

Abstract
There is an urgent need for rapid and highly sensitive detection of pathogen-derived DNA in a point-of-care (POC) device for diagnostics in hospitals and clinics. This device needs to work in a ‘sample-in-result-out’ mode with minimum number of steps so that it can be completely integrated into a cheap and simple instrument. We have developed a method that directly detects unamplified DNA, and demonstrate its sensitivity on realistically sized 5 kbp target DNA fragments of Micrococcus luteus in small sample volumes of 20 μL. The assay consists of capturing and accumulating of target DNA on magnetic beads with specific capture oligonucleotides, hybridization of complementary fluorescently labeled detection oligonucleotides, and fluorescence imaging on a miniaturized wide-field fluorescence microscope. Our simple method delivers results in less than 20 minutes with a limit of detection (LOD) of ~5 pM and a linear detection range spanning three orders of magnitude.
Nucleic acid sequences are commonly identified by well-established DNA hybridization based techniques1,2. Improvements in specificity and detection sensitivity have led to the development of various applications including the diagnosis of genetic and infectious diseases3,4 and the investigation of expression levels of genes in cells5,6. However, clinical diagnostics requiring rapid, sensitive, and quantitative detection of specific pathogens or characterization of microbes remains challenging. Sensitive pathogen detection and identification is of crucial importance as it allows early medical treatment and thus increases the chances of success7 and reduces follow-up costs8,9.
Although polymerase chain reaction (PCR) and the cultivation of bacterial strains are used as standard methods, they also have disadvantages that complicate their implementation in point-of-care (POC) systems. On the one hand, bacterial cultivation is - presupposing a cultivable microbe - very time consuming requiring several hours to days, especially when used for slow-growing microorganisms. On the other hand, amplification of contaminants or unspecific primer hybridization can cause PCR errors resulting in false positive signals and wrong identification. Furthermore, formation of primer-dimers and other PCR-derived artifacts limit the multiplexing capacity to 10–20 targets10,11,12. Finally, the amplification required for PCR adds additional time to the assay (at least 10–20 minutes), when for POC applications, the time-to-result is of critical importance for clinical benefit13. The ideal POC assay delivers results in <1 h, allowing patients to collect the results and any appropriate medication at an early stage. Therefore, the development of new refined and fast on-site diagnostic systems is of utmost importance for the early-stage detection and treatment of infectious diseases. Especially the problem of multi resistant strains caused by a negligent antimicrobial drug policy can be limited by early-stage identification of pathogens and diligent treatment with antibiotics14.
In order to provide a fast, reliable and comparably easy method the process for on-site quantitative pathogen identification has to be simplified, minimizing the number of error-prone assay steps to enable integration in a POC device. Concerning improvements in DNA detection specificity and sensitivity, several novel methods for direct specific detection of DNA or RNA have been developed in the last years. They are based on confocal15,16,17 or wide-field18 fluorescence microscopy as well as on electrochemical19,20 and other physical techniques21,22,23,24. Despite impressive improvements made regarding ultrasensitive DNA detection, e.g. at the single-molecule level16,17,25,26, the issue is still the development and refinement of methods that are able to combine high detection sensitivity and specificity with the desired processing speed and applicability for use as a reliable tool for miniaturized in vitro diagnostic (IVD) systems.
The critical evaluation of different published methods identifies also obvious disadvantages that prevent their efficient integration in lab-on-a-chip (LOC) assemblies. Beside complex multistep assay procedures and long processing times, most methods require highly complex and expensive hardware assemblies that are difficult to miniaturize. Furthermore, often the sensitivity of single-molecule detection techniques is demonstrated using synthetic short single-stranded DNA target sequences with a length of a few tens of nucleotides. This is, unfortunately, far away from real-world patient DNA samples consisting typically of double-stranded DNA samples with a length of a few hundred to thousands of nucleotides.
Here we introduce a very simple and robust but sensitive fluorescence method for the quantification of realistic long double-stranded DNA fragments based on a new bead fluorescence assay and wide-field microscopy using a miniaturized low cost optical detection system. We demonstrate the potential of the method by quantifying 5 kbp DNA fragments containing the entire glyoxalase gene of the organism M. luteus. We show that it is possible to detect and quantify DNA targets in the nanomolar to picomolar concentration range without using mechanical supporting tools or complex optical applications like flow cytometry27,28 or single-molecule fluorescence detection. Our assay relies on efficient binding of the target sequence on paramagnetic beads by specific capture oligonucleotides (capture probes) and fluorescence detection by the hybridization of a second fluorescently labeled complementary detector oligonucleotide (detection probe) (Fig. 1). The method works with 20 μL sample volumes and requires a total assay time of only 20 min to accumulate enough fluorescence signal on the bead surface for the unequivocal identification of a positive signal.
Figure 1. Principle of bead-based DNA quantification.

(a) Target DNA fragments are captured by beads functionalized with specific complementary oligonucleotides (capture probe) and labeled by reporter oligonucleotides with a fluorophore attached (detection probe). (b) Fluorescence of the beads is measured on a compact, robust and simplified wide-field fluorescence microscope. (c–e) Data analysis. (c) Beads are excited with different excitation intensities dependent on their position in the field of view. (d) Principles of data processing. The image is corrected for the variation in excitation across the image and then single beads are selected for analysis. (e) Dependency between mean bead intensity and position in the field of view.  Raw and
Raw and  profile corrected intensity values.
profile corrected intensity values.
