacted with triphenylphosphine in refluxing benzene32 to give the respective (phenoxyalkyl)triphenylphosphonium bromides 9a, 9b and 9c in very good yields (80–85%). Treatment of 9a–9c with potassium bis(trimethylsilyl)amide and coupling of the generated phosphoranes with 1-(3,5-dimethoxyphenyl)cyclopentanecarboxaldehyde24,25,27,30 (10) at 0°C produced intermediate alkenes 11a–11c (91–95%), bearing five, six and seven carbon atom side chains, respectively. Based on 1H NMR analysis, these Wittig olefination reactions afforded exclusively the Z olefins with J2′H-3′H = 11.3 Hz. Catalytic hydrogenation of 11a–11c led to the respective resorcinol dimethyl ethers 12a–12c in 94–96% yields. Exposure of 12a–12c to boron tribromide in methylene chloride cleaved all three ether groups and introduced the C5′, C6′ and C7′ bromo groups for 13a, 13b and 13C respectively.32 Coupling with terpene acetates 3 in the presence of p-TSA (50–54% yield) and exposure of the resulting norpinanones 14a–14c to trimethylsilyl triflate gave 9-keto-hexahydrocannabinols 15a–15c (73–75% yield). Treatment of bromide 15a with sodium cyanide in dimethyl sulfoxide produced the respective side chain cyano-substituted analog 16 in 64% yield.
Scheme 2.

Reagents and conditions: (a) Ph3P, C6H6, reflux, 2 days, 80–85%; (b) (Me3Si)2N−K+, THF, 0°C, 10 min, then 1-(3,5-dimethoxyphenyl)cyclopentanecarboxaldehyde (10), 20 min, 91–95%; (c) H2, Pd/C, AcOEt, room temperature, overnight, 94–96%; (d) BBr3, CH2Cl2, −78°C to room temperature, 12 hours, 89–93%; (e) 3, p-TSA, CHCl3, 0°C to room temperature, 3 days, 50–54 %; (f) TMSOTf, CH2Cl2/CH3NO2 (3:1), 0°C to room temperature, 3 h, 73–75%; (g) NaCN, DMSO, room temperature, 20 h, 64%.
The C1′-cyclobutyl analogs 22, 23 and 24 were synthesized in a similar fashion from 1-(3,5-dimethoxyphenyl)cyclobutanecarboxaldehyde (17, Scheme 3), which was in turn obtained from commercially available 2 in five steps by a methodology developed in our laboratories.24,25,27,30 Thus, combination of 17 and the ylide derived from (4-phenoxybutyl)triphenylphosphonium bromide (9b) and potassium bis(trimethylsilyl)amide, resulted in the exclusive formation of the Z isomer 18 in 91% yield (J2′H-3′H = 11.1 Hz). Catalytic hydrogenation of 18 led to resorcinol dimethyl ether 19 which was treated with boron tribromide to give the corresponding resorcinol 20 in quantitative yield. Condensation of 20 with terpene acetates 3 afforded norpinanone 21 (46% yield) which upon treatment with catalytic trimethylsilyl triflate gave the hexahydrocannabinol 22 in 70% isolated yield. Sodium borohydride reduction of ketone 22 in methanol (−40°C) gave a mixture of isomeric alcohols 23 and 24 in a 97:3 ratio respectively (by 1H NMR). Pure equatorial alcohol 23 was obtained by chromatographic purification in 85% yield. Reduction of 22 with potassium tri-sec-butyl borohydride33 (K-selectride) at −78°C was very selective and gave the axial alcohol 24 exclusively (70% yield). The n-pentyl-HHC 1H31 (Table 1) which carry the side chain of the natural cannabinoid (−)-Δ9-THC was synthesized similarly by coupling commercially available olivetol with diacetates 3.
Scheme 3.

Reagents and conditions: (a) PhO(CH2)4P+ PPh3 Br−, (Me3Si)2N−K+, THF, 0°C, 10 min, then addition of 17, 20 min, 91%; (b) H2, Pd/C, EtOH, room temperature, 50 psi, 10 h, quantitative; (c) BBr3, CH2Cl2, −78°C to room temperature, 12 hours, quantitative; (d) 3, p-TSA, CHCl3, 0°C to room temperature, 3 days, 46%; (e) TMSOTf, CH2Cl2/CH3NO2 (3:1), 0°C to room temperature, 3 h, 70%; (f) NaBH4, MeOH, −40°C, 1.5 h, 85%; (g) K-Selectride, THF, −78°C, 2 h, 70%.
Results and Discussion
Receptor Binding Studies
The abilities of 1H, 6a–6e, 15a–15c, 16, 22, 7c-β, 7e-β, 23 and 24 to displace radiolabeled CP-55,940 from purified rat forbrain synaptosomes and mouse spleen synaptosomes were determined, as described in the Experimental Section. Ki values calculated from the respective displacement curves are listed in Table 1 and reflect the affinities of these hexahydrocannabinol analogs for the rat CB1 (rCB1) and mouse CB2 (mCB2) receptors.
The compounds included in this study are 9-keto- and 9-hydroxyl-HHC analogs in which the six- to eight-atom long side chain pharmacophore incorporates cyclic substituents at the C1′ position. As can be observed in Table 1, the range of Ki values of the C1′-ring substituted analogs, included in this study, spans over two orders of magnitude. This indicates that the size and the nature of the C1′-ring substituent along with the length of the side chain can have a profound effect on the affinities of HHC analogs for both the CB1 and CB2 receptors.
A comparison of the binding data of n-pentyl HHC 1H and its C1′-dimethylheptyl analog 1b suggests that presence of the two C1′-methyl groups enhances the ligand’s affinity for both the CB1 and CB2 receptors. This interaction is optimized when the geminal dimethyl substitution is modified into a larger and sterically more confined five-membered carbocyclic ring as seen in the cyclopentane analog 6b. However, the affinity is reduced when the C1′-cyclopentyl group in 6b is substituted with the bulkier dithiolane ring in analog 6c. This analog now has a 4-fold lower affinity for CB1 and an even lower affinity (44-fold) for CB2. Interestingly, this reduction in affinity is more accentuated in CB2. Examination of the Ki values of the C1′-cyclopentyl analogs 6b and 6a indicates that while introduction of a C2′–C3′ Z-double bond can be tolerated at the CB1 receptor, it seems to somewhat reduce the side chain’s optimal interaction with CB2. Thus, analog 6a has a 5-fold lower affinity for CB2 when compared to 6b.
As seen in analog 16, replacement of the two terminal carbon atoms of the heptyl side chain (6b) with a cyano group is well tolerated in both the CB1 and CB2 receptors. Also, substitution of the terminal side chain methyl group in 6b with a bromine atom (analog 15b) maintains affinity for both receptors. For ω-bromo side chains (analogs 15a, 15b, 15c), extension of the 7-atom chain length to eight (15c) retains practically all affinity for CB1 and CB2. However, the shorter, six-atom long, side chain leads to an analog (15a) with a significant reduction in affinity for both the CB1 and CB2 (13-fold and 5-fold respectively).
In summary, within the 9-keto HHC’s the C1′-cyclopentyl substituted analogs appear to exhibit no significant preference for either of the two receptors. Conversely, reduction of the size of the C1′-ring substituent to a four membered cyclobutane ring is accompanied by a preference for CB1. This is seen in analog 22 in which the end carbon of the side chain carries a bromine atom, and more so in 6d where a C2′–C3′ Z-double bond is added. Significantly, the 11-fold CB1 selectivity for the heptenyl side chain analog 6d is further enhanced in its heptyl counterpart 6e where the CB1 selectivity is now 16-fold.
A comparison of the binding affinities of the C9 equatorial hydroxyl analogs 7c-β, 7e-β and 23 with their 9-keto counterparts 6c, 6e and 22 respectively, indicates that conversion of the 9-keto group to 9β-hydroxyl group enhances the ligand’s affinity for both the CB1 and CB2 receptors. The most successful analog from this modification is the C1′-cyclobutyl derivative 7e-β with a Ki of 0.16 nM for CB1. Importantly, 7e-β exhibits a 26-fold selectivity for CB1 over CB2. Finally, a comparison of the equatorial and the axial alcohols 23 and 24, respectively, shows that the relative stereochemistry of the hydroxyl group at C9 does not affect the ligand’s affinity and selectivity for CB1 and CB2. This is an apparent incongruence in previously reported in vivo data where the 9α-OH hexahydrocannabinol exhibitted reduced potency compared to its 9β-OH isomer.22
In conclusion, 9-keto-hexahydrocannabinol analogs bearing seven-atom long side chains substituted with a cyclopentyl ring at C1′ position have very high affinities for both CB1 and CB2 (0.97 nM<Ki<5.25 nM), with no preference for either of the two receptors. However, presence of the smaller cyclobutyl group at C1′ position of HHC’s leads to an optimal affinity and selectivity interaction with CB1. The above conclusion is strongly reinforced by our results with the C1′-cyclobutyl analogs 6e and 7e-β. These compounds with Ki values of 0.84 nM and 0.16 nM for CB1 respectively are cannabinergic probes with one of the highest CB1 binding affinities reported to date. In addition, compounds 6e and 7e-β shown a 16- and a 26-fold respectively selectivity for CB1 over CB2.
It is especially worthy of note that the side chain SAR trends observed in this study parallel those of (−)-Δ8-THC analogs25,27 suggesting that THC’s and HHC’s share key binding motifs and support the hypothesis for a subsite23–27 within CB1 and CB2 binding domain at the level of the benzylic side chain carbon in the tetrahydrocannabinol and hexahydrocannabinol series.
The rat,3 mouse,5,9 and human CB1 (hCB1) receptors4 have 97–99% sequence identity across species, and are not expected to exhibit variations in their Ki values. However, mouse CB210,11 (mCB2) exhibits only 82% sequence identity with the human clone2 (hCB2). This divergent nature of mCB2 and hCB2 receptors could possibly result in species-based differences in affinity.34,35 For this reason, representative HHC analogs were assayed using membranes from HEK293 cells expressing hCB2 and the results are listed in Table 2. We observe that the tested compounds exhibit similar binding affinities for both the mouse and the human CB2.
Table 2.
Affinities (Ki) of Hexahydrocannabinol Analogs for hCB2 Cannabinoid Receptors (95% Confidence Limits).
| compd | hCB2 (Ki, nM)a |
|---|---|
| 6a | 7.02 ± 1.62 |
| 6b | 3.34 ± 1.31 |
| 6c | 32.6 ± 7.1 |
| 6d | 15.1 ± 4.3 |
| 6e | 11.9 ± 2.4 |
| 15b | 1.32 ± 0.27 |
| 15c | 3.02 ± 0.73 |
| 22 | 5.88 ± 1.73 |
| 7e-β | 5.13 ± 1.27 |
| 23 | 1.62 ± 0.45 |
| 24 | 1.63 ± 0.37 |
Affinities for hCB2 were determined using membranes from HEK293 cells expressing human CB2 and [3H]CP-55,940 as the radioligand following previously described procedures.33 Data were analyzed using nonlinear regression analysis. Ki values were obtained from three independent experiments run in duplicate and are expressed as the mean of the three values.
Molecular Modeling of 7e-β
A conformational search of 7e-β in implicit water was carried out as described in the Experimental section and found the global energy minimum to be as shown in Figure 2. While the global minimum conformer is not necessarily identical to that found in the receptor binding site, it is likely that the hexyl moiety is approximately perpendicular to the tricyclic ring system. In this conformation the cyclobutane ring can engage in optimal interactions at the putative receptor-binding subsite.
Figure 2.

Accessible conformational space for the C1′-cyclobutylheptyl substituent of 7e-β using an energy window of 5 kcal mol−1. Heavy atoms are shown in line representation with the cyclobutane ring highlighted in cyan. The minimum energy conformer of 7e-β is shown in stick representation with the cyclobutylheptyl moiety highlighted in yellow.
Functional Characterization of 7e-β
The functional potency of AM2389 for the CB1 receptor was obtained by measuring the decrease in foskolin-stimulated cAMP, as described in the Experimental section, and shown to be a full agonist with EC50 = 1.5±0.3 nM.
In Vivo Behavioral Characterization of 7e-β
We have explored the in vivo potency of our more CB1 selective high affinity analog 7e-β in the hypothermia and analgesia tests (detailed procedures are given under the Experimental section). Body temperature was measured in isolated rats over a 6 hour period following drug injection. 7e-β decreased core body temperature in a dose-dependent manner, with a dose of 0.1 mg/kg reducing body temperature up to 4.2±0.5°C from an average baseline of 38.48±0.06°C. At this dose, the onset of drug effect occurred within 60–90 min after injection, although peak effects were not obtained until 300 min after injection. Antinociception was measured using a tail-flick procedure over a 6 hour period following drug injection. Prior to drug administration, the average baseline tail-flick latency was 2.1±0.1 sec. Compound 7e-β increased tail-flick latency over the same dose range as in the hypothermia measurements and with a similar time course (Figure 3). Doses of 0.01–0.3 mg/kg 7e-β had significant antinociceptive effects, with a mean (±95%CL) ED50 value of 0.026 mg/kg (0.020, 0.034). The onset of the antinociceptive effects occurred between 60–120 min after injection and these effects generally increased over the 6 hour test period (Figure 4). In comparison, the mean (±95%CL) ED50 value of morphine was 2.8 mg/kg (2.0, 3.8) with peak effects occurring at 30–120 min after injection and decreasing by 3 hours after injection.
Figure 3.

Effects of 7e-β (■) and morphine (○) on antinociception (top graph) and effects of 7e-β on body temperature (bottom graph). Symbols represent the group mean ± sem (n=5–6 rats). Abscissa: dose, in mg/kg; top ordinate: Percentage of the maximum possible antinociceptive effect, bottom ordinate; change in body temperature. Asterisks indicate effects that are significantly different from vehicle, **p<0.01, *** p<0.001.
Figure 4.

Effects of 7e-β at different times after injection on antinociception (top graph) and body temperature (bottom graph). Abscissa: time (in minutes) after injection; top ordinate: Percentage of the maximum possible antinociceptive effect, bottom ordinate; change in body temperature. Asterisks indicate effects that are significantly different from vehicle, *p<0.05, **p<0.01, *** p<0.001.
The antinociceptive effects of 7e-β were subsequently redetermined in a different group of animals; in these animals 0.1 mg/kg 7e-β alone produced 100% of the maximum possible effect, and this was reduced to 85.4±5.8% or 48.0±10.9% by a 30-min pretreatment with 1.0 or 3.0 mg/kg rimonabant (SR141716A), respectively.
Our in vivo experiments show that 7e-β has a slow onset and long duration of action and has potent activity in assays of hypothermia and antinociception. This compound produced full antinociceptive responses in a tailflick assay. These effects were suppressed by the CB1-selective antagonist rimonabant thus, supporting a CB1-based mechanism for the analgesic action of 7e-β. Although, similar antinociceptive effects were obtained with morphine, these were approximately 100-fold lower than 7e-β. Furthermore, the antinociceptive effects of 7e-β were longer lasting than were effective doses of morphine. The hypothermic effects of 7e-β were also profound, and peak hypothermic and antinociceptive effects of 7e-β occurred over the same range of doses and over similar time courses. However, small yet significant antinociceptive effects of 7e-β occurred at low doses that did not significantly lower body temperature.
