

Ndufaf5 is a conserved protein mutated in patients with mitochondrial complex I (CI) disease. A Dictyostelium model lacking functional Ndufaf5 provides new insights into the cytopathology of the disease, including a specific CI deficiency, AMPK-independent defects in growth and development, and a connection with autophagy.
Abstract
Ndufaf5 (also known as C20orf7) is a mitochondrial complex I (CI) assembly factor whose mutations lead to human mitochondrial disease. Little is known about the function of the protein and the cytopathological consequences of the mutations. Disruption of Dictyostelium Ndufaf5 leads to CI deficiency and defects in growth and development. The predicted sequence of Ndufaf5 contains a putative methyltransferase domain. Site-directed mutagenesis indicates that the methyltransferase motif is essential for its function. Pathological mutations were recreated in the Dictyostelium protein and expressed in the mutant background. These proteins were unable to complement the phenotypes, which further validates Dictyostelium as a model of the disease. Chronic activation of AMP-activated protein kinase (AMPK) has been proposed to play a role in Dictyostelium and human cytopathology in mitochondrial diseases. However, inhibition of the expression of AMPK gene in the Ndufaf5-null mutant does not rescue the phenotypes associated with the lack of Ndufaf5, suggesting that novel AMPK-independent pathways are responsible for Ndufaf5 cytopathology. Of interest, the Ndufaf5-deficient strain shows an increase in autophagy. This phenomenon was also observed in a Dictyostelium mutant lacking MidA (C2orf56/PRO1853/Ndufaf7), another CI assembly factor, suggesting that autophagy activation might be a common feature in mitochondrial CI dysfunction.
INTRODUCTION
Mitochondrial diseases are complex disorders caused by mutations in either nuclear or mitochondrially encoded proteins. The pathological outcome varies in severity and includes a wide range of neurodegenerative and muscular disorders, including blindness, deafness, epilepsy, heart and muscle disease, diabetes, and cancer, among others (Wallace, 2010). A large proportion of mitochondrial diseases affect the first and largest complex of the respiratory chain, mitochondrial complex I or NADH:ubiquinone oxidoreductase (CI). This huge multimeric protein structure is formed by 44 subunits in mammalian cells and couples the oxidation of NADH to reduction of ubiquinone and the transport of four protons from the matrix side to the intermembrane space (Fearnley et al., 2007). The resulting proton gradient is also contributed to by complexes III and IV and allows the synthesis of ATP in complex V (Koopman et al., 2010).
Prokaryotes present a comparatively simple CI composed of 14 core subunits, which are able to perform the essential biochemical reactions (Efremov et al., 2010). The presence of additional accessory subunits in higher organisms might reflect the presence of additional unknown functions (Hirst, 2011). Pathogenic mutations have been described affecting all core and some accessory subunits. The assembly of such a large structure is only partially understood and involves the function of many assembly factors (McKenzie and Ryan, 2010; Nouws et al., 2012). Of interest, nine of them have been found to be mutated in CI disease: NDUFAF1–F4, C8orf38, ACAD9, FORXRED1, NUBPL, and Ndufaf5 (Ogilvie et al., 2005; Vogel et al., 2005; Pagliarini et al., 2008; Sugiana et al., 2008; Gerards et al., 2009; Hoefs et al., 2009; Saada et al., 2009, 2012; Calvo et al., 2010; Fassone et al., 2010; Haack et al., 2010; Nouws et al., 2010). Other CI assembly factors have been described, but their involvement in human mitochondrial disease has not yet been documented, including Ecsit (Vogel et al., 2007), MidA (also known as Ndufaf7, C2orf56, or PRO1853; Torija et al., 2006; Carilla-Latorre et al., 2010), and TMEM126B (Heide et al., 2012). Remarkably, CI dysfunction has also been associated with neurodegenerative diseases such as Parkinson and Alzheimer diseases (Janssen et al., 2006; Lazarou et al., 2009; Sharma et al., 2009). Despite all these connections and importance in human pathology, many questions remain to be answered as to how such a variety of associated phenotypes arise and what their underlying cytopathological mechanisms might be.
The complexity of mitochondrial pathology has prompted the use of simple model systems. Dictyostelium, due to its similarities to mammalian cells and the presence of diverse, consistent, and readily assayed phenotypes, has been developed as a model for mitochondrial diseases and other pathologies (Escalante, 2011; Francione et al., 2011). Dictyostelium is a social amoeba with two distinctive lifestyles: a motile unicellular stage, and a multicellular developmental stage, including relatively complex processes of cell differentiation and morphogenesis. The multicellular slug form is phototactic and thermotactic and after a variable period of migration gives rise to the terminal fruiting body composed of spores aloft a cellular stalk (Escalante and Vicente, 2000; Annesley and Fisher, 2009a).
