

Abstract
Conditioning strategies that deplete host lymphocytes have been shown to enhance clinical responses to some adoptive T-cell therapies. However, host T cells are capable of eliminating tumor cells upon the relief of immunosuppression, indicating that lymphodepletion prior to T-cell transfer may reduce optimal tumor protection elicited by cell treatments that are capable of shaping host immunity. In this study, we show that adoptively transferred T cells bearing a chimeric antigen receptor (CAR) harness endogenous T cells for optimal tumor elimination and the development of a tumor-specific memory T cell response. Mice bearing ID8 ovarian cancer cells were treated with T cells transduced with a NKG2D-based CAR. CAR-expressing T cells increased the number of host CD4+ and CD8+ T cells at the tumor site in a CXCR3-dependent manner and increased the number of antigen-specific host CD4+ T cells in the tumor and draining lymph nodes. In addition, the administration of CAR-expressing T cells increased antigen presentation to CD4+ T cells, and this increase was dependent on interferon γ and granulocyte-macrophage colony-stimulating factor produced by the former. Host CD4+ T cells were sufficient for optimal tumor protection mediated by NKG2D CAR-expressing T cells, but they were not necessary if CD4+ T cells were adoptively co-transferred. However, host CD4+ T cells were essential for the development of an antigen-specific memory T-cell response to tumor cells. Moreover, optimal tumor elimination as orchestrated by NKG2D CAR-expressing T cells was dependent on host CD8+ T cells. These results demonstrate that adoptively transferred T cells recruit and activate endogenous T-cell immunity to enhance the elimination of tumor cells and the development of tumor-specific memory responses.
Keywords: adoptive T cell therapy, CD4+ T cells, CD8+ T cells, chNKG2D, ID8, immunotherapy, NKG2D, ovarian cancer, tumor immunology
Introduction
Many protocols for adoptive T-cell therapy include host preconditioning strategies to deplete lymphocytes prior to T-cell transfer. Total body irradiation and high-dose chemotherapy regimens not only relieve immunosuppression, but also increase the persistence of transferred T cells in the host.1 These preconditioning regimens appear to correlate with improved clinical responses following adoptive T-cell transfer in melanoma.2 However, they can also result in the death of healthy cells and reduce the quality of life for patients. In addition, some clinical trials based on pre-conditioning strategies in combination with adoptive T-cell transfer have shown tumor persistence and relapse as a result of the outgrowth of antigenic variants.3 Harnessing endogenous lymphocytes is one method to broaden the tumor-specific T-cell repertoire and eliminate tumor cells that have lost the antigen targeted by adoptive T-cell therapy.4,5 Recent evidence suggests that adoptively transferred T cells may eradicate tumors without the need for pre-conditioning, indicating that T cell-based therapies can enhance tumor elimination by relieving immunosuppression and promoting endogenous immunity.6
Studies on the role of T cells in tumor elimination mediated by adoptive T-cell transfer have primarily focused on CD8+ T cells. CD8+ T cells possess the ability to recognize and kill tumor cells. Many tumor cells do not express MHC Class II molecules, preventing them from being direct recognized by CD4+ T cells. Thus, CD4+ T cells mediate immune response against MHC Class II-negative tumors indirectly, by regulating CD8+ T-cell recruitment, proliferation and effector functions through the CD40/CD40L-dependent licensing of antigen-presenting cells (APCs).7-9 In addition, CD4+ T cells promote antitumor effects by activating macrophages to kill malignant cells.10-12 Although CD4+ T cells are dispensable for tumor elimination mediated by many treatments, signals from CD4+ T cells are often necessary to license APCs for the generation of protective T cell-mediated immunity.13,14
Chimeric antigen receptor (CAR)-transduced T cells have been shown to constitute an effective means to eliminate tumors and increase patient survival.15-17 The ligands for NKG2D, an activating receptor expressed primarily on natural killer (NK) cells, NKT cells and CD8+ T cells, are expressed on primary tumors of diverse origin, yet they are absent (or transiently expressed at low levels) on healthy tissues.18-23 T cells transduced to express a chimeric NKG2D-based CAR (chNKG2D), consisting of the NKG2D receptor fused to the CD3ζ cytoplasmic domain, kill tumor cells and secrete T-cell effector cytokines that promote endogenous antitumor immunity.24-27 Interferon γ (IFNγ) and granulocyte-macrophage colony-stimulating factor (GM-CSF) secreted by NKG2D CAR-expressing T cells have been shown to activate APCs to induce a tumor-specific host T-cell response.24,28 In contrast with other adoptive T-cell therapies, cyclophosphamide-based lymphodepleting regimens administered prior to the infusion of CAR-expressing T cells did not improve the therapeutic efficacy of NKG2D CAR-expressing T-cell therapy.29 Lymphodepletion reduces the host immune responses that are activated by CAR-expressing T-cell infusion, and this therapy does not function as effectively after pre-conditioning. In immunocompetent mice bearing ID8 ovarian cancer cells, the administration of chNKG2D-expressing T cells leads to long-term tumor-free survival and the development of antitumor immunity.17
However, the effects of host T cells on the antitumor response following the infusion of CAR-expressing T cells remain unclear. Here, we investigated how NKG2D CAR-expressing T-cell transfer regulates the endogenous T-cell response, and explored the role of endogenous T lymphocytes in tumor elimination and the development of an immunological memory. Our findings demonstrate the necessity of both host CD4+ and CD8+ T cells for optimal tumor elimination and the development of a tumor-specific memory response following the adoptive transfer of NKG2D CAR-expressing T cells.
Results
Treatment with NKG2D CAR-expressing T cells increases the number of antigen-specific CD4+ T cells at the tumor site
CAR-expressing T cells can attack tumor cells and secrete effector cytokines that shape the tumor microenvironment and promote host antitumor immunity. In addition, NKG2D CAR-expressing T-cell therapy is impaired in mice lacking T and B cells, indicating that this immunotherapeutic approach requires host lymphocytes for optimal therapeutic efficacy.24 To address how CAR-expressing T-cell treatment affects the recruitment of antigen-specific CD4+ T cells in vivo, OT-II T cells were injected i.v. into mice bearing ID8-GFP-tOVA ovarian cancer cells one hour prior to the injection of chNKG2D-expressing T cells or cells expressing wild-type NKG2D (wtNKG2D) i.p. The treatment of mice with chNKG2D-expressing T cells increased the number of OT-II T cells in the peritoneum and draining lymph nodes as compared with the administration of wtNKG2D-expressing T cells or PBS (Fig. 1A). To determine if the increase in OT-II cell recruitment was antigen-specific, mice bearing ID8-GFP-derived tumors were treated with chNKG2D- or wtNKG2D-expressing T cells and injected with OT-II cells i.v. The administration of chNKG2D-expressing T cells did not increase the number of antigen-specific T cells in the draining lymph nodes of ID8-GFP tumor-bearing mice. The number of OT-II cells in the peritoneum of mice bearing ID8-GFP-tOVA cells (that express a truncated version of OVA that cannot be secreted) was higher than in ID8-GFP tumor-bearing mice (Fig. 1B). These data indicate that NKG2D CAR-expressing T cells increase the number of antigen-specific CD4+ T cells at the tumor site. This increase may result from increased T-cell trafficking, survival and/or proliferation at the tumor site and in draining lymph nodes.
