

Abstract
Mouse models have informed us that p63 is critical for normal epidermal development and homeostasis. The p53/p63/p73 family is expressed as multiple protein isoforms due to a combination of alternative promoter usage and C-terminal alternative splicing. These isoforms can mimic or interfere with one another, and their balance ultimately determines biological outcome in a context-dependent manner. While not frequently mutated, p63, and in particular the ΔNp63 subclass, is commonly overexpressed in human squamous cell cancers. In vitro keratinocytes and murine transgenic and transplantation models have been invaluable in elucidating the contribution of altered p63 levels to cancer development, and studies have identified the roles for ΔNp63 isoforms in keratinocyte survival and malignant progression, likely due in part to their transcriptional regulatory function. These findings can be extended to human cancers; for example, the novel recognition of NFκB/c-Rel as a downstream effector of p63 has identified a role for NFκB/c-Rel in human squamous cell cancers. These models will be critical in enhancing the understanding of the specific molecular mechanisms of cancer development and progression.
1. Introduction
p53 is a tumor suppressor that is upregulated and activated across organ systems as a tissue protective stress response mechanism [1]. p63 is a member of the p53 gene family which also includes p73. In contrast to p53, both p63 and p73 exhibit cell-type-specific expression patterns and exert tissue-specific functions [2, 3]. Relevant to this review, p63 plays an essential role in the development and maintenance of normal stratified squamous epithelium. All p53 family members encode multiple protein isoforms that act in overlapping or opposing manners both within and across family members. Given the complexity of the p53 family and the potential for the different family members to mimic or interfere with each other, the balance of p53 family isoforms in a given cellular context can impact the biological outcome. In this review, we highlight how information derived from mouse models has provided insight into molecular mechanisms of normal keratinocyte growth regulation and human cancer pathogenesis. In particular, we focus on the p63 gene, the role of its gene products in normal epidermal development and homeostasis, and how dysregulation of p63 protein expression, which is tightly controlled under normal conditions, contributes to squamous carcinogenesis, not only of the skin, but also in other squamous epithelial cancers such as those of the head and neck.
2. Overview of p63 Structure/Function
Members of the p53 family were identified based on shared homology within their major functional domains: transactivation (TA), DNA binding (DBD), and oligomerization (OD); and exist as multiple protein isoforms due to a combination of alternate promoter usage and alternative splicing [4, 5]. Use of alternative promoters gives rise to isoforms of two classes: TA and ΔN. The TAp63 and TAp73 isoforms possess a transactivation domain with homology and function similar to that of p53, while the ΔNp63 and ΔNp73 isoforms lack this domain and can act to block TAp53-, TAp63-, and TAp73-mediated transcription [4] via the mechanisms discussed below. However, this does not imply that ΔNp63 isoforms lack transcriptional activation activity as alternate transactivation domains have been described both within the N-terminus of the ΔNp63 isoforms [6, 7] and in exons 11 and 12 of the C-terminus (transactivation domain 2 (TA2)) [8]. Further analysis has suggested that the second region is unlikely to be an independent activation domain [7]. Refined mapping studies indicate that this domain instead serves to modulate transcriptional activities associated with ΔNp63 isoforms [7]. Correspondingly, many positive transcriptional targets of ΔNp63 have been identified, which are discussed in this review.
All TA and ΔNp63 isoforms contain the DBD and OD domains but differ at the C-termini. This additional complexity is conferred on these proteins due to C-terminal alternative splicing, which in the case of p63 gives rise to TA and ΔN subclasses of p63α, β, γ, δ, and ε isoforms [4, 9] (Figure 1). Of these isoforms, α is the longest and contains a sterile alpha motif (SAM) protein-protein interaction domain [10] and a transcriptional inhibition domain (TID) [11]. The TID comprises 2 subdomains, one of which binds and masks the TA domain of TAp63α and the other, which is subject to sumoylation resulting in decreased intracellular p63α concentration and correspondingly to decreased activity [11–13]. Degradation of p63α is also promoted by the E3 ubiquitin ligase ITCH via ubiquitylation at the N-terminal border of the SAM domain of the p63α isoforms [14]. Regarding the other p63 C-terminal splice variants, exon 13 is spliced out of the β-isoform, which thus also lacks the SAM and TID domains. Both the α and β isoforms of p63 contain a phosphodegron motif utilized by Fbw7 E3 ubiquitin ligase in MDM2-mediated degradation [15]. The γ, δ, and ε isoforms all truncate shortly after the oligomerization domain, with each containing a unique C-terminal sequence [9]. Thus, all three isoforms lack the SAM and TID domains (Figure 1).
Figure 1.
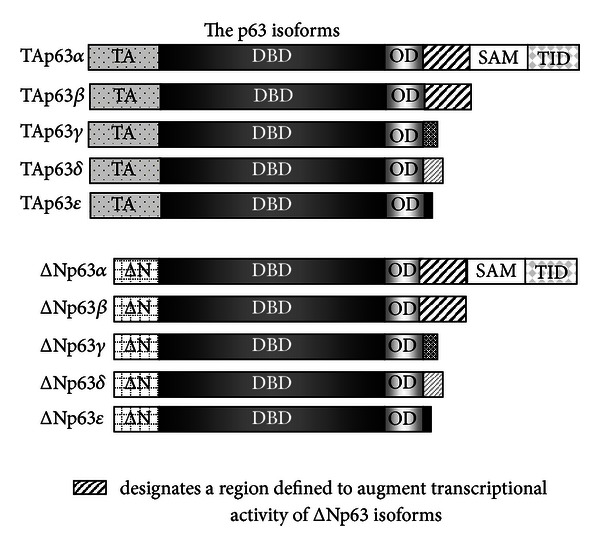
Use of alternative splicing gives rise to p63 isoforms of 2 subclasses: TAp63 and ΔNp63. Within each of these subclasses, C-terminal alternative splicing gives rise to α, β, γ, δ, and ε isoforms. The isoforms share homology in certain protein domains: TA (transactivation domain), ΔN, DBD (DNA binding domain), OD (oligomerization domain), SAM (sterile alpha motif domain), and TID (transactivation inhibition domain).
Like p53, the p63 and p73 proteins function as tetramers via their oligomerization domains. The oligomerization domains of p63 and p73, due to the presence of an additional α-helix, are more similar to one another than to that of p53 [16]. p63 and p73 were not observed to interact with p53 through their oligomerization domains but strongly interact with one another through this domain, with the p63/p73 heterotetramers exhibiting enhanced stability over homotetramers [16]. While p53 does not interact with p63/p73 through the oligomerization domain, WTp53 has been shown to target ΔNp63α for caspase-mediated degradation via interactions between the DNA binding domain of each protein [17], and mutated p53 has been shown to interact with the core DNA binding domains of p63 and p73, thereby impairing DNA binding and transactivation [18].
DNA binding is an area in which p53 family members can mimic or compete with each other. While p63 has been shown to bind to p53 responsive consensus sequences, distinct p63 responsive elements have also been identified [19–21]. It has been reported that the global DNA binding pattern of p73 does not differ from that of p63, but intensity of binding at given sites does vary depending on the cell type profiled [22]. This suggests that competition between the homo- and heterotetramers of different isoforms of p63 and p73, which are subject to cellular context, may define site occupancy. Beyond interactions with one another via their oligomerization domains or their DNA binding domains, and their competition at DNA response elements, p53 family members have been shown to be involved in feedback loops with one another that impact expression levels [23, 24]. Thus, at many levels dysregultion of any one family member may impact the fine balance that is involved in maintaining normal epidermal homeostasis.
3. p63 and Normal Skin Biology
p63 is critical for normal epidermal morphogenesis [3, 25]. In the mature epidermis, the predominant p63 isoform expressed is ΔNp63α, and expression of this isoform is associated with the proliferative compartment [26]. Expression of this isoform is critical for the maintenance of the mature epidermis [27]. However, it is also the ΔNp63α isoform that is overexpressed in many squamous cell cancers [28]. As a starting point to understand how overexpression of a single isoform with ensuing disruption of the balance of p53 family members might contribute to squamous cancer pathogenesis, it is important to first understand the role of p63 both in normal epidermal morphogenesis and in homeostasis of the mature epidermis. Significant insight has been obtained through the use of mouse models outlined below.
3.1. Role of p63 in Mouse Models of Epidermal Morphogenesis and in Human Ectodermal Dysplasias
3.1.1. Mouse Models of Epidermal Morphogenesis
The criticality of p63 to normal epidermal development was highlighted by simultaneously published mouse models lacking functional p63 [3, 25]. The mice were developed using distinct molecular strategies and upon gross phenotypic examination appeared similar; however, in-depth analysis of the epidermal phenotype gave rise to alternate hypotheses as to the role of p63 in epidermal development: epidermal progenitor cell maintenance versus commitment to stratification. In the mice developed by Yang et al., exons 6–8 corresponding to p63's DBD were replaced with the neomycin resistance gene [3]. In these mice, patches of disorganized epithelial cells positive for late markers of keratinocyte differentiation and negative for keratin 5 were evident, suggestive of a role for p63 in maintaining epidermal progenitor cells. Mice generated by Mills et al. were derived using an insertional gap repair mechanism [25]. Two strains generated by this approach, Brdm1 (truncating within exon 6) and Brdm2 (truncating after exon 10), appeared macroscopically identical, and thus, the strains were not distinguished in subsequent experiments in the seminal paper. Microscopic analysis of these mice revealed a layer of flattened cells expressing keratin 14 at low levels with no evidence of stratification or differentiation marker expression, suggestive of a failure to commit to a stratified epidermis. While no mRNA transcripts were detected from these mice by northern blotting, the transcript in the Brdm2 mouse model, which truncates after exon 10 [25], could in theory give rise to shortened ΔNp63 transcripts, similar to those described by Mangiulli et al. [9]. A recent recharacterization of a line of Brdm2 mice by Wolff et al. [29] revealed patches of keratinizing epidermis expressing truncated p63 at levels similar to wild type with stratification overlaying hair follicles. Based on further studies in embryos, the authors proposed these patches to be remnants of a more developed E15 epidermis 3–5 layers thick containing terminally differentiated epithelium that was transient in nature due to mechanical stress at birth, and suggested that the Brdm2 mice were equivalent to p63α/β knockout mice [29]. This observation and ensuing studies generated much controversy, as to whether the recharacterized mice were the same as those used by others or if perhaps a spontaneous genetic event might be at play [29–33], which to date remains unresolved.
