

Abstract
Over the lifespan of therapeutic proteins, from the point of biosynthesis to the complete clearance from tested subjects, they undergo various biological modifications. Therapeutic influences and molecular mechanisms of these modifications have been well appreciated for some while remained less understood for many. This paper has classified these modifications into multiple categories, according to their processing locations and enzymatic involvement during the trafficking events. It also focuses on the underlying mechanisms and structural-functional relationship between modifications and therapeutic properties. In addition, recent advances in protein engineering, cell line engineering, and process engineering, by exploring these complex cellular processes, are discussed and summarized, for improving functional characteristics and attributes of protein-based biopharmaceutical products.
1. Introduction
Nature has evolved complex biological processes that enable mammalian cells to conduct sophisticated physiological activities for living and adaptation. Primary structure of a protein obtained from genome projects serves as a nice starting point for the understanding of biological complexity but not sufficient to explain various functions and regulations. Majority of proteins from eukaryotic cells are subjected to certain kind of covalent modifications either during or after their ribosomal synthesis. An increasing appreciation of these cellular homeostatic modifications, which have been shown to initiate various biological functions and regulation mechanisms, should contribute to unfolding detailed knowledge about biological networks and living systems.
One of the earliest pieces of evidences for the existence of posttranslational modifications can be traced back to the finding of protein phosphorylation published in 1883 [1, 2], in which protein casein was found containing stoichiometric amount of phosphate. One of the earliest modified proteins to be studied, the first glycoprotein, is likely the glycogen of liver which the French physiologist Claude Bernard famously identified as “glycogenous matter” in 1855 [3]. During the biosynthesis of all proteins, the building blocks are 19 standard amino acids (plus selenocysteine for a few selenoproteins) and one imino acid. Nonetheless upon hydrolysis, close to 200 different amino acids have been identified, indicating a significant posttranslational modification of the originally encoded sequences [4]. Over the years, more than 200,000 reported modification events have been unveiled by a PubMed analysis [5]. Several hundred modifications have been characterized to date [4, 6, 7]. Some of these modifications are spontaneous reactions, while most of them involve specific enzymes and pathways. Certain structural determinants within the primary sequences of polypeptides are recognized by cellular machineries, and are carefully regulated by time and space. Efficiency of these modifications varies according to cell types, availability of substrates and cofactors, and biological conditions.
Most therapeutic proteins approved or in development bear at least one or more of posttranslational modifications [8–10]. Majority of these proteins are originally synthesized in endoplasmic-reticulum-(ER-) bound ribosomes, translocated across ER membranes through translocon, and transported through the secretory pathway into extracellular space. A subset of modifications accompany along with this biological process, and additional modifications occur during in vitro manipulations, that is, purification, formulation, storage, and injection into test subjects. These modifications and their underlying molecular mechanisms form the main focus of this review. Other modifications that are characteristics of intracellular proteins, such as acetylation, ADP ribosylation, sumoylation, and ubiquitination, can be found in several recent reviews [5–7, 11–14] and therefore not considered in this paper.
It is obvious that posttranslational modifications affect structural and functional aspects of therapeutic proteins. The effects can be detrimental, that is, heterogeneity [15] and immunogenicity [16], even though the modification may originally be required for functional activity of the polypeptide. A better understanding of the relationship between the primary sequences of therapeutic proteins and cellular machineries can allow developers to avoid unwanted side effects of these modifications. Such knowledge can also help improve protein efficacy and quality through protein engineering, cell line engineering, and process engineering. This review classifies these modifications into four major categories, three of which are based on the locations where these modifications take place within the trafficking events: ER, Golgi, and Exocellular space (Figure 1). N-linked glycosylation modification involves both ER and Golgi compartments, therefore being categorized as a separated group. In each of these categories, molecular mechanism and specific pathways for each modification are described (Table 1). The purpose of the review is restricted to modifications that may be found on therapeutic proteins secreted from animal cells. Therapeutic protein examples bearing the corresponding modification are discussed, and their effects on physiological and biophysical properties are analyzed. In the last part of the paper, recent progresses on protein engineering, cell line engineering, and process engineering, by utilizing posttranslational modification pathways, are considered and summarized (Table 2).
Figure 1.
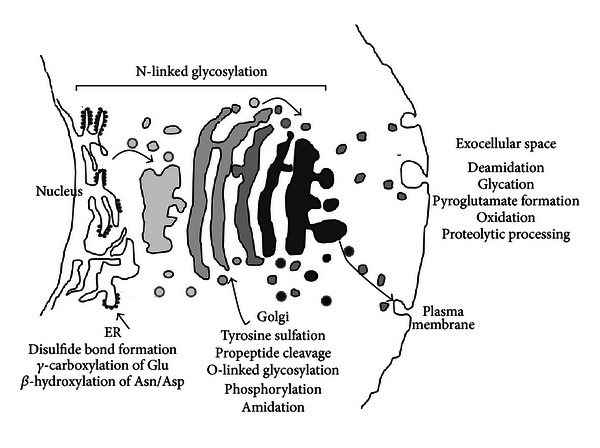
Classification of therapeutic protein modifications along trafficking pathways.
Table 1.
Consensus motifs and enzymes responsible for therapeutic modifications.
| Modification type | Linkage | Consensus sequence | Enzymes |
|---|---|---|---|
| N-glycosylation | GlcNAc-Asn | N-X-(S/T), X≠Pro | OST |
| Mucin-type O-glycosylation |
GalNAc-Ser/Thr | No consensus, Pro favorable | ppGalNAcT |
| Disulfide bond | –S–S– | Cysteine pairs | Ero1-PDI |
| γ-carboxylation | Glu→Gla | Mediated by adjacent propeptide | GGCX-VKOR |
| β-hydroxylation | Asn→Hyn; Asp→Hya | C-X-D/N-X-X-X-X-F/Y-X-C-X-C but not sufficient |
β-hydroxylase |
| Tyrosine sulfation | O4-sulfate ester | No simple consensus site Glu/Asp around Tyr favorable |
TPST1/TPST2 |
| Propeptide cleavage | R/K ↓ | With single or pair of basic amino acids | PC family members |
| Phosphorylation | pS, pT | S-x-E/pS or Ser/Thr | FAM20C or four-jointed |
| Amidation | C-terminal carboxyl→amide | C-terminal glycine | PAM |
| Deamidation | Asn→Asp; Gln→Glu | Asn-Gly most susceptible | Unknown or none |
| Glycation | Ketoamine or Amadori product | N-terminal primary amine Or amino group of lysine side chain |
Unknown or none |
| Pyroglutamate | Pyrrolidone carboxylic acid | N-terminal Gln or Glu | Unknown or none |
| Oxidation | Met→Met sulfoxide | Met (Trp, Cys, Tyr, His) | Unknown or none |
| Proteolytic processing | Arg ↓ or ↓ Lys | Basic amino acid | APC, carboxypeptidase B |
Table 2.
Biopharmaceutical applications of therapeutic protein modifications.
| Applications | Function/gene | Mechanism of action | Reference |
|---|---|---|---|
| (1) Protein engineering | |||
|
| |||
| Disulfide bond introduction | Cysteine engineering | Stabilizing FVIIIa | [46] |
| Stabilizing scFV | [47] | ||
| N-linked site introduction | Half-life extension of EPO | Reducing renal clearance | [48] |
| IgG framework/CDR | Improving solubility | [49, 50] | |
| Poly O-linked sites | Fusion of CTP of gonadotropin | half-life extension | [51, 52] |
| Engineering N-terminal Gln | Forming pyroglutamate | Preventing N-terminal degradation | [53] |
| Protease resistant mutant | Mutating Arg336 & 562 of FVIII | Eliminating two APC sites | [54] |
|
| |||
| (2) Cell-line engineering | |||
|
| |||
| Overexpressing GnTIV, GnTV, ST3GalIV, and ST6GalI |
Increasing sialic acid content | See text | |
| Half-life extension | CMP-sialic acid transporter | [55, 56] | |
| CMP-sialic acid synthase, GNE | [57, 58] | ||
| Knockdown sialidase | Preventing sialic acid degradation | [59, 60] | |
| Reducing immunogenicity | knockdown CMP-sialic acid hydroxylase |
Preventing conversion of Neu5Ac to Neu5Gc | [61] |
| E-selectin interaction | Overexpressing C2GnT, Fut6 Knockdown ST3Gal |
Enhancing sLex formation | [62, 63] |
| Modulating effector function | Knockout or knockdown | Fucose removal | [64, 65] |
| Fut8, GMD, overexpressing GnTIII | [66, 67] | ||
| Lysosomal targeting | Inactivating GnTI | Oligomannose formation | [68, 69] |
| Improving ADCC | Overexpressing sialidase | Reducing sialylation | [70] |
| Improving carboxylation | Overexpressing VKOR | Facilitating vitamin K reduction | [71] |
| VKOR ↑ calumenin ↓ | Increasing carboxylation of FVII | [72] | |
|
| |||
| (3) Process engineering | |||
|
| |||
| Modulating glycosylation | Serum content, pH, dissolved oxygen, Temperatures, ammonium, salt concentration |
Bioprocessing parameters | See text |
| Glucosamine/uridine | Enhancing glycan antennarity | Elevating intracellular UDP-GlcNAc | [73] |
| Lipid supplement | Maintaining site occupancy | [74] | |
| Decreasing temperature/adding butyrate |
Enhancing glycan occupancy | Increasing site exposure time to OST | [75] |
| Addition of ManNAc | Improving sialylation | CMP-Neu5Ac precursor | [76] |
| Adding manganese/iron | Enhancing glycan occupancy | Increasing OST activity | [75] |
| Addition of kifunensine | Oligomannose formation | α-mannosidase I inhibitor | [77] |
| Lowering glucose | Reducing heterogeneity | Reducing glycation | [78] |
2. ER Modifications
During biogenesis in eukaryotic cells, precursor forms of therapeutic proteins are translated in ribosomes, which are targeted to rough ER through a hydrophobic signal sequence bound by a signal recognition particle ribonuclear protein complex [17]. Depending on the overall amino acid content of the protein, the translocation across ER membranes can occur co- or posttranslationally, both of which require protein translocon, the Sec61 complex [18]. In ER lumen, the protein has its signal peptide removed via membrane-bound signal peptidase, gains proper folding through ER luminal folding machinery, is subjected to ER quality control process, and transports to next secretory compartment. Several modifications take place for therapeutic proteins when they transit through this compartment. These modifications include disulfide bond formation, gamma carboxylation, beta hydroxylation, and N-linked glycosylation which is discussed in the latter section.
2.1. Disulfide Bond Formation
Disulfide bond formation is one of the most common modifications found in extracellular proteins. Therapeutic proteins, such as antibodies, coagulation factors, and insulin, contain both interchain and intrachain disulfide bonds. Disulfide bond formation helps stabilize tertiary structures and is important for assembly and maintenance of protein structural integrity. Without disulfide bonds, native conformations for these proteins are not obtained. For multi-unit proteins such as antibody, interchain disulfide bonds help connect multiple protein subunits together during polypeptide assembly.
Disulfide bond formation takes place in ER lumen where it is much more oxidizing than cytosol as measured by reduced-oxidized glutathione couple [19]. A highly specific and targeted redox protein network maintains this redox homeostasis and ensures proper disulfide bond formation [20, 21]. To attain a given conformation, native and nonnative disulfides are transiently formed and reduced. A precise equilibrium between oxidation and reduction reactions in ER is required to make certain that these covalent links remain dynamic until folding is completed. Either a reducing ER, preventing disulfide formation, or an overoxidizing ER, stabilizing nonnative bonds, can trigger ER stress responses [22].
Molecular pathways for disulfide bond formation have been discovered in eukaryotes and prokaryotes [21, 23]. Both systems share a common design, including the transfer of thiols between membrane-associated oxidoreductases to soluble oxidoreductases and direct introduction of disulfide bonds into substrate proteins. In mammalian cells, membrane-associated flavoprotein Ero1 utilizes the oxidizing power of molecular oxygen, coupled with its flavin cofactor, to generate disulfide bonds within itself, which are transferred to protein disulfide isomerase (PDI). Then PDI transfers its disulfide bonds directly to secretory proteins in ER. Both human and yeast cells contain multiple PDI-like proteins. These PDI homologs may contribute to discrete protein maturation pathways in different cell types [21]. Besides the Ero1-dependent pathway, alternative oxidative folding pathways, such as the quiescin sulphydryl oxidase/Erv superfamily and vitamin K epoxide reductase, also contribute to the cellular disulfide bond formation process [24].
Incomplete formation of disulfide bonds can cause issues like heterogeneity and disulfide bond scrambling for therapeutic proteins [15]. It has been reported that a significant fraction of free sulfhydryl and reactive cysteine residues is found in recombinant antibodies of IgG1, IgG2, and IgG4 [25]. Interchain disulfide bonds between heavy chains or between heavy chain and light chain are susceptible to reduction [26], while intrachain disulfide bonds within each IgG domain can only form free sulfhydryl groups due to incomplete bond formation during biosynthesis. This formation of free sulfhydryl group can increase molecular mass by 2 Da and result in size heterogeneity. Presence of incomplete disulfide bonding can induce disulfide bond scrambling and cause structural disturbances. Human IgG2 antibodies contain three types of disulfide isoforms classified by the disulfide links between Fab arms and heavy chain hinge region, contributing to IgG2 heterogeneity [27]. Human IgG4 is also known to have nonclassic disulfide bond heterogeneous structure, with an equilibrium between forming two inter heavy chain disulfide bonds and forming two intrachain disulfide bonds of half-molecules [26]. Trisulfides, rare variants of disulfides that contain an extra sulfur atom, are reported to be detected in IgG antibodies [26, 28]. The formation mechanism is unclear, possibly due to in vitro protein manipulation as a result of the reaction of an intact disulfide bond with dissolved hydrogen sulfide [26, 28].
2.2. Gamma Carboxylation of Glutamate Residues
γ-carboxylation is a type of modification with a conversion of target glutamate (Glu) residues to γ-carboxyglutamate (Gla), first discovered in 1974 in Prothrombin [29, 30]. This process is undertaken by a vitamin K dependent carboxylase (GGCX), an ER bound integral membrane protein with at least five transmembrane domains [31, 32]. In the presence of CO2, O2, and vitamin K hydroquinone, GGCX converts glutamate residues to Gla residues, yielding vitamin K epoxide and H2O. The vitamin K epoxide is subsequently reduced to regenerate vitamin K hydroquinone by vitamin K epoxide reductase (VKOR) [33].
