

Abstract
Vertebrate photoreceptors contain a unique tetraspanin protein known as retinal degeneration slow (RDS). Mutations in the RDS gene have been identified in a variety of human retinal degenerative diseases and more than 70% of these mutations are located in the second intradiscal (D2) loop, highlighting the importance of this region. Here we examined conformational and thermal stability properties of the D2 loop of RDS, as well as interactions with RDS’ non-glycosylated homologue ROM-1. The RDS D2 loop was expressed in E. coli as a fusion protein with maltose binding protein (MBP). The fusion protein, referred to as MBP-D2, was purified to homogeneity. Circular dichroism (CD) spectroscopy showed that the wild-type (WT) D2 loop consists of ~21% α-helix, ~20% β-sheet and ~59% random coil. D2 loop fusion proteins carrying disease-causing mutations in RDS (e.g R172W, C214S, N244H/K) were also examined, and conformational changes were observed (compared to WT D2). In particular, C150S, C214S, and N244H showed significant reductions in the α-helicity. Nevertheless, the thermal stability of the mutants was unchanged compared to WT, and all mutants were all capable of interacting with ROM-1, indicating that this functional aspect of the isolated D2 loop remained intact in the mutants despite the observed conformational changes. An I-TASSER model of the RDS D2 loop predicted structure consistent with the CD experiments and with the structure of the conserved region of the D2 loop of other tetraspanin family members. These results provide significant insight into the mechanism of RDS complex formation and the disease process underlying RDS-associated retinal degeneration.
Keywords: RDS, tetraspanin, CD spectroscopy, macular dystrophy, retinitis pigmentosa
Retinal degeneration slow (RDS) is a photoreceptor-specific glycoprotein required for the formation and maintenance of the outer segment (OS) rim region in rods and cones [1, 2]. The importance of RDS for OS morphogenesis is supported by the observation that rds−/− mice lack OSs [3] and exhibit no retinal function [4]. As a member of the tetraspanin protein superfamily, RDS exhibits several highly conserved structural characteristics including four membrane spanning domains, small (D1 or EC1 or LEL1) and large (D2 or EC2 or LEL2) extracellular/intradiscal loops, and cytoplasmic N- and C-termini [5, 6].
In general, tetraspanins form large functional membrane microdomains assembled via protein-protein interactions between tetraspanins and interactions with other proteins [7]. RDS is known to interact with itself and its non-glycosylated homologue rod outer segment membrane protein 1 (ROM-1). Both in vivo and in vitro studies have demonstrated that non-covalent interactions between RDS and ROM-1 result in the formation of homo- and hetero-tetrameric complexes [8, 9], and in the OS, these tetramers are linked together through intermolecular disulfide bonds to form octamers and higher-order oligomers that are crucial for disc rim formation [10–12].
RDS has two main functional domains which have been studied thus far: the C-terminal domain and the large D2 loop. The cytoplasmic C-terminal is highly variable among tetraspanin proteins and is often very short. The RDS C-terminal domain is intrinsically disordered [13] and is thought to play a role in OS targeting [14], membrane fusion [15], and interaction with binding partners such as calmodulin and melanoregulin [16, 17].
The tetraspanin D2 loop, which is extracellular in most cases but either extracellular (cone) or intradiscal (rods) in the case of RDS and ROM-1, is divided into a highly conserved region and a hypervariable region [18, 19]. The D2 loop of RDS consists of 142 amino acids; the highly conserved region corresponds approximately to residues 125-167 and residues 250-263, while the intervening amino acids (168-249) comprise the hypervariable region [19, 20]. The D2 loop is known to play an important role in protein-protein interactions necessary for disc formation and stabilization [8, 21, 22], and has specifically been mapped as the region responsible for interactions between molecules of RDS and interactions between RDS and ROM-1 [21]. Tetraspanins exhibit highly conserved D2 loop cysteines which are involved in the formation of intramolecular disulfide bonds. RDS has six such cysteines, C165, C166, C213, C214, C222 and C250, and mutagenesis studies [22] suggest that mutation of any of these six cysteines results in a failure to form normal complexes, probably due to improper folding. RDS also contains a seventh cysteine in the D2 loop (C150) which is important for the formation of the intermolecular disulfide bonds and assembly of large complexes described above. In a heterologous expression system and transgenic animal model, C150S RDS was unable to form intermolecular disulfide bonds [10, 22]. Animals which expressed only C150S RDS (no WT RDS) were incapable of forming OSs, and the mutation caused a dominant negative (i.e. even in the presence of WT RDS) degeneration in cones [10, 23, 24] underscoring the importance of this cysteine in RDS function and the differential role of RDS in rods vs. cones.
Mutations in RDS cause a variety of retinal degenerations including rod-dominant autosomal dominant retinitis pigmentosa (ADRP) and multiple classes of cone-dominant macular dystrophy (MD) [25, 26]. Over 70% of these mutations are located in the D2 loop (http://www.retina-international.org/files/sci-news/rdsmut.htm), highlighting the functional importance of this region. Evidence from in vitro and animal studies has led to the hypothesis that mutations associated with rod-defect are likely to be null alleles and result in disease via haploinsufficiency while cone-dominant disease mutations are likely to be gain-of-function alleles [10, 24, 27–29]. However, the mechanism underlying the development of cell-type specific disease in the case of RDS mutations is not understood. The goal of the present study was to understand the effects of various disease-causing mutations on the structure of the D2 loop. We examine C214S- and N244K-RDS which cause ADRP in patients [30, 31], R172W- and N244H-RDS which cause MD in patients [26, 32], and the C150S-RDS which does not form intermolecular disulfide bonds (Table 1). We show that these mutations do not alter the thermal stability of the D2 loop, but do result in substantial changes in the secondary structure of the D2 loop. Interestingly, we report that the pattern of these changes does not correlate with the cell-type targeted by the mutations.
Table 1.