Multicellular development in Dictyostelium takes place in the absence of nutrients, and thus autophagy, a “self-eating” mechanism to recycle the cell's material, is essential for cellular homeostasis and development in this system as in all eukaryotes (Calvo-Garrido et al., 2010). There are several types of autophagy, but the best known and the most conserved is macroautophagy (called just autophagy hereafter, for simplicity). Mechanistically, autophagy begins with the formation of a double-membrane vesicle called an autophagosome, which engulfs organelles or parts of the cytoplasm. This is followed by fusion of the autophagosome with lysosomes and the subsequent degradation of the vesicle and its contents. The simple molecular constituents released during this degradation process can be recycled or used for ATP production. Autophagy is not only triggered by starvation conditions such as nutrient or growth factor depletion, but it is also induced in circumstances requiring, for example, the elimination of protein aggregates or defective organelles or in response to bacterial pathogens (Ravikumar et al., 2010). Mitophagy—the specific degradation of mitochondria—has been implicated in diverse pathologies such as Parkinson disease (Rochet et al., 2012).
Different patterns of aberrant phenotypes occur as a result of mitochondrial dysfunction in Dictyostelium, including growth defects, developmental abnormalities, and defects in phototaxis and thermotaxis (Bokko et al., 2007; Francione et al., 2011). Of interest, the Dictyostelium model has shown that several of these phenotypes arise from abnormal signaling rather than from ATP insufficiency. Bokko et al. (2007)provided the first genetic evidence that the energy stress–sensing protein AMP-activated protein kinase (AMPK) is chronically activated in a Dictyostelium strain deficient in the mitochondrial protein chaperonin 60 (Hsp60), a protein mutated in hereditary spastic paraplegia (Hansen et al., 2002). Inhibition of AMPK expression in the Hsp60-deficient strain resulted in a suppression of the aforementioned phenotypic defects. In addition, the growth, developmental, and photo/thermotaxis defects observed in this strain were phenocopied by overexpressing a constitutively active form of AMPK.
The consequences of a CI deficiency in Dictyostelium have been investigated in a MidA loss-of-function strain. MidA is the homologue of the human C2orf56 (also known as PRO1853), a conserved protein of unknown function. Carilla-Latorre et al. (2010) showed that Dictyostelium and human cells deficient in this protein have a specific defect in CI activity and assembly. This mitochondrial deficiency caused the expected defects in phototaxis and thermotaxis that could be rescued by AMPK inhibition but also displayed additional defects, which were AMPK independent, including impaired phagocytosis, pinocytosis, and growth. This highlights the complexity of the mitochondrial cytopathology and more specifically that of CI disease (Francione et al., 2011).
Mutations in the assembly factor Ndufaf5 cause different mitochondrial syndromes. A family with homozygous missense mutation L229P was reported to suffer a lethal neonatal form of CI deficiency (Sugiana et al., 2008). Another pathogenic amino acid substitution (L159F) was found to cause Leigh syndrome in a Moroccan family (Gerards et al., 2009). Recently a new mutation (G250V) causing Leigh syndrome was described (Saada et al., 2012). All of these mutations decrease CI activity and assembly, but little is known about the function of the protein and the mechanisms of the disease. Ndufaf5 has a predicted S-adenosylmethionine (SAM)–dependent methyltransferase domain, and thus a possible role of methylation in CI assembly has been proposed but not yet tested experimentally.
Here we describe the first Ndufaf5 model of the disease in a simple experimental system. We generated a Ndufaf5-null mutant in Dictyostelium discoideum and investigated the relationship between the resulting phenotypes and AMPK in this model. Furthermore, site-directed mutagenesis in a critical amino acid from the predicted SAM-binding domain strongly suggests the presence of a functional methyltransferase domain. Recreation of pathogenic mutations in Dictyostelium Ndufaf5 resulted in a loss-of-function of the protein, reinforcing the usefulness of this model. Finally, autophagy was tested and found to be overactivated in Ndufaf5- and MidA-null mutants, suggesting that mitochondrial complex I dysfunction has a wider effect on cellular homeostasis than expected.
RESULTS
Generation of an insertional mutant in Dictyostelium Ndufaf5
Mutations causing mitochondrial complex I disease have been described in core and accessory subunits, as well as in a number of complex I assembly factors. One of these assembly factors, Ndufaf5, is an uncharacterized protein conserved during evolution from bacteria to humans (Supplemental Figure S1 and Supplemental Table S2). The phylogenetic profile of this protein resembles that of other complex I proteins, being present in alpha proteobacteria, the closest living organisms to the putative precursors of eukaryote mitochondria, and in most eukaryotes, with the remarkable exception of the fermentative yeasts Saccharomyces cerevisiae and Schizosaccharomyces pombe (Supplemental Table S2; Pagliarini et al., 2008). The Dictyostelium genome codes for a protein highly similar to the human Ndufaf5 (dictyBase ID: DDB_G0287769; Supplemental Figure S1 and Supplemental Table S2), with a Blast E-value of 6e-62. The National Center for Biotechnology Information conserved-domain database (Marchler-Bauer et al., 2011) predicts a SAM-dependent methyltransferase motif in both the Dictyostelium and human proteins. Gene disruption in Dictyostelium was achieved by insertional mutagenesis (Figure 1A). The resulting insertion of a blasticidin-resistance cassette interrupted the gene in nucleotide 206 of the coding sequence at amino acid 69. As described in Figure 1, B and C, genomic PCR and reverse transcription-PCR (RT-PCR) analysis confirmed the isolation of disruptant strains, which showed no expression of ndufaf5 mRNA. All strains showed similar phenotypes, and one of these clonal isolates (ndufaf5−) was selected for the following studies.