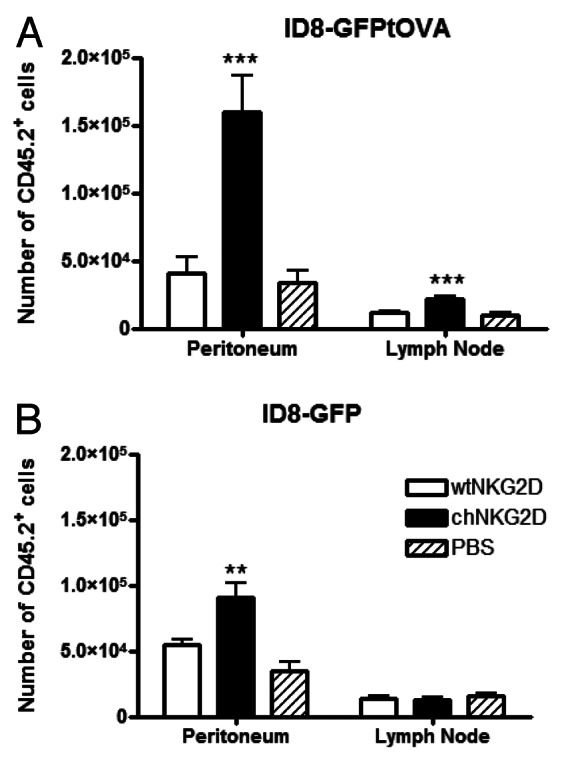
Figure 1. The administration of chNKG2D-expressing T cells increases antigen-specific CD4+ T cells in the peritoneum and tumor-draining lymph nodes. (A and B) Mice bearing ID8-GFP-tOVA- (A) or ID8-GFP-derived (B) tumors were injected with chNKG2D-expressing, wtNKG2D-expressing T cells or PBS i.p. One hour before local T-cell injection, CD45.2+ OT-II cells were injected i.v. Three days after T-cell transfer, the absolute number of CD45.2+ cells in the peritoneum and mediastinal lymph nodes was determined by flow cytometry. The average of each group and SD (n = 8) is shown (**p < 0.01; ***p < 0.001 as compared with animals receiving wtNKG2D-expressing cells).
IFNγ and GM-CSF secreted by NKG2D CAR-expressing T cells induce antigen-specific proliferation of host CD4+ T cells
To determine if the adoptive transfer of NKG2D CAR-expressing T cells enhances host antigen-specific CD4+ T-cell proliferation, ID8-GFP-tOVA tumor-bearing mice were treated with chNKG2D-expressing T cells or wtNKG2D-expressing control T cells. Some groups of mice were treated with NKG2D CAR-expressing T cells deficient in the effector cytokines IFNγ or GM-CSF, to determine the role of CAR-expressing T cell-derived cytokines on host CD4+ T-cell proliferation. Spleen and tumor-draining lymph node cells isolated from mice treated with chNKG2D-expressing T cells enhanced OT-II T-cell proliferation as compared with tissues obtained from wtNKG2D-expressing T cell-treated mice (Fig. 2A and B). The increase in CD4+ T-cell proliferation was dependent on both IFNγ and GM-CSF secreted by adoptively transferred CAR-expressed T cells. To determine if such an increase in OT-II cell proliferation was antigen-specific, mice bearing ID8-GFP tumors were treated with wtNKG2D-expressing or chNKG2D-expressing T cells. Spleen and lymph node cells isolated from mice treated with chNKG2D-expressing T cells induced somewhat more OT-II cell proliferation than similar cells from wtNKG2D-expressing T-cell treated or naïve mice. However, the increase in OT-II cell proliferation obtained with cells from mice bearing tumors that did not express OVA was lower as compared with that promoted by lymph node or spleen cells isolated from mice bearing ID8-GFP-tOVA tumors. These data demonstrate that chNKG2D-expressing T cells enhance the proliferation of antigen-specific CD4+ T cell in secondary lymphoid organs, and that this relies on their production of IFNγ and GM-CSF.
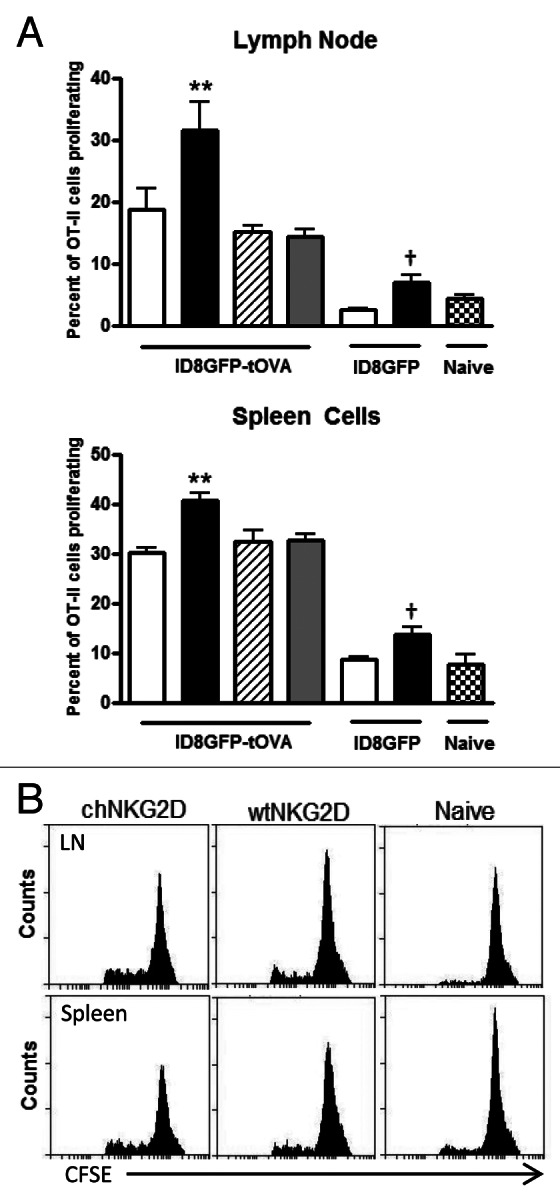
Figure 2. chNKG2D-expressing T cells enhance MHC Class II antigen presentation and the proliferation of CD4+ T cells. (A and B) ID8-GFP or ID8-GFP-tOVA tumor-bearing mice were injected with wtNKG2D-expressing (open), chNKG2D-expressing (filled), interferon γ (IFNγ)-deficient chNKG2D-expressing (hatched), or granulocyte-macrophage colony-stimulating factor (GM-CSF)-deficient chNKG2D-expressing T cells (dotted) i.p. (A) Seven days after T-cell transfer, mediastinal lymph node and spleen cells were isolated and cultured with CFSE-labeled OT-II cells. After 4 d, T-cell proliferation was assessed by flow cytometry. The proliferation of T cells cultured with cells from naïve mice is shown (checked). (B) Representative OT-II CFSE dilution flow cytometry plots as induced by lymph node and spleen cells isolated from naïve mice or from mice bearing ID8-GFP-tOVA tumors treated with wtNKG2D-expressing or chNKG2D-expressing T cells. Data are representative of two individual experiments. The average of each group and SD (n = 8) are shown (**p < 0.01 as compared with ID8-GFP-tOVA tumor-bearing animals receiving wtNKG2D-expressing T cells; †p < 0.05 as compared with ID8-GFP tumor-bearing animals receiving wtNKG2D-expressing T cells).
Treatment with chNKG2D-expressing T cells increases the number of endogenous CD4+ and CD8+ T cells at the tumor site in a CXCR3-dependent mechanism
IFNγ induces the production of the chemokines CXCL9 and CXCL10, which preferentially recruit CXCR3+ TH1 T cells.30 To determine if IFNγ produced by chNKG2D-expressing T cells increases the amount of CXCL9 and CXCL10 in the tumor microenvironment, ID8 tumor-bearing mice were treated with wtNKG2D-expressing T cells, chNKG2D-expressing T cells, or chNKG2D-expressing T cells deficient in IFNγ, and chemokine expression was assessed. The administration of chNKG2D-expressing T cells increased CXCL9 and CXCL10 expression by peritoneal cells as compared with that of wtNKG2D-expressing T cells, and such an increase in chemokine production was dependent on IFNγ produced by adoptively transferred cells (Fig. 3A). Because F4/80+ macrophages makeup a large percentage of leukocytes in the tumor microenvironment, we quantified their secretion of CXCL9 and CXCL10.31 Both the F4/80+ and F4/80− cells isolated from chNKG2D-expressing T-cell treated mice produced higher amounts of CXCL9 and CXCL10 than their counterparts isolated from wtNKG2D-expressing T-cell treated mice, and F4/80+ and F4/80− cells mixed together produced more of both chemokines than either cell population cultured alone (Fig. 3B). To determine if increased levels of CXCR3 ligands regulate the recruitment of endogenous T cells to the tumor site, C57BL/6 or CXCR3-deficient mice bearing ID8 tumors were treated with chNKG2D- or wtNKG2D-expressing T cells. CXCR3-deficient hosts exhibited lower numbers of both CD4+ and CD8+ T cells at the tumor site as compared with C57BL/6 hosts upon the administration of chNKG2D-expressing T cells (Fig. 3C). In contrast, C57BL/6 and Cxcr3−/− mice treated with wtNKG2D-expressing T cells had equal numbers of intratumoral CD4+ and CD8+ T cells. These data demonstrate that the administration of chNKG2D-expressing T cells induced the secretion of CXCL9 and CXCL10 by host macrophages and suggest that these chemokines increase the endogenous T-cell recruitment at the tumor site.