Studies of the p63-deficient mouse lines provided strong evidence for the critical nature of the p63 gene; however, interpretation of p63 function is confounded by the existence of multiple p63 protein isoforms. Therefore, single isoform knock-in mouse models have been developed on a p63 null background to elucidate the role of specific p63 isoforms. These models have also generated controversy. Reconstitution of different p63 isoforms in the Brdm2 mice using tissue-specific inducible mouse models generated by separate groups gave rise to opposing conclusions as to the role of ΔNp63 in initiating stratification of simple epithelium [34, 35]. In one model, TAp63α, but not ΔNp63α, was found to drive stratification and keratin 5/keratin 14 expression of the simple lung epithelium [34], while in the other model, ΔNp63α or ΔNp63β caused stratification and keratin 5/keratin 14 expression in the simple lung epithelium [35]. With respect to the epidermis, differential results were also obtained by these two groups. In the first model, keratin 14-driven expression of TAp63α resulted in a severely hyperplastic epidermis exhibiting delayed differentiation [34], and based on their data the authors concluded that TAp63α is the initiating switch for epidermal stratification. In contrast, in the second model, expression of ΔNp63α or ΔNp63β under the control of the keratin 5 promoter did not result in complete restoration of epithelial integrity, but it did result in several areas of stratified epidermis, which expressed differentiation markers, indicating that the ΔNp63α and ΔNp63β can act to initiate stratification [35]. Further support for ΔNp63α as an initiator of stratification comes from other genetic complementation studies in which ΔNp63α or TAp63α, both under the keratin 5 promoter, was introduced into the p63(−/−) mice from Yang et al. [3, 36]. In these studies, ΔNp63α was able to partially restore the epidermal basal layer, but not differentiation marker expression, whereas TAp63α reconstitution resulted in a phenotype similar to p63(−/−) mice [36]. Reconstitution of a combination of ΔNp63α and TAp63α resulted in a more complete epidermis formation containing patches with a more organized structure that expressed markers of differentiation [36]. It is possible that differences with respect to the differentiation status of the epidermis generated by reconstitution of ΔNp63α in the later two studies could be due to the mouse model used, but in contrast to the first model discussed, partial epidermal restoration by ΔNp63α is a common feature of both.
Finally, subclass-specific knockout mice have been developed as a means of exploring functions attributable to the TAp63 or ΔNp63 subclasses in the presence of wild-type expression levels of the opposing subclass. Germline ablation of TAp63 did not impact normal epidermal morphogenesis in the presence of ΔNp63 isoforms [37]. In contrast, mice in which ΔN exon was replaced with GFP appeared phenotypically similar to p63(−/−) mice, and, like the p63(−/−) mice generated by Yang et al. [3], retained only disorganized patches of keratinocytes expressing terminal markers of differentiation [38]. However, in contrast to the mice generated by Yang et al. [3], these mice coexpressed keratin 5 along with the markers of terminal differentiation. Furthermore, expression of the basal transcription factor AP-2α indicated that in the absence of ΔNp63, basal patches can form, but these were observed to have decreased Ki67 staining. Taken together, this is suggestive of a role for ΔNp63 during epidermal morphogenesis in both progenitor cell maintenance and in epidermal commitment, closing the gap between the original interpretations of the pan-p63 mouse models [38].
3.1.2. p63 and Human Genetic Syndromes
In humans, heterozygous mutations in p63 are linked to genetic syndromes that include ectodermal dysplasia as part of the disease phenotype [39]. Distinct phenotypes are associated with mutations in specific p63 domains, providing clues to structure/function relationships. For example, ankyloblepharon-ectodermal defects-cleft lip/palate (AEC) syndrome is associated with mutations in the SAM domain [40], while ectodermal dysplasia and cleft/lip palate (EEC) syndrome is associated with mutations within the DBD [41, 42]. Not all of the syndromes include skin involvement. Of the syndromes, skin involvement is most pronounced and severe in AEC, which is linked to missense mutations in the SAM domain [40, 43] and therefore implicates the α isoforms.
Unlike normal skin where ΔNp63 expression is associated with the basal proliferative compartment, in AEC patient skin samples, nuclear p63 expression extends beyond the basal layer to the terminally differentiating cells and is accompanied by coexpression of differentiation markers [40]. In vitro studies have shown that SAM domain mutations, as seen in AEC, block interaction between p63α and mRNA splicing/processing proteins critical to direct splicing of FGFR-2 to the isoform required for normal epithelial differentiation [44]. Characterization of the AEC L514F ΔNp63α mutant in stable cell lines revealed that activation of a cryptic splice site due to loss of these interactions resulted in production of a C-terminally truncated form of ΔNp63α exclusively located in the nucleus and resistant to proteosome degradation [45]. Mouse models have helped to further define the contribution of this genetic alteration to the phenotype of this syndrome. A knock-in mouse model developed for the L514F mutation recapitulates the expected AEC phenotype including hypoplastic and fragile skin with a transient reduction in proliferation during embryonic development [46]. Skin fragility in these mice, and in humans with AEC syndrome, was associated with altered desmosome gene expression mediated by mutant p63 [47]. To gain insight into further pathways impacted, intact and eroded AEC syndrome skin and normal skin were compared by microarray analysis. The findings revealed changes in expression of genes associated with epidermal adhesion, skin barrier formation, and hair follicle biology, all consistent with the clinical presentation [48]. Thus, SAM domain mutations highlight the importance of p63 and in particular ΔNp63α to normal epidermal morphogenesis/homeostasis.
3.2. p63 in Normal Epidermal Homeostasis
Maintenance of normal epidermal homeostasis involves mediation of processes including proliferation, differentiation, stem cell maintenance, senescence, viability, and cell adhesion. Evidence suggests that each of these is impacted by p63 protein expression (Figure 2). In vivo, in the adult human epidermis, p63 is highly expressed in the basal cells with proliferative potential [26] and is downregulated in the suprabasal layers [4]. In vitro depletion of p63 in human regenerating organotypic cultures resulted in hypoproliferation and a lack of stratification and differentiation [49]. These effects were found to be mainly due to the ΔNp63α isoform. In a mouse model, specific knockdown of ΔNp63α in the mature epidermis resulted in severe skin fragility with erosion [27]. A multitude of studies, primarily in vitro, focused on the downstream targets mediated by ΔNp63α have shed light on the network of target genes implicated in these ΔNp63α-mediated biological processes. While an extensive cataloguing of all of these studies is beyond the scope of this review, some of these studies, with a focus on those performed in keratinocytes, are discussed here to highlight the potential impact of dysregulated ΔNp63α on signaling pathways that may be assessed using mouse models of the skin.
Figure 2.
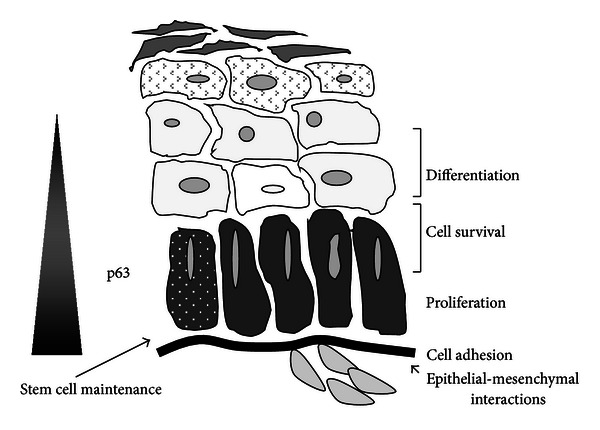
p63 impacts multiple biological endpoints involved in normal epidermal homeostasis. Overexpression of ΔNp63α impacts pathways that can contribute to cancer development.
3.2.1. Cell Cycle Regulation
Numerous examples serve to illustrate how perturbation of ΔNp63 expression could result in altered biological outcome. In vitro, in developmentally mature murine keratinocytes, we and others demonstrated that ΔNp63α is associated with maintenance of proliferative capacity [49–53]. Mimicking overexpression of ΔNp63α seen in squamous cell carcinomas blocks the normal growth arrest and induction of the cyclin-dependent kinase inhibitor p21WAF1 in response to elevated Ca2+ conditions [50, 51] and correspondingly suppresses the differentiation markers keratin 10 and filaggrin, but not keratin 1. The α-tail of ΔNp63 is required for its suppressive effect on differentiation but not for the aberrant growth arrest response [51], which may be mediated at least in part by transcriptional repression of p21WAF1 by binding of ΔNp63α to its promoter [52]. Regulation of p21WAF1 is further impacted by crosstalk between p63 and Notch 1, whereby Notch 1 is negatively regulated by ΔNp63α in cells of high renewal potential but synergizes with ΔNp63α during early differentiation to induce keratin 1. Subsequently, Notch 1 downregulates ΔNp63α to permit the expression of the late differentiation marker involucrin [54]. This context-dependent crosstalk is implicated in maintaining the balance between keratinocyte growth arrest and differentiation.
In addition to the factors regulating p21WAF1 described above, a balance exists between ΔNp63α and many other target genes involved in proliferation and differentiation that are critical for maintenance of or for the switch between the states. For example, the cell cycle inhibitor PTEN is negatively regulated by ΔNp63α. Depletion of either ΔNp63α or PTEN alone had opposite effects on colony growth in colony forming assays, but depletion of both ΔNp63α and PTEN at the same time had no impact, implying the balance between the two is critical to biological outcome [55]. Another example focuses on the epidermis of mice with mutant IRF6, which is hyperproliferative and fails to undergo differentiation [56]. This has been attributed to a failure of a feedback loop with ΔNp63α that controls ΔNp63α expression, thereby regulating the switch between proliferation and differentiation. In this feedback loop, IRF6 is a direct transcriptional target of ΔNp63α, which when upregulated induced proteasome-mediated degradation of ΔNp63α allowing for keratinocytes to exit the cell cycle [57]. In addition to regulation of levels of ΔNp63α impacting biological outcome as exemplified by the previous two examples, Runx1, a transcription factor involved both in keratinocyte proliferation and differentiation, is directly differentially regulated by ΔNp63α in proliferating versus differentiating keratinocytes by binding to distinct DNA binding sites on the Runx1 promoter. This represents a different mechanism of regulation [58], however, one that also could be perturbed by altered expression of ΔNp63α.
