

Abstract
Protein toxins are important virulence factors contributing to neonatal sepsis. The major pathogens of neonatal sepsis, group B Streptococci, Escherichia coli, Listeria monocytogenes, and Staphylococcus aureus, secrete toxins of different molecular nature, which are key for defining the disease. Amongst these toxins are pore-forming exotoxins that are expressed as soluble monomers prior to engagement of the target cell membrane with subsequent formation of an aqueous membrane pore. Membrane pore formation is not only a means for immediate lysis of the targeted cell but also a general mechanism that contributes to penetration of epithelial barriers and evasion of the immune system, thus creating survival niches for the pathogens. Pore-forming toxins, however, can also contribute to the induction of inflammation and hence to the manifestation of sepsis. Clearly, pore-forming toxins are not the sole factors that drive sepsis progression, but they often act in concert with other bacterial effectors, especially in the initial stages of neonatal sepsis manifestation.
1. Introduction
The birth canal, that is, the area between the fully dilated uterus and the outside of the vagina, harbours a polymicrobial community that may fulfil the definition of a biofilm [1]. Next to apathogenic species, such as Lactobacillus spp., potentially pathogenic bacteria including group B streptococci (GBS), Escherichia coli (E. coli), Listeria monocytogenes (L. monocytogenes), and Staphylococcus aureus (S. aureus) are found in the vagina of up to 20% of women. During birth, the fetus needs to pass from the sterile uterus through these bacteria. Accordingly, aspiration of bacteria during birth is regarded as a major cause of neonatal sepsis in the first three to seven days of life (early-onset sepsis). In line with this model, early-onset sepsis is predominantly caused by GBS, E. coli, L. monocytogenes, and S. aureus. Yet most infants successfully control the bacteria at the mucocutaneous surfaces.
Subsequent to aspiration, bacteria like GBS can proliferate to striking densities in the neonatal lung, as shown in newborn primates with neonatal GBS pneumonia (109–1011 colony-forming units (CFUs)/g lung tissue, [2]). The antimicrobial quality of the local pulmonary environment, for example, the concentration of surfactant, may be important for the metabolic activity in the bacterial community and therefore for the expression of bacterial virulence factors such as bacterial toxins [3].
Sepsis imposes a major threat to newborn infants. It is estimated that sepsis causes over half a million neonatal deaths annually, thereby accounting for about 15% of all neonatal deaths worldwide [4]. Whereas sepsis causes approximately 2.5% of infant deaths in developed countries, it is responsible for up to 50% of neonatal deaths in developing countries [5, 6]. Moreover, neonatal sepsis often occurs as meningoencephalitis, which leaves almost 50% of affected patients with lifelong disabilities [7]. On the other hand, GBS, E. coli, and S. aureus are normal components of the mucocutaneous microbiome, and it is impossible to predict the risk to an individual baby.
2. Bacterial Membrane-Damaging Toxins
The first membrane-damaging bacterial toxin was described by Paul Ehrlich in 1898 [8], who found that Clostridium tetani extracts lyse erythrocytes. Today, three different mechanisms of membrane damage by proteinaceous agents can be delineated. First, toxins can solubilise target membranes acting essentially as amphiphilic surfactants. δ-Toxins from various staphylococcal species [9, 10] and the cyclolipopeptides from Bacillus subtilis are prominent examples [11] (see Figure 1). Second, toxins can act as phospholipases and damage membranes by enzymatic hydrolysis of phospholipid ester bonds. β-Hemolysin from S. aureus, for instance, is a sphingomyelin-specific phospholipase, which cleaves sphingomyelin to ceramide and phosphorylcholine. However, the large majority of membrane damaging proteins belong to the class of pore-forming proteins/toxins (PFTs). PFTs, which make up approximately 30% of all protein toxins in pathogenic bacteria [12], have evolved in all domains of life. They are secreted as water-soluble proteins and subsequently integrate into foreign membranes.
Figure 1.
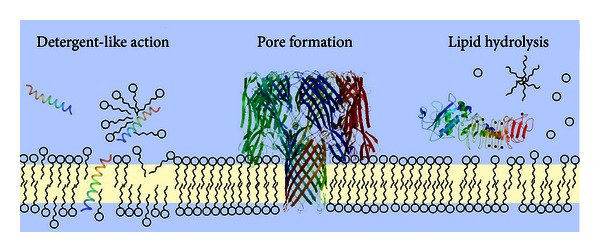
Ways to damage a lipid membrane. There are various mechanisms of membrane damage by protein toxins. Amphiphilic toxins can integrate into the membrane and essentially solubilise the lipid membrane like a detergent (structure: S. aureus δ-toxin, PDB ID 2KAM). Similarly, the membrane lipids can be hydrolysed by phospholipases also resulting in the destruction of the membrane (structure: Clostridium, perfringens α-toxin, PDB ID 1KHO, [165]). By far the largest class of membrane damaging toxins is that of the pore-forming toxins (structure: α-toxin from S. aureus, PDB ID 7AHL, [100]). These toxins integrate as stable channels into the lipid bilayer, thus creating an aqueous connection between the cytosol and the extracellular space of the target cell.
3. Mechanism of Membrane Pore Formation by Pore-Forming Toxins
Common structural themes of protein/membrane association are insertion of transmembrane α-helices or β-sheet barrel arrangements, anchoring by prosthetic glycolipids, or direct linkage to hydrophobic lipid tails, such as myristic or palmitic acid. Most pores or channels allowing for communication across biological membranes are formed by integral membrane proteins spanning the lipid bilayer. However, PFTs form pores by acting initially extraneously of the lipid bilayer. They start out as soluble molecules and then turn themselves into integral membrane proteins, with a membrane-spanning region that defines the pore. Pore formation is a dynamic process with structurally and functionally distinct states (see Figure 2). Initial binding to the membrane, for example, to a membrane lipid or protein receptor, is followed by homotypic oligomerisation to a prepore state on the membrane surface (Figure 2, arrows 1 and 2). In this state, the protomer configuration resembles that of the soluble monomer, and the whole oligomer still stands prone to the membrane with an intact lipid bilayer beneath the assembled ring. In Figure 3(a1), this is depicted for pneumolysin from Streptococcus pneumoniae, a close homologue of listeriolysin from L. monocytogenes and, interestingly, also of perforin secreted by cytotoxic T cells [13], and of the complement membrane attack complex [14, 15], which indicates that bacterial attack and immune defence employ the same mechanisms. This prepore state then undergoes drastic conformational rearrangements to be inserted as a stable pore into the membrane (see Figure 2, arrow 3). This rearrangement can even involve the refolding of α-helices in the soluble state to β-sheets in the membrane-inserted form [16]. While this general mechanism of pore formation can be proposed for nearly all PFTs, the structural changes, which the individual proteins undergo, remain largely elusive. Detailed mechanistic models on how hydrophilic proteins can suddenly change their solubility and integrate into biological membranes are available only for a few PFTs involved in neonatal sepsis (see Figure 3). Not surprising when considering that one not only needs to be able to study the solution state in sufficient structural detail, for example, via X-ray crystallography, but the structure of the membrane state also needs to be resolved. PFTs are classically divided into two main groups based on the structural motifs that form the pore [17–19]. Pores can be formed by α-helices, α-PFTs or by β-sheets, β-PFTs. For certain pore-forming proteins, it was recently proposed that lipids might play a direct role in pore formation, but our understanding of how this can be achieved is limited by the available structural data to date [20–22]. The structures, where known, of the soluble and membrane states of the PFTs discussed in this review are displayed in Figure 3. However, not all membrane pores are equal. The pore diameter, for instance, of the α-hemolysin membrane heptamer is considerably smaller than that of listeriolysin with up to 50 protomers (cf. Figures 3(a) and 3(c)). Clearly, the size of the membrane pore has important consequences for the targeted cell, as a large pore diameter is not selective for what it can conduct across a membrane, potentially mediating diffusion of larger molecules, such as ATP or even small proteins.
Figure 2.
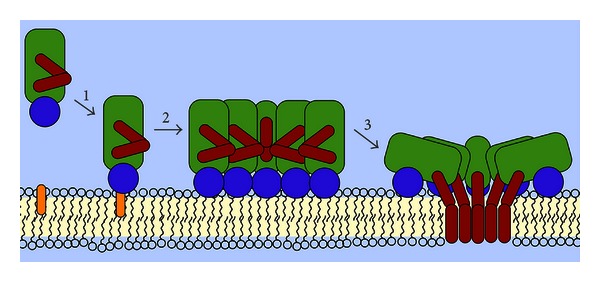
Pore formation is a dynamic process with structurally and functionally distinct states. Distinct molecular states exist on the path to membrane pore formation by PFTs. The toxin is secreted by the bacterial pathogen into the extracellular medium in a water-soluble form, usually as a monomer. Upon engagement of the membrane via binding to a receptor (step 1), for example, a membrane lipid or protein, the monomers assemble to a prepore oligomer (step 2). The membrane beneath the prepore oligomer remains intact and is only punctured once the prepore refolds to the membrane-inserted pore oligomer (step 3). This step usually goes along with considerable structural rearrangements.
Figure 3.

Structures of PFTs that are important for neonatal sepsis. Panel (a) shows available structures of cholesterol-dependent cytolysins to illustrate listeriolysins' mechanism of pore formation. (a1) displays the crystal structure of the soluble, monomeric form of perfringolysin from Clostridium perfringens (left, PDB ID 1PFO, [57]). The cryo-electron microscopy (cryo-EM) reconstruction of the prepore (EM databank: 1106) of the listeriolysin homologue pneumolysin from Streptococcus pneumoniae displayed on the right revealed that the protomer configuration in the prepore resembles that of the soluble monomer [16]. Lipid membrane is coloured yellow. Molecular modeling of the protomer fitted into the cryo-EM pore structure below (EM databank: 1107) revealed the considerable structural rearrangements that accompany membrane pore formation. The α-helices that refold into β-sheets are coloured in red. Panel (b) shows the different structures available for ClyA from E. coli, (b1) the soluble state (PDDid 1QOY, [148]) monomer and (b2) a protomer from the dodecameric pore state, which is shown as side and top view on the right (PDB ID 2WCD, [149]). Pore-lining α-helices are in red and the β-tongue in yellow. Panel (c) shows the PFTs from S. aureus. (c1) shows from top to bottom LukF (PDB ID 1LKF, [113]), LukF-PV (PDB ID 1PVL, [114]), and LukS-PV (PDB ID 1T5R, [115]). (c2) shows the octameric pore structure of γ-hemolysin (PDB ID 3B07, [116]), protomer on the left, side and top views on the right. (c3) displays the heptameric pore structure of the AFT pore (PDB ID 7AHL, [100]), individual protomer, side and tops views. The β-stem that unfolds into the membrane lining, extended β-hairpin is shown in red.