γ-carboxylation is a characteristic of a group of proteins termed vitamin K dependent proteins that play roles in blood coagulation and bone metabolism [34]. They contain this so-called Gla domain that possesses 9–12 Gla residues in a roughly 45 amino acids peptide sequence. Gla residues provide the calcium binding ability to the Gla-domain-containing proteins. This activity induces conformation changes for interacting with membrane phospholipids. Undercarboxylation of these proteins can severely impair their biological activities. Gla residue is not reused for protein synthesis but is excreted in urine for mammals [34]. Carboxylation of Gla domain is not dependent on a specific consensus sequence, but mediated by an immediately adjacent propeptide region. The propeptide contributes to docking protein substrate to GGCX and may also serve as an allosteric effector in trans [34].
Currently three therapeutic proteins are γ-carboxylated: factor IX, VIIa, and activated protein C. Another γ-carboxylated protein thrombin is used as a topical treatment for breeding control [35]. The recombinant factor IX contains 12 glutamic acids that can be carboxylated. High-level expression of factor IX in Chinese Hamster Ovary (CHO) cells decreases the carboxylation rate and therefore results in a much lower specific activity when compared to the natural human plasma-derived protein [36]. Factor X was found only 30% fully carboxylated when expressed in human embryonic kidney cells [37], likely due to the saturation of GGCX enzyme. Interestingly, replacing factor X propeptide with that of prothrombin, a 43-fold lower affinity substrate for GGCX, results in 90–95% of factor X being fully carboxylated. Factor VIIa contains 10 carboxylation sites that are important for its biological activities [38], and the recombinant version with 9 out of 10 sites carboxylated retains full activity. Activated protein C that is used to treat severe sepsis has 9 carboxylation sites, and full carboxylation of all sites is required for its biological activity [39].
2.3. Beta Hydroxylation of Aspartate and Asparagine
β-hydroxylation was first discovered in early 1980s from bovine protein C [40]. It is undertaken by hydroxylase enzymes that convert target aspartate (Asp) residues to β-hydroxyaspartate (Hya) or asparagine (Asn) residues to β-hydroxyasparagine (Hyn) [41–43]. This modification has also been shown to bind calcium and to be involved in protein-protein interaction.
β-hydroxylation takes place in ER and requires 2-ketoglutarate and Fe2+ [41, 43]. This modification is catalyzed by a type II membrane protein β-hydroxylase with a C-terminal catalytic domain in ER lumen [42]. The reaction does not require vitamin K or an adjacent propeptide. A consensus β-hydroxylation site (Cys-X-Asp/Asn-X-X-X-X-Phe/Tyr-X-Cys-X-Cys) found in EGF domain was proposed [44]; however, proteins like factor VII with such a site is not hydroxylated, suggesting that further requirements are needed [38].
β-hydroxylation is found in epidermal growth factor (EGF) precursor and in some calcium-binding EGF-like domain receptors, ligands, extracellular matrix proteins, complement proteins, and vitamin K dependent homeostatic proteins [35]. It occurs in therapeutic protein factor IX and protein C. Both plasma-derived factor IX and recombinant factor IX from CHO cells are similarly hydroxylated at Asp64 (~30–40%) [45]. Hydroxylation seems to be nonessential for the clotting activity. On the other hand, hydroxylation of activated protein C at Asp71 is essential in maintaining its biological activity in factor Va and factor VIIIa inactivation [10, 39].
3. Golgi Modifications
Golgi complex is well established as a central processing and sorting station for protein trafficking [79] and as an organelle with multiple complex functions [80]. In mammalian cells, Golgi stacks are laterally linked to form a ribbon-like membrane system. When neosynthesized and properly folded soluble therapeutic proteins exit ER, membrane vesicles containing these proteins fuse with a network of vesicular tubular clusters known as ER-Golgi intermediate compartment that sorts proteins to cis-Golgi or carries them back to ER. While the Golgi-arrived proteins transverse Golgi cisternae and sort further at trans-Golgi network to various cellular destinations, they are subjected to a number of modifications.
3.1. Tyrosine Sulfation
Sulfation addition to tyrosine of secretory proteins as an O4-sulfate ester takes place in trans-Golgi [81]. This modification was first discovered in 1954 in a bovine fibrinogen peptide [82]. Since then hundreds of tyrosine-sulfated proteins have been reported [83–86]. The modification is mediated by two closely related integral membrane protein tyrosylprotein sulfotransferases (TPST1 and TPST2) [87–89], with the catalytic domain orientated in the lumen of trans-Golgi [90, 91]. Orthologues of TPST1 and TPST2 have been found in both vertebrate and invertebrate [84], supporting functional importance of this modification, although no tyrosine sulfation occurs in yeast or prokaryotes.
The reaction catalyzed by TPSTs employs 3′-phosphoadenosine 5′-phosphosulfate (PAPS) as sulfate donor [83], in which the sulfate from 5′-phosphosulfate group of PAPS is transferred to the phenol of a tyrosine residue to form an O-sulfated tyrosine and adenosine 3′,5′-diphosphate [84]. There is no simple consensus sequence for tyrosine sulfation, although certain common features have been identified based on the sequence analysis with known sulfated tyrosines that seem to favor this modification. One of the major characteristics is the presence of acidic amino acids, such as aspartic or glutamic acid, adjacent to tyrosine at N-terminal end [81, 87, 92]. Turn-induced amino acids around tyrosine residue, as well as lack of cysteine and glycosylation sites, favor sulfation of nearby tyrosine residues [81, 92].
For therapeutic proteins, tyrosine sulfation is required for factor VIII's full procoagulant activity, including conversion of factor VIII to VIIIa and optimal binding to von Willebrand factor [36]. Tyrosine sulfation at 155 in factor IX influences factor IX's in vivo recovery [36]. The functional importance of tyrosine sulfation appears to be modulating biological activity and binding affinities by affecting protein-protein interaction [86]. Many chemokine receptors, G-protein-coupled receptors of complement proteins, phospholipids, and glycoprotein hormone receptors, have been shown to be tyrosine sulfated, which play an important role in their high affinity ligand binding and subsequent receptor activation. C-terminal tyrosine sulfated hirudin displays tenfold higher affinity to thrombin than the unsulfated version [93–95]. Tyrosine sulfation also appears to affect protein stability, as human secreted fizzled-related protein-1 is less stable with the presence of sulfation in its tyrosines 34 and 36 [96].
3.2. Propeptide Processing
Many bioactive proteins and peptides are synthesized as an inactive precursor which is activated by limited proteolysis. The specific processing of these proproteins and prohormone peptides occurs at relatively conserved sites often with single or pairs of basic amino acids [97, 98]. A special family of proteases, termed preprotein convertases (PCs), process these inactive precursors while they traffick through the secretory pathway. Seven members of this PC family are basic amino acid specific mammalian proteases that are known as PC1/3, PC2, Furin, PC4, PC5/6, PACE4, and PC7 [98]. The eighth member of the family is SKI-1 that cleaves substrates at the motif (R/K)-X (hydrophobic)-X↓ [99]. The latest member of the family is PCSK9 that autocatalytically cleaves its propeptide at VFAQ152↓ [100].
All these convertases themselves are synthesized as inactive zymogens. Through an autocleavage event in ER or in immature secretory granules (for PC2 only), an inactive heterodimer of inhibitory propeptide associated with the rest of the protein is formed. After exiting ER, a second autocatalytical event occurs on the prosegment at specific subcellular compartments along the secretory pathway to liberate the active enzyme free from the inhibitory propeptide [98]. This activation mechanism ensures that these PCs are only active at specific intracellular sites. SKI-1, Furin, PACE4, PC5/6, and PC7 are active at the Golgi complex, PC1/3 and PC2 are maximally active in secretory granules, while PCSK9 is the only PC that is secreted as catalytically inactive heterodimer [98].
A number of therapeutic proteins or peptides are synthesized as proproteins. Proinsulin is processed by either PC1 or PC2 [97]. Bone morphogenetic protein-2 precursor [101] is specifically processed by PC5/6 [102]. The presegments of both Pro-factor VII [97] and Pro-factor IX [103] are processed by Furin. Failure to remove the propeptide from Pro-factor IX can result in a secretable molecule that is defective in phospholipid interaction and displays reduced γ-carboxylation of glutamic acid residues [104]. Factor VIII is processed at arginine 1313 and 1648 of its B-domain which has consensus site for Furin, though the exact PC members responsible for this processing remain unidentified [105].
3.3. O-Linked Glycosylation
In contrast to N-linked glycosylation which initiates cotranslationally in ER lumen with a transfer of a core glycan chain from dolichol-pyrophosphate, O-linked glycosylation in mammalian cells begins in Golgi apparatus with addition of single monosaccharide from sugar nucleotide precursor [106, 107]. Several types of O-linked glycosylation can be classified. Mucin-type glycans have N-acetylgalactosamine (GalNAc) attached to hydroxyl group of Ser or Thr, which are found on many secreted and membrane-bound glycoproteins. No particular sequence motif has been identified for mucin-type O-linked glycosylation, except with proline favorable and charged amino acids interfering nearby specific sites [106, 107]. Elongation of this GalNAc generates at least eight different core structures. Some other specific types of O-linked glycosylation have also been identified. O-linked fucoses and glucoses are found on a specific consensus sequence in EGF protein domains [108]. One special O-linked glycosylation is galactose (Gal) on hydroxylysines of collagen in the sequence of Gly-Xaa-Hyl-Gly- [109]. Another special kind of O-linked glycans is glycosaminoglycans (GAGs) that are typically found in proteoglycans produced virtually by all mammalian cells [110]. GAGs are linked to serine residues in core protein by xylose. Xylosyltransferase initiates this modification in Golgi at specific sites defined by Ser-Gly flanked with one or two acidic residues.
Mucin-type of O-linked glycosylation can be often found in therapeutic proteins. The first step for this modification is the transfer of GalNAc to Ser or Thr, catalyzed by a polypeptide-N-acetyl-galactosaminyltransferase (ppGalNAcT). There are at least 21 ppGalNAcT homologs expressed in eukaryotic organisms [111]. ppGalNAcT expression and its Golgi sublocalization vary considerably between cell types and tissues, contributing partly to heterogeneity of O-linked glycans. Further elongation and termination of O-linked glycans after addition of the first GalNAc can be synthesized by various glycosyltransferases. Expansion with galactose, GlcNAc, or GalNAc produces eight different core structures. The glycans can be additionally modified by sialylation, fucosylation, sulphatation, methylation, or acetylation, which lead to a large number of oligosaccharide structures [107, 111].
Like N-glycans, O-linked glycans play a role in maintaining structures of fully folded proteins, modulating aggregation, maintaining protein stability, and conferring protease and heat resistance. O-linked glycan modifications have been found in hinge region and possibly Fc region for a number of antibodies [15]. The hinge region modification can render protease resistant for antibody. Examples include modifications at the hinge regions of IgG2b [112], IgA1, and IgD [107, 113]. O-linked modification in human erythropoietin (EPO) is not essential for its activity [114]. O-linked glycans are also found in the activation peptides of factor V and VIII and in the EGF domain of factor IX, VII [36], tissue plasminogen activator [115], and recombinant human urokinase [116]. The functional significance of these glycans remains unclear. One of the adverse effects of O-linked glycans on therapeutic proteins is heterogeneity [15, 107]. Recently O-linked glycans are found inhibiting the binding activity and efficacy of a therapeutic peptide-antibody bispecific fusion by blocking its interaction with cytokine IL17A [53].
3.4. Phosphorylation
Intracellular protein phosphorylation has been well documented as a fundamental mechanism regulating many essential cellular processes; yet physiological importance and functional regulation of extracellular phosphorylation on extracellular proteins remain obscure comparatively. Recent proteomic and computational analysis has revealed a large number of extracellular phosphorylated proteins and phosphorylated sites [117]. It also provides evidence that extracellular phosphorylation plays a role in various physiological functions, such as blood coagulation, formation of neuronal networks, immune cell activation, and biomineralization [2, 117, 118].
It has been long assumed that the concentration of ATP is not sufficiently high in extracellular environment, even though constitutive release of ATP extracellularly has been observed in many cell types [119–121]. The most recent proof for extracellular phosphorylation events is the identification of two Golgi-localized ectokinases, four-jointed [122], and FAM20C [2, 118]. Both proteins contain an N-terminal signal sequence and a kinase domain. FAM20C has been demonstrated to be the long-sought “Golgi-enriched fraction casein kinase” (GEF-CK) that phosphorylates many extracellular proteins. The consensus site recognized by GEF-CK is S-x-E/pS, where X can be any amino acid and E/pS can be Glu or phosphoserine [2, 118]. Another FAM20 member, FAM20B, has been identified as an ectokinase that phosphorylates xylose within GAG core linker [124]. These findings are consistent with the notion that extracellular phosphorylation mainly takes place in the Golgi compartment, where sufficiently high concentration of luminal ATP provides phosphorylation substrates and is the source of constitutively released ATP into the extracellular space of many cell types [121].
A number of therapeutic products or candidates have been found phosphorylated. Two well-known examples are coagulation factors. Plasma-derived factor IX is fully phosphorylated at serine 158, but recombinant factor IX is not phosphorylated [125], suggesting that ectokinases in CHO cells may be insufficiently expressed. Although the absence of phosphorylation modification does not affect in vitro clotting activity, the effect on the in vivo clotting activity remains unknown. Factor VIII is also found phosphorylated when exposed to activated platelets [126]. Similar data was also obtained for factor V. Phosphorylation of these two cofactors seems to downregulate their activities, since partially phosphorylated factor Va appears to be more sensitive to activated protein C inactivation [36]. Biologically active peptide hormones, such as adrenocorticotropin [127] and progastrin [128], are found phosphorylated. It would be of interest to know if FAM20C or four-jointed is the right ectokinase responsible for phosphorylating these proteins in vivo.
3.5. Amidation
Amidation is a replacement of C-terminal carboxyl group of a protein with an amide group. This is a typical modification of many bioactive peptides for full activity [129, 130]. Amidation is ubiquitously found in higher eukaryotes but not in yeast or prokaryotes. Peptide hormones are produced as glycine-extended intermediates, and then the glycine N–C alpha bond is oxidatively cleaved to form active amidated hormones.
Amidation reaction is catalyzed by two enzymes: peptidylglycine α-hydroxylating monooxygenase (PHM) and peptidyl-α-hydroxyglycine α-amidating lyase (PAL). In mammals and higher organisms, these enzymes are expressed as a single bifunctional enzyme, peptidylglycine alpha-amidating monooxygenase (PAM) [130]. The PHM catalyzes the reduction of molecular oxygen for the hydroxylation of the glycine α-carbon of glycine-extended substrates, with two redox-active coppers reduced by ascorbate. The PAL produces α-amidated peptide product and glyoxylate by cleaving the glycine N–C alpha bond from the peptidyl α-hydroxyglycine intermediate. The PAM enzyme localizes in the distal trans-Golgi network through its cytosolic domain [131].