Results
Design of the bead-based assay with compact fluorescence microscope
The basic principle of the developed quantitative DNA bead-based assay is depicted in Fig. 1. Briefly, an inverse wide-field fluorescence microscope is used to excite and detect the bead fluorescence on a charge-coupled device (CCD) camera. In terms of cost reduction and on-site applicability, all components used in the fluorescence microscope were chosen to be robust and as simple and low-priced as possible. The abandonment of high-numerical aperture immersion objectives and CCD cameras with high quantum yield comes along with lower collection efficiency and hence detection sensitivity. To demonstrate the impact of objective and CCD camera quality on detection sensitivity we performed comparative assays detecting 150 bp target fragments using (i) a 1.4 NA oil-immersion objective in combination with a CCD camera with ~30% quantum yield (CoolSnap HQ, Roper Scientific) and (ii) a 0.65 NA air-objective with a CCD camera with ~20% quantum efficiency (Pixelfly VGA, PCO). Figure 2a clearly demonstrates that the reduction in detection efficiency sustains a loss in sensitivity of up to 90% dependent on the quality of the microscope equipment. This result indicates that the use of a CCD camera with higher quantum yield (e.g. an electron-multiplying- (EM) CCD camera) can further facilitate the detection of low target concentrations.
Figure 2. Detection efficiency is controlled by microscope equipment and probe characteristics.
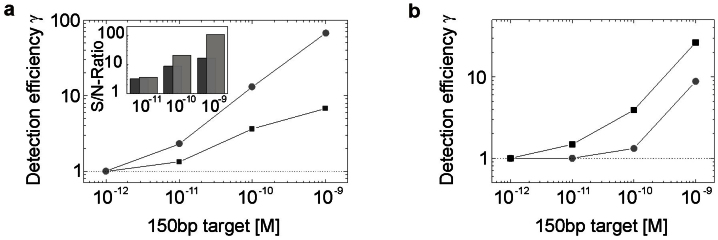
(a) Dependency of the detection sensitivity on microscope equipment. Target fragment denaturation and oligonucleotide hybridization was achieved by heating the sample for 2 minutes at 95°C with subsequent annealing to room temperature. Capturing of target DNA fragments was attained by incubating beads for 60 minutes at room temperature.  High-end fluorescence microscope,
High-end fluorescence microscope,  miniaturized optical breadboard. (b) The substitution of
miniaturized optical breadboard. (b) The substitution of  DNA capturing probes by
DNA capturing probes by  LNA modified strands increases the detection sensitivity. Dotted lines display the mean blank sample intensity level, the detection efficiency γ is defined as the ratio of specific to unspecific fluorescence signal. Error bars representing the standard error of mean bead intensities (SEM, n = 60–89 beads) of acquired images are smaller than data points.
LNA modified strands increases the detection sensitivity. Dotted lines display the mean blank sample intensity level, the detection efficiency γ is defined as the ratio of specific to unspecific fluorescence signal. Error bars representing the standard error of mean bead intensities (SEM, n = 60–89 beads) of acquired images are smaller than data points.
Data analysis
Since the assay procedure is developed for LOC end-to-end diagnostic applications all steps of data processing have to be fully automated. For quantification of the fluorescence intensity of beads the sample has to be either homogeneously illuminated in the entire sampling area or the fluorescence intensity measured for each bead has to be corrected by the laser beam profile. In order to get rid of the dependency between bead fluorescence intensity and its spatial coordinate all images acquired were corrected for inhomogeneous excitation using a homogeneously fluorescing sample (e.g. a fluorophore solution) (Figs. 1c–1e). Moreover it is important to define which beads are selected by the automatic routine and thus contribute to the mean fluorescence intensity value used as quantification parameter. Here, beads showing blurred images, e.g. due to movement, or clustered beads (paramagnetic beads have a pronounced aggregation tendency dependent on their pre-treatment) are discarded from further analysis (Supplementary Fig. 1).
Optimizing detection efficiency
The coating of beads with capture probes also has a significant influence on the detection sensitivity achievable. Here different factors play an important partly converse role. On the one hand, the efficiency of target DNA accumulation on the bead surface is controlled by the density of capture probes per bead. Hence, higher capture probe densities result in higher capturing efficiency and increased detection sensitivity. On the other hand, the accessibility of capture probes decreases with increasing capture probe density due to steric hindrance and Coulombic repulsion29 causing a lower hybridization efficiency30. Another issue is related to the affinity of capture probes which strongly depends on the length of oligonucleotides. It is known from DNA microarray investigations that the length of DNA probes affects the possible density of surface functionalization31. That is, the optimal capture probe density on the bead surface is influenced by both accessibility and affinity of capture probes. These necessities afford a compromise in assay design between length and density of capture probes. In order to increase the affinity of capture probes avoiding extension of the capture sequence we used a chimeric DNA/LNA (locked nucleic acid) capture probe (Table 1) because DNA-LNA duplexes exhibit increased stability32 (Fig. 2b). Furthermore, we selected coupling conditions that yield a capture probe density of around 8 × 1012 molecules cm−2 (data not shown), in accordance with literature values for optimal coating densities29,30.
