

SUMMARY
In bacteria, two signal sequence dependent secretion pathways translocate proteins across the cytoplasmic membrane. While the mechanism of the ubiquitous general secretory pathway (SEC) is becoming well understood, that of the twin-arginine translocation pathway (TAT), responsible for translocation of folded proteins across the bilayer, is more mysterious. TatC, the largest and most conserved of three integral membrane components, provides the initial binding site of the signal sequence prior to pore assembly. Here, we present two crystal structures of TatC from the thermophilic bacteria Aquifex aeolicus at 4.0Å and 6.8Å resolution. The novel membrane architecture of TatC includes a glove-shaped structure with a lipid-exposed pocket predicted by molecular dynamics to distort the membrane. Correlating the biochemical literature to these results suggests that the signal sequence binds in this pocket leading to structural changes that facilitate higher order assemblies.
Keywords: Protein secretion, membrane protein, X-ray crystallography, molecular dynamics, TAT pathway
INTRODUCTION
Prokaryotic organisms secrete protein via two main pathways: the general secretory pathway (SEC), where the Sec translocon facilitates passage of unfolded protein across the bilayer, and the twin arginine translocation (TAT) pathway, which is involved in targeting and translocation of fully folded proteins across the inner membrane of bacteria (for recent reviews: SEC pathway (Park and Rapoport, 2012) and TAT pathway (Frobel et al., 2012b; Palmer and Berks, 2012)). TAT pathway substrates are characterized by a critical pair of arginines in a consensus sequence (Berks, 1996; Chaddock et al., 1995). The components of the pathway were identified in the thylakoid membrane of the chloroplast (Settles et al., 1997) and in E. coli (Sargent et al., 1998; Weiner et al., 1998). While the TAT system is broadly conserved, it is not essential for viability under standard lab conditions in bacteria (Bogsch et al., 1998; Jongbloed et al., 2004).
In TAT pathway containing organisms, approximately 10% of the total secretome are TAT substrates. The most significant exceptions are halophilic archaea, in which the majority of secreted proteins appear to utilize the TAT pathway (Bolhuis, 2002; Rose et al., 2002; Thomas and Bolhuis, 2006) and the pathway is required for viability (Dilks et al., 2005). Most TAT substrates are complex, containing co-factors and/or oligomeric assemblies, and must be correctly folded and assembled in the cytoplasm prior to translocation, necessitating a large pore that can translocate a diversity of folded proteins. Example secretion substrates include respiratory redox enzymes, bacterial virulence factors (Kassem et al., 2011; van der Ploeg et al., 2011), lipoproteins (Shruthi et al., 2010) and proteins involved in maintaining cell wall integrity and cell motility (Stanley et al., 2001). Additionally, some inner membrane proteins have been found that can be inserted via this pathway (Hatzixanthis et al., 2003; Heikkila et al., 2001; Ochsner et al., 2002; Schaerlaekens et al., 2001).
The TAT pathway, as described in E. coli and chloroplasts, minimally requires three membrane proteins: TatA, TatB and TatC (Bogsch et al., 1998; Sargent et al., 1998; Weiner et al., 1998), which can all be purified at variable ratios in a complex (Bolhuis et al., 2001). TatA and TatB contain a single N-terminal transmembrane helix (TM) while TatC contains six TMs (Behrendt et al., 2004; Gouffi et al., 2002). TatC has the highest conservation, performing the crucial role of recognition and initial binding of the signal sequence at the N-terminal end of the pre-protein substrate (Allen et al., 2002; Jongbloed et al., 2000). TatB and TatC form a stable complex predicted to contain up to eight copies of each protein in a size range of 360–700 kDa (Bolhuis et al., 2001; de Leeuw et al., 2002; Kneuper et al., 2012; Lee et al., 2006; Mcdevitt et al., 2005; Tarry et al., 2009) in a possible 1:1 stoichiometric ratio (Alami et al., 2003; Cline, 2001). This complex binds signal sequences that contain the TAT motif (S/T-R-R-x-F-L-K) and transfers the substrate to a TatA complex (Alami et al., 2003). TatA is predicted to serve as the protein-conducting translocation channel forming a modular homo-oligomeric ring-like pore for secretion of various sized substrates (Gohlke et al., 2005; Sargent et al., 2001). Translocation is powered by the proton motive force (PMF) and can be blocked PMF inhibitors (Bageshwar and Musser, 2007; Gérard and Cline, 2007; Kwan et al., 2008; Panahandeh et al., 2008). TatA and TatB perform distinct functions in E. coli (Sargent et al., 1998; Sargent et al., 1999); yet, sequence conservation suggests they are derived from a common ancestor, as some TAT containing bacteria do not appear to contain a TatB, with TatA taking on a dual role (Dilks et al., 2003; Wu et al., 2000; Yen et al., 2002).
Extensive genetic and biochemical studies have been performed to understand the interaction of TatC with the signal sequence and TatA and TatB; however, the aggregate of the data has led to a variety of very different models. The central role of TatC and its high conservation suggests that a structure of this protein will provide a wealth of information towards understanding the TAT pathway.
Here we present a structure of TatC from the thermophile Aquifex aeolicus in two different crystal forms at resolutions of 4.0Å and 6.8Å. The structure reveals a membrane protein that is shaped like a baseball glove with the concave pocket exposed to the bilayer. We used molecular dynamics to look at this unusual architecture in a bilayer demonstrating the flexible parts of the protein and a water funnel that lines the pocket. We use computational docking to suggest possible dimerization interfaces. Finally, we correlate these results with existing biochemical data to develop a model for signal sequence binding where the signal sequence docks into the groove. This provocative solution to substrate recognition would allow for subsequent conformational changes required for further complex assembly.
RESULTS AND DISCUSSION
Crystallization and structure determination of Aquifex aeolicus TatC
Genes for TatC from 26 eubacteria, archaea and a mitochondrion (Malawimonas jakobiformis) were either codon optimized for E. coli and synthesized based on their protein sequence or amplified from genomic DNA, and then cloned into an inducible expression vector. To prevent co-purification of native E. coli TAT components, the operon for TatABCD followed by the gene for TatE, a TatA paralog, were deleted from the strain BL21(DE3)GOLD to generate the new strain CJMS2. Using this strain, each clone was tested for expression in a variety of conditions. Ultimately, TatC from the hyperthermophile Aquifex aeolicus (AaTatC) proved to give the best expression and was used for structural studies. This protein was well behaved by gel filtration in a variety of detergents including dodecyl-maltoside (DDM) and di-heptanoyl phosphatidylcholine (DHPC).
The wild-type AaTatC crystallized in DHPC resulted in diffraction with reflections visible at ~10Å resolution after some optimization. To improve the diffraction, mutants were generated to reduce surface entropy with one combination (K40A, E41A with a C-terminal truncation) resulting in well-formed crystals that diffracted to 7.5Å. By visual inspection, it became clear that two different kinds of crystal morphology were growing in the same drop. The less frequent of the two crystal forms diffracted better and was used for seeding into clear drops, resulting in only this form appearing. A single crystal of this form that diffracted to high resolution was used to collect a native dataset at 4.0Å resolution in the space group P4122 with cell dimension a=b=110.43Å c=107.42Å referred to here as AaDHPC.
An alternative approach to obtaining crystals was to generate a lysozyme-AaTatC fusion similar to that used for other membrane protein crystallization (Rosenbaum et al., 2007). We added a codon optimized T4 lysozyme at the C-terminus of AaTatC. This fusion was well behaved by gel filtration and crystallized in DDM. Refinement of these conditions resulted in a 6.8Å data set in the space group I4122 with cell dimension a=b=142.015Å c=251.748Å referred to here as AaDDM.
Although many approaches were attempted, we were unable to obtain phases for either crystal form by isomorphous replacement or related methods; therefore, the recently deposited TatC structure from Berks, Lea and co-workers from Aquifex aeolicus was used as a molecular replacement (MR) search model (PDBID 4B4A referred to here as AaMNG as the protein was crystallized in a maltose-neopentyl glycol detergent). For the AaDHPC crystals, a single molecule was found in the asymmetric unit, while two were identified in the AaDDM asymmetric unit (Fig. S1C & D). For the latter, we were unable to identify a lysozyme in the resulting maps.
