

Abstract
Evolutionary aspects of the genetic architecture of common human diseases remain enigmatic. The results of more than 200 genome-wide association studies published to date were compiled in a catalog (http://www.genome.gov/26525384/). We used cataloged data to determine whether derived (mutant) alleles are associated with higher risk of human disease more frequently than ancestral alleles. We placed all allelic variants into ten categories of population frequency (0%–100%) in 10% increments. We then analyzed the relationship between allelic frequency, evolutionary status of the polymorphic site (ancestral versus derived), and disease risk status (risk versus protection). Given the same population frequency, derived alleles are more likely to be risk associated than ancestral alleles, as are rarer alleles. The common interpretation of this association is that negative selection prevents fixation of the risk variants. However, disease stratification as early or late onset suggests that weak selection against risk-associated alleles is unlikely a major factor shaping genetic architecture of common diseases. Our results clearly suggest that the duration of existence of an allele in a population is more important. Alleles existing longer tend to show weaker linkage disequilibrium with neighboring alleles, including the causal alleles, and are less likely to tag a SNP-disease association.
Keywords: Genome-wide association studies, ancestral allele, derived allele, minor allele frequency
1. Introduction
Genome-wide association studies (GWASs) are a powerful tool for uncovering the genetic architecture of complex human diseases.1–3 The results of more than 200 GWASs have been published to date.3 Although the ultimate goal of each individual GWAS is to identify single-nucleotide polymorphisms (SNPs) associated with increased risk, the results of GWASs can be used to address a broader range of questions, such as those related to the evolution of the genetic architecture of common human diseases. A recent article by Lachance4 demonstrated that GWAS-identified disease-associated alleles are enriched with derived low-frequency variants. We and other researchers5–7 interpreted this association as indicating that weak negative selection may hold the risk alleles at low frequencies, increasing their proportion among rare alleles.
In this study, we used recent GWAS data to address the question of whether ancestral or derived (mutant) alleles are associated with a greater risk (i.e. serve as a risk allele) randomly. The answer to this question is important for understanding the evolutionary history of the genetic control of human diseases.
We conducted separate analyses of early-onset (potentially undergoing negative selection) and late-onset (likely to be evolutionarily neutral) diseases. Contrary to the currently accepted point of view, our results suggest that negative selection is unlikely a major factor underlying the association between allelic frequency and the probability of the allele to be a risk variant. We hypothesized that the duration of existence of the allele (i.e., the time since its origin) has a profound effect on the detection of an association between a SNP and a disease. An original linkage disequilibrium (LD) block associated with a novel allele breaks down as the allele ages, making it more difficult to detect an association by using tagging SNPs.
2. Materials and Methods
We accessed an open-access database of GWAS results3 on February 9, 2011. Information on the ancestral allele of the SNPs was retrieved from the dbSNP. All alleles of SNPs shown to be significant in a GWAS were placed into 10 categories according to their population frequency (from 0% to 100%, in 10% increments). We separately estimated the proportions of risk alleles in each category, further stratified the alleles as ancestral or derived, and estimated the proportions of risk alleles in these two groups. Next we stratified the diseases as early or late onset and separately analyzed them. An early-onset disease was defined as one with a typical onset before the age of 30 years. We chose this threshold because disease onset before 30 years may affect the survival and fertility of the carrier. The early-onset diseases include a number of autoimmune diseases, such as celiac disease, Crohn disease, inflammatory bowel disease, Kawasaki disease, psoriasis, type 1 diabetes, and asthma. The late-onset diseases include most cancers, Alzheimer disease, coronary disease, and rheumatoid arthritis.
We used computer simulation to estimate the existence time of alleles from different frequency categories. Our simulation model was described in detail earlier.8 Assuming a diploid Wright–Fisher population of a constant size of 1000, we simulated the age of derived alleles conditioning on their frequencies. A derived allele was either neutral or under a positive or purifying selection with selection coefficient s. We used an additive model and assigned fitness values 1, 1-s, 1-2s for genotypes AA, Aa, and aa, where a is derived. We simulated the age of alleles with frequencies from 0 to 1 at an interval of 0.01, each with 100 replicates, and grouped the results into bins of 0.1.
For statistical analysis, we used SAS statistical software (SAS Institute, Inc., Cary, North Carolina, USA).
