

Abstract
Using quantum mechanics/molecular mechanics calculations and the 1.9-Å crystal structure of Photosystem II [Umena Y, Kawakami K, Shen J-R, Kamiya N (2011) Nature 473(7345):55–60], we investigated the H-bonding environment of the redox-active tyrosine D (TyrD) and obtained insights that help explain its slow redox kinetics and the stability of TyrD•. The water molecule distal to TyrD, located ∼4 Å away from the phenolic O of TyrD, corresponds to the presence of the tyrosyl radical state. The water molecule proximal to TyrD, in H-bonding distance to the phenolic O of TyrD, corresponds to the presence of the unoxidized tyrosine. The H+ released on oxidation of TyrD is transferred to the proximal water, which shifts to the distal position, triggering a concerted proton transfer pathway involving D2-Arg180 and a series of waters, through which the proton reaches the aqueous phase at D2-His61. The water movement linked to the ejection of the proton from the hydrophobic environment near TyrD makes oxidation slow and quasiirreversible, explaining the great stability of the TyrD•. A symmetry-related proton pathway associated with tyrosine Z is pointed out, and this is associated with one of the Cl− sites. This may represent a proton pathway functional in the water oxidation cycle.
Keywords: oxygen-evolving complex, proton-coupled electron transfer, reaction center evolution, controlling electron transfer rate, hydrogen bond direction switching
The heart of the Photosystem II (PSII) reaction center consists of the D1 and D2 subunits. These form a quasi-symmetrical complex that contains cofactors arranged to span the transmembrane protein in two branches. From the luminal side to the stromal side of the complex, the following cofactors are present: an overlapping pair of chlorophyll a (Chla) molecules (PD1/PD2), two monomeric Chla molecules (ChlD1/ChlD2), two pheophytins, and two quinone molecules (the most recent crystal structure is described in ref. 1). Excitation of Chla leads to charge separation on the D1 branch and formation of the cationic [PD1/PD2]•+ state (reviewed in refs. 2, 3). Extending the symmetry to the luminal side, there are two redox-active tyrosine residues (4–6), D1-Tyr161 [tyrosine Z (TyrZ)] and D2-Tyr160 [tyrosine D (TyrD)], that can provide electrons to [PD1/PD2]•+. TyrZ, which has D1-His190 as an H-bond partner, is the kinetically competent tyrosine that mediates proton-coupled electron transfer from Mn4CaO5 to [PD1/PD2]•+ (P680•+). The TyrD is not kinetically competent and plays no obligatory role in enzyme function; indeed, when TyrD is replaced by Phe, enzyme function appeared to be unaffected (5, 6). Nevertheless, TyrD is likely to play specific roles beneficial for PSII function (reviewed in ref. 7). TyrD oxidizes Mn4CaO5 in the S0 state to form S1 (8) and oxidizes overreduced states of Mn4CaO5 (9), and this may be relevant to the oxidative assembly of the Mn4CaO5 cluster (6, 8, 10). It has also been proposed that TyrD has an electrostatic influence on energetics of the cationic [PD1/PD2]•+ state (2, 7, 11).
The neutral radical, TyrD-O•, is formed on oxidation of TyrD-OH by [PD1/PD2]•+ (7, 12–14), and this occurs on a time scale in the tens of milliseconds (11, 15), so slowly that it does not compete with rapid electron donation from TyrZ (which occurs on a time scale in the tens to hundreds of nanoseconds). In the functional enzyme, TyrD oxidation occurs when the reversible intermediates of the water-splitting cycle, the so-called “S2 or S3 states,” equilibrate with TyrZ-O•, and thence [PD1/PD2]•+, allowing the slow electron donation from the TyrD, forming the stable radical TyrD-O•. At higher pH values (pKa 7.6), TyrD oxidation rate speeds up dramatically, with TyrD-O• formation occurring at rates comparable to those seen for TyrZ-O• (11). Remarkably, under these high-pH conditions, TyrD oxidation can occur at the temperature of liquid helium, indicating a barrier-less proton transfer (16, 17).
Once formed, TyrD-O• is very stable for many hours under physiological conditions, giving rise to the term “Signal II slow/dark” describing the EPR signal from TyrD-O• (7, 12, 13). In contrast, TyrZ-O• is reduced by Mn4CaO5 on a time scale in the tens of microseconds to milliseconds (12, 13). It has been suggested that the proton released from TyrD-OH on oxidation remains near TyrD-O• (e.g., ref. 7), and the crystallographic models showing a hydrophobic environment appear to be consistent with that suggestion (1, 18, 19). It had been generally assumed that both TyrZ and TyrD underwent oxidation with the simultaneous transfer of the phenolic proton to a base, D1-His190 and D2-His189, respectively, as originally suggested by Debus et al. (5). However, FTIR studies suggested a proton carrier other than D2-His189 (20) could play a role in the redox properties of TyrD and water could be the proton acceptor from TyrD (21). Exchangeable protons near TyrD-O• have also been detected by advanced EPR methods (e.g., refs. 22, 23). The TyrD-O• EPR signal was lost and/or significantly modified and PSII photochemistry was perturbed when D2-Arg180 was mutated, and this led Manna et al. (24) to propose that D2-Arg180 could accept or stabilize a proton from TyrD.