Experimental section
Chemistry
All reagents and solvents were purchased from Aldrich Chemical Company, unless otherwise specified, and used without further purification. All anhydrous reactions were performed under a static argon or nitrogen atmosphere in flame-dried glassware using scrupulously dry solvents. Flash column chromatography employed silica gel 60 (230–400 mesh). All compounds were demonstrated to be homogeneous by analytical TLC on pre-coated silica gel TLC plates (Merck, 60 F245 on glass, layer thickness 250 µm), and chromatograms were visualized by phosphomolybdic acid staining. Melting points were determined on a micro-melting point apparatus and are uncorrected. IR spectra were recorded on a Perkin Elmer Spectrum One FT-IR spectrometer. NMR spectra were recorded in CDCl3, unless otherwise stated, on a Varian Mercury-300 (1H at 300 MHz, 13C at 75 MHz) or on a Bruker Ultra Shield 400 WB plus (1H at 400 MHz, 13C at 100 MHz) or on a Bruker DMX-500 (1H at 500 MHz, 13C at 125 MHz) or on a Bruker Ultra Shield 700 WB plus (1H at 700 MHz, 13C at 175 MHz) spectrometers and chemical shifts are reported in units of δ relative to internal TMS. Multiplicities are indicated as br (broadened), s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet) and coupling constants (J) are reported in hertz (Hz). Low and high-resolution mass spectra were performed in School of Chemical Sciences, University of Illinois at Urbana-Champaign. Mass spectral data are reported in the form of m/z (intensity relative to base = 100). Elemental analyses were obtained in Baron Consulting Co, Milford, CT, and were within ± 0.4% of the theoretical values (see supporting information). Purities of the tested compounds were determined by elemental analysis and were > 95%.
(4R)-4-{4-[1-(1,2-cis-Hexen-1-yl)-cyclopentyl]-2,6-dihydroxy-phenyl}-6,6-dimethyl-2-norpinanone (5a)
To a degassed solution of 4a27 (1.25 g, 4.81 mmol) and diacetates 331 (2.14 g, ca. 75% pure by 1H NMR, 8.99 mmol) in CHCl3 (48 mL) at 0°C, under an argon atmosphere was added p-toluenesulfonic acid monohydrate (1.28 g, 6.73 mmol). The mixture was warmed to room temperature and stirred for 3 days to ensure complete formation of the product. The reaction mixture was diluted with diethyl ether and washed sequentially with water, saturated aqueous NaHCO3, and brine. The organic phase was dried over MgSO4 and the solvent removed under reduced pressure. The residue was chromatographed on silica gel (43% diethyl ether in hexane) and fractions containing almost pure product (TLC) were combined and evaporated. Further purification by recrystallization from CHCl3 and hexane gave 5a as a white crystalline solid (1.01 g, 53% yield). m p 171–173°C; 1H NMR (500 MHz, CDCl3) δ 6.31 (s, 2H, ArH), 5.63 (dt, J = 11.3 Hz, J = 1.5 Hz, 1H, 2′-H), 5.27 (dt, J = 11.3 Hz, J = 7.5 Hz, 1H, 3′-H), 4.92 (br s, 2H, OH), 3.94 (t, J = 8.2 Hz, 1H, 4-H), 3.49 (dd, J = 18.8 Hz, J = 7.8 Hz, 1H, 3α-H), 2.60 (dd, J = 18.9 Hz, J = 8.7 Hz, 1H, 3β-H), 2.58 (t, J = 5.1 Hz, 1H, 1-H), 2.49 (m, 1H, 7α-H), 2.46 (d, J = 10.5 Hz, 1H, 7β-H), 2.28 (t, J = 5.2 Hz, 1H, 5-H), 1.95-1.85 (m, 4H of the cyclopentane ring), 1.76-1.65 (m, 6H, 4H of the cyclopentane ring, 4′-H), 1.36 (s, 3H, 6-Me), 1.11-1.07 (m, 4H, 5′-H, 6′-H), 0.99 (s, 3H, 6-Me), 0.75 (t, J = 6.7 Hz, 3H, 7′-H); 13C NMR (CDCl3) δ 216.0 (C-2), 153.5 (ArC-2, ArC-6), 148.1 (ArC-4), 137.2 (>C=C<), 131.0 (>C=C<), 112.3 (ArC-1), 106.4 (ArC-3, ArC-5), 56.8, 50.5, 45.8, 41.1, 39.7, 36.8, 30.3, 28.4, 27.2, 25.1, 23.3, 22.5, 21.2, 21.0, 12.8; mass spectrum m/z (relative intensity) 396 (M+, 73), 381 (10), 379 (14), 353 (29), 313 (100), 273 (21), 229 (20), 203 (27), 175 (24), 109 (16), 83 (34). Exact mass calculated for C26H36O3, 396.2664; found, 396.2667. Anal. (C26H36O3) C, H.
(4R)-4-[4-(1-Hexyl-cyclopentyl)-2,6-dihydroxy-phenyl]-6,6-dimethyl-2-norpinanone (5b)
The synthesis was carried out as described for 5a starting from 4b25 (1.9 g, 7.25 mmol), diacetates 331 (4.31 g, ca. 75% pure by 1H NMR, 13.57 mmol), and p-toluenesulfonic acid monohydrate (1.93 g, 10.15 mmol) in CHCl3 (73 mL) and gave 1.68 g (58% yield) of 5b as a white crystalline solid. m p 187–188°C (from CHCl3-hexane); IR (neat) 3330, 3089, 2958, 2924, 2873, 1676 (s, >C=O), 1616, 1583, 1417, 1372, 1343, 1262, 1189, 1051, 1019, 971, 841 cm−1; 1H NMR (700 MHz, CDCl3) δ 6.22 (s, 2H, ArH), 4.97 (br s, 2H, OH), 3.94 (t, J = 8.1 Hz, 1H, 4-H), 3.50 (dd, J = 18.8 Hz, J = 7.8 Hz, 1H, 3α-H), 2.62 (dd, J = 18.8 Hz, J = 8.6 Hz, 1H, 3β-H), 2.59 (t, J = 5.0 Hz, 1H, 1-H), 2.51 (m, 1H, 7α-H), 2.47 (d, J = 10.6 Hz, 1H, 7β-H), 2.31 (t, J = 5.3 Hz, 1H, 5-H), 1.84-1.75 (m, 2H of the cyclopentane ring), 1.73-1.57 (m, 6H of the cyclopentane ring), 1.50-1.46 (m, 2H, 2′-H), 1.36 (s, 3H, 6-Me), 1.26-1.11 (m, 6H, 4′-H, 5′-H, 6′-H), 1.02-0.95 (m, 5H, 3′-H, 6-Me, especially 0.99, s, 3H, 6-Me), 0.83 (t, J = 6.9 Hz, 3H, 7′-H); 13C NMR (CDCl3) δ 217.22 (C-2), 154.55 (ArC-2, ArC-6), 149.22 (ArC-4), 113.39 (ArC-1), 107.60 (ArC-3, ArC-5), 57.96 (C-1), 50.56 (C-1′), 46.80 (C-5), 42.19 (C-6), 41.83 (C-2′), 37.97 (C-3), 37.58 (C-8′, C-11′), 31.82 (C-5′), 30.00 (C-4′), 29.52 (C-4), 26.20 (6-CH3), 25.23 (C-3′), 24.47 (C-7), 23.32 (C-9′, C-10′), 22.72 (C-6′), 22.18 (6-CH3), 14.09 (C-7′); mass spectrum m/z (relative intensity) 398 (M+, 33), 383 (7), 381 (8), 355 (13), 315 (43), 275 (17), 203 (19), 149 (28), 111 (38), 83 (66), 57 (100). Exact mass calculated for C26H38O3, 398.2821; found, 398.2822. Anal. (C26H38O3) C, H.
(4R)-4-[4-(2-Hexyl-1,3-dithiolan-2-yl)-2,6-dihydroxy-phenyl]-6,6-dimethyl-2-norpinanone (5c)
The synthesis was carried out as described for 5a starting from 4c23 (879 mg, 2.95 mmol), diacetates 331 (1.54 g, ca. 80% pure by 1H NMR, 5.16 mmol), and p-toluenesulfonic acid monohydrate (785 mg, 4.13 mmol) in CHCl3 (30 mL) and gave 451 mg (35% yield) of 5c as a white solid. m p 160–161°C (from CHCl3-hexane); IR (neat) 3329, 2926, 2856, 1683 (s, >C=O), 1616, 1585, 1417, 1371, 1303, 1266, 1207, 1051, 1022, 919, 843, 793 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.68 (s, 2H, ArH), 5.02 (br s, 2H, OH), 3.95 (t, J = 8.2 Hz, 1H, 4-H), 3.44 (dd, J = 19.5 Hz, J = 7.8 Hz, 1H, 3α-H), 3.37-3.30 (m, 2H, -S(CH2)2S-), 3.25-3.18 (m, 2H, -S(CH2)2S-), 2.60 (dd, J = 19.5 Hz, J = 8.5 Hz, 1H, 3β-H), 2.58 (t, J = 4.7 Hz, 1H, 1-H), 2.53-2.49 (m, 1H, 7α-H), 2.44 (d, J = 10.8 Hz, 1H, 7β-H), 2.30 (t, J = 5.2 Hz, 1H, 5-H), 2.26-2.22 (m, 2H, 2′-H), 1.36 (s, 3H, 6-Me), 1.27-1.19 (m, 8H, 3′-H, 4′-H, 5′-H, 6′-H), 0.99 (s, 3H, 6-Me), 0.85 (t, J = 6.5 Hz, 3H, 7′-H); mass spectrum m/z (relative intensity) 434 (M+, 18), 391 (2), 349 (100), 239 (9). Exact mass calculated for C24H34O3S2, 434.1949; found, 434.1948. Anal. (C24H34O3S2) C, H.
(4R)-4-{4-[1-(1,2-cis-Hexen-1-yl)cyclobutyl]-2,6-dihydroxy-phenyl}-6,6-dimethyl-2-norpinanone (5d)
The synthesis was carried out as described for 5a starting from 4d27 (710 mg, 2.89 mmol), diacetates 331 (1.5 g, ca. 80% pure by 1H NMR, 5.06 mmol), and p-toluenesulfonic acid monohydrate (770 mg, 4.05 mmol) in CHCl3 (29 mL) and gave 264 mg (24% yield) of 5d as a white crystalline solid. m p 174–175°C (from CHCl3-hexane); IR (neat) 3280, 2955, 2926, 2870, 1674 (s, >C=O), 1615, 1581, 1417, 1374, 1348, 1301, 1263, 1192, 1053, 1016, 969, 920, 834, 754, 704 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.31 (s, 2H, ArH), 5.73 (d, J = 11.0 Hz, 1H, 2′-H), 5.30 (dt, J = 11.0 Hz, J = 7.5 Hz, 1H, 3′-H), 4.85 (br s, 2H, OH), 3.95 (t, J = 8.1 Hz, 1H, 4-H), 3.49 (dd, J = 18.7 Hz, J = 7.8 Hz, 1H, 3α-H), 2.60 (dd, J = 18.7 Hz, J = 8.5 Hz, 1H, 3β-H), 2.58 (t, J = 5.1 Hz, 1H, 1-H), 2.50 (m, 1H, 7α-H), 2.46 (d, J = 10.6 Hz, 1H, 7β-H), 2.43-2.38 (m, 2H of the cyclobutane ring), 2.35-2.30 (m, 2H of the cyclobutane ring), 2.29 (t, J = 5.3 Hz, 1H, 5-H), 1.99-1.92 (m, 1H of the cyclobutane ring), 1.90-1.85 (m, 1H of the cyclobutane ring), 1.84-1.78 (m, 2H, 4′-H), 1.36 (s, 3H, 6-Me), 1.22-1.16 (m, 4H, 5′-H, 6′-H), 1.00 (s, 3H, 6-Me), 0.81 (t, J = 6.7 Hz, 3H, 7′-H); mass spectrum m/z (relative intensity) 382 (M+, 100), 367 (16), 354 (18), 339 (29), 299 (27), 271 (43), 188 (41), 83 (44). Exact mass calculated for C25H34O3, 382.2508; found, 382.2507. Anal. (C25H34O3) C, H.
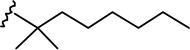
(4R)-4-[4-(1-Hexyl-cyclobutyl)-2,6-dihydroxy-phenyl]-6,6-dimethyl-2-norpinanone (5e)
The synthesis was carried out as with 5a by using 4e27 (514 mg, 2.07 mmol), diacetates 331 (1.077 g, ca. 80% pure by 1H NMR, 3.62 mmol), and p-toluenesulfonic acid monohydrate (551 mg, 2.9 mmol) in CHCl3 (21 mL). Yield 49% (390 mg); white crystalline solid; m p 194–196°C (from CHCl3-hexane); IR (neat) 3315, 3080, 2944, 2930, 2855, 1675 (s, >C=O), 1615, 1582, 1417, 1373, 1345, 1262, 1190, 1053, 1039, 1018, 968, 837, 761 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.06 (s, 2H, ArH), 4.85 (br s, 2H, OH), 3.95 (t, J = 8.1 Hz, 1H, 4-H), 3.49 (dd, J = 18.7 Hz, J = 7.8 Hz, 1H, 3α-H), 2.61 (dd, J = 18.7 Hz, J = 8.6 Hz, 1H, 3β-H), 2.58 (t, J = 5.0 Hz, 1H, 1-H), 2.51 (m, 1H, 7α-H), 2.47 (d, J = 10.6 Hz, 1H, 7β-H), 2.30 (t, J = 5.4 Hz, 1H, 5-H), 2.26-2.20 (m, 2H of the cyclobutane ring), 2.04-1.92 (m, 3H of the cyclobutane ring), 1.81-1.75 (m, 1H of the cyclobutane ring), 1.69-1.66 (m, 2H, 2′-H), 1.36 (s, 3H, 6-Me), 1.27-1.16 (m, 6H, 4′-H, 5′-H, 6′-H), 1.03-0.96 (m, 5H, 3′-H, 6-Me, especially 0.99, s, 3H, 6-Me), 0.85 (t, J = 6.9 Hz, 3H, 7′-H); mass spectrum m/z (relative intensity) 384 (M+, 100), 367 (9), 341 (31), 313 (48), 301 (27), 273 (47), 176 (39), 83 (46). Exact mass calculated for C25H36O3, 384.2664; found, 384.2662. Anal. (C25H36O3) C, H.
