

Abstract
Purpose
Alterations in FOXC1 dosage lead to a spectrum of highly penetrant, ocular anterior segment dysgenesis phenotypes. The most serious outcome is development of glaucoma, but this only occurs in 50–75% of patients. Therefore, the need to identify specific pathways and genes that interact with FOXC1 to promote glaucoma is great. In this study, we investigated loss of foxC1 in the zebrafish to characterize phenotypes and gene interactions that may impact glaucoma pathogenesis.
Methods
Morpholino knockdown in zebrafish, RNA and protein marker analyses, transgenic reporter lines, and angiography, along with histology and transmission electron microscopy were used to study foxC1 function and gene interactions.
Results
Zebrafish foxC1 genes were expressed dynamically in the developing vasculature and periocular mesenchyme during development. Multiple ocular and vascular defects were found after knockdown of foxC1. Defects in the hyaloid vasculature, arterial-venous malformations, and coarctation of the aorta were observed with maximal depletion of foxC1. Partial loss of foxC1 resulted in CNS and ocular hemorrhages, defects in intersegmental vessel patterning, and increased vascular permeability. To investigate the basis for these disruptions, ultrastructure of foxC1-depleted hyaloid vascular cells was studied. These experiments, along with Laminin-111 immunoreactivity, revealed disruptions in basement membrane integrity. Finally, co-depletion of laminin alpha-1 and foxC1 uncovered a genetic interaction between these genes during development.
Conclusions
Genetic interactions between FOXC1 and basement membrane components influence vascular stability and may impact glaucoma development and increase stroke risk in FOXC1 patients.
INTRODUCTION
Gene dosage changes in FOXC1 result in a spectrum of ocular anterior segment dysgeneses (ASD), including Axenfeld Rieger Anomaly and iridogoniodysgensis.1–8 All disease forms are developmental, dominantly inherited, and highly penetrant. The most debilitating outcome for ASD patients is the development of glaucoma, which occurs in 50–75% of cases and is often refractive to treatment. 9–11
Glaucoma is a progressive blinding disease which results from atrophy of the optic nerve and death of retinal ganglion cells. In ASD patients, raised intraocular pressure (IOP) is the greatest risk factor for developing glaucoma.10 IOP is regulated by aqueous humor flow in the anterior segment of the eye and most typically, elevated IOP is caused by reduced aqueous humor drainage. The precise mechanistic relationship between elevated IOP and glaucoma is still unknown. In ASD, development of the outflow structures is compromised, and it is hypothesized that defects in these structures lead to elevated IOP and glaucoma. However, in FOXC1 disease, it is not understood why glaucoma occurs only in some patients despite the high penetrance of ASD phenotypes.
The role of FOXC1 in development has been studied in a number of experimental systems including cell culture, mice, zebrafish and frogs. FoxC1 is widely expressed during embryogenesis, but is not ubiquitous. Mice with null Foxc1 mutations show defects in differentiation of chondrocytes, the renal system, the eyes and vascular system.2, 12, 13 In addition to null mutations, a hypomorphic allele revealed later developmental requirements of FOXC1.14 Later on-set defects include cortical dyslamination and skull anomalies. Interestingly, the cortical defects were suggested to be secondary to altered meningeal differentiation. Haploinsufficiency of Foxc1 in mice has also been used to model human disease. Interestingly, ASD phenotypes were found only in specific mouse strains, while other genetic backgrounds did not show observable defects.15 These observations reinforce the role of genetic modifiers on Foxc1-dependent phenotypes.
A similar distribution of phenotypic defects was found with loss of the closely related gene, Foxc2. FOXC1 and FOXC2 share nearly identical DNA binding domains and developmental expression patterns. Compound mutant analysis in mice revealed redundant functions between these factors.15–18 Vascular phenotypes of FoxC1/FoxC2 double knockout mice included arterial-venous fate defects and vessel morphogenesis and remodeling anomalies.17, 19–21 In the zebrafish, foxC1 is duplicated and there is no evidence for a foxC2 homolog.22 However, the combined expression of foxC1a and foxC1b during zebrafish embryogenesis is similar to that of FoxC1 and FoxC2 in higher vertebrates 22, 23, suggesting that zebrafish foxC1b is the functional homolog of mammalian FoxC2.
In this study, we explore the mechanisms of the eye and vascular phenotypes associated with loss of foxC1 function in zebrafish. Overall, depletion of foxC1 resulted in multiple, dose-sensitive vascular phenotypes. In particular, we found that loss of foxC1 results in defects to vascular basement membrane integrity. These studies provide insight into the mechanisms for how altered FoxC1 function can affect glaucoma and suggest that FOXC1 mutations in humans may also increase the risk of stroke.
MATERIALS AND METHODS
Specimens
Wild-type zebrafish (Danio rerio) of the AB/AB and LF/LF backgrounds were reared under standard conditions with a light cycle of 14h light/10h dark. Prior to experimental manipulation or tissue fixation, fish were anesthetized in 0.2 mg per ml of ethyl 3-aminobenzoate methanesulfonate (tricane). All experiments were performed in compliance with the ARVO statement for use of animals in vision research.
In situ Hybridization
Probes against zebrafish foxC1a (foxC1.1), foxC1b (foxC1.2), and flt4 were produced as previously described.22, 24 Antisense RNA probes were produced and whole-mount in situ hybridization was conducted as previously described.25, 26 10–15 embryos, treated with 0.003% phenylthiourea (PTU), were analyzed in each in situ experiment. PTU inhibits melanin synthesis. For post-hybridization sectioning, embryos were fixed in 4% paraformaldehyde/PBS and infiltrated with 15% sucrose, 30% sucrose and then 100% Tissue-Tek OCT (Miles Inc., Elkhart, IN, USA). Embryos were oriented in a freezing mold and 10 μm sections were cut on a cryostat and mounted on gelatin-coated glass slides.
Morpholino Knockdown
Previously described anti-sense morpholinos (MO) used for knockdown were ordered from Gene Tools (Philomath, OR), resuspended in sterile water and injected into 1–4 cell stage embryos. MOs used were (Start codon underlined): foxC1a MO2 (GeneID 79374): 5′-CCTGCATGACTGCTCTCCAAAACGG -3′; foxC1b MO1 (GeneID 79375): 5′-GCATCGTACCCCTTTCTTCGGTACA-3′ 27; laminin alpha-1 MO1 (GeneID 77993333): 5′-ATAAAGCTAAAGCTGTGCTGAAATC-3′ 28, 29 ; Control MO: 5′-CCTCTTACCTCAGTTACAATTTATA-3′. foxC1a MO2 and foxC1b MO1 were injected together to achieve foxC1 dMO morphant zebrafish. Other foxC1 MOs used to verify phenotypes were: foxC1a MO1 5′-GTCAAGAAGACTGAAGCAATCCACA -3′; foxC1b MO1: 5′-AAGTGAAATGAAGACTATGCAGACG -3′ 27
Transmission Electron Microscopy (TEM) and Histological Analysis
Histological specimens for light microscopy were processed as previously described.30 In brief, embryos were fixed in primary fixative [2% paraformaldehyde, 2.5% glutaraldehyde, 3% sucrose, 0.06% phosphate buffer (pH 7.4)] at 4°C for 24 h and then washed in 0.1 M PBS, dehydrated through an ethanol series and propylene oxide and then infiltrated with EMbed-812/Araldyte resin mixture. Semi-thin plastic sections were cut with a glass knife on a JB4 microtome and stained with 1% Toluidine blue in 1% Borax. For TEM, an additional fixation in 1.0% Osmium was included, followed by dehydration in MeOH/Aralhydrate. Embryos were then embedded in EMBed-812/DER 736. Ultrathin sections (60–70 nm) were collected on coated grids and stained with uranyl acetate and lead citrate for contrast. Images were captured using Hitachi H600 TEM.