Interestingly, clearing of toxin pores from host membranes, a mechanism that is of marked importance when considering the amounts of toxin that are produced during sepsis, seems also to be size dependent. Cholesterol-dependent cytolysins, such as listeriolysin, can induce Ca2+-dependent resealing of membrane pores by induction of endocytosis [23–25] but α-hemolysin from S. aureus cannot [26]. This is counterintuitive, as van der Goot and coworkers nicely state [27] that small pores are harder to repair than larger ones.
4. Bacterial Pore-Forming Toxins in Pathogens Causing Neonatal Sepsis
In the major pathogens isolated from newborn infants with sepsis, PFTs are key virulence factors. They initiate a multitude of events ranging from direct necrotic cell deaths to the induction of signalling cascades, for instance, Ca2+-mediated signalling [27]. Prominent PFTs in the context of neonatal sepsis are listeriolysin O from L. monocytogenes, β-hemolysin/cytolysin from GBS (Streptococcus agalactiae), α-hemolysin and cytolysin A from E. coli, and α-hemolysin, γ-hemolysin, and the leukocidins from S. aureus. It has long been appreciated that PFTs are especially important during initiation of bacterial infections through induction of necrosis and apoptosis of host epithelial and endothelial cells, which promotes microbial invasion and subverts defence mechanisms. However, PFTs may also contribute to sepsis by receptor mediated or membrane damaging mechanisms in immune cells, which respond with the formation of inflammatory mediators, as was shown for β-hemolysin/cytolysin from GBS [28] and listeriolysin O [29, 30] and α-hemolysin and Panton-Valentine leukocidins from S. aureus [31, 32].
5. β-Hemolysin/Cytolysin from Group B Streptococci
Streptococcus agalactiae (group B streptococci, GBS) are the major cause of sepsis and meningitis in newborn infants without underlying disease in the western world. In addition, they are a significant cause of invasive infections in pregnant woman and immuneocompromised patients [33, 34]. The pore-forming toxin β-hemolysin/cytolysin is one of the main virulence factors of GBS. It has been implicated in the pathogenesis of early- [35] and late-onset neonatal sepsis, although its role in both cases remains controversial. Rabbits infected with wild-type GBS had significantly higher bacterial blood counts than those infected with GBS mutants lacking the β-hemolysin/cytolysin [35]; mortality also increased dramatically. Similarly, in a neonatal rat model of meningitis wild-type GBS induced more neuronal damage in the cortex and the hippocampus than cytolysin-deficient mutants [36]. The clinical outcome score, assessed in this study by weighted changes and motor activity, decreased profoundly upon presence of the cytolysin. The β-hemolysin/cytolysin lytic protein agent is thought to be encoded by the cylE gene of the cyl operon [37], since expression of cylE induces β-hemolytic activity in nonhemolytic E. coli. However, the exact molecular nature of the protein component responsible for hemolysis and pore formation remains obscure, as it has evaded purification to homogeneity as of yet. Interestingly, β-hemolysin/cytolysin is also necessary for the synthesis of an orange carotenoid pigment [38], with which it also associates [39]. Hemolytic activity of partially purified β-hemolytic activity containing the carotenoid pigment could be inhibited by addition of the lipid dipalmitoylphosphatidylcholine (DPPC), the major component of surfactant in the lung. Accordingly, surfactant deficiency may explain in part the particular susceptibility of preterm infants to GBS sepsis and meningitis [40, 41]. As outlined above, breaching of epithelial barriers by GBS is the first step in sepsis pathogenesis. Accordingly, it appears to be important that β-hemolysin/cytolysin mediates not only injury of lung epithelial [40] and of lung microvascular endothelial cells [42] and invasion of brain endothelial cells [43] but also injury of professional phagocytes [39] and neurons [36]. Moreover, GBS mutants lacking clyE were more readily cleared from mouse and human blood [39]. Interestingly, Rubens and colleagues initially proposed that β-hemolysin/cytolysin was no longer needed for systemic disease manifestation once the epithelial barriers have been breached [44]. However, pro- and anti-inflammatory activity of β-hemolysin/cytolysin in macrophages was demonstrated, indicating more profound immunomodulatory functions of the cytolysin [45, 46]. One direct or indirect molecular β-hemolysin/cytolysin target with important implications for mononuclear phagocyte activation is the NLPRP3 inflammasome. Activation of the NLRP3 inflammasome requires GBS expressing β-hemolysin/cytolysin. This pathway is essential in a mouse model of GBS sepsis, where deficiency in NLRP3 or its signalling partners apoptosis-associated speck-like protein and caspase-1 increases lethality and bacterial dissemination [28]. Yet direct evidence for binding, engagement, and activation of TLRs by the β-hemolysin/cytolysin is not available.
A second pore-forming toxin of GBS is the CAMP factor that has long been used for microbiological identification of GBS, since it characteristically synergizes with secreted sphingomyelinase of S. aureus to lyse erythrocytes on blood agar plates [47–49]. However, its role in neonatal sepsis is not clear, as it was not required for systemic infection in a mouse model of GBS infection [50].
6. Listeriolysin O from Listeria monocytogenes
Listeria monocytogenes (L. monocytogenes) is a Gram-positive bacterium that causes early- and late-onset neonatal sepsis and meningitis. L. monocytogenes has the capacity to breach the intestinal barrier, thereby causing food-borne listeriosis, the blood-brain barrier, causing meningitis, and the maternal-placental barrier, causing early-onset listeriosis. Listeriolysin O (LLO), a member of the PFT class of cholesterol-dependent cytolysins (CDCs), is a major virulence factor of L. monocytogenes with multivalent functions [51]. In the late 1980s, Kathariou et al. and Portnoy et al. reported that L. monocytogenes mutants lacking functional LLO were avirulent in mice [52, 53]. Furthermore, LLO mutants did not induce secretion of cytokines such as TNF-α, IL-1β, or IFN-γ, when injected intravenously into C57BL/6 mice [54]. Recently, a single-gene signature-tag-based approach was used to assess the contribution of individual amino acids of LLOs to its virulence in mice [55].
Based on structural homology with other toxins such as pneumolysin from Streptococcus pneumoniae [56] and perfringolysin from Clostridium perfringens [57], common pore-forming properties can be proposed [58] (see Figure 3(a)). LLO engages cholesterol as a native membrane receptor in dependence on the two amino acids threonine 515 and leucine 516, oligomerises to a prepore complex of up to 50 monomers, and forms a membrane pore in a concerted refolding step with each protomer contributing two beta-hairpins to the membrane-spanning β-barrel, which originates from five α-helices in the soluble state [16, 59–61]. The allosteric monomer assembly and prepore refolding process were recently shown to rely on an undecapeptide sequence (483-ECTGLAWEWWR-493), which was originally thought to be soley responsible for cholesterol binding [62]. The exact nature of the membrane pore remains controversial, as arciform pores, that is, membrane pores with a seemingly incomplete protein ring lining the aqueous membrane hole, are often observed in electron microscopic and atomic force microscopic imaging of various CDCs [22, 63–65]. Moreover, LLO does not lose its membrane targeting properties after incubation with cholesterol [66].
Listeria are classical intracellular pathogens [67] and LLO pore formation was traditionally thought to only mediate escape of Listeria from the phagolysosome [68]. This concept was based on the finding that LLO was active only at acidic and not at neutral pH, which is found in the maturing phagolysosome [58]. However, host factors also play an important part in regulating the activity of LLO in the phagolysosome. LLO hijacks the reductive capacity of the γ-interferon-inducible lysosomal thiol reductase GILT to maintain cysteine 484 of LLO in its reduced thiol state [69], thus greatly increasing bacterial escape from the phagolysosome. Intriguingly, CDCs were originally, that is, before the identification of cholesterol as a membrane receptor, termed sulfhydryl-activated [70] or thiol-activated, oxygen-labile cytolysins [71], as chemical reduction activated the toxins towards hemolysis of red blood cells. Additionally, the cystic fibrosis transmembrane conductance regulator (CFTR), which transports chloride not only across the apical plasma membrane of epithelial cells in the lung but also into the phagolysosomes of macrophages, potentiates LLO oligomerisation on the phagosomal membrane and its lytic activity and phagolysosomal escape of L. monocytogenes [72]. However, the role of LLO extends beyond mediating phagosomal escape. LLO reduces formation of reactive oxygen species (ROS) by inhibiting the NADPH oxidase NOX2 in RAW 264.7 macrophages [73]. This activity seems to rely on pore formation in the phagosomal membrane and prevents degradation of bacteria inside the phagosome. Pore formation at the plasma membrane of target cells induces the dynamin-/F-actin-dependent but clathrin-independent uptake of L. monocytogenes into HepG2 cells [74]. This finding questions the traditional model of LLO pore-forming activity being strictly dependent on the low phagosomal pH [58], whereby premature lysis of target cells by (secreted) LLO is prevented [75]. Residual lytic activity and structural integrity of LLO at neutral pH [74, 76, 77] are in line with LLO-mediated calcium influx into epithelial Hep-2 [78] and HEK 293 cells [79] along with concomitant L. monocytogenes uptake. Indeed LLO seems to form pores at neutral pH in the plasma membrane, which do not result in lysis of the target cell but rather in uptake of the pathogen [74]. As pneumolysin from S. pneumoniae can replace LLO in the uptake of L. monocytogenes into HepG2 cells, CDCs from other bacterial pathogens may similarly induce cellular uptake. The contribution of TLR signalling in response to CDC has been subject of several studies. As examples, the LLO homologues anthrolysin (Bacillus anthracis) and pneumolysin can signal via TLR4 [80, 81]. On the other hand, LLO induces an inflammatory cellular response in a TLR-independent fashion [82]. Moreover, LLO activity at the plasma membrane induces clustering of lipid rafts [83], suppression of antigen-induced T-cell activation [84], inflammasome activation, and histone H3 dephosphorylation [30], all of which might contribute to sepsis progression either at the stage of heightened inflammation or at later stages of immune suppression.
