

Introduction
Disc degeneration is a multifaceted chronic process that alters the structure and function of intervertebral discs [1]. Degenerated discs occur in 40% of individuals under 30 years of age and greater than 90% of those over 50 [2]. Degeneration can lead to herniations, radiculopathy, myelopathy, spinal stenosis, and degenerative spondylolisthesis, which can cause acute or chronic pain; however, degeneration does not always lead to painful conditions, making painful degeneration a difficult and subjective topic to study and motivating more precise definitions of degeneration [3–9]. Low back pain is one of the most frequent conditions requiring medical care and work disability, and 70–85% of all people have back pain at some point in their life [10]. Over 90% of surgical spine procedures are performed as a result of conditions associated with degeneration [9]. If we can better understand the pathophysiology of degeneration, we can work to directly prevent or slow the process, rather than treat the plethora of disabling conditions it creates [8].
Discs separate spinal vertebrae and distribute pressure while maintaining height against gravity, allowing motion, and providing structural stiffness during loading [1]. The inner nucleus pulposus (NP) is a gel-like structure primarily composed of proteoglycans (PG), water, and collagens. The PG, predominantly aggrecan, is hydrophilic, providing osmotic pressurization that resists compressive loads [11, 12]. The collagen, mainly type II, provides structural support. The outer annulus fibrosus (AF) has a fibrocartilaginous structure made of tough concentric lamellae rich in collagen [7]. Collagen in the AF is mainly type I, and is important for resisting tension, maintaining structure and keeping the NP constrained within the AF [13].
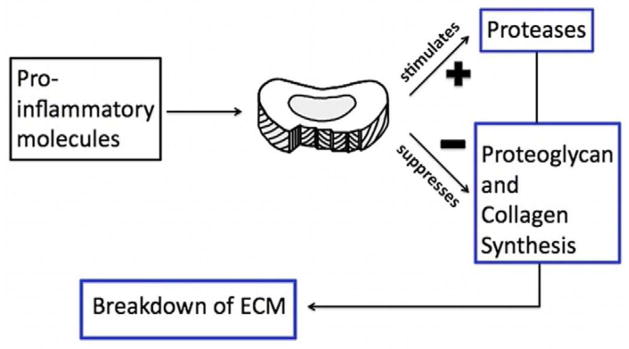
The pathophysiology of degeneration is not completely understood but the consensus is that degeneration begins as early as the second decade of life [14]. When degeneration occurs, the structure and function of the disc are compromised. In early stages of degeneration, the cells make collagen and PG in attempt to repair [15] but as degeneration proceeds, synthesis slows and disc components break down [16]. A decrease in PG synthesis leads to a loss of water, height and hydrostatic pressure. Over time this can cause decreased flexibility and strength. The degeneration process also involves an increase in the synthesis of catabolic molecules such as matrix metalloproteinase (MMP), which cause further degradation (Figure 1) [17, 18].
Figure 1.
Genetic polymorphisms may explain why some individuals are at a higher risk for degeneration. These polymorphisms are abundant in the genome, relatively frequent in the population and some have significant biological effects; thus they are helpful to study. The exact role of most genetic polymorphisms remains to be unraveled. Elucidating the genetic components that are associated with degeneration could provide insights into the mechanism of the process. Eventually, it is hoped that genetic information may be used for identification of patients at risk of early structural changes but more importantly that it may identify those patients that will become symptomatic as a result of this degeneration process.
A significant limiting factor in our understanding of degeneration is the lack of a universally accepted definition for degenerative disc disease and its phenotype [19, 20]. The definition used in published studies is largely determined by characteristics on MRIs and patient symptoms [5]. Common structural changes visualized on MRIs include radial fissures, radial bulging of the annulus, reduced disc height, osteophyte formation, reduced signal intensity (reflecting disc dehydration) and endplate defects [1, 13, 21, 22]. These phenotypic alterations occur sometimes but not always, making it hard to know which are necessary to define clinically relevant degeneration [23]. Using a MRI-based summary score that includes most of these phenotypes markers may dilute associations [23]. Studies underscore the heterogeneity of MRI traits and recommend using these phenotypes separately, because each may represent different aspects of the pathological process [24]. Initiation and the progression of degeneration are not well understood so it is difficult to know what MRI trait is most relevant during each stage of the disease [25]. And there may be patients with abnormal MRIs but no symptoms, further confusing the definition [26, 27].
The purpose of this review is to provide an up-to-date review of gene polymorphisms and their relationship to disc degeneration that also includes functional roles of these polymorphisms. Several good review articles exist [20, 28–31] and this article expands on those existing studies by including a larger list of genes and several associations only recently described. This review also highlights the functional role of the genes and their polymorphisms in the pathogenesis of disc degeneration.
Methods
The Pubmed database was searched by the first author for the following keywords: (disc degeneration OR degenerative disc disease) AND (genetic OR polymorphisms OR gene). All relevant articles in English were reviewed. There were no cutoff dates utilized. References were also obtained based on known literature by all co-authors and any applicable secondary references that were cited in papers that the co-authors read. Our initial search produced 638 articles that were narrowed by reading article titles and abstracts and focusing on studies that specifically related to the topic of this review: genetic polymorphisms and their associations with human disc degeneration.
The co-authors narrowed and finalized the list to 38 studies on 20 genes that are discussed in this review with the goal of including potential genetic associations with disc degeneration. Disc degeneration was defined as a chronic, slow process of structural breakdown that occurs as individuals age; in those with degenerative disc disease, these changes are accelerated. However most of the studies in the literature do not make such distinction so this review included papers that involve individuals with either condition. Papers were excluded if they focused exclusively on low back pain without assessing the imaging-based phenotypic characteristics of degeneration because there are many conditions that can cause low back pain and the focus of this review is on disc degeneration. Papers were also excluded if they evaluated individuals exclusively with herniations alone without a broader description of disc degeneration because the focus of this review is on a chronic not acute process. The co-authors all agreed on these inclusion and exclusion criteria as well as the final paper selection.
Identified genetic risk factors were grouped into five categories based on their function in the disc: structural, catabolic, anti-catabolic, inflammatory and other. Where possible, the associated polymorphisms are uniformly referenced according to their dbSNP rsID (http://www.ncbi.nlm.nih.gov/projects/SNP/).
Results
Epidemiology
Genetics is an important factor in determining individual risk for developing disc degeneration. The traditional view that dominated much of the last century was that occupation [32], physical activity [33], force-injuries [34], smoking [35], repetitive loading [33, 36], gender and exposure to vibration were the major risk factors for accelerated degeneration [37]. The influence of genetics was uncertain [36]. However, many twin studies have identified positive familial aggregation, suggesting some degree of genetic influence. In an article by Simmons et al. [38], 44.6% of patients who had surgery for degenerated discs had a positive family history of degeneration compared to 25.4% who did not. A study by Sambrook et al [11] involving 172 monozygotic and 154 dizygotic twins found similar results. A summary degeneration score, which included height, signal intensity, bulging and osteophyte formation, found heritability estimates up to 74% for lumbar spine and 73% for cervical spine after adjusting for age, weight, height, smoking, occupation and physical activity [11]. A “severe disease score”, which excluded minor grades, estimated heritability to be 64% at lumbar and 79% at cervical spine. This suggests that in addition to environmental risk factors, genetics is also an important factor in determining variation in disc degeneration.
Further epidemiological twin studies have assessed the relative importance of genetics compared to other risk factors. In one of the first studies, Battie et al. [39] looked at disc bulging, narrowing and decreased signal intensity in 115 pairs of male monozygotic twins. Physical loading explained 7% of the variance in degeneration scores, age an additional 9% and familial aggregation an additional 61% in the T12-L4 region [39]. In the lower lumbar levels, physical loading explained 2% of the variability, age an additional 7% and familial aggregation an additional 34% [39]. These results suggested that genetics and unidentified factors, likely complex multi-variable interactions, may primarily explain the variance in disc degeneration. A more recent article by Battie et al. [40] summarized the Twin Spine Study, which looked at the major determinants of disc degeneration, including occupation, vibration exposure, smoking, body weight and genetic contributions. The results suggested that while environmental risk factors do influence degeneration, genetics factors are a larger determinant of disc degeneration [40].
It is now understood that the development of disc degeneration is likely determined by a complex combination of factors with gene-environment and gene-gene interactions that uniquely determine the progression of degeneration in each individual [29]. For example, Solovieva et al. [41] showed that a polymorphism in type IX collagen and obesity acted synergistically to increase the risk of DD. Another study by Solovieva et al. [25] found gene-gene interactions; the polymorphism in type IX collagen increased the risk of DD only in the absence of another polymorphism in interleukin-1β. In an association study by Videman et al. [42] in a Finnish population (n=588), 12 of 99 variants of 25 genes were found to have significant associations with disc signal intensity in magnetic resonance imaging (MRI), underscoring the polygenic nature of disc degeneration.
Further complicating this topic, evidence suggests that as DD progresses, heritability of MRI-defined traits changes. In a recent study by Williams et al. [24] looking at 234 pairs of twins (90 monozygotic pairs and 144 dizygotic same-sex pairs) in the UK and Australia with a 10-year follow-up, the degree of heritability of MRI-determined traits varied between traits as well as age. Change in disc height was not heritable at any age, change in disc signal intensity and anterior osteophytes were heritable only in those under 50 years of age, and the presence of disc bulges was heritable for all age groups, especially in those over 60 years of age. This suggests that genetics may be important not only in determining the risk for developing DD but also the progression of the disease pathology.
Genetic Polymorphisms
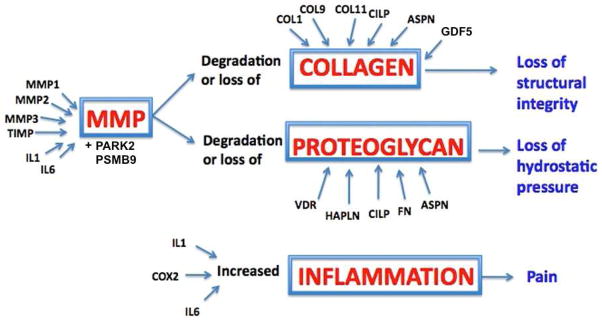
Genetic polymorphisms in 20 genes have been analyzed in association with disc degeneration. In this review, the proteins encoded by these genes are grouped into five categories based on their potential function in the disc: structural, catabolic, anti-catabolic, inflammatory and other (Table 1, Figure 2). Vitamin D receptor and growth differentiation factor 5 (GDF5) make up the other category. Aggrecan, collagen types I, IX and XI, fibronectin, hyaluronan and proteoglycan link protein 1 (HAPLN1), thrombspondin, cartilage intermediate layer protein 1 (CILP1) and asporin are grouped together because they are, or contribute to components of the extracellular matrix. Matrix metalloproteinase (MMP) 1, 2 and 3, parkinson protein 2 E3 ubiquitin protein ligase (PARK2) and proteosome subunit β type 9 (PSMB9) are catabolic molecules and tissue inhibitor of metalloproteinases (TIMP) is an anti-catabolic molecule. Cyclooxygenase-2 (COX-2), interleukin-1 (IL-1) and interleukin-6 (IL-6) are inflammatory cytokines. This review focuses on polymorphisms and their putative role in the pathophysiology of degeneration (Table 2). These genes were integrated into a conceptual model explaining how polymorphisms can lead to loss of structural integrity, loss of hydrostatic pressure or enhanced pro-inflammatory states that can cause potentially painful conditions (Figure 3).
Table 1. Gene Categories.
Genes of interest are separated into five groups based on the functional role of the protein in intervertebral discs. Full names and abbreviations are listed in this table.
| Other | Vitamin D Receptor | VDR |
| Growth Differentiation Factor 5 | GDF5 | |
|
| ||
| Structural | Aggrecan | ACAN |
| Type I Collagen | COL1 | |
| Type IX Collagen | COL9 | |
| Type XI Collagen | COL11 | |
| Fibronectin | FN | |
| Hyaluronan and proteoglycan link protein 1 | HAPLN1 | |
| Thrombospondins | THBS | |
| Cartilage Intermediate Layer Protein | CILP | |
| Asporin | ASPN | |
|
| ||
| Catabolic | Matrix Metalloproteinase 1 | MMP1 |
| Matrix Metalloproteinase 2 | MMP2 | |
| Matrix Metalloproteinase 3 | MMP3 | |
| Parkinson protein 2 E3 ubiquitin protein ligase= | PARK2 | |
| Proteosome subunit β type 9 | PSMB9 | |
|
| ||
| Anti-Catabolic | Tissue inhibitors of metalloproteinase | TIMP |
|
| ||
| Inflammatory | Interleukin 1 | IL1 |
| Interleukin 6 | IL6 | |
| Cyclooxygenase 2 | COX2 | |
Figure 2.
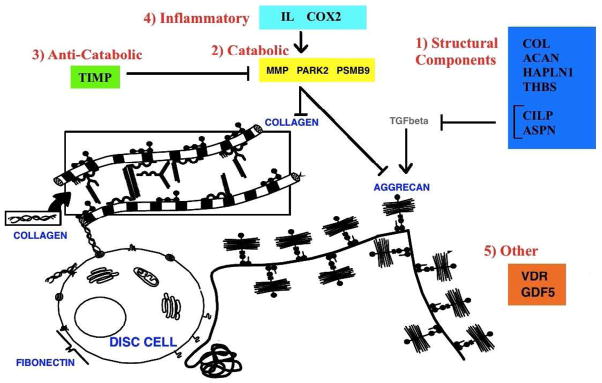
Table 2. Genetic Polymorphisms.