The recent resolved crystal structure showed the presence of a cluster of water molecules near TyrZ, but no corresponding water cluster was seen near TyrD (1). Instead, only a single water molecule was identified near TyrD. Curiously, this water molecule occupies two different positions, proximal (H2Oprox) and distal (H2Odist) to the TyrD and separated by 1.8 Å with B-factors of 20.1 and 19.3, respectively (Fig. 1). The proximal position is at H-bonding distance with the phenolic O atom of TyrD (OTyrD−OH2Oprox = 2.73 Å), whereas the distal position is beyond H-bonding distance from TyrD (OTyrD−OH2Odist = 4.30 Å) but is instead at H-bonding distance with the guanidinium N atom of D2-Arg180 (OH2Odist−ND2-Arg180 = 3.01 Å). The reason for the density of this water molecule being shared over two locations is not known. In the present study, we used a large-scale quantum mechanics/molecular mechanics (QM/MM) approach to investigate the chemical properties of the water molecule in these two locations and their relationship to the redox state of TyrD. We go on to investigate the potential role of D2-Arg180 and a chain of waters reported in the crystal structure that may be involved in an exit/inlet channel to/from the luminal surface. From our findings, we propose a mechanistic model to explain the chemical and physical properties of TyrD.
Fig. 1.

H-bond network around TyrD (Left) and TyrZ (Right). Red and green spheres indicate water O and Cl atoms, respectively.
Results
Neutral Radical TyrD-O• in the Presence of H2O at the Distal Site.
In the QM/MM calculations, the H-bond geometry of the water in the distal position (H2Odist) was obtained when TyrD was taken as a deprotonated, neutral radical (TyrD-O•) (Table 1). In the presence of TyrD-O•, the water molecule would not remain at the H-bonding, proximal position (H2Oprox); instead, it moved to the distal position even if it initially donated an H-bond to the phenolic O atom of TyrD-O• (OTyrD). The calculated OTyrD−Nε,His189 distance was 2.78 Å, practically identical to the distance of 2.74 Å in the original PSII crystal structure (PSII monomer unit “A”) (1) (Table 1). Although both the neutral TyrD radical and the neutral His radical can be in equilibrium (Table 1), the potential-energy profile indicates that the neutral TyrD radical is energetically more relevant to the H-bond geometry in the existing PSII crystal structure (Fig. 2, Right). Fig. 3 (Right) illustrates the structure in the presence of TyrD-O•.
Table 1.
H-bond distances for TyrD in QM/MM optimized geometries in the PSII protein environment (measured in angstroms)
| Redox/protonation state | OTyrD−Nε,His | OTyrD–H | H–Nε,His | OTyrD−OH2O | Nδ,His−NArg |
| Original (1.9-Å structure) | 2.74 | — | — | Distal, 4.30; proximal, 2.73 | 2.81 |
| TyrD-OH (Fig. 2, Left) | |||||
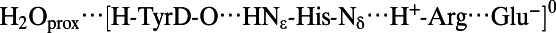
|
2.78 | 1.77 | 1.02 | 2.78 | 2.89 |
| TyrD-O• (Fig. 2, Right) | |||||
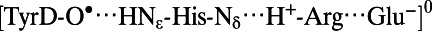
|
2.78 | 1.77 | 1.02 | 4.23 | 2.90 |
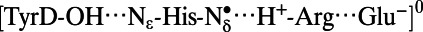
|
2.71 | 0.99 | 1.72 | 4.13 | 2.92 |
Note that the 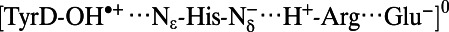 state was energetically very unstable and that only the
state was energetically very unstable and that only the 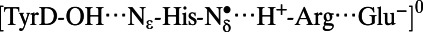 state was possible. Arg, D2-Arg294; Glu, CP47-Glu364; His, D2-His189; —, not applicable.
state was possible. Arg, D2-Arg294; Glu, CP47-Glu364; His, D2-His189; —, not applicable.
Fig. 2.

Potential-energy profiles along proton transfer coordinate for an H-bond donor-acceptor pair, TyrD and D2-His189. The proton to be released is colored purple. (Left) TyrD-OH is shown as an H-bond donor to a water molecule at the proximal position (black triangle curve). H atoms moving along the TyrD and D2-His189 moieties are indicated underlined in bold. (Right) TyrD-O• is shown with a water molecule at the distal position (red circle curve). PCET, proton coupled electron transfer.
Fig. 3.

H-bond patterns and concomitant water molecule positions (blue) for TyrD-OH (Left) and TyrD-O• (Right) species. The proton to be released is colored purple.
Protonated TyrD-OH in the Presence of the Proximal Water.
The distance between the phenolic oxygen of TyrD and the oxygen of the proximal water, OTyrD−OH2Oprox, is 2.73 Å in the PSII crystal structure (1). This strongly suggests that the proximal water forms an H-bond with OTyrD. However, in most of the cases investigated, the water would not stay at the proximal position; instead, it moved to the distal position (Table 1). Indeed, we found that a water molecule was stabilized at the proximal position only when TyrD accepts an H-bond from D2-His189 while simultaneously donating an H-bond to the water molecule (Fig. 3, Left). QM/MM calculations resulted in an OTyrD−OH2O distance of 2.78 Å in the H-bond pattern [H2O···H-TyrD-O···HNε-His-Nδ] where the key protons are indicated in bold face (Table 1), confirming that this is the relevant H-bond geometry for the proximal water in the PSII crystal structure.
It is of note that the distance from the phenolic O of TyrD to the nearest N of D2-His189 (OTyrD−Nε,His189) was identical (2.78 Å), irrespective of whether the water was in the distal or proximal position. This explains why the original PSII structure possesses two distinct positions of a single water molecule but only a single position for TyrD, D2-His189, and the other residues involved in the same H-bond network.
Stability of H2O at the Proximal and Distal Sites.