3.2.2. Differentiation
The mouse models described in Section 3.1.1 support a role for ΔNp63α not only in the maintenance of epidermal progenitor cells, but also, in the commitment to stratification. At a molecular level, ΔNp63α synergizes with Notch 1 to induce keratin 1 expression during differentiation, and the ΔNp63α target gene IKKα is necessary for epidermal differentiation [54, 59–63]. However, overexpression of ΔNp63α in primary murine keratinocyte cultures blocks expression of keratin 10 and filaggrin, but not keratin 1 [50] implying that a fine balance in levels of ΔNp63α is required for complete differentiation. Some examples of transcription factors which based on in vitro studies are thought to interplay with ΔNp63α during differentiation follow.
Basally expressed keratin 14 is a known direct transcriptional target of ΔNp63α [64]. The transcription factor Skn1a (Oct11) blocks ΔNp63α induction of the keratin 14 promoter and promotes keratin 10 upregulation [65]. There is also evidence for ΔNp63α both blocking and inducing transcription factors that promote differentiation in a cell-context-dependent manner. For instance, ΔNp63α directly represses high-mobility group box protein 1 (HBP1), a transcription factor necessary for stratification of organotypic cultures [66]. In contrast, in differentiating keratinocytes ΔNp63α induces ZNF750, a transcription factor that is required for terminal epidermal differentiation [67]. Interestingly, ZNF750 is bound by ΔNp63α in both proliferating and differentiating keratinocytes, but expression is only induced in differentiating cells, suggesting that additional cofactors are involved for distinct biological endpoints. The contribution of cofactors is further exemplified by the case of Alox12, a granular layer protein important for epidermal barrier formation, which is induced by ΔNp63α only in differentiating keratinocytes [68].
ΔNp63α expression can be regulated at the transcriptional level as well as by altered protein stability, as noted above. Another means of controlling levels of ΔNp63α is by microRNAs, short RNA molecules that act as posttranscriptional regulators. They recognize seed sequences in the 3′UTR and serve to block protein translation or decrease RNA stability. Such an interaction was identified as part of a feedback loop between p63 and iASPP, an inhibitory member of the apoptosis stimulating protein of p53 family, critical for epidermal homeostasis [69]. In this loop, iASPP is a direct transcriptional target of p63 that positively regulates ΔNp63 via the repression of miRs 754-3p and 720 to allow for proliferation. Blocking iASPP expression allows for differentiation via upregulation of miRs 754-3p and 720, which downregulate ΔNp63α. Other examples include miR203, which directly targets p63 through its 3′UTR for degradation and promotes differentiation by restricting proliferative potential and promoting cell cycle exit [70]. miRs are also regulated by ΔNp63α. miR-34a and miR-34c, associated with cell cycle withdrawal, are negatively regulated by ΔNp63α [71]. In contrast, miR17, miR20b, miR30a, miR106a, miR143, and miR 455-3p are positively regulated by p63 and critical for the onset of keratinocyte differentiation via modulation of the MAPKs [72].
3.2.3. Epidermal-Dermal Interface and Adhesiveness and Viability
Adhesiveness and cell viability are two additional properties positively impacted by ΔNp63α. Epidermal-specific knockdown of ΔNp63 in mature keratinocytes in mice resulted in impaired differentiation and compromised basement membranes [63]. In an in vitro model, Fras1, which encodes for an extracellular matrix protein, was identified as a ΔNp63α regulated gene important for maintaining the epidermal-dermal interface integrity [63]. To maintain this interface, p63 prevents nonepidermal gene expression in keratinocytes via positive regulation of bone morphogenetic protein- (BMP-) 7 [73]. The importance of ΔNp63 in maintaining epithelial-mesenchymal crosstalk was highlighted by the discovery of interleukin-1 α (Il-1α) as a p63 target gene. IL-1α induces growth factors in fibroblasts that can bind to receptors on the basal keratinocytes to promote proliferation [74]. Cell-cell adhesiveness was found to be mediated by p63 via Perp, which is a critical desmosomal component for cell-cell adhesion in normal development and in wound healing [75, 76]. With respect to apoptosis, the proapoptotic protein, IGFBP3, is directly repressed by ΔNp63 in both normal and SCC cells [77]. Similarly, downregulation of p63 in primary human foreskin keratinocytes was found to induce apoptosis and to reduce both β1 and β4 integrin expression [78], linking adhesiveness with viability.
Taken together, the data presented in Sections 3.2.1–3.2.3 demonstrate that the network of genes regulated by ΔNp63α is large and perturbation of the balance between ΔNp63α and members of this network could have adverse biological consequences.
3.3. Stem Cell Maintenance and Senescence
The proliferative lifespan of cells is limited by replicative senescence during which the cells permanently withdraw from the cell cycle, yet remain viable [79, 80]. This phenomenon is associated with the normal ageing process of renewable tissues such as the epidermis. p63 has been proposed as a marker of human epidermal keratinocyte stem cells that is downregulated when keratinocytes become transient amplifying cells [81]. Consistent with these data and the hypothesis that epidermal progenitor cell exhaustion occurs in p63(−/−) mice, depletion of p63 in immature human epidermal keratinocytes resulted in reduced clonal growth [82]. Regulation of replicative senescence in human epidermal keratinocytes involves miRs-138, 181a, 181b, and 130b which promote senescence by targeting ΔNp63α and Sirt1 for degradation. However, in a feedback loop, these miRs are themselves targets of negative regulation by ΔNp63α [83]; thus, overexpression of ΔNp63α could also perturb senescence.
Mouse models provide support for a role for p63 in the maintenance of stem cell proliferative capacity. Characteristics of accelerating ageing were noted in p63(+/−) mice observed for extended periods generated by two groups using the mice developed by both Yang et al. and Mills et al. [3, 25, 84, 85]. Germline or somatic p63 depletion under control of the keratin 5 promoter gave rise to enhanced senescence marker expression [84], suggesting a role for p63 in the negative regulation of senescence. Indeed, overexpression of ΔNp63α in primary mouse keratinocytes overcame replicative senescence in association with delayed and diminished induction of INK4/p16 and Arf/p19 [86]. Consistent with these findings, crossing of p63(−/−) mice developed by Yang et al. [3] with INK4/p16(−/−) or Arf/p19(−/−) mice was able to partially rescue the proliferation and differentiation defects observed in p63(−/−) mice [87], reinforcing a role for p63 in blocking senescence. While these mice display reepithelialization, skin from p63(−/−) mice crossed with Ink4a/p16(−/−) or Arf/p19(−/−) mice is fragile and easily detachable, suggestive of defective adhesion, which also can be attributed to p63.
In the mouse models described above, all p63 isoforms were knocked down. A TAp63-specific knockdown mouse model that supports a role for TAp63 in adult stem cell maintenance was generated by crossing TAp63 floxed mice with germline-specific promoter cre or keratin 14-cre mice [37]. TAp63(−/−) mice exhibited signs of premature ageing. Interestingly, overexpression of ΔNp63α under control of the keratin 14 promoter resulted in a phenotype similar to that reported in mice lacking TAp63 [88]. Skin-derived precursor (SKP) cells are multipotent precursor cells derived from the dermis that can differentiate into mesodermal and neural cells [89]. In the TAp63(−/−) mice, SKP cells proliferate more rapidly than wild-type SKP cells, and thus, undergo senescence more rapidly. As adult stem cell populations are not immortal, this enhanced proliferation in TAp63(−/−) cells would be expected to lead to stem cell exhaustion, which is associated with accelerated ageing.
4. p63 and Neoplasia
4.1. Observational Studies of Human Tumors
The p53 tumor suppressor gene is commonly mutated in human cancer [1]. Due to the similarity of the TAp63 isoforms with p53, it was hypothesized that mutation of p63 could provide a mechanistic explanation for tumors in which p53 was not mutated. It was found, instead, that mutation of p63 is a rare event in human cancer cell lines [90], but that p63 overexpression is seen in human squamous cell cancers including esophageal squamous cell carcinoma [91, 92], nasopharyngeal carcinoma [93], and squamous cell carcinoma of the skin [94, 95]. Overexpression of the ΔNp63 protein in primary squamous cell carcinomas (SCCs) of the head, neck, and lung correlates with amplification of the p63 gene locus, which occurs frequently in these cancers [28, 96]. While there is agreement that ΔNp63α is overexpressed in lung SCCs, conflicting results have been published as to whether this correlates with prognosis [96, 97].
In squamous cell carcinomas of the skin, a significant increase in p63 expression, both in terms of intensity and distribution, is seen relative to normal skin, as the proliferative fraction is expanded in tumors [26, 95]. Examination of skin lesions ranging from keratoacanthoma to a grade IV spindle cell carcinoma revealed very strong p63 immunoreactivity in grade 3 SCC with decrease in a single grade IV spindle SCC. In these tumors, carcinoma in situ was characterized by p63 immunoreactivity in all layers [94]. While ΔNp63α was shown to be the most overexpressed isoform in squamous cell tumors, careful characterization of the TA and ΔN isoforms from different tissue and tumor types revealed that individual isoforms are differentially expressed in the neoplastic transformation of different tissue types [98], implying specific contributions of the isoform expressed in a context-dependent manner. While ΔNp63α is overexpressed in primary skin tumors, expression of TAp63 is not a common event but has been reported to be downregulated relative to normal skin using PCR-based methods [99]. It is clear that ΔNp63α is overexpressed in skin SCCs, however, whether it actively plays a role in tumor formation or is a bystander has been unclear. Further insight into this question has been gained by in vitro and in vivo studies, as discussed below.
4.2. In Vitro/Molecular Studies with Human Cancer Cell Lines and Primary Keratinocytes
4.2.1. Impact on Signaling Pathways
In particular, in vitro studies in SCC cells have provided insight into the potential signaling pathways impacted by p63 dysregulation in squamous cell carcinoma. As discussed previously, IRF6 is involved in a negative feedback loop with ΔNp63α that is necessary for the downregulation of ΔNp63α seen with differentiation [57], and an appropriate balance between these factors is required for the switch between proliferation and differentiation in the normal epidermis [100]. Correspondingly, expression of IRF6 was found to be strongly downregulated in human SCC [101]. Reexpression of IRF-6 in the context of primary human keratinocytes expressing both ΔNp63α and a mutant v-ras 12 oncogene was found to abolish the ability of ΔNp63α to promote colony growth and restore oncogene induced senescence [101], supporting a role for IRF6 in regulating ΔNp63α as part of its tumor suppressor function. In other studies, ΔNp63α has been shown to upregulate Hsp70, a protein colocalized with ΔNp63 in primary SCCs of the head and neck (HNSCCs) that is associated with proliferation and viability of HNSCC [102]. Likewise, accumulation of β-catenin in the nucleus and activation of downstream signaling pathways common to many cancers are induced by ΔNp63α in HNSCC cells [103].