Figure 3. The administration of chNKG2D-expressing T cells increases the number of host T cells at the tumor site in a CXCR3-dependent mechanism. (A and B) Mice bearing ID8-GFP tumors were injected with wtNKG2D-expressing (open), chNKG2D-expressing (filled) or interferon γ (IFNγ)-deficient chNKG2D-expressing T cells (hatched). (A) Peritoneal cells isolated 3 d after T-cell transfer were assessed for CXCL9 and CXCL10 expression by quantitative RT-PCR. (*p < 0.05 as compared with peritoneal cells from animals receiving wtNKG2D-expressing T cells). (B) F4/80+ cells were isolated 3 d after T-cell transfer and cultured for 24 h. Cell-free culture media from the F4/80+ fraction, the F4/80− fraction, and the combined F4/80+ and F4/80− fractions (1:1) were assessed for CXCL9 and CXCL10 production by multiplex protein analysis (*p < 0.05; ***p < 0.001 as compared with cells from animals receiving wtNKG2D-expressing T cells; †p < 0.05 as compared with F4/80+ and F4/80− cells from animals receiving chNKG2D-expressing T cells). (C) C57BL/6 or Cxcr3−/− mice were injected with ID8-GFP tumor cells and treated with CD45.1+ chNKG2D-expressing T cells 5 weeks later. Three days after T-cell transfer, a peritoneal wash was performed and the absolute numbers of CD4+ and CD8+ host T cells were determined. The average of each group and SD (n = 8) are shown. Data are representative of two independent experiments (*p < 0.05; **p < 0.01 as compared with C57BL/6 mice receiving wtNKG2D-expressing cells; †p < 0.05 as compared with C57BL/6 mice receiving chNKG2D-expressing T cells).
CD4+ T cells are necessary for optimal tumor elimination
To determine the role of CD4+ T cells in tumor protection as mediated by CAR-expressing T-cell transfer, tumor-bearing mice were treated with CD4-depleting antibodies and then with chNKG2D- or wtNKG2D-expressing T cells. CD4 depleting antibodies eliminated both host and transferred CD4+ T cells. Mice injected with CD4-depleting antibodies and treated with chNKG2D-expressing T cells had a higher number of solid tumors and tumor cells within ascites as compared with mice treated with chNKG2D-expressing T cells and Hank’s balanced salt solution (HBSS) (Fig. 4A). However, mice treated with CD4-depleting antibodies and chNKG2D-expressing T cells had lower tumor burden than mice receiving control T cells only. CD4-depletion itself had no effect on tumor growth, since mice treated with control T cells and CD4-depleting antibodies had a similar tumor burden than mice treated with control T cells and HBSS. In addition, the depletion of CD4+ T cells resulted in a lower percentage of host CD8+ T cells producing IFNγ following the administration of chNKG2D-expressing T cells, as well as in a decreased amount of IFNγ produced by peritoneal and spleen cells (Fig. 4B and C). These results demonstrate that CD4+ T cells are critical for optimal tumor elimination and host CD8+ T-cell IFNγ production.

Figure 4. CD4+ T cells are necessary for optimal tumor protection. (A and B) Tumor-bearing mice were injected with anti-CD4 depleting antibodies (GK1.5) on day 33, 39 and 45 and treated with CD45.1+ wtNKG2D-expressing or chNKG2D-expressing T cells on day 35. (A) Eight weeks after tumor-cell injection, the number of solid tumors on the peritoneal wall and number of tumor cells in the peritoneal wash was assessed. (B) Spleen and peritoneal cells were isolated from mice 8 weeks after tumor cell injection and cultured for 24 h. Cell-free supernatants were then assessed for the presence of interferon γ (IFNγ). (C) Tumor bearing mice were injected with anti-CD4 depleting antibodies on day 33 and then treated with T cells on day 35. Peritoneal cells were harvested 7 d after T-cell transfer and CD45.2+CD8b+ cells were assayed for IFNγ expression by flow cytometry. Cumulative data of two independent experiments are shown. The average of each group and SD (n = 8) are shown (*p < 0.01; ***p < 0.001 as compared with mice receiving wtNKG2D-expressing T cells; †p < 0.05 as compared with non-depleted mice receiving chNKG2D-expressing T cells).
Host CD4+ T cells are sufficient for tumor elimination in the absence of transferred CD4+ T cells
Both CD8+ and CD4+ T cells are adoptively transferred during the infusion of NKG2D-expressing CAR T cells. Adoptively transferred CD4+ T cells have been shown to mediate antitumor immune responses in other studies, but the specific role of host CD4+ T cells in NKG2D CAR-expressing T-cell transfer is unclear.32-34 In addition, the administration of anti-CD4 antibodies removes both transferred and host CD4+ T cells. To determine whether host CD4+ T cells are sufficient for tumor protection in the absence of transferred CD4+ chNKG2D-expressing T cells, tumor-bearing mice were treated with wtNKG2D-expressing T cells, total chNKG2D-expressingT cells, purified CD8+ chNKG2D-expressing T cells and purified CD4+ chNKG2D-expressing T cells. Total chNKG2D-expressing T cells consist of 10–20% CD4+ cells, although few of the CD4+ T cells express NKG2D because they lack DAP10, which is required for NKG2D expression at the cell surface (Fig. 5A). Mice treated with purified CD8+ chNKG2D-expressing T cells had fewer solid tumors on the peritoneal wall and fewer free tumor cells as compared with control T-cell treated mice, indicating that purified CD8+ chNKG2D-expressing T cells mediated antitumor effects similar to total chNKG2D-expressing T cells (Fig. 5B). Mice treated with purified CD4+ chNKG2D-expressing T cells had no reduction in the number of solid tumors or free tumor cells in ascites as compared with control T-cell treated mice. Taken together with the results depicted in Figure 4, these data demonstrate that host CD4+ T cells are sufficient for tumor elimination as mediated by NKG2D CAR-expressing T cells. To determine whether CD4+ T cells from the CAR-expressing T-cell population respond to NKG2D ligand-positive tumor cells, control, total and purified CD4+ and CD8+ chNKG2D-expressing T cells were stimulated with ligand-positive and -negative tumor cells in vitro. CD4+ chNKG2D-expressing T cells produced low amounts of IFNγ when stimulated with ID8 tumor cells or RMA-Rae1 lymphoma cells, which express the NKG2D ligand Rae-1 (Fig. 5C). Conversely, total chNKG2D-expressing T cells and purified CD8+ chNKG2D-expressing T cells produced similar amounts of IFNγ in response to ID8 and RMA-Rae1 cells, but did not respond when stimulated with RMA tumor cells, which lack the expression of NKG2D ligands.
