

Abstract
Despite extensive experimental and theoretical studies, the detailed catalytic mechanism of orotidine 5′-monophosphate decarboxylase (ODCase) remains controversial. In particular simulation studies using high level quantum mechanics have failed to reproduce experimental activation free energy. One common feature of many previous simulations is that there is a water molecule in the vicinity of the leaving CO2 group whose presence was only observed in the inhibitor bound complex of ODCase/BMP. Various roles have even been proposed for this water molecule from the perspective of stabilizing the transition state and/or intermediate state. We hypothesize that this water molecule is not present in the active ODCase/OMP complex. Based on QM/MM minimum free energy path simulations with accurate density functional methods, we show here that in the absence of this water molecule the enzyme functions through a simple direct decarboxylation mechanism. Analysis of the interactions in the active site indicates multiple factors contributing to the catalysis, including the fine-tuned electrostatic environment of the active site and multiple hydrogen-bonding interactions. To understand better the interactions between the enzyme and the inhibitor BMP molecule, simulations were also carried out to determine the binding free energy of this special water molecule in the ODCase/BMP complex. The results indicate that the water molecule in the active site plays a significant role in the binding of BMP by contributing ∼ -3 kcal/mol to the binding free energy of the complex. Therefore, the complex of BMP plus a water molecule, instead of the BMP molecule alone, represents much better the tight binding transition state analogue of ODCase. Our simulation results support the direct decarboxylation mechanism and highlight the importance of proper recognition of protein bound water molecules in the protein-ligand binding and the enzyme catalysis.
Keywords: Enzyme proficiency, QM/MM, decarboxylation, confined water, transition state analogue, hydrophobic interaction
As one of the most proficient enzymes known, 1 orotidine-5′-monophosphate decarboxylase (ODCase) catalyzes the decarboxylation reaction converting orotidine-5′-monophosphate (OMP) to uridine 5′-monophosphate (UMP). (Fig. 1) An interesting observation of catalytic promiscuity has also been reported recently. 2 The rate of uncatalyzed decarboxylation of 1-methyl orotate molecule in solution (knon) was determined to be 2.8 × 10−16 s−1, 1 while the rate of the OMP decarboxylation in ODCase (kcat) is 39 s−1. 3 The rate enhancement by ODCase is thus 1.4 × 1017, and the catalytic proficiency of ODCase, defined as (kcat/Km)/knon, is ∼ 1023 M−1. Since it has been realized that this remarkable proficiency arises mostly from the difficulty of the reaction taking place in water solution, how ODCase hurdles the reaction barrier, without the assistance of any metal ions or cofactors, 4 becomes an interesting and important question to the understanding of the origin of catalytic power of the enzyme. 5
Figure 1.

Reaction scheme of OMP decarboxylation.
The active form of ODCase is a dimer, but there is no evidence indicating that the chemical events of the two active sites are coupled. Four residues in the active site were found to be critical for the catalytic activity, including two Lys and one Asp from the same monomer, and an Asp from the other monomer. 6, 7 The four residues are conserved upon different organisms, with their positions in the enzyme/substrate structures being conserved too. 8-11 Mutation of any of the four residues leads to significant, or some times complete, loss of the activity and often the affinity for the substrate as well. This observation suggests that it might be more appropriate to model the side chains of the four residues at the same level of theory when the enzymatic reaction is simulated. The phosphate and ribose groups of the substrate OMP have also been found to contribute significantly to both the binding and the catalysis of the substrate. 12-15
Several catalytic mechanisms of ODCase have been proposed and examined in detail theoretically and experimentally. 8, 16-21 Even though theoretical study has suggested that protonations of the pyrimidine ring can lower the reaction barrier substantially, 19, 20 this proposal has not gained much support from experiments. One of the major challenges for mechanistic schemes involving protonated-pyrimidine ring is that available enzyme structures do not reveal good and definite candidates for a proton donor. A recent structural study of human ODCase in complex with substrate, product and several inhibitors has raised the topic for a covalent mechanism, 22 however most experimental studies have so far supported a simple mechanism of direct decarboxylation. 5 In this scheme, the breaking of C-C bond to form the CO2 molecule determines the reaction rate; the proton transfer step, required for the formation of the product UMP molecule, may be in concert with or after the C-C bond dissociation. 23-25
In the direct decarboxylation mechanism, the enzyme neither forms a covalent bond with the substrate, nor chemically modifies the substrate (e.g., general acid or base catalysis). Compared with the uncatalyzed solution reaction, the stabilization of the enzymatic transition state (TS) thus mostly originates from the different non-covalent interactions between the enzyme and the TS of the substrate. It has been questioned whether the non-covalent interactions alone can be responsible for such a large degree of stabilization inferred by the classical transition state theory. In attempt to clarifying this issue, extensive theoretical simulations have been carried out, including pure classical molecular mechanical (MM) simulations, 26 quantum mechanical (QM) calculations of gas-phase model systems, 20 and combined quantum mechanics/molecular mechanics (QM/MM) simulations. 10, 19, 27-30 Some simulations have supported the direct decarboxylation mechanism, while some have not been able to reach a definite conclusion. Even for those simulations that supported the direct decarboxylation mechanism, the causes for the mechanism, i.e., the driving force for catalysis, have not been agreed on. For example, whether the electrostatic stress interaction or a desolvation effect drives the catalytic reaction has been under extensive debate. 10, 20, 27, 31
One additional fact responsible for the controversy is that several calculations employing density functional theory (DFT) and/or other ab initio QM methods have predicted barriers significantly higher than the experimental value. 19, 28-30 Particularly, simulations combining Car-Parrinello molecular dynamics 32 with the Jarzynski non-equilibrium method 33 yielded a barrier of 21.5 kcal/mol for direct decarboxylation and 33 kcal/mol for C6-protonation assisted decarboxylation. 29 Even though this work provided the strongest theoretical evidence for the direct decarboxylation mechanism, the computed barrier is still too high. Recently, this result was challenged by Houk and coworkers 30 whose simulations again combined DFT with a meta-dynamics sampling method. 34 In this work, simulations using two differently sized QM subsystems both yielded barriers significantly higher than experimental data. Many literatures have shown that applications of DFT in the study of reaction processes have been very successful, despite the well known difficulty of proper theoretical treatment of the electron exchange and correlation. 35, 36 Thus, one wonders if the direct decarboxylation mechanism is not the correct scenario, or if something else has not been correctly captured in previous simulations.
Mechanistic hypothesis
As a common practice, simulations of the enzyme-catalyzed reaction often start from the structures of enzyme bound with an inhibitor. For ODCase, structures bound with inhibitors such as 6-hydroxyUMP (BMP) and 6-AzaUMP have been used. The sizes of BMP and 6-AzaUMP are smaller than that of the substrate OMP, thus not surprisingly water molecules are often found in the binding pocket of the pyrimidine ring. Because of the stronger binding affinity and a negative change on the pyrimidine ring, BMP is thought to be a good transition state analogue for ODCase. BMP differs from OMP only at the place of the leaving group, which is a negatively charged -CO2− group for OMP and an O− atom (O6) for BMP. Using the ODCase/BMP structure as the initial model for the reaction simulations has been deemed to require the least modification for the construction of the structure of OMP.