(6aR, 10aR)-3-[1-(1,2-cis-Hexen-1-yl)-cyclopent-1-yl]-6,6a,7,8,10,10a-hexahydro-1-hydroxy-6,6-dimethyl-9H-dibenzo[b,d]pyran-9-one (6a)
The synthesis was carried out analogous to the preparation of 6b (see text below) using 5a (467 mg, 1.18 mmol) and trimethylsilyl trifluoromethanesulfonate (1.2 mL, 0.3 M solution in CH3NO2, 0.36 mmol) in anhydrous CH2Cl2/CH3NO2 (3:1 mixture, 24 mL); yield: 61% (284 mg); white foam. IR (neat) 3266 (br, OH), 2953, 2871, 1694 (s, >C=O), 1619, 1574, 1415, 1336, 1258, 1203, 1183, 1135, 1093, 1041, 966, 835 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.42 (d, J = 1.5 Hz, 1H, Ar-H), 6.31 (d, J = 1.5 Hz, 1H, Ar-H), 5.95 (br s, 1H, OH), 5.63 (d, J = 11.2 Hz, 1H, 2′-H), 5.25 (dt, J = 11.2 Hz, J = 7.5 Hz, 1H, 3′-H), 3.97 (ddd, J = 15.0 Hz, J = 3.5 Hz, J = 2.0 Hz, 1H, 10eq-H), 2.87 (m as td, J = 12.0 Hz, J = 3.4 Hz, 1H, 10a-H), 2.63-2.59 (m, 1H, 8eq-H), 2.48-2.40 (m, 1H, 8ax-H), 2.18-2.12 (m, 2H, 10ax-H, 7eq-H), 1.99-1.92 (m, 3H, cyclopentane ring, 6a-H), 1.91-1.83 (m, 2H, cyclopentane ring), 1.77-1.65 (m, 6H, 4′-H, cyclopentane ring), 1.57-1.45 (m, 4H, 7ax-H, 6-Me, especially 1.47, s, 6-Me), 1.12-1.04 (m, 7H, 5′-H, 6′-H, 6-Me, especially 1.11, s, 6-Me), 0.73 (t, J = 6.8 Hz, 3H, 7′-H); mass spectrum m/z (relative intensity) 396 (M+, 39), 381 (3), 379 (4), 353 (16), 339 (13), 313 (44), 302 (22), 220 (16), 205 (56), 83 (100). Exact mass calculated for C26H36O3, 396.2664; found, 396.2669. Anal. (C26H36O3) C, H.
(6aR, 10aR)-3-(1-Hexyl-cyclopent-1-yl)-6,6a,7,8,10,10a-hexahydro-1-hydroxy-6,6-dimethyl-9H-dibenzo[b,d]pyran-9-one (6b)
To a stirred solution of 5b (550 mg, 1.38 mmol) in anhydrous CH2Cl2/CH3NO2 (3:1 mixture, 28 mL) at 0°C, under an argon atmosphere was added trimethylsilyl trifluoromethanesulfonate (1.4 mL, 0.3M solution in CH3NO2, 0.42 mmol). Stirring was continued for 3 hours while the temperature was allowed to rise to 25°C. The reaction was quenched with saturated aqueous NaHCO3/brine (1:1), and diethyl ether was added. The organic phase was separated, the aqueous phase was extracted with diethyl ether, and the combined organic phase was washed with brine and dried over MgSO4. Solvent evaporation and purification by flash column chromatography on silica gel (47% diethyl ether-hexane) afforded 359 mg (65% yield) of the title compound 6b as white foam. IR (neat) 3292 (br, OH), 2953, 2927, 2870, 1695 (s, >C=O), 1620, 1574, 1414, 1344, 1259, 1202, 1184, 1137, 1093, 1038, 838 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.33 (d, J = 1.5 Hz, 1H, ArH), 6.19 (d, J = 1.5 Hz, 1H, ArH), 5.50 (br s, 1H, OH), 3.93 (ddd, J = 15.0 Hz, J = 3.5 Hz, J = 2.0 Hz, 1H, 10eq-H), 2.88 (m as td, J = 12.0 Hz, J = 3.5 Hz, 1H, 10a-H), 2.62-2.58 (m, 1H, 8eq-H), 2.48-2.41 (m, 1H, 8ax-H), 2.19-2.13 (m, 2H, 10ax-H, 7eq-H), 1.97 (m as td, J = 12.1 Hz, J = 2.7 Hz, 1H, 6a-H), 1.84-1.79 (m, 2H, cyclopentane ring), 1.74-1.45 (m, 12H, cyclopentane ring, 7ax-H, 2′-H, 6-Me, especially 1.47, s, 6-Me), 1.22-1.10 (m, 9H, 4′-H, 5′-H, 6′-H, 6-Me, especially 1.13, s, 6-Me), 1.01-0.91 (m, 2H, 3′-H), 0.82 (t, J = 6.8 Hz, 3H, 7′-H); mass spectrum m/z (relative intensity) 398 (M+, 57), 383 (4), 381 (3), 356 (8), 314 (100), 302 (22), 220 (5), 204 (22), 83 (39). Exact mass calculated for C26H38O3, 398.2821; found, 398.2822. Anal. (C26H38O3) C, H.
(6aR, 10aR)-3-(2-Hexyl-1,3-dithiolan-2-yl)-6,6a,7,8,10,10a-hexahydro-1-hydroxy-6,6-dimethyl-9H-dibenzo[b,d]pyran-9-one (6c)
The synthesis was carried out analogous to the preparation of 6b using 5c (180 mg, 0.41 mmol) and trimethylsilyl trifluoromethanesulfonate (0.4 mL, 0.3M solution in CH3NO2, 0.12 mmol) in anhydrous CH2Cl2/CH3NO2 (3:1 mixture, 8.2 mL); yield: 67% (121 mg); white foam. IR (neat) 3252 (br, OH), 2925, 2854, 1693 (s, >C=O), 1616, 1574, 1411, 1332, 1259, 1203, 1182, 1136, 1093, 1044, 968 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.76 (d, J = 1.8 Hz, 1H, ArH), 6.64 (d, J = 1.8 Hz, 1H, ArH), 5.85 (br s, 1H, OH), 3.94 (m as d, J = 14.6 Hz, 1H, 10eq-H), 3.36-3.30 (m, 2H, -S(CH2)2S-), 3.28-3.21 (m, 2H, -S(CH2)2S-), 2.87 (m as td, J = 11.9 Hz, J = 3.3 Hz, 1H, 10a-H), 2.63-2.59 (m, 1H, 8eq-H), 2.48-2.40 (m, 1H, 8ax-H), 2.27 (m, 2H, 2′-H), 2.18-2.14 (m, 2H, 10ax-H, 7eq-H), 1.96 (m as td, J = 11.7 Hz, J = 2.0 Hz, 1H, 6a-H), 1.56-1.45 (m, 4H, 7ax-H, 6-Me, especially 1.48, s, 6-Me), 1.26-1.19 (m, 8H, 3′-H, 4′-H, 5′-H, 6′-H), 1.12 (s, 3H, 6-Me), 0.84 (t, J = 7.0 Hz, 3H, 7′-H); mass spectrum m/z (relative intensity) 434 (M+, 6), 398 (2), 349 (43), 264 (20), 222 (29), 194 (13), 152 (100), 137 (67), 83 (8). Exact mass calculated for C24H34O3S2, 434.1949; found, 434.1949. Anal. (C24H34O3S2) C, H.
(6aR, 10aR)-3-[1-(1,2-cis-Hexen-1-yl)-cyclobut-1-yl]-6,6a,7,8,10,10a-hexahydro-1-hydroxy-6,6-dimethyl-9H-dibenzo[b,d]pyran-9-one (6d)
The synthesis was carried out analogous to the preparation of 6b using 5d (140 mg, 0.37 mmol) and trimethylsilyl trifluoromethanesulfonate (0.37 mL, 0.3M solution in CH3NO2, 0.11 mmol) in anhydrous CH2Cl2/CH3NO2 (3:1 mixture, 7.5 mL); yield: 51% (72 mg); white foam. IR (neat) 3254 (br, OH), 2954, 2931, 2870, 1694 (s, >C=O), 1617, 1574, 1414, 1337, 1259, 1202, 1183, 1135, 1093, 1039, 962, 835 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.45 (d, J = 1.5 Hz, 1H, ArH), 6.23 (d, J = 1.5 Hz, 1H, ArH), 5.74 (d, J = 11.1 Hz, 1H, 2′-H), 5.34 (br s, 1H, OH), 5.27 (dt, J = 11.1 Hz, J = 7.5 Hz, 1H, 3′-H), 3.90 (ddd, J = 15.0 Hz, J = 3.5 Hz, J = 2.0 Hz, 1H, 10eq-H), 2.87 (m as td, J = 12.5 Hz, J = 3.6 Hz, 1H, 10a-H), 2.62-2.56 (m, 1H, 8eq-H), 2.48-2.38 (m, 3H, 8ax-H, cyclobutane ring), 2.34-2.28 (m, 2H, cyclobutane ring), 2.18-2.12 (m, 2H, 10ax-H, 7eq-H), 1.98-1.90 (m, 2H, 6a-H, cyclobutane ring), 1.89-1.80 (m, 3H, 4′-H, cyclobutane ring), 1.54-1.49 (m, 1H, 7ax-H), 1.47 (s, 3H, 6-Me), 1.20-1.15 (m, 4H, 5′-H, 6′-H), 1.11 (s, 3H, 6-Me), 0.79 (t, J = 6.7 Hz, 3H, 7′-H); mass spectrum m/z (relative intensity) 382 (M+, 100), 354 (31), 339 (14), 311 (17), 272 (26), 201 (19). Exact mass calculated for C25H34O3, 382.2508; found, 382.2511. Anal. (C25H34O3) C, H.
(6aR, 10aR)-3-(1-Hexyl-cyclobut-1-yl)-6,6a,7,8,10,10a-hexahydro-1-hydroxy-6,6-dimethyl-9H-dibenzo[b,d]pyran-9-one (6e)
The synthesis was carried out analogous to the preparation of 6b using 5e (218 mg, 0.57 mmol) and trimethylsilyl trifluoromethanesulfonate (0.57 mL, 0.3M solution in CH3NO2, 0.17 mmol) in anhydrous CH2Cl2/CH3NO2 (3:1 mixture, 12 mL); yield: 64% (140 mg); white foam. IR (neat) 3247 (br, OH), 2925, 2856, 1694 (s, >C=O), 1619, 1575, 1415, 1358, 1259, 1203, 1183, 1138, 1093, 1053, 970, 839 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.19 (d, J = 1.4 Hz, 1H, ArH), 6.02 (d, J = 1.4 Hz, 1H, ArH), 5.34 (br s, 1H, OH), 3.90 (ddd, J = 15.0 Hz, J = 3.5 Hz, J = 2.0 Hz, 1H, 10eq-H), 2.87 (m as td, J = 12.4 Hz, J = 3.4 Hz, 1H, 10a-H), 2.62-2.57 (m, 1H, 8eq-H), 2.48-2.40 (m, 1H, 8ax-H), 2.29-2.21 (m, 2H, cyclobutane ring), 2.19-2.13 (m, 2H, 10ax-H, 7eq-H), 2.10-1.97 (m, 4H, 6a-H, cyclobutane ring), 1.82-1.73 (m, 1H, cyclobutane ring), 1.69-1.64 (m, 2H, 2′-H), 1.57-1.46 (m, 4H, 7ax-H, 6-Me, especially 1.47, s, 6-Me), 1.27-1.15 (m, 6H, 4′-H, 5′-H, 6′-H), 1.13, s, 6-Me), 1.05-0.96 (m, 2H, 3′-H), 0.84 (t, J = 6.8 Hz, 3H, 7′-H); mass spectrum m/z (relative intensity) 384 (M+, 100), 356 (38), 341 (9), 313 (27), 274 (30), 203 (18). Exact mass calculated for C25H36O3, 384.2664; found, 384.2655. Anal. (C25H36O3) C, H.
(6aR, 9R, 10aR)-3-(2-Hexyl-1,3-dithiolan-2-yl)-6a,7,8,9,10,10a-hexahydro-6,6-dimethyl-6H-dibenzo[b,d]pyran-1,9 diol (7c-β)
To a solution of 6c (26 mg, 0.06 mmol) in anhydrous methanol (1.2 mL) at −40°C, under an argon atmosphere was added NaBH4 (8 mg, 0.21 mmol). The reaction mixture was stirred for 1.5 hours at −40°C and then quenched by the addition of saturated NaCl solution. The mixture was warmed to room temperature, diluted with H2O, and extracted with ethyl acetate. The organic layer was washed with brine, dried (MgSO4) and evaporated to a semisolid material 7c. On the basis of 1H NMR analysis, 7c is a mixture of two isomeric alcohols 7c-α (Rf = 0.27, 45% AcOEt in hexane) and 7c-β (Rf = 0.25, 45% AcOEt in hexane) in a ratio 4:96, respectively. Purification of this material by flash column chromatography on silica gel (50% AcOEt in hexane) gave, in order of elution, 3 mg of a mixture of 7c-α and 7c-β (1:2 ratio by 1H NMR) and 20 mg of pure 7c-β as a glassy substance (m p = 91–93°C). Overall yield for 7c-β: 85% (22 mg). Compound 7c-β: IR (neat) 3246 (br, OH), 2927, 2857, 1615, 1573, 1411, 1364, 1332, 1274, 1140, 1044, 820 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.70 (d, J = 1.5 Hz, 1H, ArH), 6.59 (d, J = 1.5 Hz, 1H, ArH), 5.98 (br s, 1H, OH), 3.87 (dddd, J = 15.5 Hz, J = 15.0 Hz, J = 4.5 Hz, J = 4.5 Hz, 1H, 9ax-H, peak half-width = 21 Hz), 3.50 (m as br d, J = 11.0 Hz, 1H, 10eq-H), 3.37-3.29 (m, 2H, -S(CH2)2S-), 3.27-3.18 (m, 2H, -S(CH2)2S-), 2.47 (ddd, J = 12.0 Hz, J = 11.5 Hz, J = 2.5 Hz, 1H, 10a-H), 2.27-2.23 (m, 2H, 2′-H), 2.17 (m as br d, J = 10.6 Hz, 1H, 8eq-H), 1.91-1.85 (m as br d, J = 14.0 Hz, 1H, 7eq-H), 1.49 (ddd, J = 12.0 Hz, J = 11.5 Hz, J = 2.5 Hz, 1H, 6a-H), 1.45-1.37 (m and s overlapping, 4H, 8ax-H, 6-Me, especially 1.38, s, 6β-Me), 1.30-1.09 (m, 10H, 3′-H, 4′-H, 5′-H, 6′-H, 7ax-H, 10ax-H), 1.06 (s, 3H, 6-Me), 0.84 (t, J = 6.8 Hz, 3H, 7′-H); mass spectrum m/z (relative intensity) 436 (M+, 17), 351 (100), 205 (5). Exact mass calculated for C24H36O3S2, 436.2106; found, 436.2107. Anal. (C24H36O3S2) C, H. Compound 7c-α: 4.32 (m, 1H, 9eq-H, peak half-width = 10.5 Hz).