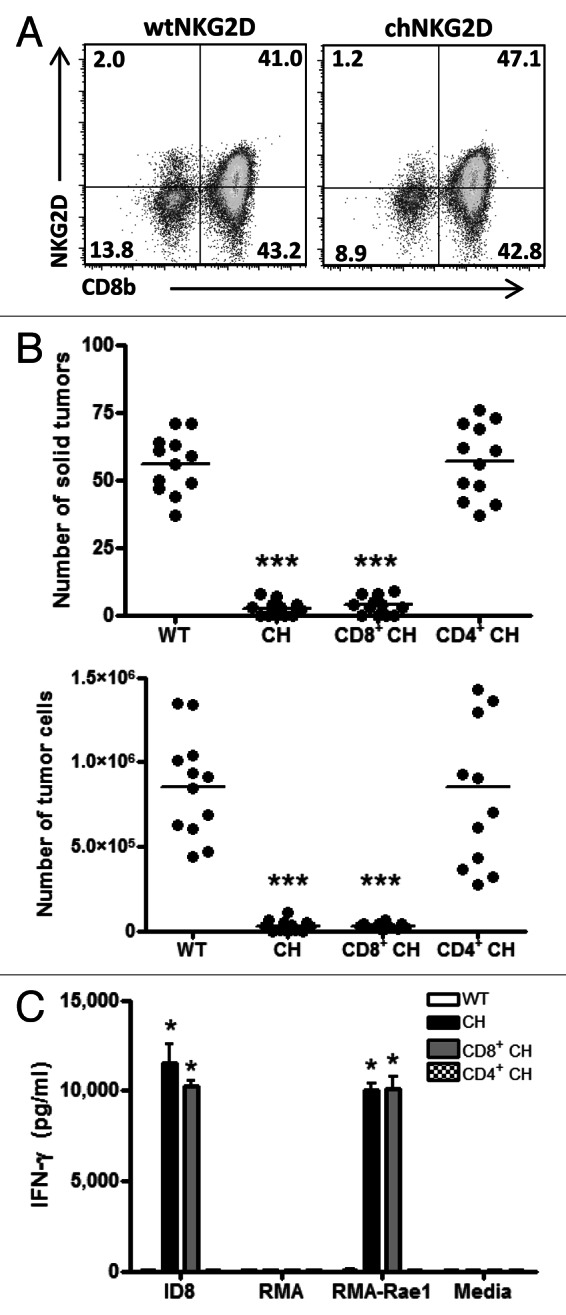
Figure 5. Adoptively transferred CD8+ T cells are sufficient for tumor elimination in the presence of host CD4+ T cells. (A–C) Mice bearing 5-week ID8 tumors were treated with wtNKG2D-expressing, chNKG2D-expressing, purified CD8+ chNKG2D-expressing or purified CD4+ chNKG2D-expressing T cells. (A) chNKG2D-expressing T cells gated on CD3e+ cells were assayed for NKG2D and CD8b expression by flow cytometry. (B) Eight weeks after tumor cell injection, the number of solid tumors on the peritoneal wall and number of tumor cells in the peritoneal wash was assessed. The average of each group and SD (n = 12) are shown. (C) WtNKG2D-expressing, chNKG2D-expressing, CD8+ chNKG2D-expressing and CD4+ chNKG2D-expressing T cells were cultured in standard conditions or together with RMA, RMA-Rae1 or ID8 tumor cells. Cell-free supernatants were assayed for interferon γ (IFNγ) production after 24 h. (*p < 0.01; ***p < 0.001 as compared with wtNKG2D-expressing T cells)
Host CD4+ T cells are necessary for the development of a memory response but not for tumor elimination
To determine if host CD4+ T cells are required for complete tumor elimination after the administration of CAR-expressing T cells, C57BL/6 or MHC Class II-deficient tumor-bearing mice were treated with chNKG2D-expressing or wtNKG2D-expressing T cells, and the number of solid tumors and free tumor cells in ascites was determined. Unlike CD4-deficient mice, MHC Class II-deficient mice lack all Class II-reactive T cells. Hosts deficient in MHC Class II molecules treated with chNKG2D-expressing T cells had as few solid tumors as MHC Class II-wildtype hosts (Fig. 6A). However, the lack of MHC Class II molecules in the host resulted in a low number of free tumor cells in ascites upon the treatment with chNKG2D-expressing T cells. Such a reduction in free tumor cells may be due to a lack of CD4+ T regulatory cells or to elevated numbers of CD8+ T cells in MHC Class II-deficient animals.35 In addition, it is possible that transferred CD4+ T cells promote tumor elimination in the absence of host CD4+ T cells. To get further insights into this issue, MHC Class II-deficient hosts were treated with wtNKG2D-expressing T cells, total chNKG2D-expressing T cells or purified CD8+ chNKG2D-expressing T cells. MHC Class II-deficient hosts treated with CD8+ chNKG2D-expressing T cells exhibited a reduced tumor burden as compared with control T-cell treated mice, but increased tumor burden as compared with MHC Class II-deficient mice treated with total chNKG2D-expressing T cells (Fig. 6B). The transfer of chNKG2D-expressing T cells resulted in lower tumor burden in the absence of host and transferred CD4+ T cells than the administration of wtNKG2D-expressing T cells. These data demonstrate that transferred CD4+ T cells promote optimal tumor elimination by chNKG2D-expressing T-cell therapy in the absence of host CD4+ T cells. Taken together, these results indicate that host CD4+ T cells are sufficient, but not necessary, for optimal tumor elimination as mediated by the adoptive transfer of NKG2D CAR-expressing T cells.

Figure 6. Host CD4+ T cells are required for the development of a tumor-specific memory T-cell response, but not for tumor protection mediated by chNKG2D-expressing T cells. (A) C57BL/6 and MHC Class II-deficient mice bearing 5-week ID8 tumors were treated with wtNKG2D-expressing T cells or chNKG2D-expressing T cells. Eight weeks after tumor cell injection, the number of solid tumors on the peritoneal wall and number of tumor cells in the peritoneal wash was assessed. Cumulative data of two independent experiments are shown. The average of each group and SD (n = 8) are shown (**p < 0.01; ***p < 0.001 as compared with C57BL/6 mice receiving wtNKG2D-expressing T cells; †p < 0.05 as compared with C57BL/6 mice receiving chNKG2D-expressing T cells). (B) MHC Class II-deficient mice bearing ID8 tumors were treated with wtNKG2D-expressing, chNKG2D-expressing or purified CD8+ chNKG2D-expressing T cells 5 weeks after tumor inoculation. The number of solid tumors on the peritoneal wall and number of tumor cells in the peritoneal wash was assessed. Cumulative data of two independent experiments are shown. The average of each group and SD (n = 8) are shown. (***p < 0.001 as compared with mice receiving wtNKG2D-expressing T cells; †p < 0.05 as compared with mice receiving chNKG2D-expressing T cells). (C) C57BL/6 or MHC Class II-deficient mice bearing ID8 tumors were treated with chNKG2D-expressing T cells and control C57BL/6 mice were treated with Hank’s balanced salt solution (HBSS) 1, 2 and 3 weeks after tumor-cell inoculation. The survival of the mice was measured (n = 8–10 mice) (*p < 0.01 as compared with HBSS-treated mice). (D) Two-hundred days after tumor-cell injection, spleen cells from surviving or naïve mice were cultured with ID8 cells, RMA tumor cells or medium alone. Cell-free supernatants were analyzed for interferon γ (IFNγ) production (***p < 0.001 as compared with spleen cells from naïve mice; †p < 0.01 vs. as compared with spleen cells from MHC Class II-deficient mice). (E) Spleen cells from tumor-surviving mice (day 200) or from naïve mice were stimulated with ID8 tumor cells and IFNγ production by CD8b+ T cells was assessed by intracellular flow cytometry (*p < 0.05 as compared with spleen cells from naïve mice).