7. α-Hemolysin, γ-Hemolysin, and Leukocidins from Staphylococcus aureus
Staphylococcus aureus (S. aureus) is well recognised as a significant cause of neonatal sepsis [85]. Around ten bacteria are sufficient to colonise the umbilical cord. After birth, S. aureus can colonise the upper respiratory tract in up to 40% of infants [86]. S. aureus produces a number of PFTs with distinct specificity for target cell membranes. Although most clinical isolates produce the PFTs α-hemolysin, bicomponent γ-hemolysins, and bicomponent leukocidins, none of these toxins was found to be a necessary and sufficient virulence determinant of neonatal sepsis. In contrast, other factors such as the antigenic, peptidoglycan-associated protein A [87], superantigens [88], and sphingomyelinase C (β-hemolysin, [89]) contribute to host invasion, subversion of the immune system, and sepsis manifestation. However, there is evidence that pore formation by α-hemolysin (α-toxin, AFT) contributes to the pathogenesis of sepsis [90]. As an example, erythrocyte lysis by AFT could be directly imaged [91]. Downregulation of AFT expression in vivo clearly reduces virulence of S. aureus [92, 93]. In a model using C57BL/6J mice, AFT activates the NLRP3 inflammasome, thereby promoting necrotising pneumonia [94]. Moreover, monoclonal antibodies to AFT are protective against staphylococcal pneumonia [95]. Indeed, a nonhemolytic variant of AFT was used to vaccinate rabbits, and antisera could be used to passively immunize mice against an otherwise lethal challenge with wild-type S. aureus [96]. In a mouse model of mastitis, coagulase and AFT proved to be the primary virulence determinants [97]. Heat-inactivated S. aureus and an AFT mutant greatly reduced the bacterial burdens in a mouse brain abscess model and attenuated the expression of inflammatory mediators [98]. In an in vivo model of corneal virulence, AFT also proved to be a decisive virulence factor [99].
AFT is one of the best-studied PFTs to date. It was the first toxin of which the membrane structure was solved by X-ray crystallography (see Figure 3(c3), [100]). AFT consists of 293 amino acids and oligomerises on the plasma membrane of target cells to a heptamer (potentially a hexamer) prior to membrane insertion and pore formation. It was shown to have an important role in bacterial pathogenesis, especially by its ability to induce necrotic cell death [90], by which it can cause vascular leakage when perfused into the lung [101]. Recently the metalloprotease ADAM10 was identified as the membrane receptor of AFT [102]. At low toxin concentrations, ADAM10 is required to mediate the cytotoxic effects of AFT. Interestingly, binding of AFT to ADAM10 resulted in the upregulation of ADAM10 in alveolar epithelial cells and concomitant cleavage of E-cadherin. This leads to epithelial barrier disruption thereby aggravating staphylococcal pneumonia in mice [103]. Moreover, activation of the NLRP3-inflammasome by AFT might contribute to later stages of sepsis, although the molecular mechanism underlying inflammasome activation remains elusive at this stage [31]. In this respect, it is interesting to note that, whereas direct TLR activation has not been demonstrated for AFT, NOD2-dependent sensing of S. aureus was dependent on AFT [104].
Next to AFT, S. aureus expresses the bicomponent cytotoxins leukocidins (Luk) and the γ-hemolysins (Hlg). Bicomponent implies that class S toxins (LukS-PV, LukE, HlgA, HlgC, and LukS) have to associate with class F toxins (LukF, LukD, LukF-PV, and HlgB) in a 1 : 1 stoichiometric ratio to form a functional oligomer before insertion into the membrane (see Figure 3(c)). Pathogenic S. aureus can produce several different bicomponent toxin pores, among the most prominent are LukE/LukD [105], Panton-Valentine leukocidin LukS-PV/LukF-PV, γ-hemolysins LukF/HlgA [106], HlgA/HlgB, HlgB/HlgC, and the M/F-PV-like leukocidins, all of which might be expressed at different stages during sepsis. The Panton-Valentine leukocidin (PVL), which was first isolated from furuncles in 1936 [107], is probably the most widely studied member. In a mouse model, secreted PVL promotes tissue invasion and causes necrotizing pneumonia, via mechanisms including upregulation of protein A and other adhesins [108]. PVL is an important factor in the early stages of skin infection, as shown in a rabbit skin infection model [109]. Association of pvl and spa (protein A) genes seems to be an important virulence determinant in methicillin-resistant S. aureus (MRSA). Moreover, PVL was reported to directly bind to TLR2 and induce inflammation in the mouse lung [110]. LukE/LukD promotes systemic bacterial growth in vivo by specifically targeting neutrophils [111]. Interestingly, via engagement of their native receptor CCR5 (CC-motif-chemokine-receptor type 5), LukE/LukD toxin pores clear antigen-presenting cells (macrophages and dendritic cells) and S. aureus-specific CCR5-positive Th1/Th17 cells [112], thus greatly contributing to the spread of S. aureus in the host.
AFT, Hlg, and the leukocidins belong to the class of β-pore-forming toxins [17]. Despite their moderate sequence identity (around 30% for the pairwise alignment with AFT), they share a common structural fold (see Figure 3(c)). The structures of the soluble, monomeric LukF (Figure 3(c1), top, [113]), LukF-PV (Figure 3(c1), middle, [114]), LukS-PV (Figure 3(c1), bottom, [115]), and the octameric, membrane complex of Hlg (LukF/HlgA, Figure 3(c2), [116]) are available. A common molecular mechanism could be proposed in which a prestem, triple stranded β-sheet in the soluble monomer, refolds to a double, stranded, membrane-inserted β-sheet (Figures 3(c2) and 3(c3), left), resulting in a hexadeca- or tetradecastranded β-barrel that penetrates the membrane.
The target membrane specificities of S. aureus PFTs hint towards their role in neonatal sepsis. Whereas AFT has a broad specificity and might thus be involved at the initial stages of sepsis manifestation, where the lung epithelium needs to be breached, bicomponent leukocidins and Hlgs mainly attack polymorphonuclear neutrophils, macrophages and lymphocytes [117, 118]. Bearing in mind that PVL does not attack lymphocytes and Hlg can be hemolytic, at least in vitro, their restricted but highly directed mode of attack predisposes the bicomponent leukocidins to be important factors for the subversion of the immune system once the initial barrier has been breached.
8. α-Hemolysin and Cytolysin A from Escherichia coli
Pathogenic Escherichia coli (E. coli) cause around 25% of invasive neonatal sepsis [119], and antibiotic resistance is an emerging threat in this context [120]. Generally, E. coli can persist in the intestine as a normal constituent of the intestinal microbiota. However, extraintestinal pathogenic E. coli (ExPECs) are the most common gram-negative bacterial species isolated from neonates with bacterial infections, and neonatal mortality from gram-negative sepsis remains high [121]. Urinary tract infections of pregnant women can lead to aspiration of ExPECs during partition with subsequent uncontrolled growth in the lung of the newborn and potential progression to a systemic infection. Pathogenic E. coli often secrete the pore-forming toxin α-hemolysin (HlyA, CylA), a 107 kDa member of the RTX class of toxins [122], which is usually associated with strains causing uropathogenic infections [123]. Deletion of HlyA in E. coli greatly reduced mortality and cytokine production as compared to the isogenic wild type bacteria in an intravenous infection model [124]. HlyA furthermore induced hemorrhagic bleeding of bladder tissue and exfoliation of urothelium when pathogenic bacteria were administered into the urethra [125]. HlyA is encoded by at least 50% of all ExPEC clinical isolates [126]. Around 80% of meningitis- and sepsis-associated E. coli belong to the K1 serotype [127, 128]. The E. coli K1 strain RS218 expressed HlyA in a zebrafish model of systemic infection [126] and the hlyA gene was present in more than 40% of E. coli from the genital tracts of pregnant women [129]. As a member of the repeats in toxin (RTX) family of hemolysins, the hlyA gene is part of the chromosomal hlyCABD operon, which also encodes a type 1 ABC transporter for secretion of the PFT. The amino acid toxin repeats, which are located in the C-terminal portion of the protein, are composed of the sequence GGXGCDXUX (with U being a large hydrophobic residue). These repeats are responsible for Ca2+ binding, which is a prerequisite for membrane association and pore formation by the N-terminal, hydrophobic, and acylated domain of HlyA [130]. Interestingly, other pore-forming proteins, such as the human cytotoxic lymphocyte encoded perforin, also require binding of Ca2+ for membrane association and pore formation [131]. HlyA oligomerises on the plasma membrane of target cells, where it accumulates in cholesterol-rich microdomains [132, 133]. However, rather than cholesterol being a direct, lipid membrane receptor as in the case of the CDCs listeriolysin or pneumolysin, cholesterol seems to contribute to the physicochemical environment necessary for HlyA membrane engagement. The exact nature of the pore formation mechanism is under considerable debate. It seems now accepted that HlyA forms membrane pores as an oligomer, at least in artificial membrane mimics [134–136], albeit possibly heterogeneous in size [137]. In this respect, it is intriguing that the P2X7 receptor and pannexin 1 were found to mediate HlyA-dependent pore formation [138, 139]. Sublytic doses of HlyA initiate degradation of paxillin and proteolytic cascades inside epithelial cells and macrophages, thus attenuating the inflammatory host response and promoting epithelial exfoliation [140]. Similarly, HlyA inhibits epithelial cytokine production potentially promoting epithelial invasion of E. coli [141].