Table 1. Sequences of the used oligonucleotides. Letters in bold indicate LNA nucleotides.
| Description/Name | Sequence from 5′ to 3′ |
|---|---|
| Capture Strand | Amino modifier C6-AAAAAAAAAA- CCGGCGAGGAGTTCGGCCACTACCG |
| Detection Strand | Fluorophore-GATGATCACCAAGGACGGCGCCTCC |
| Blocking Oligomer 1 | GATCCCCCGCCTGGA |
| Blocking Oligomer 2 | TCGACTACACCGCCCCCGACTTCGC |
| Blocking Oligomer 3 | CCGCCAGCGGGCCTTCTACGAGGCC |
| Blocking Oligomer 4 | CTTTTCGGTTGGACCTTCACCGACT |
| Blocking Oligomer 5 | CGCCCCGCCCGCCGCGTGGAC |
| 150 bp target fragment | GCGCCGCCCACGGAGGCGCCGTCCTTGGTGATCATCCGGTAGTGGCCGAACTCCTCGCCGGAGTCGGTGAAGGTCCAACCGAAAAGGGCCTCGTAGAAGGCCCGCTGGCGGGCGAAGTCGGGGGCGGTGTAGTCGATCCAGGCGGGGGATC |
Even though at first glance the detection sensitivity seems to be mainly controlled by the hardware properties, i.e. photon collections and detection efficiency (Fig. 2a), the autofluorescence of the beads and non-specific binding of detection probes, i.e. the background signal, also significantly influence the achievable detection limit. For example, the autofluorescence of different commercially-available magnetic beads upon excitation at 640 nm varies dramatically (see Supplementary Fig. S2). As a consequence, the signal which is not generated by specific binding events of target-detection DNA complexes determines the number of labeled targets that can be detected above this background signal. Non-specific binding of detection probes to beads can have three possible causes: (i) non-specific or partial hybridization of capture and detection probes on the target sequence, (ii) cross-hybridization of detection and capture probes (Supplementary Fig. S3) and (iii) non-specific adsorption of detection probes on the bead surface (Supplementary Fig. S4). While non-specific hybridization can be largely suppressed by the choice of appropriate DNA sequences (Supplementary Fig. S3), adsorption on the bead surface is partly controlled by the hydrophobicity of the fluorophore structure (Supplementary Fig. S2), the accessibility of the surface, or in other words the capture probe density (Supplementary Fig. S5), and the passivation of the bead surface. Figure 3 demonstrates that adsorption of free Alexa Fluor 647 dye shows an approx. 200-fold lower non-specific background signal than Atto 647N under otherwise identical conditions. This result emphasizes also the importance of efficient removal of free fluorophores from the detection probe sample. The background signal resulting from non-specific adsorption decreases when fluorophores are conjugated to the detection probe oligonucleotide but is still significantly different for different fluorophores (Supplementary Fig. S6); even the lowest non-specific background signal achieved is significantly higher than the autofluorescence of blank beads (Fig. 4). Furthermore, the concentration of detection probes used in the experiments influences the non-specific background intensity (Supplementary Fig. S7). According to these experiments we selected paramagnetic 4.7-μm Varian beads, LNA-modified capture probes and Alexa Fluor 647 labeled detection probes at a concentration of 1 μM in the following assay experiments.
Figure 3. Nonspecific adsorption of fluorophores (1 μM) on beads.
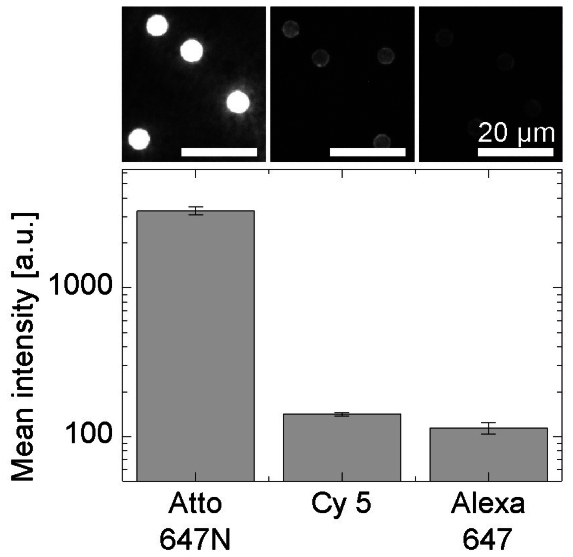
The fluorophores used in this study were obtained as N-hydroxysuccinimidyl esters. When dissolved in water, N-hydroxysuccinimidyl esters are hydrolyzed to carboxylic acids within a few hours. Beads exposed to Atto 647N, Cy5 and Alexa Fluor 647 were imaged with integrations times of 10, 100 and 100 ms, respectively. Error bars display the standard error of mean bead intensities (SEM) with n = 50.
Figure 4. Fluorescence intensity at the lowest detected target concentration.