3. Results
Overall, 351 SNPs were used as being significant in at least one GWAS. Figure 1 shows the proportions of risk alleles in the 10 frequency categories. The proportion was highest among rare alleles and decreased as the population frequency increased. Note that the curve has left–right and top–bottom symmetry. This is because for biallelic SNPs, the alleles are complementary, so that if allele 1 is a derived, risk-associated allele with a frequency of 0.2, the alternative allele will be ancestral and protective, with a frequency of 0.8. The right side of the distribution, therefore, can be deduced from the left side. However, we show the whole spectrum because it provides a better picture of the relationships between allele frequency and risk. We found that among minor alleles (defined as those with a frequency of <0.5), the proportion of risk alleles was 245/351 = 0.70 ± 0.02, whereas the proportion of risk-associated variants among major alleles was 106=351 = 0.3 ± 0.02.
Fig. 1.

Proportions of the risk alleles in the different allele-frequency groups. Error bars = standard error.
When we analyzed ancestral and derived alleles separately, we found that 0.96 ± 0.01 of derived alleles with a frequency ≤0.1 were risk associated, whereas the proportion of risk alleles among rare ancestral variants was only 0.67 ± 0.04 (Fig. 2). Among the minor alleles, the mean proportions of the risk variants were 0.84 ± 0.05 for the derived and 0.63 ± 0.02 for the ancestral alleles.
Fig. 2.
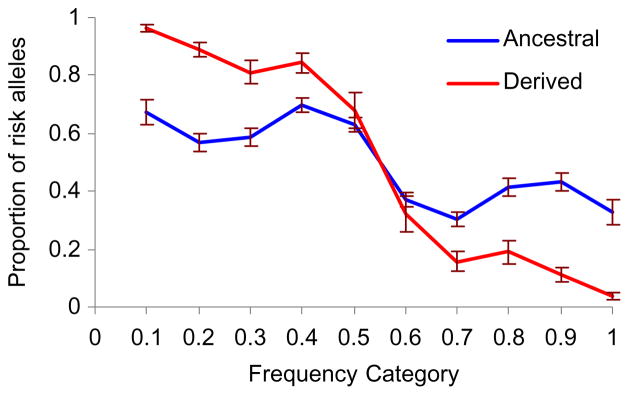
Proportions of the risk alleles in the different allele-frequency groups of the ancestral and derived alleles. Error bars = standard error.
The analysis of early- versus late-onset diseases (Fig. 3) showed that early-onset diseases have a larger difference between ancestral and derived alleles. For the late-onset diseases, the proportion of the risk alleles drops faster when frequency increases, compared to the proportion for the early-onset diseases. The less dramatic overall drop in the proportion of the risk alleles as frequency increases in early-onset diseases is driven by ancestral alleles for which the trend is opposite to derived alleles.
Fig. 3.
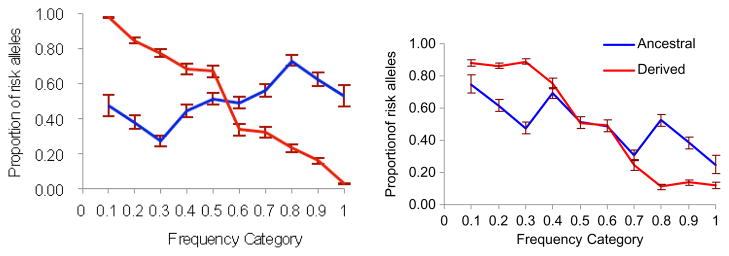
The proportions of risk variants in different frequency groups. Left panel, early-onset diseases; right panel, late-onset diseases. Error bars = standard error.
4. Discussion
Our analyses showed that risk alleles are mostly derived, low-frequency (i.e. minor) variants. These results are consistent with other studies, both theoretical and experimental, that have demonstrated that derived alleles generally have a lower frequency than ancestral variants.9–12 The analysis stratified by ancestral/derived status and frequency demonstrated that rare derived alleles have a higher proportion of risk variants compared to ancestral alleles.
Mutations causing early-onset diseases are subject to negative selection. Such mutations, however, can reach substantial population frequency because of the effects of random factors like genetic drift and founder and bottleneck effects. It is generally accepted that for late-onset diseases, the negative selection will less likely affect allelic frequency, though some studies13,14 demonstrate that negative selection may also affect allele frequencies in late-onset diseases.
If we assume that mutations causing late-onset diseases are mostly neutral, they will have stochastic dynamics and may completely replace ancestral alleles, leading to a situation in which the risk allele is the derived (common) one and the protective allele is the ancestral (rare) one. In this case, the proportion of risk alleles is expected to be the same (or similar) for ancestral and derived variants as was observed in our analysis (Fig. 3). The proportion of the disease-associated variants for the late-onset diseases was notably higher for rare alleles regardless of their status as ancestral or derived.