In all ODCase/BMP crystal structures (PDB ID: 1DQX, 1EIX, 1LOR, 1X1Z, and 3BBG) but one (1JJK), the O6 atom of the BMP molecule binds a water molecule in the vicinity of the assumed binding pocket for the leaving CO2 molecule. (Fig. 2) (Thereby this water molecule is termed as “BMP bound water molecule” to avoid confusion). Previous reaction simulations started from the ODCase/BMP structure have always included this water molecule but have treated it at different level of theories. Some roles have also been proposed for this water molecule from the standpoint of stabilizing the transition state and/or intermediate state. Nevertheless, to our knowledge the existence of this water molecule in the OMP/ODCase complex has never been confirmed experimentally, including a recently solved mutant structure of ODCase. 22 Simply considering the fact that the decarboxylation reaction proceeds extremely slowly in water solution, one wonders why and how ODCase utilizes this water molecule in the catalytic process. If stabilizing the transition state requires some charged groups, direct interactions between the reacting moieties of OMP and those charged groups of enzyme would be more effective than the indirect interaction through this water molecule. Furthermore, the position of this water molecule is in fact obstructive to the catalytic process. In ODCase/BMP structure, this water is very close to a hydrophobic pocket, presumably a site to which the leaving CO2 molecule will bind favorably. The existence of this water molecule would thus only increase the difficulty of CO2 leaving as it probable will jam the exit path. The existence of this water may also disturb the hydrogen-bond network of Lys-Asp-Lys-Asp(B) and consequently destabilizes the transition state. From these two considerations, we hypothesize that this water molecule may in fact appear only as a cofactor for the binding of the BMP molecule or in some cases the side reactions catalyzed by ODCase, 2 and should not appear in the normal course of the catalytic process of OMP decarboxylation.
Figure 2.

Superposition of OMP/BMP X-ray structures showing the position of the water molecule.
To examine the validity of this hypothesis, we carried out accurate QM/MM simulations on the direct decarboxylation processes of OMP in solution and in ODCase without the presence of the BMP bound water molecule. The simulations were performed with the recently developed ab initio QM/MM minimum free energy path (QM/MM-MFEP) method 37-39 which allows the use of very large basis sets and accurate but costly ab initio QM methods. The results, showing good convergence with basis sets and level of theories, agreed well with experimental measurements and thus supported the mechanism of direct decarboxylation without the BMP bound water molecule in the active site. Furthermore, to illustrate the important role this water molecule plays in the tight binding of BMP and ODCase, we carried out free energy simulations to determine the binding affinity of this BMP bound water molecule in the BMP/ODCase complex. The results indicated that this water molecule contributes ∼ -3.1 kcal/mol to the total binding free energy of BMP/ODCase complex. Therefore, the combination of BMP and the water molecules, instead of the BMP molecule alone, may resemble more closely the transition state of ODCase.
Computational Details
QM/MM-MFEP method
The details of the QM/MM minimum free energy path method have been discussed in details in previous publications. 37-39 Here we only briefly review the method. The key of the QM/MM-MFEP method is to optimize the geometry of the QM subsystem on a potential of mean force (PMF) surface of QM conformations, i.e.,
| (1) |
where is the total energy of the entire system expressed as a function of the coordinates of the QM and MM subsystems, rQM and rMM, respectively. Instead of absolute values, in simulations the QM PMF is often determined as relative values through free energy perturbation (FEP) of QM geometries. 40, 41 Very conveniently, the gradient of the PMF is the ensemble average of the QM gradient
| (2) |
which can be obtained from molecular dynamics (MD) simulations of the MM atoms with the QM atoms frozen. For the consideration of computational efficiency, and without much loss of accuracy, the total energy is often recast with an electrostatic potential (ESP) fitted charge approximation for the QM atoms. 37, 38, 41, 42 That is, the quantum mechanical energy of the QM system in the presence of the MM electrostatic potential can be expressed as
| (3) |
where Heff is the effective QM Hamiltonian including the MM electrostatic potential, is defined as the QM internal energy polarized by the MM electrostatic potential, and is the electrostatic interaction between the QM and MM subsystems which can explicitly include the polarization effect on the QM system. 42 Once the QM ESP charge model is built, acts together with other classical MM potentials to drive the dynamics of the MM atoms and perform FEP calculations without the need for carrying out expensive QM calculations at every MD step. 37-39
To optimize the reaction path on the PMF surface, chain-of-conformations algorithms such as the nudged-elastic-band 43, quadratic string, 44, 45 and sequential quadratic programming methods 46 can be employed. Usually the complete QM degrees of freedom are used to construct a discrete reaction path without explicitly specifying a reaction coordinate such as bonds, bond angles, or dihedrals. The use of full QM degrees of freedom avoids the complication of computing the Jacobian term which is required in the blue-moon type general sampling methods. 47 This method has been shown to be equally applicable to the simulation of reactions in solution and in enzymes, especially after the development of an efficient sequential iterative sampling and optimization algorithm. 37 The new sequential iterative optimization algorithm allows iterative QM optimization in a fixed and finite MM ensemble, thus reduces significantly the needed MM sampling and also circumvents the challenge of generating a smooth QM free energy surface for QM optimization.
Gas-phase and solution reactions
For the decarboxylation reactions in the gas phase, we used 1-methyl orotate ion as the model compound. The reaction process was modeled by the reaction-coordinate driving method. Specifically, the C6-C7 bond was stretched from 1.55 Å to 4.05 Å at 0.1 Å per step, unless otherwise noted. In each step, the molecular geometry was optimized with the bond length of C6-C7 fixed. All calculations were conducted with the Gaussian03 program. 48 To examine the convergence of the calculations, this process was repeated with different basis sets and/or level of theories, including HF/6-31+G*, B3LYP/3-21+G*, B3LYP/6-31+G*, B3LYP/6-311+G*, B3LYP/3-21+G*/BSSE, B3LYP/6-31+G*/BSSE, B3LYP/6-311+G*/BSSE, B3LYP/aug-cc-pvdz, B3LYP/aug-cc-pvtz, B3LYP/aug-cc-pvqz, MP2/6-311+G*, and MP2/aug-cc-pvtz. The basis set superposition error (BSSE) corrections were made with the counterpoise method 49, 50 provided in Gaussian03. Because of the extremely high computational costs, the calculations with B3LYP/aug-cc-pvqz and MP2/6-311+G* were only carried out for a few selected conformational states of the bond dissociation process using geometries optimized with smaller basis sets, B3LYP/aug-cc-pvtz and MP2/6-31+G*, respectively. Functionals other than B3LYP 51, 52 were tested and did not show significant differences, thus their results will not be reported here.
For the solution reaction process, the QM/MM-MFEP method implemented in the Sigma program was employed to determine the reaction path, 37, 38 as well as the reaction free energies. The 1-methyl orotate ion was treated by QM, while 3,583 water molecules in a cubic box of 48 × 48 × 48 Å3 were simulated by the TIP3P model. 53 All the path optimizations were carried out at the level of B3LYP/6-31+G*, while the final free energies were computed with B3LYP/6-311+G*. A dual cutoff of 10 and 15 Å was used for the MD simulations and FEP calculations. The integration time step was 1 femtosecond. The long-range forces were updated every 4 integration steps with a multiple-timestep algorithm for MD integration. 54, 55 The nonbond pairlists were updated every 16 integration steps. The temperature was held at 300K through Berendsen thermostat. 56 In each optimization cycle of the QM/MM-MFEP simulation, 128 ps MD simulations were carried out in which the first 16 ps were discarded as equilibration.