We investigated the potential energy profiles of H2O between the phenolic O of TyrD and guanidinium N atom of D2-Arg180. We confirmed that distal and proximal water sites are energetically the most stabilized positions for TyrD-O• and TyrD-OH, respectively (Fig. 4). H2O can be stabilized by two H-bonds at the proximal site in the [H2Oprox···H-TyrD-O···HNε-His-Nδ] state, where TyrD-OH donates an H-bond to the water, whereas the backbone carbonyl group of D2-Phe169 accepts an H-bond from water (Fig. 3, Left). Thus, the energy profiles of the H2O located between TyrD and D2-Arg180 provide the following findings: (i) H2O can be located at the proximal position only when TyrD-OH is present; it should be noted, however, that in the presence of TyrD-OH, a minor fraction of the H2O may also be located at the distal position due to the presence of an energy barrier between the two positions (Fig. 4); (ii) on formation of TyrD-O•, H2O is energetically less favorable at the proximal position; and (iii) thus, if H2O is located initially at the proximal position, it will move to the distal position when TyrD undergoes oxidation (Fig. 4).
Fig. 4.

Potential-energy profiles for a water molecule between OTyrD and NArg180 in the presence of TyrD-OH (black curve) and TyrD-O• (red curve). The horizontal axis indicates the OTyrD−OH2O bond distance, which practically describes movement of a water molecule (H2O) between TyrD and Arg180 moieties. Vertical dotted lines indicate the H2Odist and H2Oprox positions in the 1.9-Å structure. Solid blue circles represent the H2O molecule (1). H2O can be originally located at the proximal position in the presence of TyrD-OH (black triangle curve). Note that it can also be located at the distal position (2). On oxidation, the energy curve for the H2O molecule switches to TyrD-O• (red circle curve) (3). The energy minimum for a water molecule in the presence of TyrD-O• is located at the distal position. If H2O is originally located at the proximal position, it moves to the distal position. Note that the energy minimum is set to 0 kcal/mol for each curve.
Proton Transfer Pathway from TyrD.
The predicted displacement of the water on TyrD oxidation (Fig. 4) represents a functional link between TyrD and D2-Arg180 that is mediated by a single mobile water molecule acting as a proton carrier. Examination of the crystal structure (1) shows that an H-bond network is present beyond D2-Arg180, extending out to D2-His61 near the luminal bulk surface via a series of water molecules (Fig. 5). Indeed previous electrostatic calculations indicated that the protonation states of D2-Arg180 and D2-His61 were likely to be linked (25). Here, QM/MM calculations based on the more recent crystal structure (1) show that the proton released from TyrD was transferred via the mobile water and D2-Arg180 all the way to D2-His61 through a concerted single-step proton transfer process (Fig. 6). The proton did not go back to the TyrD/D2-His189 moiety but went through D2-Arg180 in an energetically favorable process irrespective of the presence of positively charged D2-Arg180. Clearly, there was no energy barrier for the proton transfer at D2-Arg180 (discussed below). As soon as the proton approached an -NH2 group of D2-Arg180, the NH-bond stretched toward the next water molecule, W480. Synchronizing the bond stretch, an OH bond of W480 also stretched toward W373. Similar bond stretching occurred at W373 and W783. The proton was finally stabilized at D2-His61. Although the H-bond network terminates at W354 in the 1.9-Å crystal structure (1), the proton relay may continue further, releasing the proton into bulk water via W354. Overall, the calculation showed that the proton was transferred to D2-His61 along a proton transfer path involving several OH (and NH) bond stretches toward acceptor moieties, without explicitly forming H3O+.
Fig. 5.

Overview of the H-bond network proceeding from the TyrD/D2-His189 moiety toward the luminal bulk surface (Left) and the H-bond donor-acceptor distances measured in angstroms (Right).
Fig. 6.

H-bond patterns of the proton transfer (PT) pathway from TyrD observed before (Left) and after (Right) proton transfer in QM/MM calculations.
Discussion
Movement of a Water Molecule on Redox Change of TyrD.
The present study indicates that in the 1.9-Å crystal structure (1), (i) the presence of water at the distal site, ∼4 Å away from the phenolic O atom of TyrD, corresponds to the presence of the oxidized radical, TyrD-O• and (ii) the presence of water at the proximal site, which is within H-bonding distance to the phenolic O of TyrD, corresponds to the presence of the unoxidized TyrD-OH (Fig. 3 and Table 1). This result is consistent with FTIR studies by Takahashi et al. (21) indicating the presence of a water molecule that interacts directly with the phenolic O atom of TyrD-OH rather than TyrD-O•.
This assignment of the water positions to specific TyrD redox states is a clear indication that the water acts as the proton acceptor on TyrD oxidation and that the water movement is involved in this reaction. To move from the proximal site to the distal site, a water molecule must reorient at D2-Phe169 (Fig. 3); this was manifest in the energy curve in Fig. 4 as a discontinuity occurring when the water is ∼3.9 Å away from TyrD, the point at which the TyrD-OH…OH2O bond breaks, although remaining H-bonded to the backbone carbonyl of D2-Phe169 (Fig. 3).
On the basis of the FTIR data, Hienerwadel et al. (20) proposed the presence of a proton carrier other than D2-His189 playing a role in modifying the properties of TyrD. The proximal/distal position change in response to the TyrD-OH/TyrD-O• redox transition observed in the present study (Fig. 3) probably represents the proposed proton carrier, tuning the TyrD polarity and acting as the proton acceptor on TyrD oxidation.
Differences in the H-Bond Geometries for TyrZ and TyrD.
In the 1.9-Å crystal structure (1), the H-bond from TyrZ-OH to D1-His190 (OTyrZ−ND1-His190) is unusually short, 2.46 Å. QM/MM calculations have reproduced the OTyrZ−ND1-His190 distance (2.47 Å) (26). Remarkably, the potential-energy profile indicated that this H-bond is a barrier-less, single-well (ionic) H-bond, an unusually short H-bond that can be formed only when the pKa difference between donor and acceptor is nearly zero (27–30) (Fig. S1). The results indicated that the cluster of waters near TyrZ was responsible for tuning the crucial pKa values (26).