Treatment chNKG2D T cells has been shown to induce host memory responses against tumor antigens and to result in long-term tumor-free survival, and CD4+ T cells are known to be involved in the development and maintenance of immunological memory.17,36 To test whether CD4+ T lymphocytes are involved in long-term protective immunity, B6 and MHC Class II-deficient mice were analyzed for the development of tumor-specific memory after the administration of CAR-expressing T cells. The treatment of tumor-bearing C57BL/6 or MHC Class II-deficient mice with chNKG2D-expressing T cells increased survival as compared with HBSS-treated, control mice, and the survival of MHC Class II-deficient mice was similar to that of C57BL/6 mice (Fig. 6C). To determine if host CD4+ T cells are necessary to develop a memory response to ID8 tumor cells, spleen cells from long-term surviving mice (day 200) were cultured in the presence of ID8 cells, RMA cells or culture medium only. Spleen cells from long-term surviving mice lacking MHC Class II molecules produced much lower amounts of IFNγ when cultured with ID8 tumor cells than spleen cells isolated from long-term surviving MHC Class II-wildtype (C57BL/6) mice (Fig. 6D). Spleen cells from both long-term surviving C57BL/6 and MHC Class II-deficient mice did not produce IFNγ in response to RMA cells or media alone, demonstrating tumor specificity. A higher percentage of CD8+ T cells from long-term surviving C57BL/6 mice treated with chNKG2D-expressing T cells produced IFNγ in response to ID8 re-stimulation as compared with T cells from long-term surviving MHC Class II-deficient or naïve control mice (Fig. 6E). These data demonstrate that the adoptive transfer of chNKG2D-expressing T cells induces long-term tumor-free survival even in the absence of endogenous CD4+ T cells, but that host CD4+ T cells contribute to the generation of an optimal memory CD8+ T-cell response.
Host CD8+ are necessary for tumor elimination
CD4+ T cells mediate antitumor immunity against MHC Class II-negative tumor cells by regulating CD8+ T-cell responses.37 To determine the role of host CD8+ T cells in this setting, CD8-deficient tumor-bearing mice were treated with chNKG2D- or wtNKG2D-expressing T cells and tumor burden was assessed. Wild-type C57BL/6 mice had a lower number of solid tumors and tumor cells in ascites as compared with CD8-deficient tumor-bearing hosts treated with chNKG2D-expressing T cells (Fig. 7B). Even so, CD8-deficient hosts treated with chNKG2D-expressing T cells had a significantly reduced tumor burden as compared with mice receiving wtNKG2D-expressing T cells, demonstrating that chNKG2D-expressing T cells have a substantial effect on tumor growth even in the absence of host CD8+ T cells. CD8 deficiency impaired local and systemic antitumor immunity, since spleen and peritoneal cells from CD8-deficient mice treated with NKG2D CAR-expressing T cells produced lower amounts of IFNγ as compared with the same cells obtained from CD8-wildtype mice receiving CAR-expressing T cells (Fig. 7B). However spleen and peritoneal cells from CD8-deficient mice treated with CAR-expressing T cells produced higher amounts of IFNγ than cells from mice treated with wtNKG2D-expressing T cells. These data demonstrate that host CD8+ T cells contribute to antitumor immune responses and are necessary for the optimal inhibition of tumor growth as mediated by NKG2D CAR-expressing T cells.
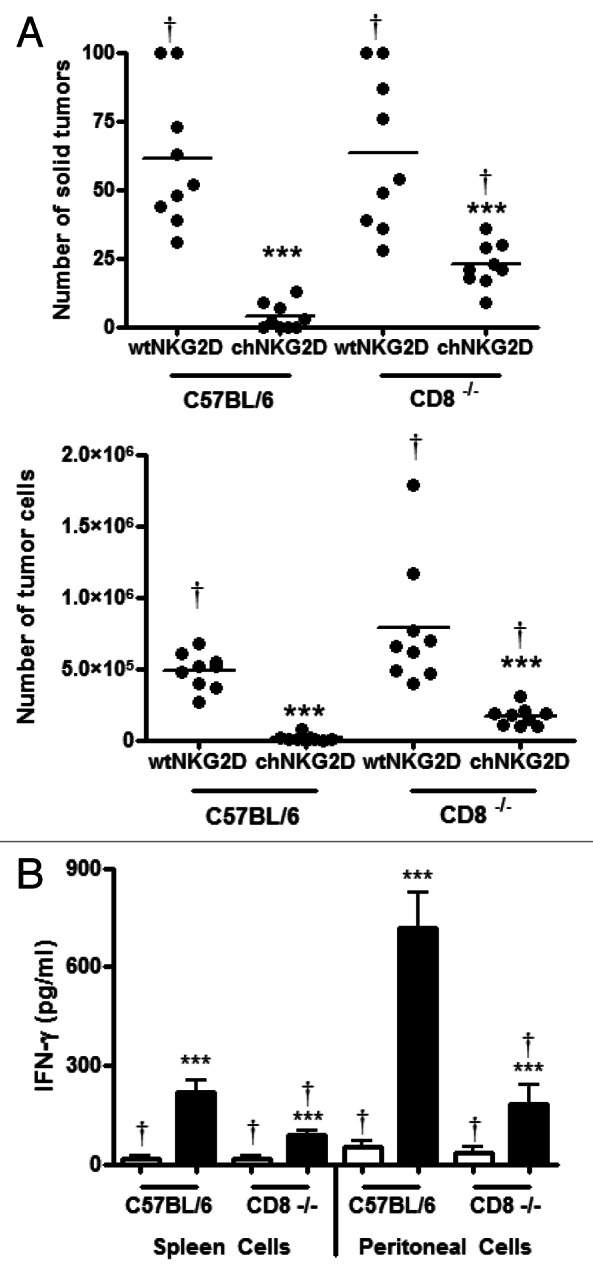
Figure 7. Host CD8+ T cells are necessary for complete tumor elimination mediated by chNKG2D-expressing T cells. (A and B) C57BL/6 and Cd8−/− mice bearing 5-week ID8 tumors were treated with wtNKG2D-expressing or chNKG2D-expressing T cells. (A) Eight weeks after tumor-cell inoculation, the number of solid tumors on the peritoneal wall and number of tumor cells in the peritoneal wash was assessed. (B) Spleen and peritoneal cells were isolated from mice 8 weeks after tumor cell injection and cultured for 24 h. Cell-free supernatants were then assessed for interferon γ (IFNγ) production. The average of each group and SD (n = 8) are shown (***p < 0.001 as compared with C57BL/6 mice receiving wtNKG2D-expressing cells; †p < 0.05 as compared with C57BL/6 mice receiving chNKG2D-expressing cells).
Discussion
The two main goals of anticancer immunotherapy are tumor eradication and the establishment of long-term tumor-free survival. Harnessing the host immune system is one way to promote tumor destruction and achieve these goals. Although reports indicate that host pre-conditioning strategies such as lymphodepletion are important for the success of adoptive T-cell transfer, the present study demonstrates that treatment with NKG2D CAR-expressing T cells requires host T cells for complete tumor eradication and the development of a tumor-specific memory CD8+ T-cell response after tumor clearance. Pretreatment with cyclophosphamide to deplete host lymphocytes neither improved nor worsened the efficacy of NKG2D CAR-expressing T cell-based therapy in a myeloma model.29 However, tumor elimination as mediated by NKG2D CAR-expressing T cells is impaired in the absence of host T cells.24 These findings indicate that the administration of cyclophosphamide prior to the infusion of NKG2D CAR-expressing T cells may exert antitumor benefits, but the immunostimulatory effects of NKG2D CAR-expressing T cell-based therapy are diminished in lymphodepleted hosts.
The administration of CAR-expressing T cells increased antigen-specific T-cell proliferation and T-cell recruitment to the tumor site. However, some non-specific CD4+ T-cell proliferation and recruitment was also detected upon the transfer of chNKG2D-expressing T cells. The increase in proliferation of OT-II cells as induced by lymph node and spleen cells upon chNKG2D-expressing T-cell transfer may be induced by γ chain cytokines, which promote homeostatic T-cell proliferation in the absence of antigens. In addition, inflammation as induced by chNKG2D-expressing T-cell transfer may promote the recruitment of T cells to the tumor site in an antigen-independent manner, through the increased expression of chemokines and adhesion molecules. CAR-expressing T cell-derived IFNγ and GM-CSF were required for increased antigen-specific T-cell proliferation. These cytokines are known to induce the maturation of dendritic cells and their subsequent trafficking to the tumor-draining lymph nodes, where they initiate endogenous immunity. The increased number of antigen-specific T cells in draining lymph nodes and at the tumor site in mice treated with NKG2D CAR-expressing T cells is due to increased T-cell proliferation and trafficking.