The exact biological roles of α-amidation remain to be fully elucidated. This modification prevents ionization of the C-terminus of peptides. It may therefore render it more hydrophobic and better bind to its receptor [130]. It may also contribute to peptide stability. Recombinant salmon calcitonin that regulates serum calcium and phosphate levels is amidated [132]. The product (Forcaltonin) is for the treatment of Paget's disease and hypercalcaemia. It is produced in E. coli as a fusion protein and needs downstream in vitro modification with a recombinant α-amidating enzyme. It is known that CHO, COS-7, and NIH-3T3 can allow amidation of secretory products with an engineered C-terminal glycine [133, 134]. Therefore recombinant bioactive peptides and their fusion proteins could presumably be produced with an in vivo approach.
4. N-Linked Glycosylation
N-linked glycosylation is the most common posttranslational modification found in eukaryotic cells, contributing to many crucial biological and physiological roles [111, 135]. The sophisticated modification process involves both compartments of ER and Golgi. The consensus site for N-linked glycosylation is the sequon of Asn-X-Ser/Thr where X can be any amino acid except proline [136, 137]. Acidic amino acids such as aspartate and glutamate can also reduce efficiency. Glycosylation at Asn-Ala-Cys has also been reported [138]. Glycosylation efficiency of these Thr, Ser, and Cys containing sequon is very different with an order of Thr>Ser>Cys [139]. Some nonstandard sequons have been recently identified for N-linked oligosaccharide modification in a small percentage of human recombinant antibodies [140].
N-linked oligosaccharides are added to proteins en bloc in the lumen of ER as presynthesized core units of 14 saccharides (Glc3Man9GlcNAc2) in virtually all eukaryotes [137, 141]. This core glycan is the product of a biosynthesis pathway in which monosaccharides are added to a lipid carrier (dolichol pyrophosphate) on both sides of ER membranes by monosaccharyltransferases in the membranes. These enzymes are called ALG (for Altered in glycosylation), identified from studies of yeast mutants. The sugar moiety is translocated from cytosolic side to the luminal side of ER by an ATP-independent bidirectional flippase [142]. The oligosaccharyltransferase (OST) then scans the emerging polypeptide from translocon complex for glycosylation sequon and adds the core glycan unit to the side chain nitrogen of the Asn residue by N-glycosidic bond [141, 143]. Three oligosaccharyltransferase complexes have been identified in mammals; each contains one of two Stt3 proteins, the presumed catalytic units. After the core glycan is added to the growing nascent polypeptide chain, the oligosaccharide portion is modified by a series of glycosidases. Glucosidase I and II, located in the ER, remove all three glucose residues from the core unit to produce a Man9GlcNAc2 high mannose structure.
This ER modification process choreographs with protein folding cycle of many glycoproteins, in which glycans serve as sorting signals reflecting the folding status of the protein. The monoglucosylated glycans after the action of glucosidase I and II are bound to two ER lectin chaperones, calnexin or calreticulin, which prevents protein aggregation and ER exit of partially folded proteins [136]. This binding exposes the glycoprotein to the binding of ERp57, the protein disulfide-isomerase A3, for protecting from nonproductive disulfide bonding. When glucosidase II removes the remaining glucose residue, the glycoprotein no longer binds to the lectins and is free to exit ER unless recognized by UDP-Glc:glycoprotein glucosyltransferase (GT). GT can reglucosylate incompletely folded glycoproteins and reinitiate the lectin chaperone binding. The repeated cycles of GT and glucosidase II facilitate efficient folding of newly synthesized glycoprotein. The unsalvageable misfolded proteins are subjected to the ER-associated degradation process [136]. For correctly folded-glycoproteins, many of them are further acted by ER α-mannosidase I, before exiting ER, to have terminal mannose removed from the central arm of Man9GlcNAc2.
In the cis-Golgi, α-1,2 mannosidase IA, IB, IC further trim 4–6 terminal mannose to produce a Man5GlcNAc2, a key intermediate for hydrid and complex N-glycans [141]. An N-acetylglucosaminyltransferase named GnTI in medial-Golgi adds a GlcNAc residue to C-2 of the mannose α1-3 in the core of Man5GlcNAc2. After this step, majority of N-glycans are trimmed by α-mannosidase II to form GlcNAcMan3GlcNAc2. Then a second GlcNAc is added by the action of GnTII to yield the precursor for all biantennary complex N-glycans. Those glycans not digested by α-mannosidase II become precursors of hybrid N-glycans. A “bisecting” GlcNAc can be added to the core β-mannose by GnTIII enzyme for both complex and hybrid N-glycans. Further sugar additions and maturation, most of which occur in the trans-Golgi, add fucose, galactoses, GlcNAc, sialic acids, and sulfate to convert limited glycan repertoire into an extensive array of mature complex oligosaccharides [111].
N-glycans are most commonly found in therapeutic proteins [9, 10]. The presence and nature of the oligosaccharides clearly affect these proteins' folding, stability, trafficking, immunogenicity, and their primary activities. It is well known that the N-glycans at Asn297 in Fc region of recombinant antibodies and Fc fusion proteins are critical to the activation of downstream effector mechanisms. The Fc oligosaccharides are different from those typical N-glycans and are predominantly fucosylated nongalactosylated diantennary oligosaccharides, with a small portion of galactosylated or sialylated diantennary oligosaccharides. The difference may be due to the inaccessibility of Fc N-glycan for further modification, as the N-glycans at the Fc regions are integral to the IgG structure and have a defined conformation [144]. Besides Fc core glycans, about 30% of polyclonal human IgG molecules contain N-linked oligosaccharides within the IgG-Fab region [144]. Therapeutic antibody cetuximab has an N-linked glycan at Asn88 of the heavy chain variable region and an unoccupied N-linked motif at Asn41 of the light chain variable region [145]. Fab oligosaccharides are more a normal type of N-glycans with heterogeneous complex diantennary and hybrid oligosaccharides with sialic acids and galactoses.
N-glycans also affect many nonimmunoglycoproteins such as growth factors, cytokines, hormones, and therapeutic enzymes. Removal of either two or all three N-linked sites of human EPO results in poor product secretion [146]. Inhibiting glycosylation decreases biological activity and circulatory half-life of interferon (IFN)-β and IFN-γ, even though glycosylation is not essential for INFs protein efficacy or safety [147]. Oligosaccharide structures of follicle-stimulating hormone heterodimer play an important role in its biosynthesis, secretion, metabolic fate, and functional potency [148]. The glycans at each subunit seem to exhibit distinct roles, with those in α subunit being critical for dimer assembly, signal transduction, and secretion, and those in β subunit being more crucial for circulation clearance. In addition, many therapeutics enzymes such as recombinant human glucocerebrosidase for Gaucher disease [68] are glycoproteins and N-glycosylation is important for targeting and functional activities. Factors V, VIII, IX, and X contain N-linked glycosylation [36]. Removing N-glycans from recombinant factor IX drastically increases its specific activity. N-linked glycosylation is essential for the secretion of both factors V and VIII.
5. Exocellular Modifications
Soon after therapeutic proteins complete the trafficking pathways and reach cell surface, they are released into extracellular medium environment and incubated there during the production period. Then they are purified, formulated, and stored. They can go into test subjects almost exclusively through parenteral routes such as intravenous, subcutaneous, or intramuscular injection [149]. Systemic circulation allows these proteins to be limitedly distributed into various tissues. Therapeutic proteins are then metabolized and eventually cleared out of the test subjects. A number of modifications occur to proteins when they are in the extracellular media, during in vitro manipulation, and in vivo circulation.
5.1. Deamidation
Nonenzymatic deamidation of asparagine and glutamine residues to aspartate and glutamate is a common modification for proteins [150, 151]. It is a hydrolytic reaction with water to form products through the formation of succinimide intermediate [151, 152]. Deamidation of Gln is slower than that of Asn. The deamidation rate is affected by conditions of pH, temperature, and ionic strength [151]. The rate also depends on the sequence around Asn and Gln. Polar residues preceding Asn and Gln, neighboring with Ser and Thr, can increase deamidation rates. Asn followed by Gly is found to be the most susceptible site [152]. Secondary, tertiary, and quaternary structures also influence deamidation reaction.
Deamidation of Asn and Gln contributes to charge heterogeneity of therapeutic proteins, since more acidic species are introduced by adding one negative charge and by decreasing isoelectric point [15, 151]. It has been shown that deamidation of Asn and Gln determines the irreversible thermal denaturation of proteins at acidic and neutral pH. Deamidation also affects protein crystallizability and X-ray diffraction due to microheterogeneity. Deamidation regulates the rate of protein breakdown, and could shorten in vivo half-life [151]. Both human insulin and growth hormone have been found to deamidate slowly in different formulations, affecting biological potency [151]. Deamidation commonly occurs for recombinant monoclonal antibodies under various conditions and at various domain regions such as CDRs, CH1-3, and CL [15], contributing to heterogeneity and instability, and presumable efficacy.
5.2. Glycation
Glycation is a nonenzymatic condensation reaction between a carbonyl group on reducing sugars and N-terminal primary amine or the amine group of lysine side chains [153]. Glycation can occur to physiological proteins in human body where it is implicated in the pathogenesis of multiple chronic diseases [154, 155].
It has been shown that glycation can happen to recombinant antibodies during cell culture [156, 157], which generates product heterogeneity and may affect product stability [158]. Glycation to various antibodies can also take place in vitro, such as storage with lactose [159]. Glycation can disguise positive charges of N-terminal primary amino group or side chains of lysine residues, making the antibody more acidic [15]. It can reduce immunoreactivity of antibodies [160]. It has been reported that lowering the concentration of glucose in the fedbatch culture can control the glycation level of a recombinant antibody in CHO cells [78].
5.3. N-Terminal Pyroglutamate Formation
It has been well known that N-terminal Gln residue can readily cyclize with its own terminal amino group to form pyrrolidone carboxylic acid [161]. It remains uncertain whether or not the conversion reaction is nonenzymatic [162, 163]. Pyroglutamate is frequently detected in heavy chain and light chain of antibodies. N-terminal residues of human IgG heavy chains typically start with either glutamine or glutamate [164], while Lambda light chains begin with glutamine occasionally with serine, arginine, or leucine [165] and Kappa light chains with glutamate or aspartate [166–168]. The N-terminal glutamate can also be cyclized to form pyroglutamate.
The formation of N-terminal pyroglutamate from Gln and Glu decreases molecular mass by 17 and 18 Da. It also causes a loss of positive charge at neutral pH from a loss of the N-terminal primary amine, making the antibody more acidic [15]. Antibody with pyroglutamate elutes earlier than those with glutamine on cation exchange column. Pyroglutamate formation is found resistant to amino peptidases and therefore is proposed as a stabilization mechanism [169]. It has been recently demonstrated that engineering an N-terminal pyroglutamate to a peptide-antibody bispecific genetic fusion molecule can restore its IL17A neutralizing activity in a cell-based assay by preventing N-terminal degradation [53].
5.4. Oxidation
Methionine, one of the most susceptible residues for oxidation, can be oxidized to methionine sulfoxide for therapeutic proteins such as antibodies [15] and growth factors [170]. This increases the mass by 16 Da and makes the side chain of Met more polar. Met255 and Met431 in the Fc region of human IgG1 are two most susceptible sites for oxidation when incubated at elevated temperatures [171]. These two residues are close to the CH2-CH3 interface [172], making them more susceptible. Antibodies with oxidized Met elute earlier relative to nonoxidized molecule on HIC column [171].
In addition to Met residues, Trp and Cys amino acids are also susceptible to oxidation. Oxidized Trp residues have been reported to be located in the CDRs regions for several antibodies [173–175]. Cys residues of antibodies are typically involved in disulfide bonding, but oxidation of unpaired Cys of a murine antibody is reported after a long storage [176]. Oxidation of histidine and tyrosine is also reported [177].
5.5. Proteolytic Processing
Proteolytic processing is a common modification in the extracellular space. Besides the nonspecific protein degradation mediated by various plasma and tissue proteases, some proteolytic processings are explicitly required for enzyme activation. Native protein of recombinant activated protein C, Xigris, is produced and circulated as a zymogen which is activated by thrombin in complex with thrombomodulin [178]. In plasma, factor VIII is activated by thrombin by the cleavage initially after Arg740 and then after Arg372 and Arg1689 to produce a heterotrimer [36]. It can be inactivated by activated protein C at Arg336 and Arg562. Factor V is activated by Thrombin at Arg739, Arg1018, and Arg1545, and inactivated by activated protein C at Arg306 and Arg506 [36]. Factor IX is activated by factor XIa to remove activation peptide. Factor X is activated by the cleavage of the tissue factor/factor VIIa complex or the factor VIIIa and factor IXa complex.
C-terminal lysine removal of antibody heavy chain is a common modification of recombinant antibodies. It decreases the molecular mass by 128 Da and 1 unit of positive charge. Mammalian cell lines such as CHO, NSO, and SP2/0, produce an endogenous carboxypeptidase B that can result in this processing [179]. Partial removal of this residue can cause heterogeneity.
6. Biopharmaceutical Applications of Protein Modifications
Posttranslational modifications have been utilized as strategies for therapeutic improvements [9, 10, 144]. A better understanding of structure and function relationship as well as molecular pathways has facilitated the development of next generation of biotherapeutic products. The following summarizes the recent advances in protein engineering, cell line engineering, and process engineering, for improving functional characteristics and attributes of biopharmaceutical products (Table 2).
6.1. Applications in Protein Engineering
Engineering disulfide bonds by introducing cysteines into proteins have long been a protein engineering strategy for stabilizing small proteins and multiunit proteins [180–183]. In order to prolong the activity of factor VIIIa, Tyr664 and Thr1826 are mutated to Cys to introduce a disulfide bond between A2 and A3 domains [46]. The resulting protein significantly prolongs factor VIIIa stability and activity. A similar result is obtained for factor Va when a disulfide bond between its A2 and A3 domain is introduced [184]. Disulfide bond has also been introduced to framework region of antibody Fv fragments to improve stability [47]. It improves stability of IgG-like bispecific scFv antibody [185]. Also an in vivo cleavable disulfide linker has been designed for in vivo release of granulocyte colony-stimulating factor from its transferrin fusion [186].
Interchain disulfide bonds of IgG1 can be distinguished by chemical reduction and oxidation method [187]. Cytotoxic drugs are therefore conjugated to antibodies through these activated cysteine sulfhydryl groups [188]. To resolve the issue of heterogeneity and to improve therapeutic index (defined as the lethal dose of a drug for 50% of the population divided by the minimum effective dose for 50% of the population), cysteine substitutions at positions of light chain and heavy chain have been applied successfully for site-specific conjugation [189, 190].