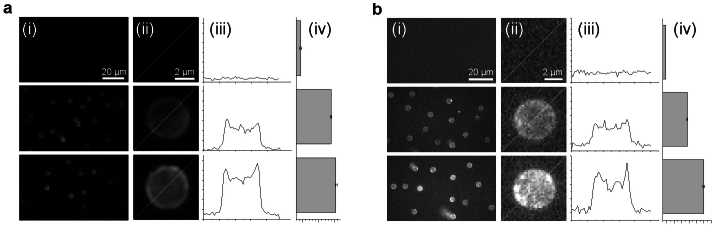
Images obtained using (a) Atto 647N labeled detection probes (100 pM [target]) and (b) Alexa Fluor 647 labeled detection probes (10 pM [target]). First row: autofluorescence; second row: unspecific signal; third row: specific signal for 5 kbp target quantification. (i) Fluorescence images of beads. (ii) Expanded image of a selected bead. (iii) Fluorescence intensity profile plot of the bead shown in (ii). (iv) Mean fluorescence intensity and SEM of beads (n = 100).
The detection efficiency γ of target DNA strands can be determined as the normalized specific signal which is the quotient of specific to unspecific fluorescence intensity. The detection efficiency describes the binding events detected above unspecific background and results in γ-values >1 for positive results. Moreover this value enables the comparison of intensity increase upon target binding between different measurements with varying non-specific background (Supplementary Figs. S4 and S6). In addition it nicely demonstrates that the detection efficiency can be enhanced by either increasing the specific or decreasing the non-specific signal e.g. by the use of higher-affinity capture probes or less hydrophobic detection probes.
Capturing of DNA samples
In order to provide an efficient DNA quantification method applicable for on-site LOC measurements all procedure steps have to be as simple as possible. Therefore the use of magnetic beads is advantageous because they can be easily manipulated in microfluidic devices. Experimentally-complex steps such as separation of labeled target complexes from unbound detector probes, exchanges of reaction buffers or movement of particles can be easily accomplished by applying an external magnetic field. These benefits enable an assay procedure completed in only three processing steps (Fig. 5a). In addition, the utilization of magnetic beads as solid phase also facilitates other assay steps.
Figure 5. Procedure scheme for DNA quantification.
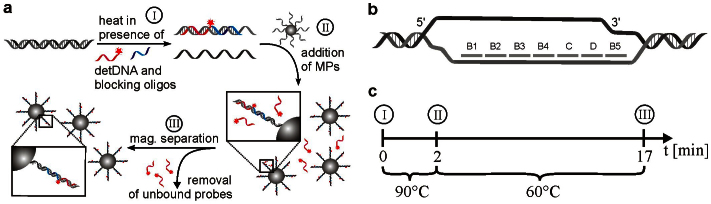
(a) Overview of processing steps. (MPs: microparticles) (b) Oligonucleotide hybridization sequences. Blocking oligonucleotides (B1-5) assure the prevention of re-hybridization and thus the accessibility of capture probe (C) and detection probe (D) binding sequences on the target strand. (c) Sweep line. The whole assay procedure is accomplished in only three procedure steps in just 17 minutes processing time.
For example, the hybridization efficiency can be enhanced substantially33 by active movement of particles since the reaction radius of target DNA molecules is independent of Brownian motion. In combination with LNA as capture probes the processing time of the entire assay can be drastically shortened as compared to other DNA assay procedures21,25,26 and can be completed in only 17 minutes (Fig. 5c). Mixing also increases the local concentration of target DNA molecules in close vicinity to the bead and thus supports efficient capturing and accumulation on the surface. This enables high detection sensitivities comparable to single-molecule detection schemes using sample volumes of only 20 μL.
The next critical step of the assay procedure is the efficient hybridization of long (5–7 kbp) realistic target fragments which can be obtained for example from an upstream disposed LOC fragmentation structure34. Because the DNA fragments are generated as double-strands, efficient denaturation and prevention of re-hybridization is of utmost importance in order to guarantee optimal hybridization of capture and detection probes. Therefore, the samples were heated to 95°C in the presence of a large excess (1 μM) of short blocking oligonucleotides (Table 1). The high temperature ensures efficient thermal denaturation35 while the presence of blocking oligonucleotides which bind next to probe binding regions guarantees accessibility for probes and prevents re-hybridization during annealing26,36, respectively (Fig. 5b).
Detection sensitivity
Application of the optimized assay procedure enables the specific detection and quantification of 5 kbp target DNA fragments down to the picomolar concentration range (Fig. 6). Especially minimization of non-specific binding of detection probes is a key step to improve detection sensitivity, affording a ten-fold increase in detection sensitivity between Atto 647N- and Alexa Fluor 647-labeled detection probes under otherwise identical experimental conditions (Fig. 6a).
Figure 6. Quantification of 5 kbp DNA fragments.
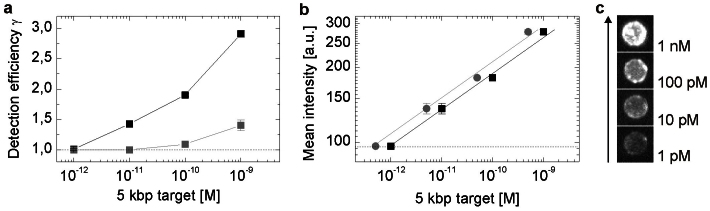
(a) Comparison of detection efficiencies between differently labeled detection probes.  Alexa Fluor 647,
Alexa Fluor 647,  Atto 647N. (b) Mean fluorescence intensity values of 5 kbp DNA fragment using Alexa 647 labeled detection probes.