Frozen Section Immunohistochemistry
Wild-type and morphant embryos were fixed in 4 % paraformaldehyde/PBS and infiltrated with 2-hour steps of 15 % sucrose, 30 % sucrose and 100 % Tissue-Tek OCT. Embryos were orientated, sectioned, and mounted on gelatin-coated glass slides. Antibodies that recognize Laminin -111 (Laminin-1 epitope, Sigma L9393; 1:200 dilution) were used in a 5 % Goat Serum/PBT blocking solution. This antibody was raised against purified basement membranes and affinity purified against entire Laminin-111 heterotrimer. Immunoreactivity was detected with fluorescent a secondary antibody and imaged with a Nikon C1 confocal microscope.
Microangiography
Fluorescently labeled 2MDa dextrans were injected into the tail vein or sinus venosus of 2–5dpf, PTU treated, anesthetized Tg(fli1a:GFP) zebrafish embryos as previously described.31 Embryos were mounted in 1% low-melt agarose and injections were done using a Drummond Nanoject (Drummond Scientific Co, Broomall, PA) with micropipettes cut to a bore size of 5–10 μM. Distribution of dextrans throughout the vasculature was monitored through a fluorescent dissecting microscope and imaging was done with a Nikon C1 confocal microscope.
RESULTS
Zebrafish foxC1a and foxC1b are expressed in the periocular mesenchyme and developing vasculature
Expression of foxC1a and foxC1b were assessed during development by in situ hybridization. As previously reported, we found both genes expressed in the paraxial mesoderm, head mesenchyme, and vascular system before 24 hours post fertilization (hpf) (data not shown; 22). By 24 hfp, strong expression of foxC1a was found in the periocular mesenchyme (Figure 1A,C). Levels in the periocular mesenchyme peaked between 24 and 36 hpf, and then gradually decreased over time (Figure 1A,C). In the developing hyaloid vasculature, levels of foxC1a expression correlated with expression in the periocular mesenchyme (Figure 1C). Expression of foxC1a was also maintained in endothelial cells of the developing trunk vasculature (Figure 1D). From 72–96 hpf, strongest expression of foxC1a was found in the branchial arches (Figure 1A,C). foxC1b showed both overlapping and distinct patterns and timing of expression as compared to foxC1a. In the periocular mesenchyme, expression lagged behind foxC1a, with levels not detectable until 36 hpf, but then persisting later during development (Figure 1 B,C). Expression in trunk vascular endothelial cells was evident at all times assessed, though levels in the hyaloid were below detection by whole mount in situ hybridization (Figure 1 B,C,D). Overall, strongest expression of foxC1b was in the branchial arch region and the fin buds (Figure 1B,C).
Figure 1. Expression of foxC1a and foxC1b.

A. Whole mount expression of foxC1a during development. Strong expression was found in the periocular mesenchyme surrounding the eye at 24–36 hpf. Expression was observed in the branchial arch region (arrows) and in the periocular mesenchyme at 36, 48 and 96hpf. B. Whole mount expression of foxC1b during development. High levels were found in the branchial arches (arrows) throughout development and in the fin buds (arrow heads) at 24–48 hpf. Periocular mesenchyme expression was absent at 24 hpf, but increased thereafter. C. Sections through the eyes showed dynamic expression in the periocular mesenchyme for both foxC1a and foxC1b. Expression of foxC1a was also found in the hyaloid vasculature (arrows). D. Schematic of the zebrafish trunk vasculature (left) and expression of foxC1a and foxC1b in vascular endothelial cells of the dorsal aorta and the cardinal vein at 36 hpf. SC = Spinal cord, NC = Notocord, DA = Dorsal aorta, CV = Cardinal vein.
Loss of foxC1 results in vascular defects
To address loss of function phenotypes during zebrafish development, morpholinos were used to knock down both foxC1 genes. The morpholinos used were originally described to assess phenotypes during early somitogenesis (<18 hpf).27 For both foxC1a and foxC1b, two independent morpholinos were designed to specifically target each gene. As both genes contain only one exon, morpholinos were designed to target the ATG start codon and 5′ untranslated regions. For both genes, injection of either morpholino resulted in similar phenotypes. The morpholinos targeting the ATG start codons were effective at lower doses and these were used for the remainder of our studies. Phenotypes were unchanged when a morpholino targeting p53 was co-injected, indicating the morpholinos do not promote phenotypes through off-target mediated cell death.32 Knock-down of foxC1a resulted in a number of gross phenotypes, including hydrocephaly, heart edema, a small eye, and variable hemorrhage within the CNS and eyes (Figure 2A). When foxC1b was targeted, no obvious phenotypes were observed except a slight developmental delay which is common with morpholino injections (Figure 2A). Based on gene similarity, we hypothesized the lack of gross phenotypes, may be due to compensation by foxC1a. This also raised the possibility that foxC1b may compensate to some degree for loss of foxC1a. To address this, morpholinos targeting both genes were co-injected and compared to embryos injected with the individual morpholinos plus control morpholino to maintain equal total morpholino concentrations. No additional phenotypes were observed as compared to foxC1a morphants, but the severity of phenotypes was increased (Figure 2A). The remaining studies were done with a combination of morpholinos targeting both foxC1 genes (foxC1dMOs).
Figure 2. Gross Phenotypes of foxC1 Morphants.