(6aR, 9R, 10aR)-3-(1-Hexyl-cyclobut-1-yl)-6a,7,8,9,10,10a-hexahydro-6,6-dimethyl-6H-dibenzo[b,d]pyran-1,9 diol (7e-β)
The synthesis was carried out analogous to the preparation of 7c-β using 6e (300 mg, 0.78 mmol) and NaBH4 (104 mg, 2.74 mmol) in anhydrous methanol (15.6 mL). On the basis of 1H NMR analysis, the crude material (7e) obtained after workup, is a mixture of two isomeric alcohols 7e-α (Rf = 0.28, 40% AcOEt in hexane) and 7e-β (Rf = 0.25, 40% AcOEt in hexane) in a ratio 3:97, respectively. Purification by flash column chromatography on silica gel (45% AcOEt in hexane) gave, in order of elution, 36 mg of a mixture of 7e-α and 7e-β (1:3 ratio by 1H NMR) and 260 mg of pure 7e-β as a glassy substance (m p = 79–81°C). Overall yield for 7e-β: 95% (287 mg). Compound 7e-β: IR (neat) 3277 (br, OH), 2924, 2855, 1620, 1572, 1454, 1414, 1360, 1271, 1185, 1140, 1052, 972, 836 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.15 (d, J = 1.5 Hz, 1H, ArH), 5.97 (d, J = 1.5 Hz, 1H, ArH), 5.24 (br s, 1H, OH), 3.86 (dddd, J = 15.5 Hz, J = 15.0 Hz, J = 4.5 Hz, J = 4.5 Hz, 1H, 9ax-H, peak half-width = 22 Hz), 3.48 (m as br d, J = 7.0 Hz, 1H, 10eq-H), 2.48 (ddd, J = 12.0 Hz, J = 11.5 Hz, J = 2.5 Hz, 1H, 10a-H), 2.30-2.21 (m, 2H, cyclobutane ring), 2.17 (m as br d, J = 10.5 Hz, 1H, 8eq-H), 2.03-1.93 (m, 3H, cyclobutane ring), 1.92-1.86 (m as br d, J = 14.0 Hz, 1H, 7eq-H), 1.82-1.72 (m, 1H, cyclobutane ring), 1.70-1.64 (m, 2H, 2′-H), 1.51 (ddd, J = 12.0 Hz, J = 11.5 Hz, J = 2.0 Hz, 1H, 6a-H), 1.46-1.36 (m and s overlapping, 4H, 8ax-H, 6-Me, especially 1.39, s, 6-Me), 1.29-1.09 (m, 8H, 4′-H, 5′-H, 6′-H, 7ax-H, 10ax-H), 1.08 (s, 3H, 6-Me), 1.04-0.96 (m, 2H, 3′-H), 0.84 (t, J = 7.0 Hz, 3H, 7′-H); mass spectrum m/z (relative intensity) 386 (M+, 100), 358 (42), 315 (22), 297 (19), 274 (15), 203 (11), 83 (16). Exact mass calculated for C25H38O3, 386.2821; found, 386.2823. Anal. (C25H38O3) C, H. Compound 7e-α: 4.25 (m, 1H, 9eq-H, peak half-width = 10.5 Hz).
(3-Phenoxypropyl)triphenylphosphonium bromide (9a)
A mixture of 3-phenoxypropyl bromide (8a) (12 g, 55.8 mmol) and triphenylphosphine (15.35 g, 58.6 mmol) in anhydrous benzene (100 mL) was refluxed with vigorous stirring, for two days under argon. The reaction mixture was cooled to room temperature, and the precipitating product (9a) was isolated by filtration under reduced pressure as a white microcrystalline solid (m p 154–156°C) in 80% yield (21.3 g). 1H NMR (500 MHz, CDCl3) δ 7.87 (dd, J = 12.1 Hz, J = 8.0 Hz, 6H, -PPh3), 7.79 (td, J = 8.0 Hz, J = 1.4 Hz, 3H, -PPh3), 7.69 (td, J = 8.0 Hz, J = 3.5 Hz, 6H, -PPh3), 7.24 (t, J = 8.1 Hz, 2H, 3-H, 5-H, -OPh), 6.92 (t, J = 8.1 Hz, 1H, 4-H, -OPh), 6.85 (d, J = 8.1 Hz, 2H, 2-H, 6-H, -OPh), 4.33 (t, J = 5.4 Hz, 2H, -CH2OPh), 4.10 (dt, J = 12.4 Hz, J = 8.0 Hz, 2H, -CH2PPh3), 2.27-2.19 (m, 2H).
(4-Phenoxybutyl)triphenylphosphonium bromide (9b)
The synthesis was carried out analogous to the preparation of 9a using 4-phenoxybutyl bromide (8b) (22.0 g, 96 mmol) and triphenylphosphine (26.2 g, 100 mmol) in anhydrous benzene (100 mL); yield: 85% (40.0 g); white solid, m p 185–186°C. 1H NMR (500 MHz, CDCl3) δ 7.96 (dd, J = 12.0 Hz, J = 8.0 Hz, 6H, -PPh3), 7.88 (td, J = 8.0 Hz, J = 1.5 Hz, 3H, -PPh3), 7.80 (td, J = 8.0 Hz, J = 3.5 Hz, 6H, -PPh3), 7.25 (t, J = 7.9 Hz, 2H, 3-H, 5-H, -OPh), 6.92 (t, J = 7.9 Hz, 1H, 4-H, -OPh), 6.82 (d, J = 7.9 Hz, 2H, 2-H, 6-H, -OPh), 4.09 (t, J = 5.5 Hz, 2H, -CH2OPh), 4.01 (dt, J = 12.6 Hz, J = 8.1 Hz, 2H, -CH2PPh3), 2.25 (qt, J = 6.4 Hz, 2H), 1.93-1.85 (m, 2H).
(5-Phenoxypentyl)triphenylphosphonium bromide (9c)
The synthesis was carried out analogous to the preparation of 9a using 5-phenoxypentyl bromide (8c) (6.96 g, 28.63 mmol) and triphenylphosphine (8.25 g, 31.5 mmol) in anhydrous benzene (70 mL); yield: 83% (12 g); white solid, m p 174–176°C. 1H NMR (500 MHz, CDCl3) δ 7.86 (dd, J = 12.1 Hz, J = 8.0 Hz, 6H, -PPh3), 7.78 (td, J = 8.0 Hz, J = 1.5 Hz, 3H, -PPh3), 7.68 (td, J = 8.0 Hz, J = 3.5 Hz, 6H, -PPh3), 7.24 (t, J = 7.9 Hz, 2H, 3-H, 5-H, -OPh), 6.91 (t, J = 7.9 Hz, 1H, 4-H, -OPh), 6.78 (d, J = 7.9 Hz, 2H, 2-H, 6-H, -OPh), 3.92 (t, J = 5.6 Hz, 2H, -CH2OPh), 3.85 (dt, J = 12.5 Hz, J = 8.0 Hz, 2H, -CH2PPh3), 1.90-1.79 (m, 4H), 1.77-1.69 (m, 2H).
3,5-Dimethoxy-1-[1-(1,2-cis-4-phenoxybuten-1-yl)cyclopentyl]benzene (11a)
To a stirred suspension of (3-phenoxypropyl)triphenylphosphonium bromide (9a) (17.2 g, 36 mmol) in dry THF (180 mL) at 0°C, under an argon atmosphere, was added potassium bis(trimethylsilyl)amide (7.16 g, 36 mmol). The mixture was stirred for 10 min, and a solution of aldehyde 1025,27 (1.68 g, 7.2 mmol) in anhydrous THF (10 mL) was added dropwise. The reaction was stirred for an additional 20 min and upon completion (TLC) was quenched by the addition of saturated aqueous NH4Cl (40 mL). The reaction mixture was warmed to room temperature, the organic layer was separated, and the aqueous phase was extracted with diethyl ether. The combined organic layer was washed with brine and dried over MgSO4, and the solvent evaporated under reduced pressure. The residue was chromatographed through a short column of silica gel (10% diethyl ether in hexane) to give compound 11a as a colorless liquid in 91% yield (2.3 g). 1H NMR (300 MHz, CDCl3) δ 7.24 (t, J = 7.9 Hz, 2H, OPh), 6.91 (t, J = 7.9 Hz, 1H, OPh), 6.77 (d, J = 7.9 Hz, 2H, OPh), 6.54 (d, J = 2.4 Hz, 2H, ArH), 6.28 (t, J = 2.4 Hz, 1H, ArH), 5.86 (d, J = 11.3 Hz, 1H, 2′-H), 5.45 (dt, J = 11.3 Hz, J = 7.3 Hz, 1H, 3′-H), 3.76 (s, 6H, OMe), 3.70 (t, J = 6.8 Hz, 2H, 5′-H), 2.23 (q, J = 6.8, 2H, 4′-H), 2.10-1.89 (m, 4H of the cyclopentane ring), 1.79-1.65 (m, 4H of the cyclopentane ring); mass spectrum m/z (relative intensity) 352 (M+, 35), 258 (15), 231 (29), 205 (100), 152 (29), 77 (31). Exact mass calculated for C23H28O3, 352.2038; found, 352.2038.
3,5-Dimethoxy-1-[1-(1,2-cis-5-phenoxypenten-1-yl)cyclopentyl]benzene (11b)
The synthesis was carried out as described for 11a using (4-phenoxybutyl)triphenylphosphonium bromide (9b) (21 g, 42.8 mmol) in anhydrous THF (100 mL), potassium bis(trimethylsilyl)amide (8.52 g, 42.8 mmol) and a solution of 1025,27 (2 g, 8.7 mmol) in anhydrous THF (10 mL). The crude product obtained after work up was purified by flash column chromatography on silica gel (10% diethyl ether in hexane) to give compound 11b as a colorless liquid in 94% yield (2.99 g). 1H NMR (500 MHz, CDCl3) δ 7.25 (t, J = 8.2 Hz, 2H, OPh), 6.90 (t, J = 8.2 Hz, 1H, OPh), 6.80 (d, J = 8.2 Hz, 2H, OPh), 6.53 (d, J = 2.2 Hz, 2H, ArH) 6.27 (t, J = 2.2 Hz, 1H, ArH), 5.77 (d, J = 11.3 Hz, 1H, 2′-H), 5.31 (dt, J = 11.3 Hz, J = 7.5 Hz, 1H, 3′-H), 3.76 (s, 6H, OMe), 3.65 (t, J = 6.8 Hz, 2H, 6′-H), 2.03-1.97 (m, 2H of the cyclopentane ring), 1.96-1.89 (m, 4H, 4′-H and cyclopentane ring, overlapping), 1.76-1.65 (m, 4H of the cyclopentane ring), 1.64-1.55 (m, 2H); mass spectrum m/z (relative intensity) 366 (M+, 57), 272 (23), 246 (36), 231 (67), 205 (100), 189 (17), 177 (23), 151 (43), 77 (33). Exact mass calculated for C24H30O3, 366.2195; found, 366.2193.
3,5-Dimethoxy-1-[1-(1,2-cis-6-phenoxyhexen-1-yl)cyclopentyl]benzene (11c)
The synthesis was carried out as described for 11a using (5-phenoxypentyl)triphenylphosphonium bromide (9c), (11.60 g, 22.95 mmol) in anhydrous THF (50 mL), potassium bis(trimethylsilyl) amide (4.56 g, 22.95 mmol) and a solution of 1025,27 (1.07 g, 4.59 mmol) in anhydrous THF (10 mL). The crude product obtained after work up was purified by flash column chromatography on silica gel (10% diethyl ether in hexane) to give compound 11c as a colorless liquid in 95% yield (1.66 g). 1H NMR (500 MHz, CDCl3) δ 7.26 (t, J = 8.0 Hz, 2H, OPh), 6.91 (t, J = 8.0 Hz, 1H, OPh), 6.83 (d, J = 8.0 Hz, 2H, OPh), 6.52 (d, J = 2.1 Hz, 2H, ArH), 6.26 (t, J = 2.1 Hz, 1H, ArH), 5.74 (d, J = 11.3 Hz, 1H, 2′-H), 5.29 (dt, J = 11.3 Hz, J = 7.5 Hz, 1H, 3′-H), 3.77-3.73 (t and s overlapping, 8H, OMe and 7′-H), 2.04-1.97 (m, 2H of the cyclopentane ring), 1.94-1.88 (m, 2H of the cyclopentane ring), 1.80 (q, J = 7.0 Hz, 2H, 4′-H), 1.75-1.65 (m, 4H of the cyclopentane ring), 1.58-1.52, (m, 2H), 1.29 (qt, J = 7.5 Hz, 2H); mass spectrum m/z (relative intensity) 380 (M+, 29), 287 (14), 262 (7), 246 (24), 231 (26), 205 (100), 191 (22), 177 (15), 152 (27), 77 (34). Exact mass calculated for C25H32O3, 380.2351; found, 380.2353.
3,5-Dimethoxy-1-[1-(4-phenoxybutyl)cyclopentyl]benzene (12a)
To a solution of 11a (704 mg, 2 mmol) in EtOAc (20 mL) was added 10% Pd/C (106 mg) and the suspension stirred vigorously under hydrogen atmosphere, overnight at room temperature. The catalyst was removed by filtration through celite, and the filtrate was evaporated under reduced pressure to give the product 12a as a colorless liquid, in 96% yield (680 mg) which was used in the next step without further purification. 1H NMR (300 MHz, CDCl3) δ 7.25 (t, J = 8.1 Hz, 2H, 3-H, 5-H, OPh), 6.91 (t, J = 8.1 Hz, 1H, 4-H, OPh), 6.83 (d, J = 8.1 Hz, 2H, 2-H, 6-H, OPh), 6.43 (d, J = 3.0 Hz, 2H, 2-H, 6-H, ArH) 6.29 (t, J = 3.0 Hz, 1H, 4-H, ArH), 3.83 (t, J = 7.2 Hz, 2H, 5′-H), 3.78 (s, 6H, OMe), 1.95-1.51 (m, 12H, 8H of the cyclopentane ring and 4H of the 4-phenoxybutyl group), 1.21-1.11 (m, 2H of the 4-phenoxybutyl group); mass spectrum m/z (relative intensity) 354 (M+, 32), 270 (13), 261 (22), 206 (100), 177 (9), 165 (20), 151 (30), 107 (16), 77 (20). Exact mass calculated for C23H30O3, 354.2195; found, 354.2198.
3,5-Dimethoxy-1-[1-(5-phenoxypentyl)cyclopentyl]benzene (12b)
The synthesis was carried out as described for 12a using 11b (2.35 g, 6.42 mmol) and 10% Pd/C (360 mg) in EtOAc (60 mL) to give the product 12b as a colorless liquid in 94% yield (2.22 g) which was used in the next step without further purification. 1H NMR (500 MHz, CDCl3) δ 7.26 (t, J = 7.9 Hz, 2H, 3-H, 5-H, OPh), 6.91 (t, J = 7.9 Hz, 1H, 4-H, OPh), 6.84 (d, J = 7.9 Hz, 2H, 2-H, 6-H, OPh), 6.43 (d, J = 2.2 Hz, 2H, 2-H, 6-H, ArH) 6.29 (t, J = 2.2 Hz, 1H, 4-H, ArH), 3.85 (t, J = 6.5 Hz, 2H, 6′-H), 3.79 (s, 6H, OMe), 1.92-1.85 (m, 2H of the cyclopentane ring), 1.80-1.74 (m, 2H of the cyclopentane ring), 1.73-1.61 (m, 6H, 4H of the cyclopentane ring and 2H of the 5-phenoxypentyl group, overlapping), 1.60-1.54 (m, 2H, 2′-H), 1.31 (qt, J = 7.7 Hz, 2H of the 5-phenoxypentyl group), 1.10-1.02 (m, 2H of the 5-phenoxypentyl group); mass spectrum m/z (relative intensity) 368 (M+, 21), 275 (7), 248 (15), 206 (100), 165 (17), 151 (24), 77 (13). Exact mass calculated for C24H32O3, 368.2351; found, 368.2350.