Phenotypes of RDS mutations characterized in this study.
| Mutation | Patient Phenotype | Refs | In Vitro/Murine Phenotype | Refs |
|---|---|---|---|---|
| C150S | N/A | No intermolecular disulfide bonds No formation of higher-order oligomeric complexes Does not interact with ROM-1 in cones Dominant-negative structural and functional degeneration in cones |
[10, 22] | |
| R172W | Macular Dystrophy, Central Areolar Choroidal Dystrophy | [38, 39, 50] | Forms intermolecular disulfide bonds Formation of normal complement of RDS complexes in vivo Normal interactions with ROM-1 Dominant-negative structural and functional degeneration in cones. |
[27, 28, 44] |
| C214S | Autosomal Dominant Retinitis Pigmentosa | [30] | Forms aggregates in vitro Does not interact with ROM-1 in vitro or in vivo. Causes haploinsufficiency-like phenotype in vivo |
[22, 29, 44] |
| N244H | Macular Dystrophy | [32] | Forms intermolecular disulfide bonds Normal interactions with ROM-1 Not studied in vivo |
[43] |
| N244K | Autosomal Dominant Retinitis Pigmentosa, Bull’s Eye Maculopathy, Cone-Rod Dystrophy | [32, 40] | Forms aggregates in vitro Does not bind ROM-1 Not studied in vivo |
[43] |
Results
Expression of the MBP fusion proteins
A fusion protein comprising MBP and the wild-type (WT) RDS D2 loop (Figure 1A–B) was generated and used as a base to create several MBP-D2 loop mutants. Purified MBP protein migrated to a position corresponding to ~37 kDa, while the MBP-D2 (WT) fusion protein migrated to ~56 kDa which is comparable to the predicted size of 37 + 19 kDa (Figure 1C). Western blot analysis confirmed that our polyclonal antibody against the RDS-D2 loop recognized the fusion proteins but not MBP (Figure 1D). MBP-D2 loop fusion proteins carrying mutations (C214S, C150S, R172W, N244H, and N244K, Table 1) were also expressed and purified, and migrated equivalently to MBP-D2 (WT) (Figure 1E). Occasionally a small amount of free MBP was detected in the fusion protein preparations, which may reflect either partial degradation during the purification procedure or formation of an early truncated product. The small amounts of these degradation/truncation products represent <10% of the total protein, and thus do not significantly affect the results reported in this study.
Figure 1. Generation and purification of RDS D2 loop fusion proteins.

(A) Structural features of RDS protein. RDS contains four transmembrane domains, two intradiscal loops (D1 and D2), cytoplasmic N- and C- termini, and a small cytoplasmic loop (C1). (B) Schematic representation of MBP-fusion protein. The full-length D2 loop of RDS consisting of residues Phe120-Asn256 corresponds to the red bar labeled D2-RDS. Purified MBP (middle lane) and MBP-D2 (right lane) were subjected to (C) SDS-PAGE and stained with Coomassie brilliant blue, and (D) western blot analysis using polyclonal anti-D2 (RDS) antibody. (E) Different mutants of RDS D2 (C214S, C150S, R172W, N244H and N244K) were expressed and purified as MBP fusion proteins. Purified proteins were subjected to SDS-PAGE and stained with Coomassie brilliant blue to check the purity. The mutant proteins were also highly expressed in this system and migrated to the same position as MBP-D2 -WT in the gel.
MBP-D2 interacts with Rom-1
Since the D2 loop is isolated from the rest of the RDS polypeptide when expressed as an MBP fusion protein; we wanted to confirm that the fusion protein exhibits similar functional characteristics to the endogenous protein. As RDS interacts with itself and ROM-1 through non-covalent interactions in the D2 loop, we examined the interaction between MBP-D2 and ROM-1/RDS from freshly prepared retinal extract. Immunoprecipitation with antibodies against the C-terminal of ROM-1 (ROM1-CT) showed that MBP-D2 (WT) retained the ability to bind endogenous ROM-1 (Figure 2A). Similarly, IP with antibodies against the C-terminal of RDS (RDS-CT) also brought down MBP-D2 (Figure 2B). Importantly, these results were recapitulated when mutant MBP-D2 proteins were incubated with retinal extracts and immunoprecipitated with anti-ROM1-CT indicating that the mutations did not affect this protein-protein interaction (Figure 2C), and suggesting that the MBP-D2 fusion proteins preserve the ability to interact with native proteins. Although our data suggesting that MBP-D2-C214S can bind ROM-1 seems to contradict previous evidence showing that full-length C214S-RDS does not have the ability to bind ROM-1, this discrepancy likely arises due to the nature of the MBP-D2 fusion proteins. The MBP-D2 proteins used here for study of secondary structure are soluble (rather than membrane bound as full-length RDS is), and the C214S mutation likely induces aggregation of the full-length membrane-bound protein to a much greater extent than in the MBP fusion protein due to interactions of the misfolded C214S D2 domain (in the full length protein) with the membrane.
Figure 2. MBP-D2 fusion proteins retain the ability to interact with endogenous RDS and ROM-1.

MBP-D2 (WT) and the D2 mutants interact with RDS and ROM-1 from retinal extract. (A) Co-immunoprecipitation using anti-ROM-1 shows that ROM-1 from retinal extract interacts with MBP-D2 (WT). The blot was probed with anti-MBP to recognize MBP-D2. (B) Co-immunoprecipitation using anti-RDS-CT (left panel) and anti-ROM-1 (right panel) shows that RDS and ROM-1 from retinal extract interacts with MBP-D2. (C) Co-immunoprecipitation using anti-ROM-1 shows that ROM-1 from retinal extract interacts with MBP-D2 (WT) and also with the mutants C150S, R172W, C214S, N244H and N244K, but not with MBP alone. The blot was probed with anti-MBP (top) and with ROM-1 (bottom).
Secondary structures of MBP-D2 fusion proteins
CD spectra were obtained for MBP and the MBP fusion proteins (Figure 3A–B). The CD spectra are typical of proteins containing mixed α-helical and β-sheet secondary structures. The mutant MBP-D2 proteins yield CD signals that vary by differing extents compared to the WT spectra (Figure 3A), indicating conformational changes between the mutant versus WT fusion proteins. As all of the MBP fusion proteins are readily purified using the amylose resin (Figure 1E), and are recognized by anti-MBP (Figure 1D), we infer that the conformational changes occur in the D2 portion of the fusion proteins. To analyze the secondary structures of the WT and mutant D2 loop proteins, each of the MBP-D2 spectra were subtracted from an MBP only spectra (shown for MBP-D2 in Figure 3B), to generate the spectra corresponding to the D2 loop portion of the fusion protein (green line in Figure 3B). The average (N=3–5) subtracted CD spectra for the WT and mutant D2 loops were analyzed using two different algorithms, SELCON3 and CONTILL (CD Pro software), to calculate the secondary structure content of each protein. The distribution of α-helix and β-sheet in each D2 loop are presented in Figure 3C. The WT-D2 protein was ~ 21% α-helix, ~20% β-sheet and ~59% random coil. Several of the mutants, including C150S, C214S, and N244H, showed a significant decrease in the α-helicity in comparison to D2 (WT). Interestingly, N244K and R172W did not show as large of a conformational change compared to the WT as the other mutants. In most, but not all cases, a loss in α-helicity was reflected in an increase in the percentage of β-sheet.