Another PFT of E. coli is ClyA (also termed hemolysin-E or SheA), which is expressed by various pathogenic and nonpathogenic E. coli including K12 strains, that are also found in clinical isolates of neonatal meningitis [142], bacteremia [143, 144], and neonatal sepsis [145]. However, three neonatal meningitis K1 strains isolated by Ludwig and colleagues harboured deletion mutations at the clyA gene locus [146]. Nevertheless, a synergistic enhancement of extraintestinal infection was recently reported between nonpathogenic E. coli K12 and pathogenic ExPEC strains in a mouse model of septicaemia [147], which hints towards a potential involvement of ClyA. ClyA is a 34 kDa protein belonging to the class of α-PFTs. The mechanism of pore formation is well understood, as crystal structures for the soluble [148] and membrane state [149] are available (see Figure 3(b)). Upon association with the membrane, insertion of the so-called β-tongue induces a series of substantial structural rearrangements in the now membrane-anchored monomer, resulting in a perpendicular position to membrane with the amphiphatic helix a1 now lying along its surface. After oligomerisation to a dodecamer, helices a1 become inserted into the membrane forming a 130 Å hollow cylinder with a 30 Å aperture protruding through the membrane.
9. Synopsis and Medical Outlook
Neonatal sepsis is a syndrome caused by systemic inflammation and defined by clinical criteria such as tachycardia, respiratory distress, temperature instability, and unusually high amount of immature immune cells in the blood. Pathogenic bacteria that are aspirated by the fetus in the birth canal during parturition can cause neonatal sepsis if their infection is not controlled locally by the innate defence mechanisms of the respiratory and alveolar epithelia. Toxins, either proteinaceous or of other molecular nature, are important factors contributing to neonatal sepsis. While endotoxins such as the lipopolysaccharide (LPS) from Gram-negative bacteria contribute to the sepsis phenotype by activating monocytes and macrophages via Toll-like receptor 4 binding, pore-forming proteinaceous exotoxins act by permeabilising target membranes of host cells. Whereas the membrane targeting effects of PFTs, that is, engagement of membrane, oligomerisation, and pore formation, are well defined, their secondary downstream effects are manifold, owing to the fact that defined ionic and molecular gradients across cellular membranes modulate a diverse set of signalling cascades. Unregulated cell death and its consequences are important in neonatal sepsis [150]. The PFTs described above can cause direct necrotic cell death, which in the case of E. coli, GBS, and S. aureus contributes to overcoming the epithelial and endothelial barriers in the lung. LLO of L. monocytogenes is critical for cell invasion and cell-to-cell spread of this intracellular pathogen and thus also contributes to breaching the epithelial barriers of the host. Necrotic cell death can release proinflammatory cytokines from leukocytes thus contributing to the inflammatory storm during sepsis. Interestingly, several PFTs discussed above can induce the inflammasome: AFT [31], β-hemolysin/cytolysin [28], LLO [29, 151], HlyA [140], and leukocidins [32, 152]. Inflammasome activation ultimately may contribute to hyperinflammation in newborn infants similar to what has been shown in GBS and E. coli sepsis in mice [28, 153]. PFTs can also elicit and alter apoptosis of host immune cells, that is, AFT via caspase-2 [154, 155], leukocidins via activation of caspases 3 and 9 [156], LLO via release of cytochrome C from mitochondria [157], β-hemolysin/cytolysin independently of caspase activation [36, 39], HlyA [158], and ClyA [159], thus potentially contributing to the apparent immunodeficiency of the patient that is characteristic during stages of sepsis [160]. Despite the formation of hydrophilic channels by all PFTs, the ways in which they induce the inflammasome are varied, hinting towards the fact that functions of PFTs at cellular membranes are more subtle than might be expected from their general mode of action. In this respect, effects of sublytic concentrations of PFTs have recently been explored. For instance, sublytic doses of LLO induce mitochondrial network disorganisation with transient alteration of the metabolic state of the target cell, thus weakening the cell for L. monocytogenes entry without destroying it [161]. Moreover, targeting of organs by PFTs might also contribute to the septic phenotype. GBS β-hemolysin/cytolysin, for example, had marked effects on cardiomyocyte contractility and viability [162].
Due to their role in neonatal sepsis and bacterial infection in general, PFTs present attractive therapeutic targets. In cases where membrane receptors have been defined, specific inhibitors, akin to viral entry inhibitors, might prevent membrane binding and pore formation. Monoclonal antibodies against PFTs that prevent membrane binding and/or refolding to the pore state could be a way of neutralising the toxin, at least in the blood stream. Moreover, vaccines based on PFTs may be used for immunizing women and thereby protecting newborn infants through placental transfer of specific immunoglobulins, given the fact that pneumolysin from S. pneumoniae is considered a vaccine candidate [163]. Indeed, novel vaccines based on AFT are currently developed [164]. As PFTs elicit specific cellular responses, it might, however, also be promising to designing therapeutics based on the pathways that the toxins induce in the target cell, as has been proposed for p38 MAPK and β-hemolysin/cytolysin from GBS [45]. In any case, it is exactly these cellular responses towards PFTs that need to be investigated in the context of neonatal sepsis in the future to improve therapeutic strategies.
Acknowledgment
This work was supported by a Grant from the German Bundesministerium für Bildung und Forschung (BMBF 01 EO 0803).
References
- 1.Verstraelen H, Swidsinski A. The biofilm in bacterial vaginosis: implications for epidemiology, diagnosis and treatment. Current Opinion in Infectious Diseases. 2013;26(1):86–89. doi: 10.1097/QCO.0b013e32835c20cd. [DOI] [PubMed] [Google Scholar]
- 2.Rubens CE, Raff HV, Jackson JC, Chi EY, Bielitzki JT, Hillier SL. Pathophysiology and histopathology of group B streptococcal sepsis in Macaca nemestrina primates induced after intraamniotic inoculation: evidence for bacterial cellular invasion. The Journal of Infectious Diseases. 1991;164(2):320–330. doi: 10.1093/infdis/164.2.320. [DOI] [PubMed] [Google Scholar]
- 3.Herting E, Gan X, Rauprich P, Jarstrand C, Robertson B. Combined treatment with surfactant and specific immunoglobulin reduces bacterial proliferation in experimental neonatal group B streptococcal pneumonia. American Journal of Respiratory and Critical Care Medicine. 1999;159(6):1862–1867. doi: 10.1164/ajrccm.159.6.9810047. [DOI] [PubMed] [Google Scholar]
- 4.Black RE, Cousens S, Johnson HL, et al. Global, regional, and national causes of child mortality in 2008: a systematic analysis. The Lancet. 2010;375(9730):1969–1987. doi: 10.1016/S0140-6736(10)60549-1. [DOI] [PubMed] [Google Scholar]
- 5.Kochanek KD, Xu J, Murphy SL, Mininö AM, Kung H-C. Deaths: final data for 2009. National Vital Statistics Reports. 2012;60(3):1–95. [PubMed] [Google Scholar]
- 6.Stoll BJ. The global impact of neonatal infection. Clinics in Perinatology. 1997;24(1):1–21. [PubMed] [Google Scholar]
- 7.Libster R, Edwards KM, Levent F, et al. Long-term outcomes of group B streptococcal meningitis. Pediatrics. 2012;130(1):8–15. doi: 10.1542/peds.2011-3453. [DOI] [PubMed] [Google Scholar]
- 8.Ehrlich P. Diskussion waehrend der Gesellschaft der Charite Aerzte. Klinische Medizin. 1898;35, article 273 [Google Scholar]
- 9.Pokorny A, Birkbeck TH, Almeida PFF. Mechanism and kinetics of δ-lysin interaction with phospholipid vesicles. Biochemistry. 2002;41(36):11044–11056. doi: 10.1021/bi020244r. [DOI] [PubMed] [Google Scholar]
- 10.Freer JH, Arbuthnott JP. Toxins of Staphylococcus aureus . Pharmacology and Therapeutics. 1982;19(1):55–106. doi: 10.1016/0163-7258(82)90042-0. [DOI] [PubMed] [Google Scholar]
- 11.Bernheimer AW, Rudy B. Interactions between membranes and cytolytic peptides. Biochimica et Biophysica Acta. 1986;864(1):123–141. doi: 10.1016/0304-4157(86)90018-3. [DOI] [PubMed] [Google Scholar]
- 12.Alouf JE. Bacterial protein toxins. An overview. Methods in Molecular Biology. 2000;145:1–26. doi: 10.1385/1-59259-052-7:1. [DOI] [PubMed] [Google Scholar]
- 13.Law RHP, Lukoyanova N, Voskoboinik I, et al. The structural basis for membrane binding and pore formation by lymphocyte perforin. Nature. 2010;468(7322):447–451. doi: 10.1038/nature09518. [DOI] [PubMed] [Google Scholar]
- 14.Hadders MA, Beringer DX, Gros P. Structure of C8α-MACPF reveals mechanism of membrane attack in complement immune defense. Science. 2007;317(5844):1552–1554. doi: 10.1126/science.1147103. [DOI] [PubMed] [Google Scholar]
- 15.Rosado CJ, Buckle AM, Law RHP, et al. A common fold mediates vertebrate defense and bacterial attack. Science. 2007;317(5844):1548–1551. doi: 10.1126/science.1144706. [DOI] [PubMed] [Google Scholar]
- 16.Tilley SJ, Orlova EV, Gilbert RJC, Andrew PW, Saibil HR. Structural basis of pore formation by the bacterial toxin pneumolysin. Cell. 2005;121(2):247–256. doi: 10.1016/j.cell.2005.02.033. [DOI] [PubMed] [Google Scholar]
- 17.Gouaux E. Channel-forming toxins: tales of transformation. Current Opinion in Structural Biology. 1997;7(4):566–573. doi: 10.1016/s0959-440x(97)80123-6. [DOI] [PubMed] [Google Scholar]