3,5-Dimethoxy-1-[1-(6-phenoxyhexyl)cyclopentyl]benzene (12c)
The synthesis was carried out as described for 12a using 11c (1.08 g, 2.84 mmol) and 10% Pd/C (162 mg) in EtOAc (25 mL) to give the product 12c as a colorless liquid in 95% yield (1.03 g) which was used in the next step without further purification. 1H NMR (500 MHz, CDCl3) δ 7.26 (t, J = 7.8 Hz, 2H, 3-H, 5-H, OPh), 6.90 (t, J = 7.8 Hz, 1H, 4-H, OPh), 6.83 (d, J = 7.8 Hz, 2H, 2-H, 6-H, OPh), 6.43 (d, J = 1.9 Hz, 2H, 2-H, 6-H, ArH) 6.29 (t, J = 1.9 Hz, 1H, 4-H, ArH), 3.88 (t, J = 6.4 Hz, 2H, 7′-H), 3.78 (s, 6H, OMe), 1.93-1.87 (m, 2H of the cyclopentane ring), 1.80-1.73 (m, 2H of the cyclopentane ring), 1.72-1.60 (m, 6H, 4H of the cyclopentane ring and 2H of the 6-phenoxyhexyl group, overlapping), 1.59-1.53 (m, 2H, 2′-H), 1.39-1.20 (m, 4H of the 6-phenoxyhexyl group), 1.09-1.01 (m, 2H of the 6-phenoxyhexyl group); mass spectrum m/z (relative intensity) 382 (M+, 23), 306 (4), 289 (12), 262 (17), 220 (100), 179 (27), 165 (30), 77 (15). Exact mass calculated for C25H34O3, 382.2508; found, 382.2505.
5-[1-(4-Bromobutyl)cyclopentyl]resorcinol (13a)
To a stirred solution of 12a (650 mg, 1.84 mmol) in dry CH2Cl2 (20 mL), at −78°C, under an argon atmosphere, was added boron tribromide (6 mL, 1M solution in CH2Cl2). Following the addition, the reaction mixture was gradually warmed to room temperature and the stirring was continued at that temperature until completion of the reaction (12 h). Unreacted boron tribromide was destroyed by the addition of methanol and ice at 0°C. The mixture was warmed to room temperature and volatiles were removed in vacuo. The residue was diluted with ethyl acetate and washed with saturated NaHCO3 solution, water and brine. The organic layer was dried over MgSO4, filtered and concentrated under reduced pressure. Purification by flash column chromatography on silica gel (50% diethyl ether in hexane) afforded 535 mg (93% yield) of 13a as a slightly brown viscous oil. 1H NMR (500 MHz, CDCl3) δ 6.32 (d, J = 2.3 Hz, 2H, ArH), 6.17 (t, J = 2.3 Hz, 1H, ArH), 4.72 (br s, 2H, OH), 3.30 (t, J = 6.6 Hz, 2H, 5′-H), 1.91-1.85 (m, 2H of the cyclopentane ring), 1.78-1.59 (m, 8H, 6H of the cyclopentane ring and 2H of the 4-bromobutyl group, overlapping), 1.57-1.51 (m, 2H, 2′-H), 1.17-1.09 (m, 2H, 4-bromobutyl group); mass spectrum m/z (relative intensity) 314 (M++2, 5), 312 (M+, 5), 271 (3), 233 (27), 191 (7), 177 (100), 161 (8), 149 (16), 137 (39), 123 (62), 67 (39). Exact mass calculated for C15H21BrO2, 312.0725; found, 312.0734.
5-[1-(5-Bromopentyl)cyclopentyl]resorcinol (13b)
The synthesis was carried out as described for 13a using 12b (2.0 g, 5.43 mmol) and boron tribromide (18 mL, 1M solution in CH2Cl2) in anhydrous CH2Cl2 (50 mL). The crude product obtained after work up was purified by flash column chromatography on silica gel (50% diethyl ether in hexane) to give pure 13b as a slightly brown viscous oil in 89% yield (1.58 g). 1H NMR (500 MHz, CDCl3) δ 6.32 (d, J = 2.1 Hz, 2H, ArH), 6.16 (t, J = 2.1 Hz, 1H, ArH), 4.66 (br s, 2H, OH), 3.32 (t, J = 6.8 Hz, 2H, 6′-H), 1.90-1.79 (m, 2H of the cyclopentane ring), 1.78-1.59 (m, 8H, 6H of the cyclopentane ring and 2H of the 5-bromopentyl group, overlapping), 1.54-1.49 (m, 2H, 2′-H), 1.29 (qt, J = 7.5 Hz, 2H, 5-bromopentyl group), 1.05-0.96 (m, 2H, 5-bromopentyl group); mass spectrum m/z (relative intensity) 328 (M++2, 8), 326 (M+, 7), 247 (13), 232 (6), 230 (6), 217 (7), 191 (4), 177 (100), 161 (7), 149 (14), 137 (16), 123 (43), 67 (24). Exact mass calculated for C16H23BrO2, 326.0881; found, 326.0875.
5-[1-(6-Bromohexyl)cyclopentyl]resorcinol (13C)
The synthesis was carried out as described for 13a using 12c (1.0 g, 2.62 mmol) and boron tribromide (9 mL, 1M solution in CH2Cl2) in anhydrous CH2Cl2 (20 mL). The crude product obtained after work up was purified by flash column chromatography on silica gel (50% diethyl ether in hexane) to give 13C as a slightly brown viscous oil in 93% yield (830 mg). 1H NMR (500 MHz, CDCl3) δ 6.32 (d, J = 1.9 Hz, 2H, ArH), 6.17 (t, J = 1.9 Hz, 1H, ArH), 4.91 (br s, 2H, OH), 3.35 (t, J = 6.8 Hz, 2H, 7′-H), 1.89-1.80 (m, 2H of the cyclopentane ring), 1.79-1.59 (m, 8H, 6H of the cyclopentane ring and 2H of the 6-bromohexyl group, overlapping), 1.54-1.48 (m, 2H, 2′-H), 1.32 (qt, J = 7.8 Hz, 2H, 6-bromohexyl group), 1.17 (qt, J = 7.7 Hz, 2H, 6-bromohexyl group), 1.04-0.95 (m, 2H, 6-bromohexyl group); mass spectrum m/z (relative intensity) 342 (M++2, 6), 340 (M+, 6), 261 (12), 246 (6), 244 (6), 219 (4), 191 (4), 177 (100), 161 (5), 149 (11), 137 (17), 123 (46), 67 (29). Exact mass calculated for C17H25BrO2, 340.1038; found, 340.1031.
(4R)-4-{4-[1-(4-Bromobutyl)cyclopentyl]-2,6-dihydroxy-phenyl}-6,6-dimethyl-2-norpinanone (14a)
The synthesis was carried out as described for 5a using 13a (450 mg, 1.44 mmol), diacetates 331 (730 mg, ca. 75% pure by 1H NMR, 2.3 mmol), and p-toluenesulfonic acid monohydrate (548 mg, 2.3 mmol) in CHCl3 (15 mL); yield: 54% (350 mg); white solid, m p 180–182°C (from CHCl3-hexane); IR (neat) 3348, 2938, 2869, 1676 (s, >C=O), 1618, 1582, 1417, 1372, 1265, 1188, 1051, 1016, 920, 845 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.22 (s, 2H, ArH), 4.95 (br s, 2H, OH), 3.94 (t, J = 8.2 Hz, 1H, 4-H), 3.48 (dd, J = 18.9 Hz, J = 7.6 Hz, 1H, 3α-H), 3.31 (t, J = 7.0 Hz, 2H, 5′-H), 2.61 (dd, J = 18.9 Hz, J = 8.7 Hz, 1H, 3β-H), 2.59 (t, J = 5.0 Hz, 1H, 1-H), 2.52 (m, 1H, 7α-H), 2.46 (d, J = 10.6 Hz, 1H, 7β-H), 2.31, (t, J = 5.3 Hz, 1H, 5-H), 1.87-1.79 (m, 2H of the cyclopentane ring), 1.76-1.57 (m, 8H, 6H of the cyclopentane ring and 2H of the 4-bromobutyl group, overlapping), 1.54-1.48 (m, 2H, 2′-H), 1.36 (s, 3H, 6-Me), 1.17-1.10 (m, 2H, 4-bromobutyl group), 1.00 (s, 3H, 6-Me). mass spectrum m/z (relative intensity) 450 (M++2, 15), 448 (M+, 14), 433 (17), 407 (15), 405 (15), 379 (16), 365 (77), 314 (43), 285 (27), 259 (24), 243 (33), 203 (81), 189 (29), 149 (34), 123 (56), 83 (100). Exact mass calculated for C24H33BrO3, 448.1613; found, 448.1604. Anal. (C24H33BrO3) C, H.
(4R)-4-{4-[1-(5-Bromopentyl)cyclopentyl]-2,6-dihydroxy-phenyl}-6,6-dimethyl-2-norpinanone (14b)
The synthesis was carried out as described for 5a using 13b (1 g, 3.06 mmol), diacetates 331 (1.55 g, ca. 75% pure by 1H NMR, 4.90 mmol), and p-toluenesulfonic acid monohydrate (930 mg, 4.9 mmol) in CHCl3 (30 mL); yield: 53% (750 mg); white solid, m p 152–154°C (from CHCl3-hexane); IR (neat) 3307, 2926, 2866, 1673 (s, >C=O), 1618, 1582, 1417, 1373, 1301, 1264, 1206, 1052, 1020, 922, 836, 728 cm−1; 1H NMR (400 MHz, CDCl3) δ 6.21 (s, 2H, ArH), 5.03 (br s, 2H, OH), 3.95 (t, J = 8.2 Hz, 1H, 4-H), 3.48 (dd, J = 18.8 Hz, J = 7.8 Hz, 1H, 3α-H), 3.32 (t, J = 7.2 Hz, 2H, 6′-H), 2.61 (dd, J = 18.8 Hz, J = 8.7 Hz, 1H, 3β-H), 2.58 (t, J = 5.1 Hz, 1H, 1-H), 2.52 (m, 1H, 7α-H), 2.47 (d, J = 10.6 Hz, 1H, 7β-H), 2.31, (t, J = 5.2 Hz, 1H, 5-H), 1.88-1.60 (m, 10H, 8H of the cyclopentane ring and 2H of the 5-bromopentyl group, overlapping), 1.53-1.47 (m, 2H, 2′-H), 1.36 (s, 3H, 6-Me), 1.29 (qt, J = 9.3 Hz, 2H, 5-bromopentyl group), 1.09-0.98 (m and s overlapping, 5H, especially 1.00, s, 3H, 6-Me); mass spectrum m/z (relative intensity) 464 (M++2, 16), 462 (M+, 17), 447 (15), 421 (14), 419 (13), 393 (12), 381 (55), 379 (57), 341 (23), 339 (24), 314 (100), 299 (12), 271 (13), 243 (32), 203 (67), 189 (24), 149 (34), 83 (74). Exact mass calculated for C25H35BrO3, 462.1770; found, 462.1760. Anal. (C25H35BrO3) C, H.
(4R)-4-{4-[1-(6-Bromohexyl)cyclopentyl]-2,6-dihydroxy-phenyl}-6,6-dimethyl-2-norpinanone (14c)
The synthesis was carried out as described for 5a using 13C (800 mg, 2.35 mmol), diacetates 331 (1.2 g, ca. 75% pure by 1H NMR, 3.76 mmol), and p-toluenesulfonic acid monohydrate (887 mg, 3.76 mmol) in CHCl3 (25 mL); yield: 50% (560 mg); white solid, m p 156–158°C (from CHCl3-hexane); IR (neat) 3308, 3109, 2939, 2865, 1674 (s, >C=O), 1618, 1583, 1462, 1418, 1373, 1347, 1301, 1262, 1206, 1189, 1052, 1019, 921, 841, 725 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.22 (s, 2H, ArH), 5.01 (br s, 2H, OH), 3.95 (t, J = 8.2 Hz, 1H, 4-H), 3.49 (dd, J = 18.7 Hz, J = 7.5 Hz, 1H, 3α-H), 3.36 (t, J = 6.8 Hz, 2H, 7′-H), 2.61 (dd, J = 18.7 Hz, J = 8.6 Hz, 1H, 3β-H), 2.59 (t, J = 5.1 Hz, 1H, 1-H), 2.52 (m, 1H, 7α-H), 2.46 (d, J = 10.6 Hz, 1H, 7β-H), 2.31, (t, J = 5.3 Hz, 1H, 5-H), 1.85-1.74 (m, 4H, 2H of the cyclopentane ring and 2H of the 6-bromohexyl group, overlapping), 1.72-1.60 (m, 6H of the cyclopentane ring), 1.52-1.46 (m, 2H, 2′-H), 1.38-1.30 (s and m overlapping, 5H, especially 1.36, s, 3H, 6-Me), 1.17 (qt, J = 7.8 Hz, 2H, 6-bromohexyl group), 1.04-0.96 (s and m overlapping, 5H, especially 1.00, s, 3H, 6-Me); mass spectrum m/z (relative intensity) 478 (M++2, 17), 476 (M+, 17), 461 (18), 435 (16), 433 (16), 407 (16), 395 (67), 393 (69), 355 (24), 353 (25), 314 (91), 271 (16), 243 (35), 203 (84), 189 (32), 149 (34), 123 (23), 83 (100). Exact mass calculated for C26H37BrO3, 476.1926; found, 476.1931. Anal. (C26H37BrO3) C, H.
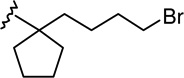
(6aR,10aR)-3-[1-(4-Bromobutyl)cyclopentyl]-6,6a,7,8,10,10a-hexahydro-1-hydroxy-6,6-dimethyl-9H-dibenzo[b,d]pyran-9-one (15a)
The synthesis was carried out analogous to the preparation of 6b using 14a (100 mg, 0.23 mmol) and trimethylsilyl trifluoromethanesulfonate (0.23 mL, 0.3M solution in CH3NO2, 0.07 mmol) in anhydrous CH2Cl2/CH3NO2 (3:1 mixture, 5 mL); yield: 75% (75 mg); white foam. IR (neat) 3279 (br, OH), 2952, 2869, 1694 (s, >C=O), 1620, 1576, 1414, 1346, 1257, 1184, 1093, 1037, 838; 1H NMR (500 MHz, CDCl3) δ 6.33 (d, J = 1.0 Hz, 1H, ArH), 6.21 (d, J = 1.1 Hz, 1H, ArH), 5.76 (br s, 1H, OH), 3.96 (m as br d, J = 15.1 Hz, 1H, 10eq-H), 3.29 (t, J = 6.7 Hz, 2H, 5′-H), 2.88 (m as td, J = 12.5 Hz, J = 3.6 Hz, 1H, 10a-H), 2.66-2.57 (m, 1H, 8eq-H), 2.50-2.42 (m, 1H, 8ax-H), 2.20-2.13 (m, 2H, 10ax-H, 7eq-H), 1.97 (m as td, J = 12.0 Hz, J = 1.7 Hz, 1H, 6a-H), 1.91-1.83 (m, 2H of the cyclopentane ring), 1.78-1.43 (m, 14H, 6H of the cyclopentane ring, 2′-H, 4′-H, 7ax-H, 6-Me, overlapping, especially 1.48, s, 6-Me), 1.19-1.02 (m and s overlapping, 5H, 3′-H, 6-Me, especially 1.13, s, 6-Me); mass spectrum m/z (relative intensity) 450 (M++2, 16), 448 (M+, 16), 435 (4), 433 (5), 407 (5), 369 (93), 314 (100), 286 (17), 273 (20), 259 (17), 245 (19), 229 (8), 204 (52), 189 (17), 163 (14), 149 (18), 123 (22), 83 (23), 69 (47). Exact mass calculated for C24H33BrO3, 448.1613; found, 448.1607. Anal. (C24H33BrO3) C, H.