Figure 3. CD spectral analysis indicates that disease causing mutations alter the secondary structure of the D2 loop.

Far UV circular dichroism (CD) spectroscopy experiments of purified MBP-D2 and MBP were performed using a JASCO J715 spectropolarimeter with a PTC-348WI peltier temperature controller. (A) Representative CD spectra of WT (red), N244K (periwinkle), N244H (yellow-brown), C214S (light blue), R172W (purple), C150S (green) and MBP (blue) are presented here. (B) Representative CD spectra of MBP-D2 (red), MBP (blue), and the calculated spectrum of D2 (green) are shown. The latter spectrum was obtained by subtraction of the MBP spectrum from the MBP-D2 spectrum. (C) Quantitative analyses of the percent of α-helix and β-sheet were performed using the algorithms SELCON3 and CONTINLL based on subtracted spectra obtained as in (B).
Conformational changes in D2 loop as a function of temperature
Given the observed structural changes of the D2 mutants relative to WT, we next examined the effects of the mutations on the stability of the D2 loop. To analyze thermal stability, we investigated changes in the CD signal over a wide temperature range (10°C–80°C) at 220 nm. Changes in molar ellipticity of D2 (WT) at 10°C increments are shown in Figure 4A. It is evident that by 80°C, the majority of secondary structure has been lost (purple, Figure 4A). Molar ellipticity is plotted as a function of temperature in Figure 4B, and the midpoint of the transition (Tm) for the denaturation of MBP alone was ~50°C (blue, Figure 4B). In contrast, MBP-D2 (WT) showed two separate broad transitions, one at ~50°C and a second at ~60°C (red, Figure 4B). Given that the first transition coincides with the denaturation of MBP, it is likely that the second transition is due to unfolding of the D2 loop. Notably, all of the mutants show similar thermal denaturation curves to the WT D2 loop (Figure 4C). Thus, although several of the mutations affect the secondary structure of the D2 loop, the mutants still retain a folded core structure, which shows similar thermal stability to the WT D2 loop. Interestingly, the two unfolding transitions are more apparent for some of the mutants, including the R172W, N244H, and N244K mutants. This may indicate the loss of an intermediate in the unfolding pathway, resulting in more cooperative unfolding transitions. Further studies will be necessary to elucidate the unfolding pathways of WT and mutant D2 proteins.
Figure 4. Common disease causing mutations in the RDS D2 loop do not affect thermal stability.

(A) To indicate the overall changes in the CD spectra as a function of temperature, wavelength scans were obtained every 10°C during a thermal denaturation experiment of MBP-D2 (WT). (B) Thermal denaturation curves of MBP (blue) and MBP-D2 (red) were obtained by monitoring the CD signal at 220 nm from 10°C to 80°C. Denaturation curves are in units of molar ellipticity vs temperature. (C) Thermal denaturation curves of WT and mutant MBP-D2 proteins.
Molecular Modeling
We used computational methods, in this case the I-TASSER server [33], to predict and model the three dimensional structure of the D2 loop. First the secondary structure was predicted using the programs Jpred 3 [34] and GOR4 [35]. Second, the three-dimensional structure was predicted using I-TASSER (Iterative threading assembly refinement) [33]. Jpred 3 predicts α–helices between amino acid residues (123-141), (150-164) and (251-254) in D2-WT and mutant proteins. With GOR4, the D2-WT was predicted to contain α-helix content of ~25%, β-sheet of ~23% and coil content of ~53%. These predictions are consistent with the secondary structure content determined by CD spectroscopy. A representative model is shown in Figure 5. To generate this model, distance constraints were used to keep the 3 different highly conserved intramolecular disulfide bonds within range. However, regardless of whether distance constraints were used, the high scoring models (with Z values > −1.5 [33]) consisted of a similar orientation of the first 60 N-terminal residues (120-180). This region is predicted to consist of three α-helical segments that include residues 121-145, 149-163, and 172-181 as helices 1 (green), 2 (yellow), and 3 (orange), shown in Figure 5. This N-terminal region is shown in Figure 5 as green, yellow, and orange helices. Furthermore, the I-TASSER-generated models of the D2 loop showed the C-terminal 10-15 residues of the D2 loop consistently oriented in close proximity to helix 1. This resulted in the N- and C-terminal residues extending in similar directions, as would be expected for anchoring of the D2 loop to the membrane. C150, R172, and N244 are represented as red and blue stick models in Figure 5. Notably, C150 is located near the turn between helices 1 and 2, in a position that appears to be accessible for formation of an intermolecular disulfide bond. In addition, R172 and N244 are located on the same face of the protein, suggesting that they could be located in the same intermolecular interface with a potential binding partner. The remainder of the D2 structure is not reliably predicted by the computational methods used. Overall, we show here a conservative estimate of the major structural features of the D2 loop that are consistently predicted by computational methods. Such a model will be useful in future studies to determine structure-function correlations of the D2 loop of RDS.
Figure 5. Three dimensional model of the RDS D2 loop.