The effects of the polymorphism on gene and protein level, and how they contribute to the degeneration process.
| Gene | Function | Genetic Functional Change | Protein Functional Change (contributing to degeneration) |
|---|---|---|---|
| VDR | Modulate sulfate concentration which is critical for sulfation of proteoglycan |
Fok1: decreased transcription factor binding results in decreased functional VDR Taq1: increased decay of VDR results in decreased VDR product |
Decreased amount of sulfate for proteoglycan results in decreased functional proteoglycan |
| GDF5 | Promotes growth and repair | Decreased transcription | Decreased growth and repair |
| ACAN | Structure, Maintains pressure | Shorter allele binds to less chondroitin sulphate | Decreased functional proteoglycan |
| COL1 | Structure | Increase transcription factor binding results in increased collagen a1(I)/a2(I) product ratio | Impaired COL1 protein |
| COL9 | Structure | Addition of Tryptophan- large, hydrophobic, less soluble amino acid that is rare in collagen | Decreased interactions with other matrix molecules |
| COL11 | Structure | Unstable transcripts?1 | Decreased functional collagen |
| FN | Structure | Fragments | Decreased aggrecan, Increased MMP |
| HAPLN1 | Structure, Up-regulate aggrecan and type II collagen synthesis | Altered transcriptional regulation? | Decreased amount of HAPLN1 results in decreased Aggrecan stability? |
| THBS | Structure, Regulate MMP | Altered transcriptional regulation? | Modulate amount MMP |
| CILP | Structure, Inhibit TGFβ | Increased binding to and inhibition of TGFβ2 | Inhibit aggrecan and type II collagen synthesis |
| ASPN | Structure, Inhibit TGFβ | Increased binding to and inhibition of TGFβ | Inhibit aggrecan and type II collagen synthesis |
| MMP1 | Catabolic | Linkage Disequilibrium with other MMPs? | ? |
| MMP2 | Catabolic | Increased transcription factor binding | Increased MMP |
| MMP3 | Catabolic | Increased promoter activity | Increased MMP |
| PARK2 | Catabolic | Decreased transcription? | Decreased proteosomal degradation? |
| PSMB9 | Catabolic | ? | ? |
| TIMP | Anti-Catabolic | ? | Decreased TIMP? |
| IL1 | Pro-inflammatory cytokine | Increased transcription | Increased IL1 and inflammation |
| IL6 | Pro and anti-inflammatory cytokine | ? | Increased IL6 and inflammation |
| COX2 | Synthesis of prostanoids (pain sensation) | ? | Increased COX2, PGE23, pain and inflammation |
?, Unknown information;
TGF-β, Transforming growth factor beta;
PGE-2, Prostaglandin-E2.
Figure 3.
In 1998, Jones et al. [43] and Videman et al [23] were the first to publish on this topic with a focus on vitamin D receptor. There are many studies prior to 1998 that discuss the pathophysiology of the proteins involved in the degeneration process, but Jones and Videman were among the first to look at genetic polymorphisms as risk factors for degeneration.
Other
Vitamin D Receptor (VDR)
The association between VDR and degenerative disc disease is the first and most robust because it has been validated in large populations of diverse ethnic backgrounds, including Australian, English, Chinese, Japanese and Finnish populations. Vitamin D is mediated by VDR and regulates calcium homeostasis, bone mineralization and remodeling [23]. Two VDR polymorphisms, FokI and TaqI, have been studied extensively with degeneration and are associated with other common bone disorders, including osteoporosis and osteoarthritis [44, 45].
Fok1 (rs2228570) is a polymorphism (ATG to ACG) at the first of two potential translation initiation sites in exon 2 of VDR. Individuals with the T allele, the risk allele (designated f), initiate translation at the first start site, producing a full-length VDR protein [46]. Those with the C allele (designated F) initiate translation at the second start site, and produce a shorter protein [46]. However, the F allele (shorter allele) interacts more efficiently with the transcription factor and shows relatively increased function compared to the f allele (longer allele) [46]. Videman et al. [23] found that in a cohort of 170 men, those with an ff genotype had a signal intensity 9.3% lower and a summary degeneration score (based on signal intensity, bulging and disc height) 6.9% more severe than men with the FF genotype. For this study and all subsequent studies, refer to the table for summaries (Table 3a–b). In a recent cohort of 300 individuals, the ff genotype was statistically significantly associated with severe forms of DD [47]. These studies suggest that the longer f allele, which seems to cause decreased VDR function, increases the risk of degeneration. The exact mechanism of decreased VDR function leading to degeneration is not well understood. Studies suggested that Vitamin D might modulate sulfate concentrations and determine the degree of PG sulfation, which is critical for disc stability [48, 49]. The f allele, with decreased VDR function, might contribute to less than optimal disc structure that is prone to early degeneration.
Table 3a. Studies on Structural Genetic Polymorphisms and Disc Degeneration.
Summary of Studies on Genetic Polymorphisms and Disc Degeneration.This table summarizes the important details of studies on genetic polymorphisms and degeneration. The ages of subjects are outlined in this table as a mean, range or cut-off point, in years. The phenotypes listed in the table are the MRI-defined traits other than frequently used summation scores, which are MRI-based grading schemes that combine a few traits into a total degeneration score.
| Gene | Polymorphism | Author, Year | Study Population | Phenotype | Association? | Ref.1 | |||
|---|---|---|---|---|---|---|---|---|---|
| Ethnicity | Gender | Num. | Age | ||||||
| ACAN | VNTR (variable number of tandem repeats) | Roughley, 2006 | Canadian | M+F | 44 | 55 | No | [59] | |
| Kawaguchi, 1999 | Japanese | F | 64 | 20–29 | Yes | [62] | |||
| Kim, 2011 | Korean | M+F | 104 | <40 | Yes | [63] | |||
| Solovieva, 2007 | Finnish | M | 132 | 25–49 | SI2, DB3, DH4 | Yes | [64] | ||
| rs1042631, rs1516797 | Videman, 2009 | Finnish | M | 588 | 35–70 | SI, DB, DH | Yes | [42] | |
| COL1 | rs1800012 | Plujm, 2004 | Dutch | M+F | 517 | >65 | Yes | [68] | |
| Tilkeridis, 2005 | Greek | M | 24 | 29 | Yes | [69] | |||
| rs2075555, rs1007086 | Videman, 2009 | Finnish | M | 588 | 35–70 | SI | Yes | [42] | |
| COL9 | Trp2 | Annunen, 1999 | Finnish | M+F | 157 | 44 | Yes | [76] | |
| Jim, 2005 | Chinese | M+F | 804 | 18–55 | AT5, DH, SN6 | Yes, if <49yo7 | [77] | ||
| Higashino, 2007 | Japanese | M+F | 84 | <40, >40 | Yes, if >40yo | [78] | |||
| Kales, 2004 | Greek | M+F | 105 | <60 | No | [79] | |||
| Wrocklage, 2000 | German | M+F | 250 | 71 | Yes, if >60yo | [80] | |||
| COL9A2 | Seki, 2006 | Japanese | M+F | 470 | 35 | Yes, if <40yo | [81] | ||
| Trp3 | Karppinen, 2003 | Finnish | M+F | 153 | Yes | [83] | |||
| Solovieva, 2006 | Finnish | M | 135 | 40–45 | SI, DB, DH | Yes, without IL1 polym.8 | [25] | ||
| Jim, 2005 | Chinese | M+F | 804 | 18–55 | AT, DH, SN | No | [77] | ||
| Higashino, 2007 | Japanese | M+F | 84 | <40; >40 | No | [78] | |||
| Kales, 2004 | Greek | M+F | 105 | <60 | No | [79] | |||
| Paassilta, 2001 | Finnish | M+F | 171 | 45 | Yes | [82] | |||
| COL9A1: rs696990 | Videman, 2009 | Finnish | M | 588 | 35–70 | SI2, DB | Yes | [42] | |
| COL9A2: rs7533552, COL9A3 | SI SI, DB, DH |
Yes No |
|||||||
| COL11 | COL11A2: rs1800587 | Noponen, 2003 | Finnish | M+F | 29 | 59 | Yes | [74] | |
| COL11A2: intron 6; intron 9 | Solovieva, 2006 | Finnish | M | 135 | 40–45 | SI, DB, DH | Yes | [25] | |
| COL11A1: rs1337185, rs1463035; | Videman, 2009 | Finnish | M | 588 | 35–70 | DB | Yes | [42] | |
| COL11A2: rs2072915, rs9277933, rs2076311 | SI | Yes | |||||||
| FN | alternative splicing: EDA, EDB and V | Oegema, 2000 | Pathology samples | M+F | 11 | 56 | Yes | [94] | |
| Anderson, 2010 | Surgical samples | Yes | [92] | ||||||
| HAPLN1 | rs179851 | Urano, 2010 | Japanese | F | 622 | 67 | SN, OP9, DN10 | Yes | [21] |
| THBS | intron 3 C/T; A386G; CD36 | Valdes, 2005 | English | F | 720 | 74 | OP, DN | Yes | [51] |
| CILP | rs2033711 | Seki, 2005 | Japanese | M+F | 467 | 35 | Yes | [108] | |
| Virtanen, 2007 | Finnish; Chinese | M+F | 243; 348 | No | [109] | ||||
| Min, 2009 | Japanese | M | 89 | 20 | Yes | [111] | |||
| Min, 2010 | Japanese | M+F | 601 | 20 | Yes, only in men | [110] | |||
| ASPN | number of repeats | Song, 2008 | Chinese; Japanese | M+F | 527; 745 | 20–49 | Yes | [114] | |
Ref., reference;
SI, signal intensity;
DB, disc bulges;
DH, disc height;
AT, annular tears;
SN, schmorl nodes;
yo, years old;
polym., polymorphism;
OP, osteophytes;
DN, disc narrowing.
Table 3b. Studies on Catabolic, Anti-catabolic, Inflammatory and Other Genetic Polymorphisms and Disc Degeneration.
Summary of Studies on Genetic Polymorphisms and Disc Degeneration.This table summarizes the important details of studies on genetic polymorphisms and degeneration. The ages of subjects are outlined in this table as a mean, range or cut-off point, in years. The phenotypes listed in the table are the MRI-defined traits other than frequently used summation scores, which are MRI-based grading schemes that combine a few traits into a total degeneration score.
| Gene | Polymorphism | Author, Year | Study Population | Phenotype | Association? | Ref.1 | |||
|---|---|---|---|---|---|---|---|---|---|
| Ethnicity | Gender | Num. | Age | ||||||
| VDR | Fok1 | Videman, 1998 | Finnish | M | 170 | 49 | SI2, DB3, DH4 | Yes | [23] |
| Taq1: rs731236 | Videman, 1998 | Finnish | M | 170 | 49 | SI, DB, DH | Yes | [23] | |
| Jones, 1998 | Australian | M+F | 282 | >60 | OP5, DN6, DB | Yes | [43] | ||
| Cheung, 2006 | Chinese | M+F | 804 | 18–55; <40 | DB, AT7, SN8 | Yes, especially if <40yo9 | [50] | ||
| Valdes, 2005 | English | F | 720 | 74 | OP, DN | Yes | [51] | ||
| Videman, 2001 | Finnish | M | 142 | 49 | SI, DB, DH, AT, OP, H10 | Yes | [52] | ||
| GDF5 | rs143383 | Williams, 2011 | Northern European | F | 5,259 | 19–90 | DH, OP | Yes | [118] |
| MMP1 | rs1799750 | Song, 2008 | Chinese | M+F | 478 | 18–55 | DB, SN, AT | Yes, especially >40yo | [128] |
| MMP2 | rs243865 | Dong, 2007 | Chinese | M+F | 162 | 25 | Yes | [129] | |
| MMP3 | number of adenine repeats | Takahashi, 2001 | Japanese | M+F | 103 | 21; 73 | Yes, only in elderly | [131] | |
| Yuan, 2010 | Chinese | M+F | 178 | Yes | [37] | ||||
| Noponen, 2003 | Finnish | 29 | 59 | No | [74] | ||||
| Valdes, 2005 | English | F | 720 | 74 | OP, DN | Yes | [51] | ||
| PARK2 | Williams, 2012 | Northern European | M+F | 4683 | 57.7 | DH, OP | Yes | [133] | |
| PSMB9 | Williams, 2012 | Northern European | M+F | 4683 | 57.7 | DH, OP | Yes | [133] | |
| TIMP | C124T | Valdes, 2005 | English | F | 720 | 74 | OP, DN | Yes | [51] |
| IL1 | IL1α: rs1800587 | Eskola, 2010 | Danish | F | 352 | 13 | AT, DB, SI | Yes, only girls | [161] |
| Virtanen, 2007 | Finnish | M | 150 | 38–56 | Yes, especially with vibration | [165] | |||
| Solovieva, 2004 | Finnish | M | 45 | 40–45 | SI, DB, DH | Yes, especially with workload | [166] | ||
| rs2071375 | Videman, 2009 | Finnish | M | 588 | 35–70 | SI | Yes | [42] | |
| IL1β: rs1143634 | Solovieva, 2006 | Finnish | M | 135 | 40–45 | SI, DB, DH | Yes, with Trp3 polym11 | [25] | |
| Videman, 2009 | Finnish | M | 588 | 35–70 | SI, DB, DH | No | [42] | ||
| rs1800587, rs1143634 | Noponen, 2005 | Finnish | M+F | 155 | 44 | No | [167] | ||
| IL6 | rs1800797, rs1800796, rs1800795; rs1800797, rs1800796, rs1800795, rs13006435 | Eskola, 2010 | Danish | M+F | 352 | 13 | AT, DB, SI | Yes, only in girls No |
[161] |
| rs13006435, rs1800796, rs1800795, rs13006435 | Noponen, 2005 | Finnish | M+F | 155 | 44 | Yes | [167] | ||
| COX2 | rs5277 | Valdes, 2005 | English | F | 750 | 74 | OP, DN | Yes | [51] |
Ref., reference;
SI, signal intensity;
DB, disc bulges;
DH, disc height;
OP, osteophytes;
DN, disc narrowing;
AT, annular tears;
SN, schmorl nodes;
yo, years old;
H, herniation;
polym, polymorphism
The second polymorphism of the VDR gene, Taq1 (rs731236), is found in a non-coding region of exon 9 [50]. The risk allele, the t allele, has been shown to increase decay of mRNA by 30% compared to the normal T allele, which may lead to impaired sulfation of glycosaminoglycan (GAG) during PG synthesis [48, 49]. Since PG is integral to disc structure, this polymorphism directly contributes to the breakdown of structure characteristic of disc degeneration (DD). Alternatively, this variant in VDR may be in linkage disequilibrium with a true disease predisposing locus nearby, such as in the COL2A1 gene, and thus may be a proxy marker for DD rather than a true genetic risk marker [50]. A study by Jones et al. [43] on 282 elderly Australians was the first to show an association between DD and VDR; individuals homozygous for the risk allele (tt genotype) were associated with an increased risk of osteophytes and disc narrowing compared to the normal, TT genotype. A subsequent study on 720 English women confirmed this result [51, 52]. In two studies, Videman et al. [23] showed that signal intensity was lower (12.9%) and the number annular tears was higher, in men with the tt genotype compared to the TT genotype. In one of the largest genetic studies involving 804 Chinese individuals, the t allele was significantly associated with DD (OR=2.61, p=0.041) [50]. In those less than 40 years old, the association was even stronger (OR=5.97, p=0.002) [50]. This age correlation is consistent with the findings in a Japanese population [53]. The frequency of the t allele in the Chinese population was only 3.7%, while it was 13% in the Japanese and 33% in the Finnish population. This suggests that the polymorphism may have more of an influence on disc degeneration in certain populations [50].