(6aR,10aR)-3-[1-(5-Bromopentyl)cyclopentyl]-6,6a,7,8,10,10a-hexahydro-1-hydroxy-6,6-dimethyl-9H-dibenzo[b,d]pyran-9-one (15b)
The synthesis was carried out analogous to the preparation of 6b using 14b (200 mg, 0.43 mmol) and trimethylsilyl trifluoromethanesulfonate (0.43 mL, 0.3M solution in CH3NO2, 0.13 mmol) in anhydrous CH2Cl2/CH3NO2 (3:1 mixture, 10 mL); yield: 73% (146 mg); white foam. IR (neat) 3282 (br, OH), 2933, 2869, 1695 (s, >C=O), 1620, 1575, 1414, 1345, 1257, 1184, 1093, 1037, 837; 1H NMR (500 MHz, CDCl3) δ 6.32 (d, J = 1.1 Hz, 1H, ArH), 6.21 (d, J = 1.1 Hz, 1H, ArH), 5.80 (br s, 1H, OH), 3.96 (ddd, J = 15.0 Hz, J = 3.5 Hz, J = 2.0 Hz, 1H, 10eq-H), 3.29 (t, J = 6.8 Hz, 2H, 6′-H), 2.88 (m as td, J = 12.6 Hz, J = 3.6 Hz, 1H, 10a-H), 2.65-2.57 (m, 1H, 8eq-H), 2.49-2.40 (m, 1H, 8ax-H), 2.19-2.12 (m, 2H, 10ax-H, 7eq-H), 1.97 (m as td, J = 12.0 Hz, J = 1.8 Hz, 1H, 6a-H), 1.87-1.79 (m, 2H of the cyclopentane ring), 1.77-1.58 (m, 8H, 6H of the cyclopentane ring and 2H of the 5-bromopentyl group, overlapping), 1.57-1.45 (m and s overlapping, 6H, 7ax-H, 2′-H, 6-Me, especially 1.47, s, 6-Me), 1.28 (qt, J = 7.5 Hz, 2H, 5-bromopentyl group), 1.12 (s, 3H, 6-Me), 1.07-0.98 (m, 2H, 5-bromopentyl group); mass spectrum m/z (relative intensity) 464 (M++2, 10), 462 (M+, 10), 449 (3), 447 (3), 383 (14) 353 (10), 314 (100), 299 (7), 273 (4), 245 (8), 204 (27), 189 (8), 175 (5), 149 (13), 83 (11), 69 (37). Exact mass calculated for C25H35BrO3, 462.1770; found, 462.1767. Anal. (C25H35BrO3) C, H.
(6aR,10aR)-3-[1-(6-Bromohexyl)cyclopentyl]-6,6a,7,8,10,10a-hexahydro-1-hydroxy-6,6-dimethyl-9H-dibenzo[b,d]pyran-9-one (15c)
The synthesis was carried out analogous to the preparation of 6b using 14c (150 mg, 0.31 mmol) and trimethylsilyl trifluoromethanesulfonate (0.30 mL, 0.3 M solution in CH3NO2, 0.09 mmol) in anhydrous CH2Cl2/CH3NO2 (3:1 mixture, 7 mL); yield: 74% (111 mg); white foam. IR (neat) 3292 (br, OH), 2932, 2869, 1695 (s, >C=O), 1620, 1574, 1414, 1345, 1264, 1184, 1092, 1037, 838; 1H NMR (500 MHz, CDCl3) δ 6.32 (d, J = 1.4 Hz, 1H, ArH), 6.23 (d, J = 1.4 Hz, 1H, ArH), 5.99 (br s, 1H, OH), 3.99 (m as br d, J = 15.1 Hz, 1H, 10eq-H), 3.33 (t, J = 6.9 Hz, 2H, 7′-H), 2.89 (m as td, J = 12.6 Hz, J = 3.5 Hz, 1H, 10a-H), 2.65-2.57 (m, 1H, 8eq-H), 2.49-2.41 (m, 1H, 8ax-H), 2.19-2.13 (m, 2H, 10ax-H, 7eq-H), 1.97 (m as td, J = 12.0 Hz, J = 1.6 Hz, 1H, 6a-H), 1.87-1.80 (m, 2H of the cyclopentane ring), 1.79-1.60 (m, 8H, 6H of the cyclopentane ring and 2H of the 6-bromohexyl group, overlapping), 1.59-1.45 (m and s overlapping, 6H, 7ax-H, 2′-H, 6-Me, especially 1.47, s, 6-Me), 1.32 (qt, J = 7.5 Hz, 2H, 6-bromohexyl group), 1.16 (qt, J = 7.6 Hz, 2H, 6-bromohexyl group), 1.12 (s, 3H, 6-Me), 1.08-0.99 (m, 2H, 6-bromohexyl group); mass spectrum m/z (relative intensity) 478 (M++2, 5), 476 (M+, 5), 465 (3), 463 (3), 447 (4), 397 (10), 381 (14), 379 (15), 341 (6), 339 (7), 314 (100), 299 (4), 220 (7), 205 (42), 189 (15), 149 (18), 83 (28), 69 (43). Exact mass calculated for C26H37BrO3, 476.1926; found, 476.1918. Anal. (C26H37BrO3) C, H.
(6aR,10aR)-3-[1-(4-Cyanobutyl)cyclopentyl]-6,6a,7,8,10,10a-hexahydro-1-hydroxy-6,6-dimethyl-9H-dibenzo[b,d]pyran-9-one (16)
To a stirred solution of 15a (50 mg, 0.11 mmol) in DMSO (2 mL), at room temperature, under an argon atmosphere, was added NaCN (27 mg, 0.55 mmol). After stirring at the same temperature for 20 hours, the reaction mixture was cooled to 0°C and diluted with water. The mixture was extracted with diethyl ether and the organic layer was washed with brine, dried over MgSO4 and the solvent was removed in vacuo. The residue was purified by flash column chromatography on silica gel (70% EtOAc in hexane) to give 16 as a white foam in 64% yield (28 mg). IR (neat) 3376, 2946, 2869, 2243 (w, CN), 1695 (s, >C=O), 1621, 1575, 1509, 1453, 1385, 1355, 1258, 1184, 1136, 1093, 1037, 948, 839; 1H NMR (500 MHz, CDCl3) δ 6.32 (d, J = 1.5 Hz, 1H, ArH), 6.22 (d, J = 1.5 Hz, 1H, ArH), 5.99 (br s, 1H, OH), 3.97 (ddd, J = 15.0 Hz, J = 3.5 Hz, J = 2.0 Hz, 1H, 10eq-H), 2.88 (m as td, J = 12.7 Hz, J = 3.5 Hz, 1H, 10a-H), 2.65-2.57 (m, 1H, 8eq-H), 2.50-2.41 (m, 1H, 8ax-H), 2.23 (t, J = 7.2 Hz, 2H, 5′-H), 2.21-2.12 (m, 2H, 10ax-H, 7eq-H), 1.98 (m as td, J = 12.2 Hz, J = 1.6 Hz, 1H, 6a-H), 1.91-1.82 (m, 2H of the cyclopentane ring), 1.75-1.61 (m, 6H of the cyclopentane ring), 1.56-1.46 (m, 8H, 2′-H, 4′-H, 7ax-H, 6-Me, overlapping, especially 1.48, s, 6-Me), 1.21-1.11 (m and s overlapping, 5H, 3′-H, 6α-Me, especially 1.13, s, 6-Me); mass spectrum m/z (relative intensity) 395 (M+, 59), 380 (13), 368 (29), 353 (6), 327 (7), 314 (100), 285 (13), 272 (8), 245 (47), 227 (6), 215 (9), 203 (37), 177 (12), 149 (16), 115 (11), 91 (17), 69 (53). Exact mass calculated for C25H33NO3, 395.2460; found, 395.2456. Anal. (C25H33NO3) C, H, N.
3,5-Dimethoxy-1-[1-(1,2-cis-5-phenoxypenten-1-yl)cyclobutyl]benzene (18)
The synthesis was carried our as described for 11a using (4-phenoxybutyl)triphenylphosphonium bromide (9b) (33.13 g, 67.42 mmol) in anhydrous THF (375 mL), potassium bis(trimethylsilylamide) (13.40 g, 67.19 mmol) and a solution of aldehyde 1727 (4.95 g, 22.47 mmol) in anhydrous THF (75 mL). The crude product obtained after work up was purified by flash column chromatography on silica gel (4% diethyl ether in hexanes) to give compound 18 as a colorless liquid in 91% yield (7.2 g). 1H NMR (500 MHz, CDCl3) δ 7.25 (t, J = 8.2 Hz, 2H, 3-H, 5-H, OPh), 6.91 (t, J = 8.2 Hz, 1H, 4-H, OPh), 6.80 (d, J = 8.2 Hz, 2H, 2-H, 6-H, OPh), 6.50 (d, J = 2.2 Hz, 2H, 2-H, 6-H, ArH) 6.28 (t, J = 2.2 Hz, 1H, 4-H, ArH), 5.88 (d, J = 11.1 Hz, 1H, 2′-H), 5.31 (dt, J = 11.1 Hz, J = 7.5 Hz, 1H, 3′-H), 3.77 (s, 6H, OMe), 3.74 (t, J = 6.8 Hz, 2H, 6′-H), 2.52-2.46 (m, 2H of the cyclobutane ring), 2.39-2.33 (m, 2H of the cyclobutane ring), 2.05-2.00 (tdd, J = 7.5 Hz, J = 7.5 Hz, J = 1.5 Hz, 2H, 4′-H), 1.99-1.83 (m, 2H of the cyclobutane ring), 1.72-1.66 (quintet, J = 7.0 Hz, 2H, 5′-H); mass spectrum m/z (relative intensity) 352 (M+, 71), 324 (9), 259 (19), 231 (58), 217 (45), 203 (70), 189 (100), 177 (14), 151 (32), 77 (23). Exact mass calculated for C23H28O3, 352.2038; found, 352.2031.
3,5-Dimethoxy-1-[1-(5-phenoxypentyl)cyclobutyl]benzene (19)
To a solution of alkene 18 (7.9 g, 22.41 mmol) in absolute EtOH (80 mL) was added Pd (10% on activated carbon, 1.4 g) and the resulting suspension was hydrogenated under pressure (50 psi), at room temperature for 10 hours. The catalyst was removed by filtration through Celite and the filtrate was evaporated under vacuum to give 7.88 g (quantitative yield) of liquid alkane 19, which was used in the next step without further purification. 1H NMR (500 MHz, CDCl3) δ 7.24 (t, J = 7.5 Hz, 2H, 3-H, 5-H, OPh), 6.90 (t, J = 7.5 Hz, 1H, 4-H, OPh), 6.84 (d, J = 7.5 Hz, 2H, 2-H, 6-H, OPh), 6.27-6.25 (m, 3H, 2-H, 4-H, 6-H, ArH), 3.86 (t, J = 7 Hz, 2H, 6′-H), 3.76 (s, 6H, OMe), 2.34-2.28 (m, 2H of the cyclobutane ring), 2.09-1.98 (m, 3H of the cyclobutane ring), 1.83-1.74 (m, 3H, 1H of the cyclobutane ring and 2′-H), 1.69 (quintet, J = 7.5 Hz, 2H, 5′-H), 1.35 (quintet, J = 7.5 Hz, 2H, 4′-H), 1.12-1.06 (m, 2H, 3′-H); mass spectrum m/z (relative intensity) 354 (M+, 82), 326 (14), 277 (10), 261 (12), 233 (45), 205 (100), 192 (42), 178 (53), 165 (29), 151 (39), 77 (12). Exact mass calculated for C23H30O3, 354.2195; found, 354.2190.
5-[1-(5-Bromopentyl)cyclobutyl]resorcinol (20)
The synthesis was carried our as described for 13a using 19 (7.59 g, 21.41mmol) and boron tribromide (94 mL, 1M solution in CH2Cl2) in anhydrous CH2Cl2 (350 mL). The crude product obtained after work up was purified by flash column chromatography on silica gel (25% ethyl acetate in hexane) to give 20 as a slightly brown viscous oil in quantitative yield (6.7 g). 1H NMR (500 MHz, CDCl3) δ 6.15 (s, 3H, ArH), 4.70 (br s, 2H, OH), 3.33 (t, J = 6.5 Hz, 2H, 6′-H), 2.30-2.24 (m, 2H of the cyclobutane ring), 2.06-2.00 (m, 3H of the cyclobutane ring), 1.84-1.76 (m, 3H, 2H of 5-bromopentyl group and 1H of cyclobutane ring), 1.75-1.71 (m, 2H, 2′-H), 1.33 (quintet, J = 7.0 Hz, 2H, 5-bromopentyl group), 1.06-0.99 (m, 2H, 5-bromopentyl group); Exact mass calculated for C15H21BrO2, 312.0725; found, 312.0731.
(4R)-4-{4-[1-(5-Bromopentyl)cyclobutyl]-2,6-dihydroxy-phenyl}-6,6-dimethyl-2-norpinanone (21)
The synthesis was carried out as described for 5a using 20 (5.8 g, 18.5 mmol), diacetates 331 (6.86 g, ca. 90% pure by 1H NMR, 28.8 mmol), and p-toluenesulfonic acid monohydrate (4.93 g, 25.9 mmol) in CHCl3 (370 mL); yield: 46% (3.8 g); white solid, m p 164–165°C (dec.) (from CHCl3-hexane); IR (neat) 3368 (OH), 2936 (CH aromatic), 1684(>C=O), 1620, 1585 cm−1. 1H NMR (500 MHz, CDCl3) δ 6.06 (s, 2H, 3-H, 5-H, ArH), 5.04 (br s, 2H, OH), 3.95 (t, J = 8.1 Hz, 1H, 4-H), 3.49 (dd, J = 18.7 Hz, J = 7.8 Hz, 1H, 3α-H), 3.35 (t, J = 6.5 Hz, 2H, 6′-H), 2.61 (dd, J = 18.7 Hz, J = 8.6 Hz, 1H, 3β-H), 2.59 (t, J = 5.0 Hz, 1H, 1-H), 2.52 (m, 1H, 7α-H), 2.47 (d, J = 10.6 Hz, 1H, 7β-H), 2.31 (t, J = 5.4 Hz, 1H, 5-H), 2.28-2.20 (m, 2H of the cyclobutane ring), 2.06-1.95 (m, 3H of the cyclobutane ring), 1.84-1.74 (m, 3H, 5′-H, cyclobutane ring), 1.72-1.67 (m, 2H, 2′-H), 1.37 (s, 3H, 6-Me), 1.34 (quintet, J = 8.0 Hz, 2H, 4′-H), 1.08-1.01 (m, 2H, 3′-H), 1.00 (s, 3H, 6-Me); 13C NMR (175 MHz, CDCl3) δ 217.75 (>C=O), 154.95 (ArC-2, ArC-6), 150.44 (ArC-4), 113.49 (ArC-1), 106.49 (ArC-3, ArC-5), 58.11 (C-1), 46.95 (C-5), 46.17 (C-1′), 42.38 (C-6 or C-2′), 42.34 (C-2′ or C-6), 38.09 (C-3), 34.22 (C-6′), 32.93 (cyclobutane ring or C-5′), 32.85 (cyclobutane ring or C-5′), 32.81 (cyclobutane ring or C-5′), 29.67 (C-4), 28.76 (C-4′), 26.35 (6-Me), 24.62 (C-7), 24.04 (C-3′), 22.34 (6-Me), 15.95 (cyclobutane ring); mass spectrum m/z (relative intensity) 450 (M++2, 55), 448 (M+, 55), 69 (100). Exact mass calculated for C24H33BrO3, 448.1613; found, 448.1621.