MD simulation of enzyme complex
To simulate the decarboxylation reaction of OMP catalyzed by ODCase, the recently determined high-resolution structure of BMP-bound ODCase from Methanobacterium thermoautotrophicum (PDB ID 1X1Z) was used to construct the initial structure. 2 The structure has a resolution of 1.45 Å and it clearly shows that no good candidates can be found for providing protons for the pyrimidine ring. The two BMP molecules were replaced by OMP for our reaction simulations. Two BMP bound water molecules in the crystal structure were deleted for reasons aforementioned.
After hydrogen atoms were added using the website MolProbity, 57 the dimer was solvated in a rectangular box of 72 × 72 × 100 Å3. The final system contained 6642 protein atoms and 14,169 water molecules, described by CHARMM force field 58 and TIP3P water model, 53 respectively. The system was first energy minimized and then equilibrated through a series of restrained MD simulations in which positions of selective sets of atoms were restrained by a harmonic force of 40 kcal/mol/Å2. The restrained sets started from all the heavy atoms, then were reduced to all the backbone heavy atoms, and finally were left only to the Cα atoms. Afterwards, a 3.5 ns MD simulation was carried out for the entire system without any restraint. Using the multiple-timestep algorithm, the integration step sizes were 2 fs for short-range forces, 4 fs for medium-range forces, and 8 fs for long-range electrostatic forces. The PME method was used for computing the long-range electrostatic interactions. 59 All bonds were constrained by the SHAKE algorithm. 60 An 8 and 12 Å dual cutoff was employed to generate the nonbond pairlists, which were updated every 16 MD steps. The temperature and pressure of the system were maintained at 300K and 1 bar with the Berendsen thermostat and manostat. 56
QM/MM-MFEP simulation of ODCase catalysis
As the initial structure for QM/MM simulations, the final structure of the MD simulations of the ODCase/OMP complex was selected. Even though there are two active sites present in the dimer structure of ODCase/OMP, only the first active site was selected for simulating the catalytic event. The QM subsystem included the side-chain Cβ-CH2-CH2-CH2-NH3 group of Lys42 and Lys72, the side-chain Cα-CH2-COO− group of Asp70 and Asp75(B), and the OMP molecule except for the terminal -PO42− group. (Fig. 3). The total number of QM atoms is 69. The boundary atoms between the QM and MM subsystems were modeled by the pseudo-bond method. 61 All the geometry optimizations were carried out at the level of B3LYP/6-31+G*, while B3LYP/6-311+G* was used in the final free energy calculation for the optimized reaction path. The ESP charges were computed with a recently developed scheme which provided improved numerical stability with respect to the molecular geometry. 62
Figure 3.
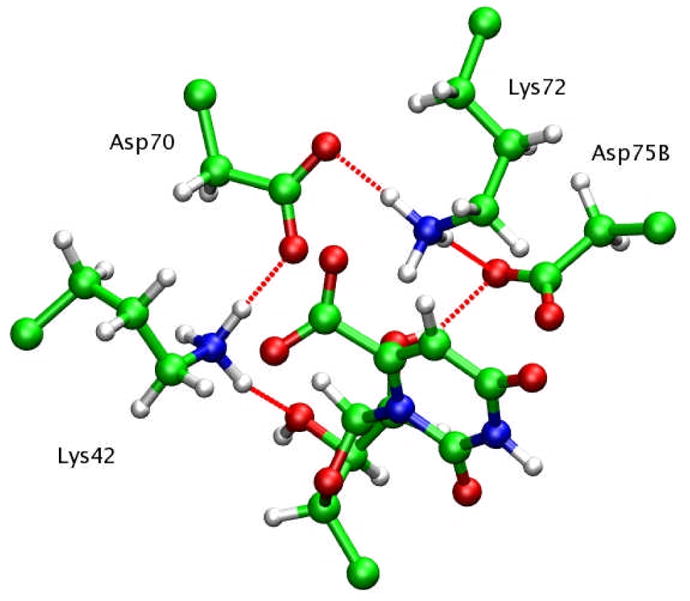
Active site atoms selected as QM subsystem.
The optimization of the reaction path and calculation of free energies were carried out with the QM/MM-MFEP method. The length of the MD sampling used for performing the QM free energy perturbation and free energy gradient calculation was 80 ps for the initial stage and was increased to 160 ps in the later stage. A dual cutoff of 10 and 15 Å was used for all QM/MM-MFEP calculations. SHAKE was only applied to water molecules. The integration time steps were 1 fs for short-range forces, 4 fs for medium-range forces, and 8 fs for long-range electrostatic forces.
Energy decomposition analysis
In the QM/MM-MFEP method, an approximate decomposition of the electrostatic and van der Waals interactions is straightforward. The QM internal energy, is defined as
| (4) |
which characterizes the internal QM energy of the QM system with the polarization due to the electrostatic potentials of the MM environment. The pairwise electrostatic interactions between the QM subsystem and MM groups can be computed just like the term , specifically,
| (5) |
where Qi is the point charge of QM atoms from fitting the QM electrostatic potentials, 62 and qj is the point charge of MM atoms usually taken from MM force fields.
As such an energy decomposition can shed very useful light into the site-specific contribution of the enzyme groups to the catalytic process, it was carried out for the reactant and transition states of the enzymatic process using structures optimized with the QM/MM-MFEP method. For each state, the entire simulation system was subjected with a 640 ps QM/MM-MFEP sampling, while the electrostatic and van der Walls interactions between the QM subsystem and each residue of the enzyme were computed and recorded for averaging.
ODCase/BMP simulation
To understand the interactions that are responsible for the tight binding of the BMP molecule to ODCase, we carried out free energy simulations to determine exactly how much the water molecule contributes to the binding affinity of BMP. The crystal structure of ODCase/BMP complex (1X1Z) was used as the initial model without any modifications. The setup of the simulation was identical to the simulation of the ODCase/OMP complex. The absolute binding free energy of the water molecule was determined through two steps of free energy simulations. (Scheme I) In the first step, the water molecule was transferred from the bound state in the BMP/ODCase complex into the gas phase but with a set of restraints to keep it staying in the nearby space, i.e., ΔG1. The free energy cost for restraining the water molecule in gas phase, a pure entropic contribution, was determined in the second step of simulations by gradually removing the restraints, i.e., ΔG2. This restrain-release approach has been previously developed for computing the absolute binding free energy and entropic contribution in ligand binding and enzyme catalysis. 63-65 The sum of the free energies from the two steps of simulation minus the free energy of transferring one water molecule from bulk state into gas phase, i.e., -ΔG3, yields the binding free energy of the BMP bound water molecule. The latter term is known as the excess free energy of liquid water, which has been previously determined for several popular water models, including the TIP3P model employed in this study.