In contrast, the corresponding H-bond between TyrD and D2-His189 is of standard length: 2.74 Å (for TyrD-OH and TyrD-O•). The potential-energy profiles for the OTyrD−ND2-His189 H-bond (Fig. 2, Left and Right) resemble those of standard, asymmetrical, double-well H-bonds (Fig. S1). The energy minimum is located at the D2-His189 moiety, suggesting that the pKa of the Nε site of D2-His189 is high comparable to that of TyrD. There are two main factors that are likely responsible for this difference. First, there is no corresponding water cluster near TyrD-OH; instead, a single water (H2O prox) is present acting as an H-bond acceptor from the TyrD-OH. Second, D2-His189 acts as an H-bond donor to the TyrD-OH (rather than the other way around in D1, where D1-His190 acts as an H-bond acceptor from the TyrZ-OH). This occurs because there is an arginine (D2-Arg294) rather than an asparagine (D1-Asn298) in the distal H-bond partner for the D2-His189. The H-bond from the positively charged Arg prevents protonation at the Nδ site of the D2-His189, leading to its proximal Nε site being protonated and thus unable to act as an H-bond acceptor from TyrD-OH (25).
Energetics of the Proton Transfer Pathway Proceeding from TyrD.
A concerted single-step proton transfer occurring over ∼13 Å between TyrD and D2-His61 (Fig. 6) requires (i) a well-arranged H-bond network (e.g., with appropriate donor-acceptor distances for all donor-acceptor pairs; Figs. 5 and 6) and (ii) a sufficient driving force. For a proton moving along the pathway, the energy profile indicated that the reaction is sufficiently downhill from the TyrD-associated water to D2-His61, even including the D2-Arg180 moiety (Fig. 7). The driving force of the proton transfer toward the bulk surface appears to disfavor protons returning to TyrD-O•, thus stabilizing the radical.
Fig. 7.

Energy profiles obtained for a proton translocation from H2Odist to D2-His61. The entire QM regions, including all groups in the proton transfer pathway (Figs. 5 and 6), are fully QM/MM optimized (□); alternately, only the H atoms are QM/MM optimized, whereas the heavy atom positions are fixed to those in the original 1.9-Å structure (■).
There are two key factors that facilitate proton transfer, even with the (positively charged) D2-Arg180 in the proton transfer pathway. First, TyrD is located in a hydrophobic environment with less chance of solvation for a charged species. The absence of a cluster of water molecules near TyrD seems to be associated directly or indirectly with the presence of several phenylalanines: D2-Phe168, D2-Phe184, D2-Phe185, D2-Phe188, CP47-Phe362, and CP47-Phe363 near TyrD, which block much of the space occupied by the water cluster (and also by the Mn4CaO5 cluster) at the TyrZ site. Thus, the presence of H3O+ near TyrD would be energetically unstable due to the lack of solvation relative to the region near the protein bulk surface. Second, D2-Arg180 forms a salt-bridge with D2-Asp333. The proton accepter for the distal water is thus not a simple arginine base but a neutralized ion pair [D2-Arg180/D2-Asp333]0. In addition, to maintain its charge neutrality, the ion pair releases its own proton as soon as it is approached by another proton.
As far as we are aware, involvement of D2-His61 in the proton transfer path from TyrD has not been reported. The electrostatic link between D2-Arg180 and D2-His61, as previously suggested in electrostatic calculations, will likely be functionally relevant (25). Remarkably, the H-bond donor acceptor distances of W783 near D1-His61 are very short in the 1.9-Å crystal structure (1), namely, OW783 − OW354 = 2.51 Å and OW783 − ND2-His61 = 2.58 Å (Fig. 5). In QM/MM calculations, these distances were 2.55 Å and 2.53 Å in the presence of protonated D2-His61 (i.e., after proton transfer), distances that are close to the original geometry of the crystal structure, whereas longer values were obtained (2.69 Å and 2.71 Å, respectively) in the presence of deprotonated D2-His61 (i.e., before proton transfer). Because a standard (asymmetrical double-well) H-bond possesses O−O distances of ∼2.8 Å (31, 32), the very short H-bond distances between W783 and His61 may be an indication of a proton being on D2-His61 or possibly shared with W783, which would thus be functioning as part of the proton transfer pathway from TyrD.
Implications from the “Old” Proton Transfer Pathway in D2.
On the D1 side, the 1.9-Å structure showed a number of ionizable residues and water molecules near Mn4CaO5. With so many possibilities, it is difficult to identify unambiguously the actual components of the proton transfer pathways involved in water oxidation. Umena et al. (1) proposed that the proton transfer pathways for proton-coupled electron transfer involving TyrZ and D1-His190 (33) may proceed from D1-Asn298 via several water molecules and residues, including CP43-Ala411, D1-Asn322, and PsbV-Tyr137, toward the luminal surface.
Considering the presence of the redox-active tyrosines TyrZ/TyrD in symmetrical positions on the D1/D2 heterodimer subunits, it has been suggested that an ancestral homodimer was able to oxidize tyrosines rapidly on both sides of the reaction center (e.g., in a D1/D1 homodimer PSII) (34, 35). This led to the suggestion that in an ancestral homodimer, there was once an active Mn cluster in the cavity adjacent to TyrD (34, 35). It seems possible then that the D2-Arg180–mediated proton pathway (Figs. 5 and 6) may represent a vestigial proton transfer pathway that once functioned in water oxidation. From this evolutionary point of view, we looked for a corresponding pathway in D1 as a way of defining a particular pathway among the numerous ionizable residues and water molecules near TyrZ.