(6aR,10aR)-3-[1-(5-Bromopentyl)cyclobutyl]-6,6a,7,8,10,10a-hexahydro-1-hydroxy-6,6-dimethyl-9H-dibenzo[b,d]pyran-9-one (22)
The synthesis was carried out analogous to the preparation of 6b using 21 (2.0 g, 4.45 mmol) and trimethylsilyl trifluoromethanesulfonate (4.45 mL, 0.3 M solution in CH3NO2, 1.34 mmol) in anhydrous CH2Cl2/CH3NO2 (3:1 mixture, 90 mL); yield: 70% (1.4 g); white foam; m p 69–70°C; IR (neat) 3292 (OH), 2932 (CH aromatic), 1693(>C=O), 1619, 1575 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.17 (d, J = 1.5 Hz, 1H, ArH), 6.03 (d, J = 1.5 Hz, 1H, ArH), 5.69 (br s, 1H, OH), 3.95 (ddd, J = 15.0 Hz, J = 3.5 Hz, J = 2.0 Hz, 1H, 10eq-H), 3.33 (t, J = 7.0 Hz, 2H, 6′-H), 2.88 (ddd, J = 14.0 Hz, J = 13.5 Hz, J = 3.5 Hz, 1H, 10a-H), 2.65-2.58 (m, 1H, 8eq-H), 2.45 (ddd, J = 15.5 Hz, J = 13.5 Hz, J = 7.5 Hz, 1H, 8ax-H), 2.32-2.23 (m, 2H, cyclobutane ring), 2.21-2.13 (m, 2H, 10ax-H, 7eq-H), 2.22-1.92 (m, 4H, 6a-H, cyclobutane ring), 1.82-1.74 (m, 3H, 5′-H, cyclobutane ring), 1.73-1.68 (m, 2H, 2′-H), 1.52 (dddd, J = 15.0 Hz, J = 13.5 Hz, J = 13.0 Hz, J = 5.0 Hz, 1H, 7ax-H), 1.48 (s, 3H, 6-Me), 1.32 (quintet, J = 7.5 Hz, 2H, 4′-H), 1.13 (s, 3H, 6-Me), 1.08-1.00 (m, 2H, 3′-H); 13C NMR (100 MHz, CDCl3) δ 214.93 (>C=O), 155.43 (ArC), 154.53 (ArC), 151.43 (ArC), 107.96 (ArC), 107.20 (ArC-2 or ArC-4), 105.57 (ArC-4 or ArC-2), 47.76 (C-6a), 46.54 (C-1′), 45.35 (C-10), 42.48 (C-2′), 41.18 (C-8), 35.16 (C-10a), 34.38 (C-6′), 33.12 (cyclobutane ring or C-5′), 33.07 (cyclobutane ring or C-5′), 32.96 (cyclobutane ring or C-5′), 28.95 (C-4′), 28.21 (6-CH3), 27.25 (C-7), 24.14 (C-3′), 19.23 (6-CH3), 16.12 (cyclobutane ring); mass spectrum m/z (relative intensity) 450 (M++2, 77), 448 (M+, 77), 422 (33), 420 (33), 369 (82), 313 (78), 203 (55), 69 (100). Exact mass calculated for C24H33BrO3, 448.1613; found, 448.1606. Anal. (C24H33BrO3) C, H.
(6aR, 9R, 10aR)-3-[1-(5-Bromopentyl)cyclobutyl]-6a,7,8,9,10,10a-hexahydro-6,6-dimethyl-6H-dibenzo[b,d]pyran-1,9 diol (23)
The synthesis was carried out analogous to the preparation of 7c-β using 22 (200 mg, 0.445 mmol) and NaBH4 (59 mg, 1.56 mmol) in anhydrous methanol (10 mL). On the basis of 1H NMR analysis, the crude material obtained after the workup is a mixture of two isomeric alcohols 24 (Rf = 0.30, 40% AcOEt in hexane) and 23 (Rf = 0.27, 40% AcOEt in hexane) in a ratio 3:97, respectively. Purification by flash column chromatography on silica gel (45% AcOEt in hexane) gave, in order of elution, 24 mg of a mixture of 24 and 23 (1:3 ratio by 1H NMR) and 153 mg of pure 23 as a glassy substance (m p = 59–60°C). Overall yield for 23: 85% (171 mg); IR (neat) 3328 (OH), 2932 (CH aromatic), 1622, 1574 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.15 (d, J = 2.0 Hz, 1H, ArH), 5.97 (d, J = 2.0 Hz, 1H, ArH), 5.09 (br s, 1H, ArOH), 3.86 (dddd, J = 15.5 Hz, J = 15.0 Hz, J = 4.5 Hz, J = 4.5 Hz, 1H, 9ax-H, peak half-width = 22 Hz), 3.47 (dddd, J = 12.0 Hz, J = 4.5 Hz, J = 2.5 Hz, J = 2.0 Hz, 1H, 10eq-H), 3.35 (t, J = 7.0 Hz, 2H, 6′-H), 2.48 (ddd, J = 12.0 Hz, J = 11.5 Hz, J = 2.5 Hz, 1H, 10a-H), 2.32-2.23 (m, 2H, cyclobutane ring), 2.17 (m as br d, J = 9.0 Hz, 1H, 8eq-H), 2.04-1.93 (m, 3H, cyclobutane ring), 1.90 (dddd, J = 13.5 Hz, J = 3.5 Hz, J = 2.5 Hz, J = 2.0 Hz, 1H, 7eq-H), 1.82-1.74 (m and quintet overlapping, 3H, 5′-H, cyclobutane ring, especially 1.78, quintet, J = 7.0 Hz, 5′-H), 1.69 (m, 2H, 2′-H), 1.51 (ddd, J = 12.0 Hz, J = 11.5 Hz, J = 2.5 Hz, 1H, 6a-H), 1.42 (dddd, J = 15.5 Hz, J = 15.0 Hz, J = 13.0 Hz, J = 3.5 Hz, 1H, 8ax-H), 1.39 (s, 3H, 6-Me), 1.32 (quintet, J = 8.0 Hz, 2H, 4′-H), 1.16 (dddd, J = 13.5 Hz, J = 13.0 Hz, J = 12.0 Hz, J = 3.5 Hz, 1H, 7ax-H), 1.12 (ddd, J = 15.5 Hz, J = 12.0 Hz, J = 12.0 Hz, 1H, 10ax-H), 1.08 (s, 3H, 6-Me), 1.07-1.00 (m, 2H, 3′-H); 13C NMR (100 MHz, CDCl3) δ 154.92 (ArC), 154.43 (ArC), 150.86 (ArC), 109.52 (ArC), 108.05 (C-2 or C-4), 106.84 (C-4 or C-2), 71.43 (C-9), 48.47 (C-6a), 46.22 (C-6), 41.72 (C-2′), 38.61 (C-10), 35.98 (C-8), 34.26 (C-6′), 34.21 (C-1′), 33.41 (C-10a), 33.02 (C-5′), 32.65 (cyclobutane ring), 29.32 (C-4′), 27.73 (6-Me), 26.22 (C-7), 24.03 (C-3′), 19.23 (6-Me), 15.92 (cyclobutane ring); mass spectrum m/z (relative intensity) 452 (M++2, 45), 450 (M+, 45), 422 (23), 406 (119), 315 (57), 189 (38), 62 (100). Exact mass calculated for C24H35BrO3, 450.1770; found, 450.1775. Anal. (C24H35BrO3) C, H.
(6aR, 9S, 10aR)-3-[1-(5-Bromopentyl)cyclobutyl]-6a,7,8,9,10,10a-hexahydro-6,6-dimethyl-6H-dibenzo[b,d]pyran-1,9 diol (24)
To a stirred solution of ketone 22 (703 mg, 1.56 mmol) in anhydrous THF (80 mL) was added K-Selectride (4.69 mL, 1M solution in THF), under an argon atmosphere, at −78°C. Stirring was continued at that temperature for 2 hours and then the reaction was quenched by the addition of brine. The organic phase was separated and the pH of the aqueous phase was adjusted to 4–5 using a 5% solution of hydrochloric acid. The aqueous phase was extracted with EtOAc and the combined organic layer was washed with brine, dried over MgSO4 and evaporated to give an oily residue, which was further purified by Biotage column chromatography (30% ethyl acetate in hexanes) to afford 490 mg (70% yield) of 24 as a white solid; m p 41–43°C. IR (neat) 3337 (OH), 2930 (CH aromatic), 1620, 1568 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.14 (br s, 1H, ArOH), 6.14 (d, J = 1.5 Hz, 1H, ArH), 6.10 (d, J = 1.5 Hz, 1H, ArH), 4.30 (m as br s, 1H, 9eq-H, peak half-width = 10.5 Hz), 3.33 (t, J = 7.0 Hz, 2H, 6′-H), 3.25 (ddd, J = 14.5 Hz, J = 3.0 Hz, J = 2.5 Hz, 1H, 10eq-H), 2.95 (ddd, J = 12.0 Hz, J = 11.0 Hz, J = 2.5 Hz, 1H, 10a-H), 2.30-2.21 (m, 2H, cyclobutane ring), 2.02-1.92 (m, 4H, 8eq-H, cyclobutane ring), 1.82-1.72 (m, 3H, 5′-H, cyclobutane ring), 1.71-1.60 (m, 4H, 2′-H, 8ax-H, 7-H), 1.57-1.49 (m, 2H, 6a-H, 7-H), 1.42-1.26 (m, s and quintet overlapping, 6H, 6-Me, 4′-H, 10ax-H, especially 1.37, s, 6-Me and 1.30, quintet, J = 7.5 Hz, 4′-H), 1.08-0.98 (m and s overlapping, 5H, 6-Me, 3′-H, especially 1.04, s, 6-Me); 13C NMR (175 MHz, CDCl3) δ 154.80 (ArC), 154.39 (ArC), 150.46 (ArC), 109.46 (ArC), 107.85 (C-2 or C-4), 105.96 (C-4 or C-2), 67.33 (C-9), 49.46 (C-6a), 46.29 (C-6), 42.34 (C-2′), 36.48 (C-10), 34.23 (C-6′), 33.47 (C-8), 32.93 (C-5′, C-1′), 32.68 (cyclobutane ring), 29.00 (C-10a), 28.78 (C-4′), 27.69 (6-Me), 23.95 (C-3′), 22.84 (C-7), 19.12 (6-Me), 15.93 (cyclobutane ring); mass spectrum m/z (relative intensity) 452 (M++2, 53), 450 (M+, 53), 422 (18), 406 (15), 389 (12), 371 (54), 315 (42), 189 (31), 149 (40), 62 (100). Exact mass calculated for C24H35BrO3, 450.1770; found, 450.1770. Anal. (C24H35BrO3) C, H.
Radioligand Binding Assays
Forebrain synaptosomal membranes were prepared from frozen rat brains by the method described by Dodd et al.36 and were used to assess the affinities of the novel analogs for the CB1 binding sites, while affinities for the CB2 sites were measured using a membrane preparation from frozen mouse spleen using a similar procedure.37 Membrane preparations from HEK293 cells expressing human CB2 (hCB2) receptor were used to assess the affinities of representative analogs for hCB2.33 The displacement of specifically tritiated CP-55,940 from these membranes was used to determine the IC50 values for the test compounds. The assay was conducted in a 96-well microfilter plate. The samples were filtered using a Packard Filtermate Harvester and Whatman GF/B unifilter-96 plates, and 0.5% BSA was incorporated into the wash buffer. Radioactivity was detected using MicroScint 20 scintillation cocktail added to the dried filter plates and was counted using a Packard Instruments Top Count. Data were collected from three independent experiments between 100% and 0% specific binding for [3H]CP-55,940, determined using 0 and 100 nM CP-55,940. The normalized data from three independent experiments were combined and analyzed using a four-parameter logistic equation to yield IC50 values that were converted to Ki values using the assumptions of Cheng and Prussoff.38
Molecular Modeling
Compound 7e-β underwent torsional sampling using the MCMM (Monte Carlo multiple minimum) protocol39,40 in MacroModel.41The search was conducted using the OPLS_2005 force field42 in a GB/SA water model43 with a constant dielectric of 1.0 and an extended cutoff. An energy window of 21.0 kJmol−1 (5 kcal mol−1) was employed and redundant conformers were eliminated using a RMSD cutoff of 0.5 Å for all atoms.
cAMP Assay
HEK293 cells stably expressing rCB1 receptor were used for the studies. The cAMP assay was carried out using PerkinElmer’s Lance ultra cAMP kit following the protocol of the manufacturer. Briefly, the assay was carried out in a 384-well plate using 1000 cells/well. The cells were harvested with non-enzymatic cell dissociation reagent Versene and they were washed once with HBSS and resuspended in the stimulation buffer. The various concentrations of 7e-β (5 µl) in forskolin (2 µM final concentration) containing stimulation buffer were added to the plate followed by the cell suspension (5 µl). The cells were stimulated for 30 min at room temperature. Then Eu-cAMP tracer working solution (5 µl) and Ulight-anti-cAMP working solution (5 µl) were added to the plate and incubated at room temperature for 60 minutes. The plate was read on a PerkinElmer Envision instrument. The EC50 was determined by non-linear regression analysis using GraphPad Prism software (GraphPad Software, Inc., San Diego, CA).
Methods for In Vivo Behavioral Characterization of 7e-β
Subjects
Female Sprague-Dawley rats (n=5–6/group), weighing between 200 and 350g (Charles River, Wilmington MA). Rats were tested repeatedly with at least three days intervening between drug sessions. Experiments occurred at roughly the same time (10:00 am–5:00 pm) during the light portion of the daily light/dark cycle. Outside of experimental sessions rats were group housed (2/cage) in a climate controlled vivarium, and they had unrestricted access to food and water except during the 6hr test sessions.
Procedure
Temperature was recorded using a thermistor probe (Model 401, Measurement Specialties, Inc., Dayton, OH) inserted to a depth of 7.5cm and secured to the tail with micropore tape. Rats were minimally restrained and isolated in 38×50×10cm plastic stalls. Temperature was read to the nearest 0.01°C using a Thermometer (Model 4000A, Measurement Specialties, Inc.). Two baseline temperature measures were recorded at 15 min intervals, and drugs were injected immediately after the second baseline was recorded. After injection, temperature was recorded every 30 min for three hours and every hour thereafter for a total of six hours. The change in temperature was determined for each rat by subtracting temperature readings from the average of the two baseline measures.