Atto 647N. (b) Mean fluorescence intensity values of 5 kbp DNA fragment using Alexa 647 labeled detection probes.  Concentration of target fragments in spiked samples introduced in the assay,
Concentration of target fragments in spiked samples introduced in the assay,  final concentration while incubation phase. (c) Visualization of increasing bead brightness with ascending target concentration using the Atto 647N label. Dotted lines display the mean blank sample intensity level; error bars represent the standard error of mean bead intensities (SEM, n = 53–97). Solid lines are guides to the eye. Error bars of (a) indicate the standard error of mean intensity (SEM) between three independent measurements demonstrating the reliability of the parameter.
final concentration while incubation phase. (c) Visualization of increasing bead brightness with ascending target concentration using the Atto 647N label. Dotted lines display the mean blank sample intensity level; error bars represent the standard error of mean bead intensities (SEM, n = 53–97). Solid lines are guides to the eye. Error bars of (a) indicate the standard error of mean intensity (SEM) between three independent measurements demonstrating the reliability of the parameter.
Although we find a broad distribution of fluorescence intensity values for single beads with a relative standard deviation (S.D.) of 2–12%, the mean intensity value is very robust using a sample size of n > 50 beads represented in relative standard errors of mean values (SEM) of only 1–6%. The reliability of the mean bead intensity value as a robust quantification parameter is further emphasized by its small relative standard error (3.5–5.1%) corresponding to the standard error of mean values between three independent measurements (Supplementary Fig. S8) and by its high fluorescence intensity stability related to axial focus variations (Supplementary Fig. S9). With the mean bead intensity value defined as quantification parameter the limit of 5 kbp DNA fragment quantification is 5 × 10−12 M (i.e. 0.1 fmol) using the Alexa Fluor 647 detection probe (Fig. 6b). Importantly, this value refers to the target concentration present in the sample at the beginning of the assay and not the concentration of single-stranded target DNA available for hybridization which is often referred to in other DNA detection assays.
To test the specificity of the method we performed experiments in the presence of a large excess (2000-fold) of non-target double-stranded DNA fragments of similar length (7.5 kbp) (Supplementary Fig. S10). Using a target DNA concentration of 10 pM, the detection efficiency only decreased by 22%, clearly demonstrating the high sequence specificity of the probes used. Despite the small variations in the mean bead fluorescence intensity in the absence and presence of an excess of non-target DNA, our results demonstrate the potential of the bead-based assay for quantification of pathogen DNA at picomolar concentrations in complex, real-world patient samples.
Simultaneous detection of multiple targets (multiplexing)
The described method allows for detection of one type of pathogen DNA. It is however desirable to be able to simultaneously detect several different pathogens in one sample. This can be achieved by employing unique capture beads and detection probes for all tested pathogen DNAs, each detection probe with a distinct fluorescent label. The images of the beads incubated with the sample are then recorded in two or more spectral ranges matched to the emission spectra of the fluorophores, and the relative intensities of a bead in all detection channels uniquely identify the pathogen.
The principle is demonstrated in Fig. 7 on a model system consisting of beads labeled with two fluorescent dyes, Atto 488 and Atto Rh6G. The fluorescence images of the two detection channels were combined by calculating the log2 of the ratio of the intensities in both channels on a pixel-by-pixel basis, resulting in a pseudocolor image in which beads with different emission spectra (spectral code) can be clearly distinguished. Furthermore, it should be possible to create additional spectral codes by combining two or more fluorophores, thus increasing the multiplexing capability. This is exemplified by beads labeled with the two fluorophores at the ratio 1:5 (Atto 488:Atto Rh6G) in Fig. 7.
Figure 7. Multiplexing.

Demonstration of multiplexed detection of fluorescently labeled beads by spectrally splitting the image into two channels: green, centered at 535 nm (a) and red, centered at 620 nm (b). Two fluorophores were used to label Dynabeads with a diameter of 2.8 μm: Atto 488 (emission maximum at 523 nm) and Atto Rh6G (emission maximum at 560 nm). Four types of beads are present in the images: unlabeled beads and three color codes: with Atto 488 only, with Atto Rh6G only and with a mixture of the two fluorophores at 1:5 ratio. (c) False color image displaying the logarithm r of the green-to-red ratio, clearly identifying all three types of labeled beads and the weakly autofluorescent unlabeled beads.
Discussion
We present a new method for the specific detection and quantification of double-stranded DNA fragments of 5–7 kbp length in small sample volumes (20 μL) using a miniaturized and robust wide-field fluorescence microscope. With the possibility for simple automatic alignment and the usage of low cost components (overall costs in the range of a few thousand Euros) the setup exhibits the required cost-efficiency for general applicability in diagnostics. The diligent consideration and optimization of several key parameters e.g. number, size and autofluorescence of beads, bead functionalization density, incubation time and technique, the utilization of blocking oligonucleotides and especially the precise development of data analysis software enables the loss in detection sensitivity caused by capturing a 5 kbp dsDNA target and detection system miniaturization to be surmounted.
A further combination of a precise selection of binding regions for capture and detector probes on the target sequence and the use of LNA-modified capture probes increases the affinity of the binding process and paves the way for highly rapid and sensitive LOC DNA quantification assays. The use of magnetic beads allows active mixing to increase the hybridization and capturing efficiency and thus the detection of target DNA fragments with a processing time of only 17 minutes with a LOD of 5–10 pM (0.1–0.2 fmol or approximately 7.8 log10 copies) and quantification over a linear detection range spanning three orders of magnitude. Furthermore, magnetic beads can be easily handled in microfluidic chips by means of permanent magnets and thus are ideally suited as mobile solid phase in LOC assays.