Scheme I.

Two-step free energy simulation for the binding affinity of water in the ODCase/BMP/water complex.
The slow growth method was employed for all free energy simulations. 66, 67 Each forward and backward transformation spans for 640 ps, with another 40 ps free MD simulation between two processes for equilibration of the system. A total of 15 forward-backward cycles were carried out.
Results
Gas phase reaction
In previous simulations, various QM methods, ranging from semi-empirical AM1, EVB, to more accurate DFT and MP2 methods, have been employed. How the calculation results may depend on the employed QM theory and/or basis set thus does become a key question that must be addressed before any further QM/MM studies.
Fig. 4 depicts the energy changes computed with different QM methods. Surprisingly to some extent DFT methods show considerable basis-set dependence. Significant energetic differences were observed when the size of the basis sets was increased from 3-21+G*, to 6-31+G*, and finally to 6-311+G* whose results converged to the results of the basis sets of aug-cc-pvtz and aug-cc-pvqz. Correction of BSSE had the largest effect on the smallest basis sets, i.e., 3-21+G*, but a small to an insignificant effect on 6-31+G* and 6-311+G*. The different BSSE effects may be due to the fact that this process is a bond dissociation process without forming a new bond. At the basis sets of 6-311+G* or aug-cc-pvtz, B3LYP results agreed very well with MP2 results, which provided the justification for using B3LYP in the QM/MM simulations of enzyme catalysis.
Figure 4.

Energy change of the decarboxylation of 1-methyl orotate in gas phase computed with various methods and basis sets. The methods used include: B3LYP/3-21+G* (black line); B3LYP/3-21+G*/BSSE (black cross); B3LYP/6-31+G* (red line); B3LYP/6-31+G*/BSSE (red cross); B3LYP/6-311+G* (blue line); B3LYP/6-311+G*/BSSE (blue cross); B3LYP/aug-cc-pvtz (cyan line); B3LYP/aug-cc-pvqz (yellow line); HF/6-31+G* (violet line); MP2/6-311+G* (magenta diamond); MP2/aug-cc-pvtz (green diamond).
The observation of basis-set dependence in DFT calculations also poses an important challenge to a common approach in the QM modeling of reaction processes. That is, to save computational cost, it is a common practice to start from a low level theory and/or small basis set for geometry optimizations and switch to a high level theory and/or large basis set for energy calculations. From many prior experiences, this approach usually works very well and provides a considerable saving of computational costs. Nevertheless, such an approach may not be suitable for simulating the decarboxylation reaction of ODcase. To illustrate this point, the geometry of 1-methyl orotate ion was optimized with B3LYP functional and different basis sets. Fig. 5 shows the optimized geometries. The B3LYP/3-21+G* geometry shows a CO2 group in parallel with the ring, while B3LYP/6-31+G* and B3LYP/6-311+G* yielded almost identical structures with the CO2 group twisted from the parallel orientation of the ring. The latter two structures are consistent with the X-ray structures in which the CO2 group has been observed to be unparallel and slightly tilted with respect to the ring. 7 This structural difference between different QM methods suggests that for this decarboxylation process, 6-31+G* is the least basis set needed for geometry optimization, and 6-311+G* might be needed for energy calculations.
Figure 5.

Structure of 1-methyl orotate in gas phase optimized with different basis sets. (a) B3LYP/3-21+G*; (b) B3LYP/6-31+G*; (c) B3LYP/6-311+G*.
Solution reaction
The results of the solution reaction process, computed with the QM/MM-MFEP method at the B3LYP/6-311+G* level, are plotted in Fig. 6. The free energy barrier for this decarboxylation process was 40.2 kcal/mol, in good agreement with the experimental measurement of 38.7 kcal/mol. Part of the difference might be contributed by the fact that in the current simulations no attempts were made to identify the proton donor necessary for the final formation of 1-methyluracil. Another contribution to the difference is the translational and rotational entropy of the free CO2 molecule in solution which would further stabilize the product and likely lower the free energy profile. The good agreement between our QM/MM calculations with the experimental results on the solution reaction provides support for the reliability of our method, considering that our calculations do not use any adjustable parameter, apart from the van der Waals parameters from the standard MM force field.
Figure 6.

Potential of mean force of the decarboxylation of 1-methyl orotate in solution.
Enzyme reaction
The PMF curve of the direct decarboxylation of OMP catalyzed by ODCase is shown in Fig. 7. The optimized path is shown in a movie provided in supplementary material. The activation barrier was estimated to be 16.5 kcal/mol, in good agreement with the experimental activation free energy of 15.2 kcal/mol. 3 The initial stage of the reaction process was mainly controlled by the rotation of the ε-NH3 group of Lys72, while the C6-C7 bond length only slightly increased. Once an N-H bond pointed close to the C6 atom, the C6-C7 bond started significant bond dissociation. This result of stepwise geometrical change once again demonstrated the complexity of the reaction process as well as the difficulty of choosing an appropriate reaction coordinate in the simulation of enzymatic reaction processes, an argument we and others have raised before. 38
Figure 7.

Potential of mean force of the catalyzed decarboxylation reaction in ODCase.
The observation of the rotation of the ε-NH3 group of Lys72 in the early stage of the direct decarboxylation process may naturally make one wonder whether one of the protons can concurrently transfer to the C6 atom of OMP, thus constituting a concerted proton-transfer-decarboxylation mechanism. Indeed this process has been simulated before and it has been concluded that the barrier is too high for the mechanism to be correct. 29
Binding free energy of water in the ODCase/BMP complex
The free energy simulations results for the binding of the water molecule in the ODCase/BMP complex are reported in Table I. Using the simulation determined excess free energy of TIP3P water model, 68 the binding free energy the special water molecule was estimated to be -3.1 kcal/mol. Thus this water molecule makes significant free energy contribution to the formation of the ODCase/BMP(/H2O) complex.
Table I.
Results of free energy simulations of the ODCase/BMP/water complex.
Numbers in the parenthesis are experimental measurement of excess free energy of water molecule and corresponding binding free energy.
Discussion
Existence of BMP bound Water Molecule
The activation barrier calculated with the QM/MM-MFEP method was obtained under the assumption that there is no such “BMP bound water molecule” in the ODCase/OMP complex. The good agreement between the calculated and experimentally measured activation barrier strongly supports this hypothesis.
Experimentally, the special water molecule has indeed been observed in quite a few X-ray structures of ODCase/ligand complexes. This perhaps contributes to the formation of a common impression that there must be a water molecule bound in the native ODCase/OMP complex. Given the conformational plasticity of the enzyme, as indicated by the large-scale conformational change of the enzyme upon binding of the ligand, 5, 9 ODCase may or may not necessarily bind a water molecule. The choice is solely determined by the combined interactions between the substrate and the enzyme, which are of course affected by the shape, size, and charge distribution of the ligand. Binding of smaller ligands, such as BMP or UMP, to ODCase, may leave a cavity large enough for the favored binding of a water molecule. Interestingly, in a recently reported X-ray structure of ODCase in which Asp312 (equivalent to Asp70 in M. thermoautotrophicum) was mutated to Asn, there was no water molecule observed in the vicinity of the CO2− group. 22 As this mutation to the largest extent preserved the volume of the side chains in the active site, the structure may reflect the real situation in the ODCase/OMP complex more closely than other mutant structures.