A pathway leading from TyrZ can indeed be distinguished. It involves [TyrZ, D1-His190] → [W3, W5, W6, W7, the backbone C = O of D1-Asp170 (a ligand of Ca and Mn4)] → [W2, a ligand of Mn4] → [W446, Cl-1, D1-Asn181] → [W442] → [D1-Asp61] (Fig. 8). This corresponds to the TyrD proton pathway defined here, which involves [TyrD, D2-His189] → [H2Odist, the backbone C = O of D2-Phe169] → [D2-Arg180, D2-Asp333] → [W480] → [W373] → [W783] → [D2-His61].
Fig. 8.

Proton transfer pathway in the D1 side (Left), deduced from the old D2-Arg180 pathway (Right).
The distal water close to TyrD forms an H-bond with the backbone carbonyl group of D2-Phe169, as does W5 near TyrZ, with the backbone carbonyl group of D1-Asp170 in this case (Figs. 1 and 8). Thus, the distal water of TyrD corresponds geometrically to W5 near TyrZ. W5 is linked with the phenolic O atom of TyrZ via W3 and W7 (36). W3 and W7 are well ordered, because W3 is ligated to the Ca atom of Mn4CaO5 and W7 is H-bonded to D1-Glu189, a ligand of the Mn1 atom of Mn4CaO5. D1-Asp61 has been proposed to be located at the entrance of the possible proton transfer pathway (e.g., ref. 37). Mutations of D1-Asp61 affected the properties of Mn4CaO5 (38) and O2 release (39). As described, D2-Arg180 not only provides the binding moiety of the distal water for TyrD-O• (Fig. 3) but serves as a proton carrier (Figs. 6 and 7). This may be associated with the fact that in PSII from Synechocystis Pasteur Culture Collection (PCC) 6803, mutations of D2-Arg180 resulted in a loss and/or serious modifications of the EPR signal from TyrD and perturbations in the PSII photochemistry (24). Intriguingly, its counterpart, D1-Asn181, provides the binding moiety of a chloride ion, Cl-1 (1), thus forming part of a possible exit pathway of a proton released from Mn4CaO5 (37, 40). The presence of Cl-1 may facilitate transient formation of H3O+ and proton transfer on the TyrZ side of PSII. Because Cl− is required to progress through the S2 to S3 transition (41, 42) and the S3 to S0 transition (43), the deduced pathway proceeding from TyrZ may be active specifically for the S2 to S3 or S3 to S0 transition.
Understanding Why TyrD Radical Formation Is Slow and Why It Is Stable.
Two of the key features that differentiate TyrD from TyrZ are its oxidation rate, which is five orders of magnitude slower than that of TyrZ, and its reduction rate, which is at least six orders of magnitude slower than that of TyrZ (7, 12, 13). The insights gained from the present study help to understand these differences in structural and mechanistic terms. The rapid oxidation rate of TyrZ is related to the short H-bond in the TyrZ-O··H··Nε-His-Nδ-H, which has a single energy well and is finely tuned (in terms of pKa values) thanks to the water cluster that is structured by the Ca in the Mn4CaO5 cluster (26). For TyrD, the H-bond situation is quite different. The Tyr-O··H··Nε-His-Nδ-H··›O=C-Asn motif (here “··›” represents the H-bond direction from the donor to the acceptor) with its short H-bond is absent, and, significantly, the H-bonding direction is reversed because of the presence of the D2-Arg294, which gives rise to the H2Oprox‹··H-TyrD-O‹··HNε-His-Nδ‹··H+-Arg motif. In this case, the crucial TyrD-OH proton is donated to the proximal water rather than to D2-His189. There are three features of our results that would contribute to TyrD having a slow oxidation rate: (i) the H-bond from TyrD-OH to the proximal water is clearly not barrier-less; (ii) not all unoxidized TyrD moieties will have the proximal water bound at a given time; and (iii) the deprotonation is associated with the water moving from proximal to distal positions, resulting in an additional reorganizational energy associated with TyrD redox chemistry (Fig. 4).
The mechanistic model for TyrD oxidation proposed here may provide insights to help understand the remarkable speed-up in the rate of TyrD oxidation at pH values higher than 7.6 (11, 16, 17). In the absence of a crystal structure at this pH, the situation will inevitably be less clear-cut. Based on the same basic structural model, we have found that deprotonation of the H-bond network around TyrD results in properties consistent with those expected for rapid activation-less oxidation of TyrD. A detailed discussion of the possible protonation states of the H-bond network of TyrD (Table S1) and the potential-energy profile (Fig. S2) at high pH is provided in SI Text.
The current data also help to understand why TyrD• is so much more stable than TyrZ•. The proton transfer pathway mediated by a single mobile water molecule in a hydrophobic pocket may be seen as a stabilizing feature (Fig. 5). The protonated water moves away from the radical, physically disconnecting from it (Fig. 3). In addition, the distal water sheds the proton to the proton transfer chain in a concerted process all the way to the aqueous surface with a significant driving force (Figs. 6 and 7). This means that reduction of TyrD• from any source of electrons [e.g., by back-reactions from the electron acceptor side (44) involving equilibrium with P680/P680•+] will be inhibited because of the lack of an available proton, with the back-reacting proton being far away, probably in the bulk water, and requiring both energy and movement of water.
Computational Methods
Coordinates and Atomic Partial Charges.