Growth differentiation factor (GDF5)
GDF5 polymorphisms have been shown to be associated with peripheral joint osteoarthritis [117] and lumbar disc degeneration [118], which is consistent with known similarities between the two conditions. The study by Williams et al. [118] evaluated five female population cohorts from Northern Europe (total n=5,259). The 5′ upstream single-nucleotide polymorphism variant (rs143383) was found to be associated with lumbar disc degeneration, as defined by disc space narrowing and osteophytes on plain radiography or MRI. GDF5 encodes a protein that is involved in promoting growth and repair. It is a pro-chondrogenic growth factor which has been shown to increase fiber size in the ligament model of knee stability in rats [118]. The C to T mutation at position +104 in the promoter region of GDF5 suggests that the variant reduces GDF5 transcription. The role of this altered protein in disc degeneration may result from direct effects on the disc itself or periarticular structures such as the longitudinal spinal ligaments, however further work is needed to better establish its functional role in the degenerative process [118].
Structural
Aggrecan (ACAN)
The NP is rich in proteoglycan (PG), mainly aggrecan, which forms large aggregates and become entrapped in a mesh of collagen fibrils [54, 55]. Aggrecan is responsible for attracting and holding water to maintain swelling pressure that can help the disc absorb shock and counter compressive loads [7, 16, 56]. As discs degenerate and as individuals age, aggrecan and water content in the NP decreases [12]. PG accounts up to 70% of dry weight of the NP in young children, but only 20% in mature adults [12, 57]. The degeneration process also causes a switch in the synthesis of PG from aggrecan to smaller proteoglycans, biglycan and decorin [58]. This switch alters the matrix physicochemical properties and may contribute to the pathogenesis of degeneration [58]. The function of aggrecan relies on the protein’s substituted chondroitin sulphate (CS1 and CS2) and keratan sulphate (KS) chains with negatively charged glycosaminoglycan (GAG) [59]. The region of ACAN that encodes for the CS1 domain exhibits variable number of tandem repeats (VNTR) ranging from 13–33 [57, 60, 61]. Studies suggest that individuals with fewer repeats may have a lower number of CS chains. This may lead to reduced pressure and increased susceptibility to premature degeneration [16, 59].
A study by Kawaguchi et al. [62] on 64 individuals was the first to analyze the association between this polymorphism and lumbar DD in humans. The authors found a significant difference between the distribution of allele sizes and the severity of MRI-defined DD. In a group of 20–29 year old Japanese women, A18 (18 repeats) and A21 (21 repeats) were over-represented in those women with multilevel and severe degeneration (p=0.008). A recent study by Kim et al. [63] on a Korean population found that the A21 allele was overrepresented in individuals with degeneration, but only in those less than 40 years old. Both studies used young subjects who are less exposed to occupational loads and other environmental stresses allowing results to reflect predominately genetic associations. A study on 132 Finnish individuals found that a medium-length, specific allele, A26, was associated with and overrepresented in those with lower back pain and decreased MRI signal intensity [64]. It is unclear why A26 had the highest risk; there may be a protective mechanism for shorter and longer genes that has yet to be uncovered. The results of this study conflict with those of other studies and the disparity can be attributed to differences in allele distributions across populations. In the Finnish population, only 2.3% had alleles with less than 26 repeats and none had two short alleles [64]. This suggests that 26 repeats may be a short fragment and thus carry an increased risk for degeneration in this population, while it is a medium-length fragment without additional risk in the other populations.
A study by Roughley et al. [59] on 102 individuals average age of 55 years, did not find such an association. These MRI-defined patients all presented with lower back pain and were candidates for surgical decompression. The authors suggested that one short allele is not sufficient to influence disc function in a detrimental manner [59]. Unfortunately, it is difficult to compare this study to Kawaguchi’s because they focused on younger and older populations, respectively.
In a recent study of cohort of 588 Finnish males from the Twin Spine Study, after controlling for age and suspected risk factors such occupation, MRI-based phenotypes were analyzed to estimate disc degeneration with certain polymorphisms [42]. This was a true association study in which candidate genes were tested for an association with disc degeneration. There was an association between one aggrecan gene polymorphism (rs1042631) and signal intensity as well as disc bulging, while another gene polymorphism (rs1516797) was found to be associated with disc height narrowing [42]. This is one of the largest studies to look at aggrecan gene polymorphisms, and the results suggest that it may be a candidate gene involved in the development of disc degeneration.
Type I Collagen (COL1)
Type I collagen is crucial for the strength of the intervertebral disc. It is primarily found in the outer AF [12]. Mice with reduced COL1 expression had mechanically inferior vertebral disc tissue compared to control animals, with decreased apparent modulus in both compression and torsion [65]. An Sp1 polymorphism (rs1800012) in the promoter region of COL1A1 is associated with many bone related conditions such as osteoporosis, and increased fracture risk [66, 67]. Studies suggest that the T allele, the risk allele, increases Sp1 transcription factor binding, leading to increased collagen α1(I) chain and collagen α1(I)/α2(I) ratio. The relative increase of collagen α1(I) chain has been associated with impaired bone strength and may similarly impair disc structure [68].
In a study of 517 elderly Dutch individuals, those homozygous for the risk allele (TT genotype) were associated with an increased risk of degeneration compared to GT and GG genotypes (OR 3.6) [68]. This study rightly acknowledged its limitations, particularly the fact that disc degeneration was defined based on lateral radiographs. Interestingly, another study found the association to hold true in 24 young Greek soldiers [69]. The TT genotype was more likely to have MRI-evaluated degeneration than the GT and GG genotypes (33% and 0% respectively) [69]. However, this cohort is very small so it is difficult to draw conclusions.
In a recent study on 588 Finnish men from the Twin Spine Study, disc signal intensity was associated with allelic variants of COL1A1 gene (rs2075555 and rs1007086) [42]. This study was done on a large population, which helps to strengthen the support for COL1A1 as an important gene in the development of disc degeneration.
Type IX Collagen (COL9)
Type IX collagen is a small structural component of the NP, AF and endplates of the disc and thus disruption of it is likely to cause degeneration [7]. Collagen IX is a heterotrimer of three α chains (α1, α2, α3) that encode for genes COL9A1, COL9A2 and COL9A3 [70]. The function is not totally understood, but type IX collagen provides mechanical support to the disc [71]. Mutations in the COL9 are known to affect disc degeneration in murine models as well as humans. Transgenic mice with an over-expression of a mutant α1(IX) collagen and mice with an inactivated COL9A1 gene were found to have accelerated disc degeneration and more herniations than the age-matched control mice [72, 73]. There are two polymorphisms, Trp2 (Gln326Trp) and Trp3 (Arg103Trp), found in COL9A2 and COL9A3 respectively, that have been studied in relation to human disc degeneration. The Trp2 and Trp3 alleles have been associated with degenerative lumbar spinal stenosis [74] as well as symptomatic spinal stenosis [75]. Thus, they are candidate polymorphisms to study in relation to disc degeneration.
Annunen et al. [76] was the first to show an association between the Trp2 allele and disc degeneration in 157 individuals. The Trp2 allele was found in six of 157 individuals with sciatica, a characteristic symptom of herniation but none of 174 controls. 73% of the sciatic patients had degeneration detectable on MRI [76]. Linkage analysis showed that the Trp2 allele co-segregated with the disease phenotype in all four of the families studied [76]. A large cohort study of 804 Chinese individuals confirmed this finding [77]. The Trp2 allele was associated with a 4-fold increased risk of annular tears in individuals aged 30–39 and a 2.4-fold increase in MRI-defined degeneration and endplate herniations in those 40–49 years of age [77]. Trp2 was found in 20% of the Chinese population [77]. Another study found the Trp2 allele in 21.3% of the Japanese population [78]. The study also found Trp2 to be an age-dependent risk factor, this time in younger patients [78]. Trp2 increased the risk of severe degeneration 6-fold in patients less than 40 years old with symptomatic herniated lumbar discs. There was no association with patients over the age of 40, which differs from the trends of the Chinese study. Interestingly there were no Trp2 alleles found in 105 Greek individuals [79]. The association between Trp2 and disc degeneration did not hold true in a German population of 250 surgically treated patients [80] nor in a second Japanese population [81]. The study on the Japanese population did find another susceptibility allele in COL9A2 that was associated with disc degeneration (p=0.025) and even more with severe degeneration (p=0.011) [81]. The discrepancy in Trp2 associations to disc degeneration can be likely attributed to the different allele frequencies in the various ethnic groups.
The Trp3 allele in COL9A3 showed a 3-fold increased risk of disc degeneration in the two different Finnish populations. The Trp3 allele was found in 24% of patients and 9% of controls in one study [76], and 12.3% of 171 patients compared to 4.7% of 186 controls in the other study (p=0.000013) [82]. Karppinen et al. [83] found that individuals with at least one Trp3 allele had an increased number of degenerated discs compared to controls. Interestingly, the Trp3 allele appeared to be absent in Chinese [77] and Japanese patients [78]. Solovieva et al. [25] confirmed the Trp3 association with disc degeneration, and also noted a gene-gene interaction with an IL1β polymorphism (rs1143634). Those with the Trp3 allele without the IL1β polymorphism had an increased risk of signal intensity changes (OR=7.0), but there was no effect with the IL1β polymorphism [25]. This significant odds ratio suggests that Trp3 is modified by this additional and seemingly unrelated polymorphism or that the IL1β polymorphism is a negative confounder, with an unknown single complementary third factor [25].
The Trp associations are among the most strongly correlated relationships with degenerative disc disease, with replicated associations in more than two different ethnic populations [28]. This increased risk may be explained by the addition of a big hydrophobic and less soluble Tryptophan, which is rarely found in collagenous domains. Tryptophan may affect interactions with other matrix molecules, disrupt the triple helix and weaken disc resistance to compressive loads [31, 80, 84]. In a biomechanical study, Trp2+ nondegenerated discs had a mean swelling pressure and compressive modulus value significantly lower that of Trp2- nondegenerated samples. This study suggests that the Trp2 allele negatively influences disc mechanics, which may ultimately explain the association with disc degeneration [85].
The large study on 588 Finnish males from the Twin Spine Study found a COL9A1 polymorphism (rs696990) strongly associated with disc intensity and bulging on MRI as well as a COL9A2 polymorphism (rs7533552) also associated with disc intensity, but no associations with polymorphisms of COL9A3 [42]. This study was controlled for age and suspected risk factors such as occupation.
Type XI Collagen (COL11)
Collagen XI is a quantitatively minor collagen in the intervertebral disc, but is essential for the maintaining the interactions between PG and collagens [86–88]. It is a heterotrimeric triple-helix molecule made of three α chains - α1(XI), α2(XI), and α3(XI) [89]. There are several different COL11 single nucleotide polymorphisms (SNPs) that have been associated with degeneration, however none have so far been replicated in other populations. Noponen et al. [74] found that those heterozygous for the risk T allele of a single nucleotide polymorphism in intron 6 of COL11A2 (rs1800587) had an increased risk of developing degenerative lumbar spinal stenosis, which may relate to underlying degeneration. Solovieva et al. [25] studied a sequence variation in intron 9 (A/G) of COL11A2 and found that in 135 Finnish men, those with at least one G allele were associated with an increased risk of bulges compared to those without this polymorphism (OR 2.1). A large study by Videman et al. [42] on 588 Finnish men found that two COL11A1 polymorphisms (rs1337185, rs1463035) and three COL11A2 polymorphisms (rs2072915, rs9277933, rs2076311) were associated with MRI-defined disc bulging and signal intensity respectively. The functional roles in these polymorphisms are not yet known - they may produce unstable transcripts of the disease-associated allele. Instability would cause decreased functional collagen and subsequent degeneration [90].
Fibronectin (FN)
Fibronectin is an extracellular glycoprotein that helps organize and maintain homeostasis of the extracellular matrix [91]. Fibronectin binds to collagen, functions in cell adhesion, migration and differentiation and regulates cell-matrix and matrix-matrix interactions [92]. Alternative splicing at three major sites, EDA, EDB and V, produces up to 20 different FN variant fragments [92, 93]. Evidence is growing to suggest that these isoforms affect fibronectin function [94, 95]. In cartilage, FN fragments cause chondrocytes to decrease aggrecan and increase MMP synthesis, leading to articular cartilage degradation [96–98]. Due to the similarity of cellular and extracellular matrix properties between cartilage and intervertebral discs, FN fragments may also play a role in disc degeneration [99, 100].
A study by Oegema et al. [94], found an association between FN fragments and MRI-defined degeneration, but did not specify the size of the fragment. FN fragments may stimulate MMP production and contribute to degeneration in the disc, as done in chondrocytes [99, 100]. A recent study that looked at FN splice variants found higher levels of EDB+ in degenerated discs compared to controls, suggesting its potential contribution to degeneration [92]. However these studies may not explain a genetic relationship with degeneration because many non-genetic components could affect or regulate the gene splicing. Therefore, further studies need to look carefully at these various fragments and splicing mechanisms as well as other potential polymorphisms.