For the AaDHPC crystals, refinement proceeded through normal processes. In the final model, residues 5 to 232 had continuous density except for the residues 133 to 140 in the second periplasmic loop, which were disordered and given occupancies of zero. The final AaDHPC model gave an R/R-free of 28.8/32.3% while the lower resolution AaDDM gave a final R/R-free of 34.4/42.9%. Complete crystallographic statistics are found in Table 1. Unless noted, the figures and description of AaTatC will use the higher resolution AaDHPC crystal structure and not the lysozyme fusion.
Table 1.
Crystallographic statistics
| AaDHPC | AaDDM | |
|---|---|---|
| Wavelength (Å) | 1.08 | 1.08 |
| Resolution range (Å) | 29.9–4.0 (4.5–4.0)a | 30.0–6.80 (7.0–6.8) |
| Space group | P4122 | I4122 |
| Cell dimensions: a=b, c (Å) | 109.8, 107.0 | 142.0, 251.8 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 |
| Unique reflections | 5906 (1627) | 2281 (220) |
| Completeness (%) | 99.8 (100.0) | 95.9 (96.9) |
| Redundancy | 5.8 (6.0) | 6.8 (7) |
| Rmergeb | 0.127 (0.918) | 0.048 (0.79) |
| Mean I/σ(I) | 6.6 (1.7) | 22.45 (2.70) |
|
| ||
| Refinement | ||
| Reflections: work/free | 5885/567 | 2164/98 |
| Rwork/Rfreec | 0.288/0.323 | 0.344/0.429 |
| No. protein atoms | 1819 | 3608 |
| Protein B-factors (Å2) | 247.76 | 474.5 |
| RMSD bond lengths (Å) | 0.003 | 0.004 |
| RMSD bond angles (°) | 0.882 | 0.95 |
| Ramachandran outliers | 0.4% | 4% |
Values in parentheses are for the highest resolution shell
Rmerge = Σhkl Σi |Ii(hkl) − 〈I(hkl)〉|/ Σhkl ΣiIi(hkl), where Ii(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations i of reflection hkl.
R = Σ (| Fobs − Fcalc |)/Σ| Fobs |, where Fobs and Fcalc are observed and calculated structure factors amplitudes, respectively
The structure of TatC
The majority of TatC homologs are expected to have six membrane spanning helices (Behrendt et al., 2004; Ki et al., 2004; Punginelli et al., 2007) consistent with the structure, where all the TMs are visible (Fig. 1A). TatC spans the membrane with the longest dimension, ~55Å, essentially the same as the width of the bilayer resulting in very little of the protein exposed outside the membrane. Viewed from the cytoplasm (Fig. 1B), the longest dimensions result in a length and width of approximately 35 by 20Å. For reference, residues will be noted using E. coli numbering signified by Ec followed by the equivalent A. aeolicus number in italics unless noted (Fig. 1H for reference).
Figure 1. Overview of the structure of AaTatC.

(A) Cartoon diagram of AaDHPC viewed from in the plane of the membrane color-ramped from N- to C-terminus (blue to red) from the front and rotated 90°. TMs and loops are numbered (1–6) according to the text. The amphipathic helix in Per1 is labeled as 1A. The bilayer is represented in the back as a rectangle with the most hydrophobic portions in gray and the hydrophilic head groups in blue. Dimensions are shown for the whole protein and the narrowest point in the glove.
(B) As in A viewed from the cytoplasm.
(C–E) Front molecular surface representations: (C) colored as in A; (D) electrostatic surface potential from negative −6 kB/e (red) to positive +6 kB/e (blue); (E) percent conservation based on an alignment of TatC Pfam seed sequences from 10 (white) to 90% (red). See also Figure S1.
(F) Cytoplasmic views of E.
(G) Sequence alignment of TatC homologs. The species are E. coli, A. aeolicus, Campylobacter jejuni, Thermus thermophilus and Staphylococcus aureus. Alignment and residue coloring are based on ClustalX output (Larkin et al., 2007). Secondary structure features are highlighted above the sequence with helices (rectangles) colored as in A. Sequence numbering is below the alignment for E. coli and A. aeolicus. Residues that are disordered in the crystal structure have red numbering. Residues mutated in the AaDHPC are underlined in black. Residues highlighted in Fig. 3 are shown above the sequence with boxes colored according to the type of mutation (see labeling below where Xlink indicates a signal sequence cross-link and SS Supp are signal sequence suppressor mutants). X indicates residues involved in crosslinks while other letters indicate specific mutations. The dashed line marks a salt bridge between K190 and E221.
Starting with the N-terminus in the cytoplasm, TM1 is roughly perpendicular to the membrane ending in a sharp turn that continues as an amphipathic helix (H1A) curling under TM2 to form a large part of the periplasmic face of TatC (Fig. 1A–C). The rest of the first periplasmic loop (Per1) continues as a structured loop. This is followed by TM2 that starts angled relative to TM1 until a conserved proline (Ec85/78) generates a kink and the remaining cytoplasmic half of the helix forms a parallel interface with TM1. TM3 is connected to TM2 by a short loop (Cyt1) then forms a long steeply angled helix that makes contacts across the back to TM2, TM4 and TM6. This is reminiscent of the TM of SecE in the SecY translocon suggesting a role in stabilization of the overall complex (van den Berg et al., 2004). The following periplasmic loop (Per2) runs below TM5 and TM6 and is partially disordered in the structure. TM4 has similar features and is parallel to TM2 including a highly conserved proline kink (Ec172/167). In addition, a highly conserved glycine (Ec166/161) forms a tight interface with a reciprocal conserved glycine (Ec121/114) in TM3. This is connected via a short loop (Cyt2) to TM5 that begins with a steep angle out from the core of the protein then sharply kinks making contacts to TM4. TM5 ends with a highly conserved proline turn (Per3) that lies just within the hydrophobic core of the bilayer. TM6 has a fairly shallow angle on the backside of the protein making contacts to TM3, TM4 and TM5 with the C-terminus of the protein ending in the cytoplasm.
Surface properties of TatC
Overall, the most noticeable feature of TatC is that the kinked helices are arranged in a manner where they are perpendicular to the membrane in the cytoplasmic leaflet then kinked into an angle in the periplasmic leaflet. This results in a membrane exposed concave surface reminiscent of a baseball glove (Fig. 1A–C). TMs 1, 2, 4 and 5 line the pocket with the base made by the angled parts of TM2 and TM4. H1A, TM3 and TM6 line the back of the glove forming contacts to multiple helices. The bulk of this pocket lies deep within the lipid bilayer.
For many membrane proteins, mapping the hydrophobic parts of the surface helps to estimate the orientation of the protein in the membrane. As expected, much of the surface buried in the membrane is hydrophobic (SFig. 1B); however, there are a number of polar groups within the hydrophobic part of the membrane including in the pocket. This is further illustrated when the electrostatic surface potential is mapped to the exposed surface. Here, we solved the Poisson-Boltzmann equation using the dielectric value of water (Fig. 1D and SFig. 1C). This highlights a number of charged patches that would be buried in the membrane including a negative charge deep within the pocket.
Another useful visualization is to look at the conservation of residues exposed on the surface. Pfam is a curated database that clusters proteins into family groups based on homology across all of the available genomes (Punta et al., 2012). For TatC, Pfam PF00902, 2560 TatC homologs have been identified across bacteria, archaea and eukaryotes (chloroplast and a few mitochondria) with a generated seed group (144 homologs) representative of sequence diversity. Taking the seed sequences, along with the sequence for AaTatC, an alignment was performed using ClustalX (Larkin et al., 2007) that was then mapped onto the surface of TatC (Fig. 1E, F and SFig. 1D). Despite many of the most conserved residues being structural, such as helix breaking prolines, a few hot spots stand out. The highest concentration of conserved residues is found in a grouping around the N-terminus and Cyt1 mostly on the cytoplasmic and groove surfaces. Per3 is buried in the membrane and highly conserved, as is a groove on the back between TM3, 4 and 6. Adjacent to this groove is EcE170/165 that is buried in the pocket providing the negative charge there (Fig. 1D). This residue is conserved in over 80% of the Pfam seed group (which includes very divergent sequences) as a glutamate or glutamine.
The structure of TatC in a lipid bilayer
Of the three crystal forms, AaMNG and the two described here, there is close agreement in overall structure despite differences in purification methods, detergent, pH and space groups. Compared to AaDHPC, AaMNG has an RMSD of 0.623Å and AaDDM has an RMSD of 0.963Å (SFig. 2A). The only significant region of difference is in Per3 and an additional turn of helix at the C-terminus suggesting TatC has a generally rigid structure that would be maintained in the membrane. To test this, we used a MD simulation to study the protein in a lipid bilayer.