The transfer of chNKG2D-expressing T cells increased the number of endogenous CD4+ and CD8+ T cells at the tumor site in a CXCR3-dependent manner. However, it also reduced the numbers of CD4+ T cells at the tumor site as compared with the administration of wtNKG2D-expressing T cells. The transfer of chNKG2D-expressing T cells has been shown to reduce the number of CD4+FOXP3+ regulatory T cells (Tregs) at the tumor site in a perforin-dependent mechanism.31 This may account for the reduced numbers of CD4+ T cells at the tumor site in mice treated with chNKG2D-expressing T cells.
IFNγ produced by CD8+ chNKG2D-expressing T cells presumably acts in a direct fashion on leukocytes at the tumor site to induce the production of CXCL9 and CXCL10. F4/80+ and F4/80− cells at the tumor site produced higher amounts of CXCL9 and CXCL10 upon the transfer of CAR-expressing T cells. Intratumoral F4/80+ cells express higher amounts of IFNγ receptor upon chNKG2D-expressing T-cell administration, which may enhance their ability to respond to CAR-expressing T cell-derived IFNγ 28. Because macrophages cannot induce the autocrine production of CXCL9 and CXCL10, IFNγ as secreted by T and NK cells in the F4/80− cell fraction may be responsible for the elevated amount of CXCR3 ligands that we observed when F4/80+ and F4/80− cells were co-cultured. In summary, chNKG2D-expressing T cell-derived effector cytokines regulate the activation of F4/80+ cells, which subsequently promote the recruitment of endogenous T cells to the tumor site through the secretion of CXCL9 and CXCL10.
The administration of GK1.5 CD4-depleting antibodies impaired tumor elimination as mediated by NKG2D CAR-expressing T cells. CD4+ T cells can mediate antitumor immunity against MHC Class II-negative cancer cells by regulating CD8+ T-cell effector functions.36,38 In the context of NKG2D CAR-expressing T-cell transfer, the depletion of CD4+ cells impaired IFNγ production by host CD8+ T cells indicating that CD4+ T-cell help is crucial for the acquisition of optimal effector functions by host CD8+ T cells. The transfer of purified CD8+ chNKG2D-expressing T cells mediated tumor protection as well as that of total chNKG2D-expressing T cells, demonstrating that host CD4+ T cells are sufficient for tumor protection as mediated by T cell therapy in the absence of transferred CD4+ T cells. However, tumor elimination was not impaired in MHC Class II-deficient mice, indicating that endogenous CD4+ T cells are sufficient, but not necessary, for tumor elimination. In fact, MHC Class II-deficient hosts treated with chNKG2D-expressing T cells had fewer tumor cells compared with similarly treated C57BL/6 mice. The enhanced elimination of tumor cells in MHC Class II-deficient hosts may be due to increased numbers of CD8+ T lymphocytes or to a lack of regulatory T cells in these animals.35 However, it has previously been shown that CD4+FOXP3+ Tregs at the tumor site express NKG2D ligands, and that the administration of chNKG2D-eexpressing T cells reduces the number of intratumoral Tregs in a perforin-dependent mechanism.31 In addition, adoptively transferred CD4+ T cells appear to be in sufficient numbers to act as a surrogate for host CD4+ T cells following adoptive transfer.
The administration of NKG2D CAR-expressing T cells has been shown to induce tumor-specific memory and long-term tumor-free survival in multiple tumor models.17,29,39 Transferred chNKG2D-expressing T cells do not persist in the host for a long period, and endogenous tumor-specific CD4+ and CD8+ T cells mediate protection against tumor re-challenges.17 In this study, we demonstrated that host CD4+ T cells are involved but not necessary for tumor elimination. The lack of survival benefits in MHC Class II-wildtype hosts as compared with MHC II-deficient animals demonstrates that adoptively transferred CD4+ T cells are sufficient for tumor elimination in the absence of host CD4+ T cells. Yet, the impaired T-cell recall response in MHC Class II-deficient hosts demonstrates that host CD4+ T cells promote the development of tumor-specific immunological memory. The defect in the memory T-cell response observed in the absence of host CD4+ T cells is due to impaired memory development and/or maintenance. Transferred NKG2D CAR-expressing CD4+ T cells do not persist in the host for extended periods. Because cytokines secreted by memory CD4+ T cells promote the longevity of memory T-cell populations, the short lifespan of transferred CD4+ T cells in vivo, in the absence of host CD4+ T cells, may result in the erosion of memory T cells. Thus, host CD4+ T cells may be the source of homeostatic cytokines that are required for the maintenance of the memory T-cell population. It is also possible that host CD4+ T-cell help is required during the primary response for the development of a memory T-cell population, as it has previously been shown in other models that a primary CD8+ response can remain intact in the absence of CD4+ T cells, while a memory recall response becomes impaired.36,40 Thus, the reduced number of adoptively transferred CD4+ T cells coupled to their short half-life in vivo, may result in the provision of insufficient signals to promote the initial development of a memory CD8+ T-cell population.
In conclusion, the data presented in this study demonstrate that endogenous T cells are critical for complete tumor elimination and the development of a tumor-specific recall response following adoptive T-cell therapy. Our results demonstrate unique roles for host CD4+ and CD8+ T lymphocytes in the primary and memory antitumor immune responses following adoptive T-cell therapy with CAR-expressing T cells, and how effector cytokines secreted by adoptively transferred T cells shape host T-cell proliferation and trafficking. The potential antitumor immune response mediated by endogenous T lymphocytes and the mechanisms through which adoptively transferred T cells regulate endogenous T-cell responses should be considered for the optimization of adoptive T-cell therapies to enhance tumor eradication and long-term tumor-free survival.
Materials and Methods
Mice
C57BL/6 and B6-Ly5.2Cr (CD45.1+) mice were obtained from The Jackson Laboratory (Bar Harbor, ME) or the National Cancer Institute (Frederick, MD). B6.129P2-Cxcr3tm1Dgen/J (Cxcr3−/−), B6.129S7-Ifngtm1Agt/J (IFNγ-deficient), B6.129S2-H2dlAb1-Ea/J (MHC Class II-deficient) and B6.129S2-CD8atm1Mak/J (Cd8−/−) mice were obtained from The Jackson Laboratory. GM-CSF-deficient mice on a C57BL/6 background were provided by Dr. Jeff Whitsett (University of Cincinnati, Cincinnati, OH). Mice of 7 to 10 weeks of age were used for experimental determinations. All animal work was performed in the Dartmouth Medical School Animal Facility (Lebanon, NH) in accordance with institutional guidelines.
Injection of ID8 cells and treatment of mice with genetically modified T cells
Mouse splenocytes were stimulated with 1 μg/mL concanavalin A for 18 h and transduced with chNKG2D- or wtNKG2D-coding viruses, as previously described.24,26 Two days after viral transduction, T cells were selected in medium containing 0.5 mg/mL G418 and 25 U/mL IL-2 for 3 d. Viable cells were isolated by Histopaque-1083 (Sigma Aldrich) and expanded for 2 d without G418. In some experiments, CD4+ chNKG2D-expressing T cells were purified by positive selection using anti-FITC magnetic beads, according to the manufacturer’s protocol (Miltenyi). The purity of the CD4+ cell product was invariably greater than 95%. The negative fraction was collected after CD4+ T-cell purification and over 98% of these cells were CD8+. ID8-GFP cells (2 × 106) were administered to mice i.p. on day 0. WtNKG2D-expressing or chNKG2D-expressing T cells (5 × 106) were transferred i.p. several weeks after tumor injection, as indicated in figure legends. Mice were sacrificed and peritoneal washes were performed using 10 mL PBS. Red blood cells in the peritoneal washes were lysed with the ACK lysis buffer, the number of leukocytes determined, and other cells and solid tumors analyzed as indicated.