Hyaluronan and proteolgycan link protein 1 (HAPLN1)
HAPLN1 binds to aggrecan on the hyaluronic chain, stabilizing the aggregates and thus increasing compression resistance and shock absorption [101]. HAPLN1 also functions as a growth factor, up-regulating aggrecan and type II collagen synthesis in cartilage [102]. It seems to be an essential part of cartilage homeostasis and may also contribute to homeostasis in the disc tissue. In a recent study of 622 postmenopausal Japanese women, the TT genotype in comparison to CT or CC genotypes of a single nucleotide polymorphism in intron 2 (rs179851) was found to be significantly associated with discs narrowing and osteophyte formation (O.R=1.83 and OR =2.12 respectively) [21]. The mechanism through which this single nucleotide polymorphism confers increased risk is not well understood. It may have altered the transcriptional regulation of the gene or may be linked with disease-causing variants in nearby genes [21]. This large study offers promising data that requires follow up studies in other populations.
Thrombospondins (THBS)
Thrombospondins are a class of extracellular glycoproteins that bind to collagen and participate in cell-to-cell and cell-to-matrix communication during tissue genesis and repair [103]. They have diverse functions related to the extracellular matrix and help regulate the levels of MMP2 and MMP9, which are important in the pathogenesis of degeneration [104, 105]. Mice deficient in thrombospondin-2 show abnormal curvature [106] and knockout mice show increased levels of MMP2 after injury [107]. In a study by Valdes et al. [51], polymorphisms in both THBS2 (intron 3 C>T) and COMP (A386G), a subgroup of thrombospondins, were associated with a change in the Kellgren-Lawrence degeneration grade, which is a summary grade based on osteophyte formation, joint space, sclerosis and bone contour deformities. Another polymorphism, this one in the thrombospondin receptor, CD36, in the 5′ UTR, was associated with a change in osteophytes and disc height, but not with the Kellgren-Lawrence degeneration score [51]. These studies suggest that thrombospondins may play an important role in MMP homeostasis and disc degeneration. These are, to our knowledge, the only studies looking at such an association, and this area warrants further investigation.
Cartilage Intermediate Layer Protein (CILP)
CILP encodes a protein that is widely expressed in the disc with increased expression in degeneration [108]. CILP co-localizes with TGFβ. In vitro studies suggest that CILP may act as a negative regulator of TGFβ, potentially down regulating aggrecan and collagen II [108]. The negative effect of CILP on TGFβ signaling increases with degeneration, suggesting that CILP could be a factor in the pathogenesis of disc degeneration [108].
In an amino acid substitution polymorphism (Ille395Thr) in exon 8 (rs2033711), functional analysis shows that the risk allele, the C allele, results in increased binding and inhibition of TGFβ [108]. In a study comparing 467 Japanese patients with symptomatic degeneration to 654 controls, this CILP polymorphism was identified as a risk factor for degeneration (OR 1.61) [108]. However, this association was not found in other ethnic populations, including 243 Finnish and 348 Chinese subjects [109]. The lack of an association does not negate the results of the Japanese study, especially given its high power, but suggests that there may be unique genetic risk factors in different ethnic populations, as it may be the case with the Trp2 and Trp3 polymorphisms in COL9A2 and COL9A3, respectively. Two studies by Min et al. [110, 111] supported the association between the CILP functional single nucleotide polymorphism and degeneration in male Japanese judo athletes. These athletes experienced above normal stress on the lumbar spine, which increases the risk of degeneration. Male athletes with a C allele were more likely to have disc degeneration than those with a T allele, suggesting that the product of the risk allele may enhance the degenerative progress. Interestingly, the C allele did not affect the occurrence of degeneration in Japanese female athletes [110]. The gender discrepancy may be explained by weight or occupational differences, or by the fact that this CILP polymorphism is a gender-dependent risk factor [110]. More studies need to further analyze this association.
Asporin (ASPN)
Asporin is a member of the family of small leucine-rich proteoglycans, most closely related to decorin and biglycan [112]. It is associated with the extracellular matrix in cartilage, meniscus and several other tissues [112]. It functions to provide functional support as well as regulate signaling molecules. A typical allelic product has 13 aspartic acid repeats at the N-terminus, but a polymorphic allele at this site can contain 9–20 aspartate repeats [112]. Thee polymorphic allele with 14 aspartate repeats (D14) has been shown to be associated with osteoarthritis [113]. Interestingly, Song et al. [114] found this same allele, the D14 allele, to be significantly associated with disc degeneration in Chinese and Japanese individuals (p=0.000013) from a meta-analysis. Degeneration was measured using a sliding-window method, which adjusts degeneration scores to an age-specific median score [115]. They also found that asporin expression levels increased with age and degeneration, along with a decrease in collagen II and aggrecan [114]. Thus, increased binding to TGFβ, which is seen in D14 products compared to D13 products, may decrease the collagen II and aggrecan levels more significantly, leading to increased degeneration [113, 116]. While a few studies have looked at this association, it has replicated across two populations, and thus could be a promising new genetic risk factor for degenerative disc disease.
Catabolic
Matrix Metalloproteinase (MMP)
A delicate balance between degradation and synthesis maintains disc integrity. MMPs are key proteins involved in the breakdown of the extracellular matrix. There are currently at least 18 members of the MMP family that are involved in the degradation of different matrix components [119]. Some of these MMPs also indirectly degrade the matrix by activating other latent MMPs [119]. MMP production can be up-regulated by pro-inflammatory cytokines, IL1 [119] and TNFα [120]. MMP expression has been shown to increase with age, which correlates with an associated increase in degradation [121]. Many MMPs are expressed in both degenerated and non-degenerated discs, and thus have a role in homeostasis and degradation. In degraded tissue, there is an increase in many MMPs, including MMP1, 2, 3, 7, 9, 13, and 28, often according to the severity of degeneration [13, 122–127]. However, only polymorphisms in MMP1, MMP2, and MMP3 have been studied as risk factors for disc degeneration.
MMP1
A guanine (G) insertion in the promoter region of MMP1 (rs1799750) creates a core-binding site for Ets transcription factors, and results in increased promoter activity [120]. Thus, one would expect that individuals with the insertion have an increased risk of degeneration due to increased MMP1 production. However, a study by Song et al. [128] found just the opposite - individuals without the guanine insertion had a 1.5 times increased risk of degeneration compared to those with insertion. Authors explained that this apparent contradiction may be due to linkage disequilibrium, with MMP1 located close to other MMPs [128]. Alternatively, the allele without the guanine insertion may be a common ancestor allele (high frequency in African and Caucasian populations) that is in linkage disequilibrium with an unknown disease-causing polymorphism within the same gene [128].
MMP2
A single nucleotide polymorphism in MMP2 (rs243865), disrupts the Sp1 transcription factor binding site and results in decreased transcriptional activity [129]. Individuals homozygous for the risk allele, with a CC genotype, have higher MMP2 levels than those with a TT or CT genotype [129, 130]. Not surprisingly, the study found that young individuals with the CC genotype had a nearly a 3-fold increased risk of degeneration and more severe degeneration compared to those with at least one T allele [129]. This suggests that the C allele in this MMP2 polymorphism may be a risk factor for early degeneration [129].
MMP3
Takahashi et al. [131] showed an association with an MMP3 polymorphism and degenerative disc disease. This promoter region polymorphism produces alleles with 5 or 6 repeats of adenine (5A and 6A respectively) [108]. Compared to the 6A allele, the 5A allele had twice the promoter activity, which results in more protein produced and a potential mechanism for increased degeneration [127, 132]. Not surprisingly, in the Japanese population, having either one or two copies of the 5A allele was associated with increased degeneration [131]. This association held true in the elderly population, but there was no significant difference in the young population [131]. The discrepancy may be explained by a small sample size (49 elderly and 59 young subjects), different techniques used to define degeneration (MRI in the younger population but radiographs in the elderly population) or it may be an age-dependent risk factor [51]. Another study of 720 English women found the 5A allele to be associated with lumbar disc degeneration based on osteophytes and disc narrowing on radiographs. Disc degeneration was not significantly associated with the Kellgren-Lawrence summary score, which suggests that summary scores may mask true associations [51]. Another study confirmed the above results; there was a 1.96-fold increased risk of lumbar disc degeneration with the 5A allele compared to 6A allele in 178 Chinese individuals [37]. This study also found a synergistic gene-environment interaction between the 5A allele and vibration exposure, bending and twisting [37]. A small Finnish study did not show an association between the 5A allele and degeneration. These results may be due to the small sample size or because they focused on a distinct subgroup of degeneration causing spinal stenosis [74].
Parkinson protein 2, E3 ubiquitin protein ligase (PARK2)
In a recent publication using a large meta-analysis of five Northern European cohorts (n=4,683) a variant in the PARK2 gene on chromosome 6 was found to be significantly associated with disc degeneration (p<5×10−8) [133]. Single nucleotide polymorphism rs926849 lies within an intron in the PARK2 gene; it encodes a base change from T to C. Hypermethylation at one of the CpG sites on the PARK2 promoter was associated with MRI-defined lumbar disc degeneration. The PARK2 gene encodes a protein called parkin, which is a component of the multiprotein E3 ubiquitin ligase complex, which tags unwanted or damaged proteins with ubiquitin ultimately for proteosomal degradation. Mutations in PARK2 are known in autosomal recessive juvenile Parksinson’s disease, Alzheimer’s disease, diabetes mellitus and several solid tumors [134]. This paper is the first to suggest the possibility that methylation of the PARK2 promoter may influence degeneration of the intervertebral disc. Further functional studies of the disc are needed to determine the relationship between this gene and degeneration.
Proteosome subunit β type 9 (P) SMB9
The recent study by Williams et al. [133] also identified two single nucleotide polymorphisms (rs2187689 and rs7767277) in strong linkage disequilibrium with PSMB9, which is located in the class II region of the major histocompatibility complex (MHC) and in association with disc degeneration (p<5×10−8). The study performed a meta-analysis of five Northern European cohorts (n=4,683). The protein encoded by PSMB9 is a proteosome that functions to degrade unneeded or damaged proteins by cleaving peptides in an ATP/ubiquitin-dependent process in a non-lysosomal pathway [133]. As this is a novel association between this gene and disc degeneration, the mechanism of this protein as it relates to degeneration still remains to be determined.
Anti-Catabolic
Tissue inhibitors of metalloproteinases (TIMP)
TIMP is a group of protease inhibitors that act on specific MMPs and A Disintegrin and Metalloproteinase with Thrombospondin Motifs (ADAMTS), a family of peptidases [135]. The balance between these catabolic and anti-catabolic enzymes is a delicate balance and a slight increase in catabolic or decrease in anti-catabolic enzymes could tip the balance in favor of degeneration. Valdes et al. [51] looked at radiographs of 750 women and found a single nucleotide polymorphism in TIMP1, C124T, which was significantly associated with a change in osteophyte grade (p=0.006), but not with a change in the Kellgren-Lawrence summary grade or disc narrowing. This has been the only study thus far to look at TIMP polymorphisms in association with degenerative disc disease and more MRI-defined phenotypes in others populations need to be analyzed.
Inflammatory
Interleukins
It is well known that the immune system and inflammation cascades play important roles in disc degeneration [136, 137] and herniation [138]. These important factors may have a strong genetic component; polymorphisms in a number if interleukins have been associated with radiculopathy [123], osteoarthritis [139, 140], rheumatoid arthritis [141], lower back pain [142], disc degeneration, herniation and sciatica [18, 138, 143]. Interleukins are cytokines that respond to stress and cause a range of systemic responses [144]. Specifically in the intervertebral disc, pro-inflammatory interleukins stimulate the production of MMP and inhibit TIMP, thereby inducing a catabolic environment [145, 146]. Studies have found significantly increased levels of cytokines in degenerated, herniated and aged discs including IL1 (α, β, receptor), -2, -4, -6, -10, and -12, TNFα, and prostaglandin-2 [18, 136, 147–149]. Although only IL1 and IL6 have known polymorphisms associated with degeneration, many of the others are studied as targets for possible therapeutic interventions.
Interleukin-1 (IL1)
IL1 has an important role in the degradation of matrix proteins [143, 144, 150–152]. While TNFα and IL1 are both pro-inflammatory cytokines, compared to TNFα, IL1 has a higher level of expression in normal discs, suggesting its importance in regulating normal function as well as a greater increased expression in degenerated discs, suggesting its importance in the degeneration process [153]. In normal discs, IL1 has a negative feedback mechanism involving IL1 receptor antagonists, which help regulate the level of IL1. In degeneration, the negative feedback cycle is not significant because IL1 expression increases while IL1-receptor antagonist does not [18, 154]. This results in a loss of equilibrium between IL1 and antagonists and an unchecked increased IL1 activity. Enhanced IL1 can result in increased expression of IL6 [155] and prostaglandin-2 [143] as well as matrix degrading enzymes including MMPs and ADAMTS [18, 154, 156, 157]. Consequently, IL1 leads to decreased levels of matrix components including aggrecan, COL1 and COL2 [18, 139, 158, 159]. IL1 also causes a switch in synthesis from type II to type I collagen which may contribute to decreased functionality of the disc as it degenerates [160]. As discs age, sensitivity to IL1 also increases, because of changes in the number or ligand-binding affinities of receptors, which could contribute to degeneration given its close relationship with age [150].
Eskola et al. [161] showed that young Danish girls, aged 12–14, with one or more risk alleles (T allele) in the IL1α single nucleotide polymorphism (rs1800587), had an increased risk of disc degeneration (OR=2.85). Interestingly, there appears to be a gender effect as it this polymorphism was not associated with an increased risk for boys [162, 163]. However, the cohort size of the study was relatively small, and may account for this difference. Functionally, this polymorphism is associated with an increased transcription of IL1a in the TT genotype compared to CC, which could explain the increased risk for degeneration [164]. Studies by Virtanen et al. [165, 166] and Solovieva et al. [166] on Finnish train engineers, paper mill workers, machine drivers, carpenters and office workers also confirmed the association between the IL1a T allele and an increased risk of degeneration. Individuals with the TT genotype had a 2-fold increased risk of bulges compared to the CC genotype [166]. Both studies also showed gene-environment interactions with a negative effect of vibration and physical workload on disc structure [165, 166].
Another polymorphism on IL1, this time in IL1β (rs1143634) was studied in relation to disc degeneration. In an interesting gene-gene effect, the genetic interaction between the polymorphism in IL1β and the Trp3 allele of COL9A3 was shown to determine the risk of disc degeneration [25]. This association was discussed in the Type IX Collagen section. However, in a study by Videman et al. [42] on 588 Finnish men, this IL1β polymorphism was not found to be associated with MRI-defined degenerative phenotypes while another IL1α polymorphism (rs2071375) was found to be associated with changes in disc signal intensity.
