

Abstract
The cell secretome is a collection of proteins consisting of transmembrane proteins (TM) and proteins secreted by cells into the extracellular space. A significant portion (~13-20%) of the human proteome consists of secretory proteins. The secretory proteins play important roles in cell migration, cell signaling and communication. There is a plethora of methodologies available like Serial Analysis of Gene Expression (SAGE), DNA microarrays, antibody arrays and bead-based arrays, mass spectrometry, RNA sequencing and yeast, bacterial and mammalian secretion traps to identify the cell secretomes. There are many advantages and disadvantages in using any of the above methods. This review aims to discuss the methodologies available along with their potential advantages and disadvantages to identify secretory proteins. This review is a part of a Special issue on The Secretome.
Keywords: Secretome, Secretory protein, Signal peptides
1. Introduction
The repertoires of proteins that are secreted externally by the cells constitute the cell secretomes. The secretory proteins are important for maintaining cell-cell communication and proliferation. Examples of secretory proteins include hormones, digestive enzymes, cytokines, chemokines, interferons (IFNs), colony-stimulating factors (CSFs), growth factors, and Tumor necrosis factors (TNFs). Secretory proteins are encoded on the outside of the Rough Endoplasmic Reticulum (RER) which have ribosomes attached to it (Fig. 1).
Fig. 1. Translation of secretory proteins.
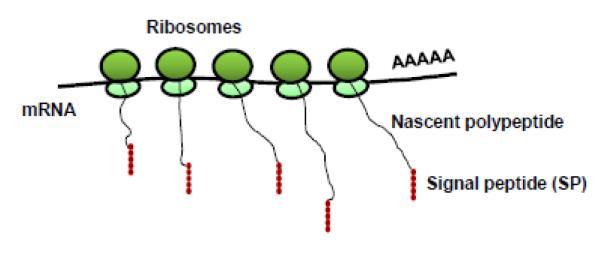
Schematic representation of nascent polypeptide containing a signal peptide (SP) being translated from ribosomes.
The secretory proteins contain an amino terminal signal peptide (SP) which targets them to enter the endoplasmic reticulum (ER). The SPs are 16-45 amino acids long and have a three-domain structure (Fig. 2). They have a short, positively charged amino terminal domain which contains 1-2 basic amino acids. This region is followed by the hydrophobic core consisting of 7-15 hydrophobic α-helix forming amino acids. Then there is the polar C-terminus with small amino acids, followed by the signal peptidase cleavage site. The SP is cleaved by signal peptidase during translation and the mature protein is released into the ER lumen at the end of synthesis. The SPs are highly variable in length and have no obvious sequence homology [1].
Fig. 2. Structure of signal peptide.

Schematic representation of a signal peptide (SP) showing basic N-terminus, hydrophobic core and polar C-terminus followed by the signal peptidase cleavage site.
An in-depth study of the cell secretomes can lead to the development of new therapeutics and discovery of biomarkers for cancer and other diseases like Type II diabetes. Secretome analysis can also be useful to study disease progression from normal cells to the metastatic state in different types of cancer including breast [2] and lung [3]. There is a vast array of techniques currently available to unfold the secretome of various cell types in different organisms. They all have their share of merits and demerits. Here we summarize the techniques available to date to study cell secretomes.
2. Methodologies available to study cell secretomes
There is a vast array of methodologies available to study cell secretomes of plant cells, mammalian cells, stem cells and cancer cells (Fig. 3) [4]. There are many advantages and disadvantages in using any of these methods to study the secretome of various cell types (Table. 1). Depending on whether the secretory protein sequence (with or without SP) is known or unknown these methods can be used accordingly (Fig.4). Conditioned media has largely been used to study secretory proteins and identify cancer biomarkers [5, 6]. Secreted proteins are present in very low concentrations due to dilution of culture media. Proteins secreted by dead cells in the media and serum proteins can mask and contaminate the secreted proteins making its isolation difficult [7]. Hence serum deprived media can be used [2, 8]. Also serum-deprivation can cause differential responses in different cell types [9]. The different methods have been outlined in this review for secretome identification.
Fig.3. Methods to study secretomes.

Schematic representation of methodologies (SAGE, DNA microarray, antibody arrays, bead arrays, Mass spectrometry, secretion traps, bioinformatics and RNA sequencing) available to study cell secretome.
Table. 1.
Methodologies to study cell secretomes with their potential advantages and disadvantages.
| Methodologies | Advantages | Disadvantages |
|---|---|---|
| SAGE |
|
|
| DNA microarray |
|
|
| Antibody and bead arrays |
|
|
| Mass spectrometry 2-DE DIGE ICAT iTRAQ SILAC SELDI-TOF |
|
|
| RNA sequencing |
|
|
| Yeast Secretion Trap (YST) |
|
|
| Computation algorithms |
|
|
Fig.4. Secretome analysis using different methods.

Schematic representation of methodologies used to analyze secretome based on known or unknown secretory protein sequence with or without signal peptides (SP).
2.1. Serial Analysis of Gene Expression (SAGE)
SAGE is a technique which measures global gene expression patterns [10]. It is a sequence-based method and relies on a short (9-10bp) tag to identify a gene. Linking (concatenation) these short sequences increases the efficiency of identifying unknown transcripts in a serial manner [11]. In this method, first RNA is isolated from an input sample and then converted to double stranded cDNA with the help of biotinylated oligo(dT) primers. The cDNA is digested with an anchoring enzyme and then bound to Streptavidin beads. The reaction mixture is divided into half and linkers are ligated. Then it is cleaved with tagging enzyme creating blunt ends. Again it is ligated and amplified with specific primers. This is cleaved with anchoring enzyme and ditags are isolated. Finally these tags are concatenated followed by cloning [12]. These tags are then sequenced using high-throughput sequencers.
SAGE has been applied to identify the rust secretome and it was shown that transcripts encoding secretory proteins were regulated in a stage-specific manner [13]. Although the method has been widely used there are certain advantages and disadvantages.