FIGURE 1:

ndufaf5 disruption and loss of expression. (A) Scheme of the disruption vector and the disrupted gene after homologous recombination. The BsR cassette interrupted the gene at the nucleotide 206 of the coding sequence and deleted the intron (indicated as a V-shaped line). Open boxes represent ndufaf5 exons. Solid boxes represent the disruption vector with the position of the N and C-terminal fragments used to allow homologous recombination. The BsR cassette is located between the fragments and is not depicted in scale to allow the correct overlapping with the scheme of ndufaf5 locus. The arrowheads with numbers indicate the different oligonucleotides used for verification of the disruptant strains (Supplemental Table S1). (B) Genomic verification. DNA extracted from wild-type and knockout strains was subjected to PCR using oligos 1 and 2. The amplification band indicates that homologue recombination has taken place in the expected region. The unrelated gene (dictyBase ID: DDB_G0268840) was used as a PCR control. (C) Transcriptomic verification. RNA extracted from wild-type and knockout strains at 14 h of development was subjected to RT-PCR using oligos 3 and 4. The absence of amplification in the mutant strains indicates that the transcript has been disrupted. Differences in the size of the bands between DNA and RNA templates are due to the presence of a small intron in both ndufaf5 and the control (DDB_G0268840) genes. M, DNA marker.
The Ndufaf5-null mutant has pleiotropic defects in growth and development
Previous studies showed that mitochondrial defects in Dictyostelium result in defects in growth and development (Bokko et al., 2007; Carilla-Latorre et al., 2010). We first analyzed these phenotypes in the ndufaf5− strain. The null strain showed a significant defect in growth, both in association with bacteria (Figure 2A) and in HL5 axenic medium. Generation time increased in axenic media to 14 h, compared with 10 h in the parental AX4 strain. For developmental analysis, cells grown in axenic media were deposited onto nitrocellulose filters (Figure 2B). The ndufaf5− cells showed a delay in development. At 24 h wild-type strains formed early culminants, whereas ndufaf5− structures remained at the finger/slug stage. After a longer incubation (48 h) the mutant eventually culminated, but the fruiting bodies were smaller than those of the wild type. Moreover, spores taken from the fruiting bodies after 10 d, a time necessary for full maturation, showed abnormal morphology under phase contrast microscopy, suggesting a defect in differentiation or maturation (Figure 2B). To confirm this hypothesis, we performed viability experiments by counting colony-forming units of spores plated in association with bacteria. The experiments showed a near-complete loss of spore viability (<1%), in contrast with wild-type spores, which showed 90–100% viability.
FIGURE 2:
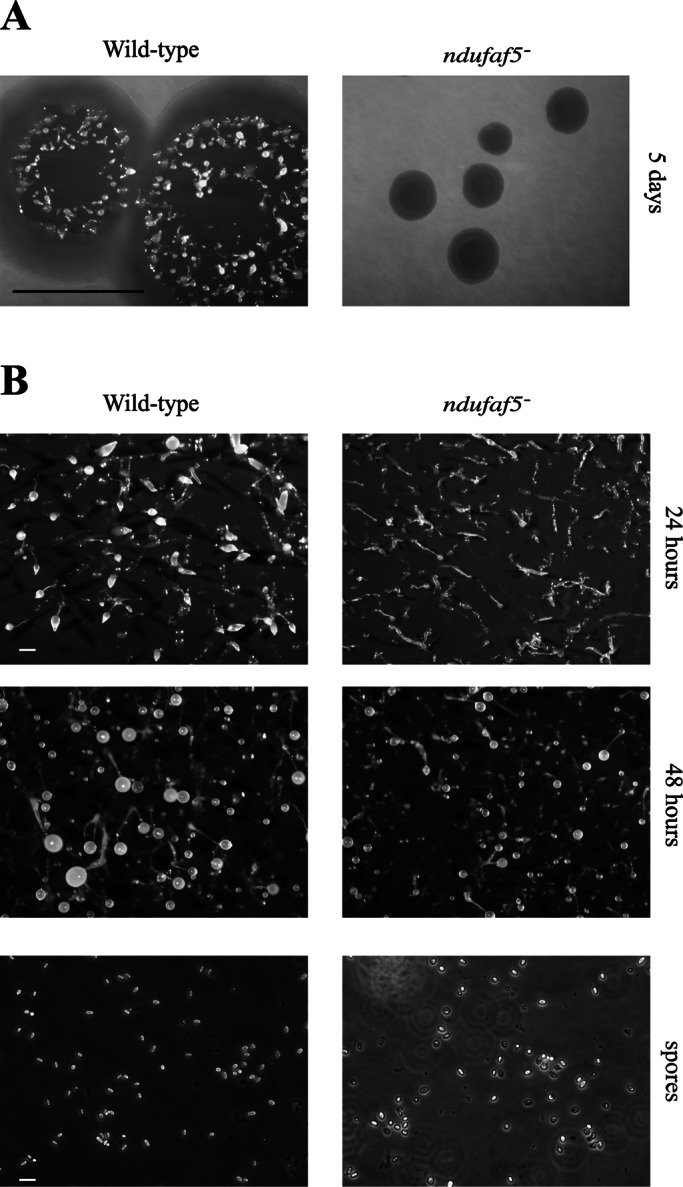
Analysis of growth and development in ndufaf5−. (A) Wild-type and ndufaf5− cells were seeded on SM plates in association with K. aerogenes and grown for 5 d. The ndufaf5− strain showed a strong defect in the diameter of the clearing zone. Bar, 1 cm. (B) Cells grown exponentially in axenic medium were harvested, washed with PDF buffer, and deposited onto nitrocellulose filters according to Materials and Methods. Photographs were taken at the indicated times. The mutant strain showed a delay at the finger-slug stage. Structures culminated after 48 h, and the fruiting bodies were smaller than those of wild type. Bar, 2 mm. Wild-type and mutant spores were taken from 10-d-old fruiting bodies and observed under phase contrast microscopy. Mutant spores were rounder and less refractive, suggesting loss of viability. Bar, 10 μm.