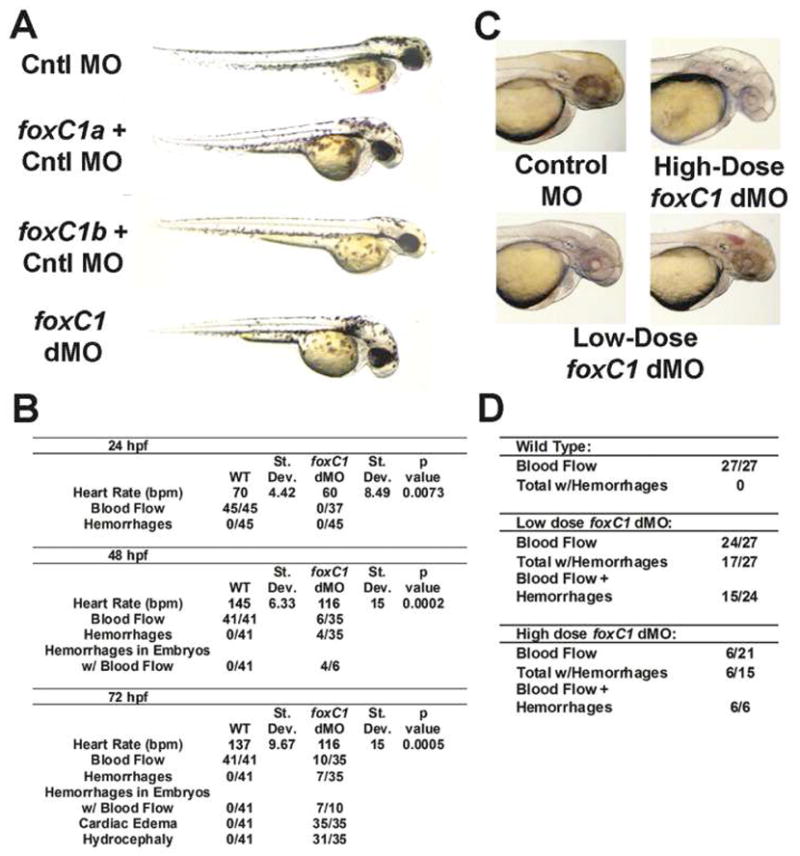
A. Comparison of Control (Cntl) MO, foxC1a + Cntl MO, foxC1b + Cntl MO and foxCa+b double morphants (foxC1dMO) at 48 hpf. Hydrocephaly, heart edema and a small eyes were found in foxC1a morphants. Only a slight delay was found with injection of foxC1b morpholino. Severity of phenotypes found in foxC1a morphants increased in foxC1dMO. B. Quantification of phenotypes after injection of high dose foxC1 morpholinos. Heart rates represent an average of 5 embryos, rounded to the nearest integer. Blood flow indicates that blood cells were directly observed flowing throughout the vasculature of the embryo C. Dosage-dependent phenotypes of foxC1dMO in 48 hpf PTU treated embryos: one control morphant (upper, left), one high-dosage foxC1dMO embryo (3.25 ng of each morpholino) (upper, right), and two lower-dose foxC1dMO embryos (1.63ng of each morpholino) At high doses, blood flow and hemorrhaging were only occasionally observed. At lower doses, both blood flow and CNS or ocular hemorrhaging was found in the majority of embryos (arrowheads). D. Quantification of observed phenotypes with changing dosages of foxC1 morpholino.
To explore the hemorrhage phenotypes in more detail, we tested several doses of foxC1dMOs and quantified the phenotypes. In our initial studies of foxC1dMO, we found that ocular and CNS hemorrhaging phenotypes presented with variable penetrance. Interestingly, the number of embryos with hemorrhages in foxC1dMO embryos decreased as compared to embryos injected with foxC1a MO alone. The reduction of hemorrhages in foxC1dMO morphants correlated with a lack of blood flow which was noted in the majority of these embryos (Figure 2B,C,D). Blood cells were present in foxC1dMO embryos, but most often accumulated in the tail veins (Figure 2B). Lowering the dosage of foxC1dMO (1.65ng of each morpholino/embryo as compared to 3.25 ng/embryo), restored blood flow in the majority of embryos, but also increased the number of hemorrhages (Figure 2B,D). This result is consistent with the requirement of blood flow in the development of vascular hemorrhaging following loss of foxC1 function.
As the majority of hemorrhages in foxC1 morphants were confined to the CNS and eyes, we examined semi-thin histological sections of these regions to evaluate gross vessel morphology. Dilated, hydrocephalic ventricles were apparent in the CNS, but no gross differences in vessels were noted (Figure 3A). As described for other species, the eyes were small and lacked proper anterior segment development (Figure 3A). Specifically, the cornea was thickened with decreased corneal endothelial cells and poorly condensing corneal stroma (Figure 3A). Cells within the developing iridiocorneal angle, as well as periocular cells overall, appeared loosely organized and undifferentiated (Figure 3A) To further assess the state of periocular cells, which arise from migrating neural crest cells, we examined foxC1 knockdown in a transgenic zebrafish line which has a subset of neural crest cells labeled with GFP (Tg(foxd3:GFP)zf15).48 Analysis through development shows neural crest cells in foxC1dMO embryos do migrate around to the anterior eye, but are fewer in number and retain an immature morphology as compared to controls (Figure 3B, Supplemental Figure 1). Assessment of fully formed anterior segment structures was not possible as maturation occurs after 72 hpf, when morpholinos no longer have a primary affect on protein expression. Examination of the retina in foxC1dMOs also revealed defects. At 48 hpf, lamination of the neural retina was slightly delayed in foxC1dMO embryos and eye size was reduced overall (Figure 3A, B). Striking differences in the morphology of the hyaloid vessels were also noted. In control eyes, multiple, small vessels were found between the lens and the retina (Figure 3A, right panels). In contrast, one or two large, dilated vessels were observed in foxC1dMO morphant eyes at the same position between the lens and retina (Figure 3A and Figure 6A–C). In addition, there were often undifferentiated cells behind the lens in foxC1dMO morphant eyes (Figure 6E). A slight delay in lens development was also noted, but cellular morphology judged by semi-thin histology and TEM did not reveal defects (Figure 3A, Figure 6A–E).
Figure 3. Assessment of Ocular and CNS development in foxC1 dMO.
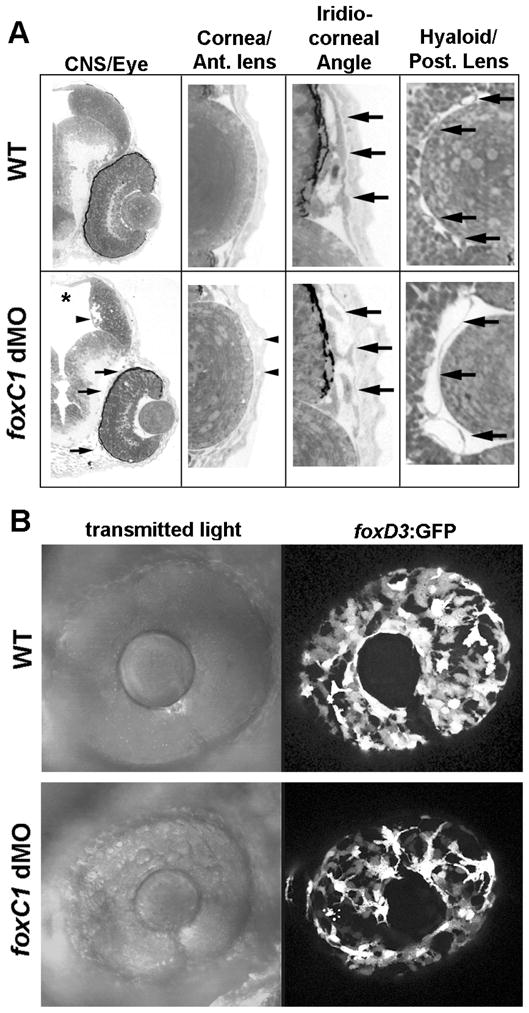
A. Histological Examination of 48hpf WT and foxC1 dMO. Sagittal sections of the brain and eye show expanded ventricles of the brain (askerisk) and the loosely organized periocular mesenchyme (arrows) in foxC1 dMOs. Areas of cell loss are also noted along the ventricles within the brain (arrow head). High magnification of the cornea and anterior lens shows the corneal endothelium is absent and defects in corneal stroma (arrow heads) are found in foxC1 dMOs. High magnification of the iridio-corneal angle shows undifferentiated and poorly organized periocular cells (arrows) in foxC1 dMOs. High magnification of the posteror lens and hyaloid vasculature shows fewer, dilated hyaloid vessels behind the lens (arrows) in foxC1 dMOs. B. Analysis of foxD3 positive neural crest cells in 48 hpf foxC1 dMOs. Fewer foxD3:GFP positive cells were found migrating to the anterior of the eye. Cells that have migrated show abnormal morphology as compared to controls.