The advantages include: 1) It is a direct and quantitative method for measuring gene expression. No prior knowledge of sequences to be analyzed is required [14]. 2) Both known and unknown transcripts can be identified. 3) No hybridization is involved as compared to DNA microarrays, hence no bias. There are many disadvantages in using this method: 1) It is a high – throughput but time consuming method as compared to microarrays and RNA sequencing [15, 16]. 2) Same tag can be present on multiple genes and the same gene that is alternatively spliced can have different tags at the ends’ 3 making identification difficult [17]. 3) mRNA levels may not represent protein levels as in microarrays. 4) There can be sequencing errors and quantitation biases. 5) Restriction site is a determining factor. 6) Limited by the total number of tags that can be sequenced. 7) Low abundance transcripts are difficult to identify.
2.2. DNA Microarray
DNA microarray is used to measure differential gene expression in different tissues, in normal and diseased states, during development and/or under different conditions. Microarrays can be of two types: oligonucleotide arrays (GeneChips) and cDNA arrays (spotted array). In the oligonucleotide arrays (Affymetrix, Nimblegen), oligos/probes (~25bp) are synthesized directly on the chip in high density. Labeled RNA molecules are applied on these arrays. A fluorescent signal is generated if hybridization occurs. One color detection method is used to measure gene expression with these arrays. In the spotted array, cDNA probes are synthesized and then spotted on glass slides. RNA molecules labeled with Cy3 and Cy5 are hybridized to the array. Two color detection method is used and gene expression is measured [18].
DNA microarray has been utilized in secretome studies also. Gene expression studies were performed with microarrays and 18 novel secreted proteins were identified in differentiating human adipocytes [19]. Recently, microarray and bioinformatics was applied in human omental vs. subcutaneous adipose tissue to identify secretory proteins [20]. Secretome analysis by microarray in human fat cells revealed 8 novel adipokines involved in adipogenesis [21].
There are many advantages and disadvantages in using DNA microarray [22]. Advantages include: 1) High-throughput, quantitative method to measure abundance. 2) Less expensive compared to RNA sequencing [23]. There are also multiple disadvantages: 1) Incomplete coverage. 2) Require prior knowledge of sequences to be analyzed. 3) Proteins that are not differentially expressed at the mRNA level cannot be identified. 4) Gene expression may not necessarily correlate with changes in protein expression. 5) Biases are introduced during labeling and hybridization of microarrays. 6) It has a smaller dynamic range. 7) Identification of rare transcripts is difficult.
2.3. Protein Microarray
2.3.1. Antibody arrays
Protein/antibody arrays are used in a high-throughput manner for protein expression profiling in various tissues and under different conditions [24]. Proteins are immobilized on glass, plastic or nitrocellulose slides and different capture molecules (label-based or sandwich) are used to study expression. There are commercial arrays available (Cell Signaling, Sigma) which contains antibodies printed on slides against specific known secretory proteins. Signal is detected by fluorescence or chemiluminescence [24].
Antibody arrays have been used as a complementary method with mass spectrometry for secretory protein identification. Adipokine arrays were used to validate proteins secreted in low amounts during adipogenesis in humans [25]. Cytokine arrays have been used to identify secretory proteins from mesenchymal stem cells [26].
2.3.2. Bead based array
In the bead based arrays, antibodies are immobilized on color-coded beads instead of slides. The arrays are then incubated with the sample and detection is via fluorescence Bead-based arrays have been used in a multiplexed format to detect secretory proteins. The multiplexed bead assay was used in fibroblast cells and cardiomyocytes to identify secretory proteins [27].
These protein chip based methods although has been popularly used they are fraught with many merits and demerits. There are many advantages in using protein microarrays. 1) It is a very sensitive method to detect low abundance proteins [25]. 2) It is a highly specific process and has broad reproducibility [24]. Disadvantages include: 1) Cost is a very limiting factor. 2) Depends on the availability of high-affinity and specific antibodies for capture and detection. 3) The antibodies can bind non-specifically to other proteins. 4) Some secretory proteins are prone to fast denaturation (disulfide bond cleavage) [28] which can affect the binding.
2.4. Mass spectrometry
Liquid chromatography tandem mass spectrometry (LC-MS/MS) is an analytical method where proteins are first separated by liquid chromatography and detected by tandem mass spectrometry based on mass to charge ratio. There are two methods to identify secretory proteins: the gel-based methods and the gel-independent techniques. The gel-based methods include two-dimensional gel electrophoresis (2DE) and differential gel electrophoresis (DIGE) which is followed by mass spectrometry. Gel-independent techniques like SILAC (Stable isotope labeling by amino acids in cell culture), iTRAQ (Isobaric tag for relative and absolute quantization) and ICAT (Isotope-coded affinity tag) are often followed by LC-MS/MS or SELDITOF MS (Surface Enhanced Laser Desorption Ionization-Time of Flight mass spectrometry).
LC-MS/MS has been used to identify secretomes in various cell types. One dimensional SDS-PAGE followed by LC-MS/MS is the most widely used technology to identify cell secretomes in various cell types of both plants and mammals [29, 30]. Novel proteins secreted by astrocytes were identified from conditioned media of murine astrocyte culture by a proteomics approach [31]. The secretome of adipose stem cells induced by TNFα was elucidated by a proteomics approach [32]. Secretory proteins from skeletal muscle, stimulated by insulin were also identified by LC-MS/MS [33] .
2.4.1. Gel-based methods
Two-dimensional gel electrophoresis (2-DE)
2-DE is the method where proteins are separated on the basis of their net charge (isoelectric focusing, IEF) in first dimension then separated by mass (SDS-Polyacrylamide gel electrophoresis, SDS-PAGE) in the second dimension [34, 35]. They are resolved as discrete spots and are subjected to mass spectrometry to identify the proteins [36].