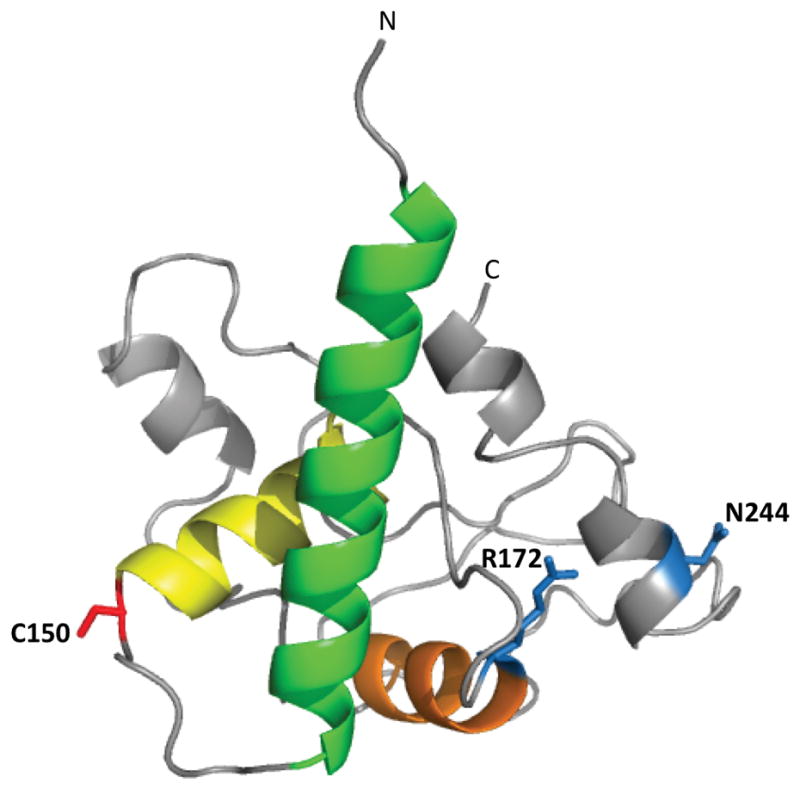
The three dimensional molecular model of the D2 loop was predicted by a protein threading method (see Materials and Methods). Three α-helixes predicted with high confidence are represented as green, yellow and orange. The location of C150 is represented by red sticks, and the locations of R172 and N244 by blue sticks. The N-terminal and C-terminal ends of the D2 loop are labeled.
Discussion
Here we used CD spectroscopy and bioinformatics to analyze the RDS-D2 loop sequence and generate models of the structure of this critical region of the protein. The only tetraspanin whose D2 loop has a solved crystal structure is CD81 [36] (a cell surface receptor for hepatitis C) however, comparative analysis and structural predictions of a wide array of tetraspanins have indicated that some features of the CD81 D2 loop crystal structure are likely to be common across members of the protein family [19]. Specifically, the CD81 D2 loop exhibits 2 α-helices in the N-terminal portion of the D2 loop (termed A and B) and one α-helix in the C-terminal portion of the D2 loop (termed E) that are predicted to be conserved throughout all tetraspanins [19, 36]. In addition, the CD81 D2 loop has two additional alpha helices (C and D) that are predicted to be conserved in some tetraspanins [19]. The tetraspanin D2 loop is thought to have a mushroom-like structure with the highly conserved helices A and E (which are antiparallel) serving as the “stalk” while the remainder of the D2 loop serves as the “cap” [36]. The majority of this cap region corresponds to the hypervariable region of the D2 loop and is thought to confer protein specific function. Although the RDS D2 loop is significantly larger than the CD81 D2 loop (88 vs. 142 amino acids) this difference is entirely in the hypervariable region. Our predicted model of the RDS D2 loop (Figure 5) exhibits secondary structure which correlates well with CD81; namely helices 1-3 in Figure 5 (green, yellow, orange) correspond in approximate size and location with helices A-C of CD81, and our structure identifies two helical regions in the C-terminal portion of the D2 loop which are similar in size and location to helices D and E of CD81.
These similarities might be thought to have significant implications for the mechanism of RDS complex assembly. In common with most other tetraspanins (including RDS), CD81 forms homodimers. The crystal structure of dimerized CD81 D2 loops predicts that the two subunits are held together by interactions between the two opposing A helices and by interactions between several residues in the B helix and several residues in the E helix of the opposing subunit [36]. However, our previous work has indicated that the region required for assembly of RDS homodimers is C165 to N182 [21], a region corresponding to helix 3 (orange, Figure 5) in RDS and roughly to helix C in CD81. ROM-1/RDS hetero-interactions have been mapped to a region incorporating Y140 to N182, roughly corresponding to helices 2–3 (yellow and orange, Figure 5). These data suggest that while the structure of the conserved region of the RDS D2 loop is consistent with that known or predicted for other tetraspanins [19, 36], the orientation and assembly of RDS complexes in the membrane diverges from other family members. This may arise due to the different nature of RDS/ROM-1 complexes compared to other tetraspanins complexes. For many tetraspanins, CD81 included, the core unit is a homo or heterodimer which serves to anchor the rest of the tetraspanin microdomain [19, 36, 37]. In contrast, evidence indicates that the core unit of RDS complexes is tetrameric in nature [11, 12]. The difference between RDS complex assembly and that of other tetraspanins is further emphasized by the presence of C150 in RDS. RDS and ROM-1 alone among tetraspanins form D2 loop mediated intermolecular disulfide bonds as part of their normal complex assembly.
More than 70% of RDS disease causing mutations occur in the D2 loop and there are a wide variety of associated degenerative phenotypes. Many of these mutations have been examined in heterologous expression systems and animal models, and have led to the hypothesis that mutations can be grouped into those which result in null alleles and are associated with rod-dominant disease (such as ADRP), and those which result in gain-of-function alleles and are associated with cone-dominant disease (such as MD). While this hypothesis may have some utility in guiding our understanding of genotype-phenotype correlations, our data here, along with the variation in patient phenotypes, suggests that such a dichotomous interpretation is too simplistic. Although some patients exhibit disease which truly targets rods or cones, many others exhibit disease which targets both cell types such as cone-rod or rod-cone dystrophy. Here we see that there is no clear correlation between the effects of mutations on secondary structure and the target cell type. R172W and N244H are both associated with MD or cone-rod dystrophy (cone-targeted) [32, 38, 39], but our results indicate that the secondary structure of D2-R172W is similar to D2-WT while D2-N244H exhibits reduced α-helicity compared to WT. Similarly the fraction of α-helix in D2-N244K is similar to D2-WT while the fraction in D2-C214S is reduced even though both cause ADRP or rod-cone dystrophy [30, 40]. The only other study which has examined the secondary structure of RDS [41] reported very little change in secondary structure in the case of the P216L ADRP mutation [25], although that study examined the secondary structure of the entire RDS polypeptide (not just the D2 loop) so it is difficult to directly compare results. These results suggest that assessing each mutation independently may be a better way to approach understanding RDS-associated disease, and it is likely that for many mutations disease arises due to a combination of molecular defects.