Consistent with a role in promoting adhesion, ΔNp63 is negatively regulated by the epithelial-to-mesenchymal transition (EMT) promoting transcription factors snail and slug, and this association is observed in primary human cervical, head and neck, and esophageal SCCs. This decrease in ΔNp63α is associated with increased migration in SCC cell lines [104]. ΔNp63α also physically sequesters YB-1, a positive translational mediator of snail, thereby preventing both enhanced snail activity and YB1's function in actin cytoskeleton reorganization, both of which lead to cancer cell migration and invasion [105]. Another direct transcriptional target of ΔNp63α is the vitamin D receptor (VDR) [106], which is induced by multiple p63 isoforms. Downregulation of VDR expression results in increased cell migration of A431 epidermoid carcinoma cells, which can be rescued by ΔNp63α or VDR [107]. A role for ΔNp63α in preventing metastasis is further supported by the finding that antagonism of ΔNp63α by mutant-p53/Smad complex allows TGF-β to convert from a tumor suppressor role to a role in promoting metastases [108]. In line with this, knockdown of p63 in squamous cancer cell lines, in which the predominant isoform expressed was ΔNp63, led to an increase of mesenchymal and neural markers and upregulation of genes associated with invasion and motility [109].
Based on the data, it is enticing to contemplate that ΔNp63α plays a role in cancer development by promoting proliferation and viability at earlier stages, while it may need to be downregulated during progression to allow for the necessary enhanced motility, invasiveness, and EMT [110] that allow metastases to form.
4.2.2. Altered Responsiveness to Genotoxic Stress
ΔNp63α can impact cellular response to genotoxic stress. A mouse model in which ΔNp63 was overexpressed under control of the loricrin promoter showed that downregulation of ΔNp63 is required for UVB-induced apoptosis of the epidermis [111]. Mechanistically, degradation of ΔNp63α in keratinocytes exposed to apoptotic doses of UV was shown to be mediated by p38 MAPK, which phosphorylates ΔNp63α. This led to its detachment from p53-dependent promoters and results in apoptosis induction [112]. Consistent with this report, occupancy of binding sites involved in cell cycle arrest and apoptosis switched following adriamycin or UV treatment of human epidermal keratinocytes from ΔNp63α to p53 occupancy, which would be expected to result in increased apoptosis or cell cycle arrest [113].
Many therapeutic agents used in cancer treatment promote genotoxic stress as a means to reduce or control tumor growth. Expression of high levels of ΔNp63α predicts responsiveness of primary HNSCC to platinum-based therapies [114]. Upon exposure to cisplatin, ΔNp63α is proteosomally degraded via stratifin-mediated nuclear export and Rack1 targeting [114, 115]. An interaction between the p63 proteins and the NF-κB pathway also plays a role in responsiveness to chemotherapeutics. In JHU-022 oral cavity SCC cells, IKKβ, a known activator of RelA, promotes ΔNp63α degradation in response to cisplatin [116]. In this cell line, cisplatin treatment resulted in a physical interaction between RelA and ΔNp63α that abrogates ΔNp63α mediated p21WAF1 promoter repression and targets ΔNp63α for proteosomal degradation [117]. The presence of c-Abl, which has been implicated as an oncogene, in HNSCC cells treated with cisplatin stabilizes ΔNp63α expression. This stabilization of ΔNp63α leads to enhanced cell viability [118], which could be anticipated to result in clinical consequences.
Survival of HNSCC cells that overexpress ΔNp63α is dependent on the presence of ΔNp63α, which functions by blocking TAp73-driven apoptosis both via promoter binding and physical interaction with p73 in a p53-independent manner [119]. TAp73 and ΔNp63α are engaged in a feedback loop involving miR-193a-5p, which is repressed by ΔNp63α and activated by TAp73 and targets the p73 UTR. Cisplatin treatment results in ΔNp63α degradation and TAp73-mediated activation of miR-193a-5p, limiting TAp73's pro-apoptotic effects and chemosensitivity [23]. Reimplantation in the presence or absence of a miR-193a-5p antagomir of disaggregated cells from primary mouse SCCs generated by a chemical carcinogenesis protocol revealed that knockdown of this miR resulted in reduced tumor formation and enhanced chemosensitivity [23], indicating that a strategy targeting both ΔNp63α and miR-193a-5p might be more effective in this scenario. HNSCC cells can circumvent the requirement for ΔNp63α expression for survival by the overexpression of Bcl2 [119]. In addition to blocking p73 to promote survival of HNSCC, ΔNp63α associates with histone deacetylase 1 and 2 forming an apoptotic transcriptional repressor complex. This complex is sensitive to breakdown by cisplatin and HDAC inhibitors, in the presence of low, but not high, levels of endogenous Bcl-2 indicating once again that the context of the tumor impacts the success of chemotherapy [120]. Unlike the case of TAp73 described above, in HaCaT cells in response to chemotherapy, ΔNp63α is involved in an antiapoptotic feedback loop in which it, as well as mutant p53, induces ΔNp63α [24]. Thus, response to genotoxic stress is another biological endpoint that can be impacted by dysregulated ΔNp63α.
4.3. Modeling Human Cancers in Mouse to Assess the Contribution of p63 to Neoplasia
Cancer arises as a multistep process that can be reiterated in well-established mouse models in a controlled fashion [121]. Results of the studies presented above suggest that TAp63 would harbor tumor suppressor properties and overexpressed ΔNp63 would harbor oncogenic properties. In this section, we highlight the use of mouse models to dissect out how altered p63 levels contribute biologically to prevention or development of cancer, either alone or in altered balance with other family members or other oncogenic pathways. Approaches discussed utilize mice with a heterozygous null mutation in p63 on a background of wild type, p53 (+/−) and/or p73 (+/−); TA-isoform-specific knockout mice; and mouse models where the elevated levels of ΔNp63α observed in human SCC are mimicked in cultured keratinocytes and transplanted to nude mice.
4.3.1. TAp63 as a Tumor Suppressor
The potential role for physiological levels of p63 acting as a tumor suppressor with respect to spontaneous tumor development was explored by two groups in the context of alteration of other p53 family members [85, 122]. In a mouse model in which the p63 genotype was contributed by mice developed by Yang et al. [3], mice heterozygous for a null mutation in both p63 and p73 displayed a higher incidence of spontaneous tumor formation relative to wild-type mice. Furthermore, mice heterozygous for p53, p63, and p73 developed a higher incidence and formed more aggressive tumors than mice heterozygous for the p53 null mutation alone. These findings suggest that p63 and p73 share a tumor suppressor role as has been long established for p53 [1, 85]. In the absence of additional genetic mutations, these p63(+/−) mice developed squamous cell carcinomas (10%), adenomas (15%), and histiocytic sarcomas (20%) at 10%, 15%, and 20% greater rates, respectively, than wild type. In contrast, in a study using mice with a p63 genotype contributed by the mice developed by Mills et al. [25], p63(+/−)/p53(+/−) mice were found to be less prone to spontaneous tumors than p53 +/− mice alone. Additionally, these p63(+/−) mice were shown to have decreased susceptibility to chemically induced carcinogenesis, suggesting that p63 does not contribute a tumor suppressor activity in cancer. To date, this controversy remains unresolved.
In the mouse models described above, all p63 isoforms were targeted. TAp63-subclass-specific knockdown mice allow distinction between the TA and ΔN subclass properties. Following observation for 2.5 years, an enhanced incidence of carcinoma, including SCC of the skin and sarcoma development, was observed in TAp63(+/−) and TAp63(−/−) mice relative to wild-type mice [123], again supporting a tumor suppressive role for TAp63. It was noted that tumors from the TAp63(+/−) and TAp63(−/−) mice were highly metastatic, and at a mechanistic level TAp63 was found to positively regulate Dicer, a protein critical for miR processing, and miR 130b. Reexpression of both Dicer and miR130b in TAp63(−/−) MEFs decreased invasiveness of these cells, suggesting that TAp63's tumor suppressor role could be mediated at least in part through Dicer and miR130b [123]. As mentioned previously, miR130b targets ΔNp63α for degradation [83].
4.3.2. Overexpressed ΔNp63α Facilitates Tumor Progression
The mouse models described above focused on the TAp63 isoforms and were performed at wild-type or decreased levels of endogenous p63. However, overexpression of ΔNp63α is a common event in squamous cancers. Two independent studies have used similar approaches to mimic this overexpression with the goal of examining the in vivo functional consequences of ΔNp63α overexpression in the epidermis. Results from both lab groups support a contributory role for ΔNp63α in the cancer phenotype with mechanistic distinctions. In studies performed in our laboratory, wildtype primary murine keratinocytes were transduced with retrovirus encoding a v-rasHA oncogene in combination with a lentivirus encoding either a control GFP construct or ΔNp63α and grafted onto the dorsum of nude mice in combination with primary dermal fibroblasts [86]. This model allows growth of normal keratinocytes as well as benign and malignant tumor phenotypes in the graft site. Mice were observed up to a month following cell grafting for tumor formation. No lesions were observed in graft sites following transplantation of keratinocytes expressing only GFP or ΔNp63α alone. Grafting of keratinocytes expressing v-RasHA + GFP resulted, as expected, in the formation of well differentiated papillomas, while grafting of keratinocytes expressing v-RasHA + ΔNp63α resulted in 100% malignant conversion to carcinoma [86]. Although elevated levels of ΔNp63α alone are insufficient to confer a tumor phenotype in vivo, we found that ΔNp63α blocks oncogene-induced senescence by inhibiting p16ink4a/p19arf pathways and cooperates with oncogenic v-RasHA to enhance malignant conversion in vivo. This study supports a contributory role for ΔNp63α in cancer pathogenesis and a mechanistic link to cell survival by overriding oncogene-induced senescence through inhibition of p16ink4a and p19arf, key mediators of cellular senescence.