This method has been applied to a vast majority of cell types to decipher the secretomes of skeletal muscle cells [37], adipose stem cells (ASCs) [38], astrocytes [39], fibroblast feeder cells [40-42], arterial smooth muscle cells [43] and choroidal epithelial cells [44]. In conjunction with mass spectrometry, 2-DE was utilized to identify the secretome of leptin induced breast cancer cells [45].
Difference gel electrophoresis (DIGE)
The DIGE method is used to measure differential protein expression. In the DIGE method, before running the gel in first dimension, proteins are labeled with different fluorescent dyes (Cy2, Cy3 and Cy5). The single gel is then run in both dimensions (net charge and mass) and detection is by fluorescence imaging followed by superimposition of the images [46].
DIGE has been applied to kidney cells where substrates for the secreted protease ADAMTS1 was identified [47]. DIGE was also applied to human colon carcinoma cells to identify cancer biomarkers regulated by Smad4 [48].
Although many secretory proteins are identified using this technology there are many advantages and disadvantages. The advantages in using the gel-based assays are: 1) Unknown secretory proteins can be identified. 2) The methods have a high resolving power. The disadvantages are manifold: 1) Low abundance proteins cannot be detected. 2) They have a limited reproducibility and false positives are often encountered. 3) They have a limited dynamic range. 4) Low-throughput method.
2.4.2. Gel-independent methods
Isotope-coded affinity tag (ICAT)
In this method, two protein samples are first labeled with light and heavy ICAT reagent. The reagent binds to cysteine residues, has heavy and light isotopes (linkers) and has a biotin tag. Samples are mixed, digested and recovered via the biotin tag by affinity chromatography followed by LC-MS/MS [49]. A method named cleavable-ICAT (cICAT) was also introduced which uses a acid-cleavable linker and 12C or 13C isotopes [50].
Both ICAT and cICAT have been applied to study the secretome profile of various cell types. Using cICAT, secretome analysis of adipocytes with or without insulin stimulation revealed 240 new secretory proteins [51]. Secretory proteins from the tumor microenvironment of human glioma cells were identified by ICAT technology [52].
Isobaric tag for relative and absolute quantization (iTRAQ)
In this method relative abundance of proteins from four samples can be compared in a single mass spectrometric experiment. iTRAQ uses four tag reagents which binds to the N-terminus of the peptide. Hence, iTRAQ labels the primary amines of peptides and proteins. Each sample is digested separately and mixed with the specific iTRAQ tag followed by LC-MS/MS [53].
This method has been applied to decipher the cell secretome in influenza A virus-infected human primary macrophages [54]. This method has also been applied for secretory protein identification in Saccharomyces cerevisiae [55] and marine bacterium [56].
Stable isotope labeling by amino acids in cell culture (SILAC)
Cell culture media from two samples lacking amino acids are labeled with light and heavy isotopes of essential amino acids. They are combined, digested with trypsin and then used for LC-MS/MS [57].
Identifying the secretome of various cell types in plants and mammals have largely depended on the SILAC method. Novel secretory proteins were identified from the pancreatic cancer cell secretome using this method [58]. In esophageal squamous cell carcinoma, various known and novel biomarkers were identified using SILAC [59].
SELDI-TOF MS
SELDI-TOF MS is a high-throughput method based on direct separation of protein on SELDI protein chips followed by mass spectrometric detection. This method has been applied in stromal cells to detect the secretome [60].
The gel-free methods also have some advantages and disadvantages. The advantages include: 1) Low abundance proteins can be detected. 2) The SILAC method can label all peptides isotopically, hence improving sequence coverage. 3) SELDI-TOF MS is a high-throughput method needing small amount of samples and highly reproducible. The disadvantages are: 1) ICAT is only applicable to cysteine containing proteins. 2) SILAC cannot be applied to clinical protein samples in vivo. 3) In SELDI-TOF MS, identification of potential biomarkers for cancer secretome is difficult.
2.5. RNA sequencing
RNA sequencing is a high-throughput method for analyzing the transcriptome profiles of an organism [61-63]. Complementary DNA (cDNA) is made from the mRNA, fragmented and sequencing is performed using different technologies (Illumina, Roche 454, Solid etc). Once reads are obtained, they are aligned to the reference genome or assembled de novo and a transcriptome map is generated. The reads can be single end or paired end. Typically 30-400bp reads can be obtained. Single base resolution can be obtained with this method and it requires very low amounts of RNA. The background noise is very low as well as compared to other methods like DNA microarray. RNA sequencing is a very powerful tool to quantify mRNA abundance [64], map transcription start site [65], analyze splicing patterns [66] and measure gene expression changes during development and under different conditions [67, 68]. RNA sequencing can also be applied to unravel the secretome profile of a particular cell type in an organism. After the reads are aligned and sequences obtained, they can be analyzed by SignalP 4.0 that can predict whether the protein has a signal peptide and the cleavage site [69].
There are many merits and demerits of using RNA sequencing [70, 71]. The advantages are as follows: 1) No prior knowledge of genome sequence is necessary. 2) Non-model organisms can be used. Disadvantages in using RNA sequencing: 1) Very large number of sequences/reads are obtained hence it has to be sieved through by prediction algorithms to identify the secretory proteins. Storage of large amounts of data also poses a problem 2) Bioinformatics remains a challenging task. 3) It is a structural assay, only sequence information can be obtained. 4) Majority of reads represent common RNA. There can be an over-representation of most abundant RNAs, leading to identification of most common secretory proteins. 5) Rare transcripts may be under-representated. For identifying rare transcripts sequencing depth is necessary. 6) There is strand bias and strand specific libraries are difficult to produce. 7) Artifacts are introduced during amplification, selection and hybridization.