- 18.Anderluh G, Lakey JH. Disparate proteins use similar architectures to damage membranes. Trends in Biochemical Sciences. 2008;33(10):482–490. doi: 10.1016/j.tibs.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 19.Parker MW, Feil SC. Pore-forming protein toxins: from structure to function. Progress in Biophysics and Molecular Biology. 2005;88(1):91–142. doi: 10.1016/j.pbiomolbio.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 20.Gilbert RJC. Pore-forming toxins. Cellular and Molecular Life Sciences. 2002;59(5):832–844. doi: 10.1007/s00018-002-8471-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anderluh G, Dalla Serra M, Viero G, Guella G, Maček P, Menestrina G. Pore formation by equinatoxin II, a eukaryotic protein toxin, occurs by induction of nonlamellar lipid structures. The Journal of Biological Chemistry. 2003;278(46):45216–45223. doi: 10.1074/jbc.M305916200. [DOI] [PubMed] [Google Scholar]
- 22.Gilbert RJC. Inactivation and activity of cholesterol-dependent cytolysins: what structural studies tell us. Structure. 2005;13(8):1097–1106. doi: 10.1016/j.str.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 23.Idone V, Tam C, Goss JW, Toomre D, Pypaert M, Andrews NW. Repair of injured plasma membrane by rapid Ca2+ dependent endocytosis. Journal of Cell Biology. 2008;180(5):905–914. doi: 10.1083/jcb.200708010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Corrotte M, Fernandes MC, Tam C, Andrews NW. Toxin pores endocytosed during plasma membrane repair traffic into the lumen of MVBs for degradation. Traffic. 2012;13(3):483–494. doi: 10.1111/j.1600-0854.2011.01323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tam C, Idone V, Devlin C, et al. Exocytosis of acid sphingomyelinase by wounded cells promotes endocytosis and plasma membrane repair. Journal of Cell Biology. 2010;189(6):1027–1038. doi: 10.1083/jcb.201003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Husmann M, Dersch K, Bobkiewicz W, Beckmann E, Veerachato G, Bhakdi S. Differential role of p38 mitogen activated protein kinase for cellular recovery from attack by pore-forming S. aureus α-toxin or streptolysin O. Biochemical and Biophysical Research Communications. 2006;344(4):1128–1134. doi: 10.1016/j.bbrc.2006.03.241. [DOI] [PubMed] [Google Scholar]
- 27.Bischofberger M, Iacovache I, van der Goot FG. Pathogenic pore-forming proteins: function and host response. Cell Host & Microbe. 2012;12(3):266–275. doi: 10.1016/j.chom.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 28.Costa A, Gupta R, Signorino G, et al. Activation of the NLRP3 inflammasome by group B streptococci. Journal of Immunology. 1950;188(4):1953–1960. doi: 10.4049/jimmunol.1102543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mariathasan S, Weiss DS, Newton K, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440(7081):228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 30.Hamon MA, Cossart P. K+ efflux is required for histone H3 dephosphorylation by Listeria monocytogenes listeriolysin o and other pore-forming toxins. Infection and Immunity. 2011;79(7):2839–2846. doi: 10.1128/IAI.01243-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Craven RR, Gao X, Allen IC, et al. Staphylococcus aureusα-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS ONE. 2009;4(10) doi: 10.1371/journal.pone.0007446.e7446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holzinger D, Gieldon L, Mysore V, et al. Staphylococcus aureus Panton-Valentine leukocidin induces an inflammatory response in human phagocytes via the NLRP3 inflammasome. Journal of Leukocyte Biology. 2012;92(5):1069–1081. doi: 10.1189/jlb.0112014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fluegge K, Supper S, Siedler A, Berner R. Serotype distribution of invasive group B streptococcal isolates in infants: results from a nationwide active laboratory surveillance study over 2 years in Germany. Clinical Infectious Diseases. 2005;40(5):760–763. doi: 10.1086/427942. [DOI] [PubMed] [Google Scholar]
- 34.Henneke P, Berner R. Interaction of neonatal phagocytes with group B streptococcus: recognition and response. Infection and Immunity. 2006;74(6):3085–3095. doi: 10.1128/IAI.01551-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hensler ME, Liu GY, Sobczak S, Benirschke K, Nizet V, Heldt GP. Virulence role of group B Streptococcus β-hemolysin/cytolysin in a neonatal rabbit model of early-onset pulmonary infection. The Journal of Infectious Diseases. 2005;191(8):1287–1291. doi: 10.1086/428946. [DOI] [PubMed] [Google Scholar]
- 36.Reiss A, Braun JS, Jäger K, et al. Bacterial pore-forming cytolysins induce neuronal damage in a rat model of neonatal meningitis. The Journal of Infectious Diseases. 2011;203(3):393–400. doi: 10.1093/infdis/jiq047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pritzlaff CA, Chang JCW, Kuo SP, Tamura GS, Rubens CE, Nizet V. Genetic basis for the β-haemolytic/cytolytic activity of group B streptococcus. Molecular Microbiology. 2001;39(2):236–247. doi: 10.1046/j.1365-2958.2001.02211.x. [DOI] [PubMed] [Google Scholar]
- 38.Tapsall JW. Pigment production by Lancefield-group-B streptococci (Streptococcus agalactiae) Journal of Medical Microbiology. 1986;21(1):75–81. doi: 10.1099/00222615-21-1-75. [DOI] [PubMed] [Google Scholar]
- 39.Liu GY, Doran KS, Lawrence T, et al. Sword and shield: linked group B streptococcal β-hemolysin/cytolysin and carotenoid pigment function to subvert host phagocyte defense. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(40):14491–14496. doi: 10.1073/pnas.0406143101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nizet V, Gibson RL, Chi EY, Framson PE, Hulse M, Rubens CE. Group B streptococcal beta-hemolysin expression is associated with injury of lung epithelial cells. Infection and Immunity. 1996;64(9):3818–3826. doi: 10.1128/iai.64.9.3818-3826.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nizet V, Gibson RL, Rubens CE. The role of group B streptococci β-hemolysin expression in newborn lung injury. Advances in Experimental Medicine and Biology. 1997;418:627–630. doi: 10.1007/978-1-4899-1825-3_146. [DOI] [PubMed] [Google Scholar]
- 42.Gibson RL, Nizet V, Rubens CE. Group B streptococcal β-hemolysin promotes injury of lung microvascular endothelial cells. Pediatric Research. 1999;45(5, part 1):626–634. doi: 10.1203/00006450-199905010-00003. [DOI] [PubMed] [Google Scholar]
- 43.Nizet V, Kim KS, Stins M, et al. Invasion of brain microvascular endothelial cells by group B streptococci. Infection and Immunity. 1997;65(12):5074–5081. doi: 10.1128/iai.65.12.5074-5081.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rubens CE, Wessels MR, Kuypers JM, Kasper DL, Weiser JN. Molecular analysis of two group B streptococcal virulence factors. Seminars in Perinatology. 1990;14(4, supplement 1):22–29. [PubMed] [Google Scholar]
- 45.Bebien M, Hensler ME, Davanture S, et al. The pore-forming toxin β hemolysin/cytolysin triggers p38 MAPK-dependent IL-10 production in macrophages and inhibits innate immunity. PLoS Pathogens. 2012;8(7) doi: 10.1371/journal.ppat.1002812.100281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ring A, Depnering C, Pohl J, Nizet V, Shenep JL, Stremmel W. Synergistic action of nitric oxide release from murine macrophages caused by group B streptococcal cell wall and β-hemolysin/cytolysin. The Journal of Infectious Diseases. 2002;186(10):1518–1521. doi: 10.1086/344895. [DOI] [PubMed] [Google Scholar]
- 47.Lang S, Palmer M. Characterization of Streptococcus agalactiae CAMP factor as a pore-forming toxin. The Journal of Biological Chemistry. 2003;278(40):38167–38173. doi: 10.1074/jbc.M303544200. [DOI] [PubMed] [Google Scholar]
- 48.Lang S, Xue J, Guo Z, Palmer M. Streptococcus agalactiae CAMP factor binds to GPI-anchored proteins. Medical Microbiology and Immunology. 2007;196(1):1–10. doi: 10.1007/s00430-006-0021-2. [DOI] [PubMed] [Google Scholar]
- 49.Christie R, Atkins NE, Munch-Petersen E. A note on a lytic phenomenon shown by group B streptococci. The Australian Journal of Experimental Biology and Medical Science. 1944;22:197–200. doi: 10.1038/icb.1945.30. [DOI] [PubMed] [Google Scholar]
- 50.Hensler ME, Quach D, Hsieh CJ, Doran KS, Nizet V. CAMP factor is not essential for systemic virulence of Group B Streptococcus. Microbial Pathogenesis. 2008;44(1):84–88. doi: 10.1016/j.micpath.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hamon MA, Ribet D, Stavru F, Cossart P. Listeriolysin O: the Swiss army knife of Listeria. Trends in Microbiology. 2012;20(8):360–368. doi: 10.1016/j.tim.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 52.Portnoy DA, Jacks PS, Hinrichs DJ. Role of hemolysin for the intracellular growth of Listeria monocytogenes . Journal of Experimental Medicine. 1988;167(4):1459–1471. doi: 10.1084/jem.167.4.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kathariou S, Metz P, Hof H, Goebel W. Tn916-induced mutations in the hemolysin determinant affecting virulence of Listeria monocytogenes . Journal of Bacteriology. 1987;169(3):1291–1297. doi: 10.1128/jb.169.3.1291-1297.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Poston RM, Kurlander RJ. Cytokine expression in vivo during murine listeriosis: infection with live, virulent bacteria is required for monokine and lymphokine messenger RNA accumulation in the spleen. Journal of Immunology. 1992;149(9):3040–3044. [PubMed] [Google Scholar]
- 55.Melton-Witt JA, McKay SL, Portnoy DA. Development of a single-gene, signature-tag-based approach in combination with alanine mutagenesis to identify listeriolysin O residues critical for the in vivo survival of Listeria monocytogenes . Infection and Immunity. 2012;80(6):2221–2230. doi: 10.1128/IAI.06196-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rossjohn J, Gilbert RJC, Crane D, et al. The molecular mechanism of pneumolysin, a virulence factor from Streptococcus pneumoniae . Journal of Molecular Biology. 1998;284(2):449–461. doi: 10.1006/jmbi.1998.2167. [DOI] [PubMed] [Google Scholar]