One of the best known examples making use of protein modification, the so-called glycoengineering, is the half-life extension of Aranesp [48, 191]. After introducing 48 N-linked positions and evaluating 62 constructs, the terminal half-life of Aranesp with two additional N-glycans is approximately 3-fold longer than that of wild-type Epoetin Alfa when administered intravenously. This is due to the fact that glycosylation can shield nonspecific proteolytic degradation, reduce renal clearance by negatively charged sialic acids at glycan chain termini, and decrease kidney glomerular filtration by increased molecular weight and hydrodynamic radius of the glycoprotein.
Glycoengineering has also been applied to improve the solubility and physicochemical properties of therapeutic proteins such as antibodies. Introducing N-linked glycans into antibody framework regions can increase protein solubility by more than 50-fold [49]. Similar results are obtained when an N-linked site is introduced to the CDR region [50].
O-linked glycan has also been applied to protein engineering. The carboxyl-terminal peptide (CTP) of human chorionic gonadotropin-β-subunit [52] contains several proline and serine residues and four O-linked oligosaccharides sites. When this CTP sequence is fused to human follitropin [192], erythropoietin [51], or human growth hormone [193], it increases the half-life of the fusion proteins in the circulation. This is also presumably due to the negatively charged sialic acids on the O-linked glycans that decrease renal clearance and glomerular filtration.
Modifying proteolytic processing is another strategy for improving therapeutic efficacy. Engineering an N-terminal glutamine to form pyroglutamate has been successfully applied to restore cellular activity of a peptide-antibody fusion by preventing N-terminal protein degradation [53]. Modifying proteolytic specificity of activated protein C toward serine protease inhibitor family proteins has helped improve their pharmacokinetic profiles [194]. Pipe and Kaufman generated an protease resistant FVIIIa by eliminating two APC cleavage sites at Arg336 and Arg562 [54].
6.2. Applications in Cell Line Engineering
Genetic engineering of host cell lines has proven to be a practical tool to obtain therapeutic proteins with improved properties. Due to the importance and prevalence of glycosylation on therapeutic proteins, a significant progress in cell line engineering by utilizing glycosylation pathways has been made for the past two decades [9, 10, 144, 195, 196].
Terminal capping of glycans with sialic acids improves in vivo circulation half-life of glycoproteins because of negative charge and unrecognization of asialoglycoproteins and mannose receptors. A number of cell line engineering efforts have been put in place. Overexpressing GnTIV and GnTV have been found to increase glycan proportion with triantennary and tetra-antennary structures [197, 198]. Further overexpressing ST3 β-galactoside α-2,6-sialyltransferase IV (ST3GalIV) and/or ST6 β-galactoside α-2,6-sialyltransferase I (ST6GalI) increases sialylation up to 80%. ST6GalI, which transfers sialic acid in an α2,6-linkage, is not expressed in CHO. Overexpressing this gene increases the α-2,6-linked sialic acid over α-2,3-linked sialic acid, which is reported to improve circulation half-life [199, 200]. Overexpression of both β-1,4-galactosyltransferase and ST3Gal also improves sialylation [58, 201].
Sialidase can degrade sialic acid when released into the culture due to cell lysis [202, 203]. Sialidase knockdown with stable antisense or siRNA in CHO is found to increase sialic acid content of recombinant glycoproteins [59, 60]. Overexpressing CMP-sialic acid transporter is shown to improve IFN-γ sialylation [56]. Overexpressing a key enzyme GNE for sialic acid synthesis can also improve sialylation of recombinant EPO [57]. A combinatorial approach with ST3Gal, CMP-sialic acid synthase, and CMP-sialic acid transporter, is found to better improve sialylation than overexpressing individual gene alone [55, 58].
On the other end, to generate antibody with reduced sialylation, the catalytic domain of the arthrobacter ureafaciens sialidase is stably expressed in mammalian cells [70]. The soluble sialidase A is secreted into medium where it is capable of removing sialic acid thoroughly from antibodies. The low sialylated antibody possesses improved antibody-dependent cellular cytotoxicity (ADCC) activity.
N-acetylneuraminic acid (Neu5Ac) can be converted into N-glycolylneuraminic acid (NeuGc) by CMP-sialic acid hydroxylase. NeuGc is not produced by human and therefore immunogenic, antisense knockdown of CMP-sialic acid hydroxylase has been engineered in CHO to reduce NeuGc down to 1% [61].
O-linked glycan such as sialyl Lewis X (sLex) in E-Selectin protein is important for mediating cell adhesion. This glycan formation can be promoted by overexpressing β1,6-N-acetyl-glucosaminyl-transferase (C2GnT) and fucosyltransferase 6 (Fut6), as well as downregulating ST3Gal [62, 63].
Another well-known direction of cell line engineering in glycosylation is modulating antibody effector function [144, 204]. N-glycans in the Fc-region play a critical role in ADCC activity. Absence of a core α-1, 6-linked fucose improves binding to FcγRIII and the in vitro ADCC activity [205, 206]. Defucosylated anti-CCR4 antibody mogamulizumab (KW0761) has been approved in Japan for relapsed adult T-cell leukemia-lymphoma [123]. Knockingdown α-1,6-fucosyltransferase (Fut8) [65] and GDP-mannose 4,6-dehydratase (GMD) with siRNAs [64], homologous recombinant fut8 knockout [67], zinc-finger nucleases fut8 knockout [207], or overexpressing N-acetyl-glucosaminyltransferase III (GnTIII) [66] have been utilized for modifying fucosylation of IgG Fc glycans. Glycoengineered anti-CD20 antibody obinutuzumab (GA101) is currently used in clinical studies [208]. Inactivating GnTI in CHO cells (Lec1) also generates a fucose-free oligomannose glycan with enhanced ADCC and decreased complement-dependent cytotoxicity (CDC) activities for IgG [69]. Oligomannose glycan generated by the Lec1 mutant has been employed for targeted delivery to disease-affected tissues for the treatment of lysosomal storage diseases [68].
Besides glycosylation, cell line engineering has been applied for improving carboxylation modification. In order to improve carboxylation rate of factor X, a HEK293 line with overexpressing vitamin K epoxide reductase is constructed [71]. It has been shown that factor X in such a cell line is 92% carboxylated, up from control 52%. Interestingly, overexpressing the GGCX, the carboxylase, has no improvement for the modification. It is presumably due to the fact that the reduction of vitamin K epoxide to vitamin K and the conversion of vitamin K to vitamin K hydroquinone are the rate-limiting steps under the culture condition. Similarly, overexpressing VKOR in HEK293 cells together with shRNA knockdown of calumenin (an inhibitor of VKOR and γ-carboxylase) increases the fraction of bioactive factor VII by sevenfold [72]. Recombinant factor IX is produced by a CHO line engineered to coexpress Furin protein [209]. This modification enhances propeptide removal and consequentially the factor IX activity, as failure to remove propeptide prevents the Gla domain from adopting proper calcium-induced conformation.
6.3. Applications in Process Engineering
A number of bioprocessing parameters can influence protein modification such as glycosylation [210]. They include serum content, culture pH, dissolved oxygen, temperature, growth phase, ammonium concentration, and salt concentration [75, 210–218]. Glycan degradation mediated by exoglycosidases such as sialidase and galactosidase also affects product consistency through cell viability and growth condition [202, 219]. Site occupancy for N- and O-linked glycosylation varies according to culture condition. These effects on modification have been attributed to availability of dolichol phosphate and nucleotide sugars [220], glycotransferase activity [221, 222], and potential modification site competing with protein folding [223, 224]. Ammonium is a common metabolic waste product driven by a major energy source of glutamine or asparagines. Increased ammonia levels in the cell culture can increase the pH of trans-Golgi compartment, therefore compromise pH-sensitive glycotransferases activity [215, 225].
Several strategies in process engineering have been developed to improve product quality and attributes. Glucosamine and uridine supplementation into CHO cell culture has led to an elevated level of intracellular UDP-GlcNAc and enhanced glycan antennarity [73]. Addition of CMP-Neu5Ac precursor, N-acetylmannosamine (ManNAc), can improve sialylation of IFN-gamma [76]. Feeding specific lipid supplements to cultures has been found to reduce the deterioration in site occupancy of IFN-gamma during batch culture [74]. OST enzyme requires manganese for maximal activity, therefore supplement with additional manganese or iron can increase glycan site occupancy of human tissue plasminogen activator [75]. Manganese addition to the production process of human EPO can also increase galactosylation and reduce lower sialylated fraction [226]. Decreasing culture temperature from 37°C to 33°C or 31°C, and adding butyrate or thyroid hormones, can increase glycan occupancy rate. Both butyrate addition and lowering temperatures decrease cell growth and therefore slow down the protein elongation rate which increases the exposure time of glycan site to OST enzyme in ER. Trace amount of metals affects glycosylation. Addition of copper (II) chloride is found to increase sialic acid content [218, 227]. To generate nonfucosylated oligomannose modified antibody, Zhou et al. added a potent α-mannosidase I inhibitor, kifunensine, to CHO cells for 11 days in batch culture [77]. The resulting antibody shows a greater affinity to FcγRIIIA, a higher ADCC activity, a lower binding to C1q, and a lower CDC activity. Glycation is a source of heterogeneity for recombinant therapeutic proteins. Yuk and coworkers demonstrate that by lowering the concentration of glucose from 6 g/L down to 3 g/L, >40% reduction in glycation is observed [78]. A >80% reduction is further generated with a fully continuous glucose fed at ~1 g/L.
7. Conclusions and Perspectives
The importance of protein modifications and their underlying mechanisms has become a core and focus of both industrial and basic research investigations. They can potentially affect protein production level, stability, pharmacokinetics/pharmacodynamics parameters, and immunogenicity. The functional and safety consequences of any of these modifications to a therapeutic protein must be evaluated on an individual protein basis. Advances in understanding the relationship between modification structure and function have enabled the rational designs to enhance specific functional features as demonstrated by the studies previously mentioned. Also this knowledge contributes to imposing strategic bioprocessing controls to ensure consistency of product quality. While glycosylation represents the most widespread and complex modification, other protein modifications such as phosphorylation and sulfation attract increasing attentions in recent years. Knowledge gaps remain for the understanding of the biology of these modifications. A recent report on the draft genome sequence of an ancestral CHO line [228] opens a new door for feasibly manipulating gene products involved in protein modification pathways. A further fundamental understanding of therapeutic protein modifications will culminate in the development of many new generations of biopharmaceutical products with preselected modifications and enhanced properties.
Acknowledgments
The authors would like to thank Ronald Kriz and Will Somers for critical reading of the paper. They would also like to apologize to all research groups whose work could not be included due to space and scope limitations.