Using a similar approach, Keyes et al. [124] demonstrated that overexpressing ΔNp63α in keratinocytes in the presence of oncogenic ras resulted in growth of malignant carcinomas following subcutaneous injection. In this study also, the malignant phenotype was associated with a bypass of oncogene-induced senescence. Overexpression of ΔNp63α was further shown to enhance stem-like proliferation of keratinocytes and maintain survival of the keratin 15-positive stem cell population. Furthermore, chromatin-remodeling protein Lsh was identified as a new target of ΔNp63α and as an essential mediator of senescence bypass. Although p19arf was not detectable in the tumors derived from ras/ΔNp63α keratinocytes in this study, an in vitro component of the study indicated that p16ink4a and p19arf were not reduced during the initial stages of senescence bypass. Therefore, contrary to our study, it was proposed that the initiating events through which ΔNp63α inhibits senescence do not occur via p16ink4a/p19arf pathways. Although the difference in p16ink4a/p19arf between these two studies may be due to the different time courses used, it further indicates the complexity of the pathways interacting with p63 family members and underscores the need for additional studies to understand the role of p63 and its downstream effectors in tumorigenesis and senescence.
An oncogenic role for ΔNp63 is further supported by studies in a mouse model containing a dominant negative 14-3-3σ mutation (Er/+). 14-3-3σ, a protein associated with keratinocyte differentiation, is a direct target for ΔNp63α repression in undifferentiated human epidermal keratinocytes [52]. Treatment of Er/+ mice on a p63(+/+) background with a two-stage carcinogenesis protocol resulted in the formation of tumors in which ΔNp63α was strongly expressed, while loss of function of an endogenous allele of p63 in this context, which generated (Er/+/p63 +/−) mice, resulted in reduced sensitivity to this protocol, suggestive of cooperation of ΔNp63α in Ras/14-3-3σ-induced tumorigenesis [125].
A Role for NF-κB/c-Rel in ΔNp63α-Mediated Carcinogenesis. In a transcription factor profiling exercise, we identified activation of NF-κB in keratinocytes following the overexpression of ΔNp63α. The NF-κB family comprises 5 members functioning as hetero- and homodimers [126]. Only NF-κB/c-Rel was found to be modulated by ΔNp63α under these conditions, with nuclear accumulation of phosphorylated c-Rel but none of the other NFκB subunits enhanced in the presence of overexpressed ΔNp63α. NF-κB is associated with multiple human diseases, including cancer, for which therapeutics targeting its constitutive NF-κB activation are under development [127, 128]. Of the five family members, RelA, p50, and c-Rel subunits have been implicated in the maintenance of normal epidermal homeostasis [129, 130], and in SCC, the RelA/p50 heterodimer has been shown to promote or repress malignancy in a context-dependent manner [131, 132]. Nuclear c-Rel expression is associated with both solid breast tumors and hematopoietic malignancies [133]; however, it had not previously been investigated in SCC of the skin. The increase in nuclear c-Rel accumulation seen with elevated ΔNp63α levels was found to be critical to the ability of ΔNp63α overexpressing keratinocytes to proliferate under conditions that normally induce growth arrest [134]. Mechanistically, this is correlated with a physical interaction between ΔNp63α and c-Rel on the promoter of the p21WAF1 gene in these cells, both in vitro and in vivo, which represses p21WAF1 expression. These findings extended to primary human HNSCC, in which we found that ΔNp63α and c-Rel colocalized in the nuclei throughout the tumor sections, as opposed to a more restricted expression in normal tissue.
In an extension of these studies, Lu et al. characterized a dynamic mechanism whereby c-Rel, ΔNp63α, and TAp73, which are coexpressed in the nuclei of a subset of HNSCC cell lines, control expression on binding sites including p21WAF1, Noxa, and Puma [135]. Exposure of HNSCC cell line cultures to TNF-α to mimic inflammation in the tumor environment was found to induce nuclear accumulation of c-Rel. In the absence of this stimulus, ΔNp63α was found to physically interact with TAp73. Similar to our results in untreated primary keratinocytes upon overexpression of ΔNp63α, a physical interaction between endogenous ΔNp63α and c-Rel was observed following TNF-α treatment in these HNSCC cell lines. Interestingly, under conditions of c-Rel overexpression, the interaction between ΔNp63α and TAp73 was blocked and TAp73 was translocated to the cytoplasm. This suggests that c-Rel displaces TAp73. ChIP assays indicated that this regulation occurred on the promoters of genes involved in growth arrest and apoptosis, resulting in their downregulation [135].
5. Summary/Future Directions: Challenging In Vitro Findings in In Vivo Models
Primary murine cell cultures and in vivo murine models have been instrumental in elucidating the multistep nature of carcinogenesis [121] and in challenging the role of specific genetic alterations, such as those observed in p63, in cancer pathogenesis [86, 124]. The mouse models described in this review have provided us with a clear picture of the importance of p63 to normal epidermal development and homeostasis and have highlighted the roles for specific p63 isoforms in neoplasia. However, given the complexity of the p63 family members, their interactions, and the context-specific manner in which they can exert their effects, much remains to be defined.
Mouse models with molecular alterations that allow targeting of specific gene products will be indispensible in deepening our understanding of and resolving controversy related to the role of p63 both in normal tissue and in disease. Once a pathway has been implicated, primary cultures from mouse epidermis can be readily manipulated to express or eliminate a particular protein presumed active upstream or downstream to assess the impact. Applied in combination with keratinocytes from genetically altered mice, both in vitro and in vivo findings can be challenged further for the consequences of the alterations. However, it is important to remember that all models have limitations, and a deeper understanding of the role of p63 in normal epidermal homeostasis and neoplasia will ultimately be derived from an iterative process involving in vitro observations in primary cells, cell lines and primary tumors, in vivo queries of these findings, and reexamination of the outcomes in the context of human tumors. Mouse models comprise a critical component of this process.
As an example, the observation in primary mouse keratinocytes that c-Rel acts downstream of ΔNp63α in modulating keratinocyte growth regulation led to a further novel observation that c-Rel levels are enhanced in primary HNSCC of humans and links this protein accumulation to altered NFκB/c-Rel activity in human head and neck squamous cell cancer cells [134]. The requirement for c-Rel in these cancers can be tested by modulating c-Rel and ΔNp63α independently using lentiviral gene transduction followed by grafting. Long-term overexpression of ΔNp63α has been shown to support sustained high levels of nuclear c-Rel expression, and c-Rel shRNA lentiviruses are capable of depleting c-Rel in keratinocytes for extended time (unpublished observations). Assessing the impact of these modulations in vivo will clarify the interplay between these alterations and their relevance to cancer development and progression.
References
- 1.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408(6810):307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 2.Yang A, Walker N, Bronson R, et al. p73-Deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature. 2000;404(6773):99–103. doi: 10.1038/35003607. [DOI] [PubMed] [Google Scholar]
- 3.Yang A, Schweitzer R, Sun D, et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature. 1999;398(6729):714–718. doi: 10.1038/19539. [DOI] [PubMed] [Google Scholar]
- 4.Yang A, Kaghad M, Wang Y, et al. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Molecular Cell. 1998;2(3):305–316. doi: 10.1016/s1097-2765(00)80275-0. [DOI] [PubMed] [Google Scholar]
- 5.Kaghad M, Bonnet H, Yang A, et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell. 1997;90(4):809–819. doi: 10.1016/s0092-8674(00)80540-1. [DOI] [PubMed] [Google Scholar]
- 6.Dohn M, Zhang S, Chen X. p63α and ΔNp63α can induce cell cycle arrest and apoptosis and differentially regulate p53 target genes. Oncogene. 2001;20(25):3193–3205. doi: 10.1038/sj.onc.1204427. [DOI] [PubMed] [Google Scholar]
- 7.Helton ES, Zhu J, Chen X. The unique NH2-terminally deleted (ΔN) residues, the PXXP motif, and the PPXY motif are required for the transcriptional activity of the ΔN variant of p63. Journal of Biological Chemistry. 2006;281(5):2533–2542. doi: 10.1074/jbc.M507964200. [DOI] [PubMed] [Google Scholar]
- 8.Ghioni P, Bolognese F, Duijf PHG, Van Bokhoven H, Mantovani R, Guerrini L. Complex transcriptional effects of p63 isoforms: identification of novel activation and repression domains. Molecular and Cellular Biology. 2002;22(24):8659–8668. doi: 10.1128/MCB.22.24.8659-8668.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mangiulli M, Valletti A, Caratozzolo MF, et al. Identification and functional characterization of two new transcriptional variants of the human p63 gene. Nucleic Acids Research. 2009;37(18):6092–6104. doi: 10.1093/nar/gkp674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thanos CD, Bowie JU. p53 Family members p63 and p73 are SAM domain-containing proteins. Protein Science. 1999;8(8):1708–1710. doi: 10.1110/ps.8.8.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Serber Z, Lai HC, Yang A, et al. A C-terminal inhibitory domain controls the activity of p63 by an intramolecular mechanism. Molecular and Cellular Biology. 2002;22(24):8601–8611. doi: 10.1128/MCB.22.24.8601-8611.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Straub WE, Weber TA, Schäfer B, et al. The C-terminus of p63 contains multiple regulatory elements with different functions. Cell Death and Disease. 2010;1(1, article e5) doi: 10.1038/cddis.2009.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghioni P, D’Alessandra Y, Mansueto G, et al. The protein stability and transcriptional activity of p63α are regulated by SUMO-1 conjugation. Cell Cycle. 2005;4(1):183–190. doi: 10.4161/cc.4.1.1359. [DOI] [PubMed] [Google Scholar]