2.6. Secretion Traps or signal sequence traps
2.6.1. Yeast Secretion Trap (YST)
The YST methodology has been used to screen a cDNA library and identify novel secreted proteins in plants and mammals. Protease inhibitor 16 was identified as a novel protein secreted from the heart [72]. Novel secreted proteins were also identified in Arabidopsis by this method [73]. The YST system depends on the enzyme invertase encoded by the SUC2 gene in Saccharomyces cerevisiae (yeast) which functions extracellularly to catalyze the breakdown of sucrose to glucose and fructose. If the SUC2 gene is deleted or the invertase signal peptide is mutated, that prevents secretion, results in yeasts unable to grow on media containing sucrose. If the cDNA clones encode secreted proteins with a signal peptide, the yeast mutant can be rescued because the invertase fusion protein will be secreted allowing the yeasts to grow on sucrose. Here growth on sucrose is restored because the missing initiator methionine and signal peptide in the yeast mutant was provided by the fusion protein [74]. The cDNA library plasmids are transformed into a suc2 yeast mutant strain and plated on media containing sucrose. Clones are selected on their ability to grow on media containing sucrose. The yeast transformants are isolated and plasmids are purified. The plasmids are then transformed into E.coli and sequenced. To confirm the identity of the secreted proteins, computer algorithms designed to search for signal peptides are utilized (SignalP 4.0). The ability of the cDNA clones to restore invertase secretion combined with their signal peptide characteristics helps to confirm that these novel cDNAs encode genuine secretory proteins.
The advantages in using YST for identifying secretory proteins are: 1) This method can provide the gene sequence as well as the subcellular localization of the proteins. Hence, it is a functional assay as compared to RNA sequencing which provide only sequence information. The method also has certain disadvantages. These include: 1) Signal sequence of higher organism might not work in yeast [75]. Although protein folding, targeting and sorting are highly similar in yeast and higher organisms, certain pathways and proteins involved are not equal. 2) Proteins lacking a SP are difficult to identify. 3) Under-representation of low abundance protein.
2.6.2. Mammalian and bacterial secretion traps
In the mammalian secretion traps, cDNA (coding for secretory or non-secretory protein) is fused to mammalian expression vector pcDLSRa-Tac (3’). This directs the secretion of Tac (human CD25, a chain of the human interleukin-2 receptor) fusion protein which can be monitored by anti-CD25 antibody. COS-7 cells are transfected with the plasmids and secreted fusion proteins detected by immunostaining with anti-CD25 antibody [76]. The method is very laborious and not very widely used. Bacterial secretion traps has also been used with c-myc or β-lactamase as a reporter gene. Transposon assisted traps has also been done in bacteria with β-lactamase as reporter gene [77]. Although these traps have not been widely used for mammalian secretome studies, there is a potential that they can be used in place of yeast secretion traps.
2.7. Aptamer based methods
Cyclophilin B (Cyp B) has been identified as a potential pancreatic cancer cell biomarker using aptamer based methods based on molecular recognition [78]. Aptamers are synthetic oligonucleotides which bind specific targets. M9-5 aptamer was identified in this approach which bind target Cyp B and an in vitro positive/negative selection approach has been utilized to identify potential pancreatic cancer biomarkers.
2.8. Computational algorithms
Bioinformatics plays a significant role in deciphering the cell secretome of many species [79]. Novel secretory proteins were identified in the mouse secretome using bioinformatics approaches [80]. Computational analysis to predict secretory proteins based on signal peptides are used to define various secretomes. SignalP is utilized to predict secretory proteins with signal peptides traversing via the classical pathway [69]. SecretomeP is used to predict secretory proteins lacking a conventional signal peptide traveling through the non-classical pathway [81].
Although much secretory protein identification is based on computational algorithms the advantages include: 1) Prediction of secretory proteins with and without signal peptides. Disadvantages are: 1) No experimental validation, prediction only. 2) Proteins lacking signal peptide can be missed. 3) False positive and false negatives are often encountered.
3. Conclusions and further directions
Here we have discussed the vast array of methodologies that are applied to study the secretome of various cell types in different organisms including bacteria and humans. As most secretory proteins are present in very low concentrations they are difficult to isolate and analyze. Use of conditioned media often poses a problem to fully decipher the secretome profile of a cell type. New methods need to be developed to enrich for the secretory proteins in the conditioned media. Each method described above has its own merits and demerits. RNA sequencing, mass spectrometry and YST strategies are all robust methodologies that can be applied to fully decipher the cell secretome. Identifying new factors or potential biomarkers for cancer can lead to development of new therapeutics. As secretory proteins are exported in extracellular space, they will be amenable to therapeutics thereby solving many disease related questions in near future.
Highlights.
Secretome studies are significant for cell-cell signaling and biomarker discovery.
We discussed different methods to characterize the secretome of various cell types.
Merits and demerits of each method are highlighted.