To initialize the MD, TatC was placed in a model bilayer with explicit solvent in several steps that included multiple energy minimizations. The final full atomic model was used to run a 50ns MD simulation (Fig. 2A). Snapshots of the simulation were taken at 0.5ns, with the representative frame occurring at 35.5ns (i.e. Frame 71, having the lowest protein backbone RMSD to all other frames). The most dynamic regions of the protein occur at the N-terminus and Cyt1, Per1, Per2 and the cytoplasmic end of TM5 (Fig. 2B). The periplasmic loops periodically displayed additional secondary structure elements including a two-strand β-sheet between Per1 and Per2 and a short turn of α-helix in Per2 which agree with predictions based on primary sequence (Kneuper et al., 2012).
Figure 2. Molecular dynamics simulation of TatC in the lipid bilayer.

Panels A – D are oriented from the front and colored as in Fig. 1A.
(A) A sample frame from the molecular dynamics simulation. The full representative frame 71 at 35.5ns with the lowest protein RMSD to all other frames. Shown are protein as a ribbons cartoon similar to Fig. 1A with waters (6856 total) and lipids as sticks (107).
(B) A coil diagram of AaDHPC color-ramped from N- to C-terminus. The thickness of the coil is based on the average RMSD at each position across the entire MD simulation. Thicker regions have the most fluctuation.
(C) Comparison of AaDHPC (‘xtal’ lightened relative to Fig. 1A) aligned by TM3 and TM4 to Frame 71 of the MD simulation (colored as in Fig. 1A). The rotation of TM5 is highlighted by an arrow.
(D) The crystal contacts in AaTatC. The central protein is colored as in Fig. 1A. Symmetry related proteins are similarly colored except lighter. The orientations of TM1 and TM5 are highlighted by arrows pointing from the N- to C-terminus. See also Figure S2.
(E) Side view of the MD simulation frame 71 highlighting the waters. Hydrogens are removed for clarity. Protein is shown as a Cα ribbon, waters as spheres and lipids as lines. The orange arrow indicates AaE165 and the penetration of water into the bilayer. The red arrow indicates the hydration of Per3. The blue arrow highlights the lipid filled hole at the back of the protein.
The most remarkable change in the protein structure during the course of the simulation is the relaxation of TM5 which became straighter and turned in toward the pocket narrowing this groove (Fig. 2C). The sharpness of the TM5 kink in all the crystal structures stood out relative to other TM kinks throughout the molecule. For AaTatC, while the sequence at the kink does not have an expected proline, there is a proline in the equivalent E. coli position (Fig. 1G). From the Pfam seed alignment, we found a total of eight sequences with prolines near the kink in TM5, which would imply that flexibility in TM5 is evolutionarily conserved (Meruelo et al., 2011). The sharp kink is structurally inconsistent with the more typical, shallower proline kinks in nearly every helix of TatC. Looking at the crystal packing for all three forms, TM5 is involved in nearly identical contacts that involve interactions between anti-parallel TM5s (Fig. 2D & Fig. S2B–E). TM5 starts in the cytoplasm and enters the hydrophobic part of the bilayer at the kink (Fig. 1A). This hydrophobic section (Aa190–202) makes the contacts across the symmetry interface. AaY190 and AaK189 form a stabilizing hydrogen bond network to the C-terminus of the symmetry related helix (Fig. S1B) that suggests the steepness of the kink in TM5 is exaggerated by crystallographic contacts. After the movement of the helix during the MD simulation, the helix adopts a conformation more consistent with the angle of kinks in other TMs (Fig. 2C). These different conformational states point towards an inherently flexible helix.
The interaction of TatC with water and lipid showed unexpected results in the simulation. Most of the cavities on the membrane-exposed surface of TatC remain filled with lipid during the course of the simulation. An example of this is a hollow formed between 1A and TM3 (Fig. 2E, blue arrow). This is mostly true for the pocket where the lipid tails reach deep inside; however, within the pocket a funnel of water reaches into the membrane hydrating the polar AaE165 (orange arrow). This is consistent with the hydration of polar and charged residues in lipid bilayers in other model systems (MacCallum et al., 2008). The high conservation of the polar nature of this residue would suggest the deformation of the membrane due to hydration likely plays a role in TatC function; however, while mutations at this position affect insertion in vivo (Buchanan et al., 2002), this residue does not appear to be critical for signal sequence binding in vitro (Holzapfel et al., 2007). Water also intrudes into the hydrophobic part of the membrane around the polar loop between TM5 and TM6 (red arrow). This loop contains conserved prolines and an aspartate that likely play a role in flexibility.
Analysis of individual residues based on biochemistry and mutagenesis
In recent years, extensive studies of TatC have been performed that include site specific mutants and random mutagenesis to analyze the residues of TatC that are involved in binding to signal sequence and/or other TAT components and the most prominent of these are mapped onto AaTatC (Fig. 3). While there is only a consensus sequence for the recognition motif (S/TRRxFLK), the requirement for the arginine pair suggests a specific binding site. Initial mutagenesis identified the N-terminus and Cyt1 as being critical to signal sequence binding (Holzapfel et al., 2007). This is supported by work that used site-specific cross-linking to map the interaction with the signal peptide (Zoufaly et al., 2012). The strongest of these include crosslinks to the flexible N-terminus in residues that are not conserved (EcV3-D5/n/a, & I10/T4) and are disordered in equivalent positions in the crystal structure, suggesting that they are in the vicinity of the recognition site but are not required for specificity. One residue (EcL9/3) was also identified in suppressor screens for mutants that allow targeting of a modified signal sequence (Kreutzenbeck et al., 2007; Lausberg et al., 2012) that is disordered in the crystal structure. Other cross-linked positions (EcE15/9, L16/10, Y100/93, K101/S94 & E187/182) are generally highly conserved and map to the N-terminus of TM1 and Cyt1 with the exception being EcE187 that maps to TM5 (Fig. 3A & B). This is all consistent with the likely primary signal sequence-binding site localized between the N-terminus and Cyt1, which also has the highest concentration of conserved residues (Fig. 1E & F). Moreover, mutations that suppress defective signal sequences (EcL9F/3, K18E or M/Y12, N22I/I16, L99P/92, F94S/87 and W92G+P97S/85+90) all map to this region (Fig. 3B) (Kreutzenbeck et al., 2007; Lausberg et al., 2012; Strauch and Georgiou, 2007). Single alanine mutations in Cyt1 identified three residues (F94/87, Y100/93 & E103/96) that were defective in translocation (Holzapfel et al., 2007), presumably due to effects on the interaction with the signal sequence.
Figure 3. Residues correlated to TatC function based on mutagenesis or crosslinking.

(A) Ribbons diagram of AaDHPC colored similar to Fig. 1A with discussed mutants shown as sticks. Residues are colored based on identified function with A. aeolicus numbering: signal sequence crosslink (orange), signal sequence suppression mutants (red), TatC to TatC crosslink (blue), TatC to TatA or TatB crosslink (cyan) and mutants that inactivate TatC (green) (Barrett et al., 2005; Buchanan et al., 2002; Holzapfel et al., 2007; Kneuper et al., 2012; Kreutzenbeck et al., 2007; Lausberg et al., 2012; Punginelli et al., 2007; Strauch and Georgiou, 2007; Zoufaly et al., 2012). Dashed boxes indicate regions highlighted in C and D.
(B) View from the cytoplasm as in A. Labeling includes A. aeolicus numbers with E. coli numbering and mutations in parenthesis. C and D have similar schemes.
(C) Per3 in sticks highlighting the conserved hydrogen bond network stabilized by the conserved D205.
(D) Sticks view of the hydrogen bond stabilizing the kinks in TM3 and 1A facilitated by P42 and Y119.