The atomic coordinates of PSII were taken from the X-ray structure of the PSII monomer unit designated monomer A of the PSII complexes from Thermosynechococcus vulcanus at a resolution of 1.9 Å (Proton Data Bank ID code 3ARC) (1). Hydrogen atoms were generated and energetically optimized with CHARMM (45), whereas the positions of all nonhydrogen atoms were fixed, and all titratable groups were kept in their standard protonation states. For the QM/MM calculations, we added additional counter-ions to neutralize the entire system. Atomic partial charges of the amino acids were adopted from the all-atom CHARMM22 (46) parameter set. The atomic charges of Chla, pheophytin a, and quinones were taken from our previous studies on PSII (47). We considered the Mn4CaO5 cluster as the (O4)2−(O5)H− model (26) in the S1 state.
QM/MM Calculations.
We used the QSite (48) program code as utilized in previous studies (26). Owing to the large system size of PSII, we considered residues and cofactors in only subunits D1, D2, CP47, and CP43 as the protein environment. We used the restricted density functional theory (DFT) method for describing the closed-shell electronic structure and the unrestricted DFT method for the open-shell electronic structure with the B3LYP functional and LACVP∗∗+ basis sets. The geometries were refined by constrained QM/MM optimization (Dataset S1). Specifically, the coordinates of the heavy atoms in the surrounding MM region were fixed to the original X-ray coordinates, whereas those of the H atoms in the MM region were optimized using the OPLS2005 force field. All the atomic coordinates in the QM region were fully relaxed (i.e., not fixed) in the QM/MM calculation. To analyze the effects of the H-bond pattern near the TyrD moiety, the QM region was defined [TyrD (D2-Tyr160), D2-His189, D2-Arg294, CP47-Glu364, and water molecules that are within H-bond distance of these residues, namely, HOH-(chain D)-1 (distal and proximal positions, designated H2Odist and H2Oprox), -B526, -B539, -B541, -B544, -B550, -B566, and -B606], whereas other protein units and all cofactors were approximated by the MM force field. To analyze a possible pathway of the proton released from TyrD to D2-His61, the QM region was defined [D2-His61, TyrD, D2-Arg180, D2-His189, D2-Arg294, D2-Asp333, and water molecules that are within H-bond distance of these residues, namely, HOH-(chain D)-1 (distal and proximal, H2Odist and H2Oprox), -D354, -D364, -D366, -D373, -D390, -D480, and -D783]. The potential-energy profile of the H-bond was obtained as follows. First, we prepared for the QM/MM optimized geometry without constraints, and we took the resulting geometry as the initial geometry. The H atom was then moved from the H-bond donor atom (e.g., Ndonor) to the acceptor atom (e.g., Oacceptor) by 0.05 Å, after which the geometry was optimized by constraining the Ndonor−H and H−Oacceptor distances, and the energy of the resulting geometry was calculated. This procedure was repeated until the H atom reached the Oacceptor atom. To analyze the energy profile of the H2O position between the phenolic O atom of TyrD and the guanidinium N atom of D2-Arg180, D2-Arg180 was also included in the QM region. The OH2O atom was moved from OTyrD to the guanidinium N moiety of D2-Arg180 (NArg180) by 0.05 Å, after which the geometry was optimized by constraining the OTyrD−OH2O and OH2O–NArg180 distances, and the energy of the resulting geometry was calculated. This procedure was repeated until the OH2O atom reached the D2-Arg180 moiety. To analyze the energy profile of a possible proton transfer pathway from TyrD toward the luminal bulk surface, three O-H bond distances of H3O+ were constrained to avoid the proton being transferred to the D2-His61 moiety during the calculations.
Supplementary Material
Acknowledgments
We thank James Murray, Jeremy Smith, and Alain Boussac for useful discussion. This research was supported by the Japan Science and Technology Agency Precursory Research for Embryonic Science and Technology program (K.S. and H.I.); Grant-in-Aid 22740276 for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan (to K.S.); the Special Coordination Fund for Promoting Science and Technology of the MEXT (H.I.); the Takeda Science Foundation (H.I.); and a Grant for Basic Science Research Projects from The Sumitomo Foundation (to H.I.). A.W.R. is a recipient of the Wolfson Merit Award of the Royal Society.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1300817110/-/DCSupplemental.