Acknowledgements
The work of the laboratory is supported by National Institutes of Health Grant RO1CA127231 and RO1CA161879 and Albert Einstein College of medicine.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Martoglio B, Dobberstein B. Signal sequences: more than just greasy peptides. Trends Cell Biol. 1998;8:410–415. doi: 10.1016/s0962-8924(98)01360-9. [DOI] [PubMed] [Google Scholar]
- [2].Mbeunkui F, Metge BJ, Shevde LA, Pannell LK. Identification of differentially secreted biomarkers using LC-MS/MS in isogenic cell lines representing a progression of breast cancer. J Proteome Res. 2007;6:2993–3002. doi: 10.1021/pr060629m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kim JE, Koo KH, Kim YH, Sohn J, Park YG. Identification of potential lung cancer biomarkers using an in vitro carcinogenesis model. Exp Mol Med. 2008;40:709–720. doi: 10.3858/emm.2008.40.6.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sarkar P, Randall SM, Muddiman DC, Rao BM. Targeted proteomics of the secretory pathway reveals the secretome of mouse embryonic fibroblasts and human embryonic stem cells. Mol Cell Proteomics. 2012;11:1829–1839. doi: 10.1074/mcp.M112.020503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Planque C, Kulasingam V, Smith CR, Reckamp K, Goodglick L, Diamandis EP. Identification of five candidate lung cancer biomarkers by proteomics analysis of conditioned media of four lung cancer cell lines. Mol Cell Proteomics. 2009;8:2746–2758. doi: 10.1074/mcp.M900134-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wu CC, Chen HC, Chen SJ, Liu HP, Hsieh YY, Yu CJ, Tang R, Hsieh LL, Yu JS, Chang YS. Identification of collapsin response mediator protein-2 as a potential marker of colorectal carcinoma by comparative analysis of cancer cell secretomes. Proteomics. 2008;8:316–332. doi: 10.1002/pmic.200700819. [DOI] [PubMed] [Google Scholar]
- [7].Chevallet M, Diemer H, Van Dorssealer A, Villiers C, Rabilloud T. Toward a better analysis of secreted proteins: the example of the myeloid cells secretome. Proteomics. 2007;7:1757–1770. doi: 10.1002/pmic.200601024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Xiao T, Ying W, Li L, Hu Z, Ma Y, Jiao L, Ma J, Cai Y, Lin D, Guo S, Han N, Di X, Li M, Zhang D, Su K, Yuan J, Zheng H, Gao M, He J, Shi S, Li W, Xu N, Zhang H, Liu Y, Zhang K, Gao Y, Qian X, Cheng S. An approach to studying lung cancer-related proteins in human blood. Mol Cell Proteomics. 2005;4:1480–1486. doi: 10.1074/mcp.M500055-MCP200. [DOI] [PubMed] [Google Scholar]
- [9].Pirkmajer S, Chibalin AV. Serum starvation: caveat emptor. Am J Physiol Cell Physiol. 2011;301:C272–279. doi: 10.1152/ajpcell.00091.2011. [DOI] [PubMed] [Google Scholar]
- [10].Cao D, Polyak K, Halushka MK, Nassar H, Kouprina N, Iacobuzio-Donahue C, Wu X, Sukumar S, Hicks J, De Marzo A, Argani P. Serial analysis of gene expression of lobular carcinoma in situ identifies down regulation of claudin 4 and overexpression of matrix metalloproteinase 9. Breast Cancer Res. 2008;10:R91. doi: 10.1186/bcr2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Velculescu VE, Zhang L, Vogelstein B, Kinzler KW. Serial analysis of gene expression. Science. 1995;270:484–487. doi: 10.1126/science.270.5235.484. [DOI] [PubMed] [Google Scholar]
- [12].Yamamoto M, Wakatsuki T, Hada A, Ryo A. Use of serial analysis of gene expression (SAGE) technology. J Immunol Methods. 2001;250:45–66. doi: 10.1016/s0022-1759(01)00305-2. [DOI] [PubMed] [Google Scholar]
- [13].Joly DL, Feau N, Tanguay P, Hamelin RC. Comparative analysis of secreted protein evolution using expressed sequence tags from four poplar leaf rusts (Melampsora spp.) BMC Genomics. 2010;11:422. doi: 10.1186/1471-2164-11-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Patino WD, Mian OY, Hwang PM. Serial analysis of gene expression: technical considerations and applications to cardiovascular biology. Circ Res. 2002;91:565–569. doi: 10.1161/01.res.0000036018.76903.18. [DOI] [PubMed] [Google Scholar]
- [15].Ishii M, Hashimoto S, Tsutsumi S, Wada Y, Matsushima K, Kodama T, Aburatani H. Direct comparison of GeneChip and SAGE on the quantitative accuracy in transcript profiling analysis. Genomics. 2000;68:136–143. doi: 10.1006/geno.2000.6284. [DOI] [PubMed] [Google Scholar]
- [16].Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Li YJ, Xu P, Qin X, Schmechel DE, Hulette CM, Haines JL, Pericak-Vance MA, Gilbert JR. A comparative analysis of the information content in long and short SAGE libraries. BMC Bioinformatics. 2006;7:504. doi: 10.1186/1471-2105-7-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Schulze A, Downward J. Navigating gene expression using microarrays--a technology review. Nat Cell Biol. 2001;3:E190–195. doi: 10.1038/35087138. [DOI] [PubMed] [Google Scholar]
- [19].Mutch DM, Rouault C, Keophiphath M, Lacasa D, Clement K. Using gene expression to predict the secretome of differentiating human preadipocytes. Int J Obes (Lond) 2009;33:354–363. doi: 10.1038/ijo.2009.3. [DOI] [PubMed] [Google Scholar]
- [20].Hoggard N, Cruickshank M, Moar KM, Bashir S, Mayer CD. Using gene expression to predict differences in the secretome of human omental vs. subcutaneous adipose tissue. Obesity (Silver Spring) 2012;20:1158–1167. doi: 10.1038/oby.2012.14. [DOI] [PubMed] [Google Scholar]