Flow cytometry and isolation of F4/80+cells
Spleen, peritoneal and lymph node cells were incubated with anti-CD16/CD32 and mouse γ globulin (Jackson ImmunoResearch), to block unspecific binding. Cells were then stained with anti-CD45.1 (clone A20), anti-CD45.2 (clone 104), anti-F4/80 (clone BM8), anti-CD3e (clone 145-2C11), anti-CD4 (clone GK1.5), anti-CD8b (clone YTS156.7.7) or anti-CXCR3 (clone CXCR3-173) antibodies. For intracellular staining, 1.5 × 106 spleen cells, isolated from surviving mice 200 d after tumor-cell inoculation, were cultured with 7.5 × 104 ID8 tumor cells in complete medium for 24 h in a 48-well plate. Ten μg/mL brefeldin A (Sigma Aldrich) was added to the cultures for the last 5 h of incubation. Cells were stained with APC-conjugated anti-CD3e and biotin-conjugated anti-CD8b antibodies. Spleen cells were then stained with streptavidin-PerCP, fixed with 1.0% paraformaldehyde, permeabilized with 0.1% saponin, and stained with PE-conjugated anti-IFNγ or PE-conjugated isotype control antibodies. Peritoneal F4/80+ cells were isolated from tumor-bearing mice treated with wtNKG2D-expressing or chNKG2D-expressing T cells, stained with anti-F4/80-FITC and isolated by magnetic bead selection (Miltenyi), according to the manufacturer’s protocol. The purity of the F4/80+ cell fraction after isolation was > 90%.
Cytokine production in secondary cultures
Purified F4/80+ cells (2 × 105), F4/80− cells or combined F4/80+ and F4/80− (1:1) fractions from tumor-bearing mice were cultured for 24 h in complete medium. Cell-free conditioned media were assayed for CXCL9 and CXCL10 using multiplex analysis (Millipore) by DartLab at the Norris Cotton Cancer Center. To assess IFNγ production by transferred T cells, unpurified wtNKG2D-expressing or chNKG2D-expressing T cells, or purified CD4+ or CD8+ chNKG2D-expressing T cells (105) were stimulated with ID8 tumor cells (2.5 × 104), RMA lymphoma cells (105) or RMA-Rae1 (105) tumor cells. Cell-free supernatants were collected after 24 h and IFNγ production was assessed by ELISA. To assess memory recall responses, splenocytes from surviving mice were isolated on day 200. Spleen cells (6.5 × 105) were cultured with ID8 or RMA tumor cells (6.5 × 104 cells) for 24 h. Cell-free conditioned media were assayed for IFNγ by ELISA. Spleen and peritoneal cells (2 × 105) were isolated from mice 3 weeks after treatment with wtNKG2D-expressing or chNKG2D-expressing T cells and cultured for 24 h. Cell-free conditioned media were assayed for IFNγ by ELISA.
CD4+ T-cell depletion
The anti-CD4 depleting antibody GK1.5 (ATCC) was produced in bioreactors by antibody-producing hybridoma cells. Mice were injected with 250 μg anti-CD4 depleting antibody on day 33 or on days 33, 39 and 45 post-tumor inoculation, as indicated in figure legends.
Recruitment of host lymphocytes
Tumor-bearing C57BL/6 or Ccxr3−/− hosts were treated with wtNKG2D-expressing or chNKG2D-expressing T cells 5 weeks after the injection of ID8 tumor cells. Three days after T-cell transfer, peritoneal washes were performed and the number of CD8+ and CD4+ T cells in the peritoneum was assessed by flow cytometry.
In vivo OT-II T cells
CD45.1+ female mice were injected with ID8-GFP-tOVA or ID8-GFP cells (2 × 106) i.p. and treated 5 weeks later with chNKG2D-expressing T cells, wtNKG2D-expressing T cells or PBS. One h before T-cell injection, purified CD45.2+ OT-II cells (2 × 106) were injected i.v. The presence of CD45.2+ cells in the peritoneum and mediastinal lymph nodes was assessed by flow cytometry 3 d after the transfer of OT-II cells.
In vitro tumor antigen presentation
ID8-GFP-tOVA or ID8-GFP tumor-bearing mice were injected with chNKG2D-expressing or wtNKG2D-expressing T cells (5 × 106) i.p. 7 d after tumor cell injection. Spleen and lymph node cells were harvested from naïve or T cell-treated mice 7 d after T-cell injection and used as a source of APCs. CD4+ cells were purified from spleen and lymph nodes of OT-II transgenic mice by magnetic bead separation using anti-CD4 FITC antibodies (Miltenyi), according to the manufacturer’s protocol. These cells were labeled with carboxyfluorescein succinimidyl ester (CSFE) and used in vitro for co-cultures with spleen or lymph node APCs isolated from tumor-bearing or naive mice, as indicated. The proliferation of OT-II T cells was determined by flow cytometry after 96 h.
Tumor burden
ID8-GFP tumors were injected into C57BL/6, MHC Class II-deficient, Cd8−/− or Cxcr3−/− mice. After 5 weeks, chNKG2D-expressing or wtNKG2D-expressing T cells were injected into these mice i.p. Three weeks after T-cell transfer, the number of solid tumors on the peritoneal wall was counted, and a peritoneal wash was performed to assess the number of free tumor cells in the peritoneum.
Quantitative RT-PCR
Peritoneal cells were isolated, and total RNA was extracted using the RNA-Easy MiniKit (Qiagen). cDNA was synthesized using random hexamer primers (Fermentas) in accordance with the manufacturer’s protocol. cDNA (5 ng) was used as a template for quantitative RT-PCR amplification using the SYBR-Green Master Mix (Applied Biosystems) in 25 μL reaction volume, according to the manufacturer’s protocol. Statistical analysis was performed using the ΔCt values.41
Statistical analyses
Differences between groups were analyzed using a Student’s t-test or ANOVA using Prism Software (GraphPad Software). For survival studies, Kaplan-Meier curves were plotted and analyzed using the log-rank test. A Wilcoxon 2-group test was used to compare quantitative RT-PCR data. p values < 0.05 were considered as statistically significant.
Acknowledgments
This work was supported by grants from the National Institutes of Health (CA130911, T32 AI007363) and the Norris Cotton Cancer Center. The views in this paper reflect the authors’ opinions and do not necessarily reflect the opinions of the National Institutes of Health.
The authors thank DartLab for helpful assistance in Luminex analysis (Norris Cotton Cancer Center, Lebanon, NH), Drs. Mary Jo Turk and David Mullins for thoughtful suggestions on the manuscript, and the NIH Biological Resource Branch for providing recombinant human IL-2.
Disclosure of Potential Conflicts of Interest
The NKG2D CAR technology used in this paper is licensed by Celdara Medical, LLC. Dr. Sentman and Celdara are developing the technology for clinical use, for which he receives compensation. These activities are in full compliance with the policies of Dartmouth College.