The role of Ndufaf5 in CI is conserved between Dictyostelium and humans
Three different pathological mutations have been described in human ndufaf5. All of these missense mutations caused a strong defect in mitochondrial CI activity and assembly. We wanted to determine whether Dictyostelium Ndufaf5 plays a similar role in this organism and can thus be used as a valid model of the disease. We first asked about the subcellular localization of the protein in Dictyostelium cells. To this end, the green fluorescent protein (GFP) was fused at the C-terminus of Ndufaf5 (named ndufaf5− rescue) and the construct transfected in the mutant background. The clones expressing the fused protein fully complemented the phenotypes described earlier (Supplemental Figure S2). Complementation confirms that the observed defects are exclusively caused by the genetic disruption of Ndufaf5 and that the fused protein is functional. Confocal microscopy shows that Dictyostelium Ndufaf5 is localized in mitochondria as described previously for the human protein (Figure 3A; Sugiana et al., 2008). Mito Tracker Red does not stain Dictyostelium mitochondria uniformly, being concentrated in spherical structures called submitochondrial bodies (van Es et al., 2001; Gilson et al., 2003). Consequently, the merge images might show some green areas with little Mito Tracker Red staining. When the intensity of the red channel is increased the rest of the mitochondrion is visible and the colocalization appears complete, as shown in the insets of Figure 3A.
FIGURE 3:

Functional conservation of Ndufaf5 and mitochondrial phenotypes in the ndufaf5− strain. (A) Ndufaf5-GFP was expressed in the mutant background and subcellular localization of the protein analyzed by confocal microscopy. The region marked was magnified below and the color intensity adjusted to allow a clearer visualization of mitochondria. Bar, 10 μm. (B) Activities of complexes I, II, III, and IV in ndufaf5− and wild-type cells were measured by spectrophotometric analyses and corrected by protein and citrate synthase activity and finally normalized against wild-type levels. (C) Citrate synthase and cellular ATP levels. Citrate synthase activity was measured by spectrophotometric analyses and normalized as before. ATP levels were measured using a bioluminescence assay and normalized vs. wild-type levels. NS, nonsignificant. **p < 0.01.
We next measured mitochondrial respiratory chain activities in wild-type and ndufaf5− cells (Figure 3B). A significant CI-specific defect was observed (40% activity of that of the wild type), whereas the rest of respiratory activities remained normal. These experiments confirm that Dictyostelium Ndufaf5 is a mitochondrial protein required for CI function and strongly suggest functional conservation during evolution. To further characterize the mitochondrial defect in Ndufaf5-null, we measured the enzymatic activity of citrate synthase (Krebs cycle enzyme anchored to mitochondrial inner membrane). This activity has been extensively used as a marker of mitochondrial mass and was found to be increased in the mutant strain, suggesting the presence of a compensatory mechanism (Figure 3C, left). Surprisingly, we also found increased levels of total ATP in cells lacking Ndufaf5 (Figure 3C, right).