Figure 6. Ultrastructure of hyaloid vasculature in 48hpf foxC1dMO embryos.

Hyaloid vascular endothelial cells (black arrowheads) in wild-type (A, C) and foxC1dMO eyes (B, D). In some foxC1dMO embryos, undifferentiated mesenchymal cells (E, black asterisks) occupied the space where vessels normally reside. Vascular endothelial cells (black arrows) associated with the basal surface of the lens cells in wild-type (F) or foxC1dMO embryos (G). Vascular endothelial cells (black arrows) associated with the basal surface of the retina in wild-type (H) or foxC1dMO embryos (I). Black asterisk in (I) indicates undifferentiated mesenchymal cell. Boxed areas indicate magnified regions below (J–M). Basement membranes (white arrowheads) or disorganized extracellular matrix (white asterisk) associated with the lens cells (J,K) or the ILM of the retina (L,M). Note the discontinuous and disorganized basement membranes associated with foxC1dMO vascular cells. Bar equals 10 microns (A,B); 4 microns (C–E); 0.5 microns (F–I). RGC, retinal ganglion cell; ILM, inner limiting membrane of the retina; a, artifact (section fold).
Vascular endothelial fate and morphology are disrupted in foxC1 morphant embryos
To further investigate the vascular phenotypes in foxC1 morphants, knockdown experiments were done in the vascular endothelial reporter transgenic line, Tg(fli1a:EGFP)y5. This transgenic line allows visualization of the entire vasculature throughout development.33 A number of interesting phenotypes were observed with injection of the high morpholino dose described above. Confocal imaging of the dorsal aorta and cardinal vein revealed inappropriate luminal connections throughout the tail, suggestive of arterial-venous cell fate defects (Figure 4A). Consistent with this interpretation, the venous marker flt4 showed ectopic expression in vessels of the dorsal aorta (Figure 4B). Analysis of vessels in the anterior embryo also showed disruption. In this region, the paired lateral dorsal aortae normally join to form the single midline dorsal aorta (Figure 4A). Embryos depleted of foxC1 showed thin and disorganized vessels in this region (Figure 4A). The use of the Tg(fli1a:GFP) line also helped resolve defects in the hyaloid vasculature. By 48 hpf in control embryos, an organized vascular basket envelops the lens (Figure 4A; 34). In some foxC1dMO morphants, fli1a:GFP positive cells were present, but collected in an undifferentiated mass behind the lens (Figure 4A). However, in other morphant embryos, hyaloid vessel differentiation did occur, but as suggested by histology, resulted in a less branched and dilated morphology (Figure 4C). Expression of flt4 in the hyaloid was similar in foxC1dMO morphants as compared to control embryos (Figure 4B), indicating that the cellular defects were not due to altered endothelial cell fate determination and instead due to disrupted differentiation.
Figure 4. Vascular phenotypes in high-dose foxC1dMO embryos.
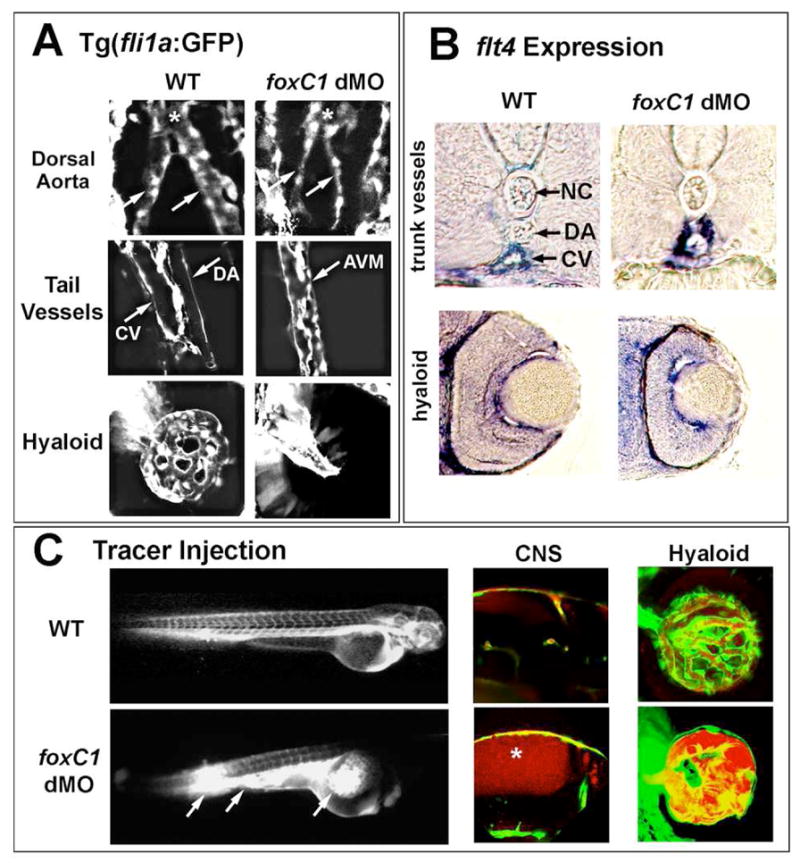
A. Knockdown of foxC1 in 48hpf Tg(fli1a:GFP) embryos. Defects in dorsal aorta development, axial vessel development and hyaloid vasculature were found in foxC1 dMOs. Dorsal aortae vessels (arrows) are dysmorphic preceding their fusion (asterisk). In the trunk region, arterial-venous malformations (AVM) are found between the cardinal vein (CV) and the dorsal aorta (DA) in foxC1dMOs. Vascular endothelial cells are present in the hyaloid of foxC1dMOs, but are less organized than wild-type vessels. B. Expression of the venous marker flt4 was found expanded into the region of the dorsal aorta in foxC1dMO embryos (upper panels). See Figure 1D for schematic of anatomy. No differences were observed in flt4 expression between foxC1dMOs and WT within the hyaloid vasculature (lower panels). C. Microangiography in foxC1dMO, Tg(fli1a:GFP) embryos revealed defects in vessel integrity, as shown by breakage of vessels and leakage of dye into the yolk (left panels, arrows). Increased permeability in CNS vessels as revealed by microangiography (center panels, asterisk). Dye also flowed behind the eye, but was encased by fli1a:GFP positive cells (right panels).
To assess blood flow and vasculature integrity in foxC1dMO morphants, microangiography with tetramethyl-rhodamine labeled 2MDa dextrans was performed in Tg(fli1a:GFP) embryos. Injection of dextrans into tail veins of foxC1dMOs showed a decreased capacity to flow throughout the vasculature as compared to controls. Injections often resulted in ruptured trunk vessels and dye accumulation into the yolk (Figure 4C). When flow of dextrans was achieved to the head, accumulation in hydrocephalic brain ventricles was observed, indicating compromised vessel integrity (Figure 4C). Within the eye, accumulation of dye was observed behind the lens, but was typically confined by fli1a:GFP positive cells when vessel formation occurred (Figure 4C).