The majority of the mutants show an increase in beta sheet content relative to the wild type protein, particularly C214S. Mutants with increased beta sheet content may increase the propensity for protein aggregation, under certain conditions, by resulting in the exposure or extension of beta sheet edges, which are normally protected in native beta-sheet rich proteins [42]. Such a propensity for increased aggregation would likely disrupt heteromeric protein-protein interactions leading to a loss of function. C214S shows the most dramatic shift in secondary structure of all the mutations we study, and previous data strongly suggests that C214S is a null allele [29, 43]. Given this alteration in structure combined with the accompanying ablation of a highly conserved disulfide bond, it is not surprising that it has been shown that full-length C214S-RDS does not bind ROM-1 in vitro or in vivo [22, 29]. In addition, it has been shown that full-length C214S does not form proper complexes, likely due to misfolding of the tertiary RDS structure as a result of ablation of a critical intramolecular disulfide bond leading to protein degradation and haploinsufficiency [22, 29]. Although we here show that the thermal stability of MBP-D2-C214S is not different from MBP-D2-WT and that MBP-D2-C214S can bind ROM-1, the significant structural changes arising from the C214S mutation may significantly impair the ability of the entire RDS protein to properly fold and embed in the membrane during synthesis in vivo thus leading to aggregation and degradation. The N244H/K mutations occur in a predicted helical region (Figure 5) and both result in the substitution of an uncharged amino acid with a positively charged amino acid. Given the similarity in charge between histidine and lysine, and the fact that both are commonly found in α-helices, it is not clear why one (D2-N244H) should result in a decrease in D2 helicity while the other (D2-N244K) is unchanged. Furthermore, this pattern of alteration in secondary structure does not follow what we have observed in other experiments. In common with C214S, heterologously expressed N244K does not bind ROM-1 or form higher order complexes (only aggregates), but in contrast to C214S, the secondary structure of N244K is relatively unchanged. Following this lack of correlation between changes in secondary structure and disease type or molecular characteristics, N244H exhibits normal complex assembly and ROM-1 binding [43] but shows a decrease in the D2 loop α-helicity.
One of the most interesting results we observe is in the case of R172W, which results in the substitution of a positively charged amino acid with a bulky hydrophobic amino acid. In our CD experiments, we used two different algorithms (CONTINLL and SELCON3) to predict the percentage of the D2 loop in α-helices or β-sheets. In most cases, the two algorithms were in fairly close agreement (observe small error bars in Figure 3C), however in the case of R172W, one algorithm predicted a substantial drop in the % as an α-helix, (16.0% in WT vs. 6.9% in R172W with CONTILL) while the other predicted virtually no change (27% in WT vs. 24% in R172W with SELCON3). The reason for this difference is not clear. R172W causes MD and butterfly-shaped pattern dystrophy in patients, and dominant, early onset, cone-specific degeneration in transgenic mice. We have observed that R172W-RDS traffics properly to the OS, retains the ability to bind ROM-1, and forms higher-order oligomeric complexes, and we have been unable to determine the mechanism underlying the development of disease.
In spite of the utility of these results, it is important to recognize limitations associated with the use of fusion constructs comprising only a portion of the protein of interest. Use of only a portion of the protein means that overall protein folding and assembly will be different for MBP-D2 fusion proteins (compared to full-length RDS). Furthermore, the fusion proteins we are using are soluble, rather than membrane bound (as is the case with full-length RDS), so mutation that cause aggregation or misfolding of the full-length protein may have a less severe effect on the soluble MBP-D2 proteinss. Although these facts may mean that the MBP system is not useful for study of RDS complex assembly (as proper folding and insertion in the membrane are necessary for oligomerization), however complex assembly has been studied in detail elsewhere [11, 12, 44]. One of the striking benefits of using the D2 loop alone for the CD experiments is that changes in secondary structure can be localized to a specific region of the protein, a result that can be difficult to achieve with the full-length protein.
In conclusion, the present study indicates that the RDS D2 loop is composed of a mix of α-helices, β-sheet, and random coil. We show that while the RDS D2 loop exhibits similar secondary and predicted tertiary structure to other tetraspanins in the highly conserved region, in contrast to other tetraspanins, this region is not where RDS-RDS and RDS-ROM-1 interactions are predicted to occur. This divergence highlights critical differences in the role of RDS/ROM-1 compared to other tetraspanins: RDS and ROM-1 may play a role in organizing a tetraspanin microdomain, but unlike most other tetraspanins, their function is primarily structural, and as a result RDS/ROM-1 complexes are much larger and of a different type (i.e. containing intermolecular disulfide bonds) than other tetraspanin dimeric complexes.
Materials and Methods
Fusion protein cloning
A maltose binding (MBP) fusion protein was generated encoding the predicted D2 loop of mouse RDS (Phe120 – Asn256) [21] joined to a MBP header via a PreScission protease site [45]. The cDNA RDS D2 loop was amplified by polymerase chain reaction from murine RDS cDNA, and the amplicon incorporated 5′ (BamH I) and 3′ (Hind III) restriction sites for subcloning. This fragment was inserted into the pMal-c2 vector (containing the PreScission site) (New England Biolabs) [45]. The C150S, R172W, C214S, N244H and N244K mutations were created via site-directed mutagenesis using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). Authenticity of the constructs was confirmed by sequence analysis.
Expression and purification of the fusion protein
Escherichia coli BL21 (DE3) cells carrying the MBP-D2 fusion protein construct were grown in Luria broth (LB) medium containing ampicillin (50μg/mL). Expression was induced and cells harvested as described previously [21, 46]. The MBP and all MBP-D2 fusion proteins were purified using amylose resin (New England Biolabs, Ipswich, MA) as described [46]. The purified protein was dialyzed overnight in buffer [20 mM Tris-HCl (pH 7.4) and 0.2 M NaCl]. Protein concentration was determined from the absorbance at 280 nm using extinction coefficients of 66.5 mM−1cm−1 for MBP; 95 mM−1cm−1 for D2-WT, -C150S, -C214S, -N244H, and -N244K; and 100.5 mM−1cm−1 for R172W. Expression and purity of the MBP and MBP-fusion D2 proteins was assessed by Coomassie blue staining and western blotting with anti-MBP (Santa Cruz Biotechnology) and anti-RDS-D2 antibodies (generated in-house and characterized previously, [21]).