Site-directed mutagenesis studies of the putative SAM-binding motif
The Conserved Domains Database (CDD) predicts a methyltransferase domain in Ndufaf5 from both humans and Dictyostelium, based on sequence and homology with other methyltransferases. Several putative catalytic residues in the SAM-binding domain are specified in the CDD prediction for Ndufaf5. The typical GxGxG sequence is conserved during evolution (Supplemental Figure S1). To advance in the knowledge of this protein, we selected the first glycine of the motif for site-directed mutagenesis (G86V) and named the resulting protein M1 (Figure 4A). Wild-type (ndufaf5− rescue) and M1 proteins were fused to GFP (for subcellular localization studies) and transfected in ndufaf5− mutant. The wild-type form of the protein was able to restore normal growth in liquid medium and SM plates (Figure 4B). However, M1 was unable to complement the growth phenotypes, which were similar to those of the ndufaf5− mutant. To ensure the lack of complementation was not due to an incorrect targeting of M1, we carried out confocal studies showing that M1, like the wild-type protein, was located in mitochondria (data not shown). The lack of complementation strongly suggests that the putative methyltransferase domain is required for Ndufaf5 function.
FIGURE 4:

Site-directed mutagenesis analyses. (A) Schematic representation of the constructs used for complementation or site-directed mutagenesis experiments. Ndufaf5 was fused to GFP and tandem affinity purification tag (TAP-tag) into the pDV-GFP-CTAP vector, where expression is directed under the control of actin-15 promoter. The V-shaped line represents an intron. M1 is the mutation in the predicted SAM-binding domain (see the text). M2 and M3 are the corresponding mutations to the pathogenic ones in human (M2 equivalent to L159F, and M3 equivalent to L229P). (B) Wild-type and mutant constructs were transfected in ndufaf5− cells. The expressed proteins colocalized with MitoTracker Red in mitochondria to the same extent as depicted in Figure 3A (data not shown). Transfected cells were spread onto SM plates to test the size of the clearing zone after 5 d. The strain transformed with the wild-type protein (ndufaf5− rescue strain) complemented the growth phenotype completely, in contrast to the mutated forms, which showed similar defects as the parental ndufaf5−. Bar, 1 cm.
Pathogenic mutations in Dictyostelium Ndufaf5 lead to a loss-of-function phenotype
Three pathogenic mutations have been described in human Ndufaf5, leading to Leigh syndrome (L159F and G250V) and lethal neonatal disease (L229P). L159 and L229 are conserved in the Dictyostelium protein (corresponding to L165 and L235), whereas the third one is not (Supplemental Figure S1). These residues and their surrounding amino acids are also conserved in bacterial homologues, suggesting that they must play a critical role in Ndufaf5 function. To test this hypothesis, we performed site-directed mutagenesis in the conserved residues homologous to those mutated in patients (L165F and L235P). The resulting mutated proteins were called M2 and M3, respectively. As for M1, both proteins were fused to GFP and the constructs transformed into ndufaf5− cells. A complete failure to rescue the null-mutant phenotype was observed (Figure 4), suggesting a loss of function of the proteins. The lack of complementation in M2 and M3 could not be the consequence of an incorrect mitochondrial location of these proteins since both colocalized with the mitochondrial marker MitoTracker Red in mitochondria (data not shown). This result further supports the functional conservation of Ndufaf5 and offers a very simple model with which to study several cytopathological aspects of Ndufaf5-associated disease.
AMPK does not govern the cytopathology of Ndufaf5-null mutant
Previous analyses performed in Dictyostelium mitochondrial diseased models (e.g., in Hsp60-deficient cells) showed that a chronic activation of AMPK was responsible for nearly all the defective phenotypes. However, not all the phenotypes are equally affected by mitochondrial dysfunction, with growth and slug phototaxis and thermotaxis being more strongly affected (Bokko et al., 2007; Francione et al., 2011). Down-regulation of AMPK by the expression of an antisense construct was shown to effectively suppress those phenotypes (Bokko et al., 2007; Francione et al., 2011). To get a better understanding of the role of AMPK in Ndufaf5 deficiency, we analyzed the consequences of down-regulating the expression of AMPK on the phenotypes associated with the lack of Ndufaf5. Therefore we tested slug phototaxis in ndufaf5− and found a clear defect in both qualitative and quantitative experiments (Figure 5), which can be rescued by the expression of Ndufaf5 (ndufaf5− rescue strain). However, in contrast with other mitochondrial defects, knockdown of AMPK did not rescue either the phototaxis (Figure 5) or the growth defects associated with Ndufaf5 deficiency (Supplemental Figure S2). These results suggest that the cytopathology of ndufaf5− is not caused by the chronic activation of AMPK as described for other mitochondrial lesions.