References
- 1.Hammarsten O. Zur Frage, ob das Casein ein einheitlicher Stoff sei. Zeitschrift für Physiologische Chemie. 1883;7:227–273. [Google Scholar]
- 2.Tagliabracci VS, Engel JL, Wen J, et al. Secreted kinase phosphorylates extracellular proteins that regulate biomineralization. Science. 2012;336:1150–1153. doi: 10.1126/science.1217817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kornfeld S. Foreword from first edition. In: Varki A, Cummings RD, Esko JD, et al., editors. Essentials of Glycobiology. 2nd edition. Cold Spring Harbor, NY, USA: Cold Spring Harbor Laboratory Press; 2009. [PubMed] [Google Scholar]
- 4.Krishna RG, Wold F. Post-translational modification of proteins. Advances in Enzymology and Related Areas of Molecular Biology. 1993;67:265–298. doi: 10.1002/9780470123133.ch3. [DOI] [PubMed] [Google Scholar]
- 5.Seo J, Lee KJ. Post-translational modifications and their biological functions: proteomic analysis and systematic approaches. Journal of Biochemistry and Molecular Biology. 2004;37(1):35–44. doi: 10.5483/bmbrep.2004.37.1.035. [DOI] [PubMed] [Google Scholar]
- 6.Higgins B. Post Translational Processing: A Practical Approach. Oxford Univesity Press; 1999. [Google Scholar]
- 7.Kannicht C. Post Translational Modification of Proteins. Humana Press; 2002. [Google Scholar]
- 8.Jenkins N. Modifications of therapeutic proteins: challenges and prospects. Cytotechnology. 2007;53(1–3):121–125. doi: 10.1007/s10616-007-9075-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walsh G. Post-translational modifications of protein biopharmaceuticals. Drug Discovery Today. 2010;15(17-18):773–780. doi: 10.1016/j.drudis.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 10.Walsh G, Jefferis R. Post-translational modifications in the context of therapeutic proteins. Nature Biotechnology. 2006;24(10):1241–1252. doi: 10.1038/nbt1252. [DOI] [PubMed] [Google Scholar]
- 11.Gibson BA, Kraus WL. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nature Reviews Molecular Cell Biology. 2012;13:411–424. doi: 10.1038/nrm3376. [DOI] [PubMed] [Google Scholar]
- 12.Mann M, Jensen ON. Proteomic analysis of post-translational modifications. Nature Biotechnology. 2003;21(3):255–261. doi: 10.1038/nbt0303-255. [DOI] [PubMed] [Google Scholar]
- 13.Perry JJP, Tainer JA, Boddy MN. A SIM-ultaneous role for SUMO and ubiquitin. Trends in Biochemical Sciences. 2008;33(5):201–208. doi: 10.1016/j.tibs.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 14.Vucic D, Dixit VM, Wertz IE. Ubiquitylation in apoptosis: a post-translational modification at the edge of life and death. Nature Reviews Molecular Cell Biology. 2011;12(7):439–452. doi: 10.1038/nrm3143. [DOI] [PubMed] [Google Scholar]
- 15.Liu H, Caza-Bulseco G, Faldu D, Chumsae C, Sun J. Heterogeneity of monoclonal antibodies. Journal of Pharmaceutical Sciences. 2008;97(7):2426–2447. doi: 10.1002/jps.21180. [DOI] [PubMed] [Google Scholar]
- 16.De Groot AS. Immunomics: discovering new targets for vaccines and therapeutics. Drug Discovery Today. 2006;11(5-6):203–209. doi: 10.1016/S1359-6446(05)03720-7. [DOI] [PubMed] [Google Scholar]
- 17.Schwartz T, Blobel G. Structural basis for the function of the β subunit of the eukaryotic signal recognition particle receptor. Cell. 2003;112(6):793–803. doi: 10.1016/s0092-8674(03)00161-2. [DOI] [PubMed] [Google Scholar]
- 18.Osborne AR, Rapoport TA, Van Den Berg B. Protein translocation by the Sec61/SecY channel. Annual Review of Cell and Developmental Biology. 2005;21:529–550. doi: 10.1146/annurev.cellbio.21.012704.133214. [DOI] [PubMed] [Google Scholar]
- 19.Hwang C, Sinskey AJ, Lodish HF. Oxidized redox state of glutathione in the endoplasmic reticulum. Science. 1992;257(5076):1496–1502. doi: 10.1126/science.1523409. [DOI] [PubMed] [Google Scholar]
- 20.Fassio A, Sitia R. Formation, isomerisation and reduction of disulphide bonds during protein quality control in the endoplasmic reticulum. Histochemistry and Cell Biology. 2002;117(2):151–157. doi: 10.1007/s00418-001-0364-0. [DOI] [PubMed] [Google Scholar]
- 21.Sevier CS, Kaiser CA. Conservation and diversity of the cellular disulfide bond formation pathways. Antioxidants and Redox Signaling. 2006;8(5-6):797–811. doi: 10.1089/ars.2006.8.797. [DOI] [PubMed] [Google Scholar]
- 22.Margittai E, Sitia R. Oxidative protein folding in the secretory pathway and redox signaling across compartments and cells. Traffic. 2011;12(1):1–8. doi: 10.1111/j.1600-0854.2010.01108.x. [DOI] [PubMed] [Google Scholar]
- 23.Frand AR, Cuozzo JW, Kaiser CA. Pathways for protein disulphide bond formation. Trends in Cell Biology. 2000;10(5):203–210. doi: 10.1016/s0962-8924(00)01745-1. [DOI] [PubMed] [Google Scholar]
- 24.Sevier CS. New insights into oxidative folding. Journal of Cell Biology. 2010;188(6):757–758. doi: 10.1083/jcb.201002114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang W, Czupryn MJ. Free sulfhydryl in recombinant monoclonal antibodies. Biotechnology Progress. 2002;18(3):509–513. doi: 10.1021/bp025511z. [DOI] [PubMed] [Google Scholar]
- 26.Liu H, May K. Disulfide bond structures of IgG molecules: structural variations, chemical modifications and possible impacts to stability and biological function. mAbs. 2012;4:17–23. doi: 10.4161/mabs.4.1.18347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu YD, Wang T, Chou R, et al. IgG2 disulfide isoform conversion kinetics. Molecular Immunology. 2013;54:217–226. doi: 10.1016/j.molimm.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 28.Gu S, Wen D, Weinreb PH, et al. Characterization of trisulfide modification in antibodies. Analytical Biochemistry. 2010;400(1):89–98. doi: 10.1016/j.ab.2010.01.019. [DOI] [PubMed] [Google Scholar]
- 29.Nelsestuen GL, Zytkovicz TH, Howard JB. The mode of action of vitamin K. Identification of γ carboxyglutamic acid as a component of prothrombin. The Journal of Biological Chemistry. 1974;249(19):6347–6350. [PubMed] [Google Scholar]
- 30.Stenflo J, Fernlund P, Egan W, Roepstorff P. Vitamin K dependent modifications of glutamic acid residues in prothrombin. Proceedings of the National Academy of Sciences of the United States of America. 1974;71(7):2730–2733. doi: 10.1073/pnas.71.7.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Furie B, Furie BC. Molecular basis of vitamin K-dependent γ-carboxylation. Blood. 1990;75(9):1753–1762. [PubMed] [Google Scholar]
- 32.Wu SM, Cheung W, F., Frazier D, Stafford DW. Cloning and expression of the cDNA for human γ-glutamyl carboxylase. Science. 1991;254(5038):1634–1636. doi: 10.1126/science.1749935. [DOI] [PubMed] [Google Scholar]
- 33.Li T, Chang CY, Jin DY, Lin PJ, Khvorova A, Stafford DW. Identification of the gene for vitamin K epoxide reductase. Nature. 2004;427(6974):541–544. doi: 10.1038/nature02254. [DOI] [PubMed] [Google Scholar]
- 34.Bandyopadhyay PK. Vitamin K-dependent γ-glutamylcarboxylation: an ancient posttranslational modification. Vitamins and Hormones. 2008;78:157–184. doi: 10.1016/S0083-6729(07)00008-8. [DOI] [PubMed] [Google Scholar]
- 35.Brown LM. Biopharmaceuticals: post-translational modification carboxylation and hydroxylation. In: Walsh G, editor. Post-Translational Modification of Protein Biopharmaceuticals. Weinheim, Germany: Wiley-VCH; 2009. [Google Scholar]
- 36.Kaufman RJ. Post-translational modifications required for coagulation factor secretion and function. Thrombosis and Haemostasis. 1998;79(6):1068–1079. [PubMed] [Google Scholar]
- 37.Camire RM, Larson PJ, Stafford DW, High KA. Enhanced γ-carboxylation of recombinant factor X using a chimeric construct containing the prothrombin propeptide. Biochemistry. 2000;39(46):14322–14329. doi: 10.1021/bi001074q. [DOI] [PubMed] [Google Scholar]
- 38.Thim L, Bjoern S, Christensen M, et al. Amino acid sequence and posttranslational modifications of human factor VIIa from plasma and transfected baby hamster kidney cells. Biochemistry. 1988;27(20):7785–7793. doi: 10.1021/bi00420a030. [DOI] [PubMed] [Google Scholar]
- 39.Yan SCB, Razzano P, Chao YB, et al. Characterization and novel purification of recombinant human protein C from three mammalian cell lines. Bio/Technology. 1990;8(7):655–661. doi: 10.1038/nbt0790-655. [DOI] [PubMed] [Google Scholar]
- 40.Drakenberg T, Fernlund P, Roepstorff P, Stenflo J. β-hydroxyaspartic acid in vitamin K-dependent protein C. Proceedings of the National Academy of Sciences of the United States of America. 1983;80(7):1802–1806. doi: 10.1073/pnas.80.7.1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gronke RS, VanDusen WJ, Garsky VM, et al. Aspartyl beta-hydroxylase: in vitro hydroxylation of a synthetic peptide based on the structure of the first growth factor-like domain of human factor IX. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(10):3609–3613. doi: 10.1073/pnas.86.10.3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jia S, VanDusen WJ, Diehl RE, et al. cDNA cloning and expression of bovine aspartyl (asparaginyl) β-hydroxylase. The Journal of Biological Chemistry. 1992;267(20):14322–14327. [PubMed] [Google Scholar]
- 43.Stenflo J, Holme E, Lindstedt S, et al. Hydroxylation of aspartic acid in domains homologous to the epidermal growth factor precursor is catalyzed by a 2-oxoglutarate-dependent dioxygenase. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(2):444–447. doi: 10.1073/pnas.86.2.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stenflo J, Lundwall A, Dahlback B. β-hydroxyasparagine in domains homologous to the epidermal growth factor precursor in vitamin K-dependent protein S. Proceedings of the National Academy of Sciences of the United States of America. 1987;84(2):368–372. doi: 10.1073/pnas.84.2.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Derian CK, VanDusen W, Przysiecki GT, et al. Inhibitors of 2-ketoglutarate-dependent dioxygenases block aspartyl β-hydroxylation of recombinant human factor IX in several mammalian expression systems. The Journal of Biological Chemistry. 1989;264(12):6615–6618. [PubMed] [Google Scholar]
- 46.Gale AJ, Pellequer JL. An engineered interdomain disulfide bond stabilizes human blood coagulation factor VIIIa. Journal of Thrombosis and Haemostasis. 2003;1(9):1966–1971. doi: 10.1046/j.1538-7836.2003.00348.x. [DOI] [PubMed] [Google Scholar]
- 47.Reiter Y, Brinkmann U, Lee B, Pastan I. Engineering antibody Fv fragments for cancer detection and therapy: disulfide-stabilized Fv fragments. Nature Biotechnology. 1996;14(10):1239–1245. doi: 10.1038/nbt1096-1239. [DOI] [PubMed] [Google Scholar]
- 48.Elliott S, Lorenzini T, Asher S, et al. Enhancement of therapeutic protein in vivo activities through glycoengineering. Nature Biotechnology. 2003;21(4):414–421. doi: 10.1038/nbt799. [DOI] [PubMed] [Google Scholar]
- 49.Pepinsky RB, Silvian L, Berkowitz SA, et al. Improving the solubility of anti-LINGO-1 monoclonal antibody Li33 by isotype switching and targeted mutagenesis. Protein Science. 2010;19(5):954–966. doi: 10.1002/pro.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu SJ, Luo J, O’Neil KT, et al. Structure-based engineering of a monoclonal antibody for improved solubility. Protein Engineering, Design and Selection. 2010;23(8):643–651. doi: 10.1093/protein/gzq037. [DOI] [PubMed] [Google Scholar]
- 51.Fares F, Havron A, Fima E. Designing a long acting erythropoietin by fusing three carboxyl-terminal peptides of human chorionic gonadotropin β subunit to the N-terminal and C-terminal coding sequence. International Journal of Cell Biology. 2011;2011:7 pages. doi: 10.1155/2011/275063.275063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fares FA, Suganuma N, Nishimori K, LaPolt PS, Hsueh AJW, Boime I. Design of a long-acting follitropin agonist by fusing the C-terminal sequence of the chorionic gonadotropin β subunit to the follitropin β subunit. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(10):4304–4308. doi: 10.1073/pnas.89.10.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhong X, Kieras E, Sousa E, et al. Pyroglutamate and O-linked glycan determine functional production of anti-IL17A and anti-IL22 peptide-antibody bispecific genetic fusions. The Journal of Biological Chemistry. 2013;288(2):1409–1419. doi: 10.1074/jbc.M112.417717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pipe SW, Kaufman RJ. Characterization of a genetically engineered inactivation-resistant coagulation factor VIIIa. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(22):11851–11856. doi: 10.1073/pnas.94.22.11851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jeong YT, Choi O, Son YD, Park SY, Kim JH. Enhanced sialylation of recombinant erythropoietin in genetically engineered Chinese-hamster ovary cells. Biotechnology and Applied Biochemistry. 2009;52(4):283–291. doi: 10.1042/BA20080044. [DOI] [PubMed] [Google Scholar]
- 56.Wong NSC, Yap MGS, Wang DIC. Enhancing recombinant glycoprotein sialylation through CMP-sialic acid transporter over expression in Chinese hamster ovary cells. Biotechnology and Bioengineering. 2006;93(5):1005–1016. doi: 10.1002/bit.20815. [DOI] [PubMed] [Google Scholar]
- 57.Bork K, Reutter W, Weidemann W, Horstkorte R. Enhanced sialylation of EPO by overexpression of UDP-GlcNAc 2-epimerase/ManAc kinase containing a sialuria mutation in CHO cells. FEBS Letters. 2007;581(22):4195–4198. doi: 10.1016/j.febslet.2007.07.060. [DOI] [PubMed] [Google Scholar]
- 58.Jeong YT, Choi O, Lim HR, Son YD, Kim HJ, Kim JH. Enhanced sialylation of recombinant erythropoietin in CHO cells by human glycosyltransferase expression. Journal of Microbiology and Biotechnology. 2008;18(12):1945–1952. [PubMed] [Google Scholar]
- 59.Ferrari J, Gunson J, Lofgren J, Krummen L, Warner TG. Chinese hamster ovary cells with constitutively expressed sialidase antisense RNA produce recombinant DNase in batch culture with increased sialic acid. Biotechnology and Bioengineering. 1998;60:589–595. [PubMed] [Google Scholar]
- 60.Ngantung FA, Miller PG, Brushett FR, Tang GL, Wang DIC. RNA interference of sialidase improves glycoprotein sialic acid content consistency. Biotechnology and Bioengineering. 2006;95(1):106–119. doi: 10.1002/bit.20997. [DOI] [PubMed] [Google Scholar]