- 57.Rossjohn J, Feil SC, McKinstry WJ, Tweten RK, Parker MW. Structure of a cholesterol-binding, thiol-activated cytolysin and a model of its membrane form. Cell. 1997;89(5):685–692. doi: 10.1016/s0092-8674(00)80251-2. [DOI] [PubMed] [Google Scholar]
- 58.Schuerch DW, Wilson-Kubalek EM, Tweten RK. Molecular basis of listeriolysin O pH dependence. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(35):12537–12542. doi: 10.1073/pnas.0500558102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gilbert RJC, Jiménez JL, Chen S, et al. Two structural transitions in membrane pore formation by pneumolysin, the pore-forming toxin of Streptococcus pneumoniae . Cell. 1999;97(5):647–655. doi: 10.1016/s0092-8674(00)80775-8. [DOI] [PubMed] [Google Scholar]
- 60.Shatursky O, Heuck AP, Shepard LA, et al. The mechanism of membrane insertion for a cholesterol-dependent cytolysin: a novel paradigm for pore-forming toxins. Cell. 1999;99(3):293–299. doi: 10.1016/s0092-8674(00)81660-8. [DOI] [PubMed] [Google Scholar]
- 61.Shepard LA, Heuck AP, Hamman BD, et al. Identification of a membrane-spanning domain of the thiol-activated pore-forming toxin Clostridium perfringens perfringolysin O: an α-helical to β-sheet transition identified by fluorescence spectroscopy. Biochemistry. 1998;37(41):14563–14574. doi: 10.1021/bi981452f. [DOI] [PubMed] [Google Scholar]
- 62.Dowd KJ, Tweten RK. The cholesterol-dependent cytolysin signature motif: a critical element in the allosteric pathway that couples membrane binding to pore assembly. PLoS Pathogens. 2012;8(7) doi: 10.1371/journal.ppat.1002787.100278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bhakdi S, Tranum-Jensen J, Sziegoleit A. Mechanism of membrane damage by streptolysin-O. Infection and Immunity. 1985;47(1):52–60. doi: 10.1128/iai.47.1.52-60.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Palmer M, Vulicevic I, Saweljew P, Valeva A, Kehoe M, Bhakdi S. Streptolysin O: a proposed model of allosteric interaction between a pore-forming protein and its target lipid bilayer. Biochemistry. 1998;37(8):2378–2383. doi: 10.1021/bi9720890. [DOI] [PubMed] [Google Scholar]
- 65.Czajkowsky DM, Hotze EM, Shao Z, Tweten RK. Vertical collapse of a cytolysin prepore moves its transmembrane β-hairpins to the membrane. EMBO Journal. 2004;23(16):3206–3215. doi: 10.1038/sj.emboj.7600350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jacobs T, Darji A, Frahm N, et al. Listeriolysin O: cholesterol inhibits cytolysis but not binding to cellular membranes. Molecular Microbiology. 1998;28(6):1081–1089. doi: 10.1046/j.1365-2958.1998.00858.x. [DOI] [PubMed] [Google Scholar]
- 67.Tilney LG, Portnoy DA. Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes . Journal of Cell Biology. 1989;109(4, part 1):1597–1608. doi: 10.1083/jcb.109.4.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gedde MM, Higgins DE, Tilney LG, Portnoy DA. Role of listeriolysin O in cell-to-cell spread of Listeria monocytogenes . Infection and Immunity. 2000;68(2):999–1003. doi: 10.1128/iai.68.2.999-1003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Singh R, Jamieson A, Cresswell P. GILT is a critical host factor for Listeria monocytogenes infection. Nature. 2008;455(7217):1244–1247. doi: 10.1038/nature07344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bernheimer HP, Tiraby JG. Inhibition of phage infection by pneumococcus capsule. Virology. 1976;73(1):308–309. doi: 10.1016/0042-6822(76)90085-4. [DOI] [PubMed] [Google Scholar]
- 71.Smyth CJ, Duncan JA. Thiol-activated cytolysins. In: Jeljaszewicz J, Wadstrom T, editors. Bacterial ToxIns and Cell Membranes. New York, NY, USA: Academic Press; 1978. pp. 129–183. [Google Scholar]
- 72.Radtke AL, Anderson KL, Davis MJ, DiMagno MJ, Swanson JA, O’Riordan MX. Listeria monocytogenes exploits cystic fibrosis transmembrane conductance regulator (CFTR) to escape the phagosome. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(4):1633–1638. doi: 10.1073/pnas.1013262108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lam GY, Fattouh R, Muise AM, Grinstein S, Higgins DE, Brumell JH. Listeriolysin O suppresses phospholipase C-mediated activation of the microbicidal NADPH oxidase to promote Listeria monocytogenes infection. Cell Host & Microbe. 2011;10(6):627–634. doi: 10.1016/j.chom.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vadia S, Arnett E, Haghighat AC, Wilson-Kubalek EM, Tweten RK, Seveau S. The pore-forming toxin listeriolysin O mediates a novel entry pathway of L. monocytogenes into human hepatocytes. PLoS Pathogens. 2011;7(11) doi: 10.1371/journal.ppat.1002356.e1002356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Glomski IJ, Gedde MM, Tsang AW, Swanson JA, Portnoy DA. The Listeria monocytogenes hemolysin has an acidic pH optimum to compartmentalize activity and prevent damage to infected host cells. Journal of Cell Biology. 2002;156(6):1029–1038. doi: 10.1083/jcb.200201081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bavdek A, Gekara NO, Priselac D, et al. Sterol and pH interdependence in the binding, oligomerization, and pore formation of listeriolysin O. Biochemistry. 2007;46(14):4425–4437. doi: 10.1021/bi602497g. [DOI] [PubMed] [Google Scholar]
- 77.Bavdek A, Kostanjšek R, Antonini V, et al. pH dependence of listeriolysin O aggregation and pore-forming ability. The FEBS Journal. 2012;279(1):126–141. doi: 10.1111/j.1742-4658.2011.08405.x. [DOI] [PubMed] [Google Scholar]
- 78.Dramsi S, Cossart P. Listeriolysin O-mediated calcium influx potentiates entry of Listeria monocytogenes into the human Hep-2 epithelial cell line. Infection and Immunity. 2003;71(6):3614–3618. doi: 10.1128/IAI.71.6.3614-3618.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Repp H, Pamukçi Z, Koschinski A, et al. Listeriolysin of Listeria monocytogenes forms Ca2+-permeable pores leading to intracellular Ca2+ oscillations. Cellular Microbiology. 2002;4(8):483–491. doi: 10.1046/j.1462-5822.2002.00207.x. [DOI] [PubMed] [Google Scholar]
- 80.Malley R, Henneke P, Morse SC, et al. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(4):1966–1971. doi: 10.1073/pnas.0435928100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jin MP, Ng VH, Maeda S, Rest RF, Karin M. Anthrolysin O and other gram-positive cytolysins are toll-like receptor 4 agonists. Journal of Experimental Medicine. 2004;200(12):1647–1655. doi: 10.1084/jem.20041215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gekara NO, Dietrich N, Lyszkiewicz M, Lienenklaus S, Weiss S. Signals triggered by a bacterial pore-forming toxin contribute to toll-like receptor redundancy in gram-positive bacterial recognition. The Journal of Infectious Diseases. 2009;199(1):124–133. doi: 10.1086/595562. [DOI] [PubMed] [Google Scholar]
- 83.Gekara NO, Jacobs T, Chakraborty T, Weiss S. The cholesterol-dependent cytolysin listeriolysin O aggregates rafts via oligomerization. Cellular Microbiology. 2005;7(9):1345–1356. doi: 10.1111/j.1462-5822.2005.00561.x. [DOI] [PubMed] [Google Scholar]
- 84.Gekara NO, Zietara N, Geffers R, Weiss S. Listeria monocytogenes induces T cell receptor unresponsiveness through pore-forming toxin listeriolysin O. The Journal of Infectious Diseases. 2010;202(11):1698–1707. doi: 10.1086/657145. [DOI] [PubMed] [Google Scholar]
- 85.McCracken GH, Jr., Shinefield HR. Changes in the pattern of neonatal septicemia and meningitis. American Journal of Diseases of Children. 1966;112(1):33–39. doi: 10.1001/archpedi.1966.02090100069006. [DOI] [PubMed] [Google Scholar]
- 86.Harrison LM, Morris JA, Telford DR, Brown SM, Jones K. The nasopharyngeal bacterial flora in infancy: effects of age, gender, season, viral upper respiratory tract infection and sleeping position. FEMS Immunology and Medical Microbiology. 1999;25(1-2):19–28. doi: 10.1111/j.1574-695X.1999.tb01323.x. [DOI] [PubMed] [Google Scholar]
- 87.Brosnahan AJ, Schlievert PM. Gram-positive bacterial superantigen outside-in signaling causes toxic shock syndrome. The FEBS Journal. 2011;278(23):4649–4667. doi: 10.1111/j.1742-4658.2011.08151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Arad G, Levy R, Nasie I, et al. Binding of superantigen toxins into the CD28 homodimer interface is essential for induction of cytokine genes that mediate lethal shock. PLoS Biology. 2011;9(9) doi: 10.1371/journal.pbio.1001149.e1001149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ramu Y, Xu Y, Lu Z. Inhibition of CFTR Cl- channel function caused by enzymatic hydrolysis of sphingomyelin. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(15):6448–6453. doi: 10.1073/pnas.0701354104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bhakdi S, Grimminger F, Suttorp N, Walmrath D, Seeger R. Proteinaceous bacterial toxins and pathogenesis of sepsis syndrome and septic shock: the unknown connection. Medical Microbiology and Immunology. 1994;183(3):119–144. doi: 10.1007/BF00196048. [DOI] [PubMed] [Google Scholar]
- 91.Plummer R, Bodkin J, Yau TW, et al. Modelling Staphylococcus aureus-induced septicemia using NMR. Magnetic Resonance in Medicine. 2007;58(4):656–665. doi: 10.1002/mrm.21392. [DOI] [PubMed] [Google Scholar]