FIGURE 5:
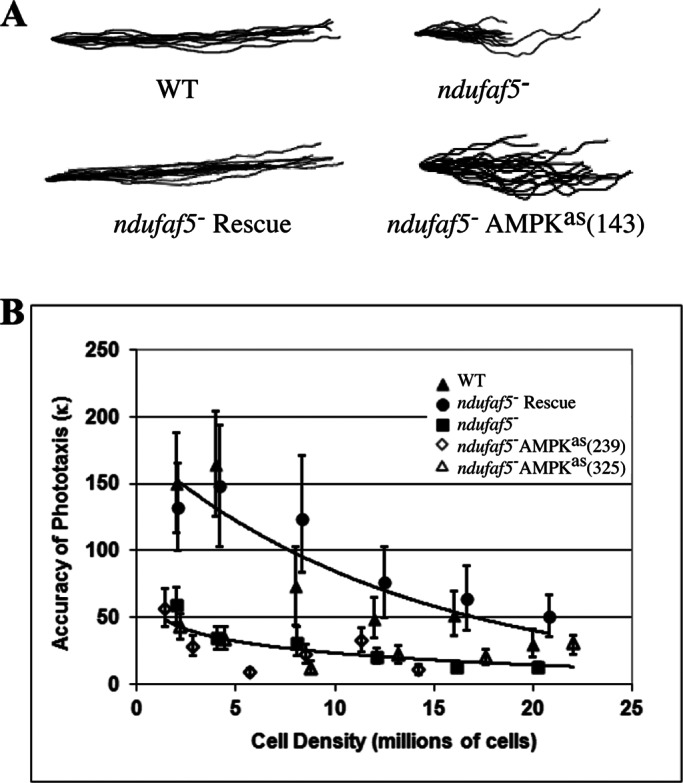
Phototaxis defects in ndufaf5− are not rescued by AMPK inhibition. (A) Qualitative experiments. Different colonies (slugs) were allowed to migrate under lateral light source. Pictures were obtained after 48 h of incubation at 21ºC and the slug trails represented (see Materials and Methods). The ndufaf5− slugs had a significant defect in phototaxis, which was rescued after transfection with the Ndufaf5 wild-type protein (ndufaf5− rescue). However, no suppression of the mutant phenotype was observed in ndufaf5− cells expressing a different number of copies of an AMPK antisense construct. A representative AMPK-inhibited strain is shown, with the number of copies between brackets. (B) Quantitative experiments. Phototaxis accuracy (κ value) was calculated for all the indicated strains at different cell densities (see Materials and Methods). A clear defect was again observed in ndufaf5− migrating slugs. This defect was complemented by overexpression of Ndufaf5 (ndufaf5− rescue) but not suppressed by antisense inhibition of AMPK (number of copies of AMPKas construct in brackets). The error bars represent 90% confidence intervals. Lines of best fit were determined by the least squares method, fitting an exponential model (wild type and ndufaf5− rescue) or logarithmic model (ndufaf5− mutant and AMPK antisense strains derived from it). The choice of model was dictated by which provided the best fit by eye.
ndufaf5− and midA− strains show strong induction of autophagy
AMPK is a major regulator of cell metabolism (Mihaylova and Shaw, 2011). Several anabolic and catabolic pathways are regulated by this kinase, including autophagy, a process by which cells degrade their components via lysosomes to maintain cellular homeostasis under stress conditions. We wanted to determine whether this major pathway of degradation is affected in ndufaf5− and compare this with cells lacking MidA, another CI assembly factor, whose deficiency leads to phenotypes that are partially dependent on AMPK, such as phototaxis and thermotaxis. To this end, we transfected wild-type, ndufaf5−, and midA− cells with the autophagic marker GFP-Atg18. This marker allows the observation of autophagosome vesicles that appear as small, bright green puncta in the cytoplasm. In wild type, the number of autophagosomes increases during starvation. Of interest, both mutants showed a strong accumulation of autophagosomes even in growth conditions (Figure 6A). Similar results were found using the autophagic marker GFP-Atg8 (data not shown). Accumulation of autophagosomes could be due to either an induction of autophagy or a block at later stages of maturation. To discriminate between these possibilities, we performed an autophagic flux assay based on the proteolytic cleavage of an autophagic substratum. The level of free GFP was measured by Western blot in wild-type, ndufaf5−, and midA− cells expressing GFP-Tkt-1 as described previously (Calvo-Garrido et al., 2011). As shown in Figure 6B, both mutants showed a significant increase of free GFP levels compared with wild type, which confirms an increase of autophagic flux.