- 61.Chenu S, Grégoire A, Malykh Y, et al. Reduction of CMP-N-acetylneuraminic acid hydroxylase activity in engineered Chinese hamster ovary cells using an antisense-RNA strategy. Biochimica et Biophysica Acta. 2003;1622(2):133–144. doi: 10.1016/s0304-4165(03)00137-5. [DOI] [PubMed] [Google Scholar]
- 62.Prati EG, Matasci M, Suter TB, Dinter A, Sburlati AR, Bailey JE. Engineering of coordinated up- and down-regulation of two glycosyltransferases of the O-glycosylation pathway in Chinese hamster ovary (CHO) cells. Biotechnology and Bioengineering. 2000;68:239–244. [PubMed] [Google Scholar]
- 63.Prati EG, Scheidegger P, Sburlati AR, Bailey JE. Antisense strategies for glycosylation engineering of Chinese hamster ovary (CHO) cells. Biotechnology and Bioengineering. 1998;59:445–450. [PubMed] [Google Scholar]
- 64.Imai-Nishiya H, Mori K, Inoue M, et al. Double knockdown of α1,6-fucosyltransferase (FUT8) and GDP-mannose 4,6-dehydratase (GMD) in antibody-producing cells: a new strategy for generating fully non-fucosylated therapeutic antibodies with enhanced ADCC. BMC Biotechnology. 2007;7, article 84 doi: 10.1186/1472-6750-7-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mori K, Kuni-Kamochi R, Yamane-Ohnuki N, et al. Engineering Chinese hamster ovary cells to maximize effector function of produced antibodies using FUT8 siRNA. Biotechnology and Bioengineering. 2004;88(7):901–908. doi: 10.1002/bit.20326. [DOI] [PubMed] [Google Scholar]
- 66.Ferrara C, Brünker P, Suter T, Moser S, Püntener U, Umaña P. Modulation of therapeutic antibody effector functions by glycosylation engineering: influence of golgi enzyme localization domain and co-expression of heterologous β1, 4-N-acetylglucosaminyltransferase III and Golgi α-mannosidase II. Biotechnology and Bioengineering. 2006;93(5):851–861. doi: 10.1002/bit.20777. [DOI] [PubMed] [Google Scholar]
- 67.Yamane-Ohnuki N, Kinoshita S, Inoue-Urakubo M, et al. Establishment of FUT8 knockout Chinese hamster ovary cells: an ideal host cell line for producing completely defucosylated antibodies with enhanced antibody-dependent cellular cytotoxicity. Biotechnology and Bioengineering. 2004;87(5):614–622. doi: 10.1002/bit.20151. [DOI] [PubMed] [Google Scholar]
- 68.Van Patten SM, Hughes H, Huff MR, et al. Effect of mannose chain length on targeting of glucocerebrosidase for enzyme replacement therapy of Gaucher disease. Glycobiology. 2007;17(5):467–478. doi: 10.1093/glycob/cwm008. [DOI] [PubMed] [Google Scholar]
- 69.Zhong X, Cooley C, Seth N, et al. Engineering novel Lec1 glycosylation mutants in CHO-DUKX cells: molecular insights and effector modulation of N-acetylglucosaminyltransferase I. Biotechnology and Bioengineering. 2012;109:1723–1734. doi: 10.1002/bit.24448. [DOI] [PubMed] [Google Scholar]
- 70.Naso MF, Tam SH, Scallon BJ, Raju TS. Engineering host cell lines to reduce terminal sialylation of secreted antibodies. mAbs. 2010;2(5):519–527. doi: 10.4161/mabs.2.5.13078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sun YM, Jin DY, Camire RM, Stafford DW. Vitamin K epoxide reductase significantly improves carboxylation in a cell line overexpressing factor X. Blood. 2005;106(12):3811–3815. doi: 10.1182/blood-2005-06-2495. [DOI] [PubMed] [Google Scholar]
- 72.Wajih N, Owen J, Wallin R. Enhanced functional recombinant factor VII production by HEK 293 cells stably transfected with VKORC1 where the gamma-carboxylase inhibitor calumenin is stably suppressed by shRNA transfection. Thrombosis Research. 2008;122(3):405–410. doi: 10.1016/j.thromres.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Baker KN, Rendall MH, Hills AE, Hoare M, Freedman RB, James DC. Metabolic control of recombinant protein N-glycan processing in NS0 and CHO cells. Biotechnology and Bioengineering. 2001;73(3):188–202. doi: 10.1002/bit.1051. [DOI] [PubMed] [Google Scholar]
- 74.Jenkins N, Castro P, Menon S, Ison A, Bull A. Effect of lipid supplements on the production and glycosylation of recombinant interferon-γ expressed in CHO cells. Cytotechnology. 1994;15(1–3):209–215. doi: 10.1007/BF00762395. [DOI] [PubMed] [Google Scholar]
- 75.Gawlitzek M, Estacio M, Fürch T, Kiss R. Identification of cell culture conditions to control N-glycosylation site-occupancy of recombinant glycoproteins expressed in CHO cells. Biotechnology and Bioengineering. 2009;103(6):1164–1175. doi: 10.1002/bit.22348. [DOI] [PubMed] [Google Scholar]
- 76.Gu X, Wang DI. Improvement of interferon-gamma sialylation in Chinese hamster ovary cell culture by feeding of N-acetylmannosamine. Biotechnology and Bioengineering. 1998;58:642–648. [PubMed] [Google Scholar]
- 77.Zhou Q, Shankara S, Roy A, et al. Development of a simple and rapid method for producing non-fucosylated oligomannose containing antibodies with increased effector function. Biotechnology and Bioengineering. 2008;99(3):652–665. doi: 10.1002/bit.21598. [DOI] [PubMed] [Google Scholar]
- 78.Yuk IH, Zhang B, Yang Y, et al. Controlling glycation of recombinant antibody in fed-batch cell cultures. Biotechnology and Bioengineering. 2011;108:2600–2610. doi: 10.1002/bit.23218. [DOI] [PubMed] [Google Scholar]
- 79.Emr S, Glick BS, Linstedt AD, et al. Journeys through the Golgi—taking stock in a new era. Journal of Cell Biology. 2009;187(4):449–453. doi: 10.1083/jcb.200909011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wilson C, Venditti R, Rega LR, Colanzi A, D’Angelo G, De MA. The Golgi apparatus: an organelle with multiple complex functions. Biochemical Journal. 2011;433(1):1–9. doi: 10.1042/BJ20101058. [DOI] [PubMed] [Google Scholar]
- 81.Huttner WB. Tyrosine sulfation and the secretory pathway. Annual Review of Physiology. 1988;50:363–376. doi: 10.1146/annurev.ph.50.030188.002051. [DOI] [PubMed] [Google Scholar]
- 82.Bettelheim FR. Tyrosine-O-sulfate in a peptide from fibrinogen. Journal of the American Chemical Society. 1954;76(10):2838–2839. [Google Scholar]
- 83.Kehoe JW, Bertozzi CR. Tyrosine sulfation: a modulator of extracellular protein-protein interactions. Chemistry and Biology. 2000;7(3):R57–R61. doi: 10.1016/s1074-5521(00)00093-4. [DOI] [PubMed] [Google Scholar]
- 84.Moore KL. The biology and enzymology of protein tyrosine O-sulfation. The Journal of Biological Chemistry. 2003;278(27):24243–24246. doi: 10.1074/jbc.R300008200. [DOI] [PubMed] [Google Scholar]
- 85.Seibert C, Sakmar TP. Toward a framework for sulfoproteomics: synthesis and characterization of sulfotyrosine-containing peptides. Biopolymers. 2008;90(3):459–477. doi: 10.1002/bip.20821. [DOI] [PubMed] [Google Scholar]
- 86.Stone MJ, Chuang S, Hou X, Shoham M, Zhu JZ. Tyrosine sulfation: an increasingly recognised post-translational modification of secreted proteins. New Biotechnology. 2009;25(5):299–317. doi: 10.1016/j.nbt.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 87.Beisswanger R, Corbeil D, Vannier C, et al. Existence of distinct tyrosylprotein sulfotransferase genes: molecular characterization of tyrosylprotein sulfotransferase-2. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(19):11134–11139. doi: 10.1073/pnas.95.19.11134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ouyang YB, Lane WS, Moore KL. Tyrosylprotein sulfotransferase: purification and molecular cloning of an enzyme that catalyzes tyrosine O-sulfation, a common posttranslational modification of eukaryotic proteins. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(6):2896–2901. doi: 10.1073/pnas.95.6.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ouyang YB, Moore KL. Molecular cloning and expression of human and mouse tyrosylprotein sulfotransferase-2 and a tyrosylprotein sulfotransferase homologue in Caenorhabditis elegans. The Journal of Biological Chemistry. 1998;273(38):24770–24774. doi: 10.1074/jbc.273.38.24770. [DOI] [PubMed] [Google Scholar]
- 90.Baeuerle PA, Huttner WB. Tyrosine sulfation is a trans-Golgi-specific protein modification. Journal of Cell Biology. 1987;105(6):2655–2664. doi: 10.1083/jcb.105.6.2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lee RWH, Huttner WB. (Glu62, Ala30, Tyr8)n serves as high-affinity substrate for tyrosylprotein sulfotransferase: a Golgi enzyme. Proceedings of the National Academy of Sciences of the United States of America. 1985;82(18):6143–6147. doi: 10.1073/pnas.82.18.6143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Niehrs C, Kraft M, Lee RWH, Huttner WB. Analysis of the substrate specificity of tyrosylprotein sulfotransferase using synthetic peptides. The Journal of Biological Chemistry. 1990;265(15):8525–8532. [PubMed] [Google Scholar]
- 93.Niehrs C, Huttner WB, Carvallo D, Degryse E. Conversion of recombinant hirudin to the natural form by in vitro tyrosine sulfation. Differential substrate specificities of leech and bovine tyrosylprotein sulfotransferases. The Journal of Biological Chemistry. 1990;265(16):9314–9318. [PubMed] [Google Scholar]
- 94.Rydel TJ, Ravichandran KG, Tulinsky A, et al. The structure of a complex of recombinant hirudin and human α-thrombin. Science. 1990;249(4966):277–280. doi: 10.1126/science.2374926. [DOI] [PubMed] [Google Scholar]
- 95.Stone SR, Hofsteenge J. Kinetics of the inhibition of thrombin by hirudin. Biochemistry. 1986;25(16):4622–4628. doi: 10.1021/bi00364a025. [DOI] [PubMed] [Google Scholar]
- 96.Zhong X, Desilva T, Lin L, et al. Regulation of secreted Frizzled-related protein-1 by heparin. The Journal of Biological Chemistry. 2007;282(28):20523–20533. doi: 10.1074/jbc.M609096200. [DOI] [PubMed] [Google Scholar]
- 97.Seidah NG, Day R, Marcinkiewicz M, Chrétien M. Precursor convertases: an evolutionary ancient, cell-specific, combinatorial mechanism yielding diverse bioactive peptides and proteins. Annals of the New York Academy of Sciences. 1998;839:9–24. doi: 10.1111/j.1749-6632.1998.tb10727.x. [DOI] [PubMed] [Google Scholar]
- 98.Seidah NG, Mayer G, Zaid A, et al. The activation and physiological functions of the proprotein convertases. International Journal of Biochemistry and Cell Biology. 2008;40(6-7):1111–1125. doi: 10.1016/j.biocel.2008.01.030. [DOI] [PubMed] [Google Scholar]
- 99.Pasquato A, Pullikotil P, Asselin MC, et al. The proprotein convertase SKI-1/S1P: in vitro analysis of Lassa virus glycoprotein-derived substrates and ex vivo validation of irreversible peptide inhibitors. The Journal of Biological Chemistry. 2006;281(33):23471–23481. doi: 10.1074/jbc.M513675200. [DOI] [PubMed] [Google Scholar]
- 100.Seidah NG, Benjannet S, Wickham L, et al. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(3):928–933. doi: 10.1073/pnas.0335507100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wozney JM, Rosen V, Celeste AJ, et al. Novel regulators of bone formation: molecular clones and activities. Science. 1988;242(4885):1528–1534. doi: 10.1126/science.3201241. [DOI] [PubMed] [Google Scholar]
- 102.Heng S, Paule S, Hardman B, et al. Posttranslational activation of bone morphogenetic protein 2 is mediated by proprotein convertase 6 during decidualization for pregnancy establishment. Endocrinology. 2010;151(8):3909–3917. doi: 10.1210/en.2010-0326. [DOI] [PubMed] [Google Scholar]
- 103.Wasley LC, Rehemtulla A, Bristol JA, Kaufman RJ. PACE/furin can process the vitamin K-dependent pro-factor IX precursor within the secretory pathway. The Journal of Biological Chemistry. 1993;268(12):8458–8465. [PubMed] [Google Scholar]
- 104.Diuguid DL, Rabiet MJ, Furie BC. Molecular basis of hemophilia B: a defective enzyme due to an unprocessed propeptide is caused by a point mutation in the factor IX precursor. Proceedings of the National Academy of Sciences of the United States of America. 1986;83(16):5803–5807. doi: 10.1073/pnas.83.16.5803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pittman DD, Tomkinson KN, Kaufman RJ. Post-translational requirements for functional factor V and factor VIII secretion in mammalian cells. The Journal of Biological Chemistry. 1994;269(25):17329–17337. [PubMed] [Google Scholar]
- 106.Brockhausen I, Schachter H, Stanley P. O-GalNAc glycans. In: Varki A, Cummings RD, Esko JD, et al., editors. Essentials of Glycobiology. Cold Spring Harbor, NY, USA: Cold Spring Harbor Laboratory Press; 2009. [PubMed] [Google Scholar]
- 107.van den Steen P, Rudd PM, Dwek RA, Opdenakker G. Concepts and principles of O-linked glycosylation. Critical Reviews in Biochemistry and Molecular Biology. 1998;33(3):151–208. doi: 10.1080/10409239891204198. [DOI] [PubMed] [Google Scholar]
- 108.Harris RJ, Spellman MW. O-Linked fucose and other post-translational modifications unique to EGF modules. Glycobiology. 1993;3(3):219–224. doi: 10.1093/glycob/3.3.219. [DOI] [PubMed] [Google Scholar]
- 109.Michaelsson E, Malmstrom V, Reis S, Engstrom A, Burkhardt H, Holmdahl R. T cell recognition of carbohydrates on type II collagen. Journal of Experimental Medicine. 1994;180(2):745–749. doi: 10.1084/jem.180.2.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Esko JD, Lindahl U. Molecular diversity of heparan sulfate. Journal of Clinical Investigation. 2001;108(2):169–173. doi: 10.1172/JCI13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Varki A, Cummings RD, Esko JD, et al. Essentials of Glycobiology. 2nd edition. Cold Spring Harbor, NY, USA: Cold Spring Harbor Laboratory Press; 2009. [PubMed] [Google Scholar]
- 112.Kim H, Yamaguchi Y, Masuda K, et al. O-glycosylation in hinge region of mouse immunoglobulin G2b. The Journal of Biological Chemistry. 1994;269(16):12345–12350. [PubMed] [Google Scholar]
- 113.Field MC, Amatayakul-Chantler S, Rademacher TW, Rudd PM, Dwek RA. Structural analysis of the N-glycans from human immunoglobulin A1: comparison of normal human serum immunoglobulin A1 with that isolated from patients with rheumatoid arthritis. Biochemical Journal. 1994;299(1):261–275. doi: 10.1042/bj2990261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wasley LC, Timony G, Murtha P, et al. The importance of N- and O-linked oligosaccharides for the biosynthesis and in vitro and in vivo biologic activities of erythropoietin. Blood. 1991;77(12):2624–2632. [PubMed] [Google Scholar]