Interleukin-6 (IL6)
IL6 is a potent pro-inflammatory cytokine that can also act as an anti-inflammatory cytokine; IL6 may play a protective role in balancing the catabolic effects of IL1β and TNFα [146]. There are many IL6 polymorphisms that have been associated with disc degeneration, specifically rs1800797, rs1800796, rs1800795 and rs13006435 that exist as a haplotype. A Finnish study that could not find any association between IL1 polymorphisms and disc disease found that the risk allele (A allele) in an IL6 single nucleotide polymorphism in exon 5 (rs13006435) was significantly association with degenerated patients compared to controls [167]. In another study analyzing the other three single nucleotide polymorphisms of IL6 (rs1800797, rs1800796, rs1800795), the GCG haplotype was associated with early disc degeneration in Danish girls (OR 6.46), but not boys [161]. The gender differences may be attributed to the small sample size. The Finnish study found that the GGGA haplotype from all four IL6 single nucleotide polymorphisms was significantly associated with degeneration and the presence of at least one GGGA allele increased the risk of degeneration by more than a factor of five [167]. In contrast, the young Danish study found the GGGA haplotype in less than 2% of the population and did not see any association with early disc degeneration [161]. The discrepancy between the Danish and Finnish populations may be related to the definition of degeneration, sample size, or, as in the case of COL9A3 and other genes, the fact that risk allele frequencies may differ significantly between populations, affecting certain populations in different ways.
Cyclooxygenase-2 (COX2)
COX2 is a gene that codes for the rate-limiting enzyme in the synthesis of prostanoids, including prostaglandins (PGE-2), prostacyclin and thromboxane [51, 168]. PGE-2 and COX2 are upregulated in herniated and degenerated discs and thus may contribute to pain sensation or mediate inflammation [168]. In a study by Valdes et al. [51] on a cohort of 750 middle-aged women, a strong association was shown between a COX2 single nucleotide polymorphism (rs5277) and the Kellgren-Lawrence degeneration grade (p<0.00002). The study used radiographs rather than MRIs, but it seems to suggest some genetic predisposition for degeneration. Unfortunately there are no large studies that have attempted to replicate this result in other populations. This may be an important gene to study in understanding why some people have painful degenerated or herniated discs and others do not.
Discussion
Studying the genetic factors implicated in disc degeneration remains challenging because of the vast number of different genes that contribute to the progression of this complex disease, which does not share a common definition or pathogenesis. Separating genetic and environmental factors requires well-defined samples in similar environments, which are often hard to isolate or characterize. There are also differences in the frequency of the genetic associations with degeneration found in various parts of the world, making replication and validation of genetic risk factors difficult across populations but necessary [20]. The lack of a clearly defined disc degeneration phenotype further confuses the genetic analysis due to the variability of phenotypes used in multiple studies on the same subject [20].
Age is also a confounding variable and other than the use of age-matched controls and age-adjusted scores, research has not sufficiently distinguished degeneration that is part of the structural alterations found with aging from degenerative disc disease or age-accelerated degeneration [12]. These two processes may have some pathophysiology in common, but likely involve different permutations that make the two outcomes unique. Most studies mentioned in this review have not distinguished between these two processes further confusing the two. Song et al. [114] made this distinction by dividing the population into two groups, those with scores at or below the median degeneration score for a given age and those above it. Age-accelerated degeneration was defined in the latter group. This sliding-window method is an important way to adjust degeneration scores for age effects and allowed this study to identify a polymorphism in asporin as a risk factor for accelerated disc degeneration [114].
The association between pain and degeneration is not clearly defined in these studies since most of them focus on objectively defined MRI phenotypes of degeneration rather than pain which is more subjectively defined. This may limit the application of these findings in preventing or alleviating chronic or acute pain that can accompany some degeneration because the genes involved in degeneration that causes pain may be different from the genes that affect asymptomatic degeneration.
The discussion of the genes analyzed above were chosen from references narrowed down by co-authors with the goal of being inclusive and focusing on chronic disc degeneration, however, our methodology could introduce more bias than a systematic review. Eskola et al. [20], recently published an extensive systematic review on the subject and also introduced protein-protein interaction analyses as a valuable method to help understand the complex genetic interactions in disc degeneration. This important paper provides perhaps the most ‘credible’ list of genetic markers and their association with lumbar disc degeneration. Our review prioritized a thorough discussion on a larger number of genes rather than the review by Eskola et al. which had a longer list of genes but only discussed a few. Our review also focused on the potential functional involvement of these genes and their polymorphisms in the pathogenesis of disc degeneration. Like our review, Eskola et al., also concluded that the phenotypic definition of disc degeneration is highly variable between studies and a large international group is necessary to help standardize these studies in an effort to isolate true genetic associations from the large amount of data that exists.
Of the collection of genes described, polymorphisms in VDR, ACAN, COL9, ASPN, MMP3, IL1 and IL6 appear to be the most promising for broad association with disc degeneration because they have been validated in more than one ethnic population and have potential functional roles that support individuals’ susceptibility with these risk alleles to disc degeneration. Each gene plays an essential role in the development and maintenance of a healthy disc matrix and each polymorphism can cause a loss of structural integrity, loss of hydrostatic pressure or an enhanced pro-inflammatory state that can lead to further catabolism and potentially painful conditions (Figure 3). The degeneration phenotype that included sciatica and herniation in the systematic review by Eskola et al., described moderate level of evidence for associations with ASPN, COL11A1, Sickle tail (SKT), THBS2 and MMP9 [20]. While ASPN is the only overlapping gene in these lists, there is overlap in the structural and catabolic categories. Advances in genetic studies and increased precision in the definition of the complex processes of disc degeneration will allow more targeted diagnosis and treatment strategies. Further analyzing the rest of the genes across various populations with robust methodologies may help to include or exclude these genes from the pool of genetic modifiers.
Conclusion
There are known associations between several genetic polymorphisms and disc degeneration. Specific physiological conditions explain why each polymorphism contributes to the degeneration process. Some associations are robust; others require follow-up studies in new populations with large sample sizes. Genetic studies are crucial for understanding the mechanism of the degeneration. A thorough understanding of the contribution of variation within the genome is also valuable. This genetic information could eventually be used as a predictive model for determining a patient’s risk for disc degeneration.
Acknowledgments
Funded by NIH grants 5R01 AR051146 and 1R01AR057397.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Adams MA, Roughley PJ. What is intervertebral disc degeneration, and what causes it? Spine (Phila Pa 1976) 2006;31(18):2151–61. doi: 10.1097/01.brs.0000231761.73859.2c. [DOI] [PubMed] [Google Scholar]
- 2.Cheung KM, Karppinen J, Chan D, et al. Prevalence and pattern of lumbar magnetic resonance imaging changes in a population study of one thousand forty-three individuals. Spine (Phila Pa 1976) 2009;34(9):934–40. doi: 10.1097/BRS.0b013e3181a01b3f. [DOI] [PubMed] [Google Scholar]
- 3.Battie MC, Videman T, Levalahti E, Gill K, Kaprio J. Heritability of low back pain and the role of disc degeneration. Pain. 2007;131(3):272–80. doi: 10.1016/j.pain.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 4.Luoma K, Riihimaki H, Luukkonen R, Raininko R, Viikari-Juntura E, Lamminen A. Low back pain in relation to lumbar disc degeneration. Spine (Phila Pa 1976) 2000;25(4):487–92. doi: 10.1097/00007632-200002150-00016. [DOI] [PubMed] [Google Scholar]
- 5.Boden SD, Davis DO, Dina TS, Patronas NJ, Wiesel SW. Abnormal magnetic-resonance scans of the lumbar spine in asymptomatic subjects. A prospective investigation. J Bone Joint Surg Am. 1990;72(3):403–8. [PubMed] [Google Scholar]
- 6.Jensen MC, Brant-Zawadzki MN, Obuchowski N, Modic MT, Malkasian D, Ross JS. Magnetic resonance imaging of the lumbar spine in people without back pain. N Engl J Med. 1994;331(2):69–73. doi: 10.1056/NEJM199407143310201. [DOI] [PubMed] [Google Scholar]
- 7.Buckwalter JA. Aging and degeneration of the human intervertebral disc. Spine (Phila Pa 1976) 1995;20(11):1307–14. doi: 10.1097/00007632-199506000-00022. [DOI] [PubMed] [Google Scholar]
- 8.Sobajima S, Shimer A, Chadderdon R, et al. Quantitative analysis of gene expression in a rabbit model of intervertebral disc degeneration by real-time polymerase chain reaction. The Spine Journal. 2005;5(1):14–23. doi: 10.1016/j.spinee.2004.05.251. [DOI] [PubMed] [Google Scholar]
- 9.An HS, Anderson PA, Haughton VM, et al. Introduction: disc degeneration: summary. Spine (Phila Pa 1976) 2004;29(23):2677–8. doi: 10.1097/01.brs.0000147573.88916.c6. [DOI] [PubMed] [Google Scholar]
- 10.Andersson GB. Epidemiological features of chronic low-back pain. Lancet. 1999;354(9178):581–5. doi: 10.1016/S0140-6736(99)01312-4. [DOI] [PubMed] [Google Scholar]
- 11.Sambrook PN, MacGregor AJ, Spector TD. Genetic influences on cervical and lumbar disc degeneration: a magnetic resonance imaging study in twins. Arthritis Rheum. 1999;42(2):366–72. doi: 10.1002/1529-0131(199902)42:2<366::AID-ANR20>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 12.Antoniou J, Steffen T, Nelson F, et al. The human lumbar intervertebral disc: evidence for changes in the biosynthesis and denaturation of the extracellular matrix with growth, maturation, ageing, and degeneration. J Clin Invest. 1996;98(4):996–1003. doi: 10.1172/JCI118884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feng H, Danfelter M, Stromqvist B, Heinegard D. Extracellular matrix in disc degeneration. J Bone Joint Surg Am. 2006;88 (Suppl 2):25–9. doi: 10.2106/JBJS.E.01341. [DOI] [PubMed] [Google Scholar]
- 14.Boos N, Weissbach S, Rohrbach H, Weiler C, Spratt KF, Nerlich AG. Classification of age-related changes in lumbar intervertebral discs: 2002 Volvo Award in basic science. Spine (Phila Pa 1976) 2002;27(23):2631–44. doi: 10.1097/00007632-200212010-00002. [DOI] [PubMed] [Google Scholar]
- 15.Nerlich AG, Schleicher ED, Boos N. 1997 Volvo Award winner in basic science studies. Immunohistologic markers for age-related changes of human lumbar intervertebral discs. Spine (Phila Pa 1976) 1997;22(24):2781–95. doi: 10.1097/00007632-199712150-00001. [DOI] [PubMed] [Google Scholar]
- 16.Cs-Szabo G, Ragasa-San Juan D, Turumella V, Masuda K, Thonar EJ, An HS. Changes in mRNA and protein levels of proteoglycans of the anulus fibrosus and nucleus pulposus during intervertebral disc degeneration. Spine (Phila Pa 1976) 2002;27(20):2212–9. doi: 10.1097/00007632-200210150-00006. [DOI] [PubMed] [Google Scholar]
- 17.Park JY, Kuh SU, Park HS, Kim KS. Comparative Expression of Matrix-associated Genes and Inflammatory Cytokines-associated Genes According to Disc Degeneration: Analysis of Living Human Nucleus Pulposus. J Spinal Disord Tech. 2010 doi: 10.1097/BSD.0b013e3181fee4df. [DOI] [PubMed] [Google Scholar]
- 18.Le Maitre CL, Freemont AJ, Hoyland JA. The role of interleukin-1 in the pathogenesis of human intervertebral disc degeneration. Arthritis Res Ther. 2005;7(4):R732–45. doi: 10.1186/ar1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Videman T, Gibbons LE, Battie MC. Age- and pathology-specific measures of disc degeneration. Spine (Phila Pa 1976) 2008;33(25):2781–8. doi: 10.1097/BRS.0b013e31817e1d11. [DOI] [PubMed] [Google Scholar]