FIGURE 6:
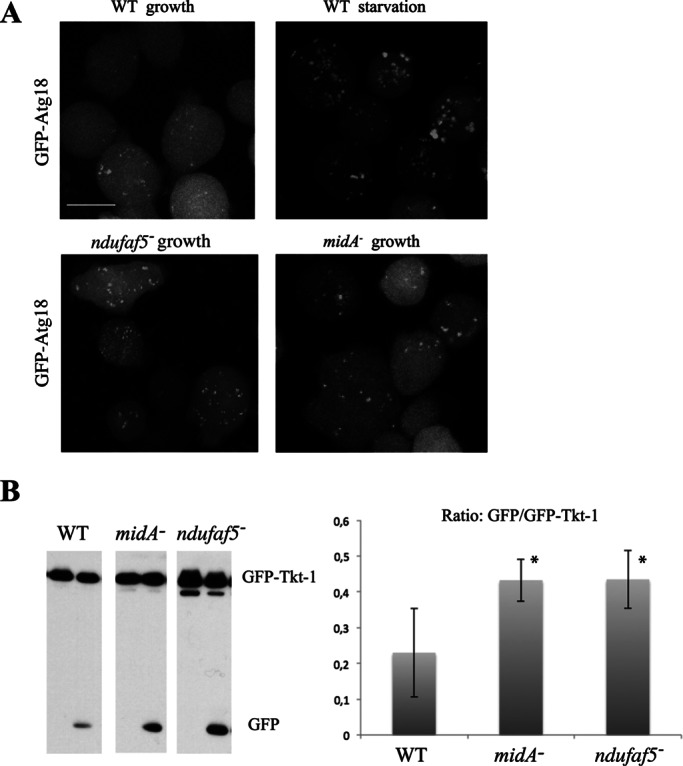
Autophagy is increased in ndufaf5− and midA− cells. (A) Labeling of autophagosomes. The indicated strains were transfected with the autophagosome marker GFP-Atg18. Cells were incubated in growth media or starvation conditions (PDF buffer). Representative images show maximum projections of confocal images covering the whole cell. Bar, 10 μm. (B) Autophagic flux. Strains transfected with the marker GFP-Tkt-1 growing in HL5 were incubated with 0 or 100 mM NH4Cl (left and right lanes, respectively) as described in Materials and Methods. Extracts containing the same protein load were subjected to SDS–PAGE, transferred by Western blot, and incubated with α-GFP antibody to detect GFP-Tkt-1 and the cleaved GFP proteins. Left, a representative image of Western blots. Right, quantification of the ratio between free GFP and GFP–Tkt-1 of three independent experiments. *p < 0.05.
Some mitochondrial defects increase specific degradation of mitochondria (so-called mitophagy; Mijaljica et al., 2010). Mitophagy events can be detected by colocalization of mitochondria with autophagic markers. Therefore wild type and ndufaf5− and midA− mitochondrial mutants transfected with the marker GFP-Atg18 were stained with MitoTracker Red and analyzed by confocal microscopy. Most of the puncta did not colocalize with mitochondria, as shown in Supplemental Figure S3. We also stained mitochondria with 4′,6-diamidino-2-phenylindole (DAPI), which is independent of mitochondrial membrane potential, and obtained similar results (Supplemental Figure S4), suggesting that the increase in autophagy is not due to an activation of mitophagy but instead to an activation of bulk autophagy.
DISCUSSION
We have shown that Dictyostelium Ndufaf5, like the human homologue, is a mitochondrial protein required for CI function, which argues for functional conservation during evolution and validates Dictyostelium as a simple model in which questions regarding the molecular function of the protein and the cytopathology of Ndufaf5 deficiency can be addressed.
The molecular function of Ndufaf5 remains elusive, and although a methyltransferase motif type I can be predicted by bioinformatics (Niewmierzycka and Clarke, 1999), no functional studies had been carried out. Our studies show that a conserved residue of the catalytic domain is essential for protein function and thus support the predicted hypothesis. Unfortunately the common methyltransferase motif type I does not give any clue about putative substrates, which could be nucleic acids, proteins, lipids, or other small metabolites (Kagan and Clarke, 1994). Ndufaf5 shows high similarity to the bacterial protein BioC (Supplemental Figure S1). Of interest, the putative SAM-binding domain and two residues that we showed to be essential for the function of the protein, which are also present in human patients (L159 and L229), are also conserved in the bacterial protein. It is thus tempting to consider the hypothesis of a functional conservation between these two proteins.
BioC is the enzyme that catalyzes the first step in biotin biosynthesis (methylation of esterified malonyl) in bacteria, using the well-characterized fatty acid synthesis pathway (FAS-II like), which is conserved from bacteria to mitochondria (Hiltunen et al., 2009, 2010). Of interest, this pathway is used in mitochondria not only for the synthesis of many different fatty acids, but also to synthesize the lipid moiety of 3-hydroxy-myristoyl-acyl carrier protein (NADH dehydrogenase [ubiquinone] complex I assembly factor [NDUFAB1], SDAP), a CI subunit. Moreover, phylogenetic studies showed that the biotin biosynthesis pathway might have existed in the proteobacterium after the endosymbiotic event, making the conservation of the pathway in actual mitochondrion possible (Gabaldon and Huynen, 2007). Similarly, the HOGENOM database includes Ndufaf5 homologues and BioC in the same gene family: HOG000265011 (Penel et al., 2009).