Our method is, however, not as sensitive as PCR-based methods but effectively designed to meet all the requirements to be successfully implemented in POC devices. If coupled with a suitable microfluidic module for nucleic acid purification and concentration for example, it already provides a sufficiently low LOD for the detection of high-load pathogens e.g. influenza (typical loads of 7.5 log10 copies of RNA per mL of nasopharyngeal aspirate37,38). Further increases in the detection sensitivity could be achieved by the use of multiple labeled detection probes, including fluorescent nanobeads. Additionally, the signal obtained could be enhanced by increasing the efficiency with which target sequences are captured; this could be achieved by making a larger surface area available for hybridization through the use of structured or partially porous microparticles, or by enhancing the affinity of the capture probes for their targets by refining the sequence design or using alternative nucleic acid analogues. Such improvements can reasonably be expected to allow rapid detection of a wide range of pathogens at the point of care, using our bead-based method integrated into a lab-on-a-chip IVD system.
Methods
Bead functionalization and assay accomplishment
In order to improve the binding affinity of target DNA on the bead surface we used capture strands modified with locked nucleic acid (LNA) nucleotides. To provide high stability and long lifetime of capturing LNA (capLNA) strands on the surface beads were covalently functionalized using a standard protocol for amide binding chemistry: 1 mg of carboxyl modified magnetic particles (4.7 μm, Varian, pt. no. PL6604-0090AB) were washed two times with 200 μL of MES buffer (2-(N-morpholino)ethansulfonic acid, 0.1 M, pH 5.75). After washing off the supernatant 1.4 nmol of amino-functionalized single stranded LNA was added as well as 30 μL EDAC (1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride) dissolved at 10 mg/mL in MES buffer. While constantly shaking at 700 rpm and 25°C, 30 μL of EDAC was added three times in steps separated by 30 minutes. Subsequently 350 μL MES buffer containing 2% Tween-20 were added to the bead suspension and incubated for another 10 minutes with constant shaking at 700 rpm and 25°C followed finally by magnetic separation. The beads were then washed five times with 1 mL PBS containing 0.05% Tween-20 and 1 mL PBS only, respectively.
Blocking oligonucleotides used to attain accessibility of the required single-stranded target sequence were added at 10 μM and detector probes labeled with ATTO 647N or Alexa Fluor 647 were added at 7 μM to 20 μL samples (50% L6 buffer39) spiked with target DNA fragments at different concentrations. After heating the samples for two minutes to 95°C, 1.3 × 103 capture probe functionalized beads were added to each target sample solution. After a second incubation phase of 15 minutes at 60°C, the beads were washed four times with hot PBS (60°C) and measured on the optical breadboard.
Data acquisition
All images were measured on a miniaturized epi-fluorescence microscope equipped with a 40× air-objective (NA 0.65, Olympus, Thorlabs). A red diode laser emitting at 640 nm (Cube; Coherent) was spectrally filtered by a clean-up filter (Z647/10; Semrock) and coupled into the microscope objective by a dichroic mirror (FF560/659-Di01, Semrock). Fluorescence emission was separated from scattered light by a bandpass filter (ET 700/75) placed in front of a CCD camera (Pixelfly VGA, PCO). All samples were measured in 1536 well microplates (Greiner bio-one, cat.-no. 783856) in a volume of 10 μL and were illuminated for 100 ms at a power density of 56 W/cm2. Data shown in Fig. 6a (Alexa Fluor 647 label) and Fig. 6b was acquired with an integration time of 200 ms and a power density of 1.98 × 102 W/cm2. The bead images demonstrating the multiplexing principle (Fig. 7) were recorded using an Ar/Kr laser (488 nm, Coherent), an oil immersion objective (Zeiss, 100×, NA = 1.46), and an emCCD camera (iXon3 DU897, Andor) with the em gain set to zero.
Data analysis
The fluorescence information is contained in 12-bit TIFF images, which were processed in three steps: (i) image normalization, (ii) identification of beads, and (iii) fluorescence information computation and data visualization (Fig. 1). Since the excitation light intensity distribution of the beam profile follows a Gaussian shape, all images have to be normalized. Images were normalized by multiplying the intensity of each pixel in the acquired image with a factor extracted from an image taken from a free dye solution at the same pixel position. For robust identification of beads a template (structuring element) containing all the characteristic parameters of an imaged bead, such as its diameter, was used to identify the center of each ring structure by morphological analysis, i.e. erosion followed by dilation40. If an identified fluorescent structure does not exhibit a perfect circular shape, as expected for a bead, it was discarded from further analysis. Furthermore, clustered or moving beads identified by their neighbor distance or the image sharpness were rejected from data processing because their mean intensity value is not representative (see Fig. S1). The final output of the processing was the mean intensity value of each bead with its corresponding statistics.