TatC forms part of a larger multimeric TatB/TatC complex. Binding of the signal sequence leads to assembly of the translocation pore presumed to be predominantly formed by TatA. Cross-linking studies have identified a number of residues that are likely components of these interfaces and some of these results are mapped onto the AaTatC structure (Fig. 3A). Based on the surface localization, many of these crosslinks are found on the same face as the pocket (Fig. 3B). For TatC/TatC interactions, strong crosslinks have been found in TM1 (EcA26/A20 & Y36/Y30), Per1 (EcD63/S56) (Zoufaly et al., 2012) and Per2 (EcG144/Q137 and S148/T141) (Punginelli et al., 2007). The distribution of these suggests multiple contact points, perhaps involving multiple TatCs. TatB cross-links have been identified at the N-terminus (EcV3/n/a, I10/T4 & I14/R8), Per2 (EcD150/S145) and Per3 (EcM205/A200) (Kneuper et al., 2012; Zoufaly et al., 2012). Again, the separation of these sites would suggest different interaction surfaces. TatA cross-links have also been found in Per2 at the same position (EcD150/145) and the additional Per3 position (EcD211/205). The latter is a highly conserved aspartate (Fig. 1F & H) that forms a hydrogen bond network in the tight turn capping the N-terminus of TM6, which lies within the bilayer (Fig. 3D). Mutation of EcQ215/209 leads to loss of function suggesting that, in general, Per3 plays a significant role in TatB binding (Buchanan et al., 2002).
A number of positions in TatC were identified as important based on general translocation defects. Of those that are unlikely to result in folding or membrane insertion defects (e.g introduction of a proline), the majority occur in the same regions that were identified in the cross-linking or suppressor studies, TM1, Cyt1 and Cyt2 (EcP48L or A/42, I60N/53, D63V/S56, F68S/L61, T70R/I63, F94S or A/86, L99P or Q/92, Y100A/93, E103A/96, Y126A/119, D150G or R/145, M205R/A200, Q215R/209) (Fig. 3A) (Holzapfel et al., 2007; Kneuper et al., 2012). This further confirms the likely direct role of these areas in TatC function. Of the remaining mutants, two positions were identified in several screens (P48/42 & Y126/119). Looking at these highly conserved residues (Fig. 1H), EcY126/119 is part of TM3 and is positioned at the kink created by EcP131/124 (Fig. 3D). The tyrosine hydroxyl forms a hydrogen bond to a backbone carbonyl exposed by the kink in 1A caused by EcP48/42, presumably stabilizing this entire region. Mutation of either residue would disrupt this leading to large effects throughout the structure. Interestingly, the EcP48A mutation causes defects in TatB/TatC complex assembly suggesting large-scale structural effects (Barrett and Robinson, 2005). This hydrogen bond is maintained throughout the entire 50ns of the MD simulation.
Dimerization of TatC
The oligomerization of TatC appears to play a substantial role in its function. In E. coli and chloroplasts, TatB/TatC complexes can be purified that contain multiple TatCs. Even in a mutant defective in TatB binding, TatC continues to form larger complexes (Barrett and Robinson, 2005) indicating that there are direct interactions between TatC monomers that do not depend on TatB. In the case of A. aeolicus, TatC has been expressed heterologously and the subsequent purification reveals a homogenous peak consistent with a larger than monomer complex by gel filtration. This was analyzed using multi-angle laser light scattering (MALLS) that allows for the measurement of the protein molecular weight independent of detergent micelle thereby obtaining stoichiometry (Fig. 4A). For AaTatC, a molecular weight of 57 kDa was obtained that would be consistent with a dimer of TatC. This fits the prediction that TatC exists minimally as a dimer as fusions of EcTatC are functional (Maldonado et al., 2011) and many of the archaeal homologues are natively fused dimers.
Figure 4. The AaTatC dimer.
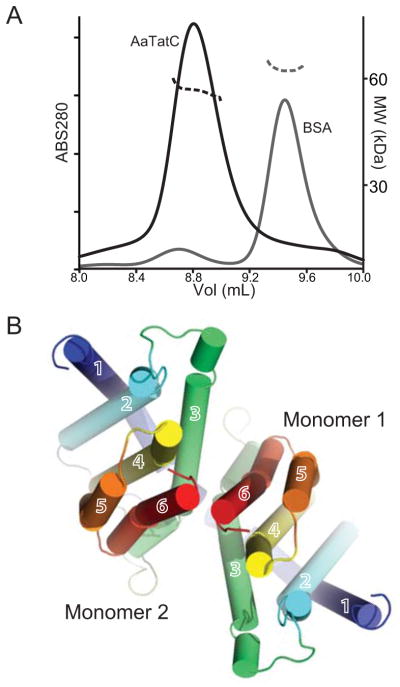
(A) Molecular weight measured by MALLS. UV traces from sizing column on left axis as thick lines. Refractive index on the right axis is shown as dashed lines. AaTatC (dimer MW 54 kDa) is in black, the control BSA (MW 66 kDa) is shown in grey.
(B) Representative dimer model viewed from the cytoplasm colored as in Fig. 1A (Model name 525 in Table S1 and Fig. S3). In this case, an MD model was used in calculating the interface. See also Figure S3.
While TM5 forms a symmetry related contact in all three crystal forms, a number of additional crystallographic contacts occur (Fig. 2D & Fig. S2C–E). There are contacts formed between symmetry related periplasmic loops and direct TM interaction. In AaDHPC and AaDDM, a contact between TM1s is stabilized by van der Waals interactions between opposing AaG26 residues (Fig. 2D, Fig. S2C & D). While these are semi-parallel, it is unlikely that this represents a dimer interface, as the angle between the symmetrical TatCs would be incompatible with the bilayer. In the AaMNG form, a contact is generated where TM1, 2 and 3 make a two-fold symmetric contact using residues in the cytoplasmic leaflet that is stabilized by a detergent molecule bound at the periplasmic leaflet interface (Fig. S2E). Although this interface is incompatible with the membrane, it is conceivable that a slight reorientation could provide a membrane bound interface. All of the contacts point to protein interaction interfaces around TM1 and TM5 consistent with the locations of TatC crosslinks to itself and other proteins (Fig. 3A).
While there are no obvious dimer interfaces, there are a number of ways to arrange TatC dimers based on the extensive crosslinks observed on all surfaces of the structure. As the biochemical evidence points to a role for a minimal dimer, plausible dimer models were generated computationally taking protein flexibility into account. Initially, two copies of either the crystal structure or each of three representative TatC conformations from the MD simulation were allowed to randomly dock to identify complementary surfaces. Subsequent dimer models were screened and kept if they were topologically compatible with the lipid bilayer. These models were scored using inter-protomer and homodimer energies (Table S2). A representative of the top scores is shown in Figure 4B. In these models, the dominant interface was a ‘back-to-back’ contact mediated predominantly by TM3 and TM6 (Fig. S2). This interaction is reminiscent of the SecY back-to-back model where a long-angled helix mediates the interface (Breyton et al., 2002; van den Berg et al., 2004). In this orientation, the two TatC molecules would work independently as predicted (Maldonado et al., 2011) and communication between the pockets could be mediated by motions in TM5 relayed through TM6. While this model is plausible, it is clear that there are additional interfaces between TatC in the higher order oligomers.
A model for TatC signal sequence recognition
Although TatC forms part of a large complex, not all of the TatC molecules bind the signal sequence (Tarry et al., 2009); therefore, it seems reasonable to presume that the signal sequence-binding site lies within a single TatC. Summarizing the structural and biochemical evidence, a model can be made for signal sequence recognition (Fig. 5). For illustration, the first 15 residues of the signal sequence for the A. aeolicus SufI protein has been modeled into the presumed binding pocket as its E. coli counterpart has been shown to be helical in a membrane-like environment (Fig. 5A & C)(San Miguel et al., 2003). We performed a similar manual docking with the crystal structure of a complex containing a Rieske iron-sulfur protein, which uses its signal sequence as a TM domain (Kurisu et al., 2003). The TM helix has a curve that could be docked in an orientation where the consensus sequence overlaid with our SufI model and the C-terminus curved to match the shape of the pocket.
Figure 5. A model for signal sequence binding by TatC.