- 20.Eskola PJ, Lemmela S, Kjaer P, et al. Genetic association studies in lumbar disc degeneration: a systematic review. PLoS One. 2012;7(11):e49995. doi: 10.1371/journal.pone.0049995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Urano T, Narusawa K, Shiraki M, et al. Single-nucleotide polymorphism in the hyaluronan and proteoglycan link protein 1 (HAPLN1) gene is associated with spinal osteophyte formation and disc degeneration in Japanese women. Eur Spine J. 2011;20(4):572–7. doi: 10.1007/s00586-010-1598-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adams MA, Freeman BJ, Morrison HP, Nelson IW, Dolan P. Mechanical initiation of intervertebral disc degeneration. Spine (Phila Pa 1976) 2000;25(13):1625–36. doi: 10.1097/00007632-200007010-00005. [DOI] [PubMed] [Google Scholar]
- 23.Videman T, Leppavuori J, Kaprio J, et al. Intragenic polymorphisms of the vitamin D receptor gene associated with intervertebral disc degeneration. Spine (Phila Pa 1976) 1998;23(23):2477–85. doi: 10.1097/00007632-199812010-00002. [DOI] [PubMed] [Google Scholar]
- 24.Williams FM, Sambrook PN. Neck and back pain and intervertebral disc degeneration: role of occupational factors. Best Pract Res Clin Rheumatol. 2011;25(1):69–79. doi: 10.1016/j.berh.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 25.Solovieva S, Lohiniva J, Leino-Arjas P, et al. Intervertebral disc degeneration in relation to the COL9A3 and the IL-1ss gene polymorphisms. Eur Spine J. 2006;15(5):613–9. doi: 10.1007/s00586-005-0988-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boden SD, McCowin PR, Davis DO, Dina TS, Mark AS, Wiesel S. Abnormal magnetic-resonance scans of the cervical spine in asymptomatic subjects. A prospective investigation. J Bone Joint Surg Am. 1990;72(8):1178–84. [PubMed] [Google Scholar]
- 27.Matsumoto M, Fujimura Y, Suzuki N, et al. MRI of cervical intervertebral discs in asymptomatic subjects. J Bone Joint Surg Br. 1998;80(1):19–24. doi: 10.1302/0301-620x.80b1.7929. [DOI] [PubMed] [Google Scholar]
- 28.Chan D, Song Y, Sham P, Cheung KM. Genetics of disc degeneration. Eur Spine J. 2006;15 (Suppl 3):S317–25. doi: 10.1007/s00586-006-0171-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalichman L, Hunter DJ. The genetics of intervertebral disc degeneration. Associated genes. Joint Bone Spine. 2008;75(4):388–96. doi: 10.1016/j.jbspin.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 30.Kalb S, Martirosyan NL, Kalani MY, Broc GG, Theodore N. Genetics of the degenerated intervertebral disc. World Neurosurg. 2012;77(3–4):491–501. doi: 10.1016/j.wneu.2011.07.014. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Y, Sun Z, Liu J, Guo X. Advances in susceptibility genetics of intervertebral degenerative disc disease. Int J Biol Sci. 2008;4(5):283–90. doi: 10.7150/ijbs.4.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luoma K, Riihimaki H, Raininko R, Luukkonen R, Lamminen A, Viikari-Juntura E. Lumbar disc degeneration in relation to occupation. Scand J Work Environ Health. 1998;24(5):358–66. doi: 10.5271/sjweh.356. [DOI] [PubMed] [Google Scholar]
- 33.Videman T, Sarna S, Battie MC, et al. The long-term effects of physical loading and exercise lifestyles on back-related symptoms, disability, and spinal pathology among men. Spine (Phila Pa 1976) 1995;20(6):699–709. doi: 10.1097/00007632-199503150-00011. [DOI] [PubMed] [Google Scholar]
- 34.Cinotti G, Della Rocca C, Romeo S, Vittur F, Toffanin R, Trasimeni G. Degenerative changes of porcine intervertebral disc induced by vertebral endplate injuries. Spine (Phila Pa 1976) 2005;30(2):174–80. doi: 10.1097/01.brs.0000150530.48957.76. [DOI] [PubMed] [Google Scholar]
- 35.Battie MC, Videman T, Gill K, et al. 1991 Volvo Award in clinical sciences. Smoking and lumbar intervertebral disc degeneration: an MRI study of identical twins. Spine (Phila Pa 1976) 1991;16(9):1015–21. [PubMed] [Google Scholar]
- 36.Battie MC, Videman T. Lumbar disc degeneration: epidemiology and genetics. J Bone Joint Surg Am. 2006;88 (Suppl 2):3–9. doi: 10.2106/JBJS.E.01313. [DOI] [PubMed] [Google Scholar]
- 37.Yuan HY, Tang Y, Liang YX, et al. Matrix metalloproteinase-3 and vitamin d receptor genetic polymorphisms, and their interactions with occupational exposure in lumbar disc degeneration. J Occup Health. 2010;52(1):23–30. doi: 10.1539/joh.l8149. [DOI] [PubMed] [Google Scholar]
- 38.Simmons ED, Jr, Guntupalli M, Kowalski JM, Braun F, Seidel T. Familial predisposition for degenerative disc disease. A case-control study. Spine (Phila Pa 1976) 1996;21(13):1527–9. doi: 10.1097/00007632-199607010-00007. [DOI] [PubMed] [Google Scholar]
- 39.Battie MC, Videman T, Gibbons LE, Fisher LD, Manninen H, Gill K. 1995 Volvo Award in clinical sciences. Determinants of lumbar disc degeneration. A study relating lifetime exposures and magnetic resonance imaging findings in identical twins. Spine (Phila Pa 1976) 1995;20(24):2601–12. [PubMed] [Google Scholar]
- 40.Battie MC, Videman T, Kaprio J, et al. The Twin Spine Study: contributions to a changing view of disc degeneration. Spine J. 2009;9(1):47–59. doi: 10.1016/j.spinee.2008.11.011. [DOI] [PubMed] [Google Scholar]
- 41.Solovieva S, Lohiniva J, Leino-Arjas P, et al. COL9A3 gene polymorphism and obesity in intervertebral disc degeneration of the lumbar spine: evidence of gene-environment interaction. Spine (Phila Pa 1976) 2002;27(23):2691–6. doi: 10.1097/00007632-200212010-00008. [DOI] [PubMed] [Google Scholar]
- 42.Videman T, Saarela J, Kaprio J, et al. Associations of 25 structural, degradative, and inflammatory candidate genes with lumbar disc desiccation, bulging, and height narrowing. Arthritis Rheum. 2009;60(2):470–81. doi: 10.1002/art.24268. [DOI] [PubMed] [Google Scholar]
- 43.Jones G, White C, Sambrook P, Eisman J. Allelic variation in the vitamin D receptor, lifestyle factors and lumbar spinal degenerative disease. Ann Rheum Dis. 1998;57(2):94–9. doi: 10.1136/ard.57.2.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uitterlinden AG, Fang Y, Bergink AP, van Meurs JB, van Leeuwen HP, Pols HA. The role of vitamin D receptor gene polymorphisms in bone biology. Mol Cell Endocrinol. 2002;197(1–2):15–21. doi: 10.1016/s0303-7207(02)00274-5. [DOI] [PubMed] [Google Scholar]
- 45.Wood RJ, Fleet JC. The genetics of osteoporosis: vitamin D receptor polymorphisms. Annu Rev Nutr. 1998;18:233–58. doi: 10.1146/annurev.nutr.18.1.233. [DOI] [PubMed] [Google Scholar]
- 46.Arai H, Miyamoto K, Taketani Y, et al. A vitamin D receptor gene polymorphism in the translation initiation codon: effect on protein activity and relation to bone mineral density in Japanese women. J Bone Miner Res. 1997;12(6):915–21. doi: 10.1359/jbmr.1997.12.6.915. [DOI] [PubMed] [Google Scholar]
- 47.Eser B, Cora T, Eser O, et al. Association of the polymorphisms of vitamin D receptor and aggrecan genes with degenerative disc disease. Genet Test Mol Biomarkers. 2010;14(3):313–7. doi: 10.1089/gtmb.2009.0202. [DOI] [PubMed] [Google Scholar]
- 48.Beck L, Silve C. Molecular aspects of renal tubular handling and regulation of inorganic sulfate. Kidney Int. 2001;59(3):835–45. doi: 10.1046/j.1523-1755.2001.059003835.x. [DOI] [PubMed] [Google Scholar]
- 49.Corvol MT, Dumontier MF, Tsagris L, Lang F, Bourguignon J. Cartilage and vitamin D in vitro (author’s transl) Ann Endocrinol (Paris) 1981;42(4–5):482–7. [PubMed] [Google Scholar]
- 50.Cheung KM, Chan D, Karppinen J, et al. Association of the Taq I allele in vitamin D receptor with degenerative disc disease and disc bulge in a Chinese population. Spine (Phila Pa 1976) 2006;31(10):1143–8. doi: 10.1097/01.brs.0000216530.41838.d3. [DOI] [PubMed] [Google Scholar]
- 51.Valdes AM, Hassett G, Hart DJ, Spector TD. Radiographic progression of lumbar spine disc degeneration is influenced by variation at inflammatory genes: a candidate SNP association study in the Chingford cohort. Spine (Phila Pa 1976) 2005;30(21):2445–51. doi: 10.1097/01.brs.0000184369.79744.a5. [DOI] [PubMed] [Google Scholar]
- 52.Videman T, Gibbons LE, Battie MC, et al. The relative roles of intragenic polymorphisms of the vitamin d receptor gene in lumbar spine degeneration and bone density. Spine (Phila Pa 1976) 2001;26(3):E7–E12. doi: 10.1097/00007632-200102010-00003. [DOI] [PubMed] [Google Scholar]
- 53.Kawaguchi Y, Kanamori M, Ishihara H, Ohmori K, Matsui H, Kimura T. The association of lumbar disc disease with vitamin-D receptor gene polymorphism. J Bone Joint Surg Am. 2002;84-A(11):2022–8. doi: 10.2106/00004623-200211000-00018. [DOI] [PubMed] [Google Scholar]
- 54.Sztrolovics R, Alini M, Roughley PJ, Mort JS. Aggrecan degradation in human intervertebral disc and articular cartilage. Biochem J. 1997;326 (Pt 1):235–41. doi: 10.1042/bj3260235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lipson SJ, Muir H. Experimental intervertebral disc degeneration: morphologic and proteoglycan changes over time. Arthritis Rheum. 1981;24(1):12–21. doi: 10.1002/art.1780240103. [DOI] [PubMed] [Google Scholar]
- 56.Urban JP, McMullin JF. Swelling pressure of the lumbar intervertebral discs: influence of age, spinal level, composition, and degeneration. Spine (Phila Pa 1976) 1988;13(2):179–87. doi: 10.1097/00007632-198802000-00009. [DOI] [PubMed] [Google Scholar]
- 57.Roughley PJ, Alini M, Antoniou J. The role of proteoglycans in aging, degeneration and repair of the intervertebral disc. Biochem Soc Trans. 2002;30(Pt 6):869–74. doi: 10.1042/bst0300869. [DOI] [PubMed] [Google Scholar]
- 58.Inkinen RI, Lammi MJ, Lehmonen S, Puustjarvi K, Kaapa E, Tammi MI. Relative increase of biglycan and decorin and altered chondroitin sulfate epitopes in the degenerating human intervertebral disc. J Rheumatol. 1998;25(3):506–14. [PubMed] [Google Scholar]
- 59.Roughley P, Martens D, Rantakokko J, Alini M, Mwale F, Antoniou J. The involvement of aggrecan polymorphism in degeneration of human intervertebral disc and articular cartilage. Eur Cell Mater. 2006;11:1–7. discussion. [PubMed] [Google Scholar]
- 60.Roughley PJ, Melching LI, Heathfield TF, Pearce RH, Mort JS. The structure and degradation of aggrecan in human intervertebral disc. Eur Spine J. 2006;15 (Suppl 3):S326–32. doi: 10.1007/s00586-006-0127-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Doege KJ, Coulter SN, Meek LM, Maslen K, Wood JG. A human-specific polymorphism in the coding region of the aggrecan gene. Variable number of tandem repeats produce a range of core protein sizes in the general population. J Biol Chem. 1997;272(21):13974–9. doi: 10.1074/jbc.272.21.13974. [DOI] [PubMed] [Google Scholar]
- 62.Kawaguchi Y, Osada R, Kanamori M, et al. Association between an aggrecan gene polymorphism and lumbar disc degeneration. Spine (Phila Pa 1976) 1999;24(23):2456–60. doi: 10.1097/00007632-199912010-00006. [DOI] [PubMed] [Google Scholar]
- 63.Kim NK, Shin DA, Han IB, Yoo EH, Kim SH, Chung SS. The association of aggrecan gene polymorphism with the risk of intervertebral disc degeneration. Acta Neurochir (Wien) 2011;153(1):129–33. doi: 10.1007/s00701-010-0831-2. [DOI] [PubMed] [Google Scholar]
- 64.Solovieva S, Noponen N, Mannikko M, et al. Association between the aggrecan gene variable number of tandem repeats polymorphism and intervertebral disc degeneration. Spine (Phila Pa 1976) 2007;32(16):1700–5. doi: 10.1097/BRS.0b013e3180b9ed51. [DOI] [PubMed] [Google Scholar]
- 65.Sarver JJ, Elliott DM. Altered disc mechanics in mice genetically engineered for reduced type I collagen. Spine (Phila Pa 1976) 2004;29(10):1094–8. doi: 10.1097/00007632-200405150-00009. [DOI] [PubMed] [Google Scholar]
- 66.Grant SF, Reid DM, Blake G, Herd R, Fogelman I, Ralston SH. Reduced bone density and osteoporosis associated with a polymorphic Sp1 binding site in the collagen type I alpha 1 gene. Nat Genet. 1996;14(2):203–5. doi: 10.1038/ng1096-203. [DOI] [PubMed] [Google Scholar]
- 67.Keen RW, Woodford-Richens KL, Grant SF, Ralston SH, Lanchbury JS, Spector TD. Association of polymorphism at the type I collagen (COL1A1) locus with reduced bone mineral density, increased fracture risk, and increased collagen turnover. Arthritis Rheum. 1999;42(2):285–90. doi: 10.1002/1529-0131(199902)42:2<285::AID-ANR10>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 68.Pluijm SM, van Essen HW, Bravenboer N, et al. Collagen type I alpha1 Sp1 polymorphism, osteoporosis, and intervertebral disc degeneration in older men and women. Ann Rheum Dis. 2004;63(1):71–7. doi: 10.1136/ard.2002.002287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tilkeridis C. Association of a COL1A1 polymorphism with lumbar disc disease in young military recruits. Journal of Medical Genetics. 2005;42(7):e44–e. doi: 10.1136/jmg.2005.033225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Paassilta P, Pihlajamaa T, Annunen S, et al. Complete sequence of the 23-kilobase human COL9A3 gene. Detection of Gly-X-Y triplet deletions that represent neutral variants. J Biol Chem. 1999;274(32):22469–75. doi: 10.1074/jbc.274.32.22469. [DOI] [PubMed] [Google Scholar]