- [21].Dahlman I, Elsen M, Tennagels N, Korn M, Brockmann B, Sell H, Eckel J, Arner P. Functional annotation of the human fat cell secretome. Arch Physiol Biochem. 2012;118:84–91. doi: 10.3109/13813455.2012.685745. [DOI] [PubMed] [Google Scholar]
- [22].Murphy D. Gene expression studies using microarrays: principles, problems, and prospects. Adv Physiol Educ. 2002;26:256–270. doi: 10.1152/advan.00043.2002. [DOI] [PubMed] [Google Scholar]
- [23].Malone JH, Oliver B. Microarrays, deep sequencing and the true measure of the transcriptome. BMC Biol. 2011;9:34. doi: 10.1186/1741-7007-9-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Gallotta A, Orzes E, Fassina G. Biomarkers quantification with antibody arrays in cancer early detection. Clin Lab Med. 2012;32:33–45. doi: 10.1016/j.cll.2011.11.001. [DOI] [PubMed] [Google Scholar]
- [25].Zhong J, Krawczyk SA, Chaerkady R, Huang H, Goel R, Bader JS, Wong GW, Corkey BE, Pandey A. Temporal profiling of the secretome during adipogenesis in humans. J Proteome Res. 2010;9:5228–5238. doi: 10.1021/pr100521c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Liu C-H, Hwang S-M. Cytokine interactions in mesenchymal stem cells from cord blood. Cytokine. 2005;32:270–279. doi: 10.1016/j.cyto.2005.11.003. [DOI] [PubMed] [Google Scholar]
- [27].LaFramboise WA, Scalise D, Stoodley P, Graner SR, Guthrie RD, Magovern JA, Becich MJ. Cardiac fibroblasts influence cardiomyocyte phenotype in vitro. Am J Physiol Cell Physiol. 2007;292:C1799–1808. doi: 10.1152/ajpcell.00166.2006. [DOI] [PubMed] [Google Scholar]
- [28].Hogg PJ. Disulfide bonds as switches for protein function. Trends Biochem Sci. 2003;28:210–214. doi: 10.1016/S0968-0004(03)00057-4. [DOI] [PubMed] [Google Scholar]
- [29].Pellitteri-Hahn MC, Warren MC, Didier DN, Winkler EL, Mirza SP, Greene AS, Olivier M. Improved mass spectrometric proteomic profiling of the secretome of rat vascular endothelial cells. J Proteome Res. 2006;5:2861–2864. doi: 10.1021/pr060287k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ul-Rehman R, Rinalducci S, Zolla L, Dalessandro G, Di Sansebastiano GP. Nicotiana tabacum protoplasts secretome can evidence relations among regulatory elements of exocytosis mechanisms. Plant Signal Behav. 2011;6:1140–1145. doi: 10.4161/psb.6.8.15750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Dowell JA, Johnson JA, Li L. Identification of astrocyte secreted proteins with a combination of shotgun proteomics and bioinformatics. J Proteome Res. 2009;8:4135–4143. doi: 10.1021/pr900248y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lee MJ, Kim J, Kim MY, Bae YS, Ryu SH, Lee TG, Kim JH. Proteomic analysis of tumor necrosis factor-alpha-induced secretome of human adipose tissue-derived mesenchymal stem cells. J Proteome Res. 2010;9:1754–1762. doi: 10.1021/pr900898n. [DOI] [PubMed] [Google Scholar]
- [33].Yoon JH, Yea K, Kim J, Choi YS, Park S, Lee H, Lee CS, Suh PG, Ryu SH. Comparative proteomic analysis of the insulin-induced L6 myotube secretome. Proteomics. 2009;9:51–60. doi: 10.1002/pmic.200800187. [DOI] [PubMed] [Google Scholar]
- [34].Klose J. Protein mapping by combined isoelectric focusing and electrophoresis of mouse tissues. A novel approach to testing for induced point mutations in mammals. Humangenetik. 1975;26:231–243. doi: 10.1007/BF00281458. [DOI] [PubMed] [Google Scholar]
- [35].O’Farrell PH. High resolution two-dimensional electrophoresis of proteins. J Biol Chem. 1975;250:4007–4021. [PMC free article] [PubMed] [Google Scholar]
- [36].Rabilloud T, Chevallet M, Luche S, Lelong C. Two-dimensional gel electrophoresis in proteomics: Past, present and future. J Proteomics. 2010;73:2064–2077. doi: 10.1016/j.jprot.2010.05.016. [DOI] [PubMed] [Google Scholar]
- [37].Gajendran N, Frey JR, Lefkovits I, Kuhn L, Fountoulakis M, Krapfenbauer K, Brenner HR. Proteomic analysis of secreted muscle components: search for factors involved in neuromuscular synapse formation. Proteomics. 2002;2:1601–1615. doi: 10.1002/1615-9861(200211)2:11<1601::AID-PROT1601>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- [38].Zvonic S, Lefevre M, Kilroy G, Floyd ZE, DeLany JP, Kheterpal I, Gravois A, Dow R, White A, Wu X, Gimble JM. Secretome of primary cultures of human adipose-derived stem cells: modulation of serpins by adipogenesis. Mol Cell Proteomics. 2007;6:18–28. doi: 10.1074/mcp.M600217-MCP200. [DOI] [PubMed] [Google Scholar]
- [39].Lafon-Cazal M, Adjali O, Galeotti N, Poncet J, Jouin P, Homburger V, Bockaert J, Marin P. Proteomic analysis of astrocytic secretion in the mouse. Comparison with the cerebrospinal fluid proteome. J Biol Chem. 2003;278:24438–24448. doi: 10.1074/jbc.M211980200. [DOI] [PubMed] [Google Scholar]
- [40].Lim JW, Bodnar A. Proteome analysis of conditioned medium from mouse embryonic fibroblast feeder layers which support the growth of human embryonic stem cells. Proteomics. 2002;2:1187–1203. doi: 10.1002/1615-9861(200209)2:9<1187::AID-PROT1187>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- [41].Prowse AB, McQuade LR, Bryant KJ, Van Dyk DD, Tuch BE, Gray PP. A proteome analysis of conditioned media from human neonatal fibroblasts used in the maintenance of human embryonic stem cells. Proteomics. 2005;5:978–989. doi: 10.1002/pmic.200401087. [DOI] [PubMed] [Google Scholar]