- 92.Ji Y, Marra A, Rosenberg M, Woodnutt G. Regulated antisense RNA eliminates alpha-toxin virulence in Staphylococcus aureus infection. Journal of Bacteriology. 1999;181(21):6585–6590. doi: 10.1128/jb.181.21.6585-6590.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kernodle DS, Voladri RKR, Menzies BE, Hager CC, Edwards KM. Expression of an antisense hla fragment in Staphylococcus aureus reduces alpha-toxin production in vitro and attenuates lethal activity in a murine model. Infection and Immunity. 1997;65(1):179–184. doi: 10.1128/iai.65.1.179-184.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kebaier C, Chamberland RR, Allen IC, et al. Staphylococcus aureus alpha-hemolysin mediates virulence in a murine model of severe pneumonia through activation of the NLRP3 inflammasome. The Journal of Infectious Diseases. 2012;205(5):807–817. doi: 10.1093/infdis/jir846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ragle BE, Bubeck Wardenburg J. Anti-alpha-hemolysin monoclonal antibodies mediate protection against Staphylococcus aureus pneumonia. Infection and Immunity. 2009;77(7):2712–2718. doi: 10.1128/IAI.00115-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Menzies BE, Kernodle DS. Passive immunization with antiserum to a nontoxic alpha-toxin mutant from Staphylococcus aureus is protective in a murine model. Infection and Immunity. 1996;64(5):1839–1841. doi: 10.1128/iai.64.5.1839-1841.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jonsson P, Lindberg M, Haraldsson I, Wadstrom T. Virulence of Staphylococcus aureus in a mouse mastitis model: studies of alpha hemolysin, coagulase, and protein A as possible virulence determinants with protoplast fusion and gene cloning. Infection and Immunity. 1985;49(3):765–769. doi: 10.1128/iai.49.3.765-769.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kielian T, Cheung A, Hickey WF. Diminished virulence of an alpha-toxin mutant of Staphylococcus aureus in experimental brain abscesses. Infection and Immunity. 2001;69(11):6902–6911. doi: 10.1128/IAI.69.11.6902-6911.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Callegan MC, Engel LS, Hill JM, O’Callaghan RJ. Corneal virulence of Staphylococcus aureus: roles of alpha-toxin and protein A in pathogenesis. Infection and Immunity. 1994;62(6):2478–2482. doi: 10.1128/iai.62.6.2478-2482.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Song L, Hobaugh MR, Shustak C, Cheley S, Bayley H, Gouaux JE. Structure of staphylococcal α-hemolysin, a heptameric transmembrane pore. Science. 1996;274(5294):1859–1866. doi: 10.1126/science.274.5294.1859. [DOI] [PubMed] [Google Scholar]
- 101.McElroy MC, Harty HR, Hosford GE, Boylan GM, Pittet JF, Foster TJ. Alpha-toxin damages the air-blood barrier of the lung in a rat model of Staphylococcus aureus-induced pneumonia. Infection and Immunity. 1999;67(10):5541–5544. doi: 10.1128/iai.67.10.5541-5544.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wilke GA, Bubeck Wardenburg J. Role of a disintegrin and metalloprotease 10 in Staphylococcus aureusα-hemolysin—mediated cellular injury. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(30):13473–13478. doi: 10.1073/pnas.1001815107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Inoshima I, Inoshima N, Wilke GA, et al. A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nature Medicine. 2011;17(10):1310–1314. doi: 10.1038/nm.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hruz P, Zinkernagel AS, Jenikova G, et al. NOD2 contributes to cutaneous defense against Staphylococcus aureus through α-toxin-dependent innate immune activation. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(31):12873–12878. doi: 10.1073/pnas.0904958106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gravet A, Colin DA, Keller D, Giradot R, Monteil H, Prévost G. Characterization of a novel structural member, LukE-LukD, of the bi-component staphylococcal leucotoxins family. FEBS Letters. 1998;436(2):202–208. doi: 10.1016/s0014-5793(98)01130-2. [DOI] [PubMed] [Google Scholar]
- 106.Cooney J, Kienle Z, Foster TJ, O’Toole PW. The gamma-hemolysin locus of Staphylococcus aureus comprises three linked genes, two of which are identical to the genes for the F and S components of leukocidin. Infection and Immunity. 1993;61(2):768–771. doi: 10.1128/iai.61.2.768-771.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wright J. Staphylococcal leucocidin (Neisser-Wechsberg type) and antileucociddin. The Lancet. 1936;227(5879):1002–1005. [Google Scholar]
- 108.Labandeira-Rey M, Couzon F, Boisset S, et al. Staphylococcus aureus Panton-Valentine leukocidin causes necrotizing pneumonia. Science. 2007;315(5815):1130–1133. doi: 10.1126/science.1137165. [DOI] [PubMed] [Google Scholar]
- 109.Lipinska U, Hermans K, Meulemans L, et al. Panton-Valentine leukocidin does play a role in the early stage of Staphylococcus aureus skin infections: a rabbit model. PLoS ONE. 2011;6(8) doi: 10.1371/journal.pone.0022864.e22864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zivkovic A, Sharif O, Stich K, et al. TLR 2 and CD14 mediate innate immunity and lung inflammation to staphylococcal panton-valentine leukocidin in vivo. Journal of Immunology. 2011;186(3):1608–1617. doi: 10.4049/jimmunol.1001665. [DOI] [PubMed] [Google Scholar]
- 111.Alonzo F, Benson MA, Chen J, Novick RP, Shopsin B, Torres VJ. Staphylococcus aureus leucocidin ED contributes to systemic infection by targeting neutrophils and promoting bacterial growth in vivo. Molecular Microbiology. 2012;83(2):423–435. doi: 10.1111/j.1365-2958.2011.07942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Alonzo F, Kozhaya L, Rawlings SA, et al. CCR5 is a receptor for Staphylococcus aureus leukotoxin ED. Nature. 2013;493(7430):51–55. doi: 10.1038/nature11724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Olson R, Nariya H, Yokota K, Kamio Y, Gouaux E. Crystal structure of staphylococcal lukF delineates conformational changes accompanying formation of a transmembrane channel. Nature Structural Biology. 1999;6(2):134–140. doi: 10.1038/5821. [DOI] [PubMed] [Google Scholar]
- 114.Pédelacq JD, Maveyraud L, Prévost G, et al. The structure of a Staphylococcus aureus leucocidin component (LukF-PV) reveals the fold of the water-soluble species of a family of transmembrane pore-forming toxins. Structure. 1999;7(3):277–287. doi: 10.1016/s0969-2126(99)80038-0. [DOI] [PubMed] [Google Scholar]
- 115.Guillet V, Roblin P, Werner S, et al. Crystal structure of leucotoxin S component: new insight into the staphylococcal β-barrel pore-forming toxins. The Journal of Biological Chemistry. 2004;279(39):41028–41037. doi: 10.1074/jbc.M406904200. [DOI] [PubMed] [Google Scholar]
- 116.Yamashita K, Kawai Y, Tanaka Y, et al. Crystal structure of the octameric pore of staphylococcal γ-hemolysin reveals the β-barrel pore formation mechanism by two components. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(42):17314–17319. doi: 10.1073/pnas.1110402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Meyer F, Girardot R, Piémont Y, Prévost G, Colin DA. Analysis of the specificity of panton-valentine leucocidin and gamma-hemolysin F component binding. Infection and Immunity. 2009;77(1):266–273. doi: 10.1128/IAI.00402-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Szmigielski S, Sobiczewska E, Prévost G, Monteil H, Colin DA, Jeljaszewicz J. Effect of purified staphylococcal leukocidal toxins on isolated blood polymorphonuclear leukocytes and peritoneal macrophages in vitro. Zentralblatt fur Bakteriologie. 1998;288(3):383–394. doi: 10.1016/s0934-8840(98)80012-1. [DOI] [PubMed] [Google Scholar]
- 119.Weston EJ, Pondo T, Lewis MM, et al. The burden of invasive early-onset neonatal sepsis in the United States, 2005–2008. The Pediatric Infectious Disease Journal. 2011;30(11):937–941. doi: 10.1097/INF.0b013e318223bad2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shane AL, Stoll BJ. Recent developments and current issues in the epidemiology, diagnosis, and management of bacterial and fungal neonatal sepsis. American Journal of Perinatology. 2013;30(2):131–141. doi: 10.1055/s-0032-1333413. [DOI] [PubMed] [Google Scholar]
- 121.Stoll BJ, Hansen N, Fanaroff AA, et al. Changes in pathogens causing early-onset sepsis in very-low-birth-weight infants. The New England Journal of Medicine. 2002;347(4):240–247. doi: 10.1056/NEJMoa012657. [DOI] [PubMed] [Google Scholar]
- 122.Wiles TJ, Mulvey MA. The RTX pore-forming toxin α-hemolysin of uropathogenic Escherichia coli: progress and perspectives. Future Microbiology. 2013;8:73–84. doi: 10.2217/fmb.12.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kaper JB, Nataro JP, Mobley HLT. Pathogenic Escherichia coli . Nature Reviews Microbiology. 2004;2(2):123–140. doi: 10.1038/nrmicro818. [DOI] [PubMed] [Google Scholar]
- 124.May AK, Sawyer RG, Gleason T, Whitworth A, Pruett TL. In vivo cytokine response to Escherichia coli alpha-hemolysin determined with genetically engineered hemolytic and nonhemolytic E. coli variants. Infection and Immunity. 1996;64(6):2167–2171. doi: 10.1128/iai.64.6.2167-2171.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Smith YC, Rasmussen SB, Grande KK, Conran RM, O’Brien AD. Hemolysin of uropathogenic Escherichia coli evokes extensive shedding of the uroepithelium and hemorrhage in bladder tissue within the first 24 hours after intraurethral inoculation of mice. Infection and Immunity. 2008;76(7):2978–2990. doi: 10.1128/IAI.00075-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wiles TJ, Bower JM, Redd MJ, Mulvey MA. Use of zebrafish to probe the divergent virulence potentials and toxin requirements of extraintestinal pathogenic Escherichia coli . PLoS Pathogens. 2009;5(12) doi: 10.1371/journal.ppat.1000697.e1000697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bingen E, Picard B, Brahimi N, et al. Phylogenetic analysis of Escherichia coli strains causing neonatal meningitis suggests horizontal gene transfer from a predominant pool of highly virulent B2 group strains. The Journal of Infectious Diseases. 1998;177(3):642–650. doi: 10.1086/514217. [DOI] [PubMed] [Google Scholar]
- 128.McCracken GH, Jr., Sarff LD, Glode MP. Relation between Escherichia coli K1 capsular polysaccharide antigen and clinical outcome in neonatal meningitis. The Lancet. 1974;2(7875):246–250. doi: 10.1016/s0140-6736(74)91413-5. [DOI] [PubMed] [Google Scholar]
- 129.Guiral E, Bosch J, Vila J, Soto SM. Prevalence of Escherichia coli among samples collected from the genital tract in pregnant and nonpregnant women: relationship with virulence. FEMS Microbiology Letters. 2011;314(2):170–173. doi: 10.1111/j.1574-6968.2010.02160.x. [DOI] [PubMed] [Google Scholar]