- 14.Rossi M, Aqeilan RI, Neale M, et al. The E3 ubiquitin ligase Itch controls the protein stability of p63. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(34):12753–12758. doi: 10.1073/pnas.0603449103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galli F, Rossi M, D’Alessandra Y, et al. MDM2 and Fbw7 cooperate to induce p63 protein degradation following DNA damage and cell differentiation. Journal of Cell Science. 2010;123(14):2423–2433. doi: 10.1242/jcs.061010. [DOI] [PubMed] [Google Scholar]
- 16.Coutandin D, Löhr F, Niesen FH, et al. Conformational stability and activity of p73 require a second helix in the tetramerization domain. Cell Death and Differentiation. 2009;16(12):1582–1589. doi: 10.1038/cdd.2009.139. [DOI] [PubMed] [Google Scholar]
- 17.Ratovitski EA, Patturajan M, Hibi K, Trink B, Yamaguchi K, Sidransky D. p53 associates with and targets ΔNp63 into a protein degradation pathway. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(4):1817–1822. doi: 10.1073/pnas.98.4.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strano S, Fontemaggi G, Costanzo A, et al. Physical interaction with human tumor-derived p53 mutants inhibits p63 activities. Journal of Biological Chemistry. 2002;277(21):18817–18826. doi: 10.1074/jbc.M201405200. [DOI] [PubMed] [Google Scholar]
- 19.Osada M, Park HL, Nagakawa Y, et al. Differential recognition of response elements determines target gene specificity for p53 and p63. Molecular and Cellular Biology. 2005;25(14):6077–6089. doi: 10.1128/MCB.25.14.6077-6089.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perez CA, Ott J, Mays DJ, Pietenpol JA. p63 consensus DNA-binding site: identification, analysis and application into a p63MH algorithm. Oncogene. 2007;26(52):7363–7370. doi: 10.1038/sj.onc.1210561. [DOI] [PubMed] [Google Scholar]
- 21.Ortt K, Sinha S. Derivation of the consensus DNA-binding sequence for p63 reveals unique requirements that are distinct from p53. FEBS Letters. 2006;580(18):4544–4550. doi: 10.1016/j.febslet.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 22.Yang A, Zhu Z, Kettenbach A, et al. Genome-wide mapping indicates that p73 and p63 Co-occupy target sites and have similar DNA-binding profiles in vivo . PLoS ONE. 2010;5(7) doi: 10.1371/journal.pone.0011572.e11572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ory B, Ramsey MR, Wilson C, et al. A microRNA-dependent program controls p53-independent survival and chemosensitivity in human and murine squamous cell carcinoma. Journal of Clinical Investigation. 2011;121(2):809–820. doi: 10.1172/JCI43897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lanza M, Marinari B, Papoutsaki M, et al. Cross-talks in the p53 family: ΔNp63 is an anti-apoptotic target for ΔNp73α and p53 gain-of-function mutants. Cell Cycle. 2006;5(17):1996–2004. doi: 10.4161/cc.5.17.3188. [DOI] [PubMed] [Google Scholar]
- 25.Mills AA, Zheng B, Wang XJ, Vogel H, Roop DR, Bradley A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature. 1999;398(6729):708–713. doi: 10.1038/19531. [DOI] [PubMed] [Google Scholar]
- 26.Parsa R, Yang A, McKeon F, Green H. Association of p63 with proliferative potential in normal and neoplastic human keratinocytes. Journal of Investigative Dermatology. 1999;113(6):1099–1105. doi: 10.1046/j.1523-1747.1999.00780.x. [DOI] [PubMed] [Google Scholar]
- 27.Koster MI, Marinari B, Payne AS, Kantaputra PN, Costanzo A, Roop DR. ΔNp63 knockdown mice: a mouse model for AEC syndrome. American Journal of Medical Genetics A. 2009;149(9):1942–1947. doi: 10.1002/ajmg.a.32794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hibi K, Trink B, Patturajan M, et al. AIS is an oncogene amplified in squamous cell carcinoma. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(10):5462–5467. doi: 10.1073/pnas.97.10.5462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolff S, Talos F, Palacios G, Beyer U, Dobbelstein M, Moll UM. The α/β carboxy-terminal domains of p63 are required for skin and limb development. New insights from the Brdm2 mouse which is not a complete p63 knockout but expresses p63 γ-like proteins. Cell Death and Differentiation. 2009;16(8):1108–1117. doi: 10.1038/cdd.2009.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mikkola ML, Costanzo A, Thesleff I, Roop DR, Koster MI. Treasure or artifact: a decade of p63 research speaks for itself. Cell Death and Differentiation. 2010;17(1):180–183. doi: 10.1038/cdd.2009.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Talos F, Wolff S, Beyer U, Dobbelstein M, Moll UM. Brdm2-an aberrant hypomorphic p63 allele. Cell Death and Differentiation. 2010;17(1):184–186. doi: 10.1038/cdd.2009.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shalom-Feuerstein R, Lena AM, Zhou H, et al. Δnp63 is an ectodermal gatekeeper of epidermal morphogenesis. Cell Death and Differentiation. 2011;18(5):887–896. doi: 10.1038/cdd.2010.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aberdam D, Mantovani R. A new p63-deficient mouse model or a fresh look at an old one? Cell Death and Differentiation. 2009;16(8):1073–1074. doi: 10.1038/cdd.2009.66. [DOI] [PubMed] [Google Scholar]
- 34.Koster MI, Kim S, Mills AA, DeMayo FJ, Roop DR. p63 is the molecular switch for initiation of an epithelial stratification program. Genes and Development. 2004;18(2):126–131. doi: 10.1101/gad.1165104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Romano RA, Ortt K, Birkaya B, Smalley K, Sinha S. An active role of the ΔN isoform of p63 in regulating basal keratin genes K5 and K14 and directing epidermal cell fate. PLoS ONE. 2009;4(5) doi: 10.1371/journal.pone.0005623.e5623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Candi E, Rufini A, Terrinoni A, et al. Differential roles of p63 isoforms in epidermal development: selective genetic complementation in p63 null mice. Cell Death and Differentiation. 2006;13(6):1037–1047. doi: 10.1038/sj.cdd.4401926. [DOI] [PubMed] [Google Scholar]
- 37.Su X, Paris M, Gi YJ, et al. TAp63 prevents premature aging by promoting adult stem cell maintenance. Cell Stem Cell. 2009;5(1):64–75. doi: 10.1016/j.stem.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Romano RA, Smalley K, Magraw C, et al. DeltaNp63 knockout mice reveal its indispensable role as a master regulator of epithelial development and differentiation. Development. 2012;139:772–782. doi: 10.1242/dev.071191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rinne T, Brunner HG, Van Bokhoven H. p63-associated disorders. Cell Cycle. 2007;6(3):262–268. doi: 10.4161/cc.6.3.3796. [DOI] [PubMed] [Google Scholar]
- 40.McGrath JA, Duijf PHG, Doetsch V, et al. Hay-Wells syndrome is caused by heterozygous missense mutations in the SAM domain of p63. Human Molecular Genetics. 2001;10(3):221–229. doi: 10.1093/hmg/10.3.221. [DOI] [PubMed] [Google Scholar]
- 41.Celli J, Duijf P, Hamel BCJ, et al. Heterozygous germline mutations in the p53 homolog p63 are the cause of EEC syndrome. Cell. 1999;99(2):143–153. doi: 10.1016/s0092-8674(00)81646-3. [DOI] [PubMed] [Google Scholar]
- 42.Van Bokhoven H, Hamel BCJ, Bamshad M, et al. p63 gene mutations in EEC syndrome, limb-mammary syndrome, and isolated split hand-split foot malformation suggest a genotype-phenotype correlation. American Journal of Human Genetics. 2001;69(3):481–492. doi: 10.1086/323123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brunner HG, Hamel BCJ, Van Bokhoven H. The p63 gene in EEC and other syndromes. Journal of Medical Genetics. 2002;39(6):377–381. doi: 10.1136/jmg.39.6.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fomenkov A, Huang YP, Topaloglu O, et al. p63α mutations lead to aberrant splicing of keratinocyte growth factor receptor in the Hay-Wells syndrome. Journal of Biological Chemistry. 2003;278(26):23906–23914. doi: 10.1074/jbc.M300746200. [DOI] [PubMed] [Google Scholar]
- 45.Huang YP, Kim Y, Li Z, Fomenkov T, Fomenkov A, Ratovitski EA. AEC-associated p63 mutations lead to alternative splicing/protein stabilization of p63 and modulation of notch signaling. Cell Cycle. 2005;4(10):1440–1447. doi: 10.4161/cc.4.10.2086. [DOI] [PubMed] [Google Scholar]
- 46.Ferone G, Thomason HA, Antonini D, et al. Mutant p63 causes defective expansion of ectodermal progenitor cells and impaired FGF signalling in AEC syndrome. EMBO Molecular Medicine. 2012;4:192–205. doi: 10.1002/emmm.201100199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ferone G, Mollo MR, Thomason HA, et al. p63 control of desmosome gene expression and adhesion is compromised in AEC syndrome. Human Molecular Genetics. 2012;22:531–543. doi: 10.1093/hmg/dds464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clements SE, Techanukul T, Lai-Cheong JE, et al. Mutations in AEC syndrome skin reveal a role for p63 in basement membrane adhesion, skin barrier integrity and hair follicle biology. British Journal of Dermatology. 2012;167:134–144. doi: 10.1111/j.1365-2133.2012.10888.x. [DOI] [PubMed] [Google Scholar]
- 49.Truong AB, Kretz M, Ridky TW, Kimmel R, Khavari PA. p63 regulates proliferation and differentiation of developmentally mature keratinocytes. Genes and Development. 2006;20(22):3185–3197. doi: 10.1101/gad.1463206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.King KE, Ponnamperuma RM, Yamashita T, et al. ΔNp63α functions as both a positive and a negative transcriptional regulator and blocks in vitro differentiation of murine keratinocytes. Oncogene. 2003;22(23):3635–3644. doi: 10.1038/sj.onc.1206536. [DOI] [PubMed] [Google Scholar]
- 51.King KE, Ponnamperuma RM, Gerdes MJ, et al. Unique domain functions of p63 isotypes that differentially regulate distinct aspects of epidermal homeostasis. Carcinogenesis. 2006;27(1):53–63. doi: 10.1093/carcin/bgi200. [DOI] [PubMed] [Google Scholar]