- [42].Buhr N, Carapito C, Schaeffer C, Hovasse A, Van Dorsselaer A, Viville S. Proteome analysis of the culture environment supporting undifferentiated mouse embryonic stem and germ cell growth. Electrophoresis. 2007;28:1615–1623. doi: 10.1002/elps.200600497. [DOI] [PubMed] [Google Scholar]
- [43].Dupont A, Corseaux D, Dekeyzer O, Drobecq H, Guihot AL, Susen S, Vincentelli A, Amouyel P, Jude B, Pinet F. The proteome and secretome of human arterial smooth muscle cells. Proteomics. 2005;5:585–596. doi: 10.1002/pmic.200400965. [DOI] [PubMed] [Google Scholar]
- [44].Thouvenot E, Lafon-Cazal M, Demettre E, Jouin P, Bockaert J, Marin P. The proteomic analysis of mouse choroid plexus secretome reveals a high protein secretion capacity of choroidal epithelial cells. Proteomics. 2006;6:5941–5952. doi: 10.1002/pmic.200600096. [DOI] [PubMed] [Google Scholar]
- [45].Perera CN, Spalding HS, Mohammed SI, Camarillo IG. Identification of proteins secreted from leptin stimulated MCF-7 breast cancer cells: a dual proteomic approach. Exp Biol Med (Maywood) 2008;233:708–720. doi: 10.3181/0710-RM-281. [DOI] [PubMed] [Google Scholar]
- [46].Unlu M, Morgan ME, Minden JS. Difference gel electrophoresis: a single gel method for detecting changes in protein extracts. Electrophoresis. 1997;18:2071–2077. doi: 10.1002/elps.1150181133. [DOI] [PubMed] [Google Scholar]
- [47].Canals F, Colome N, Ferrer C, Plaza-Calonge Mdel C, Rodriguez-Manzaneque JC. Identification of substrates of the extracellular protease ADAMTS1 by DIGE proteomic analysis. Proteomics. 2006;6(Suppl 1):S28–35. doi: 10.1002/pmic.200500446. [DOI] [PubMed] [Google Scholar]
- [48].Volmer MW, Stuhler K, Zapatka M, Schoneck A, Klein-Scory S, Schmiegel W, Meyer HE, Schwarte-Waldhoff I. Differential proteome analysis of conditioned media to detect Smad4 regulated secreted biomarkers in colon cancer. Proteomics. 2005;5:2587–2601. doi: 10.1002/pmic.200401188. [DOI] [PubMed] [Google Scholar]
- [49].Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- [50].Hansen KC, Schmitt-Ulms G, Chalkley RJ, Hirsch J, Baldwin MA, Burlingame AL. Mass spectrometric analysis of protein mixtures at low levels using cleavable 13C-isotope-coded affinity tag and multidimensional chromatography. Mol Cell Proteomics. 2003;2:299–314. doi: 10.1074/mcp.M300021-MCP200. [DOI] [PubMed] [Google Scholar]
- [51].Zhou H, Xiao Y, Li R, Hong S, Li S, Wang L, Zeng R, Liao K. Quantitative analysis of secretome from adipocytes regulated by insulin. Acta Biochim Biophys Sin (Shanghai) 2009;41:910–921. doi: 10.1093/abbs/gmp085. [DOI] [PubMed] [Google Scholar]
- [52].Khwaja FW, Svoboda P, Reed M, Pohl J, Pyrzynska B, Van Meir EG. Proteomic identification of the wt-p53-regulated tumor cell secretome. Oncogene. 2006;25:7650–7661. doi: 10.1038/sj.onc.1209969. [DOI] [PubMed] [Google Scholar]
- [53].Wiese S, Reidegeld KA, Meyer HE, Warscheid B. Protein labeling by iTRAQ: a new tool for quantitative mass spectrometry in proteome research. Proteomics. 2007;7:340–350. doi: 10.1002/pmic.200600422. [DOI] [PubMed] [Google Scholar]
- [54].Lietzen N, Ohman T, Rintahaka J, Julkunen I, Aittokallio T, Matikainen S, Nyman TA. Quantitative subcellular proteome and secretome profiling of influenza A virus-infected human primary macrophages. PLoS Pathog. 2011;7:e1001340. doi: 10.1371/journal.ppat.1001340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- [56].Evans FF, Raftery MJ, Egan S, Kjelleberg S. Profiling the secretome of the marine bacterium Pseudoalteromonas tunicata using amine-specific isobaric tagging (iTRAQ) J Proteome Res. 2007;6:967–975. doi: 10.1021/pr060416x. [DOI] [PubMed] [Google Scholar]
- [57].Mann M. Functional and quantitative proteomics using SILAC. Nat Rev Mol Cell Biol. 2006;7:952–958. doi: 10.1038/nrm2067. [DOI] [PubMed] [Google Scholar]
- [58].Gronborg M, Kristiansen TZ, Iwahori A, Chang R, Reddy R, Sato N, Molina H, Jensen ON, Hruban RH, Goggins MG, Maitra A, Pandey A. Biomarker discovery from pancreatic cancer secretome using a differential proteomic approach. Mol Cell Proteomics. 2006;5:157–171. doi: 10.1074/mcp.M500178-MCP200. [DOI] [PubMed] [Google Scholar]
- [59].Kashyap MK, Harsha HC, Renuse S, Pawar H, Sahasrabuddhe NA, Kim MS, Marimuthu A, Keerthikumar S, Muthusamy B, Kandasamy K, Subbannayya Y, Prasad TS, Mahmood R, Chaerkady R, Meltzer SJ, Kumar RV, Rustgi AK, Pandey A. SILAC-based quantitative proteomic approach to identify potential biomarkers from the esophageal squamous cell carcinoma secretome. Cancer Biol Ther. 2010;10:796–810. doi: 10.4161/cbt.10.8.12914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Sakaguchi M, Shingo T, Shimazaki T, Okano HJ, Shiwa M, Ishibashi S, Oguro H, Ninomiya M, Kadoya T, Horie H, Shibuya A, Mizusawa H, Poirier F, Nakauchi H, Sawamoto K, Okano H. A carbohydrate-binding protein, Galectin-1, promotes proliferation of adult neural stem cells. Proc Natl Acad Sci U S A. 2006;103:7112–7117. doi: 10.1073/pnas.0508793103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Andrei C, Dazzi C, Lotti L, Torrisi MR, Chimini G, Rubartelli A. The secretory route of the leaderless protein interleukin 1beta involves exocytosis of endolysosome-related vesicles. Mol Biol Cell. 1999;10:1463–1475. doi: 10.1091/mbc.10.5.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Peng Z, Cheng Y, Tan BC, Kang L, Tian Z, Zhu Y, Zhang W, Liang Y, Hu X, Tan X, Guo J, Dong Z, Bao L, Wang J. Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat Biotechnol. 2012;30:253–260. doi: 10.1038/nbt.2122. [DOI] [PubMed] [Google Scholar]