- 130.Welch RA. RTX toxin structure and function: a story of numerous anomalies and few analogies in toxin biology. Current Topics in Microbiology and Immunology. 2001;257:85–111. doi: 10.1007/978-3-642-56508-3_5. [DOI] [PubMed] [Google Scholar]
- 131.Baran K, Dunstone M, Chia J, et al. The molecular basis for perforin oligomerization and transmembrane pore assembly. Immunity. 2009;30(5):684–695. doi: 10.1016/j.immuni.2009.03.016. [DOI] [PubMed] [Google Scholar]
- 132.Bakás L, Veiga MP, Soloaga A, Ostolaza H, Goñi FM. Calcium-dependent conformation of E. coliα-haemolysin. Implications for the mechanism of membrane insertion and lysis. Biochimica et Biophysica Acta. 1998;1368(2):225–234. doi: 10.1016/s0005-2736(97)00181-8. [DOI] [PubMed] [Google Scholar]
- 133.Herlax V, Maté S, Rimoldi O, Bakás L. Relevance of fatty acid covalently bound to Escherichia coli -hemolysin and membrane microdomains in the oligomerization process. The Journal of Biological Chemistry. 2009;284(37):25199–25210. doi: 10.1074/jbc.M109.009365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Benz R, Dobereiner A, Ludwig A, Goebel W. Haemolysin of Escherichia coli: comparison of pore-forming properties between chromosome and plasmid-encoded haemolysins. FEMS Microbiology Immunology. 1992;105(1–3):55–62. doi: 10.1111/j.1574-6968.1992.tb05887.x. [DOI] [PubMed] [Google Scholar]
- 135.Benz R, Schmid A, Wagner W, Goebel W. Pore formation by the Escherichia coli hemolysin: evidence for an association-dissociation equilibrium of the pore-forming aggregates. Infection and Immunity. 1989;57(3):887–895. doi: 10.1128/iai.57.3.887-895.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ludwig A, Benz R, Goebel W. Oligomerization of Escherichia coli haemolysin (HlyA) is involved in pore formation. Molecular and General Genetics. 1993;241(1-2):89–96. doi: 10.1007/BF00280205. [DOI] [PubMed] [Google Scholar]
- 137.Bakás L, Chanturiya A, Herlax V, Zimmerberg J. Paradoxical lipid dependence of pores formed by the Escherichia coliα-hemolysin in planar phospholipid bilayer membranes. Biophysical Journal. 2006;91(10):3748–3755. doi: 10.1529/biophysj.106.090019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Skals M, Jorgensen NR, Leipziger J, Praetorius HA. α-hemolysin from Escherichia coli uses endogenous amplification through P2X receptor activation to induce hemolysis. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(10):4030–4035. doi: 10.1073/pnas.0807044106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Skals M, Leipziger J, Praetorius HA. Haemolysis induced by α-toxin from Staphylococcus aureus requires P2X receptor activation. Pflügers Archiv: European Journal of Physiology. 2011;462(5):669–679. doi: 10.1007/s00424-011-1010-x. [DOI] [PubMed] [Google Scholar]
- 140.Dhakal BK, Mulvey MA. The UPEC pore-forming toxin α-hemolysin triggers proteolysis of host proteins to disrupt cell adhesion, inflammatory, and survival pathways. Cell Host & Microbe. 2012;11(1):58–69. doi: 10.1016/j.chom.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hilbert DW, et al. Clinical Escherichia coli isolates utilize alpha-hemolysin to inhibit in vitro epithelial cytokine production. Microbes and Infection / Institut Pasteur. 2012;14(7-8):628–638. doi: 10.1016/j.micinf.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 142.Cravioto A, Gross RJ, Scotland SM, Rowe B. Mannose-resistant haemagglutination of human erythrocytes by strains of Escherichia coli from extraintestinal sources: lack of correlation with colonisation factor antigen (CFA/I) FEMS Microbiology Letters. 1979;6(1):41–44. [Google Scholar]
- 143.Bian Z, Brauner A, Li Y, Normark S. Expression of and cytokine activation by Escherichia coli curli fibers in human sepsis. The Journal of Infectious Diseases. 2000;181(2):602–612. doi: 10.1086/315233. [DOI] [PubMed] [Google Scholar]
- 144.Evans DJ, Jr., Evans DG, Hohne C. Hemolysin and K antigens in relation to serotype and hemagglutination type of M isolated from extraintestinal infections. Journal of Clinical Microbiology. 1981;13(1):171–178. doi: 10.1128/jcm.13.1.171-178.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.McCabe WR, Kaijser B, Olling S. Escherichia coli in bacteremia: K and O antigens and serum sensitivity of strains from adults and neonates. The Journal of Infectious Diseases. 1978;138(1):33–41. doi: 10.1093/infdis/138.1.33. [DOI] [PubMed] [Google Scholar]
- 146.Ludwig A, Von Rhein C, Bauer S, Hüttinger C, Goebel W. Molecular analysis of cytolysin A (ClyA) in pathogenic Escherichia coli strains. Journal of Bacteriology. 2004;186(16):5311–5320. doi: 10.1128/JB.186.16.5311-5320.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tourret J, Aloulou M, Garry L, et al. The interaction between a non-pathogenic and a pathogenic strain synergistically enhances extra-intestinal virulence in Escherichia coli . Microbiology. 2011;157, part 3:774–785. doi: 10.1099/mic.0.037416-0. [DOI] [PubMed] [Google Scholar]
- 148.Wallace AJ, Stillman TJ, Atkins A, et al. E. coli hemolysin E (Hlye, ClyA, SheA): X-ray crystal structure of the toxin and observation of membrane pores by electron microscopy. Cell. 2000;100(2):265–276. doi: 10.1016/s0092-8674(00)81564-0. [DOI] [PubMed] [Google Scholar]
- 149.Mueller M, Grauschopf U, Maier T, Glockshuber R, Ban N. The structure of a cytolytic α-helical toxin pore reveals its assembly mechanism. Nature. 2009;459(7247):726–730. doi: 10.1038/nature08026. [DOI] [PubMed] [Google Scholar]
- 150.Pinheiro Da Silva F, Nizet V. Cell death during sepsis: integration of disintegration in the inflammatory response to overwhelming infection. Apoptosis. 2009;14(4):509–521. doi: 10.1007/s10495-009-0320-3. [DOI] [PubMed] [Google Scholar]
- 151.Eitel J, Suttorp N, Opitz B. Innate immune recognition and inflammasome activation in listeria monocytogenes infection. Frontiers in Microbiology. 2010;1, article 149 doi: 10.3389/fmicb.2010.00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Perret M, Badiou C, Lina G, et al. Cross-talk between Staphylococcus aureus leukocidins-intoxicated macrophages and lung epithelial cells triggers chemokine secretion in an inflammasome-dependent manner. Cellular Microbiology. 2012;14(7):1019–1036. doi: 10.1111/j.1462-5822.2012.01772.x. [DOI] [PubMed] [Google Scholar]
- 153.Ayres JS, Trinidad NJ, Vance RE. Lethal inflammasome activation by a multidrug-resistant pathobiont upon antibiotic disruption of the microbiota. Nature Medicine. 2012;18(5):799–806. doi: 10.1038/nm.2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Imre G, Rajalingam K. Role for caspase-2 during pore-forming toxin-mediated apoptosis. Cell Cycle. 2012;11(20):3709–3710. doi: 10.4161/cc.22046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Imre G, Heering J, Takeda AN, et al. Caspase-2 is an initiator caspase responsible for pore-forming toxin-mediated apoptosis. EMBO Journal. 2012;31(11):2615–2628. doi: 10.1038/emboj.2012.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Genestier AL, Michallet MC, Prévost G, et al. Staphylococcus aureus Panton-Valentine leukocidin directly targets mitochondria and induces Bax-independent apoptosis of human neutrophils. The Journal of Clinical Investigation. 2005;115(11):3117–3127. doi: 10.1172/JCI22684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Carrero JA, Unanue ER. Mechanisms and immunological effects of apoptosis caused by Listeria monocytogenes. Advances in Immunology. 2012;113:157–174. doi: 10.1016/B978-0-12-394590-7.00001-4. [DOI] [PubMed] [Google Scholar]
- 158.Fernandez-Prada C, Tall BD, Elliott SE, Hoover DL, Nataro JP, Venkatesan MM. Hemolysin-positive enteroaggregative and cell-detaching Escherichia coli strains cause oncosis of human monocyte-derived macrophages and apoptosis of murine J774 cells. Infection and Immunity. 1998;66(8):3918–3924. doi: 10.1128/iai.66.8.3918-3924.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lai XH, Arencibia I, Johansson A, et al. Cytocidal and apoptotic effects of the ClyA protein from Escherichia coli on primary and cultured monocytes and macrophages. Infection and Immunity. 2000;68(7):4363–4367. doi: 10.1128/iai.68.7.4363-4367.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nature Reviews Immunology. 2008;8(10):776–787. doi: 10.1038/nri2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Stavru F, Bouillaud F, Sartori A, Ricquier D, Cossart P. Listeria monocytogenes transiently alters mitochondrial dynamics during infection. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(9):3612–3617. doi: 10.1073/pnas.1100126108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hensler ME, Miyamoto S, Nizet V. Group B streptococcal β-Hemolysin/cytolysin directly impairs cardiomyocyte viability and function. PLoS ONE. 2008;3(6) doi: 10.1371/journal.pone.0002446.e2446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Malley R, Anderson PW. Serotype-independent pneumococcal experimental vaccines that induce cellular as well as humoral immunity. Proceedings of the National Academy of Sciences of the United States. 2012;109(10):3623–3627. doi: 10.1073/pnas.1121383109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Adhikari RP, Karauzum H, Sarwar J, et al. Novel structurally designed vaccine for S. aureus alpha-hemolysin: protection against bacteremia and pneumonia. PloS One. 2012;7(6) doi: 10.1371/journal.pone.0038567.e38567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Justin N, Walker N, Bullifent HL, et al. The first strain of Clostridium perfringens isolated from an avian source has an alpha-toxin with divergent structural and kinetic properties. Biochemistry. 2002;41(20):6253–6262. doi: 10.1021/bi012015v. [DOI] [PubMed] [Google Scholar]