References
- 1.Umena Y, Kawakami K, Shen J-R, Kamiya N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature. 2011;473(7345):55–60. doi: 10.1038/nature09913. [DOI] [PubMed] [Google Scholar]
- 2.Diner BA, Rappaport F. Structure, dynamics, and energetics of the primary photochemistry of photosystem II of oxygenic photosynthesis. Annu Rev Plant Biol. 2002;53:551–580. doi: 10.1146/annurev.arplant.53.100301.135238. [DOI] [PubMed] [Google Scholar]
- 3.Renger G, Renger T. Photosystem II: The machinery of photosynthetic water splitting. Photosynth Res. 2008;98(1-3):53–80. doi: 10.1007/s11120-008-9345-7. [DOI] [PubMed] [Google Scholar]
- 4.Barry BA, Babcock GT. Tyrosine radicals are involved in the photosynthetic oxygen-evolving system. Proc Natl Acad Sci USA. 1987;84(20):7099–7103. doi: 10.1073/pnas.84.20.7099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Debus RJ, Barry BA, Babcock GT, McIntosh L. Site-directed mutagenesis identifies a tyrosine radical involved in the photosynthetic oxygen-evolving system. Proc Natl Acad Sci USA. 1988;85(2):427–430. doi: 10.1073/pnas.85.2.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vermass WFJ, Rutherford AW, Hansson O. Site-directed mutagenesis in photosystem II of the cyanobacterium Synechocystis sp. PCC 6803: Donor D is a tyrosine residue in the D2 protein. Proc Natl Acad Sci USA. 1988;85(22):8477–8481. doi: 10.1073/pnas.85.22.8477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rutherford AW, Boussac A, Faller P. The stable tyrosyl radical in photosystem II: Why D? Biochim Biophys Acta. 2004;1655(1-3):222–230. doi: 10.1016/j.bbabio.2003.10.016. [DOI] [PubMed] [Google Scholar]
- 8.Styring S, Rutherford AW. In the oxygen-evolving complex of photosystem II, the S0 state is oxidized to the S1 state by D+ (signal IIslow) Biochemistry. 1987;26(9):2401–2405. [Google Scholar]
- 9.Messinger J, Renger G. Generation, oxidation by the oxidized form of the tyrosine of polypeptide D2, and possible electronic configuration of the redox states S0, S-1, and S-2 of the water oxidase in isolated spinach thylakoids. Biochemistry. 1993;32(36):9379–9386. doi: 10.1021/bi00087a017. [DOI] [PubMed] [Google Scholar]
- 10.Ananyev GM, Sakiyan I, Diner BA, Dismukes GC. A functional role for tyrosine-D in assembly of the inorganic core of the water oxidase complex of photosystem II and the kinetics of water oxidation. Biochemistry. 2002;41(3):974–980. doi: 10.1021/bi011528z. [DOI] [PubMed] [Google Scholar]
- 11.Faller P, et al. Rapid formation of the stable tyrosyl radical in photosystem II. Proc Natl Acad Sci USA. 2001;98(25):14368–14373. doi: 10.1073/pnas.251382598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Babcock GT, et al. Water oxidation in photosystem II: From radical chemistry to multielectron chemistry. Biochemistry. 1989;28(25):9557–9565. doi: 10.1021/bi00451a001. [DOI] [PubMed] [Google Scholar]
- 13.Diner BA, Babcock GT. In: Oxygenic Photosynthesis: The Light Reactions. Ort DR, Yocum CF, editors. Dordrecht, The Netherlands: Kluwer; 1996. pp. 213–247. [Google Scholar]
- 14.Berthomieu C, Hienerwadel R. Vibrational spectroscopy to study the properties of redox-active tyrosines in photosystem II and other proteins. Biochim Biophys Acta. 2005;1707(1):51–66. doi: 10.1016/j.bbabio.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 15.Buser CA, Thompson LK, Diner BA, Brudvig GW. Electron-transfer reactions in manganese-depleted photosystem II. Biochemistry. 1990;29(38):8977–8985. doi: 10.1021/bi00490a014. [DOI] [PubMed] [Google Scholar]
- 16.Faller P, Rutherford AW, Debus RJ. Tyrosine D oxidation at cryogenic temperature in photosystem II. Biochemistry. 2002;41(43):12914–12920. doi: 10.1021/bi026588z. [DOI] [PubMed] [Google Scholar]
- 17.Faller P, Goussias C, Rutherford AW, Un S. Resolving intermediates in biological proton-coupled electron transfer: a tyrosyl radical prior to proton movement. Proc Natl Acad Sci USA. 2003;100(15):8732–8735. doi: 10.1073/pnas.1530926100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferreira KN, Iverson TM, Maghlaoui K, Barber J, Iwata S. Architecture of the photosynthetic oxygen-evolving center. Science. 2004;303(5665):1831–1838. doi: 10.1126/science.1093087. [DOI] [PubMed] [Google Scholar]
- 19.Loll B, Kern J, Saenger W, Zouni A, Biesiadka J. Towards complete cofactor arrangement in the 3.0 Å resolution structure of photosystem II. Nature. 2005;438(7070):1040–1044. doi: 10.1038/nature04224. [DOI] [PubMed] [Google Scholar]
- 20.Hienerwadel R, Diner BA, Berthomieu C. Molecular origin of the pH dependence of tyrosine D oxidation kinetics and radical stability in photosystem II. Biochim Biophys Acta. 2008;1777(6):525–531. doi: 10.1016/j.bbabio.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 21.Takahashi R, Sugiura M, Noguchi T. Water molecules coupled to the redox-active tyrosine YD in photosystem II as detected by FTIR spectroscopy. Biochemistry. 2007;46(49):14245–14249. doi: 10.1021/bi701752d. [DOI] [PubMed] [Google Scholar]
- 22.Tang XS, Chisholm DA, Dismukes GC, Brudvig GW, Diner BA. Spectroscopic evidence from site-directed mutants of Synechocystis PCC6803 in favor of a close interaction between histidine 189 and redox-active tyrosine 160, both of polypeptide D2 of the photosystem II reaction center. Biochemistry. 1993;32(49):13742–13748. doi: 10.1021/bi00212a045. [DOI] [PubMed] [Google Scholar]
- 23.Mino H, Satoh J-I, Kawamori A, Toriyama K, Zimmermann J-L. Matrix ENDOR of tyrosine D+ in oriented Photosystem II membranes. Biochim Biophys Acta. 1993;1144(3):426–433. [Google Scholar]