- 71.Eyre DR, Matsui Y, Wu JJ. Collagen polymorphisms of the intervertebral disc. Biochem Soc Trans. 2002;30(Pt 6):844–8. doi: 10.1042/bst0300844. [DOI] [PubMed] [Google Scholar]
- 72.Kimura T, Nakata K, Tsumaki N, et al. Progressive degeneration of articular cartilage and intervertebral discs. An experimental study in transgenic mice bearing a type IX collagen mutation. Int Orthop. 1996;20(3):177–81. doi: 10.1007/s002640050058. [DOI] [PubMed] [Google Scholar]
- 73.Boyd LM, Richardson WJ, Allen KD, et al. Early-onset degeneration of the intervertebral disc and vertebral end plate in mice deficient in type IX collagen. Arthritis & Rheumatism. 2008;58(1):164–71. doi: 10.1002/art.23231. [DOI] [PubMed] [Google Scholar]
- 74.Noponen-Hietala N, Kyllonen E, Mannikko M, et al. Sequence variations in the collagen IX and XI genes are associated with degenerative lumbar spinal stenosis. Ann Rheum Dis. 2003;62(12):1208–14. doi: 10.1136/ard.2003.008334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Matsui Y, Mirza SK, Wu JJ, et al. The association of lumbar spondylolisthesis with collagen IX tryptophan alleles. J Bone Joint Surg Br. 2004;86(7):1021–6. doi: 10.1302/0301-620x.86b7.14994. [DOI] [PubMed] [Google Scholar]
- 76.Annunen S, Paassilta P, Lohiniva J, et al. An allele of COL9A2 associated with intervertebral disc disease. Science. 1999;285(5426):409–12. doi: 10.1126/science.285.5426.409. [DOI] [PubMed] [Google Scholar]
- 77.Jim JJ, Noponen-Hietala N, Cheung KM, et al. The TRP2 allele of COL9A2 is an age-dependent risk factor for the development and severity of intervertebral disc degeneration. Spine (Phila Pa 1976) 2005;30(24):2735–42. doi: 10.1097/01.brs.0000190828.85331.ef. [DOI] [PubMed] [Google Scholar]
- 78.Higashino K, Matsui Y, Yagi S, et al. The alpha2 type IX collagen tryptophan polymorphism is associated with the severity of disc degeneration in younger patients with herniated nucleus pulposus of the lumbar spine. Int Orthop. 2007;31(1):107–11. doi: 10.1007/s00264-006-0117-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kales SN, Linos A, Chatzis C, et al. The role of collagen IX tryptophan polymorphisms in symptomatic intervertebral disc disease in Southern European patients. Spine (Phila Pa 1976) 2004;29(11):1266–70. doi: 10.1097/00007632-200406010-00017. [DOI] [PubMed] [Google Scholar]
- 80.Wrocklage C, Wassmann H, Paulus W. COL9A2 allelotypes in intervertebral disc disease. Biochem Biophys Res Commun. 2000;279(2):398–400. doi: 10.1006/bbrc.2000.3967. [DOI] [PubMed] [Google Scholar]
- 81.Seki S, Kawaguchi Y, Mori M, et al. Association study of COL9A2 with lumbar disc disease in the Japanese population. Journal of Human Genetics. 2006;51(12):1063–7. doi: 10.1007/s10038-006-0062-9. [DOI] [PubMed] [Google Scholar]
- 82.Paassilta P, Lohiniva J, Goring HH, et al. Identification of a novel common genetic risk factor for lumbar disk disease. JAMA. 2001;285(14):1843–9. doi: 10.1001/jama.285.14.1843. [DOI] [PubMed] [Google Scholar]
- 83.Karppinen J, Paakko E, Paassilta P, et al. Radiologic phenotypes in lumbar MR imaging for a gene defect in the COL9A3 gene of type IX collagen. Radiology. 2003;227(1):143–8. doi: 10.1148/radiol.2271011821. [DOI] [PubMed] [Google Scholar]
- 84.Marini JC. Genetic risk factors for lumbar disk disease. JAMA. 2001;285(14):1886–8. doi: 10.1001/jama.285.14.1886. [DOI] [PubMed] [Google Scholar]
- 85.Aladin DM, Cheung KM, Chan D, et al. Expression of the Trp2 allele of COL9A2 is associated with alterations in the mechanical properties of human intervertebral discs. Spine (Phila Pa 1976) 2007;32(25):2820–6. doi: 10.1097/BRS.0b013e31815b75c5. [DOI] [PubMed] [Google Scholar]
- 86.Tegeder I, Lotsch J. Current evidence for a modulation of low back pain by human genetic variants. J Cell Mol Med. 2009;13(8B):1605–19. doi: 10.1111/j.1582-4934.2009.00703.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Oegema TR., Jr Biochemistry of the intervertebral disc. Clin Sports Med. 1993;12(3):419–39. [PubMed] [Google Scholar]
- 88.Maeda S, Ishidou Y, Koga H, et al. Functional impact of human collagen alpha2(XI) gene polymorphism in pathogenesis of ossification of the posterior longitudinal ligament of the spine. J Bone Miner Res. 2001;16(5):948–57. doi: 10.1359/jbmr.2001.16.5.948. [DOI] [PubMed] [Google Scholar]
- 89.Trelstad RL, Amenta PS, Foran DJ, Smilow PC. The role for regional autopsy centers in the evaluation of covered deaths. Survey of opinions of US and Canadian chairs of pathology and major health insurers in the United States. Arch Pathol Lab Med. 1996;120(8):753–8. [PubMed] [Google Scholar]
- 90.Mio F, Chiba K, Hirose Y, et al. A functional polymorphism in COL11A1, which encodes the alpha 1 chain of type XI collagen, is associated with susceptibility to lumbar disc herniation. Am J Hum Genet. 2007;81(6):1271–7. doi: 10.1086/522377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Larsen M, Wei C, Yamada KM. Cell and fibronectin dynamics during branching morphogenesis. J Cell Sci. 2006;119(Pt 16):3376–84. doi: 10.1242/jcs.03079. [DOI] [PubMed] [Google Scholar]
- 92.Anderson DG, Markova D, Adams SL, Pacifici M, An HS, Zhang Y. Fibronectin splicing variants in human intervertebral disc and association with disc degeneration. Spine (Phila Pa 1976) 2010;35(17):1581–8. doi: 10.1097/BRS.0b013e3181c6ef1a. [DOI] [PubMed] [Google Scholar]
- 93.Hynes R. Molecular biology of fibronectin. Annu Rev Cell Biol. 1985;1:67–90. doi: 10.1146/annurev.cb.01.110185.000435. [DOI] [PubMed] [Google Scholar]
- 94.Oegema TR, Jr, Johnson SL, Aguiar DJ, Ogilvie JW. Fibronectin and its fragments increase with degeneration in the human intervertebral disc. Spine (Phila Pa 1976) 2000;25(21):2742–7. doi: 10.1097/00007632-200011010-00005. [DOI] [PubMed] [Google Scholar]
- 95.White ES, Baralle FE, Muro AF. New insights into form and function of fibronectin splice variants. The Journal of Pathology. 2008;216(1):1–14. doi: 10.1002/path.2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Glant TT, Hadhazy C, Mikecz K, Sipos A. Appearance and persistence of fibronectin in cartilage. Specific interaction of fibronectin with collagen type II. Histochemistry. 1985;82(2):149–58. doi: 10.1007/BF00708199. [DOI] [PubMed] [Google Scholar]
- 97.Kosher RA, Walker KH, Ledger PW. Temporal and spatial distribution of fibronectin during development of the embryonic chick limb bud. Cell Differ. 1982;11(4):217–28. doi: 10.1016/0045-6039(82)90069-0. [DOI] [PubMed] [Google Scholar]
- 98.Melnick M, Jaskoll T, Brownell AG, MacDougall M, Bessem C, Slavkin HC. Spatiotemporal patterns of fibronectin distribution during embryonic development. I. Chick limbs. J Embryol Exp Morphol. 1981;63:193–206. [PubMed] [Google Scholar]
- 99.Homandberg GA, Costa V, Wen C. Fibronectin fragments active in chondrocytic chondrolysis can be chemically cross-linked to the alpha5 integrin receptor subunit. Osteoarthritis Cartilage. 2002;10(12):938–49. doi: 10.1053/joca.2002.0854. [DOI] [PubMed] [Google Scholar]
- 100.Homandberg GA, Hui F. High concentrations of fibronectin fragments cause short-term catabolic effects in cartilage tissue while lower concentrations cause continuous anabolic effects. Arch Biochem Biophys. 1994;311(2):213–8. doi: 10.1006/abbi.1994.1229. [DOI] [PubMed] [Google Scholar]
- 101.Dudhia J, Bayliss MT, Hardingham TE. Human link protein gene: structure and transcription pattern in chondrocytes. Biochem J. 1994;303 (Pt 1):329–33. doi: 10.1042/bj3030329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.McKenna LA, Liu H, Sansom PA, Dean MF. An N-terminal peptide from link protein stimulates proteoglycan biosynthesis in human articular cartilage in vitro. Arthritis Rheum. 1998;41(1):157–62. doi: 10.1002/1529-0131(199801)41:1<157::AID-ART19>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 103.Friedl P, Vischer P, Freyberg MA. The role of thrombospondin-1 in apoptosis. Cell Mol Life Sci. 2002;59(8):1347–57. doi: 10.1007/s00018-002-8512-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bornstein P. Diversity of function is inherent in matricellular proteins: an appraisal of thrombospondin 1. J Cell Biol. 1995;130(3):503–6. doi: 10.1083/jcb.130.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bein K, Simons M. Thrombospondin type 1 repeats interact with matrix metalloproteinase 2. Regulation of metalloproteinase activity. J Biol Chem. 2000;275(41):32167–73. doi: 10.1074/jbc.M003834200. [DOI] [PubMed] [Google Scholar]
- 106.Kyriakides TR, Zhu YH, Smith LT, et al. Mice that lack thrombospondin 2 display connective tissue abnormalities that are associated with disordered collagen fibrillogenesis, an increased vascular density, and a bleeding diathesis. J Cell Biol. 1998;140(2):419–30. doi: 10.1083/jcb.140.2.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Agah A, Kyriakides TR, Bornstein P. Proteolysis of cell-surface tissue transglutaminase by matrix metalloproteinase-2 contributes to the adhesive defect and matrix abnormalities in thrombospondin-2-null fibroblasts and mice. Am J Pathol. 2005;167(1):81–8. doi: 10.1016/S0002-9440(10)62955-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Seki S, Kawaguchi Y, Chiba K, et al. A functional SNP in CILP, encoding cartilage intermediate layer protein, is associated with susceptibility to lumbar disc disease. Nature Genetics. 2005;37(6):607–12. doi: 10.1038/ng1557. [DOI] [PubMed] [Google Scholar]
- 109.Virtanen IM, Song YQ, Cheung KM, et al. Phenotypic and population differences in the association between CILP and lumbar disc disease. J Med Genet. 2007;44(4):285–8. doi: 10.1136/jmg.2006.047076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Min SK, Nakazato K, Yamamoto Y, et al. Cartilage Intermediate Layer Protein Gene Is Associated With Lumbar Disc Degeneration in Male, but Not Female, Collegiate Athletes. The American Journal of Sports Medicine. 2010;38(12):2552–7. doi: 10.1177/0363546510376714. [DOI] [PubMed] [Google Scholar]
- 111.Min SK, Nakazato K, Okada T, Ochi E, Hiranuma K. The Cartilage Intermediate Layer Protein Gene is Associated with Lumbar Disc Degeneration in Collegiate Judokas. International Journal of Sports Medicine. 2009;30(09):691–4. doi: 10.1055/s-0029-1214380. [DOI] [PubMed] [Google Scholar]
- 112.Lorenzo P, Aspberg A, Onnerfjord P, Bayliss MT, Neame PJ, Heinegard D. Identification and characterization of asporin. a novel member of the leucine-rich repeat protein family closely related to decorin and biglycan. J Biol Chem. 2001;276(15):12201–11. doi: 10.1074/jbc.M010932200. [DOI] [PubMed] [Google Scholar]
- 113.Kizawa H, Kou I, Iida A, et al. An aspartic acid repeat polymorphism in asporin inhibits chondrogenesis and increases susceptibility to osteoarthritis. Nat Genet. 2005;37(2):138–44. doi: 10.1038/ng1496. [DOI] [PubMed] [Google Scholar]
- 114.Song YQ, Cheung KM, Ho DW, et al. Association of the asporin D14 allele with lumbar-disc degeneration in Asians. Am J Hum Genet. 2008;82(3):744–7. doi: 10.1016/j.ajhg.2007.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Schneiderman G, Flannigan B, Kingston S, Thomas J, Dillin WH, Watkins RG. Magnetic resonance imaging in the diagnosis of disc degeneration: correlation with discography. Spine (Phila Pa 1976) 1987;12(3):276–81. doi: 10.1097/00007632-198704000-00016. [DOI] [PubMed] [Google Scholar]
- 116.Nakajima M, Kizawa H, Saitoh M, Kou I, Miyazono K, Ikegawa S. Mechanisms for asporin function and regulation in articular cartilage. J Biol Chem. 2007;282(44):32185–92. doi: 10.1074/jbc.M700522200. [DOI] [PubMed] [Google Scholar]
- 117.Evangelou E, Chapman K, Meulenbelt I, et al. Large-scale analysis of association between GDF5 and FRZB variants and osteoarthritis of the hip, knee, and hand. Arthritis Rheum. 2009;60(6):1710–21. doi: 10.1002/art.24524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Williams FM, Popham M, Hart DJ, et al. GDF5 single-nucleotide polymorphism rs143383 is associated with lumbar disc degeneration in Northern European women. Arthritis Rheum. 2011;63(3):708–12. doi: 10.1002/art.30169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Goupille P, Jayson MI, Valat JP, Freemont AJ. Matrix metalloproteinases: the clue to intervertebral disc degeneration? Spine (Phila Pa 1976) 1998;23(14):1612–26. doi: 10.1097/00007632-199807150-00021. [DOI] [PubMed] [Google Scholar]
- 120.Fujimoto T, Parry S, Urbanek M, et al. A single nucleotide polymorphism in the matrix metalloproteinase-1 (MMP-1) promoter influences amnion cell MMP-1 expression and risk for preterm premature rupture of the fetal membranes. J Biol Chem. 2002;277(8):6296–302. doi: 10.1074/jbc.M107865200. [DOI] [PubMed] [Google Scholar]