To examine the influences of this “fictional” water molecule on the structure and dynamics of the enzyme/substrate complex, classical MD simulations were performed for ODCase/OMP with and without this special water molecule. The Cα RMSD for both simulations is shown in Fig. 8. With respect to the initial X-ray structure of ODCase/BMP, the simulation in the absence of the BMP bound water molecule showed a slightly smaller and stable RMSD for the Cα atoms. Nonetheless, we like to emphasize here that this small difference cannot be used as evidence to support our hypothesis; instead, it merely indicates the complexity of this situation since both states showed reasonable structural fluctuations. On the other hand, diffusion of a water molecule in/out the nearby space of the carboxylate group of OMP was not observed in the complete length of our MD simulations. This observation suggests that the exchange of such a water molecule, if possible, might happen on a longer simulation timescale. Furthermore, as MD simulations indicate insignificant structural difference for the water-bound and unbound states, it is possible that both states exist for ODCase/OMP molecule, but the catalytically active form lacks this water molecule. The free energies of the two states, water-bound but catalytically inactive and water-absent but catalytically active, can be quite close. We noted in an early simulation, 69 it was observed that water molecules (modeled with MM force fields) appeared in the active site in the reactant state but diffused away in the transition state. We believe this observation in fact agrees with our hypothesis that the water molecule cannot make stabilizing contributions to the transition state. Of course, verification of this water-exchanging mechanism is beyond the scope of the current paper, thus we did not pursue on this question.
Figure 8.

Cα RMSD of ODCase dimer without (black) and with (red) the crystal water molecule.
Role of Lys72
In both mutagenesis experiments 6, 7 and computer simulations, 27, 29, 69 Lys72 has been proposed to play a vital role in the catalytic process. Our simulations revealed a vivid atomistic picture of how the ε-NH3 group of Lys72 participates in the reaction process. In our simulation, the rotation of the ε-NH3 group appears to be a prerequisite for the initialization of the decarboxylation process. The rotation apparently shortened the distance between the ε-NH3 proton and the C6 atom without the need for moving heavy atoms such as the nitrogen atom of the ε-NH3 group. This distance change is in part agreement with previous simulation which has determined a change from 4.5 Å to 2.8 Å for the distance between ε-NH3 nitrogen and C6 atom. 10, 69 Note that such a rotation can occur spontaneously in the enzyme-substrate complex, as shown by the fluctuation of the distance between the HN and C6 atoms observed in MD simulations. (Fig. 9) Some experiments 25 have suggested that a proton-involved conformational transition is responsible for the observed solvent isotope effect. From our simulations, we speculate that the rotation of the ε-NH3 group of Lys72 might be contributing to this isotope effect observed in experiments.
Figure 9.

Fluctuations of the distance between ε-H1(N) of Lys72 and C6 of OMP during MD simulations.
The current results also suggest that the ε-NH3 group of Lys72 stabilizes the carbanion intermediate state. Given the nature of this reaction, as shown by the small free energy difference between the carbanion intermediate state and the highest point on the reaction process, ε-NH3 group of Lys72 may also stabilize the transition state. This issue has been questioned before as whether the developing negative charge on the ring could be stabilized by the hydrogen-bond provided by this group. 70 Two considerations may help clarify this question. First, accompanying the leaving of the CO2 group, a negative charge will slowly develop and will distribute over the whole ring system and the surrounding groups like the ε-NH3 group of Lys72. (Table II). From the reactant state to transition state, the ESP charge of C6 atom changed from -0.047 to -0.234, while the Mulliken charges all became relatively more negative except for atom C5. Second, the pKa of the C6 position of UMP was measured to be ∼34 in solution and ≤ 22 in enzyme. 71, 72 Therefore, any nearby acidic groups, such as the ε-NH3 group of Lys72, with pKa lower than that of C6, would be able to stabilize the basic form to some extent. However, one certainly does not expect a strong acid near this active site; otherwise, the potential proton transfer between Lys and Asp, and/or between Lys and the leaving CO2 group will effectively inhibit the enzyme. This argument for the avoiding of inhibitory proton transfer reaction also suggests that there might be some tuning-up for the pKa of ε-NH3 group of Lys72 due to the fine H-bonding network of the active site.
Table II.
Atomic ESP charges of the orotidine ring and ε-NH3 group of Lys72 at reactant and transition states. Mulliken charges are also shown in parenthesis.
| atom | charge (au) | |
|---|---|---|
|
| ||
| Reactant state | Transition state | |
| Nε-Lys72 | −1.060 (−0.479) | −0.498 (−0.409) |
| Hε1-Lys72 | 0.451 (0.414) | 0.226 (0.491) |
| Hε2-Lys72 | 0.836 (0.509) | 0.996 (0.505) |
| Hε3-Lys72 | 0.786 (0.528) | 0.556 (0.511) |
| N1 | 0.115 (0.607) | −0.116 (0.458) |
| C6 | −0.047 (0.590) | −0.234 (−0.184) |
| C7 | 0.851 (−0.572) | 0.729 (0.126) |
| O71 | −0.667 (−0.206) | −0.500 (−0.174) |
| O72 | −0.737 (−0.288) | −0.522 (−0.145) |
| C2 | 0.924 (0.755) | 1.053 (0.633) |
| O2 | −0.738 (−0.328) | −0.794 (−0.360) |
| N3 | −0.787 (−0.654) | −0.842 (−0.671) |
| H3 | 0.413 (0.571) | 0.400 (0.555) |
| C4 | 0.971 (0.095) | 1.054 (0.070) |
| O4 | −0.841 (−0.609) | −0.910 (−0.652) |
| C5 | −0.623 (−0.658) | −0.662 (0.115) |
| H5 | 0.229 (0.234) | 0.185 (0.211) |
Interactions at the active site and the origin of catalysis
One reason that we believe the crystal water observed in the ODCase/BMP complex does not exist in the ODCase/OMP complex is the existence of a large hydrophobic pocket that presumably binds the leaving CO2 molecule. An interesting proposal has been that the hydrophobic interactions between the substrate and the active site groups of the enzyme provide driving force for the catalysis, as hinted by some experimental evidences. 70, 73 In fact, strong hydrophobic interactions in the CO2 side of the binding pocket have also been observed. Specifically, it has been observed that 6-thiocarboxamidouridine binds even tighter than the substrate OMP, 74 and upon binding to ODCase, the structure of 6-Cyanouridine 5′-phosphate distorted. 73 A quantitative assessment on the degree of “hydrophobicity” and associated catalytic contribution is difficult because our current knowledge about the “hydrophobic” interactions is still incomplete. This said, some information can still be inferred from careful analysis.
The contributions from “hydrophobicity” can be roughly divided into two parts by their different acting mechanisms. First, a hydrophobic active site can effectively change polarization behavior of the system and thus the height of the reaction barrier, and even the catalytic chemistry by modifying the pKa of titratable groups. Second, the van der Waals packing interactions between the reactive species and the thermally fluctuating environments could also contribute to the change of barrier height.