DNA sequences
All detection probes, capture probes and the 150 bp target are of double HPLC grade and were purchased from IBA GmbH, Göttingen Germany with the exception of the LNA-modified capture probe which was obtained from Exiqon (Vedbæk, Denmark) and purified by HPLC by the supplier. As targets served two types of dsDNA fragments: a) A recombinant plasmid containing the entire Glyoxalase/Bleomycin resistance gene of the organism Micrococcus luteus (GenBank: CP001628.1). The generated PCR product by the PwoSUPER YIELD DNA Polymerase (Roche) was cloned in the pCR®4Blunt-TOPO (Zero Blunt® TOPO® PCR Cloning Kit, Life Technologies) and transformed into competent E. coli cells (One Shot® TOP 10 Chemically Competent cells, Life Technologies). Then, in order to obtain fragments of 4818 bp length the extracted plasmid (QIAfilter Plasmid Mega Kit, Qiagen) was linearized with BamHI and purified from agarose gel using NucleoSpin® Gel and PCR Clean-up kit (Macherey-Nagel). The fragment was quantified by Quant-IT™ dsDNA BR Assay Kit (Life Technologies). b) A 150 bp target commercially synthesized by IBA GmbH, Göttingen Germany.
Non-target DNA fragments (HaloTag®-tubulin G1 vector-construct) used as background for specificity experiments were extracted from E. Coli via Miniprep (NucleoSpin® Plasmid, Macherey-Nagel) and linearized using the restriction enzyme EcoRV (Thermo Scientific). Fragments of 7.5 kbp length were subsequently purified from digestion buffer with QIAEX II Gel Extraction Kit (Qiagen) and quantified by absorption spectroscopy (NanoPhotometer® P-300, Implen).
Author Contributions
F.G., P.S., D.K. T.K. and M.S. designed and T.K., M.C., B.N., R.T.R., J.W. and Z.P. performed the experiments. T.K., M.K., B.N., R.T.R., J.W. and Z.P. analyzed the data. F.G., P.S., D.K. and M.S. supervised the project and M.S. and T.K. wrote the manuscript with contributions from F.G., R.T.R. and Z.P. All authors reviewed the manuscript.
Supplementary Material
Acknowledgments
This work was supported by the EU-FP7 project TheraEDGE (Network Contract FP7-216027), which is an industry-driven effort to accelerate the adoption of theranostic applications in Primary Care by pushing Point of Care Test (POCT) technology far beyond its current state-of-the-art by delivering clinical, analytical and operational breakthroughs.
References
- Southern E. M. Detection of specific sequences among DNA fragments feparated by Gel-Electrophoresis. Journal of Molecular Biology 98, 503–517 (1975). [DOI] [PubMed] [Google Scholar]
- Meinkoth J. & Wahl G. Hybridization of nucleic acids immobilized on solid supports. Anal. Biochem. 138, 267–284 (1984). [DOI] [PubMed] [Google Scholar]
- Relman D. A., Loutit J. S., Schmidt T. M., Falkow S. & Tompkins L. S. The agent of bacillary angiomatosis. An approach to the identification of uncultured pathogens. N. Engl. J. Med. 323, 1573–1580 (1990). [DOI] [PubMed] [Google Scholar]
- Nikkari S. et al. Broad-range bacterial detection and the analysis of unexplained death and critical illness. Emerg. Infect. Dis. 8, 188–194 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt S., Kloska S., Altmann T. & Kehr J. Using array hybridization to monitor gene expression at the single cell level. J. Exp. Bot. 53, 2315–2323 (2002). [DOI] [PubMed] [Google Scholar]
- Fan Z. H. et al. Dynamic DNA hybridization on a chip using paramagnetic beads. Anal. Chem. 71, 4851–4859 (1999). [DOI] [PubMed] [Google Scholar]
- Kumar A. et al. Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Critical care medicine 34, 1589–1596 (2006). [DOI] [PubMed] [Google Scholar]
- Mauldin P. D., Salgado C. D., Hansen I. S., Durup D. T. & Bosso J. A. Attributable hospital cost and length of stay associated with health care-associated infections caused by antibiotic-resistant gram-negative bacteria. Antimicrob. Agents Chemother. 54, 109–115 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolau D. P. Current challenges in the management of the infected patient. Curr. Opin. Infect. Dis. 24 Suppl 1, S1–10 (2011). [DOI] [PubMed] [Google Scholar]
- Broude N. E., Zhang L., Woodward K., Englert D. & Cantor C. R. Multiplex allele-specific target amplification based on PCR suppression. Proc. Natl. Acad. Sci. USA 98, 206–211 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syvanen A. C. Toward genome-wide SNP genotyping. Nat. Genet. 37 Suppl, S5–10 (2005). [DOI] [PubMed] [Google Scholar]
- Fredriksson S. et al. Multiplex amplification of all coding sequences within 10 cancer genes by Gene-Collector. Nucleic Acids Res. 35, e47 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissonnette L. & Bergeron M. G. Infectious disease management through point-of-care personalized medicine molecular miagnostic Technologies. Journal of Personalized Medicine 2, 50–70 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micek S. T. et al. Resistance to empiric antimicrobial treatment predicts outcome in severe sepsis associated with gram-negative bacteremia. Journal of Hospital Medicine 6, 405–410 (2011). [DOI] [PubMed] [Google Scholar]