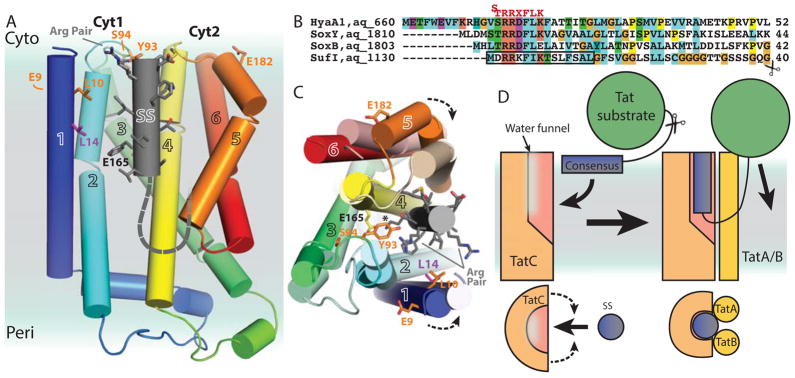
(A) AaDHPC as in Fig. 1A with an idealized helical SufI signal peptide including the first 12 amino acids (cylinder) modeled into the pocket of AaTatC with the C-terminus continuing as a dashed line. Residues identified as signal sequence crosslinks are shown as sticks (orange). L14, which crosslinks to some signal sequences, shown as sticks (magenta). The position of E165 is indicated.
(B) Representative predicted Aquifex aeolicus signal sequences aligned based on the arginine pair colored as in Fig. 1H. The consensus sequence is shown above in red. The residues used to model the signal sequence are highlighted by a black box. The alignment ends at the position of the predicted signal sequence cleavage site for SufI indicated by a scissors.
(C) Similar to A viewed from the cytoplasm with the MD frame 71 aligned as in Fig. 2C. The arginines in the consensus are highlighted and the lysine/AaE165 interaction is highlighted by a *. Arrows show predicted movement of TM1 and TM5.
(D) A cartoon model for TatC recognition of a signal sequence. The hydrophilic N-terminus of the signal sequence initially binds the membrane. The RR pair is recognized by TatC, positioning the signal sequence near the pocket. The signal sequence would enter into the membrane forming a tight interface stabilizing the pocket. This would create a looped orientation priming the substrate for translocation. The cleavage site is indicated by a scissors.
The amphipathic character of signal sequences (Klein et al., 2012) may allow them to initially interact directly with the membrane (Bageshwar et al., 2009; Molik et al., 2001; Shanmugham et al., 2006) which would allow for a two-dimensional search for the TatC recognition site. Critically, residues that are important to this recognition are found primarily on both the groove and the backside of TM1 and Cyt1. It is likely that specific recognition requires reorganizing in this region to accommodate the pairs of arginines. While one expects a clear negative pocket compatible with an arginine pair, this is not found in the structure, although the presence of several critical negative charges (e.g. EcE103/96, Fig. S1) likely contribute to this recognition. Another possible source of recognition is the presence of conserved aromatic residues that are required for binding. Of note, the conserved EcF94/87 can be modified to another aromatic (e.g. tyrosine) retaining function but modification to a hydrophobic leucine is inactive (Buchanan et al., 2002). An attractive hypothesis consistent with this would be that, in addition to the acidic side chains, the cluster of aromatics would use cation-π interactions (Gallivan and Dougherty, 1999) to bind the twin-arginine pair.
Once a substrate is recognized, the amphipathic helix would be positioned near the groove (Fig. 2F). Here, the energetically unfavorable hydration of the polar residue in the pocket could be stabilized by the signal sequence; the polar residues of the helix would line the polar face of the pocket while the hydrophobic face would contact the lipid (Fig. 5A & D). Importantly, this would not require specificity that could account for signal sequence diversity. It is likely that the shape of the pocket also contributes to excluding the lipid and the signal sequence could offset the surface tension generated at this interface. For species like AaTatC and EcTatC that contain an exposed negative charge in the pocket (Fig. 1D & G), the consensus lysine in the signal sequence would be appropriately placed to interact deep in the pocket. It is conceivable that the large cluster of aromatics in the pocket could also contribute to lysine binding. This orientation is consistent with the model of a deeply embedded signal sequence that must loop back out of the membrane (Cline and McCaffery, 2007; Di Cola and Robinson, 2005; Gérard and Cline, 2007). The signal sequence residues following the consensus are generally a mix of hydrophobic, small hydrophilic (Ser and Thr) and secondary structure breaking residues (Pro and Gly) that could snake back out of the membrane to the folded substrate (Fig. 5B & D).
The dimensions of the pocket are wider than a helix in the crystal structure; however, TM5 rotating into the pocket, as in the MD simulation, would make the size closer to that of a helix (Fig. 5C). The high conservation of Per3 likely plays a role in the flexibility of TM5 necessary for participation in substrate recognition. In TM5, EcE187/165 crosslinks to the signal sequence yet in the crystal structure it is far away from other crosslinking residues. Movement of TM5 would bring EcE187 into a position where a crosslink would seem more feasible. TM1 could also move into close contact that would be consistent with the crosslink of EcL20/14 to specific signal sequences (Fig. 5A & C) (Zoufaly et al., 2012). Movement of TM5 is likely coupled to rotation of TM6 and a salt bridge between the two (AaR188-E221) is conserved in many species that could help facilitate this coordination. The effect of this is that both helices would change conformation and a reorganized surface would be formed.
TatB and TatA both interact directly with Per3 implying that this provides a part of the binding site to TatC. TatB mutants near its periplasmic C-terminus affect binding to the signal sequence that would require interactions across the membrane (Lausberg et al., 2012). This model would suggest that, concurrent with the motion in TM5, TatB could interact with the whole of the signal sequence as it crosses the membrane. This would be consistent with the crosslinking of TatB to residues after the consensus sequence (Alami et al., 2003). These conformational changes, stimulated by signal sequence, would affect the interaction with TatB and TatA perhaps leading to oligomerization of TatA to generate a translocation pore. The final benefit of this model would be that moving the signal sequence into the membrane could be coupled to beginning the translocation process of the rest of the folded protein substrate.
Two articles appeared after the submission of this manuscript that are directly relevant to the model presented here. Müller and colleagues (Frobel et al., 2012a) presented results that demonstrate TatC is capable of translocating the signal peptidase cleavage site across the lipid bilayer independent of other factors. The cleavage requires a sufficiently long linker between the peptidase site and the mature protein so that the signal sequence can cross the membrane with enough additional unfolded sequence to loop back to the cytoplasm. This plunging of the signal sequence into the membrane exactly fits the mechanism described here. Fröbel et al. posit a model of a groove in the membrane portion of TatC that would accommodate the signal sequence. Excitingly, these results, in agreement with the TatC structure, provide evidence for transmembrane helix translocation in the absence of a pore. Mechanistically, a structured groove that facilitates TM insertion could prove to be a broader mechanism, as there are a number of these pathways that do not appear to have a pore. Example systems might include the YidC/Oxa1/Alb3 homologs (Wang and Dalbey, 2011) or the eukaryotic Get1 and Get2 complex involved in tail-anchor membrane protein insertion (Hegde and Keenan, 2011).
The independent description of the AaMNG structure referenced here by Berks, Lea and coworkers has been published (Rollauer et al., 2012). As expected, the two stories largely agree in the structural details of TatC. Despite the similarities, including the use of MD, Rollauer et al. do not predict that the signal sequence binds in the groove or the location of the region following the consensus. They do posit that the consensus sequence binds the cytoplasmic face contacting the backside of TM1 and Cyt1 based, in part, on mutations on that side that disrupt binding. As noted, residues on both sides have been implicated; therefore, the details of the model are ripe for further experimentation.
Here, we have solved a structure of TatC and used it as a template for understanding the early stages of twin-arginine translocation. The structure provides a wealth of unexpected features and a new membrane architecture. The structure and biochemical evidence is highly consistent with the signal sequence binding in the pocket formed by TatC. This model satisfies many of the requirements and will be a useful template for the design of future experiments. With the recent evidence that the TAT pathway is critical in the human pathogen Mycobacterium tuberculosis, the structure can potentially be used as a tool to design new antibiotics (Saint-Joanis et al., 2006). Moreover, the critical role of TAT in photosynthesis (Molik et al., 2001) makes this work broadly important. With this important structural step, the next stages of twin-arginine translocation research will be exciting.
EXPERIMENTAL PROCEDURES
Determination of the AaTatC crystal structure
An expression strain, CJMS2, was generated from an E. coli BL21(DE3) clone to obtain a TAT deletion strain (ΔtatABCDΔtatE) to prevent contamination of E. coli TAT components during TatC purification. Multiple TatC homologs were tested for expression in CJMS2 with the protein from Aquifex aeolicus (AaTatC) being the best behaved by gel filtration. Variants of this homolog were expressed and purified. All crystallization utilized the vapor diffusion technique. Two datasets were obtained for AaTatC from a surface entropy mutant in DHPC and a C-terminal lysozyme fusion in DDM. Diffraction data were collected at beamline 12-2 at the Stanford Synchrotron Radiation Lightsource. Phases were recovered by molecular replacement using the coordinates from PDBID 4B4A. Additional details of crystal growth, data collection, model refinement and structural analysis are provided in Supplemental Methods.