Because selection is less important in the case of late-onset diseases, there should be another reason why rare alleles have a greater chance to be risk variants. It is generally accepted that the majority of SNPs detected by GWASs are not causal variants but tagging SNPs linked to unknown/ungenotyped causal variants.15 The linkage disequilibrium (LD) between tagging and causal SNPs leads to co-segregation of causal and tagging SNPs, allowing the detection of the tagging SNP by association analysis. Rare recombination events between causal and tagging SNPs break down the LD over time, making it more difficult to detect an association. We hypothesize that the breakdown of an original LD block near a tagging SNP may explain differences in the proportions of risk-associated alleles between early- and late-onset diseases. Our computer simulation shows that the existence time increases faster for neutral alleles than for selected ones when we move from low to high frequency categories (Fig. 4). The sharper increase in existence time for neutral alleles parallels the sharper decrease in the proportion of the risk-associated alleles for the late-onset diseases, suggesting that these two observations may be mechanistically connected.
Fig. 4.

The existence time of alleles in different frequency categories. The number of generations was used as a measure of existence time. Blue line: neutral model, red line: negative selection.
If we assume that variants associated with early-onset diseases are under weak negative selection, then it takes shorter for a slightly deleterious mutation to reach the same population frequency it takes for a neutral or beneficial mutation.16 Rare derived slightly deleterious alleles exist in a population for a shorter time than neutral alleles, so LD blocks around a causal variant are better preserved and therefore may be detected more easily by a GWAS. This may be the reason why the proportion of risk alleles is high in the group of rare derived alleles. The existence time for a rare ancestral allele is longer than it is for a rare derived allele. This suggests a weak LD and, as a result, a lower probability of detection.
In conclusion, the results of our analysis suggest that the existence time of the allele is a major factor underlying the enrichment of rare variants for risk alleles. The practical outcome of this analysis is that selecting evolutionarily young allelic variants to use in GWASs may be beneficial because such variants are more likely to tag unbroken LD blocks and therefore have greater power to detect blocks with causal SNPs.
Acknowledgments
This study was supported by the David H. Koch Center for Applied Research of Genitourinary Cancers; National Institutes of Health grants R01 CA149462 and R01 AR055258 (to O.Y.G.); and NIH grant R01 CA127219 (to M.R.S.). It is also supported in part by the NIH through MD Anderson’s Cancer Center Support Grant, CA016672.
Biographies
Olga Gorlova received her Ph.D. degree in Genetics from Novosibirsk University, Russia, in 1990. Her original research interest was in the genetic control of recombination and mapping of complex human traits. In 1999, she became a postdoctoral fellow at the Department of Epidemiology at The University of Texas MD Anderson Cancer Center in Houston, Texas, where she is currently Associate Professor. Her major research interest is in uncovering epidemiological and genetic determinants of susceptibility to human diseases.
Jun Ying received her M.S. in Molecular Biology at the University of Houston and her M.S. in Biostatistics at the University of Texas School of Public Health. She joined the Department of Epidemiology at MD Anderson Cancer Center, Houston, Texas, in December 2008, and is currently a statistical analyst at the institution.
Christopher I. Amos received his Ph.D. from LSU Medical Center in New Orleans, Louisiana, in 1988, and held a position as Assistant Professor in Biostatistics and Epidemiology at Howard University Cancer Center in Washington, D.C., from 1998 to 2003 while completing fellowship training in epidemiology and genetics in the Family Studies Section at the National Institutes of Health in Bethesda, Maryland. Dr. Amos joined the faculty of MD Anderson Cancer Center in 1994, received the Margaret and James A. Elkins Jr. Faculty Achievement Award in 2000 and was honored as an Ashbel Smith Professor from 2001 to 2005. Dr. Amos directs the Human Pedigree Analysis Resource, which supports research for individuals with increased familial risk for developing cancer.
Margaret R. Spitz received her M.D. degree from the University of Witwa-tersrand Medical School in Johannesburg, South Africa, and earned her Master’s of Public Health degree from The University of Texas School of Public Health. Dr. Spitz joined the faculty of MD Anderson Cancer Center in 1981 and was named founding chair of the Department of Epidemiology in 1995, until stepping down in 2008. She joined Baylor College of Medicine in 2009 to provide strategic direction in expanding their population sciences program. Dr. Spitz has a long-standing interest in genetic susceptibility to lung cancer and has contributed to more than 400 scientific publications, with a research focus on the study of interindividual variation in susceptibility to tobacco carcinogenesis.