Because hemodynamic forces have been shown to regulate endothelial cell differentiation, we analyzed vessel formation in lower dose foxC1dMO, Tg(fli1a:GFP) embryos where blood flow was normal. In these embryos overt vascular morphology appeared normal in trunk regions and within the eye (Figure 5A). Confocal microscopy, however, revealed disorganization of endothelial cells and excess branching of trunk intersegmental vessels at 24hpf (Figure 5B). Injection of labeled 2MDa dextrans into low-dose morphant embryos at 48 and 72 hpf did not show increased overall permeability. However, by 96 hpf foxC1dMO embryos did show increased dextran permeability within the CNS and eye (Figure 5C).
Figure 5. Vascular phenotypes in low-dose foxC1dMO embryos.
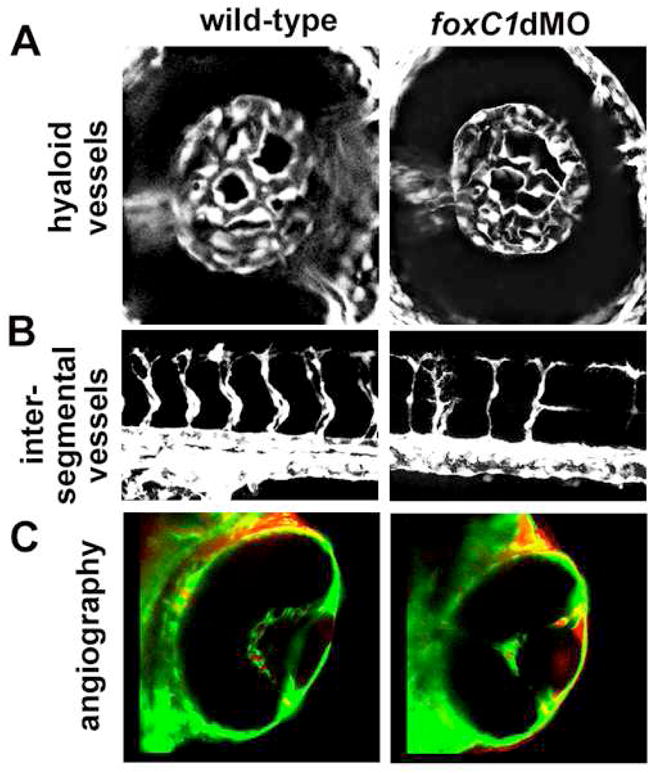
A. Hyaloid vasculature as revealed by fli1a:GFP in wild-type (left) and in low-dose foxC1dMO morphant embryos (right) at 48hpf. No differences in overall morphology was noted in low-dose morphants. C. Intersegmental vessels in wild-type (left) and low-dose foxC1dMO morphant (right) embryos at 48 hpf. D. Microangiography in wild-type (left) and low-dose foxC1dMO morphant embryos (right) at 4 dpf. Note the increased leakage of dye into the anterior chamber of foxC1dMO eyes.
Loss of foxC1 results in vascular basement membrane defects
To explore the observed vascular phenotypes in more depth, hyaloid vessels from high dose foxC1dMO embryos were analyzed by TEM. In foxC1dMO embryos endothelial cells were dilated, but showed normal morphology and cell-cell junctions as compared to controls (Figure 6). As predicted by histology, undifferentiated mesenchymal cells were also often found behind the lens of foxC1dMO embryos (Figure 6E). In addition to mesenchymal cell differentiation and vessel morphology defects in foxC1dMO eyes, TEM revealed disruptions to basement membrane present between hyaloid vessels and both the lens and the retina (Figure 6B). In controls, a smooth, continuous basement membrane was present along the entire basal surface of the lens and retina (Figure 6F,H, J, L). In foxC1dMOs, the basement membrane was disorganized and showed focal disruptions (Figure 6G, I, K, M).
To confirm the basement membrane disruptions in foxC1dMOs, immunostaining for Laminin-111 was performed. Laminins, along with type IV collagens, Nidogen and sulfated proteoglycans make up the normal components of all basement membranes. Laminin-111 is a heterotrimeric glycoprotein composed of one alpha, one beta and one gamma chain. In control embryos, a smooth, uninterrupted staining pattern was observed along both the basal surface of the retina and along the lens (Figure 7). In contrast, Laminin-111 staining was more diffuse with interruptions along the lens and retinal surfaces in foxC1dMO eyes (Figure 7).
Figure 7. Laminin-111 immunostaining in foxC1dMO eyes.
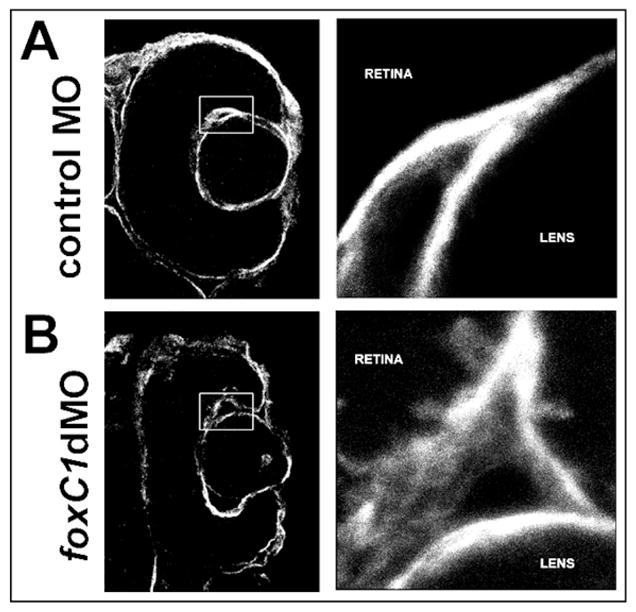
Immunostaining for Laminin-111 in the basement membranes of 48 hpf control morphant (A) and foxC1dMOs (B) eyes. Higher magnification of boxed inset shown on the right of each panel.
foxc1 and laminin alpha-1 show genetic interaction
Many of the vascular defects in foxC1dMO embryos are reminiscent of vascular anomalies caused by loss of laminin alpha-1(lama1) function. The lama1 gene encodes for the alpha subunit of the Laminin-111 complex. To investigate the relationship between foxc1 and the basement membrane component lama1, zebrafish embryos were co-injected with morpholinos targeting the foxC1 genes and lama1. lama1 has been previously investigated in the zebrafish, and homozygous mutations in this gene (bashful) or morpholino knockdown result in lens degeneration, hyaloid and trunk vasculature defects, and body truncations.28, 29, 35 When previously described concentrations of this morpholino were co-injected with either the high or low doses of foxc1 morpholinos described above, severe body truncation phenotypes were observed (data not shown). To further investigate the combined effects of foxc1 and lama1 knockdown, sub-maximal doses of all morpholinos were injected. Very mild hydrocephalic and hemorrhaging phenotypes were observed when 1.3ng of each foxC1 morpholino were injected alone (Figure 8A,B). When 3.9 ng of lama1 morpholino was co-injected with these concentrations of foxC1dMO, the incidence and severity of both hemorrhaging and hydrocephaly were increased (Figure 8A,B). This concentration of lama1 morpholino injected alone showed only occasional, mild lens and tail defects, along with a small number embryos with mild hydrocephaly and hemorrhage (Figure 8A,B). Finally, we also studied knockdown of foxC1 in homozygous bashful/lama1 mutants (bala69). Injection of sub-maximal foxC1dMO into these embryos resulted in more severe phenotypes than mutants alone, including increased severity of the tail and lens phenotypes and a more pronounced hydrocephaly (data not shown). We also tested foxC1 knockdown in embryos heterozygous for the bala69 mutation, but did not observe any differences as compared to the wild-type siblings.