- 24.Manna P, LoBrutto R, Eijckelhoff C, Dekker JP, Vermaas W. Role of Arg180 of the D2 protein in photosystem II structure and function. Eur J Biochem. 1998;251(1-2):142–154. doi: 10.1046/j.1432-1327.1998.2510142.x. [DOI] [PubMed] [Google Scholar]
- 25.Ishikita H, Knapp EW. Function of redox-active tyrosine in photosystem II. Biophys J. 2006;90(11):3886–3896. doi: 10.1529/biophysj.105.076984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saito K, Shen J-R, Ishida T, Ishikita H. Short hydrogen bond between redox-active tyrosine YZ and D1-His190 in the photosystem II crystal structure. Biochemistry. 2011;50(45):9836–9844. doi: 10.1021/bi201366j. [DOI] [PubMed] [Google Scholar]
- 27.Cleland WW, Kreevoy MM. Low-barrier hydrogen bonds and enzymic catalysis. Science. 1994;264(5167):1887–1890. doi: 10.1126/science.8009219. [DOI] [PubMed] [Google Scholar]
- 28.Frey PA, Whitt SA, Tobin JB. A low-barrier hydrogen bond in the catalytic triad of serine proteases. Science. 1994;264(5167):1927–1930. doi: 10.1126/science.7661899. [DOI] [PubMed] [Google Scholar]
- 29.Perrin CL, Nielson JB. “Strong” hydrogen bonds in chemistry and biology. Annu Rev Phys Chem. 1997;48:511–544. doi: 10.1146/annurev.physchem.48.1.511. [DOI] [PubMed] [Google Scholar]
- 30.Schutz CN, Warshel A. The low barrier hydrogen bond (LBHB) proposal revisited: the case of the Asp... His pair in serine proteases. Proteins. 2004;55(3):711–723. doi: 10.1002/prot.20096. [DOI] [PubMed] [Google Scholar]
- 31.Mikenda W. Stretching frequency versus bond distance correlation of O-D(H)...Y (Y = N, O, S, Se, Cl, Br, I) hydrogen bonds in solid hydrates. J Mol Struct. 1986;147:1–15. [Google Scholar]
- 32.Limbach H-H, et al. OHO hydrogen bond geometries and NMR chemical shifts: From equilibrium structures to geometric H/D isotope effects, with applications for water, protonated water, and compressed ice. Isr J Chem. 2009;49(2):199–216. [Google Scholar]
- 33.Hoganson CW, Babcock GT. A metalloradical mechanism for the generation of oxygen from water in photosynthesis. Science. 1997;277(5334):1953–1956. doi: 10.1126/science.277.5334.1953. [DOI] [PubMed] [Google Scholar]
- 34.Rutherford AW, Nitschke W. Photosystem II and the quinone-iron-containing reaction centers: Comparisons and evolutionary perspectives. In: Baltscheffsky H, editor. Origin and Evolution of Biological energy Conversion. New York: VCH; 1996. pp. 143–175. [Google Scholar]
- 35.Rutherford AW, Faller P. Photosystem II: Evolutionary perspectives. Philos Trans R Soc Lond B Biol Sci. 2003;358(1429):245–253. doi: 10.1098/rstb.2002.1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kawakami K, Umena Y, Kamiya N, Shen J-R. Structure of the catalytic, inorganic core of oxygen-evolving photosystem II at 1.9 Å resolution. J Photochem Photobiol B. 2011;104(1-2):9–18. doi: 10.1016/j.jphotobiol.2011.03.017. [DOI] [PubMed] [Google Scholar]
- 37.Iwata S, Barber J. Structure of photosystem II and molecular architecture of the oxygen-evolving centre. Curr Opin Struct Biol. 2004;14(4):447–453. doi: 10.1016/j.sbi.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 38.Chu HA, Nguyen AP, Debus RJ. Amino acid residues that influence the binding of manganese or calcium to photosystem II. 1. The lumenal interhelical domains of the D1 polypeptide. Biochemistry. 1995;34(17):5839–5858. doi: 10.1021/bi00017a016. [DOI] [PubMed] [Google Scholar]
- 39.Clausen J, Debus RJ, Junge W. Time-resolved oxygen production by PSII: Chasing chemical intermediates. Biochim Biophys Acta. 2004;1655(1-3):184–194. doi: 10.1016/j.bbabio.2003.06.001. [DOI] [PubMed] [Google Scholar]
- 40.Ishikita H, Saenger W, Loll B, Biesiadka J, Knapp E-W. Energetics of a possible proton exit pathway for water oxidation in photosystem II. Biochemistry. 2006;45(7):2063–2071. doi: 10.1021/bi051615h. [DOI] [PubMed] [Google Scholar]
- 41.Boussac A, Sétif P, Rutherford AW. Inhibition of tyrosine Z photooxidation after formation of the S3 state in Ca2+-depleted and Cl−-depleted photosystem II. Biochemistry. 1992;31(4):1224–1234. doi: 10.1021/bi00119a036. [DOI] [PubMed] [Google Scholar]
- 42.van Vliet P, Rutherford AW. Properties of the chloride-depleted oxygen-evolving complex of photosystem II studied by electron paramagnetic resonance. Biochemistry. 1996;35(6):1829–1839. doi: 10.1021/bi9514471. [DOI] [PubMed] [Google Scholar]
- 43.Wincencjusz H, van Gorkom HJ, Yocum CF. The photosynthetic oxygen evolving complex requires chloride for its redox state S2→S3 and S3→S0 transitions but not for S0→S1 or S1→S2 transitions. Biochemistry. 1997;36(12):3663–3670. doi: 10.1021/bi9626719. [DOI] [PubMed] [Google Scholar]
- 44.Johnson GN, Boussac A, Rutherford AW. The origin of 40–50 °C thermoluminescence bands in Photosystem II. Biochim Biophys Acta. 1994;1184:85–92. [Google Scholar]
- 45.Brooks BR, et al. CHARMM: A program for macromolecular energy minimization and dynamics calculations. J Comput Chem. 1983;4(2):187–217. [Google Scholar]
- 46.MacKerell AD, Jr, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102(18):3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 47.Saito K, et al. Distribution of the cationic state over the chlorophyll pair of the photosystem II reaction center. J Am Chem Soc. 2011;133(36):14379–14388. doi: 10.1021/ja203947k. [DOI] [PubMed] [Google Scholar]
- 48. QSite. (2012) QSite, Version 5.8 (Schrödinger, New York)
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.