- Zhang C.-Y. & Johnson L. W. Homogenous rapid detection of nucleic acids using two-color quantum dots. Analyst 131, 484–488 (2006). [DOI] [PubMed] [Google Scholar]
- Li H., Ying L., Green J. J., Balasubramanian S. & Klenerman D. Ultrasensitive coincidence fluorescence detection of single DNA molecules. Anal. Chem. 75, 1664–1670 (2003). [DOI] [PubMed] [Google Scholar]
- Yahiatene I., Doose S., Huser T. & Sauer M. Correlation-matrix analysis of two-color coincidence events in single-molecule fluorescence experiments. Anal. Chem. 84, 2729–2736 (2012). [DOI] [PubMed] [Google Scholar]
- Lehr H. P., Reimann M., Brandenburg A., Sulz G. & Klapproth H. Real-time detection of nucleic acid interactions by total internal reflection fluorescence. Anal. Chem. 75, 2414–2420 (2003). [DOI] [PubMed] [Google Scholar]
- Kavanagh P. & Leech D. Redox polymer and probe DNA tethered to gold electrodes for enzyme-amplified amperometric detection of DNA hybridization. Anal. Chem. 78, 2710–2716 (2006). [DOI] [PubMed] [Google Scholar]
- Kukol A., Li P., Estrela P., Ko-Ferrigno P. & Migliorato P. Label-free electrical detection of DNA hybridization for the example of influenza virus gene sequences. Anal. Biochem. 374, 143–153 (2008). [DOI] [PubMed] [Google Scholar]
- Nie L., Yang Y., Li S. & He N. Enhanced DNA detection based on the amplification of gold nanoparticles using quartz crystal microbalance. Nanotechnology 18, 305501 (2007). [Google Scholar]
- Cao Y. C., Jin R. & Mirkin C. A. Nanoparticles with Raman spectroscopic fingerprints for DNA and RNA detection. Science 297, 1536–1540 (2002). [DOI] [PubMed] [Google Scholar]
- Merkoçi A., Aldavert M., Tarrasón G., Eritja R. & Alegret S. Toward an ICPMS-linked DNA assay based on gold nanoparticles immunoconnected through peptide sequences. Anal. Chem. 77, 6500–6503 (2005). [DOI] [PubMed] [Google Scholar]
- Liu C.-H., Li Z.-P., Du B.-A., Duan X.-R. & Wang Y.-C. Silver nanoparticle-based ultrasensitive chemiluminescent detection of DNA hybridization and single-nucleotide polymorphisms. Anal. Chem. 78, 3738–3744 (2006). [DOI] [PubMed] [Google Scholar]
- Nam J. M., Stoeva S. I. & Mirkin C. A. Bio-bar-code-based DNA detection with PCR-like sensitivity. J. Am. Chem. Soc. 126, 5932–5933 (2004). [DOI] [PubMed] [Google Scholar]
- Hill H. D., Vega R. A. & Mirkin C. A. Nonenzymatic detection of bacterial genomic DNA using the bio bar code assay. Anal. Chem. 79, 9218–9223 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horejsh D. et al. A molecular beacon, bead-based assay for the detection of nucleic acids by flow cytometry. Nucleic Acids Res. 33, e13 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe M., Spiro A., Zhang Y.-Z. & Getts R. Multiplexed, particle-based detection of DNA using flow cytometry with 3DNA dendrimers for signal amplification. Cytometry A 60, 135–144 (2004). [DOI] [PubMed] [Google Scholar]
- Vainrub A. & Pettitt B. M. Coulomb blockage of hybridization in two-dimensional DNA arrays. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 66, 041905 (2002). [DOI] [PubMed] [Google Scholar]
- Peterson A. W., Heaton R. J. & Georgiadis R. M. The effect of surface probe density on DNA hybridization. Nucleic Acids Res. 29, 5163–5168 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steel A. B., Levicky R. L., Herne T. M. & Tarlov M. J. Immobilization of nucleic acids at solid surfaces: effect of oligonucleotide length on layer assembly. Biophys J. 79, 975–981 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vester B. & Wengel J. LNA (locked nucleic acid): high-affinity targeting of complementary RNA and DNA. Biochemistry 43, 13233–13241 (2004). [DOI] [PubMed] [Google Scholar]
- Jungell-Nortamo A., Syvanen A. C., Luoma P. & Soderlund H. Nucleic acid sandwich hybridization: enhanced reaction rate with magnetic microparticles as carriers. Mol. Cell. Probes. 2, 281–288 (1988). [DOI] [PubMed] [Google Scholar]
- Shui L., Bomer J. G., Jin M., Carlen E. T. & van den Berg A. Microfluidic DNA fragmentation for on-chip genomic analysis. Nanotechnology 22, 494013 (2011). [DOI] [PubMed] [Google Scholar]
- Archer M. J., Lin B., Wang Z. & Stenger D. A. Magnetic bead-based solid phase for selective extraction of genomic DNA. Anal. Biochem. 355, 285–297 (2006). [DOI] [PubMed] [Google Scholar]
- Minunni M. et al. Detection of fragmented genomic DNA by PCR-free piezoelectric sensing using a denaturation approach. J. Am. Chem. Soc. 127, 7966–7967 (2005). [DOI] [PubMed] [Google Scholar]
- Ngaosuwankul N. et al. Influenza A viral loads in respiratory samples collected from patients infected with pandemic H1N1, seasonal H1N1 and H3N2 viruses. Virol J 7, 75 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong M. D. et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med 12, 1203–1207 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boom R. et al. Rapid and simple method for purification of nucleic acids. J. Clin. Microbiol. 28, 495–503 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soille P. Morphological Image Analysis: Principles and Applications. (Springer-Verlag New York, Inc., 2003). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.