Antinociception was measured using modified version of the tail-flick procedure of D’Amour and Smith.44 Radiant heat from a halogen lamp was focused on the tail using a commercial tail-flick apparatus (Model#LE7106, Harvard Apparatus, Holliston, MA); movement of the tail activated a photocell, tuning off the lamp and a reaction timer. The lamp intensity was adjusted to yield baseline values of 2–3 sec, and a maximum latency of 6.0 sec was imposed to avoid damage to the tail. Two baseline tail-flick latencies were obtained in each rat at 10 min intervals, and drugs were injected immediately after the second baseline was recorded. Tail-flick responses were recorded at 30, 60, 120, 180, and 360 min after injection.
Drugs
Compound 7e-β was initially dissolved in a solution of 20% ethanol, 20% alkamuls, and 60% saline, and was further diluted with saline. Morphine sulfate was dissolved in saline. Injections were administered s.c. in a volume of 1.0 ml/kg, and drug doses are expressed as the weight of the free base.
Data Analysis
For each rat, the two baseline values recorded prior to drug injection were averaged to obtain a single baseline value. Temperatures recorded after drug injection, are expressed as a change from baseline, calculated for each animal by subtracting the baseline temperature from the temperatures recorded post-injection. Tail-flick responses are expressed as a percentage of the maximum possible effect (%MPE) calculated according to the equation: 100* (test latency–baseline latency)/(6-baseline latency); where 6 represents the cut-off latency. Dose-effect functions were constructed using the maximum effect recorded in each rat at a given dose of drug. Group means and SEM were calculated and time- effect functions were analyzed using two way repeated measures ANOVA procedures followed by Bonferroni’s post-hoc test; dose-effect functions were analyzed using one-way repeated measures ANOVA procedures followed by Dunnett’s multiple comparison t-test; p was set at <0.05 and statistical analyses were performed using GraphPad Prism 5.03 (GraphPad Software, San Diego, CA).
Supplementary Material
Acknowledgment
The authors are grateful to Pusheng Fan for the radioligand binding assays. This work was supported by grants from the National Institute on Drug Abuse, DA009158, DA003801, and DA007215.
Abbreviations
- CB1
cannabinoid receptor 1
- CB2
cannabinoid receptor 2
- SAR
structure-activity relationship
- (−)-Δ8-THC
(−)-Δ8-tetrahydrocannabinol
- PH
phenolic hydroxyl
- SC
side chain
- HHC
hexahydrocannabinol
- p-TSA
p-toluenesulfonic acid
- K-selectride
potassium tri-sec-butyl borohydride
- TMSOTf
trimethylsilyl triflate
- DMSO
dimethylsulfoxide
- HEK
human embryonic kidney
- NMR
nuclear magnetic resonance
- DEPT
Distortionless Enhancement by Polarization Transfer
- COSY
Correlation Spectroscopy
- HSQC
Heteronuclear Single Quantum Correlation
- HMBC
Heteronuclear Multiple Bond Correlation
- NOESY
Nuclear Overhauser Effect Spectroscopy.
Footnotes
Supporting Information Available
Elemental analysis results for compounds 5a–5e, 6a–6e, 14a–14c, 15a–15c, 16, 22, 7c-β, 7e-β, 23 and 24 as well as 1H NMR, 13C NMR, 13C DEPT, COSY, HSQC, HMBC, and NOESY spectra of compound 5b in CDCl3 solutions. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Devane WA, Dysarz FA, Johnson MR, Melvin LS, Howlett AC. Determination and characterization of a cannabinoid receptor in rat brain. Mol. Pharmacol. 1988;34:605–613. [PubMed] [Google Scholar]
- 2.Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 3.Matsuda LA, Lolait SJ, Brownstain MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 4.Gerard C, Mollereau C, Vassart G, Parmentier M. Nucleotide sequence of human cannabinoid receptor cDNA. Nucleic Acids Res. 1990;18:7142. doi: 10.1093/nar/18.23.7142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abood ME, Ditto KE, Noel MA, Showalter VM, Tao Q. Isolation and expression of a mouse CB1 cannabinoid receptor gene. Comparison of binding properties with those of native CB1 receptors in mouse brain and N18TG2 neuroblastoma cells. Biochem. Pharmacol. 1997;53:207–214. doi: 10.1016/s0006-2952(96)00727-7. [DOI] [PubMed] [Google Scholar]
- 6.Howlett AC. Cannabinoid inhibition of adenylate cyclase. Mol. Pharmacol. 1985;27:429–436. [PubMed] [Google Scholar]
- 7.Pertwee RG. Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol. Ther. 1997;74:129–180. doi: 10.1016/s0163-7258(97)82001-3. [DOI] [PubMed] [Google Scholar]
- 8.Howlett AC, Mukhopadhyay S. Cellular signal transduction by anandamide and 2-arachidonylglycerol. Chem. Phys. Lipids. 2000;108:53–70. doi: 10.1016/s0009-3084(00)00187-0. [DOI] [PubMed] [Google Scholar]
- 9.Chakrabarti A, Onaivi ES, Chaudhuri G. Cloning and sequencing of a cDNA encoding the mouse brain-type cannabinoid receptor protein. DNA Seq. 1995;5:385–388. doi: 10.3109/10425179509020870. [DOI] [PubMed] [Google Scholar]
- 10.Shire D, Calandra B, Delpech M, Dumont X, Kaghad M, LeFur G, Caput D, Ferrar P. Structural features of the central cannabinoid CB1 receptor involved in the binding of the specific CB1 antagonist SR141716A. J. Biol. Chem. 1996;271:6941–6946. doi: 10.1074/jbc.271.12.6941. [DOI] [PubMed] [Google Scholar]
- 11.Shire D, Calandra B, Rinaldi Carmona M, Oustric D, Pessegue B, Bonnin Cabanne O, Le Fur G, Caput D, Ferrara P. Molecular cloning, expression and function of the murine CB2 peripheral cannabinoid receptor. Biochim. Biophys. Acta. 1996;1307:132–136. doi: 10.1016/0167-4781(96)00047-4. [DOI] [PubMed] [Google Scholar]
- 12.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 13.Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, Yamashita A, Waku K. 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem. Biophys. Res. Commun. 1995;215:89–97. doi: 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]
- 14.Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminsky NE, Schatz AR, Gopher A, Almog S, Martin BR, et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- 15.Thakur GA, Nikas SP, Li C, Makriyannis A. Structural Requirements for Cannabinoid Receptor Probes. In: Pertwee RG, editor. Handbook of Experimental Pharmacology. Vol. 168. NY: Springer-Verlag; 2005. pp. 209–246. (Cannabinoids) [DOI] [PubMed] [Google Scholar]
- 16.Thakur GA, Nikas SP, Makriyannis A. CB1 cannabinoid receptor ligands. Mini Rev. Med. Chem. 2005;5:631–640. doi: 10.2174/1389557054368772. [DOI] [PubMed] [Google Scholar]
- 17.Pavlopoulos S, Thakur GA, Nikas SP, Makriyannis A. Cannabinoid receptors as therapeutic targets. Curr. Pharm. Des. 2006;12:1751–1769. doi: 10.2174/138161206776873743. [DOI] [PubMed] [Google Scholar]
- 18.Thakur GA, Duclos RI, Jr, Makriyannis A. Natural cannabinoids: Templates for drug discovery. Life Sci. 2005;78:454–466. doi: 10.1016/j.lfs.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 19.Lu D, Vemuri K, Duclos RI, Jr, Makriyannis A. The cannabinergic system as a target for anti-inflammatory therapies. Curr. Top. Med. Chem. 2006;6:1401–1426. doi: 10.2174/15680266106061401. [DOI] [PubMed] [Google Scholar]
- 20.Adams R, Harfenist M, Lowe S. New analogs of tetrahydrocannabinol. XIX. J. Am. Chem. Soc. 1949;71:1624–1628. [Google Scholar]
- 21.Archer RA, Blanchard WB, Day WA, Johnson DW, Lavagnino ER, Ryan CW, Baldwin JE. Cannabinoids. 3. Synthetic approaches to 9-ketocannabinoids. Total synthesis of nabilone. J.Org. Chem. 1977;42:2277–2284. doi: 10.1021/jo00433a020. [DOI] [PubMed] [Google Scholar]
- 22.Wilson RS, May EL, Martin BR, Dewey WL. 9-Nor-9-hydroxyhexahydrocannabinols. Synthesis, some behavioral and analgesic properties, and comparison with the tetrahydrocannabinols. J. Med. Chem. 1976;19:1165–1167. doi: 10.1021/jm00231a017. [DOI] [PubMed] [Google Scholar]
- 23.Papahatjis DP, Kourouli T, Abadji V, Goutopoulos A, Makriyannis A. Pharmacophoric requirements for cannabinoid side chains: multiple bond and C1′-substituted Δ8-tetrahydrocannabinols. J. Med. Chem. 1998;41:1195–1200. doi: 10.1021/jm970277i. [DOI] [PubMed] [Google Scholar]
- 24.Papahatjis DP, Nikas SP, Andreou T, Makriyannis A. Novel 1′,1′-chain substituted Δ8-tetrahydrocannabinols. Bioorg. Med. Chem. Lett. 2002;12:3583–3586. doi: 10.1016/s0960-894x(02)00785-0. [DOI] [PubMed] [Google Scholar]
- 25.Papahatjis DP, Nikas SP, Kourouli T, Chari R, Xu W, Pertwee RG, Makriyannis A. Pharmacophoric requirements for the cannabinoid side chain. Probing the cannabinoid receptor subsite at C1′. J. Med. Chem. 2003;46:3221–3229. doi: 10.1021/jm020558c. [DOI] [PubMed] [Google Scholar]
- 26.Papahatjis DP, Nahmias VR, Andreou T, Fan P, Makriyannis A. Structural modifications of the cannabinoid side chain towards C3-aryl and 1′,1′-cycloalkyl-1′-cyano cannabinoids. Bioorg. Med. Chem. Lett. 2006;16:1616–1620. doi: 10.1016/j.bmcl.2005.12.026. [DOI] [PubMed] [Google Scholar]
- 27.Papahatjis DP, Nahmias VR, Nikas SP, Andreou T, Alapafuja SO, Tsotinis A, Guo J, Fan P, Makriyannis A. C1′-cycloalkyl side chain pharmacophore in tetrahydrocannabinols. J. Med. Chem. 2007;50:4048–4060. doi: 10.1021/jm070121a. [DOI] [PubMed] [Google Scholar]
- 28.Durdagi S, Kapou A, Kourouli T, Andreou T, Nikas SP, Nahmias VR, Papahatjis DP, Papadopoulos MG, Mavromoustakos T. The application of 3D-QSAR studies for novel cannabinoid ligands substituted at the C1′ position of the alkyl side chain on the structural requirements for binding to cannabinoid receptors CB1 and CB2. J. Med. Chem. 2007;50:2875–2885. doi: 10.1021/jm0610705. [DOI] [PubMed] [Google Scholar]
- 29.(a) Makriyannis A, Nikas SP, Khanolkar AD. Novel bicyclic and tricyclic cannabinoids (University of CT, U.S.A) 2003/005960 A2. WO Patent Application. 2003:53.; (b) Tai S, Järbe TUC, Nikas SP, Makriyannis A. In vivo characterization of AM2389, a potent CB1R selective agonist. symposium on the cannabinoids; International Cannabinoid Research Society; Aviemore, Scotland. 2008. p. 117. [Google Scholar]
- 30.Papahatjis DP, Nikas S, Tsotinis A, Vlachou M, Makriyannis A. A new ring-forming methodology for the synthesis of conformationally constrained bioactive molecules. Chem. Lett. 2001;3:192–193. [Google Scholar]
- 31.Nikas SP, Thakur GA, Parrish D, Alapafuja SO, Huestis MA, Makriyannis A. A concise methodology for the synthesis of (−)-Δ9-tetrahydrocannabinol and (−)-Δ9-tetrahydrocannabivarin metabolites and their regiospecifically deuterated analogs. Tetrahedron. 2007;63:8112–8123. [Google Scholar]
- 32.Nikas SP, Grzybowska J, Papahatjis DP, Charalambous A, Banijamali AR, Chari R, Fan P, Kourouli T, Lin S, Nitowski AJ, Marciniak G, Guo Y, Li X, Wang C-L,J, Makriyannis A. The role of halogen substitution in classical cannabinoids: a CB1 pharmacophore model. AAPS J. 2004;6:e30. doi: 10.1208/aapsj060430. ( http://www.aapsj.org). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dixon DD, Sethumadhavan D, Benneche T, Banaag AR, Tius MA, Thakur GA, Bowman A, Wood JT, Makriyannis A. Heteroadamantyl cannabinoids. J. Med. Chem. 2010;53:5656–5666. doi: 10.1021/jm100390h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khanolkar AD, Lu D, Ibrahim M, Duclos RI, Jr, Thakur GA, Malan TP, Jr, Porreca F, Veerappan V, Tian X, George C, Parrish DA, Papahatjis DP, Makriyannis A. Cannabilactones: a novel class of CB2 selective agonists with peripheral analgesic activity. J. Med. Chem. 2007;50:6493–6500. doi: 10.1021/jm070441u. [DOI] [PubMed] [Google Scholar]
- 35.Mukherjee S, Adams M, Whiteaker K, Daza A, Kage K, Cassar S, Meyer M, Yao BB. Species comparison and pharmacological characterization of rat and human CB2 cannabinoid receptors. Eur. J. Pharmacol. 2004;505:1–9. doi: 10.1016/j.ejphar.2004.09.058. [DOI] [PubMed] [Google Scholar]
- 36.Dodd PR, Hardy JA, Oakley AE, Edwardson JA, Perry EK, Delannoy JP. A rapid method for preparing synaptosomes comparison with alternative procedures. Brain Res. 1981;226:107–118. doi: 10.1016/0006-8993(81)91086-6. [DOI] [PubMed] [Google Scholar]
- 37.Khanolkar DA, Abadji V, Lin S, Hill WA, Taha G, Abouzid K, Meng Z, Fan P, Makriyannis A. Head group analogues of arachidonylethanolamide, the endogenous cannabinoid ligand. J. Med. Chem. 1996;39:4515–4519. doi: 10.1021/jm960152y. [DOI] [PubMed] [Google Scholar]
- 38.Cheng YC, Prussoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (K50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3102. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 39.Chang G, Guida WC, Still WC. An internal-coordinate Monte Carlo method for searching conformational space. J. Am. Chem. Soc. 1989;111:4379–4386. [Google Scholar]
- 40.Saunders M, Houk KN, Wu YD, Still WC, Lipton M, Chang G, Guida WC. Conformations of cycloheptadecane. A comparison of methods for conformational searching. J. Am. Chem. Soc. 1990;112:1419–1427. [Google Scholar]
- 41.MacroModel, version 9.7. New York, NY: Schrödinger, LLC; 2009. [Google Scholar]
- 42.Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B. 2001;105:6474–6487. [Google Scholar]
- 43.Still WC, Tempczyk A, Hawley RC, Hendrickson T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc. 1990;112:6127–6129. [Google Scholar]
- 44.D’Amour FE, Smith DL. A method for determining loss of pain sensation. J. Pharmacol. Expt. Ther. 1941;72:74–79. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.