SDS-PAGE and western blot analysis
SDS-PAGE and western blot analysis were performed as described previously [11]. Blots were incubated with primary antibodies (anti-MBP, 1: 2,500 and anti-RDS-D2, 1: 1000) for 2 hours at room temperature. After washing, blots were incubated with HRP-conjugated secondary antibodies (1:25,000, KPL, Gaithersburg, MD). Blots were developed using Super Signal West Dura Extended Duration chemiluminescent substrate (Pierce, Rockford, IL), and images were captured using a Kodak Imaging Station 4000r and Kodak MI Software v.4.0.3 (Carestream Health, Rochester, NY, USA).
Immunoprecipitation
Purified MBP or MBP fusion protein (~5 μg) was incubated at 4°C with freshly prepared retinal extract (~100 μg) for 3–4 hours in the presence of 25 μl protein A-beads (Sigma-Aldrich Corp, St. Louis, MO) conjugated with anti-ROM1-CT or anti-RDS-CT (1:100 dilution, generated in house and characterized previously [11]) antibodies. After adsorption, the protein beads were washed three times with extraction buffer and proteins were eluted with Laemmli sample buffer under reducing conditions before undergoing SDS-PAGE and western blot analysis.
Circular dichroism spectroscopy
The circular dichroism (CD) spectroscopy experiments were performed using a JASCO J715 spectropolarimeter with a PTC-348WI peltier temperature controller (Jasco, Corp.; Tokyo, Japan) as described previously [47, 48]. Protein samples were dialyzed against 20 mM Tris-HCl (pH 7.4), 200 mM NaCl at 4°C overnight prior to CD spectropolarimetry analysis. Spectra were obtained for protein (at ~ 6.5 μM) with wavelengths ranging from 270 to 200 nm using a 0.1 cm cuvette path length at 25°C, and with the following parameters for the spectra measurements: 1.0 nm bandwidth, 1 nm resolution, 16 sec. response time, and 5 accumulations. The results are expressed as mean molar ellipticity. Protein secondary structural contents were calculated using the CDpro software package [49] (available at http://lamar.colostate.edu/~sreeram/CDPro). Two programs available in the CDPro package, CONTINLL and SELCON3, were executed using the same 40 - protein reference library. The values from the resulting calculations were averaged to yield the percentages of α-helix, β-sheet and random coil for each protein sample.
Thermal denaturation experiments
Thermal denaturation curves were obtained by monitoring the CD signal at 220 nm from a temperature range of 10–80°C. Thermal denaturation experiments were performed using the same protein concentrations used above for the wavelength scan and in 20 mM Tris-HCl (pH 7.4), 200 mM NaCl. The CD signal was detected at 220 nm with the following parameters: 10–80°C temperature range, 1 nm bandwidth, 0.2°C resolution and a response time of 16 sec. Tm values were obtained as previously described [48].
Molecular Modeling
Protein threading was carried out using the I-TASSER software [33]. The PDB file generated was used in PyMOL to perform three dimensional molecular modeling for the D2 loop.
Acknowledgments
We are grateful to Dr. Lori Gwyn for her help at the initial stage of this work. We thank Ruby Rahman for providing excellent technical assistance in CD spectroscopy and Seok Ho Kim for site directed mutagenesis work. This study was supported by grants from the National Institutes of Health [EY018656 & EY10609 (MIN), EY018512 (SMC)], FFB (MIN), and OCAST (MIN, KKR, SMC).
Abbreviations
- RDS
retinal degeneration slow
- ROM-1
rod outer segment membrane protein 1
- MBP
maltose binding protein
- WT
wild-type
- CD
circular dichroism
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- PMSF
phenyl methyl sulfonyl fluoride
- BSA
bovine serum albumin
- PBS
phosphate-buffered saline
References
- 1.Sanyal S, Jansen HG. Absence of receptor outer segments in the retina of rds mutant mice. Neurosci Lett. 1981;21:23–6. doi: 10.1016/0304-3940(81)90051-3. [DOI] [PubMed] [Google Scholar]
- 2.Arikawa K, Molday LL, Molday RS, Williams DS. Localization of peripherin/rds in the disk membranes of cone and rod photoreceptors: relationship to disk membrane morphogenesis and retinal degeneration. J Cell Biol. 1992;116:659–67. doi: 10.1083/jcb.116.3.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jansen HG, Sanyal S. Development and degeneration of retina in rds mutant mice: electron microscopy. J Comp Neurol. 1984;224:71–84. doi: 10.1002/cne.902240107. [DOI] [PubMed] [Google Scholar]
- 4.Reuter JH, Sanyal S. Development and degeneration of retina in rds mutant mice: the electroretinogram. Neurosci Lett. 1984;48:231–7. doi: 10.1016/0304-3940(84)90024-7. [DOI] [PubMed] [Google Scholar]
- 5.Connell G, Bascom R, Molday L, Reid D, McInnes RR, Molday RS. Photoreceptor peripherin is the normal product of the gene responsible for retinal degeneration in the rds mouse. Proc Natl Acad Sci U S A. 1991;88:723–6. doi: 10.1073/pnas.88.3.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Connell GJ, Molday RS. Molecular cloning, primary structure, and orientation of the vertebrate photoreceptor cell protein peripherin in the rod outer segment disk membrane. Biochemistry. 1990;29:4691–8. doi: 10.1021/bi00471a025. [DOI] [PubMed] [Google Scholar]
- 7.Hemler ME. Tetraspanin functions and associated microdomains. Nature Reviews: Molecular Cell Biology. 2005;6:801–11. doi: 10.1038/nrm1736. [DOI] [PubMed] [Google Scholar]