For the contribution from the first source, it is often assumed that the macroscopic dielectric constant of the active site directly affects the reaction barrier. One frequently assumed feature of a “hydrophobic” environment is low dielectric constant and low polarizability. If only considering these two properties, the best “hydrophobic” environment would be gas phase. For the OMP decarboxylation process, the barrier of the solution reaction is 40.2 kcal/mol with a C6-C7 bond length at 2.5 ∼ 2.7 Å, stretching that bond to the same distance in gas phase only costs ∼ 26 kcal/mol, or even less if entropic contribution is considered. On one hand, this result shows how unfavorable water is as a solvent for this reaction process; on the other hand, it also establishes the approximate limit of how much “hydrophobic” interaction may contribute. This analysis reiterates the conclusion that the proficiency of ODCase mostly originates from the difficulty of the solution reaction, or in other words, from water being a poor solvent for this reaction. In principle, by binding the OMP molecule and protecting the pyrimidine ring from unfavorable electrostatic interactions with the water molecule, ODCase already effectively lowers the barrier of OMP decarboxylation, not to mention the additional, even more favored, interactions with the active site Lys-Asp-Lys-Asp H-bond network.
To further lower the barrier from ∼ 26 kcal in a low dielectric environment to 15.2 kcal/mol, other interaction from the second source, such as van der Waals interactions between the leaving CO2 group and the hydrophobic side chains of enzyme may contribute. A quantitative estimation of this interaction is difficult because part of the contribution may have already been considered in our QM modeling of the side chains of four active site residues. However, experiences in physical chemistry would suggest that this term probably is small because of the weak nature of van der Waals interactions. Together with the packing interactions, interactions from the ε-NH3 group of Lys72 and other residues that make hydrogen-bonds with the pyrimidine ring should be able to further lower the barrier without much difficulty.
To assess the site-specific contributions from the enzyme, in the simulation study of enzyme catalysis, it is often thought to be helpful to determine quantitative interactions between the reacting groups and the environmental groups of enzymes. It is hoped that such an energy decomposition analysis may reveal the driving force for the catalytic power of enzyme. Even though a rigorous energy decomposition scheme is generally not possible, an MM force field inspired analysis of the nonbond interactions becomes a common practice, in which both the electrostatic and van der Waals interactions are assumed to be separable between different pairs of chemical groups. The electrostatic and van der Waals interactions between the QM subsystem and other residues were computed and plotted in Fig. 10. Somewhat as expected, those residues that form H-bond network to the pyrimidine ring, such as Met126, Ser127, and His 128, make favorable interactions to the stabilization of the transition state. Other residues interacting with these three residues, including Arg160, Arg163, Ala74(B), and Asn80(B), also make favorable contributions to the reaction process. The differences between the QM internal energy, QM/MM electrostatic energy, and QM/MM van der Waals energy of the reactant state and approximate transition state are shown in Table III. The difference of the QM internal energy, being merely a 22.6 kcal/mol, clearly indicates a significant reduction of the intrinsic barrier of the decarboxylation process in the enzyme environment. Further stabilization of the transition state was mostly achieved through the electrostatic interactions between the enzyme and the active site groups.
Figure 10.

Nonbond contributions of each residue to the QM subsystem. The energy difference is defined as ETS – ERS. a: electrostatic interactions; b: van der Waals interactions; c: total nonbond interactions. The central line denotes the separation of the two monomers of ODCase.
Table III.
Component energy contribution to the reaction barrier (in kcal/mol). ΔE1, ΔEQM/MM,ESP, and ΔEQM/MM,vdW are the difference of QM internal energy, QM/MM electrostatic energy, and QM/MM van der Waals energy, respectively. The energy difference is defined as ETS – ERS.
| Energy component | ΔE1 | ΔEQM/MM,ESP | ΔEQM/MM,vdW |
|---|---|---|---|
| 22.62 | −9.07 | 2.65 |
From these results, it becomes clear that the properly aligned structure of the active site played the most critical role in the catalysis of ODCase: the reduction of intrinsic barrier in the enzyme active site and the stabilization of the transition state from residues around the active site. This conclusion is in good coherence with previous study in which a delicate electrostatic and probable hydrophobic interaction in the ODCase active site has been discussed. 69 However, because of the difficulties of defining rigorous scheme for the decomposition of interaction energies and quantifying the degree of hydrophobicity, we will neither make attempts to classify those interactions as electrostatic or hydrophobic, nor further distinguish the two schemes as stabilization of the transition state or destabilization of the ground state.
The current reaction path determined with the QM/MM-MFEP method also brings new insight into the role of 2′-hydroxyl group on the ribose of OMP molecule. In the reactant state, 2′-OH forms H-bond with the Oδ1 atom of Asp75(B). In sync with the rotation of the ε-NH3 group of Lys72, 2′-OH switches H-bonding from Oδ1 atom of Asp75(B) to the Oδ2 atom. Obviously, replacing 2′-OH group by a hydrogen atom would create two kinds of effects. First, the precise alignment of Lys-Asp-Lys-Asp H-bonding network will be distorted; second, the ability of the rotation of the ε-NH3 group of Lys72 will be affected. For both, it is likely this replacement will reduce the enzymatic proficiency. This analysis seems to be in good agreement with experimental observations which showed that the ratio of kcat/KM of 2′-deoxyOMP is reduced by more than 2200-fold as compared with OMP. 12 Previous simulation has proposed important roles for 2′-OH too, 10, 69 however, some details are different from the current work. In previous work, there were direct hydrogen-bonding interactions between the ε-NH3 group of Lys72 and ribosyl 2′-OH observed, while in the current work, the side-chain carboxylate group of Asp75(B) bridges the two groups through H-bonds.
Transition state analogue
The free energy simulations in the current study indicate that the crystal water molecule makes significant contributions to the binding of BMP. Therefore, BMP molecule plus the water molecule, instead of the BMP molecule alone, is likely the correct transition state analogue. An interesting question arises as to why BMP binds a water molecule in the active site. On one hand, the presence of this water molecule may provide better electrostatic interactions as this water molecule can form two H-bonds, one with the O6 atom of BMP, and the other with Oδ1 atom of Asp70. One the other hand, because of the smaller size of BMP molecule, an additional water molecule may provide favorable packing interactions between the inhibitor and the enzyme.
Comparison to previous DFT simulations
Previous DFT based QM/MM simulations of direct decarboxylation process have yielded barriers significantly higher than the experimental results. 29, 30 Several factors may contribute to the high barriers obtained in those simulations. First and foremost, as we suggest in this work, the existence of the water molecule in the active site is indeed questionable. Second, the unclear protonation state of the active site histidine residue existed in ODCase from some organisms may contribute. The histidine residue is in close contacts with the ribosyl hydroxyl groups of OMP and the hydrogen bonding network of the active site. Obviously improper protonation state, or even the rotamer state, may cause a significant disturbance to the active site structure. Including or not the protonated imidazole group in the QM subsystem would predictably cause significant difference in the calculation. However, the protonation state of this histidine residue in the active site cannot be easily determined. An analysis by the X-ray crystallographic site MolProbity 57 in fact has not been able to distinguish the orientation and proton position for the crystal structure, 9 which implies that a closer examination of the structure or even diffractional data may be needed. The structure we used here has better resolution and contains no histidine residues in direct contacts to the OMP molecule. 2 The use of this particular structure indeed saved us from many technical difficulties. Third, the choice of the collective variable in previous free energy simulations may also contribute. Previous work has suggested that improper definition of reaction coordinate may lead to slow convergence and artifacts in simulation; 75 what we have shown here suggest that a good reaction coordinate for the decarboxylation process may in fact require at least two variables: rotation of the ε-NH3 group of Lys72, and the C6-C7 bond length of OMP molecule. Even though it was argued that the meta-dynamics method has less dependence on the choice of the reaction coordinate, this conclusion has yet to be examined carefully for such a complicated case.