Figure 8. Effects of lama1 loss on foxc1dMO phenotypes.
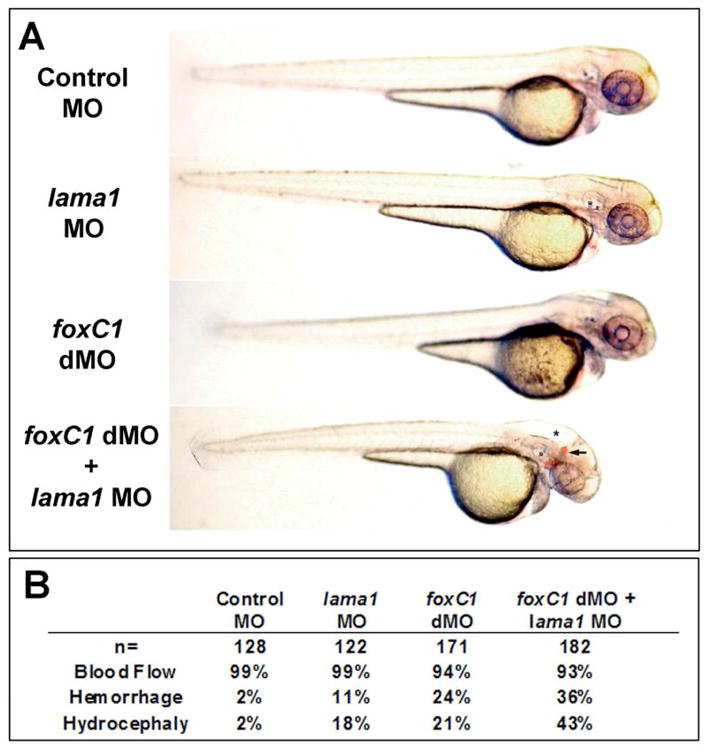
A. Mild phenotypes were found after low-dose knockdown of both foxC1 and lama1 (middle panels) as compared to controls (upper panel). Increased incidence hydrocephaly (asterisk) and hemorrhaging (arrow) was found after co-injection of these same doses of foxC1 and lama1 morpholinos (lower panel). B. Quantification of phenotypes described above. Data represents totals from three independent experiments.
DISCUSSION
In this study we describe the effects of foxC1 knockdown on zebrafish larvae. We found that the overall spectrum of phenotypes due to loss of foxC1 in the zebrafish is similar to those found with loss of FOXC1 in mammals, indicating a conservation of function across species. We focused our investigation on how loss of foxC1 impacts ocular and CNS vascular development. Knock-down of foxC1 resulted in defects in vascular endothelial cell fate and differentiation and disrupted vessel morphology and integrity. Interestingly, we found that loss of foxC1 leads to disruption of vascular basement membrane structure and foxC1 genetically interacts with lama1, which encodes a key component of the basement membrane.
In mouse, combined deletion of Foxc1 and Foxc2 result in severe vascular phenotypes including defects in arterial-venous fate decisions and concomitant arterial-venous malformations, as well as aorta coarctation.16, 18, 19 Arterial versus venous specification occurs by differential VEGF activity and subsequent reciprocal Eph/Ephrin signaling (reviewed in 36). EphrinB2 expression in arterial cells occurs by activation of the Notch pathway and Hey2 activation. FOXC1 and FOXC2 have been implicated in multiple steps in this pathway, and both have been shown to directly activate Notch target genes in response to VEGF signaling.19, 20 In our studies, we found similar arterial-venous defects in severe loss of foxC1dMO embryos. In addition to expansion of the venous marker flt4, arterial-venous malformations were reveled by fli1a:GFP. The vessel defects in the tails and the aorta coarctation of foxC1dMO embryos are similar to those described for zebrafish gridlock mutants, in which Hey2 function is disrupted.31 Defects in the trunk vessels and aorta help explain the lack of proper blood flow found in severe foxC1dMO embryos.
While foxC1 is important for arterial specification in the trunk vessels, it is unclear if this also accounts for the severe phenotypes observed in the hyaloid vasculature. In mammals, the hyaloid is an entirely arterial system and is devoid of veins.37 This led to an initial hypothesis that defects in arterial specification may cause the hyaloid defects in foxC1dMO embryos. However, in zebrafish the venous marker flt4 was expressed in both wild-type and foxC1dMO hyaloid vessels, indicating differences exist in the zebrafish hyaloid as compared to mammals. This may be indicative of the different fates these vascular beds have in the zebrafish versus mammals. The hyaloid system is a transient structure in mammals, and its regression is required for proper retinal vessel development (reviewed in 37). In contrast, the zebrafish hyaloid system does not regress, but instead migrates and grows with the retina to form the mature vitreal-retinal vascular system.34
While endothelial cell fate defects cannot explain the hyaloid defects in foxC1 morphants, clues were provided by additional experiments. Fluorescent dextrans injected into the vasculature of foxC1dMOs showed that vessel integrity was compromised and permeability was increased. We hypothesized this may be due to defects in endothelial cell-cell junction formation. Indeed, junctional complexes are disrupted in the cornea of Foxc1 knock-out mice.12 However, TEM analysis of vessels in foxC1dMOs showed morphologically normal inter-endothelial cell junctions.
Ultrastructural analyses did reveal disruptions in the basement membranes between hyaloid vessels and the lens and retina. Staining for Laminin-111 in foxC1dMO embryos confirmed basement membrane defects. As lama1 mutants show defects in the development of the hyaloid and trunk vascular system similar to those of foxC1 morphants 28, 35, we also explored the genetic interaction between foxC1 and lama1 genes. Combined sub-maximal knock-down of foxC1 and lama1 genes resulted in vascular phenotypes similar to those observed in full-dose, severe foxC1dMO embryos. These data suggest that FoxC1 regulates factor(s) essential for normal basement membrane formation or maintenance.
We posit that basement membrane defects result in the increased permeability and hemorrhaging in CNS vessels that were observed upon tracer dye injection. Basement membrane defects may also disrupt proper differentiation of vascular cells in the hyaloid, leading to the dilated vessel morphology and accumulation of undifferentiated periocular mesenchymal cells behind the lens in foxC1 morphants. The delay in retinal lamination found in foxC1dMO eyes may also be explained by the lack of a mature basement membrane on the basal surface of the retina. This is supported by the recent finding that focal disruptions in meningeal basement membrane result in lamination defects in the cortex of the mouse Foxc1 hypomorph.14 Additionally, zebrafish mutant for either pak2a or beta-pix result in hydrocephalus and CNS hemorrhaging, similar to foxC1dMO embryos.38, 39 pak2a and beta-pix function together downstream of integrin signaling and were shown to be required for proper maintenance of the basement membrane surrounding CNS vessels.38, 39 Cumulatively, these findings suggest that defects in production or maintenance of basement membrane components may underlie aspects of vascular defects found with loss of foxC1.