- 8.Goldberg AF, Molday RS. Defective subunit assembly underlies a digenic form of retinitis pigmentosa linked to mutations in peripherin/rds and rom-1. Proceedings of the National Academy of Sciences, USA. 1996;93:13726–30. doi: 10.1073/pnas.93.24.13726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldberg AF, Moritz OL, Molday RS. Heterologous expression of photoreceptor peripherin/rds and Rom-1 in COS-1 cells: assembly, interactions, and localization of multisubunit complexes. Biochemistry. 1995;34:14213–9. doi: 10.1021/bi00043a028. [DOI] [PubMed] [Google Scholar]
- 10.Chakraborty D, Ding XQ, Conley SM, Fliesler SJ, Naash MI. Differential requirements for retinal degeneration slow intermolecular disulfide-linked oligomerization in rods versus cones. Hum Mol Genet. 2009;18:797–808. doi: 10.1093/hmg/ddn406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chakraborty D, Ding XQ, Fliesler SJ, Naash MI. Outer segment oligomerization of rds: evidence from mouse models and subcellular fractionation. Biochemistry. 2008;47:1144–56. doi: 10.1021/bi701807c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loewen CJ, Molday RS. Disulfide-mediated oligomerization of Peripherin/Rds and Rom-1 in photoreceptor disk membranes. Implications for photoreceptor outer segment morphogenesis and degeneration. J Biol Chem. 2000;275:5370–8. doi: 10.1074/jbc.275.8.5370. [DOI] [PubMed] [Google Scholar]
- 13.Ritter LM, Arakawa T, Goldberg AF. Predicted and measured disorder in peripherin/rds, a retinal tetraspanin. Protein Pept Lett. 2005;12:677–86. doi: 10.2174/0929866054696217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tam BM, Moritz OL, Papermaster DS. The C terminus of peripherin/rds participates in rod outer segment targeting and alignment of disk incisures. Mol Biol Cell. 2004;15:2027–37. doi: 10.1091/mbc.E03-09-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boesze-Battaglia K, Goldberg AF, Dispoto J, Katragadda M, Cesarone G, Albert AD. A soluble peripherin/Rds C-terminal polypeptide promotes membrane fusion and changes conformation upon membrane association. Exp Eye Res. 2003;77:505–14. doi: 10.1016/s0014-4835(03)00151-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boesze-Battaglia K, Song H, Sokolov M, Lillo C, Pankoski-Walker L, Gretzula C, Gallagher B, Rachel RA, Jenkins NA, Copeland NG, Morris F, Jacob J, Yeagle P, Williams DS, Damek-Poprawa M. The tetraspanin protein peripherin-2 forms a complex with melanoregulin, a putative membrane fusion regulator. Biochemistry. 2007;46:1256–72. doi: 10.1021/bi061466i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Edrington TCt, Yeagle PL, Gretzula CL, Boesze-Battaglia K. Calcium-dependent association of calmodulin with the C-terminal domain of the tetraspanin protein peripherin/rds. Biochemistry. 2007;46:3862–71. doi: 10.1021/bi061999r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kitadokoro K, Ponassi M, Galli G, Petracca R, Falugi F, Grandi G, Bolognesi M. Subunit association and conformational flexibility in the head subdomain of human CD81 large extracellular loop. Biol Chem. 2002;383:1447–52. doi: 10.1515/BC.2002.164. [DOI] [PubMed] [Google Scholar]
- 19.Seigneuret M, Delaguillaumie A, Lagaudriere-Gesbert C, Conjeaud H. Structure of the tetraspanin main extracellular domain. A partially conserved fold with a structurally variable domain insertion. J Biol Chem. 2001;276:40055–64. doi: 10.1074/jbc.M105557200. [DOI] [PubMed] [Google Scholar]
- 20.Conley SM, Stuck MW, Naash MI. Structural and functional relationships between photoreceptor tetraspanins and other superfamily members. Cell Mol Life Sci. 2011 doi: 10.1007/s00018-011-0736-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ding XQ, Stricker HM, Naash MI. Role of the second intradiscal loop of peripherin/rds in homo and hetero associations. Biochemistry. 2005;44:4897–904. doi: 10.1021/bi048414i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goldberg AF, Loewen CJ, Molday RS. Cysteine residues of photoreceptor peripherin/rds: role in subunit assembly and autosomal dominant retinitis pigmentosa. Biochemistry. 1998;37:680–5. doi: 10.1021/bi972036i. [DOI] [PubMed] [Google Scholar]
- 23.Chakraborty D, Conley SM, Fliesler SJ, Naash MI. The Function of Oligomerization-Incompetent RDS in Rods. Adv Exp Med Biol. 2010;664:39–46. doi: 10.1007/978-1-4419-1399-9_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chakraborty D, Conley SM, Stuck MW, Naash MI. Differences in RDS trafficking, assembly and function in cones versus rods: insights from studies of C150S-RDS. Hum Mol Genet. 2010;19:4799–812. doi: 10.1093/hmg/ddq410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kajiwara K, Hahn LB, Mukai S, Travis GH, Berson EL, Dryja TP. Mutations in the human retinal degeneration slow gene in autosomal dominant retinitis pigmentosa. Nature. 1991;354:480–3. doi: 10.1038/354480a0. [DOI] [PubMed] [Google Scholar]
- 26.Wells J, Wroblewski J, Keen J, Inglehearn C, Jubb C, Eckstein A, Jay M, Arden G, Bhattacharya S, Fitzke F, et al. Mutations in the human retinal degeneration slow (RDS) gene can cause either retinitis pigmentosa or macular dystrophy. Nature Genetics. 1993;3:213–8. doi: 10.1038/ng0393-213. [DOI] [PubMed] [Google Scholar]
- 27.Conley S, Nour M, Fliesler SJ, Naash MI. Late-onset cone photoreceptor degeneration induced by R172W mutation in Rds and partial rescue by gene supplementation. Invest Ophthalmol Vis Sci. 2007;48:5397–407. doi: 10.1167/iovs.07-0663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ding XQ, Nour M, Ritter LM, Goldberg AF, Fliesler SJ, Naash MI. The R172W mutation in peripherin/rds causes a cone-rod dystrophy in transgenic mice. Hum Mol Genet. 2004;13:2075–87. doi: 10.1093/hmg/ddh211. [DOI] [PubMed] [Google Scholar]