Conclusion
Using the ab initio QM/MM-MFEP method, we showed here that ODCase catalyzes the decarboxylation of OMP through a simple direct route. Although a crystallographic water molecule was found to contribute significantly to the binding of BMP molecule, we proposed that such a water molecule cannot exist in the catalytic process of ODCase/OMP complex. Analysis of reaction energetics in gas phase, solution, and enzyme indicated that the solution reaction is most difficult, and the parallel gas phase process is substantially easier; while the internal energy barrier of the enzymatic process is of the similar height of that of the gas phase process. Thus, a combination of the site-specific H-bonding interactions and perhaps some degree of hydrophobic/desolvation effect in the enzyme active site provides the catalytic driving forces of ODCase.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health for financial support of this project. We also thank Dr. Richard Wolfenden for stimulating discussion, Dr. Ernest R. Davidson for discussion about BSSE, and Dr. Mel Levy for comments on the DFT functionals.
Footnotes
Supporting Information Available: Complete information for references 48 and 58. The MFEP optimized reaction path in a movie format. This material is free of charge via the Internet at http://pubs.acs.org.
Contributor Information
Hao Hu, Email: hao.hu@duke.edu.
Weitao Yang, Email: weitao.yang@duke.edu.
References
- 1.Radzicka A, Wolfenden R. Science. 1995;267:90–93. doi: 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]
- 2.Fujihashi M, Bello AM, Poduch E, Wei LH, Annedi SC, Pai EF, Kotra LP. J Am Chem Soc. 2005;127:15048–15050. doi: 10.1021/ja054865u. [DOI] [PubMed] [Google Scholar]
- 3.Porter DJT, Short SA. Biochemistry. 2000;39:11788–11800. doi: 10.1021/bi001199v. [DOI] [PubMed] [Google Scholar]
- 4.Miller BG, Smiley JA, Short SA, Wolfenden R. J Biol Chem. 1999;274:23841–23843. doi: 10.1074/jbc.274.34.23841. [DOI] [PubMed] [Google Scholar]
- 5.Miller BG, Wolfenden R. Annu Rev Biochem. 2002;71:847–885. doi: 10.1146/annurev.biochem.71.110601.135446. [DOI] [PubMed] [Google Scholar]
- 6.Miller BG, Snider MJ, Wolfenden R, Short SA. J Biol Chem. 2001;276:15174–15176. doi: 10.1074/jbc.M011429200. [DOI] [PubMed] [Google Scholar]
- 7.Wu N, Gillon W, Pai EF. Biochemistry. 2002;41:4002–4011. doi: 10.1021/bi015758p. [DOI] [PubMed] [Google Scholar]
- 8.Appleby TC, Kinsland C, Begley TP, Ealick SE. Proc Natl Acad Sci USA. 2000;97:2005–2010. doi: 10.1073/pnas.259441296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller BG, Hassell AM, Wolfenden R, Milburn MV, Short SA. Proc Natl Acad Sci USA. 2000;97:2011–2016. doi: 10.1073/pnas.030409797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu N, Mo YR, Gao JL, Pai EF. Proc Natl Acad Sci USA. 2000;97:2017–2022. doi: 10.1073/pnas.050417797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harris P, Poulsen JCN, Jensen KF, Larsen S. Biochemistry. 2000;39:4217–4224. doi: 10.1021/bi992952r. [DOI] [PubMed] [Google Scholar]
- 12.Miller BG, Butterfoss GL, Short SA, Wolfenden R. Biochemistry. 2001;40:6227–6232. doi: 10.1021/bi0028993. [DOI] [PubMed] [Google Scholar]
- 13.Miller BG, Snider MJ, Short SA, Wolfenden R. Biochemistry. 2000;39:8113–8118. doi: 10.1021/bi000818x. [DOI] [PubMed] [Google Scholar]
- 14.Amyes TL, Richard JP, Tait JJ. J Am Chem Soc. 2005;127:15708–15709. doi: 10.1021/ja055493s. [DOI] [PubMed] [Google Scholar]
- 15.Sievers A, Wolfenden R. Bioorg Chem. 2005;33:45–52. doi: 10.1016/j.bioorg.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 16.Silverman RB, Groziak MP. J Am Chem Soc. 1982;104:6434–6439. [Google Scholar]
- 17.Beak P, Siegel B. J Am Chem Soc. 1976;98:3601–3606. doi: 10.1021/ja00428a035. [DOI] [PubMed] [Google Scholar]
- 18.Beak P, Siegel B. J Am Chem Soc. 1973;95:7919–7920. doi: 10.1021/ja00804a088. [DOI] [PubMed] [Google Scholar]
- 19.Lee TS, Chong LT, Chodera JD, Kollman PA. J Am Chem Soc. 2001;123:12837–12848. doi: 10.1021/ja011096f. [DOI] [PubMed] [Google Scholar]
- 20.Lee JK, Houk KN. Science. 1997;276:942–945. doi: 10.1126/science.276.5314.942. [DOI] [PubMed] [Google Scholar]
- 21.Gao JL. Curr Opin Struct Biol. 2003;13:184–192. doi: 10.1016/s0959-440x(03)00041-1. [DOI] [PubMed] [Google Scholar]
- 22.Wittmann JG, Heinrich D, Gasow K, Frey A, Diederichsen U, Rudolph MG. Structure. 2008;16:82–92. doi: 10.1016/j.str.2007.10.020. [DOI] [PubMed] [Google Scholar]
- 23.Ehrlich JI, Hwang CC, Cook PF, Blanchard JS. J Am Chem Soc. 1999;121:6966–6967. [Google Scholar]
- 24.Rishavy MA, Cleland WW. J Am Chem Soc. 2000;39:4569–4574. doi: 10.1021/bi000376p. [DOI] [PubMed] [Google Scholar]
- 25.Toth K, Amyes TL, Wood BM, Chan K, Gerlt JA, Richard JP. J Am Chem Soc. 2007;129:12946–12947. doi: 10.1021/ja076222f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hur S, Bruice TC. Proc Natl Acad Sci USA. 2002;99:9668–9673. doi: 10.1073/pnas.142307099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Warshel A, Strajbl M, Villa J, Florian J. Biochemistry. 2000;39:14728–14738. doi: 10.1021/bi000987h. [DOI] [PubMed] [Google Scholar]
- 28.Lundberg M, Blomberg MRA, Siegbahn PEM. J Mole Model. 2002;8:119–130. doi: 10.1007/s00894-002-0080-2. [DOI] [PubMed] [Google Scholar]
- 29.Raugei S, Cascella M, Carloni P. J Am Chem Soc. 2004;126:15730–15737. doi: 10.1021/ja0455143. [DOI] [PubMed] [Google Scholar]