The disruption of basement membrane integrity in foxC1 morphants provides insight into FOXC1 disease phenotypes. Vascular basement membrane defects have implications for the development of glaucoma in FOXC1 patients. Integrity of retinal vessels is important to maintain the blood retinal barrier and defects affecting vascular permeability are known to impact glaucoma (reviewed in 40). Vascular tone and permeability are also under the influence of systemic factors, both environmental and genetic, and this may represent a focal point where FOXC1 disease can be modified. Finally, mutations in the basement membrane constituent Collage IV alpha 1 (COL4A1), correlate with increased incidence of stroke and are associated with Axenfeld-Reiger anomaly in humans.41–45 Indeed, one recognized cause of stroke is the localized breakdown of vessel integrity and Col4a1 knockout mice show hemorrhagic stroke and ASD phenotypes.43, 46, 47 Increased mortality in FOXC1 patients nor potential relationships between FOXC1 mutations and vascular compromise have not been reported, but, together, this data suggests that FOXC1 patients may have an increased risk for stroke.
Acknowledgments
We thank Michael Cliff for technical assistance and animal husbandry, and Clive Wells for assistance with TEM. We also gratefully acknowledge the following funding support: T32EYE014537, Research training program in vision sciences (JMS), F30AG029763 (JMS), NIH R01EY16060 (BAL), and NEI Core Facilities grant P30EY001931 to the vision research community of MCW.
References
- 1.Nishimura DY, Swiderski RE, Alward WL, et al. The forkhead transcription factor gene FKHL7 is responsible for glaucoma phenotypes which map to 6p25. Nat Genet. 1998;19:140–147. doi: 10.1038/493. [DOI] [PubMed] [Google Scholar]
- 2.Kume T, Deng KY, Winfrey V, Gould DB, Walter MA, Hogan BL. The forkhead/winged helix gene Mf1 is disrupted in the pleiotropic mouse mutation congenital hydrocephalus. Cell. 1998;93:985–996. doi: 10.1016/s0092-8674(00)81204-0. [DOI] [PubMed] [Google Scholar]
- 3.Lehmann OJ, Ebenezer ND, Jordan T, et al. Chromosomal duplication involving the forkhead transcription factor gene FOXC1 causes iris hypoplasia and glaucoma. Am J Hum Genet. 2000;67:1129–1135. doi: 10.1016/s0002-9297(07)62943-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mears AJ, Mirzayans F, Gould DB, Pearce WG, Walter MA. Autosomal dominant iridogoniodysgenesis anomaly maps to 6p25. Am J Hum Genet. 1996;59:1321–1327. [PMC free article] [PubMed] [Google Scholar]
- 5.Nishimura DY, Searby CC, Alward WL, et al. A spectrum of FOXC1 mutations suggests gene dosage as a mechanism for developmental defects of the anterior chamber of the eye. Am J Hum Genet. 2001;68:364–372. doi: 10.1086/318183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saleem RA, Banerjee-Basu S, Berry FB, Baxevanis AD, Walter MA. Analyses of the effects that disease-causing missense mutations have on the structure and function of the winged-helix protein FOXC1. Am J Hum Genet. 2001;68:627–641. doi: 10.1086/318792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saleem RA, Banerjee-Basu S, Berry FB, Baxevanis AD, Walter MA. Structural and functional analyses of disease-causing missense mutations in the forkhead domain of FOXC1. Hum Mol Genet. 2003;12:2993–3005. doi: 10.1093/hmg/ddg324. [DOI] [PubMed] [Google Scholar]
- 8.Saleem RA, Murphy TC, Liebmann JM, Walter MA. Identification and analysis of a novel mutation in the FOXC1 forkhead domain. Invest Ophthalmol Vis Sci. 2003;44:4608–4612. doi: 10.1167/iovs.03-0090. [DOI] [PubMed] [Google Scholar]
- 9.Shields MB, Buckley E, Klintworth GK, Thresher R. Axenfeld-Rieger syndrome. A spectrum of developmental disorders. Surv Ophthalmol. 1985;29:387–409. doi: 10.1016/0039-6257(85)90205-x. [DOI] [PubMed] [Google Scholar]
- 10.Strungaru MH, Dinu I, Walter MA. Genotype-phenotype correlations in Axenfeld-Rieger malformation and glaucoma patients with FOXC1 and PITX2 mutations. Invest Ophthalmol Vis Sci. 2007;48:228–237. doi: 10.1167/iovs.06-0472. [DOI] [PubMed] [Google Scholar]
- 11.Walter MA, Mirzayans F, Mears AJ, Hickey K, Pearce WG. Autosomal-dominant iridogoniodysgenesis and Axenfeld-Rieger syndrome are genetically distinct. Ophthalmology. 1996;103:1907–1915. doi: 10.1016/s0161-6420(96)30408-9. [DOI] [PubMed] [Google Scholar]
- 12.Kidson SH, Kume T, Deng K, Winfrey V, Hogan BL. The forkhead/winged-helix gene, Mf1, is necessary for the normal development of the cornea and formation of the anterior chamber in the mouse eye. Dev Biol. 1999;211:306–322. doi: 10.1006/dbio.1999.9314. [DOI] [PubMed] [Google Scholar]
- 13.Gruneberg H. Congenital hydrocephalus in the mouse, a case of spurious pleitropism. J Genet. 1943;45:1–21. [Google Scholar]
- 14.Zarbalis K, Siegenthaler JA, Choe Y, May SR, Peterson AS, Pleasure SJ. Cortical dysplasia and skull defects in mice with a Foxc1 allele reveal the role of meningeal differentiation in regulating cortical development. Proc Natl Acad Sci U S A. 2007;104:14002–14007. doi: 10.1073/pnas.0702618104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith RS, Zabaleta A, Kume T, et al. Haploinsufficiency of the transcription factors FOXC1 and FOXC2 results in aberrant ocular development. Hum Mol Genet. 2000;9:1021–1032. doi: 10.1093/hmg/9.7.1021. [DOI] [PubMed] [Google Scholar]
- 16.Kume T, Deng K, Hogan BL. Murine forkhead/winged helix genes Foxc1 (Mf1) and Foxc2 (Mfh1) are required for the early organogenesis of the kidney and urinary tract. Development. 2000;127:1387–1395. doi: 10.1242/dev.127.7.1387. [DOI] [PubMed] [Google Scholar]
- 17.Kume T, Jiang H, Topczewska JM, Hogan BL. The murine winged helix transcription factors, Foxc1 and Foxc2, are both required for cardiovascular development and somitogenesis. Genes Dev. 2001;15:2470–2482. doi: 10.1101/gad.907301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Winnier GE, Kume T, Deng K, et al. Roles for the winged helix transcription factors MF1 and MFH1 in cardiovascular development revealed by nonallelic noncomplementation of null alleles. Dev Biol. 1999;213:418–431. doi: 10.1006/dbio.1999.9382. [DOI] [PubMed] [Google Scholar]
- 19.Seo S, Fujita H, Nakano A, Kang M, Duarte A, Kume T. The forkhead transcription factors, Foxc1 and Foxc2, are required for arterial specification and lymphatic sprouting during vascular development. Dev Biol. 2006;294:458–470. doi: 10.1016/j.ydbio.2006.03.035. [DOI] [PubMed] [Google Scholar]