- [63].Pernitzsch SR, Sharma CM. Transcriptome Complexity and Riboregulation in the Human Pathogen Helicobacter pylori. Front Cell Infect Microbiol. 2012;2:14. doi: 10.3389/fcimb.2012.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Cui JY, Gunewardena SS, Yoo B, Liu J, Renaud HJ, Lu H, Zhong XB, Klaassen CD. RNA-Seq reveals different mRNA abundance of transporters and their alternative transcript isoforms during liver development. Toxicol Sci. 2012;127:592–608. doi: 10.1093/toxsci/kfs107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Yamashita R, Sathira NP, Kanai A, Tanimoto K, Arauchi T, Tanaka Y, Hashimoto S, Sugano S, Nakai K, Suzuki Y. Genome-wide characterization of transcriptional start sites in humans by integrative transcriptome analysis. Genome Res. 2011;21:775–789. doi: 10.1101/gr.110254.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Huang Q, Lin B, Liu H, Ma X, Mo F, Yu W, Li L, Li H, Tian T, Wu D, Shen F, Xing J, Chen ZN. RNA-Seq analyses generate comprehensive transcriptomic landscape and reveal complex transcript patterns in hepatocellular carcinoma. PLoS One. 2011;6:e26168. doi: 10.1371/journal.pone.0026168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Hackett NR, Butler MW, Shaykhiev R, Salit J, Omberg L, Rodriguez-Flores JL, Mezey JG, Strulovici-Barel Y, Wang G, Didon L, Crystal RG. RNA-Seq quantification of the human small airway epithelium transcriptome. BMC Genomics. 2012;13:82. doi: 10.1186/1471-2164-13-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Kim KH, Moon M, Yu SB, Mook-Jung I, Kim JI. RNA-Seq analysis of frontal cortex and cerebellum from 5XFAD mice at early stage of disease pathology. J Alzheimers Dis. 2012;29:793–808. doi: 10.3233/JAD-2012-111793. [DOI] [PubMed] [Google Scholar]
- [69].Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- [70].Baginsky S, Hennig L, Zimmermann P, Gruissem W. Gene expression analysis, proteomics, and network discovery. Plant Physiol. 2010;152:402–410. doi: 10.1104/pp.109.150433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Costa V, Angelini C, De Feis I, Ciccodicola A. Uncovering the complexity of transcriptomes with RNA-Seq. J Biomed Biotechnol. 2010;2010:853916. doi: 10.1155/2010/853916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Frost RJ, Engelhardt S. A secretion trap screen in yeast identifies protease inhibitor 16 as a novel antihypertrophic protein secreted from the heart. Circulation. 2007;116:1768–1775. doi: 10.1161/CIRCULATIONAHA.107.696468. [DOI] [PubMed] [Google Scholar]
- [73].Goo JH, Park AR, Park WJ, Park OK. Selection of Arabidopsis genes encoding secreted and plasma membrane proteins. Plant Mol Biol. 1999;41:415–423. doi: 10.1023/a:1006395724405. [DOI] [PubMed] [Google Scholar]
- [74].Lee SJ, Kim BD, Rose JK. Identification of eukaryotic secreted and cell surface proteins using the yeast secretion trap screen. Nat Protoc. 2006;1:2439–2447. doi: 10.1038/nprot.2006.373. [DOI] [PubMed] [Google Scholar]
- [75].Galliciotti G, Schneider H, Wyder L, Vitaliti A, Wittmer M, Ajmo M, Klemenz R. Signal-sequence trap in mammalian and yeast cells: a comparison. J Membr Biol. 2001;183:175–182. doi: 10.1007/s00232-001-0065-6. [DOI] [PubMed] [Google Scholar]
- [76].Tashiro K, Tada H, Heilker R, Shirozu M, Nakano T, Honjo T. Signal sequence trap: a cloning strategy for secreted proteins and type I membrane proteins. Science. 1993;261:600–603. doi: 10.1126/science.8342023. [DOI] [PubMed] [Google Scholar]
- [77].Becker F, Schnorr K, Wilting R, Tolstrup N, Bendtsen JD, Olsen PB. Development of in vitro transposon assisted signal sequence trapping and its use in screening Bacillus halodurans C125 and Sulfolobus solfataricus P2 gene libraries. J Microbiol Methods. 2004;57:123–133. doi: 10.1016/j.mimet.2003.12.002. [DOI] [PubMed] [Google Scholar]
- [78].Ray P, Rialon-Guevara KL, Veras E, Sullenger BA, White RR. Comparing human pancreatic cell secretomes by in vitro aptamer selection identifies cyclophilin B as a candidate pancreatic cancer biomarker. J Clin Invest. 2012;122:1734–1741. doi: 10.1172/JCI62385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Antelmann H, Tjalsma H, Voigt B, Ohlmeier S, Bron S, van Dijl JM, Hecker M. A proteomic view on genome-based signal peptide predictions. Genome Res. 2001;11:1484–1502. doi: 10.1101/gr.182801. [DOI] [PubMed] [Google Scholar]
- [80].Grimmond SM, Miranda KC, Yuan Z, Davis MJ, Hume DA, Yagi K, Tominaga N, Bono H, Hayashizaki Y, Okazaki Y, Teasdale RD. The mouse secretome: functional classification of the proteins secreted into the extracellular environment. Genome Res. 2003;13:1350–1359. doi: 10.1101/gr.983703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Bendtsen JD, Jensen LJ, Blom N, Von Heijne G, Brunak S. Feature-based prediction of non-classical and leaderless protein secretion. Protein Eng Des Sel. 2004;17:349–356. doi: 10.1093/protein/gzh037. [DOI] [PubMed] [Google Scholar]