Bo Peng obtained his Ph.D. degree in Biostatistics from Rice University in 2006. He joined the Department of Epidemiology at The University of Texas MD Anderson Cancer Center as a postdoctoral fellow, where he is now an instructor in the Department of Genetics. Dr. Peng is interested in computational aspects of genetic studies and has applied advanced computational techniques, especially large-scale population genetic simulations, to research topics such as the evolution and mapping of complex human diseases.
Ivan P. Gorlov received his Ph.D. in Evolutionary Genetics at the Institute of Cytology and Genetics, Novosibirsk, Russia. He continued his training in evolutionary genetics in the Institute of Evolution, Haifa University, Haifa, Israel, during 1996–1998. He also received training in epidemiology as a Research fellow in the Department of Epidemiology, The University of Texas MD Anderson Cancer Center, Houston, Texas, from 2003 to 2005. He is currently an Assistant Professor at the Department of Genitourinary Medical Oncology, MD Anderson Cancer Center. His major research interest is in application of bioinformatics in silico methods for study of genetic architecture of common human diseases.
Footnotes
Web Resources
The URLs for data presented herein are as follows.
- Catalog of published genome-wide association studies: http://www.genome.govds/26525384/
- dbSNP FTP site: ftp://ftp.ncbi.nih.gov/snp/
References
- 1.Morton NE. Into the post-HapMap era. Adv Genet. 2008;60:727–742. doi: 10.1016/S0065-2660(07)00425-7. [DOI] [PubMed] [Google Scholar]
- 2.Stein CM, Elston RC. Finding genes underlying human disease. Clin Genet. 2009;75:101–106. doi: 10.1111/j.1399-0004.2008.01083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson AD, O’Donnell CJ. An open access database of genome-wide association results. BMC Med Genet. 2009;10:6. doi: 10.1186/1471-2350-10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lachance J. Disease-associated alleles in genome-wide association studies are enriched for derived low frequency alleles relative to HapMap and neutral expectations. BMC Med Genomics. 2010;3:57. doi: 10.1186/1755-8794-3-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gorlov IP, Gorlova OY, Sunyaev SR, Spitz MR, Amos CI. Shifting paradigm of association studies: Value of rare single-nucleotide polymorphisms. Am J Hum Genet. 2008;82:100–112. doi: 10.1016/j.ajhg.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yue P, Melamud E, Moult J. SNPs3D: Candidate gene and SNP selection for association studies. BMC Bioinformatics. 2006;7:166. doi: 10.1186/1471-2105-7-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levenstien MA, Klein RJ. Predicting functionally important SNP classes based on negative selection. BMC Bioinformatics. 2011;12:26. doi: 10.1186/1471-2105-12-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peng B, Amos CI, Kimmel M. Forward-time simulations of human populations with complex diseases. PLoS Genet. 2007;3:e47. doi: 10.1371/journal.pgen.0030047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shastry BS. SNPs in disease gene mapping, medicinal drug development and evolution. J Hum Genet. 2007;52:871–880. doi: 10.1007/s10038-007-0200-z. [DOI] [PubMed] [Google Scholar]
- 10.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen H, Green RE, Paabo S, Slatkin M. The joint allele-frequency spectrum in closely related species. Genetics. 2007;177:387–398. doi: 10.1534/genetics.107.070730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fredman D, Sawyer SL, Stromqvist L, et al. Nonsynonymous SNPs: Validation characteristics, derived allele frequency patterns, and suggestive evidence for natural selection. Hum Mutat. 2006;27:173–186. doi: 10.1002/humu.20289. [DOI] [PubMed] [Google Scholar]
- 13.Pavard S, Metcalf CJ. Negative selection on BRCA1 susceptibility alleles sheds light on the population genetics of late-onset diseases and aging theory. PLoS One. 2007;2:e1206. doi: 10.1371/journal.pone.0001206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wright A, Charlesworth B, Rudan I, Carothers A, Campbell H. A polygenic basis for late-onset disease. Trends Genet. 2003;19:97–106. doi: 10.1016/s0168-9525(02)00033-1. [DOI] [PubMed] [Google Scholar]
- 15.Cantor RM, Lange K, Sinsheimer JS. Prioritizing GWAS results: A review of statistical methods and recommendations for their application. Am J Hum Genet. 2010;86:6–22. doi: 10.1016/j.ajhg.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kimura M, Ota T. Probability of gene fixation in an expanding finite population. Proc Natl Acad Sci USA. 1974;71:3377–3379. doi: 10.1073/pnas.71.9.3377. [DOI] [PMC free article] [PubMed] [Google Scholar]