- 115.Harris RJ, Leonard CK, Guzzetta AW, Spellman MW. Tissue plasminogen activator has an O-linked fucose attached to threonine-61 in the epidermal growth factor domain. Biochemistry. 1991;30(9):2311–2314. doi: 10.1021/bi00223a004. [DOI] [PubMed] [Google Scholar]
- 116.Kentzer EJ, Buko A, Menon G, Sarin VK. Carbohydrate composition and presence of a fucose-protein linkage in recombinant human pro-urokinase. Biochemical and Biophysical Research Communications. 1990;171(1):401–406. doi: 10.1016/0006-291x(90)91407-j. [DOI] [PubMed] [Google Scholar]
- 117.Yalak G, Vogel V. Extracellular phosphorylation and phosphorylated proteins: not just curiosities but physiologically important. Science Signaling. 2012;5(255):p. re7. doi: 10.1126/scisignal.2003273. [DOI] [PubMed] [Google Scholar]
- 118.Ishikawa HO, Xu A, Ogura E, Manning G, Irvine KD. The Raine syndrome protein FAM20C is a Golgi kinase that phosphorylates bio-mineralization proteins. PLoS ONE. 2012;7 doi: 10.1371/journal.pone.0042988.e42988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bodin P, Burnstock G. Purinergic signalling: ATP release. Neurochemical Research. 2001;26(8-9):959–969. doi: 10.1023/a:1012388618693. [DOI] [PubMed] [Google Scholar]
- 120.Dubyak GR, el-Moatassim C. Signal transduction via P2-purinergic receptors for extracellular ATP and other nucleotides. The American Journal of Physiology. 1993;265(3):C577–606. doi: 10.1152/ajpcell.1993.265.3.C577. [DOI] [PubMed] [Google Scholar]
- 121.Zhong X, Malhotra R, Guidotti G. ATP uptake in the Golgi and extracellular release require Mcd4 protein and the vacuolar H+-ATPase. The Journal of Biological Chemistry. 2003;278(35):33436–33444. doi: 10.1074/jbc.M305785200. [DOI] [PubMed] [Google Scholar]
- 122.Ishikawa HO, Takeuchi H, Haltiwanger RS, Irvine KD. Four-jointed is a Golgi kinase that phosphorylates a subset of cadherin domains. Science. 2008;321(5887):401–404. doi: 10.1126/science.1158159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ishida T, Joh T, Uike N, et al. Defucosylated anti-CCR4 monoclonal antibody (KW-0761) for relapsed adult T-cell leukemia-lymphoma: a multicenter phase II study. Journal of Clinical Oncology. 2012;30:837–842. doi: 10.1200/JCO.2011.37.3472. [DOI] [PubMed] [Google Scholar]
- 124.Koike T, Izumikawa T, Tamura JI, Kitagawa H. FAM20B is a kinase that phosphorylates xylose in the glycosaminoglycan-protein linkage region. Biochemical Journal. 2009;421(2):157–162. doi: 10.1042/BJ20090474. [DOI] [PubMed] [Google Scholar]
- 125.Rand MD, Kalafatis M, Mann KG. Platelet coagulation factor Va: the major secretory platelet phosphoprotein. Blood. 1994;83(8):2180–2190. [PubMed] [Google Scholar]
- 126.Kalafatis M, Rand MD, Jenny RJ, Ehrlich YH, Mann KG. Phosphorylation of factor Va and factor VIIIa by activated platelets. Blood. 1993;81(3):704–719. [PubMed] [Google Scholar]
- 127.Bennett HPJ, Browne CA, Solomon S. Biosynthesis of phosphorylated forms of corticotropin-related peptides. Proceedings of the National Academy of Sciences of the United States of America. 1981;78(8):4713–4717. doi: 10.1073/pnas.78.8.4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Dockray GJ, Varro A, Desmond H, Young J, Gregory H, Gregory RA. Post-translational processing of the porcine gastrin precursor by phosphorylation of the COOH-terminal fragment. The Journal of Biological Chemistry. 1987;262(18):8643–8647. [PubMed] [Google Scholar]
- 129.Bradbury AF, Smyth DG. Peptide amidation. Trends in Biochemical Sciences. 1991;16(3):112–115. doi: 10.1016/0968-0004(91)90044-v. [DOI] [PubMed] [Google Scholar]
- 130.Prigge ST, Mains RE, Eipper BA, Amzel LM. New insights into copper monooxygenases and peptide amidation: structure, mechanism and function. Cellular and Molecular Life Sciences. 2000;57(8-9):1236–1259. doi: 10.1007/PL00000763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Milgram SL, Eipper BA, Mains RE. Differential trafficking of soluble and integral membrane secretory granule-associated proteins. Journal of Cell Biology. 1994;124(1-2):33–41. doi: 10.1083/jcb.124.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ray MVL, Van Duyne P, Bertelsen AH, et al. Production of recombinant salmon calcitonin by in vitro amidation of an Escherichia coli produced precursor peptide. Bio/Technology. 1993;11(1):64–70. doi: 10.1038/nbt0193-64. [DOI] [PubMed] [Google Scholar]
- 133.Takahashi KI, Liu YC, Hayashi N, et al. Production of bioactive salmon calcitonin from the nonendocrine cell lines COS-7 and CHO. Peptides. 1997;18(3):439–444. doi: 10.1016/s0196-9781(96)00336-1. [DOI] [PubMed] [Google Scholar]
- 134.Wulff BS, Catipovic B, Okamoto H, Gether U, Schwartz TW, Johansen TE. Efficient amidation of C-peptide deleted NPY precursors by non-endocrine cells is affected by the presence of Lys-Arg at the C-terminus. Molecular and Cellular Endocrinology. 1993;91(1-2):135–141. doi: 10.1016/0303-7207(93)90265-l. [DOI] [PubMed] [Google Scholar]
- 135.Moremen KW, Tiemeyer M, Nairn AV. Vertebrate protein glycosylation: diversity, synthesis and function. Nature Reviews Molecular Cell Biology. 2012;13:448–462. doi: 10.1038/nrm3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Helenius A, Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annual Review of Biochemistry. 2004;73:1019–1049. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- 137.Kornfeld R, Kornfeld S. Assembly of asparagine-linked oligosaccharides. Annual Review of Biochemistry. 1985;54:631–664. doi: 10.1146/annurev.bi.54.070185.003215. [DOI] [PubMed] [Google Scholar]
- 138.Stenflo J, Fernlund P. Amino acid sequence of the heavy chain of bovine protein C. The Journal of Biological Chemistry. 1982;257(20):12180–12190. [PubMed] [Google Scholar]
- 139.Bause E, Legler G. The role of the hydroxy amino acid in the triplet sequence Asn-Xaa-Thr(Ser) for the N-glycosylation step during glycoprotein biosynthesis. Biochemical Journal. 1981;195(3):639–644. doi: 10.1042/bj1950639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Valliere-Douglass JF, Eakin CM, Wallace A, et al. Glutamine-linked and non-consensus asparagine-linked oligosaccharides present in human recombinant antibodies define novel protein glycosylation motifs. The Journal of Biological Chemistry. 2010;285(21):16012–16022. doi: 10.1074/jbc.M109.096412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Stanley P, Schachter H, Taniguchi N. N-glycans. In: Varki A, Cummings RD, Esko JD, et al., editors. Essentials of Glycobiology. Cold Spring Harbor, NY, USA: Cold Spring Harbor Laboratory Press; 2009. [PubMed] [Google Scholar]
- 142.Hirschberg CB, Snider MD. Topography of glycosylation in the rough endoplasmic reticulum and Golgi apparatus. Annual Review of Biochemistry. 1987;56:63–87. doi: 10.1146/annurev.bi.56.070187.000431. [DOI] [PubMed] [Google Scholar]
- 143.Kelleher DJ, Gilmore R. An evolving view of the eukaryotic oligosaccharyltransferase. Glycobiology. 2006;16(4):47R–62R. doi: 10.1093/glycob/cwj066. [DOI] [PubMed] [Google Scholar]
- 144.Jefferis R. Glycosylation as a strategy to improve antibody-based therapeutics. Nature Reviews Drug Discovery. 2009;8(3):226–234. doi: 10.1038/nrd2804. [DOI] [PubMed] [Google Scholar]
- 145.Qian J, Liu T, Yang L, Daus A, Crowley R, Zhou Q. Structural characterization of N-linked oligosaccharides on monoclonal antibody cetuximab by the combination of orthogonal matrix-assisted laser desorption/ionization hybrid quadrupole-quadrupole time-of-flight tandem mass spectrometry and sequential enzymatic digestion. Analytical Biochemistry. 2007;364(1):8–18. doi: 10.1016/j.ab.2007.01.023. [DOI] [PubMed] [Google Scholar]
- 146.Egrie J, Grant J, Gillies D, Aoki K, Strickland T. The role of carbohydrate on the biological activity of erythropoietin. Glycoconjugate Journal. 1993;10:p. 263. [Google Scholar]
- 147.Pestka S, Langer JA, Zoon KC, Samuel CE. Interferons and their actions. Annual Review of Biochemistry. 1987;56:727–777. doi: 10.1146/annurev.bi.56.070187.003455. [DOI] [PubMed] [Google Scholar]
- 148.Ulloa-Aguirre A, Timossi C, Damián-Matsumura P, Dias JA. Role of glycosylation in function of follicle-stimulating hormone. Endocrine. 1999;11(3):205–215. doi: 10.1385/ENDO:11:3:205. [DOI] [PubMed] [Google Scholar]
- 149.Vugmeyster Y, Xu X, Theil FP, Khawli LA, Leach MW. Pharmacokinetics and toxicology of therapeutic proteins: advances and challenges. World Journal of Biological Chemistry. 2012;3:73–92. doi: 10.4331/wjbc.v3.i4.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Manning M, Patel K, Borchardt RT. Stability of protein pharmaceuticals. Pharmaceutical Research. 1989;6(11):903–918. doi: 10.1023/a:1015929109894. [DOI] [PubMed] [Google Scholar]
- 151.Wright HT. Nonenzymatic deamidation of asparaginyl and glutaminyl residues in proteins. Critical Reviews in Biochemistry and Molecular Biology. 1991;26(1):1–52. doi: 10.3109/10409239109081719. [DOI] [PubMed] [Google Scholar]
- 152.Geiger T, Clarke S. Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. Succinimide-linked reactions that contribute to protein degradation. The Journal of Biological Chemistry. 1987;262(2):785–794. [PubMed] [Google Scholar]
- 153.Neglia CI, Cohen HJ, Garber AR, Ellis PD, Thorpe SR, Baynes JW. 13C NMR investigation of nonenzymatic glucosylation of protein. Model studies using RNase A. The Journal of Biological Chemistry. 1983;258(23):14279–14283. [PubMed] [Google Scholar]
- 154.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414(6865):813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 155.Miranda HV, Outeiro TF. The sour side of neurodegenerative disorders: the effects of protein glycation. Journal of Pathology. 2010;221(1):13–25. doi: 10.1002/path.2682. [DOI] [PubMed] [Google Scholar]
- 156.Brady LJ, Martinez T, Balland A. Characterization of nonenzymatic glycation on a monoclonal antibody. Analytical Chemistry. 2007;79(24):9403–9413. doi: 10.1021/ac7017469. [DOI] [PubMed] [Google Scholar]
- 157.Quan C, Alcala E, Petkovska I, et al. A study in glycation of a therapeutic recombinant humanized monoclonal antibody: where it is, how it got there, and how it affects charge-based behavior. Analytical Biochemistry. 2008;373(2):179–191. doi: 10.1016/j.ab.2007.09.027. [DOI] [PubMed] [Google Scholar]
- 158.Fischer S, Hoernschemeyer J, Mahler HC. Glycation during storage and administration of monoclonal antibody formulations. European Journal of Pharmaceutics and Biopharmaceutics. 2008;70(1):42–50. doi: 10.1016/j.ejpb.2008.04.021. [DOI] [PubMed] [Google Scholar]
- 159.Andya JD, Maa YF, Costantino HR, et al. The effect of formulation excipients on protein stability and aerosol performance of spray-dried powders of a recombinant humanized anti-IgE monoclonal antibody. Pharmaceutical Research. 1999;16(3):350–358. doi: 10.1023/a:1018805232453. [DOI] [PubMed] [Google Scholar]
- 160.Dolhofer R, Siess EA, Wieland OH. Nonenzymatic glycation of immunoglobulins leads to an impairment of immunoractivity. Biological Chemistry Hoppe-Seyler. 1985;366(4):361–366. doi: 10.1515/bchm3.1985.366.1.361. [DOI] [PubMed] [Google Scholar]
- 161.Melville J. Labile glutamine peptides, and their bearing on the origin of the ammonia set free during the enzymic digestion of proteins. Biochemical Journal. 1935;29:179–186. doi: 10.1042/bj0290179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Dick LW, Kim C, Qiu D, Cheng KC. Determination of the origin of the N termina pyro-glutamate variation in monoclonal antibodies using model peptides. Biotechnology and Bioengineering. 2007;97(3):544–553. doi: 10.1002/bit.21260. [DOI] [PubMed] [Google Scholar]
- 163.Liu YD, Goetze AM, Bass RB, Flynn GC. N-terminal glutamate to pyroglutamate conversion in vivo for human IgG2 antibodies. The Journal of Biological Chemistry. 2011;286(13):11211–11217. doi: 10.1074/jbc.M110.185041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Matsuda F, Ishii K, Bourvagnet P, et al. The complete nucleotide sequence of the human immunoglobulin heavy chain variable region locus. Journal of Experimental Medicine. 1998;188(11):2151–2162. doi: 10.1084/jem.188.11.2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kawasaki K, Minoshima S, Nakato E, et al. One-megabase sequence analysis of the human immunoglobulin λ gene locus. Genome Research. 1997;7(3):250–261. doi: 10.1101/gr.7.3.250. [DOI] [PubMed] [Google Scholar]
- 166.Brensing-Küppers J, Zocher I, Thiebe R, Zachau HG. The human immunoglobulin κ locus on yeast artificial chromosomes (YACs) Gene. 1997;191(2):173–181. doi: 10.1016/s0378-1119(97)00056-5. [DOI] [PubMed] [Google Scholar]
- 167.Kawasaki K, Minoshima S, Nakato E, et al. Evolutionary dynamics of the human immunoglobulin kappa locus and the germline repertoire of the Vkappa genes. European Journal of Immunology. 2001;31:1017–1028. [PubMed] [Google Scholar]
- 168.Schable KF, Zachau HG. The variable genes of the human immunoglobulin kappa locus. Biological Chemistry Hoppe-Seyler. 1993;374(11):1001–1022. [PubMed] [Google Scholar]
- 169.Rink R, Arkema-Meter A, Baudoin I, et al. To protect peptide pharmaceuticals against peptidases. Journal of Pharmacological and Toxicological Methods. 2010;61(2):210–218. doi: 10.1016/j.vascn.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 170.Teh LC, Murphy LJ, Huq NL, et al. Methionine oxidation in human growth hormone and human chorionic somatomammotropin. Effects on receptor binding and biological activities. The Journal of Biological Chemistry. 1987;262(14):6472–6477. [PubMed] [Google Scholar]
- 171.Lam XM, Yang JY, Cleland JL. Antioxidants for prevention of methionine oxidation in recombinant monoclonal antibody HER2. Journal of Pharmaceutical Sciences. 1997;86(11):1250–1255. doi: 10.1021/js970143s. [DOI] [PubMed] [Google Scholar]
- 172.Deisenhofer J. Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9- and 2.8-Å resolution. Biochemistry. 1981;20(9):2361–2370. [PubMed] [Google Scholar]
- 173.Matamoros Fernández LE, Kalume DE, Calvo L, Fernández Mallo M, Vallin A, Roepstorff P. Characterization of a recombinant monoclonal antibody by mass spectrometry combined with liquid chromatography. Journal of Chromatography B. 2001;752(2):247–261. doi: 10.1016/s0378-4347(00)00503-x. [DOI] [PubMed] [Google Scholar]