- 30.Stanton CL, Kuo IFW, Mundy CJ, Laino T, Houk KN. J Phys Chem B. 2007;111:12573–12581. doi: 10.1021/jp074858n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Warshel A, Florián J, Strajbl M, Villá J. ChemBioChem. 2001;2:109–111. doi: 10.1002/1439-7633(20010202)2:2<109::AID-CBIC109>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 32.Car R, Parrinello M. Phys Rev Lett. 1985;55:2471–2474. doi: 10.1103/PhysRevLett.55.2471. [DOI] [PubMed] [Google Scholar]
- 33.Jarzynski C. Phys Rev Lett. 1997;78:2690–2693. [Google Scholar]
- 34.Laio A, Parrinello M. Proc Natl Acad Sci USA. 2002;99:12562–12566. doi: 10.1073/pnas.202427399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mori-Sanchez P, Cohen AJ, Yang WT. J Chem Phys. 2006;125:201102. doi: 10.1063/1.2403848. [DOI] [PubMed] [Google Scholar]
- 36.Mori-Sanchez P, Cohen AJ, Yang WT. J Chem Phys. 2006;124:091102. doi: 10.1063/1.2179072. [DOI] [PubMed] [Google Scholar]
- 37.Hu H, Lu Z, Parks JM, Burger SK, Yang WT. J Chem Phys. 2008;128:034105. doi: 10.1063/1.2816557. [DOI] [PubMed] [Google Scholar]
- 38.Hu H, Lu ZY, Yang WT. J Chem Theory Comput. 2007;3:390–406. doi: 10.1021/ct600240y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu H, Yang WT. Annu Rev Phys Chem. 2008;59:573–601. doi: 10.1146/annurev.physchem.59.032607.093618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chandrasekhar J, Smith SF, Jorgensen WL. J Am Chem Soc. 1985;107:154–163. [Google Scholar]
- 41.Zhang Y, Liu H, Yang WT. J Chem Phys. 2000;112:3483–3492. [Google Scholar]
- 42.Lu Z, Yang WT. J Chem Phys. 2004;121:89–100. doi: 10.1063/1.1757436. [DOI] [PubMed] [Google Scholar]
- 43.Jónsson H, Mills G, Jacobsen KW. Nudged Elastic Band Method for Finding Minimum Energy Paths of Transitions. In: Berne BJ, Ciccotti G, Coker DF, editors. Classical and Quantum Dynamics in Condensed Phase Simulations. World Scientific; Singapore: 1998. pp. 385–404. [Google Scholar]
- 44.Burger SK, Yang WT. J Chem Phys. 2006;124:054109. doi: 10.1063/1.2163875. [DOI] [PubMed] [Google Scholar]
- 45.E W, Ren W, Vanden-Eijnden E. Phys Rev B. 2002;66:052301. [Google Scholar]
- 46.Burger SK, Yang WT. J Chem Phys. 2007;127:164107. doi: 10.1063/1.2780147. [DOI] [PubMed] [Google Scholar]
- 47.Carter EA, Ciccotti G, Hynes JT, Kapral R. Chem Phys Lett. 1989;156:472–477. [Google Scholar]
- 48.Frisch MJ, et al. Gaussian 03, C.02. Gaussian, Inc.; Wallingford, CT: 2004. [Google Scholar]
- 49.Simon S, Duran M, Dannenberg JJ. J Chem Phys. 1996;105:11024–11031. [Google Scholar]
- 50.Boys SF, Bernardi F. Mol Phys. 1970;19:553–566. [Google Scholar]
- 51.Becke AD. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 52.Lee C, Yang WT, Parr RG. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 53.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 54.Tuckerman ME, Berne BJ, Martyna GJ. J Chem Phys. 1992;97:1990–2001. [Google Scholar]
- 55.Schlick T, Skeel RD, Brünger AT, Kalé LV, Board JA, Hermans J, Schulten K. J Comput Phys. 1999;151:9–48. [Google Scholar]
- 56.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- 57.Lovell SC, Davis IW, Arendall WB, de Bakker PIW, Word JM, Prisant MG, Richardson JS, Richardson DC. Proteins: Struct, Funct, Genet. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 58.MacKerell AD, Jr, et al. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 59.Darden TA, York DM, Pedersen LG. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 60.Ryckaert JP, Ciccotti G, Berendsen HJC. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 61.Zhang Y, Lee TS, Yang WT. J Chem Phys. 1999;110:46–54. [Google Scholar]
- 62.Hu H, Lu Z, Yang WT. J Chem Theory Comput. 2007;3:1004–1013. doi: 10.1021/ct600295n. [DOI] [PubMed] [Google Scholar]
- 63.Hermans J, Wang L. J Am Chem Soc. 1997;119:2707–2714. [Google Scholar]
- 64.Strajbl M, Sham YY, Villa J, Chu ZT, Warshel A. J Phys Chem B. 2000;104:4578–4584. [Google Scholar]
- 65.Boresch S, Tettinger F, Leitgeb M, Karplus M. J Phys Chem B. 2003;107:9535–9551. [Google Scholar]
- 66.Hermans J. J Phys Chem. 1991;95:9029–9032. [Google Scholar]
- 67.Hu H, Yun RH, Hermans J. Mol Sim. 2002;28:67–80. [Google Scholar]
- 68.Hermans J, Pathiaseril A, Anderson A. J Am Chem Soc. 1988;110:5982–5986. doi: 10.1021/ja00226a009. [DOI] [PubMed] [Google Scholar]
- 69.Gao JL, Byun KL, Kluger R. Catalysis by enzyme conformational change. Orotidine Monophosphate Decarboxylase: Mechanistic Dialogue. 2004;238:113–136. [Google Scholar]
- 70.Callahan BP, Wolfenden R. J Am Chem Soc. 2004;126:14698–14699. doi: 10.1021/ja0450049. [DOI] [PubMed] [Google Scholar]
- 71.Sievers A, Wolfenden R. J Am Chem Soc. 2002;124:13986–13987. doi: 10.1021/ja021073g. [DOI] [PubMed] [Google Scholar]
- 72.Amyes TL, Wood BM, Chan K, Gerlt JA, Richard JP. J Am Chem Soc. 2008;130:1574–1575. doi: 10.1021/ja710384t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Callahan BP, Bell AF, Tonge PJ, Wolfenden R. Bioorg Chem. 2006;34:59–65. doi: 10.1016/j.bioorg.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 74.Miller BG. Insight into the catalytic mechanism of orotidine 5 ′-phosphate decarboxylase from crystallography and mutagenesis. In: Lee JKT, D J, editors. Orotidine Monophosphate Decarboxylase: Mechanistic Dialogue. Vol. 238. Springer; Berlin Heidelberg: 2004. pp. 43–62. [Google Scholar]
- 75.Bolhuis PG, Dellago C, Chandler D. Proc Natl Acad Sci USA. 2000;97:5877–5882. doi: 10.1073/pnas.100127697. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.