- 29.Stricker HM, Ding XQ, Quiambao A, Fliesler SJ, Naash MI. The Cys214-->Ser mutation in peripherin/rds causes a loss-of-function phenotype in transgenic mice. Biochem J. 2005;388:605–13. doi: 10.1042/BJ20041960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saga M, Mashima Y, Akeo K, Oguchi Y, Kudoh J, Shimizu N. A novel Cys-214-Ser mutation in the peripherin/RDS gene in a Japanese family with autosomal dominant retinitis pigmentosa. Hum Genet. 1993;92:519–21. doi: 10.1007/BF00216463. [DOI] [PubMed] [Google Scholar]
- 31.Nakazawa M, Naoi N, Wada Y, Nakazaki S, Maruiwa F, Sawada A, Tamai M. Autosomal dominant cone-rod dystrophy associated with a Val200Glu mutation of the peripherin/RDS gene. Retina. 1996;16:405–10. doi: 10.1097/00006982-199616050-00007. [DOI] [PubMed] [Google Scholar]
- 32.Nakazawa M, Kikawa E, Chida Y, Tamai M. Asn244His mutation of the peripherin/RDS gene causing autosomal dominant cone-rod degeneration. Hum Mol Genet. 1994;3:1195–6. doi: 10.1093/hmg/3.7.1195. [DOI] [PubMed] [Google Scholar]
- 33.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 5:725–38. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cole C, Barber JD, Barton GJ. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 2008;36:W197–201. doi: 10.1093/nar/gkn238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garnier J, Gibrat JF, Robson B. GOR method for predicting protein secondary structure from amino acid sequence. Methods Enzymol. 1996;266:540–53. doi: 10.1016/s0076-6879(96)66034-0. [DOI] [PubMed] [Google Scholar]
- 36.Kitadokoro K, Bordo D, Galli G, Petracca R, Falugi F, Abrignani S, Grandi G, Bolognesi M. CD81 extracellular domain 3D structure: insight into the tetraspanin superfamily structural motifs. Embo J. 2001;20:12–8. doi: 10.1093/emboj/20.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stipp CS, Kolesnikova TV, Hemler ME. Functional domains in tetraspanin proteins. Trends Biochem Sci. 2003;28:106–12. doi: 10.1016/S0968-0004(02)00014-2. [DOI] [PubMed] [Google Scholar]
- 38.Piguet B, Heon E, Munier FL, Grounauer PA, Niemeyer G, Butler N, Schorderet DF, Sheffield VC, Stone EM. Full characterization of the maculopathy associated with an Arg-172-Trp mutation in the RDS/peripherin gene. Ophthalmic Genet. 1996;17:175–86. doi: 10.3109/13816819609057891. [DOI] [PubMed] [Google Scholar]
- 39.Nakazawa M, Wada Y, Tamai M. Macular dystrophy associated with monogenic Arg172Trp mutation of the peripherin/RDS gene in a Japanese family. Retina. 1995;15:518–23. doi: 10.1097/00006982-199515060-00011. [DOI] [PubMed] [Google Scholar]
- 40.Kikawa E, Nakazawa M, Chida Y, Shiono T, Tamai M. A novel mutation (Asn244Lys) in the peripherin/RDS gene causing autosomal dominant retinitis pigmentosa associated with bull’s-eye maculopathy detected by nonradioisotopic SSCP. Genomics. 1994;20:137–9. doi: 10.1006/geno.1994.1142. [DOI] [PubMed] [Google Scholar]
- 41.Vos WL, Vaughan S, Lall PY, McCaffrey JG, Wysocka-Kapcinska M, Findlay JB. Expression and structural characterization of peripherin/RDS, a membrane protein implicated in photoreceptor outer segment morphology. Eur Biophys J. 2010;39:679–88. doi: 10.1007/s00249-009-0553-7. [DOI] [PubMed] [Google Scholar]
- 42.Richardson JS, Richardson DC. Natural beta-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc Natl Acad Sci U S A. 2002;99:2754–9. doi: 10.1073/pnas.052706099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Conley SM, Stricker HM, Naash MI. Biochemical analysis of phenotypic diversity associated with mutations in codon 244 of the retinal degeneration slow gene. Biochemistry. 2010;49:905–11. doi: 10.1021/bi901622w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goldberg AF, Molday RS. Expression and characterization of peripherin/rds-rom-1 complexes and mutants implicated in retinal degenerative diseases. Methods Enzymol. 2000;316:671–87. doi: 10.1016/s0076-6879(00)16756-4. [DOI] [PubMed] [Google Scholar]
- 45.Gwyn LM, Peak MM, De P, Rahman NS, Rodgers KK. A zinc site in the C-terminal domain of RAG1 is essential for DNA cleavage activity. J Mol Biol. 2009;390:863–78. doi: 10.1016/j.jmb.2009.05.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arbuckle JL, Fauss LA, Simpson R, Ptaszek LM, Rodgers KK. Identification of two topologically independent domains in RAG1 and their role in macromolecular interactions relevant to V(D)J recombination. J Biol Chem. 2001;276:37093–101. doi: 10.1074/jbc.M105988200. [DOI] [PubMed] [Google Scholar]
- 47.Godderz LJ, Rahman NS, Risinger GM, Arbuckle JL, Rodgers KK. Self-association and conformational properties of RAG1: implications for formation of the V(D)J recombinase. Nucleic Acids Res. 2003;31:2014–23. doi: 10.1093/nar/gkg281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arbuckle JL, Rahman NS, Zhao S, Rodgers W, Rodgers KK. Elucidating the domain architecture and functions of non-core RAG1: the capacity of a non-core zinc-binding domain to function in nuclear import and nucleic acid binding. BMC Biochem. 12:23. doi: 10.1186/1471-2091-12-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sreerama N, Woody RW. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal Biochem. 2000;287:252–60. doi: 10.1006/abio.2000.4880. [DOI] [PubMed] [Google Scholar]
- 50.Reig C, Serra A, Gean E, Vidal M, Arumi J, De la Calzada MD, Antich J, Carballo M. A point mutation in the RDS-peripherin gene in a Spanish family with central areolar choroidal dystrophy. Ophthalmic Genet. 1995;16:39–44. doi: 10.3109/13816819509056911. [DOI] [PubMed] [Google Scholar]