Multi-angle laser light scattering
Purified AaTatC in DDM (15 mg/ml) was purified on a sizing column inline with an Optilab interferometric refractometer and a quasi-elastic light-scattering instrument) (Wyatt Technologies). Data analysis was performed with the ASTRA software package. Additional details are in the Supplemental Methods.
Molecular dynamics
The TatC protein structure was aligned in a model phosphatidylcholine lipid bilayer. Standard simulation protocols were used to minimize the system. A full atomistic molecular dynamics simulation was run for 50.25 ns in 1 fs time steps. Snapshots were obtained every 10 ns and used for analysis. Full details of the MD simulation are provided in the Supplemental Methods.
Homodimer structure prediction
Homodimer prediction was done in two steps. A simplified side-chain model was used in a protein docking calculation taking either the crystal structure or a representative model from each of three MD clusters resulting in 16,000 interfaces. These were then screened for membrane compatibility resulting in 89 models. These had side-chains restored, were minimized and then ranked based on inter-protomer energy (BE) and total homodimer energy (TE) (Table S2). A full description of the docking is provided in the Supplemental Methods.
Supplementary Material
HIGHLIGHTS.
Crystal structures of TatC from the thermophile Aquifex aeolicus are presented.
The architecture of TatC generates a glove-shaped pocket buried in the bilayer.
Molecular dynamics reveal destabilization of the membrane around the pocket.
Correlation to biochemical results suggest the signal sequence binds in this pocket.
Acknowledgments
We are primarily grateful to Susan Lea and Ben Berks, both at Oxford, for collegiality and sharing their unpublished structure coordinates. We thank Justin W. Chartron for help with data processing and refinement. We thank William A. Goddard III for the use of the Materials and Process Simulation Center computing clusters for MD simulations. We are grateful to the staff at SSRL beamline 12-2 and J. Kaiser for help in data collection. This long-term project has received assistance from too many people to name specifically; therefore, we are grateful to all who have contributed to this project over the years. We thank D. Rees, S. Shan, S. Tanaka, J. Chartron, G. Lin, A. Müller, V. Somalinga, E. Chun and H. Gristick for comments on the manuscript. We thank J.T.C., O.W.C. and A.S. for support. We are grateful to Gordon and Betty Moore for support of the Molecular Observatory at Caltech. Operations at SSRL are supported by the US DOE and NIH. This work was supported by a Searle Scholar fellowship, a Burroughs-Wellcome Fund Career Award and a NIH Pioneer Award (Grant 1DP1OD008304-01) to W.M.C. Coordinates and structure factors have been deposited to the RCSB with the PDBID 4HTS for AaDHPC and 4HTT for AaDDM.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alami M, Lüke I, Deitermann S, Eisner G, Koch HG, Brunner J, Müller M. Differential interactions between a twin-arginine signal peptide and its translocase in Escherichia coli. Mol Cell. 2003;12:937–946. doi: 10.1016/s1097-2765(03)00398-8. [DOI] [PubMed] [Google Scholar]
- Allen SC, Barrett CM, Ray N, Robinson C. Essential cytoplasmic domains in the Escherichia coli TatC protein. J Biol Chem. 2002;277:10362–10366. doi: 10.1074/jbc.M109135200. [DOI] [PubMed] [Google Scholar]
- Bageshwar U, Whitaker N, Liang F, Musser S. Interconvertibility of lipid- and translocon-bound forms of the bacterial Tat precursor pre-SufI. Mol Microbiol. 2009;74:209–226. doi: 10.1111/j.1365-2958.2009.06862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bageshwar UK, Musser SM. Two electrical potential-dependent steps are required for transport by the Escherichia coli Tat machinery. J Cell Biol. 2007;179:87–99. doi: 10.1083/jcb.200702082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett CM, Mangels D, Robinson C. Mutations in subunits of the Escherichia coli twin-arginine translocase block function via differing effects on translocation activity or TAT complex structure. J Mol Biol. 2005;347:453–463. doi: 10.1016/j.jmb.2005.01.026. [DOI] [PubMed] [Google Scholar]
- Barrett CM, Robinson C. Evidence for interactions between domains of TatA and TatB from mutagenesis of the TatABC subunits of the twin-arginine translocase. FEBS J. 2005;272:2261–2275. doi: 10.1111/j.1742-4658.2005.04654.x. [DOI] [PubMed] [Google Scholar]
- Behrendt J, Standar K, Lindenstrauss U, Brüser T. Topological studies on the twin-arginine translocase component TatC. FEMS Microbiol Lett. 2004;234:303–308. doi: 10.1016/j.femsle.2004.03.048. [DOI] [PubMed] [Google Scholar]
- Berks BC. A common export pathway for proteins binding complex redox cofactors? Mol Microbiol. 1996;22:393–404. doi: 10.1046/j.1365-2958.1996.00114.x. [DOI] [PubMed] [Google Scholar]
- Bogsch EG, Sargent F, Stanley NR, Berks BC, Robinson C, Palmer T. An essential component of a novel bacterial protein export system with homologues in plastids and mitochondria. J Biol Chem. 1998;273:18003–18006. doi: 10.1074/jbc.273.29.18003. [DOI] [PubMed] [Google Scholar]
- Bolhuis A. Protein transport in the halophilic archaeon Halobacterium sp NRC-1: a major role for the twin-arginine translocation pathway? Microbiology. 2002;148:3335–3346. doi: 10.1099/00221287-148-11-3335. [DOI] [PubMed] [Google Scholar]
- Bolhuis A, Mathers JE, Thomas JD, Barrett CM, Robinson C. TatB and TatC form a functional and structural unit of the twin-arginine translocase from Escherichia coli. J Biol Chem. 2001;276:20213–20219. doi: 10.1074/jbc.M100682200. [DOI] [PubMed] [Google Scholar]
- Breyton C, Haase W, Rapoport TA, Kühlbrandt W, Collinson I. Three-dimensional structure of the bacterial protein-translocation complex SecYEG. Nature. 2002;418:662–665. doi: 10.1038/nature00827. [DOI] [PubMed] [Google Scholar]
- Buchanan G, Leeuw E, Stanley NR, Wexler M, Berks BC, Sargent F, Palmer T. Functional complexity of the twin-arginine translocase TatC component revealed by site-directed mutagenesis. Mol Microbiol. 2002;43:1457–1470. doi: 10.1046/j.1365-2958.2002.02853.x. [DOI] [PubMed] [Google Scholar]
- Chaddock AM, Mant A, Karnauchov I, Brink S, Herrmann RG, Klösgen RB, Robinson C. A new type of signal peptide: central role of a twin-arginine motif in transfer signals for the delta pH-dependent thylakoidal protein translocase. EMBO J. 1995;14:2715–2722. doi: 10.1002/j.1460-2075.1995.tb07272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CS, Haney EF, Vogel HJ, Turner RJ. Towards understanding the Tat translocation mechanism through structural and biophysical studies of the amphipathic region of TatA from Escherichia coli. Biochim Biophys Acta. 2011;1808:2289–2296. doi: 10.1016/j.bbamem.2011.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline K. Thylakoid DeltapH-dependent precursor proteins bind to a cpTatC-Hcf106 complex before Tha4-dependent transport. J Cell Biol. 2001;154:719–730. doi: 10.1083/jcb.200105149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline K, McCaffery M. Evidence for a dynamic and transient pathway through the TAT protein transport machinery. EMBO J. 2007;26:3039–3049. doi: 10.1038/sj.emboj.7601759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leeuw E, Granjon T, Porcelli I, Alami M. Oligomeric properties and signal peptide binding by Escherichia coli Tat protein transport complexes. J Mol Biol. 2002;322:1135–1146. doi: 10.1016/s0022-2836(02)00820-3. [DOI] [PubMed] [Google Scholar]