Footnotes
Previously published online: www.landesbioscience.com/journals/oncoimmunology/article/23564
References
- 1.Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005;202:907–12. doi: 10.1084/jem.20050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008;26:5233–9. doi: 10.1200/JCO.2008.16.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci U S A. 2002;99:16168–73. doi: 10.1073/pnas.242600099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boon T, Van Pel A. Teratocarcinoma cell variants rejected by syngeneic mice: protection of mice immunized with these variants against other variants and against the original malignant cell line. Proc Natl Acad Sci U S A. 1978;75:1519–23. doi: 10.1073/pnas.75.3.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.el-Shami K, Tirosh B, Bar-Haim E, Carmon L, Vadai E, Fridkin M, et al. MHC Class I-restricted epitope spreading in the context of tumor rejection following vaccination with a single immunodominant CTL epitope. Eur J Immunol. 1999;29:3295–301. doi: 10.1002/(SICI)1521-4141(199910)29:10<3295::AID-IMMU3295>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 6.Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012;119:4133–41. doi: 10.1182/blood-2011-12-400044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bos R, Sherman LA. CD4+ T-cell help in the tumor milieu is required for recruitment and cytolytic function of CD8+ T lymphocytes. Cancer Res. 2010;70:8368–77. doi: 10.1158/0008-5472.CAN-10-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ossendorp F, Mengedé E, Camps M, Filius R, Melief CJ. Specific T helper cell requirement for optimal induction of cytotoxic T lymphocytes against major histocompatibility complex class II negative tumors. J Exp Med. 1998;187:693–702. doi: 10.1084/jem.187.5.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Toes RE, Ossendorp F, Offringa R, Melief CJ. CD4 T cells and their role in antitumor immune responses. J Exp Med. 1999;189:753–6. doi: 10.1084/jem.189.5.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corthay A, Skovseth DK, Lundin KU, Røsjø E, Omholt H, Hofgaard PO, et al. Primary antitumor immune response mediated by CD4+ T cells. Immunity. 2005;22:371–83. doi: 10.1016/j.immuni.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 11.Hung K, Hayashi R, Lafond-Walker A, Lowenstein C, Pardoll D, Levitsky H. The central role of CD4(+) T cells in the antitumor immune response. J Exp Med. 1998;188:2357–68. doi: 10.1084/jem.188.12.2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, et al. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J Exp Med. 2008;205:1261–8. doi: 10.1084/jem.20080108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mackey MF, Gunn JR, Ting PP, Kikutani H, Dranoff G, Noelle RJ, et al. Protective immunity induced by tumor vaccines requires interaction between CD40 and its ligand, CD154. Cancer Res. 1997;57:2569–74. [PubMed] [Google Scholar]
- 14.Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. 1998;393:474–8. doi: 10.1038/30989. [DOI] [PubMed] [Google Scholar]
- 15.Sadelain M, Brentjens R, Rivière I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol. 2009;21:215–23. doi: 10.1016/j.coi.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sadelain M, Rivière I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer. 2003;3:35–45. doi: 10.1038/nrc971. [DOI] [PubMed] [Google Scholar]
- 17.Barber A, Zhang T, Sentman CL. Immunotherapy with chimeric NKG2D receptors leads to long-term tumor-free survival and development of host antitumor immunity in murine ovarian cancer. J Immunol. 2008;180:72–8. doi: 10.4049/jimmunol.180.1.72. [DOI] [PubMed] [Google Scholar]
- 18.Stern-Ginossar N, Gur C, Biton M, Horwitz E, Elboim M, Stanietsky N, et al. Human microRNAs regulate stress-induced immune responses mediated by the receptor NKG2D. Nat Immunol. 2008;9:1065–73. doi: 10.1038/ni.1642. [DOI] [PubMed] [Google Scholar]
- 19.Girardi M, Oppenheim DE, Steele CR, Lewis JM, Glusac E, Filler R, et al. Regulation of cutaneous malignancy by gammadelta T cells. Science. 2001;294:605–9. doi: 10.1126/science.1063916. [DOI] [PubMed] [Google Scholar]
- 20.Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol. 2003;3:781–90. doi: 10.1038/nri1199. [DOI] [PubMed] [Google Scholar]
- 21.Sentman CL, Barber MA, Barber A, Zhang T. NK cell receptors as tools in cancer immunotherapy. Adv Cancer Res. 2006;95:249–92. doi: 10.1016/S0065-230X(06)95007-6. [DOI] [PubMed] [Google Scholar]
- 22.Nice TJ, Coscoy L, Raulet DH. Posttranslational regulation of the NKG2D ligand Mult1 in response to cell stress. J Exp Med. 2009;206:287–98. doi: 10.1084/jem.20081335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Champsaur M, Lanier LL. Effect of NKG2D ligand expression on host immune responses. Immunol Rev. 2010;235:267–85. doi: 10.1111/j.0105-2896.2010.00893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barber A, Sentman CL. Chimeric NKG2D T cells require both T cell- and host-derived cytokine secretion and perforin expression to increase tumor antigen presentation and systemic immunity. J Immunol. 2009;183:2365–72. doi: 10.4049/jimmunol.0900721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barber A, Zhang T, DeMars LR, Conejo-Garcia J, Roby KF, Sentman CL. Chimeric NKG2D receptor-bearing T cells as immunotherapy for ovarian cancer. Cancer Res. 2007;67:5003–8. doi: 10.1158/0008-5472.CAN-06-4047. [DOI] [PubMed] [Google Scholar]
- 26.Zhang T, Lemoi BA, Sentman CL. Chimeric NK-receptor-bearing T cells mediate antitumor immunotherapy. Blood. 2005;106:1544–51. doi: 10.1182/blood-2004-11-4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang T, Barber A, Sentman CL. Generation of antitumor responses by genetic modification of primary human T cells with a chimeric NKG2D receptor. Cancer Res. 2006;66:5927–33. doi: 10.1158/0008-5472.CAN-06-0130. [DOI] [PubMed] [Google Scholar]
- 28.Spear P, Barber A, Rynda-Apple A, Sentman CL. Chimeric antigen receptor T cells shape myeloid cell function within the tumor microenvironment through IFN-γ and GM-CSF. J Immunol. 2012;188:6389–98. doi: 10.4049/jimmunol.1103019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barber A, Meehan KR, Sentman CL. Treatment of multiple myeloma with adoptively transferred chimeric NKG2D receptor-expressing T cells. Gene Ther. 2011;18:509–16. doi: 10.1038/gt.2010.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Groom JR, Luster AD. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol. 2011;89:207–15. doi: 10.1038/icb.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barber A, Rynda A, Sentman CL. Chimeric NKG2D expressing T cells eliminate immunosuppression and activate immunity within the ovarian tumor microenvironment. J Immunol. 2009;183:6939–47. doi: 10.4049/jimmunol.0902000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nesbeth YC, Martinez DG, Toraya S, Scarlett UK, Cubillos-Ruiz JR, Rutkowski MR, et al. CD4+ T cells elicit host immune responses to MHC class II-negative ovarian cancer through CCL5 secretion and CD40-mediated licensing of dendritic cells. J Immunol. 2010;184:5654–62. doi: 10.4049/jimmunol.0903247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, et al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med. 2010;207:637–50. doi: 10.1084/jem.20091918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thompson ED, Enriquez HL, Fu YX, Engelhard VH. Tumor masses support naive T cell infiltration, activation, and differentiation into effectors. J Exp Med. 2010;207:1791–804. doi: 10.1084/jem.20092454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Madsen L, Labrecque N, Engberg J, Dierich A, Svejgaard A, Benoist C, et al. Mice lacking all conventional MHC class II genes. Proc Natl Acad Sci U S A. 1999;96:10338–43. doi: 10.1073/pnas.96.18.10338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bevan MJ. Helping the CD8(+) T-cell response. Nat Rev Immunol. 2004;4:595–602. doi: 10.1038/nri1413. [DOI] [PubMed] [Google Scholar]
- 37.Kennedy R, Celis E. Multiple roles for CD4+ T cells in anti-tumor immune responses. Immunol Rev. 2008;222:129–44. doi: 10.1111/j.1600-065X.2008.00616.x. [DOI] [PubMed] [Google Scholar]
- 38.Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998;393:480–3. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- 39.Zhang T, Barber A, Sentman CL. Chimeric NKG2D modified T cells inhibit systemic T-cell lymphoma growth in a manner involving multiple cytokines and cytotoxic pathways. Cancer Res. 2007;67:11029–36. doi: 10.1158/0008-5472.CAN-07-2251. [DOI] [PubMed] [Google Scholar]
- 40.Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 2003;421:852–6. doi: 10.1038/nature01441. [DOI] [PubMed] [Google Scholar]
- 41.Yuan JS, Reed A, Chen F, Stewart CN., Jr. Statistical analysis of real-time PCR data. BMC Bioinformatics. 2006;7:85. doi: 10.1186/1471-2105-7-85. [DOI] [PMC free article] [PubMed] [Google Scholar]