- 52.Westfall MD, Mays DJ, Sniezek JC, Pietenpol JA. The ΔNp63α phosphoprotein binds the p21 and 14-3-3σ promoters in vivo and has transcriptional repressor activity that is reduced by Hay-Wells syndrome-derived mutations. Molecular and Cellular Biology. 2003;23(7):2264–2276. doi: 10.1128/MCB.23.7.2264-2276.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.DeYoung MP, Johannessen CM, Leong CO, Faquin W, Rocco JW, Ellisen LW. Tumor-specific p73 up-regulation mediates p63 dependence in squamous cell carcinoma. Cancer Research. 2006;66(19):9362–9368. doi: 10.1158/0008-5472.CAN-06-1619. [DOI] [PubMed] [Google Scholar]
- 54.Nguyen BC, Lefort K, Mandinova A, et al. Cross-regulation between Notch and p63 in keratinocyte commitment to differentiation. Genes and Development. 2006;20(8):1028–1042. doi: 10.1101/gad.1406006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leonard MK, Kommagani R, Payal V, Mayo LD, Shamma HN, Kadakia MP. DeltaNp63alpha regulates keratinocyte proliferation by controlling PTEN expression and localization. Cell Death & Differentiation. 2011;18:1924–1933. doi: 10.1038/cdd.2011.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Richardson RJ, Dixon J, Malhotra S, et al. Irf6 is a key determinant of the keratinocyte proliferation-differentiation switch. Nature Genetics. 2006;38(11):1329–1334. doi: 10.1038/ng1894. [DOI] [PubMed] [Google Scholar]
- 57.Moretti F, Marinari B, Iacono NL, et al. A regulatory feedback loop involving p63 and IRF6 links the pathogenesis of 2 genetically different human ectodermal dysplasias. Journal of Clinical Investigation. 2010;120(5):1570–1577. doi: 10.1172/JCI40267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Masse I, Barbollat-Boutrand L, Molina M, et al. Functional interplay between p63 and p53 controls RUNX1 function in the transition from proliferation to differentiation in human keratinocytes. Cell Death & Disease. 2012;3, article e318 doi: 10.1038/cddis.2012.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xin Y, Lu Q, Li Q. IKK1 control of epidermal differentiation is modulated by Notch signaling. American Journal of Pathology. 2011;178(4):1568–1577. doi: 10.1016/j.ajpath.2010.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu S, Chen Z, Zhu F, Hu Y. IkappaB kinase alpha and cancer. Journal of Interferon & Cytokine Research. 2012;32:152–158. doi: 10.1089/jir.2011.0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li Q, Lu Q, Hwang JY, et al. IKK1-deficient mice exhibit abnormal development of skin and skeleton. Genes and Development. 1999;13(10):1322–1328. doi: 10.1101/gad.13.10.1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Candi E, Terrinoni A, Rufini A, et al. p63 is upstream of IKKα in epidermal development. Journal of Cell Science. 2006;119(22):4617–4622. doi: 10.1242/jcs.03265. [DOI] [PubMed] [Google Scholar]
- 63.Koster MI, Dai D, Marinari B, et al. p63 induces key target genes required for epidermal morphogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(9):3255–3260. doi: 10.1073/pnas.0611376104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Romano RA, Birkaya B, Sinha S. A functional enhancer of keratin14 is a direct transcriptional target of ΔNp63. Journal of Investigative Dermatology. 2007;127(5):1175–1186. doi: 10.1038/sj.jid.5700652. [DOI] [PubMed] [Google Scholar]
- 65.Lena AM, Cipollone R, Amelio I, et al. Skn-1a/Oct-11 and ΔNp63α exert antagonizing effects on human keratin expression. Biochemical and Biophysical Research Communications. 2010;401(4):568–573. doi: 10.1016/j.bbrc.2010.09.102. [DOI] [PubMed] [Google Scholar]
- 66.Borrelli S, Candi E, Hu B, et al. The p63 target HBP1 is required for skin differentiation and stratification. Cell Death and Differentiation. 2010;17(12):1896–1907. doi: 10.1038/cdd.2010.59. [DOI] [PubMed] [Google Scholar]
- 67.Sen GL, Boxer LD, Webster DE, et al. ZNF750 is a p63 target gene that induces KLF4 to drive terminal epidermal differentiation. Developmental Cell. 2012;22:669–677. doi: 10.1016/j.devcel.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim S, Choi IF, Quante JR, Zhang L, Roop DR, Koster MI. P63 directly induces expression of Alox12, a regulator of epidermal barrier formation. Experimental Dermatology. 2009;18(12):1016–1021. doi: 10.1111/j.1600-0625.2009.00894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chikh A, Matin RN, Senatore V, Hufbauer M, Lavery D, et al. iASPP/p63 autoregulatory feedback loop is required for the homeostasis of stratified epithelia. The EMBO Journal. 2011;30:4261–4273. doi: 10.1038/emboj.2011.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yi R, Poy MN, Stoffel M, Fuchs E. A skin microRNA promotes differentiation by repressing ‘stemness’. Nature. 2008;452(7184):225–229. doi: 10.1038/nature06642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Antonini D, Russo MT, De Rosa L, Gorrese M, Del Vecchio L, Missero C. Transcriptional repression of miR-34 family contributes to p63-mediated cell cycle progression in epidermal cells. Journal of Investigative Dermatology. 2010;130(5):1249–1257. doi: 10.1038/jid.2009.438. [DOI] [PubMed] [Google Scholar]
- 72.Wu N, Sulpice E, Obeid P, et al. The miR-17 family links p63 protein to MAPK signaling to promote the onset of human keratinocyte differentiation. PLoS One. 2012;7 doi: 10.1371/journal.pone.0045761.e45761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.De Rosa L, Antonini D, Ferone G, et al. P63 suppresses non-epidermal lineage markers in a bone morphogenetic protein-dependent manner via repression of Smad7. Journal of Biological Chemistry. 2009;284(44):30574–30582. doi: 10.1074/jbc.M109.049619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Barton CE, Johnson KN, Mays DM, et al. Novel p63 target genes involved in paracrine signaling and keratinocyte differentiation. Cell Death and Disease. 2010;1(9, article e74) doi: 10.1038/cddis.2010.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ihrie RA, Marques MR, Nguyen BT, et al. Perp is a p63-regulated gene essential for epithelial integrity. Cell. 2005;120(6):843–856. doi: 10.1016/j.cell.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 76.Beaudry VG, Ihrie RA, Jacobs SB, et al. Loss of the desmosomal component perp impairs wound healing in vivo . Dermatology Research and Practice. 2010;2010:11 pages. doi: 10.1155/2010/759731.759731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Barbieri CE, Perez CA, Johnson KN, Ely KA, Billheimer D, Pietenpol JA. IGFBP-3 is a direct target of transcriptional regulation by ΔNp63α in squamous epithelium. Cancer Research. 2005;65(6):2314–2320. doi: 10.1158/0008-5472.CAN-04-3449. [DOI] [PubMed] [Google Scholar]
- 78.Carroll DK, Carroll JS, Leong CO, et al. p63 regulates an adhesion programme and cell survival in epithelial cells. Nature Cell Biology. 2006;8(6):551–561. doi: 10.1038/ncb1420. [DOI] [PubMed] [Google Scholar]
- 79.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Experimental Cell Research. 1961;25(3):585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 80.Hayflick L. The limited in vitro lifetime of human diploid cell strains. Experimental Cell Research. 1965;37(3):614–636. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- 81.Pellegrini G, Dellambra E, Golisano O, et al. p63 identifies keratinocyte stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(6):3156–3161. doi: 10.1073/pnas.061032098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Senoo M, Pinto F, Crum CP, McKeon F. p63 is essential for the proliferative potential of stem cells in stratified epithelia. Cell. 2007;129(3):523–536. doi: 10.1016/j.cell.2007.02.045. [DOI] [PubMed] [Google Scholar]
- 83.Rivetti d V, Lena AM, Nicoloso M, et al. p63-microRNA feedback in keratinocyte senescence. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:1133–1138. doi: 10.1073/pnas.1112257109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Keyes WM, Wu Y, Vogel H, Guo X, Lowe SW, Mills AA. p63 deficiency activates a program of cellular senescence and leads to accelerated aging. Genes and Development. 2005;19(17):1986–1999. doi: 10.1101/gad.342305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Flores ER, Sengupta S, Miller JB, et al. Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell. 2005;7(4):363–373. doi: 10.1016/j.ccr.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 86.Ha L, Ponnamperuma RM, Jay S, Ricci MS, Weinberg WC. Dysregulated δNp63α inhibits expression of ink4a/arf, blocks senescence, and promotes malignant conversion of keratinocytes. PLoS ONE. 2011;6(7) doi: 10.1371/journal.pone.0021877.e21877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Su X, Cho MS, Gi YJ, Ayanga BA, Sherr CJ, Flores ER. Rescue of key features of the p63-null epithelial phenotype by inactivation of Ink4a and Arf. The EMBO Journal. 2009;28(13):1904–1915. doi: 10.1038/emboj.2009.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sommer M, Poliak N, Upadhyay S, et al. ΔNp63α overexpression induces downregulation of Sirt1 and an accelerated aging phenotype in the mouse. Cell Cycle. 2006;5(17):2005–2011. doi: 10.4161/cc.5.17.3194. [DOI] [PubMed] [Google Scholar]
- 89.Toma JG, McKenzie IA, Bagli D, Miller FD. Isolation and characterization of multipotent skin-derived precursors from human skin. Stem Cells. 2005;23(6):727–737. doi: 10.1634/stemcells.2004-0134. [DOI] [PubMed] [Google Scholar]
- 90.Hagiwara K, McMenamin MG, Miura K, Harris CC. Mutational analysis of the p63/p73L/p51/p40/CUSP/KET gene in human cancer cell lines using intronic primers. Cancer Research. 1999;59(17):4165–4169. [PubMed] [Google Scholar]
- 91.Glickman JN, Yang A, Shahsafaei A, McKeon F, Odze RD. Expression of p53-related protein p63 in the gastrointestinal tract and in esophageal metaplastic and neoplastic disorders. Human Pathology. 2001;32(11):1157–1165. doi: 10.1053/hupa.2001.28951. [DOI] [PubMed] [Google Scholar]
- 92.Hu H, Xia SH, Li AD, et al. Elevated expression of p63 protein in human esophageal squamous cell carcinomas. International Journal of Cancer. 2002;102(6):580–583. doi: 10.1002/ijc.10739. [DOI] [PubMed] [Google Scholar]
- 93.Crook T, Nicholls JM, Brooks L, O’Nions J, Allday MJ. High level expression of ΔN-p63: a mechanism for the inactivation of p53 in undifferentiated nasopharyngeal carcinoma (NPC)? Oncogene. 2000;19(30):3439–3444. doi: 10.1038/sj.onc.1203656. [DOI] [PubMed] [Google Scholar]