- Di Cola A, Robinson C. Large-scale translocation reversal within the thylakoid Tat system in vivo. J Cell Biol. 2005;171:281–289. doi: 10.1083/jcb.200502067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilks K, Gimenez MI, Pohlschröder M. Genetic and biochemical analysis of the twin-arginine translocation pathway in halophilic archaea. J Bacteriol. 2005;187:8104–8113. doi: 10.1128/JB.187.23.8104-8113.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilks K, Rose RW, Hartmann E, Pohlschröder M. Prokaryotic utilization of the twin-arginine translocation pathway: a genomic survey. J Bacteriol. 2003;185:1478–1483. doi: 10.1128/JB.185.4.1478-1483.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fröbel J, Rose P, Lausberg F, Blummel AS, Freudl R, Müller M. Transmembrane insertion of twin-arginine signal peptides is driven by TatC and regulated by TatB. Nat Commun. 2012a;3:1311. doi: 10.1038/ncomms2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fröbel J, Rose P, Müller M. Twin-arginine-dependent translocation of folded proteins. Phil Trans Roy Soc of London. 2012b;367:1029–1046. doi: 10.1098/rstb.2011.0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallivan JP, Dougherty DA. Cation-π interactions in structural biology. Proc Natl Acad Sci U S A. 1999;96:9459–9464. doi: 10.1073/pnas.96.17.9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gérard F, Cline K. The thylakoid proton gradient promotes an advanced stage of signal peptide binding deep within the Tat pathway receptor complex. J Biol Chem. 2007;282:5263–5272. doi: 10.1074/jbc.M610337200. [DOI] [PubMed] [Google Scholar]
- Gohlke U, Pullan L, McDevitt CA, Porcelli I, de Leeuw E, Palmer T, Saibil HR, Berks BC. The TatA component of the twin-arginine protein transport system forms channel complexes of variable diameter. Proc Nat Acad Sci USA. 2005;102:10482–10486. doi: 10.1073/pnas.0503558102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouffi K, Santini CL, Wu LF. Topology determination and functional analysis of the Escherichia coli TatC protein. FEBS Lett. 2002;525:65–70. doi: 10.1016/s0014-5793(02)03069-7. [DOI] [PubMed] [Google Scholar]
- Hatzixanthis K, Palmer T, Sargent F. A subset of bacterial inner membrane proteins integrated by the twin-arginine translocase. Mol Microbiol. 2003;49:1377–1390. doi: 10.1046/j.1365-2958.2003.03642.x. [DOI] [PubMed] [Google Scholar]
- Hegde RS, Keenan RJ. Tail-anchored membrane protein insertion into the endoplasmic reticulum. Nat Rev Mol Cell Biol. 2011;12:787–798. doi: 10.1038/nrm3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikkila MP, Honisch U, Wunsch P, Zumft WG. Role of the Tat ransport system in nitrous oxide reductase translocation and cytochrome cd1 biosynthesis in Pseudomonas stutzeri. J Bacteriol. 2001;183:1663–1671. doi: 10.1128/JB.183.5.1663-1671.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzapfel E, Eisner G, Alami M, Barrett CM, Buchanan G, Lüke I, Betton JM, Robinson C, Palmer T, Moser M, et al. The entire N-terminal half of TatC is involved in twin-arginine precursor binding. Biochemistry. 2007;46:2892–2898. doi: 10.1021/bi062205b. [DOI] [PubMed] [Google Scholar]
- Jongbloed JD, Grieger U, Antelmann H, Hecker M, Nijland R, Bron S, van Dijl JM. Two minimal Tat translocases in Bacillus. Mol Microbiol. 2004;54:1319–1325. doi: 10.1111/j.1365-2958.2004.04341.x. [DOI] [PubMed] [Google Scholar]
- Jongbloed JD, Martin U, Antelmann H, Hecker M, Tjalsma H, Venema G, Bron S, van Dijl JM, Muller J. TatC is a specificity determinant for protein secretion via the twin-arginine translocation pathway. J Biol Chem. 2000;275:41350–41357. doi: 10.1074/jbc.M004887200. [DOI] [PubMed] [Google Scholar]
- Kassem II, Zhang Q, Rajashekara G. The twin-arginine translocation system: contributions to the pathobiology of Campylobacter jejuni. Future Microbiol. 2011;6:1315–1327. doi: 10.2217/fmb.11.107. [DOI] [PubMed] [Google Scholar]
- Ki JJ, Kawarasaki Y, Gam J, Harvey BR, Iverson BL, Georgiou G. A periplasmic fluorescent reporter protein and its application in high-throughput membrane protein topology analysis. J Mol Biol. 2004;341:901–909. doi: 10.1016/j.jmb.2004.05.078. [DOI] [PubMed] [Google Scholar]
- Klein MJ, Grage SL, Muhle-Goll C, Burck J, Afonin S, Ulrich AS. Structure analysis of the membrane-bound PhoD signal peptide of the Tat translocase shows an N-terminal amphiphilic helix. Biochim Biophys Acta. 2012;1818:3025–3031. doi: 10.1016/j.bbamem.2012.08.002. [DOI] [PubMed] [Google Scholar]
- Kneuper H, Maldonado B, Jager F, Krehenbrink M, Buchanan G, Keller R, Muller M, Berks BC, Palmer T. Molecular dissection of TatC defines critical regions essential for protein transport and a TatB-TatC contact site. Mol Microbiol. 2012;85:945–961. doi: 10.1111/j.1365-2958.2012.08151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutzenbeck P, Kröger C, Lausberg F, Blaudeck N, Sprenger GA, Freudl R. Escherichia coli twin arginine (Tat) mutant translocases possessing relaxed signal peptide recognition specificities. J Biol Chem. 2007;282:7903–7911. doi: 10.1074/jbc.M610126200. [DOI] [PubMed] [Google Scholar]
- Kurisu G, Zhang H, Smith JL, Cramer WA. Structure of the cytochrome b6f complex of oxygenic photosynthesis: tuning the cavity. Science. 2003;302:1009–1014. doi: 10.1126/science.1090165. [DOI] [PubMed] [Google Scholar]
- Kwan DC, Thomas JR, Bolhuis A. Bioenergetic requirements of a Tat-dependent substrate in the halophilic archaeon Haloarcula hispanica. FEBS J. 2008;275:6159–6167. doi: 10.1111/j.1742-4658.2008.06740.x. [DOI] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, Mcwilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Lausberg F, Fleckenstein S, Kreutzenbeck P, Frobel J, Rose P, Muller M, Freudl R. Genetic Evidence for a Tight Cooperation of TatB and TatC during Productive Recognition of Twin-Arginine (Tat) Signal Peptides in Escherichia coli. PLoS One. 2012;7:e39867. doi: 10.1371/journal.pone.0039867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PA, Orriss GL, Buchanan G, Greene NP, Bond PJ, Punginelli C, Jack RL, Sansom MS, Berks B, Palmer T. Cysteine-scanning mutagenesis and disulfide mapping studies of the conserved domain of the twin-arginine translocase TatB component. J Biol Chem. 2006;281:34072–34085. doi: 10.1074/jbc.M607295200. [DOI] [PubMed] [Google Scholar]
- MacCallum JL, Bennett WF, Tieleman DP. Distribution of amino acids in a lipid bilayer from computer simulations. Biophys J. 2008;94:3393–3404. doi: 10.1529/biophysj.107.112805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado B, Buchanan G, Muller M, Berks BC, Palmer T. Genetic evidence for a TatC dimer at the core of the Escherichia coli twin arginine (Tat) protein translocase. J Mol Microbiol Biotechnol. 2011;20:168–175. doi: 10.1159/000329076. [DOI] [PubMed] [Google Scholar]
- Mcdevitt C, Hicks MG, Palmer T, Berks B. Characterisation of Tat protein transport complexes carrying inactivating mutations. Biochem Biophys Res Commun. 2005;329:693–698. doi: 10.1016/j.bbrc.2005.02.038. [DOI] [PubMed] [Google Scholar]
- Meruelo AD, Samish I, Bowie JU. TMKink: a method to predict transmembrane helix kinks. Protein Sci. 2011;20:1256–1264. doi: 10.1002/pro.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molik S, Karnauchov I, Weidlich C, Herrmann RG, Klosgen RB. The Rieske Fe/S protein of the cytochrome b6/f complex in chloroplasts: missing link in the evolution of protein transport pathways in chloroplasts? J Biol Chem. 2001;276:42761–42766. doi: 10.1074/jbc.M106690200. [DOI] [PubMed] [Google Scholar]