- 20.Hayashi H, Kume T. Foxc transcription factors directly regulate Dll4 and Hey2 expression by interacting with the VEGF-Notch signaling pathways in endothelial cells. PLoS ONE. 2008;3:e2401. doi: 10.1371/journal.pone.0002401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayashi H, Kume T. Forkhead transcription factors regulate expression of the chemokine receptor CXCR4 in endothelial cells and CXCL12-induced cell migration. Biochem Biophys Res Commun. 2008;367:584–589. doi: 10.1016/j.bbrc.2007.12.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Topczewska JM, Topczewski J, Solnica-Krezel L, Hogan BL. Sequence and expression of zebrafish foxc1a and foxc1b, encoding conserved forkhead/winged helix transcription factors. Mech Dev. 2001;100:343–347. doi: 10.1016/s0925-4773(00)00534-7. [DOI] [PubMed] [Google Scholar]
- 23.McMahon C, Semina EV, Link BA. Using zebrafish to study the complex genetics of glaucoma. Comp Biochem Physiol C Toxicol Pharmacol. 2004;138:343–350. doi: 10.1016/j.cca.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 24.Thompson MA, Ransom DG, Pratt SJ, et al. The cloche and spadetail genes differentially affect hematopoiesis and vasculogenesis. Dev Biol. 1998;197:248–269. doi: 10.1006/dbio.1998.8887. [DOI] [PubMed] [Google Scholar]
- 25.Thisse B, Heyer V, Lux A, et al. Spatial and temporal expression of the zebrafish genome by large-scale in situ hybridization screening. Methods Cell Biol. 2004;77:505–519. doi: 10.1016/s0091-679x(04)77027-2. [DOI] [PubMed] [Google Scholar]
- 26.Thisse C, Thisse B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat Protoc. 2008;3:59–69. doi: 10.1038/nprot.2007.514. [DOI] [PubMed] [Google Scholar]
- 27.Topczewska JM, Topczewski J, Shostak A, Kume T, Solnica-Krezel L, Hogan BL. The winged helix transcription factor Foxc1a is essential for somitogenesis in zebrafish. Genes Dev. 2001;15:2483–2493. doi: 10.1101/gad.907401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Semina EV, Bosenko DV, Zinkevich NC, et al. Mutations in laminin alpha 1 result in complex, lens-independent ocular phenotypes in zebrafish. Dev Biol. 2006;299:63–77. doi: 10.1016/j.ydbio.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 29.Zinkevich NS, Bosenko DV, Link BA, Semina EV. laminin alpha 1 gene is essential for normal lens development in zebrafish. BMC Dev Biol. 2006;6:13. doi: 10.1186/1471-213X-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soules KA, Link BA. Morphogenesis of the anterior segment in the zebrafish eye. BMC Dev Biol. 2005;5:12. doi: 10.1186/1471-213X-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weinstein BM, Stemple DL, Driever W, Fishman MC. Gridlock, a localized heritable vascular patterning defect in the zebrafish. Nat Med. 1995;1:1143–1147. doi: 10.1038/nm1195-1143. [DOI] [PubMed] [Google Scholar]
- 32.Robu ME, Larson JD, Nasevicius A, et al. p53 activation by knockdown technologies. PLoS Genet. 2007;3:e78. doi: 10.1371/journal.pgen.0030078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lawson ND, Weinstein BM. In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev Biol. 2002;248:307–318. doi: 10.1006/dbio.2002.0711. [DOI] [PubMed] [Google Scholar]
- 34.Alvarez Y, Cederlund ML, Cottell DC, et al. Genetic determinants of hyaloid and retinal vasculature in zebrafish. BMC Dev Biol. 2007;7:114. doi: 10.1186/1471-213X-7-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pollard SM, Parsons MJ, Kamei M, et al. Essential and overlapping roles for laminin alpha chains in notochord and blood vessel formation. Dev Biol. 2006;289:64–76. doi: 10.1016/j.ydbio.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 36.Lamont RE, Childs S. MAPping out arteries and veins. Sci STKE. 2006;2006:pe39. doi: 10.1126/stke.3552006pe39. [DOI] [PubMed] [Google Scholar]
- 37.Saint-Geniez M, D’Amore PA. Development and pathology of the hyaloid, choroidal and retinal vasculature. Int J Dev Biol. 2004;48:1045–1058. doi: 10.1387/ijdb.041895ms. [DOI] [PubMed] [Google Scholar]
- 38.Buchner DA, Su F, Yamaoka JS, et al. pak2a mutations cause cerebral hemorrhage in redhead zebrafish. Proc Natl Acad Sci U S A. 2007;104:13996–14001. doi: 10.1073/pnas.0700947104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu J, Fraser SD, Faloon PW, et al. A betaPix Pak2a signaling pathway regulates cerebral vascular stability in zebrafish. Proc Natl Acad Sci U S A. 2007;104:13990–13995. doi: 10.1073/pnas.0700825104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Flammer J, Mozaffarieh M. What is the present pathogenetic concept of glaucomatous optic neuropathy? Surv Ophthalmol. 2007;52 (Suppl 2):S162–173. doi: 10.1016/j.survophthal.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 41.Gould DB, Phalan FC, van Mil SE, et al. Role of COL4A1 in small-vessel disease and hemorrhagic stroke. N Engl J Med. 2006;354:1489–1496. doi: 10.1056/NEJMoa053727. [DOI] [PubMed] [Google Scholar]
- 42.Plaisier E, Gribouval O, Alamowitch S, et al. COL4A1 mutations and hereditary angiopathy, nephropathy, aneurysms, and muscle cramps. N Engl J Med. 2007;357:2687–2695. doi: 10.1056/NEJMoa071906. [DOI] [PubMed] [Google Scholar]
- 43.Sibon I, Coupry I, Menegon P, et al. COL4A1 mutation in Axenfeld-Rieger anomaly with leukoencephalopathy and stroke. Ann Neurol. 2007;62:177–184. doi: 10.1002/ana.21191. [DOI] [PubMed] [Google Scholar]
- 44.Vahedi K, Boukobza M, Massin P, Gould DB, Tournier-Lasserve E, Bousser MG. Clinical and brain MRI follow-up study of a family with COL4A1 mutation. Neurology. 2007;69:1564–1568. doi: 10.1212/01.wnl.0000295994.46586.e7. [DOI] [PubMed] [Google Scholar]
- 45.Vahedi K, Kubis N, Boukobza M, et al. COL4A1 mutation in a patient with sporadic, recurrent intracerebral hemorrhage. Stroke. 2007;38:1461–1464. doi: 10.1161/STROKEAHA.106.475194. [DOI] [PubMed] [Google Scholar]
- 46.Favor J, Gloeckner CJ, Janik D, et al. Type IV procollagen missense mutations associated with defects of the eye, vascular stability, the brain, kidney function and embryonic or postnatal viability in the mouse, Mus musculus: an extension of the Col4a1 allelic series and the identification of the first two Col4a2 mutant alleles. Genetics. 2007;175:725–736. doi: 10.1534/genetics.106.064733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gould DB, Phalan FC, Breedveld GJ, et al. Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science. 2005;308:1167–1171. doi: 10.1126/science.1109418. [DOI] [PubMed] [Google Scholar]
- 48.Gilmour DT, Maischein HM, Nüsslein-Volhard C. Migration and function of a glial subtype in the vertebrate peripheral nervous system. Neuron. 2002;34:577–588. doi: 10.1016/s0896-6273(02)00683-9. [DOI] [PubMed] [Google Scholar]