- 94.Reis-Filho JS, Torio B, Albergaria A, Schmitt FC. p63 expression in normal skin and usual cutaneous carcinomas. Journal of Cutaneous Pathology. 2002;29(9):517–523. doi: 10.1034/j.1600-0560.2002.290902.x. [DOI] [PubMed] [Google Scholar]
- 95.Wrone DA, Yoo S, Chipps LK, Moy RL. The expression of p63 in actinic keratosis, seborrheic keratosis, and cutaneous squamous cell carcinomas. Dermatologic Surgery. 2004;30(10):1299–1302. doi: 10.1111/j.1524-4725.2004.30403.x. [DOI] [PubMed] [Google Scholar]
- 96.Massion PP, Taflan PM, Rahman SMJ, et al. Significance of p63 amplification and overexpression in lung cancer development and prognosis. Cancer Research. 2003;63(21):7113–7121. [PubMed] [Google Scholar]
- 97.Pelosi G, Pasini F, Stenholm CO, et al. p63 immunoreactivity in lung cancer: yet another player in the development of squamous cell carcinomas? Journal of Pathology. 2002;198(1):100–109. doi: 10.1002/path.1166. [DOI] [PubMed] [Google Scholar]
- 98.Nylander K, Coates PJ, Hall PA. Characterization of the expression pattern of p63 alpha and delta Np63 alpha in benign and malignant oral epithelial lesions. International Journal of Cancer. 2000;87:368–372. [PubMed] [Google Scholar]
- 99.Senoo M, Tsuchiya I, Matsumura Y, et al. Transcriptional dysregulation of the p73L/p63/p51/p40/KET gene in human squamous cell carcinomas: expression of δNp73L, a novel dominant-negative isoform, and loss of expression of the potential tumour suppressor p51. British Journal of Cancer. 2001;84(9):1235–1241. doi: 10.1054/bjoc.2000.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Richardson RJ, Dixon J, Malhotra S, et al. Irf6 is a key determinant of the keratinocyte proliferation-differentiation switch. Nature Genetics. 2006;38(11):1329–1334. doi: 10.1038/ng1894. [DOI] [PubMed] [Google Scholar]
- 101.Botti E, Spallone G, Moretti F, et al. Developmental factor IRF6 exhibits tumor suppressor activity in squamous cell carcinomas. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:13710–13715. doi: 10.1073/pnas.1110931108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wu G, Osada M, Guo Z, et al. ΔNp63α up-regulates the Hsp70 gene in human cancer. Cancer Research. 2005;65(3):758–766. [PubMed] [Google Scholar]
- 103.Patturajan M, Nomoto S, Sommer M, et al. ΔNp63 induces β-catenin nuclear accumulation and signaling. Cancer Cell. 2002;1(4):369–379. doi: 10.1016/s1535-6108(02)00057-0. [DOI] [PubMed] [Google Scholar]
- 104.Herfs M, Hubert P, Suarez-Carmona M, et al. Regulation of p63 isoforms by snail and slug transcription factors in human squamous cell carcinoma. American Journal of Pathology. 2010;176(4):1941–1949. doi: 10.2353/ajpath.2010.090804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Di CA, Troiano A, Di MO, et al. The p63 protein isoform DeltaNp63alpha modulates Y-box binding protein 1 in its subcellular distribution and regulation of cell survival and motility genes. The Journal of Biological Chemistry. 2012;287:30170–30180. doi: 10.1074/jbc.M112.349951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kommagani R, Caserta TM, Kadakia MP. Identification of vitamin D receptor as a target of p63. Oncogene. 2006;25(26):3745–3751. doi: 10.1038/sj.onc.1209412. [DOI] [PubMed] [Google Scholar]
- 107.Kommagani R, Leonard MK, Lewis S, Romano RA, Sinha S, Kadakia MP. Regulation of VDR by ΔNp63α is associated with inhibition of cell invasion. Journal of Cell Science. 2009;122(16):2828–2835. doi: 10.1242/jcs.049619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Adorno M, Cordenonsi M, Montagner M, et al. A mutant-p53/Smad complex opposes p63 to empower TGFβ-induced metastasis. Cell. 2009;137(1):87–98. doi: 10.1016/j.cell.2009.01.039. [DOI] [PubMed] [Google Scholar]
- 109.Barbieri CE, Tang LJ, Brown KA, Pietenpol JA. Loss of p63 leads to increased cell migration and up-regulation of genes involved in invasion and metastasis. Cancer Research. 2006;66(15):7589–7597. doi: 10.1158/0008-5472.CAN-06-2020. [DOI] [PubMed] [Google Scholar]
- 110.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. Journal of Clinical Investigation. 2009;119(6):1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Liefer KM, Koster MI, Wang XJ, Yang A, McKeon F, Roop DR. Down-regulation of p63 is required for epidermal UV-B-induced apoptosis. Cancer Research. 2000;60(15):4016–4020. [PubMed] [Google Scholar]
- 112.Papoutsaki M, Moretti F, Lanza M, et al. A p38-dependent pathway regulates ΔNp63 DNA binding to p53-dependent promoters in UV-induced apoptosis of keratinocytes. Oncogene. 2005;24(46):6970–6975. doi: 10.1038/sj.onc.1208835. [DOI] [PubMed] [Google Scholar]
- 113.Schavolt KL, Pietenpol JA. p53 and Δnp63α differentially bind and regulate target genes involved in cell cycle arrest, DNA repair and apoptosis. Oncogene. 2007;26(42):6125–6132. doi: 10.1038/sj.onc.1210441. [DOI] [PubMed] [Google Scholar]
- 114.Zangen R, Ratovitski E, Sidransky D. ΔNp63α levels correlate with clinical tumor response to cisplatin. Cell Cycle. 2005;4(10):1313–1315. doi: 10.4161/cc.4.10.2066. [DOI] [PubMed] [Google Scholar]
- 115.Fomenkov A, Zangen R, Huang YP, et al. RACK1 and stratifin target ΔNp63α for a proteasome degradation in head and neck squamous cell carcinoma cells upon DNA damage. Cell Cycle. 2004;3(10):1285–1295. doi: 10.4161/cc.3.10.1155. [DOI] [PubMed] [Google Scholar]
- 116.Chatterjee A, Chang X, Sen T, Ravi R, Bedi A, Sidransky D. Regulation of p53 family member isoform ΔNp63α by the nuclear factor-κB targeting kinase IκB kinase β . Cancer Research. 2010;70(4):1419–1429. doi: 10.1158/0008-5472.CAN-09-2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sen T, Chang X, Sidransky D, Chatterjee A. Regulation of ΔNp63α by NFκB. Cell Cycle. 2010;9(24):4841–4847. doi: 10.4161/cc.9.24.14093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yuan M, Luong P, Hudson C, Gudmundsdottir K, Basu S. C-Abl phosphorylation of Δnp63α is critical for cell viability. Cell Death and Disease. 2010;1(1, article e16) doi: 10.1038/cddis.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rocco JW, Leong CO, Kuperwasser N, DeYoung MP, Ellisen LW. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell. 2006;9(1):45–56. doi: 10.1016/j.ccr.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 120.Ramsey MR, He L, Forster N, Ory B, Ellisen LW. Physical association of HDAC1 and HDAC2 with p63 mediates transcriptional repression and tumor maintenance in squamous cell carcinoma. Cancer Research. 2011;71(13):4373–4379. doi: 10.1158/0008-5472.CAN-11-0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Frame S, Crombie R, Liddell J, et al. Epithelial carcinogenesis in the mouse: correlating the genetics and the biology. Philosophical Transactions of the Royal Society B. 1998;353(1370):839–845. doi: 10.1098/rstb.1998.0248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Keyes WM, Vogel H, Koster MI, et al. p63 heterozygous mutant mice are not prone to spontaneous or chemically induced tumors. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(22):8435–8440. doi: 10.1073/pnas.0602477103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Su X, Chakravarti D, Cho MS, et al. TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature. 2010;467(7318):986–990. doi: 10.1038/nature09459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Keyes WM, Pecoraro M, Aranda V, et al. Δnp63α is an oncogene that targets chromatin remodeler Lsh to drive skin stem cell proliferation and tumorigenesis. Cell Stem Cell. 2011;8(2):164–176. doi: 10.1016/j.stem.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Li Q, Sambandam SA, Lu HJ, Thomson A, Kim SH, et al. 14-3-3sigma and p63 play opposing roles in epidermal tumorigenesis. Carcinogenesis. 2011;32:1782–1788. doi: 10.1093/carcin/bgr207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gilmore TD. Introduction to NF-κB: players, pathways, perspectives. Oncogene. 2006;25(51):6680–6684. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- 127.Courtois G, Gilmore TD. Mutations in the NF-κB signaling pathway: implications for human disease. Oncogene. 2006;25(51):6831–6843. doi: 10.1038/sj.onc.1209939. [DOI] [PubMed] [Google Scholar]
- 128.Van Waes C. Nuclear factor-κB in development, prevention, and therapy of cancer. Clinical Cancer Research. 2007;13(4):1076–1082. doi: 10.1158/1078-0432.CCR-06-2221. [DOI] [PubMed] [Google Scholar]
- 129.Seitz CS, Lin Q, Deng H, Khavari PA. Alterations in NF-κB function in transgenic epithelial tissue demonstrate a growth inhibitory role for NF-κB. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(5):2307–2312. doi: 10.1073/pnas.95.5.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gugasyan R, Voss A, Varigos G, et al. The transcription factors c-rel and RelA control epidermal development and homeostasis in embryonic and adult skin via distinct mechanisms. Molecular and Cellular Biology. 2004;24(13):5733–5745. doi: 10.1128/MCB.24.13.5733-5745.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Loercher A, Lee TL, Ricker JL, et al. Nuclear factor-kappaB is an important modulator of the altered gene expression profile and malignant phenotype in squamous cell carcinoma. Cancer Research. 2004;64:6511–6523. doi: 10.1158/0008-5472.CAN-04-0852. [DOI] [PubMed] [Google Scholar]
- 132.Dajee M, Lazarov M, Zhang JY, et al. NF-κB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature. 2003;421(6923):639–643. doi: 10.1038/nature01283. [DOI] [PubMed] [Google Scholar]
- 133.Sovak MA, Bellas RE, Kim DW, et al. Aberrant nuclear factor-κb/Rel expression and the pathogenesis of breast cancer. Journal of Clinical Investigation. 1997;100(12):2952–2960. doi: 10.1172/JCI119848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.King KE, Ponnamperuma RM, Allen C, et al. The p53 homologue ΔNp63α interacts with the nuclear factor-κB pathway to modulate epithelial cell growth. Cancer Research. 2008;68(13):5122–5131. doi: 10.1158/0008-5472.CAN-07-6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lu H, Yang X, Duggal P, et al. TNF-alpha promotes c-REL/DeltaNp63alpha interaction and TAp73 dissociation from key genes that mediate growth arrest and apoptosis in head and neck cancer. Cancer Research. 2011;71:6867–6877. doi: 10.1158/0008-5472.CAN-11-2460. [DOI] [PMC free article] [PubMed] [Google Scholar]