- Ochsner UA, Snyder A, Vasil AI, Vasil ML. Effects of the twin-arginine translocase on secretion of virulence factors, stress response, and pathogenesis. Proc Natl Acad Sci USA. 2002;99:8312–8317. doi: 10.1073/pnas.082238299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer T, Berks BC. The twin-arginine translocation (Tat) protein export pathway. Nat Rev Microbiol. 2012;10:483–496. doi: 10.1038/nrmicro2814. [DOI] [PubMed] [Google Scholar]
- Panahandeh S, Maurer C, Moser M, DeLisa MP, Muller M. Following the path of a twin-arginine precursor along the TatABC translocase of Escherichia coli. J Biol Chem. 2008;283:33267–33275. doi: 10.1074/jbc.M804225200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E, Rapoport TA. Mechanisms of Sec61/SecY-mediated protein translocation across membranes. Ann Rev Biophys. 2012;41:21–40. doi: 10.1146/annurev-biophys-050511-102312. [DOI] [PubMed] [Google Scholar]
- Punginelli C, Maldonado B, Grahl S, Jack R, Alami M, Schröder J, Berks B, Palmer T. Cysteine scanning mutagenesis and topological mapping of the Escherichia coli twin-arginine translocase TatC Component. J Bact. 2007;189:5482–5494. doi: 10.1128/JB.00647-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, et al. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollauer SE, Tarry MJ, Graham JE, Jaaskelainen M, Jager F, Johnson S, Krehenbrink M, Liu SM, Lukey MJ, Marcoux J, et al. Structure of the TatC core of the twin-arginine protein transport system. Nature. 2012;492:210–214. doi: 10.1038/nature11683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose RW, Bruser T, Kissinger JC, Pohlschroder M. Adaptation of protein secretion to extremely high-salt conditions by extensive use of the twin-arginine translocation pathway. Mol Microbiol. 2002;45:943–950. doi: 10.1046/j.1365-2958.2002.03090.x. [DOI] [PubMed] [Google Scholar]
- Rosenbaum D, Cherezov V, Hanson MA, Rasmussen SG, Thian F, Kobilka T, Choi H, Yao XJ, Weis W, Stevens RC, et al. GPCR engineering yields high-resolution structural insights into β2-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- Saint-Joanis B, Demangel C, Jackson M, Brodin P, Marsollier L, Boshoff H, Cole ST. Inactivation of Rv2525ca substrate of the twin arginine translocation (Tat) system of Mycobacterium tuberculosis, increases beta-lactam susceptibility and virulence. J Bact. 2006;188:6669–6679. doi: 10.1128/JB.00631-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Miguel M, Marrington R, Rodger PM, Rodger A, Robinson C. An Escherichia coli twin-arginine signal peptide switches between helical and unstructured conformations depending on the hydrophobicity of the environment. FEBS J. 2003;270:3345–3352. doi: 10.1046/j.1432-1033.2003.03710.x. [DOI] [PubMed] [Google Scholar]
- Sargent F, Bogsch EG, Stanley NR, Wexler M, Robinson C, Berks BC, Palmer T. Overlapping functions of components of a bacterial Sec-independent protein export pathway. EMBO J. 1998;17:3640–3650. doi: 10.1093/emboj/17.13.3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargent F, Gohlke U, De Leeuw E, Stanley NR, Palmer T, Saibil HR, Berks BC. Purified components of the Escherichia coli Tat protein transport system form a double-layered ring structure. Eur J Biochem. 2001;268:3361–3367. doi: 10.1046/j.1432-1327.2001.02263.x. [DOI] [PubMed] [Google Scholar]
- Sargent F, Stanley NR, Berks BC, Palmer T. Sec-independent protein translocation in Escherichia coli A distinct and pivotal role for the TatB protein. J Biol Chem. 1999;274:36073–36082. doi: 10.1074/jbc.274.51.36073. [DOI] [PubMed] [Google Scholar]
- Schaerlaekens K, Schierova M, Lammertyn E, Geukens N, Anne J, Van Mellaert L. Twin-arginine translocation pathway in Streptomyces lividans. J Bacteriol. 2001;183:6727–6732. doi: 10.1128/JB.183.23.6727-6732.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanmugham A, Wong Fong Sang HW, Bollen YJ, Lill H. Membrane binding of twin arginine preproteins as an early step in translocation. Biochemistry. 2006;45:2243–2249. doi: 10.1021/bi052188a. [DOI] [PubMed] [Google Scholar]
- Shruthi H, Anand P, Murugan V, Sankaran K. Twin arginine translocase pathway and fast-folding lipoprotein biosynthesis in E. coli: interesting implications and applications. Mol Biosyst. 2010;6:999–1007. doi: 10.1039/b916510j. [DOI] [PubMed] [Google Scholar]
- Settles AM, Yonetani A, Baron A, Bush DR, Cline K, Martienssen R. Sec-independent protein translocation by the maize Hcf106 protein. Science. 1997;278:1467–1470. doi: 10.1126/science.278.5342.1467. [DOI] [PubMed] [Google Scholar]
- Stanley NR, Findlay K, Berks BC, Palmer T. Escherichia coli strains blocked in Tat-dependent protein export exhibit pleiotropic defects in the cell envelope. J Bacteriol. 2001;183:139–144. doi: 10.1128/JB.183.1.139-144.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauch EM, Georgiou G. Escherichia coli tatC mutations that suppress defective twin-arginine transporter signal peptides. J Mol Biol. 2007;374:283–291. doi: 10.1016/j.jmb.2007.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarry MJ, Schafer E, Chen S, Buchanan G, Greene NP, Lea SM, Palmer T, Saibil HR, Berks BC. Structural analysis of substrate binding by the TatBC component of the twin-arginine protein transport system. Proc Natl Acad Sci USA. 2009;106:13284–13289. doi: 10.1073/pnas.0901566106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JR, Bolhuis A. The tatC gene cluster is essential for viability in halophilic archaea. FEMS Microbiol Lett. 2006;256:44–49. doi: 10.1111/j.1574-6968.2006.00107.x. [DOI] [PubMed] [Google Scholar]
- van den Berg B, Clemons WM, Collinson I, Modis Y, Hartmann E, Harrison SC, Rapoport TA. X-ray structure of a protein-conducting channel. Nature. 2004;427:36–44. doi: 10.1038/nature02218. [DOI] [PubMed] [Google Scholar]
- van der Ploeg R, Mader U, Homuth G, Schaffer M, Denham EL, Monteferrante CG, Miethke M, Marahiel MA, Harwood CR, Winter T, et al. Environmental salinity determines the specificity and need for Tat-dependent secretion of the YwbN protein in Bacillus subtilis. PLoS One. 2011;6:e18140. doi: 10.1371/journal.pone.0018140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Dalbey RE. Inserting membrane proteins: the YidC/Oxa1/Alb3 machinery in bacteria, mitochondria, and chloroplasts. Bioch Biophys Acta. 2011;1808:866–875. doi: 10.1016/j.bbamem.2010.08.014. [DOI] [PubMed] [Google Scholar]
- Weiner JH, Bilous PT, Shaw GM, Lubitz SP, Frost L, Thomas GH, Cole JA, Turner RJ. A novel and ubiquitous system for membrane targeting and secretion of cofactor-containing proteins. Cell. 1998;93:93–101. doi: 10.1016/s0092-8674(00)81149-6. [DOI] [PubMed] [Google Scholar]
- Wu LF, Ize B, Chanal A, Quentin Y, Fichant G. Bacterial twin-arginine signal peptide-dependent protein translocation pathway: evolution and mechanism. J Mol Microbiol Biotechnol. 2000;2:179–189. [PubMed] [Google Scholar]
- Yen MR, Tseng YH, Nguyen EH, Wu LF, Saier MH., Jr Sequence and phylogenetic analyses of the twin-arginine targeting (Tat) protein export system. Arch Microbiol. 2002;177:441–450. doi: 10.1007/s00203-002-0408-4. [DOI] [PubMed] [Google Scholar]
- Zoufaly S, Frobel J, Rose P, Flecken T, Maurer C, Moser M, Muller M. Mapping precursor-binding site on TatC subunit of twin arginine-specific protein translocase by site-specific photo cross-linking. J Biol Chem. 2012;287:13430–13441. doi: 10.1074/jbc.M112.343798. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.