- 121.Arner EC, Hughes CE, Decicco CP, Caterson B, Tortorella MD. Cytokine-induced cartilage proteoglycan degradation is mediated by aggrecanase. Osteoarthritis Cartilage. 1998;6(3):214–28. doi: 10.1053/joca.1998.0114. [DOI] [PubMed] [Google Scholar]
- 122.Le Maitre CL, Pockert A, Buttle DJ, Freemont AJ, Hoyland JA. Matrix synthesis and degradation in human intervertebral disc degeneration. Biochem Soc Trans. 2007;35(Pt 4):652–5. doi: 10.1042/BST0350652. [DOI] [PubMed] [Google Scholar]
- 123.Rotshenker S, Aamar S, Barak V. Interleukin-1 activity in lesioned peripheral nerve. J Neuroimmunol. 1992;39(1–2):75–80. doi: 10.1016/0165-5728(92)90176-l. [DOI] [PubMed] [Google Scholar]
- 124.Le Maitre C, Freemont A, Hoyland J. Human disc degeneration is associated with increased MMP 7 expression. Biotechnic & Histochemistry. 2006;81(4–6):125–31. doi: 10.1080/10520290601005298. [DOI] [PubMed] [Google Scholar]
- 125.Rutges JP, Kummer JA, Oner FC, et al. Increased MMP-2 activity during intervertebral disc degeneration is correlated to MMP-14 levels. J Pathol. 2008;214(4):523–30. doi: 10.1002/path.2317. [DOI] [PubMed] [Google Scholar]
- 126.Crean JK, Roberts S, Jaffray DC, Eisenstein SM, Duance VC. Matrix metalloproteinases in the human intervertebral disc: role in disc degeneration and scoliosis. Spine (Phila Pa 1976) 1997;22(24):2877–84. doi: 10.1097/00007632-199712150-00010. [DOI] [PubMed] [Google Scholar]
- 127.Nakagawa T, Kubota T, Kabuto M, et al. Production of matrix metalloproteinases and tissue inhibitor of metalloproteinases-1 by human brain tumors. J Neurosurg. 1994;81(1):69–77. doi: 10.3171/jns.1994.81.1.0069. [DOI] [PubMed] [Google Scholar]
- 128.Song YQ, Ho DW, Karppinen J, et al. Association between promoter -1607 polymorphism of MMP1 and lumbar disc disease in Southern Chinese. BMC Med Genet. 2008;9:38. doi: 10.1186/1471-2350-9-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dong DM, Yao M, Liu B, Sun CY, Jiang YQ, Wang YS. Association between the -1306C/T polymorphism of matrix metalloproteinase-2 gene and lumbar disc disease in Chinese young adults. European Spine Journal. 2007;16(11):1958–61. doi: 10.1007/s00586-007-0454-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Price SJ, Greaves DR, Watkins H. Identification of novel, functional genetic variants in the human matrix metalloproteinase-2 gene: role of Sp1 in allele-specific transcriptional regulation. J Biol Chem. 2001;276(10):7549–58. doi: 10.1074/jbc.M010242200. [DOI] [PubMed] [Google Scholar]
- 131.Takahashi M, Haro H, Wakabayashi Y, Kawa-uchi T, Komori H, Shinomiya K. The association of degeneration of the intervertebral disc with 5a/6a polymorphism in the promoter of the human matrix metalloproteinase-3 gene. J Bone Joint Surg Br. 2001;83(4):491–5. doi: 10.1302/0301-620x.83b4.11617. [DOI] [PubMed] [Google Scholar]
- 132.Ye S, Eriksson P, Hamsten A, Kurkinen M, Humphries SE, Henney AM. Progression of coronary atherosclerosis is associated with a common genetic variant of the human stromelysin-1 promoter which results in reduced gene expression. J Biol Chem. 1996;271(22):13055–60. doi: 10.1074/jbc.271.22.13055. [DOI] [PubMed] [Google Scholar]
- 133.Williams FM, Bansal AT, van Meurs JB, et al. Novel genetic variants associated with lumbar disc degeneration in northern Europeans: a meta-analysis of 4600 subjects. Ann Rheum Dis. 2012 doi: 10.1136/annrheumdis-2012-201551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kay DM, Stevens CF, Hamza TH, et al. A comprehensive analysis of deletions, multiplications, and copy number variations in PARK2. Neurology. 2010;75(13):1189–94. doi: 10.1212/WNL.0b013e3181f4d832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wehling P, Cleveland SJ, Heininger K, Schulitz KP, Reinecke J, Evans CH. Neurophysiologic changes in lumbar nerve root inflammation in the rat after treatment with cytokine inhibitors. Evidence for a role of interleukin-1. Spine (Phila Pa 1976) 1996;21(8):931–5. doi: 10.1097/00007632-199604150-00005. [DOI] [PubMed] [Google Scholar]
- 136.Akyol S, Eraslan BS, Etyemez H, Tanriverdi T, Hanci M. Catabolic cytokine expressions in patients with degenerative disc disease. Turk Neurosurg. 2010;20(4):492–9. doi: 10.5137/1019-5149.JTN.3394-10.1. [DOI] [PubMed] [Google Scholar]
- 137.Rannou F, Corvol MT, Hudry C, et al. Sensitivity of anulus fibrosus cells to interleukin 1 beta. Comparison with articular chondrocytes. Spine (Phila Pa 1976) 2000;25(1):17–23. doi: 10.1097/00007632-200001010-00005. [DOI] [PubMed] [Google Scholar]
- 138.Gronblad M, Virri J, Tolonen J, et al. A controlled immunohistochemical study of inflammatory cells in disc herniation tissue. Spine (Phila Pa 1976) 1994;19(24):2744–51. doi: 10.1097/00007632-199412150-00002. [DOI] [PubMed] [Google Scholar]
- 139.Shinmei M, Masuda K, Kikuchi T, Shimomura Y. Interleukin 1, tumor necrosis factor, and interleukin 6 as mediators of cartilage destruction. Semin Arthritis Rheum. 1989;18(3 Suppl 1):27–32. doi: 10.1016/0049-0172(89)90081-4. [DOI] [PubMed] [Google Scholar]
- 140.Goldring MB. Anticytokine therapy for osteoarthritis. Expert Opin Biol Ther. 2001;1(5):817–29. doi: 10.1517/14712598.1.5.817. [DOI] [PubMed] [Google Scholar]
- 141.McDowell TL, Symons JA, Ploski R, Forre O, Duff GW. A genetic association between juvenile rheumatoid arthritis and a novel interleukin-1 alpha polymorphism. Arthritis Rheum. 1995;38(2):221–8. doi: 10.1002/art.1780380210. [DOI] [PubMed] [Google Scholar]
- 142.Solovieva S, Leino-Arjas P, Saarela J, Luoma K, Raininko R, Riihimaki H. Possible association of interleukin 1 gene locus polymorphisms with low back pain. Pain. 2004;109(1–2):8–19. doi: 10.1016/j.pain.2003.10.020. [DOI] [PubMed] [Google Scholar]
- 143.Takahashi H, Suguro T, Okazima Y, Motegi M, Okada Y, Kakiuchi T. Inflammatory cytokines in the herniated disc of the lumbar spine. Spine (Phila Pa 1976) 1996;21(2):218–24. doi: 10.1097/00007632-199601150-00011. [DOI] [PubMed] [Google Scholar]
- 144.Solovieva S, Kamarainen OP, Hirvonen A, et al. Association between interleukin 1 gene cluster polymorphisms and bilateral distal interphalangeal osteoarthritis. J Rheumatol. 2009;36(9):1977–86. doi: 10.3899/jrheum.081238. [DOI] [PubMed] [Google Scholar]
- 145.El-Omar EM, Carrington M, Chow WH, et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature. 2000;404(6776):398–402. doi: 10.1038/35006081. [DOI] [PubMed] [Google Scholar]
- 146.Shingu M, Nagai Y, Isayama T, Naono T, Nobunaga M. The effects of cytokines on metalloproteinase inhibitors (TIMP) and collagenase production by human chondrocytes and TIMP production by synovial cells and endothelial cells. Clin Exp Immunol. 1993;94(1):145–9. doi: 10.1111/j.1365-2249.1993.tb05992.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kang JD, Stefanovic-Racic M, McIntyre LA, Georgescu HI, Evans CH. Toward a biochemical understanding of human intervertebral disc degeneration and herniation. Contributions of nitric oxide, interleukins, prostaglandin E2, and matrix metalloproteinases. Spine (Phila Pa 1976) 1997;22(10):1065–73. doi: 10.1097/00007632-199705150-00003. [DOI] [PubMed] [Google Scholar]
- 148.Koch H, Reinecke JA, Meijer H, Wehling P. Spontaneous secretion of interleukin 1 receptor antagonist (IL-1ra) by cells isolated from herniated lumbar discal tissue after discectomy. Cytokine. 1998;10(9):703–5. doi: 10.1006/cyto.1998.0353. [DOI] [PubMed] [Google Scholar]
- 149.Doita M, Kanatani T, Harada T, Mizuno K. Immunohistologic study of the ruptured intervertebral disc of the lumbar spine. Spine (Phila Pa 1976) 1996;21(2):235–41. doi: 10.1097/00007632-199601150-00015. [DOI] [PubMed] [Google Scholar]
- 150.Maeda S, Kokubun S. Changes with age in proteoglycan synthesis in cells cultured in vitro from the inner and outer rabbit annulus fibrosus. Responses to interleukin-1 and interleukin-1 receptor antagonist protein. Spine (Phila Pa 1976) 2000;25(2):166–9. doi: 10.1097/00007632-200001150-00005. [DOI] [PubMed] [Google Scholar]
- 151.Le Maitre CL, Freemont AJ, Hoyland JA. A preliminary in vitro study into the use of IL-1Ra gene therapy for the inhibition of intervertebral disc degeneration. Int J Exp Pathol. 2006;87(1):17–28. doi: 10.1111/j.0959-9673.2006.00449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hoyland JA, Freemont AJ. Investigation of a quantitative post-hybridization signal amplification system for mRNA-oligodeoxyribonucleotide in situ hybridization. J Pathol. 1991;164(1):51–8. doi: 10.1002/path.1711640110. [DOI] [PubMed] [Google Scholar]
- 153.Le Maitre CL, Hoyland JA, Freemont AJ. Catabolic cytokine expression in degenerate and herniated human intervertebral discs: IL-1beta and TNFalpha expression profile. Arthritis Res Ther. 2007;9(4):R77. doi: 10.1186/ar2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Le Maitre CL, Hoyland JA, Freemont AJ. Interleukin-1 receptor antagonist delivered directly and by gene therapy inhibits matrix degradation in the intact degenerate human intervertebral disc: an in situ zymographic and gene therapy study. Arthritis Research & Therapy. 2007;9(4):R83. doi: 10.1186/ar2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kang JD, Georgescu HI, McIntyre-Larkin L, Stefanovic-Racic M, Donaldson WF, 3rd, Evans CH. Herniated lumbar intervertebral discs spontaneously produce matrix metalloproteinases, nitric oxide, interleukin-6, and prostaglandin E2. Spine (Phila Pa 1976) 1996;21(3):271–7. doi: 10.1097/00007632-199602010-00003. [DOI] [PubMed] [Google Scholar]
- 156.Doita M, Kanatani T, Ozaki T, Matsui N, Kurosaka M, Yoshiya S. Influence of macrophage infiltration of herniated disc tissue on the production of matrix metalloproteinases leading to disc resorption. Spine (Phila Pa 1976) 2001;26(14):1522–7. doi: 10.1097/00007632-200107150-00004. [DOI] [PubMed] [Google Scholar]
- 157.Demircan K, Hirohata S, Nishida K, et al. ADAMTS-9 is synergistically induced by interleukin-1beta and tumor necrosis factor alpha in OUMS-27 chondrosarcoma cells and in human chondrocytes. Arthritis Rheum. 2005;52(5):1451–60. doi: 10.1002/art.21010. [DOI] [PubMed] [Google Scholar]
- 158.Yu ZG, Xu N, Wang WB, Pan SH, Li KS, Liu JK. Interleukin-1 inhibits Sox9 and collagen type II expression via nuclear factor-kappaB in the cultured human intervertebral disc cells. Chin Med J (Engl) 2009;122(20):2483–8. [PubMed] [Google Scholar]
- 159.Goupille P, Jayson IVM, Valat JP, Freemont AJ. Matrix metalloproteinases: the clue to intervertebral disc degeneration? Spine. 1998;23(14):1612. doi: 10.1097/00007632-199807150-00021. [DOI] [PubMed] [Google Scholar]
- 160.Boos N, Nerlich AG, Wiest I, von der Mark K, Aebi M. Immunolocalization of type X collagen in human lumbar intervertebral discs during ageing and degeneration. Histochem Cell Biol. 1997;108(6):471–80. doi: 10.1007/s004180050187. [DOI] [PubMed] [Google Scholar]
- 161.Eskola PJ, Kjaer P, Daavittila IM, et al. Genetic risk factors of disc degeneration among 12–14-year-old Danish children: a population study. Int J Mol Epidemiol Genet. 2010;1(2):158–65. [PMC free article] [PubMed] [Google Scholar]
- 162.Kalichman L, Hunter DJ. The genetics of intervertebral disc degeneration. Familial predisposition and heritability estimation. Joint Bone Spine. 2008;75(4):383–7. doi: 10.1016/j.jbspin.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 163.Ala-Kokko L. Genetic risk factors for lumbar disc disease. Ann Med. 2002;34(1):42–7. doi: 10.1080/078538902317338634. [DOI] [PubMed] [Google Scholar]
- 164.Dominici R, Cattaneo M, Malferrari G, et al. Cloning and functional analysis of the allelic polymorphism in the transcription regulatory region of interleukin-1 alpha. Immunogenetics. 2002;54(2):82–6. doi: 10.1007/s00251-002-0445-9. [DOI] [PubMed] [Google Scholar]
- 165.Virtanen IM, Karppinen J, Taimela S, et al. Occupational and genetic risk factors associated with intervertebral disc disease. Spine (Phila Pa 1976) 2007;32(10):1129–34. doi: 10.1097/01.brs.0000261473.03274.5c. [DOI] [PubMed] [Google Scholar]
- 166.Solovieva S, Kouhia S, Leino-Arjas P, et al. Interleukin 1 polymorphisms and intervertebral disc degeneration. Epidemiology. 2004;15(5):626–33. doi: 10.1097/01.ede.0000135179.04563.35. [DOI] [PubMed] [Google Scholar]
- 167.Noponen-Hietala N, Virtanen I, Karttunen R, et al. Genetic variations in IL6 associate with intervertebral disc disease characterized by sciatica. Pain. 2005;114(1–2):186–94. doi: 10.1016/j.pain.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 168.Miyamoto H, Saura R, Doita M, Kurosaka M, Mizuno K. The role of cyclooxygenase-2 in lumbar disc herniation. Spine (Phila Pa 1976) 2002;27(22):2477–83. doi: 10.1097/00007632-200211150-00011. [DOI] [PubMed] [Google Scholar]