- 174.Wei Z, Feng J, Lin HY, et al. Identification of a single tryptophan residue as critical for binding activity in a humanized monoclonal antibody against respiratory syncytial virus. Analytical Chemistry. 2007;79(7):2797–2805. doi: 10.1021/ac062311j. [DOI] [PubMed] [Google Scholar]
- 175.Yang J, Wang S, Liu J, Raghani A. Determination of tryptophan oxidation of monoclonal antibody by reversed phase high performance liquid chromatography. Journal of Chromatography A. 2007;1156(1-2):174–182. doi: 10.1016/j.chroma.2007.01.140. [DOI] [PubMed] [Google Scholar]
- 176.Kroon DJ, Baldwin-Ferro A, Lalan P. Identification of sites of degradation in a therapeutic monoclonal antibody by peptide mapping. Pharmaceutical Research. 1992;9(11):1386–1393. doi: 10.1023/a:1015894409623. [DOI] [PubMed] [Google Scholar]
- 177.Li S, Schoneich C, Borchardt RT. Chemical instability of protein pharmaceuticals: mechanisms of oxidation and strategies for stabilization. Biotechnology and Bioengineering. 1995;48(5):490–500. doi: 10.1002/bit.260480511. [DOI] [PubMed] [Google Scholar]
- 178.Esmon CT. The protein C anticoagulant pathway. Arteriosclerosis and Thrombosis. 1992;12(2):135–145. doi: 10.1161/01.atv.12.2.135. [DOI] [PubMed] [Google Scholar]
- 179.Harris RJ. Heterogeneity of recombinant antibodies: linking structure to function. Developments in Biologicals. 2005;122:117–127. [PubMed] [Google Scholar]
- 180.Perry LJ, Wetzel R. Disulfide bond engineered into T4 lysozyme: stabilization of the protein toward thermal inactivation. Science. 1984;226(4674):555–557. doi: 10.1126/science.6387910. [DOI] [PubMed] [Google Scholar]
- 181.Sauer RT, Hehir K, Stearman RS, et al. An engineered intersubunit disulfide enhances the stability and DNA binding of the N-terminal domain of λ repressor. Biochemistry. 1986;25(20):5992–5998. doi: 10.1021/bi00368a024. [DOI] [PubMed] [Google Scholar]
- 182.Villafranca JE, Howell EE, Voet DH. Directed mutagenesis of dihydrofolate reductase. Science. 1983;222(4625):782–788. doi: 10.1126/science.6356360. [DOI] [PubMed] [Google Scholar]
- 183.Wells JA, Powers DB. In vivo formation and stability of engineered disulfide bonds in subtilisin. The Journal of Biological Chemistry. 1986;261(14):6564–6570. [PubMed] [Google Scholar]
- 184.Gale AJ, Xu X, Pellequer JL, Getzoff ED, Griffin JH. Interdomain engineered disulfide bond permitting elucidation of mechanisms of inactivation of coagulation factor Va by activated protein C. Protein Science. 2002;11(9):2091–2101. doi: 10.1110/ps.0210002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Michaelson JS, Demarest SJ, Miller B, et al. Anti-tumor activity of stability-engineered IgG-like bispecific antibodies targeting TRAIL-R2 and LTβR. mAbs. 2009;1(2):128–141. doi: 10.4161/mabs.1.2.7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Chen X, Bai Y, Zaro JL, Shen WC. Design of an in vivo cleavable disulfide linker in recombinant fusion proteins. BioTechniques. 2010;49(1):513–518. doi: 10.2144/000113450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Sun MMC, Beam KS, Cerveny CG, et al. Reduction-alkylation strategies for the modification of specific monoclonal antibody bisulfides. Bioconjugate Chemistry. 2005;16(5):1282–1290. doi: 10.1021/bc050201y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Alley SC, Okeley NM, Senter PD. Antibody-drug conjugates: targeted drug delivery for cancer. Current Opinion in Chemical Biology. 2010;14(4):529–537. doi: 10.1016/j.cbpa.2010.06.170. [DOI] [PubMed] [Google Scholar]
- 189.Junutula JR, Raab H, Clark S, et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nature Biotechnology. 2008;26(8):925–932. doi: 10.1038/nbt.1480. [DOI] [PubMed] [Google Scholar]
- 190.Shen BQ, Xu K, Liu L, et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nature Biotechnology. 2012;30:184–189. doi: 10.1038/nbt.2108. [DOI] [PubMed] [Google Scholar]
- 191.Elliott S, Egrie J, Browne J, et al. Control of rHuEPO biological activity: the role of carbohydrate. Experimental Hematology. 2004;32(12):1146–1155. doi: 10.1016/j.exphem.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 192.LaPolt PS, Nishimori K, Fares FA, Perlas E, Boime I, Hsueh AJW. Enhanced stimulation of follicle maturation and ovulatory potential by long acting follicle-stimulating hormone agonists with extended carboxyl-terminal peptides. Endocrinology. 1992;131(6):2514–2520. doi: 10.1210/endo.131.6.1446593. [DOI] [PubMed] [Google Scholar]
- 193.Fares F, Guy R, Bar-Ilan A, Felikman Y, Fima E. Designing a long-acting human growth hormone (hGH) by fusing the carboxyl-terminal peptide of human chorionic gonadotropin β-subunit to the coding sequence of hGH. Endocrinology. 2010;151(9):4410–4417. doi: 10.1210/en.2009-1431. [DOI] [PubMed] [Google Scholar]
- 194.Berg DT, Gerlitz B, Shang J, et al. Engineering the proteolytic specificity of activated protein C improves its pharmacological properties. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(8):4423–4428. doi: 10.1073/pnas.0736918100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Grabenhorst E, Schlenke P, Pohl S, Nimtz M, Conradt HS. Genetic engineering of recombinant glycoproteins and the glycosylation pathway in mammalian host cells. Glycoconjugate Journal. 1999;16(2):81–97. doi: 10.1023/a:1026466408042. [DOI] [PubMed] [Google Scholar]
- 196.Lim Y, Wong NSC, Lee YY, Ku SCY, Wong DCF, Yap MGS. Engineering mammalian cells in bioprocessing—current achievements and future perspectives. Biotechnology and Applied Biochemistry. 2010;55(4):175–189. doi: 10.1042/BA20090363. [DOI] [PubMed] [Google Scholar]
- 197.Fukuta K, Abe R, Yokomatsu T, et al. Remodeling of sugar chain structures of human interferon-γ . Glycobiology. 2000;10(4):421–430. doi: 10.1093/glycob/10.4.421. [DOI] [PubMed] [Google Scholar]
- 198.Fukuta K, Yokomatsu T, Abe R, Asanagi M, Makino T. Genetic engineering of CHO cells producing human interferon-γ by transfection of sialyltransferases. Glycoconjugate Journal. 2000;17(12):895–904. doi: 10.1023/a:1010977431061. [DOI] [PubMed] [Google Scholar]
- 199.Bragonzi A, Distefano G, Buckberry LD, et al. A new Chinese hamster ovary cell line expressing α2,6-sialyltransferase used as universal host for the production of human-like sialylated recombinant glycoproteins. Biochimica et Biophysica Acta. 2000;1474(3):273–282. doi: 10.1016/s0304-4165(00)00023-4. [DOI] [PubMed] [Google Scholar]
- 200.Chitlaru T, Kronman C, Zeevi M, et al. Modulation of circulatory residence of recombinant acetylcholinesterase through biochemical or genetic manipulation of sialylation levels. Biochemical Journal. 1998;336(3):647–658. doi: 10.1042/bj3360647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Weikert S, Papac D, Briggs J, et al. Engineering Chinese hamster ovary cells to maximize sialic acid content of recombinant glycoproteins. Nature Biotechnology. 1999;17(11):1116–1121. doi: 10.1038/15104. [DOI] [PubMed] [Google Scholar]
- 202.Gramer MJ, Goochee CF. Glycosidase activities in Chinese hamster ovary cell lysate and cell culture supernatant. Biotechnology Progress. 1993;9(4):366–373. doi: 10.1021/bp00022a003. [DOI] [PubMed] [Google Scholar]
- 203.Gramer MJ, Goochee CF, Chock VY, Brousseau DT, Sliwkowski MB. Removal of sialic acid from a glycoprotein in CHO cell culture supernatant by action of an extracellular CHO cell sialidase. Bio/Technology. 1995;13(7):692–698. doi: 10.1038/nbt0795-692. [DOI] [PubMed] [Google Scholar]
- 204.Natsume A, Niwa R, Satoh M. Improving effector functions of antibodies for cancer treatment: enhancing ADCC and CDC. Drug Design, Development and Therapy. 2009;(3):7–16. [PMC free article] [PubMed] [Google Scholar]
- 205.Niwa R, Shoji-Hosaka E, Sakurada M, et al. Defucosylated chimeric anti-CC chemokine receptor 4 IgG1 with enhanced antibody-dependent cellular cytotoxicity shows potent therapeutic activity to T-cell leukemia and lymphoma. Cancer Research. 2004;64(6):2127–2133. doi: 10.1158/0008-5472.can-03-2068. [DOI] [PubMed] [Google Scholar]
- 206.Shields RL, Lai J, Keck R, et al. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human FcγRIII and antibody-dependent cellular toxicity. The Journal of Biological Chemistry. 2002;277(30):26733–26740. doi: 10.1074/jbc.M202069200. [DOI] [PubMed] [Google Scholar]
- 207.Malphettes L, Freyvert Y, Chang J, et al. Highly efficient deletion of FUT8 in CHO cell lines using zinc-finger nucleases yields cells that produce completely nonfucosylated antibodies. Biotechnology and Bioengineering. 2010;106(5):774–783. doi: 10.1002/bit.22751. [DOI] [PubMed] [Google Scholar]
- 208.Sehn LH, Assouline SE, Stewart DA, et al. A phase 1 study of obinutuzumab induction followed by 2 years of maintenance in patients with relapsed CD20-positive B-cell malignancies. Blood. 2012;119:5118–5125. doi: 10.1182/blood-2012-02-408773. [DOI] [PubMed] [Google Scholar]
- 209.Harrison S, Adamson S, Bonam D, et al. The manufacturing process for recombinant factor IX. Seminars in Hematology. 1998;35:4–10. [PubMed] [Google Scholar]
- 210.Butler M. Animal cell cultures: recent achievements and perspectives in the production of biopharmaceuticals. Applied Microbiology and Biotechnology. 2005;68(3):283–291. doi: 10.1007/s00253-005-1980-8. [DOI] [PubMed] [Google Scholar]
- 211.Ahn WS, Jeon JJ, Jeong YR, Seung JL, Sung KY. Effect of culture temperature on erythropoietin production and glycosylation in a perfusion culture of recombinant CHO cells. Biotechnology and Bioengineering. 2008;101(6):1234–1244. doi: 10.1002/bit.22006. [DOI] [PubMed] [Google Scholar]
- 212.Andersen DC, Bridges T, Gawlitzek M, Hoy C. Multiple cell culture factors can affect the glycosylation of Asn-184 in CHO-produced tissue-type plasminogen activator. Biotechnology and Bioengineering. 2000;70:25–31. doi: 10.1002/1097-0290(20001005)70:1<25::aid-bit4>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 213.Crowell CK, Qin Q, Grampp GE, Radcliffe RA, Rogers GN, Scheinman RI. Sodium butyrate alters erythropoietin glycosylation via multiple mechanisms. Biotechnology and Bioengineering. 2008;99(1):201–213. doi: 10.1002/bit.21539. [DOI] [PubMed] [Google Scholar]
- 214.Cumming DA. Glycosylation of recombinant protein therapeutics: control and functional implications. Glycobiology. 1991;1(2):115–130. doi: 10.1093/glycob/1.2.115. [DOI] [PubMed] [Google Scholar]
- 215.Gawlitzek M, Ryll T, Lofgren J, Sliwkowski MB. Ammonium alters N-glycan structures of recombinant TNFR-IgG: degradative versus biosynthetic mechanisms. Biotechnology and Bioengineering. 2000;68:637–646. doi: 10.1002/(sici)1097-0290(20000620)68:6<637::aid-bit6>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 216.Goochee CF, Gramer MJ, Andersen DC, Bahr JB, Rasmussen JR. The oligosaccharides of glycoproteins: bioprocess factors affecting oligosaccharide structure and their effect on glycoprotein properties. Bio/Technology. 1991;9(12):1347–1355. doi: 10.1038/nbt1291-1347. [DOI] [PubMed] [Google Scholar]
- 217.Jenkins N, Parekh RB, James DC. Getting the glycosylation right: implications for the biotechnology industry. Nature Biotechnology. 1996;14(8):975–981. doi: 10.1038/nbt0896-975. [DOI] [PubMed] [Google Scholar]
- 218.Pacis E, Yu M, Autsen J, Bayer R, Li F. Effects of cell culture conditions on antibody N-linked glycosylation-what affects high mannose 5 glycoform. Biotechnology and Bioengineering. 2011;108(10):2348–2358. doi: 10.1002/bit.23200. [DOI] [PubMed] [Google Scholar]
- 219.Warner TG, Chang J, Ferrari J, et al. Isolation and properties of a soluble sialidase from the culture fluid of Chinese hamster ovary cells. Glycobiology. 1993;3(5):455–463. doi: 10.1093/glycob/3.5.455. [DOI] [PubMed] [Google Scholar]
- 220.Nyberg GB, Balcarcel RR, Follstad BD, Stephanopoulos G, Wang DI. Metabolic effects on recombinant interferon-gamma glycosylation in continuous culture of Chinese hamster ovary cells. Biotechnology and Bioengineering. 1999;62:336–347. doi: 10.1002/(sici)1097-0290(19990205)62:3<336::aid-bit10>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 221.Hendrickson TL, Imperiali B. Metal ion dependence of oligosaccharyl transferase: implications for catalysis. Biochemistry. 1995;34(29):9444–9450. doi: 10.1021/bi00029a020. [DOI] [PubMed] [Google Scholar]
- 222.Kaufman RJ, Swaroop M, Murtha-Riel P. Depletion of manganese within the secretory pathway inhibits O-linked glycosylation in mammalian cells. Biochemistry. 1994;33(33):9813–9819. doi: 10.1021/bi00199a001. [DOI] [PubMed] [Google Scholar]
- 223.Holst B, Bruun AW, Kielland-Brandt MC, Winther JR. Competition between folding and glycosylation in the endoplasmic reticulum. The EMBO Journal. 1996;15(14):3538–3546. [PMC free article] [PubMed] [Google Scholar]
- 224.Shelikoff M, Sinskey AJ, Stephanopoulos G. A modeling framework for the study of protein glycosylation. Biotechnology and Bioengineering. 1996;50:73–90. doi: 10.1002/(SICI)1097-0290(19960405)50:1<73::AID-BIT9>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 225.Schneider M, Marison IW, Von Stockar U. The importance of ammonia in mammalian cell culture. Journal of Biotechnology. 1996;46(3):161–185. doi: 10.1016/0168-1656(95)00196-4. [DOI] [PubMed] [Google Scholar]
- 226.Crowell CK, Grampp GE, Rogers GN, Miller J, Scheinman RI. Amino acid and manganese supplementation modulates the glycosylation state of erythropoietin in a CHO culture system. Biotechnology and Bioengineering. 2007;96(3):538–549. doi: 10.1002/bit.21141. [DOI] [PubMed] [Google Scholar]
- 227.Santell L, Ryll T, Etcheverry T, et al. Aberrant metabolic sialylation of recombinant proteins expressed in Chinese hamster ovary cells in high productivity cultures. Biochemical and Biophysical Research Communications. 1999;258(1):132–137. doi: 10.1006/bbrc.1999.0550. [DOI] [PubMed] [Google Scholar]
- 228.Xu X, Nagarajan H, Lewis NE, et al. The genomic sequence of the Chinese hamster ovary (CHO)-K1 cell line. Nature Biotechnology. 2011;29(8):735–741. doi: 10.1038/nbt.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]