Taken together, these evolutionary studies seem to relate Ndufaf5 and BioC, suggesting that Ndufaf5 could catalyze methyl transfer from SAM to malonyl esters or similar molecules. However, it has long been known that mammals cannot synthesize biotin (vitamin H), a final product of the FAS-II pathway. Nonetheless, this pathway might be conserved in mammals for other purposes, such as CI assembly directly through ACP acylated forms, or indirectly by synthesizing unknown fatty acids that could be required for helping CI insertion and folding into the mitochondrial inner membrane. In addition, some other CI structural subunits and CI assembly factors have been related in different ways to lipid metabolism, such as NDUFAB1, ACAD9, and C8orf38 (Nouws et al., 2010; McKenzie et al., 2011; Zurita Rendon and Shoubridge, 2012).
Ndufaf5 and MidA exhibit similarities and differences in the context of CI function in Dictyostelium. Both null mutants have a CI-specific defect, which in both cases is associated with an apparent increase in mitochondrial mass, albeit more evident for ndufaf5− (68 vs. 30% increment in midA−; Carilla-Latorre et al., 2010). Strikingly, total ATP in ndufaf5− increased up to 60% compared with the wild-type levels, whereas midA− cells showed the opposite behavior, with a decrease of 30% compared with wild type. Although normal levels of ATP have been measured in patients of some mitochondrial diseases, it is nevertheless surprising that a mitochondrial defect could lead to an increase in the levels of ATP, although this observation is not without precedent. Studies in Caenorhabditis elegans showed that mutations in the CI structural subunit NDUFB4, which render CI inactive, lead to a strong increase of cellular ATP to 300% of that of wild-type controls (Yang and Hekimi, 2010).
Several mechanisms can be envisioned to explain this compensatory phenomenon. The increase in mitochondrial mass might in part compensate for the reduced activity of CI, and the mitochondrial potential might also be supported by the entry of electrons into the respiratory chain at other points different from CI. A change in the metabolic equilibrium between anabolic and catabolic pathways might reduce the use of ATP while activating autophagy to degrade the cell's proteins, lipids, and glycogen, which can then be used to fuel ATP-generating pathways and keep energetic homeostasis within viable limits.
Despite the similarities in the phototaxis defect, which affects both Ndufaf5 and MidA mutants, the relationship of this phenotype with AMPK activity is different. Chronic activation of AMPK was previously shown to be responsible for the phototaxis and thermotaxis defects in midA− mutant, but growth defects were independent. However, AMPK is not responsible for the phototaxis or growth defects in ndufaf5− mutant, suggesting that AMPK is not activated in this background. This is in agreement with the high levels of ATP found in ndufaf5−, which should keep AMPK inactive. The differences observed between Ndufaf5- and MidA-deficient cells suggest that despite their common function in CI, they might act at different levels affecting CI activity or its regulation. Nevertheless we cannot rule out the possibility of additional, as-yet-undiscovered roles of these proteins in other mitochondrial functions.
Yeast-two hybrid screening and pull-down experiments revealed the interaction of MidA with a CI subunit (NDUFS2; Carilla-Latorre et al., 2010). Unfortunately, a similar approach did not yield reproducible results in Ndufaf5. It was proposed that Ndufaf5 might interact with the NDUFB3 subunit (Sugiana et al., 2008) since this CI structural subunit has different methylations in His residues on its N-terminal region (Carroll et al., 2005). Our studies in Dictyostelium do not support this hypothesis because phylogenetic analyses show that Ndufaf5 and NDUFB3 do not have the same phylogenetic profile: in Dictyostelium the former is conserved, whereas no homologous protein for the latter can be identified by sequence similarity in this organism. Furthermore, the CI subunit composition of Pichia pastoris has recently been characterized (Bridges et al., 2010). Both proteins have homologues in this organism, but the NDUFB3 homologue lacks the N-terminal region bearing the methylated residues in mammals. It is thus unlikely that the biochemical function of Ndufaf5 is to methylate NDUFB3. There is, however, a methylation event described in Arg-323 in the CI subunit NDUFS2 (Wu et al., 2003), a protein with a similar phylogenetic profile to Ndufaf5 and MidA. This raises the possibility that Ndufaf5 methylates NDUFS2. However, since MidA was found previously to interact with NDUFS2, it is probable that the responsible methyltransferase is MidA rather than Ndufaf5 (Carilla-Latorre et al., 2010).
In the present work, high levels of autophagy induction and accumulation of autophagosomes have been observed in both Ndufaf5- and MidA-null mutant strains. This is the first time that induction of macroautophagy has been demonstrated in a CI disease model. Besides starvation, autophagy can be activated by a variety of cellular stresses, including protein aggregation, endoplasmic reticulum stress, DNA damage, reactive oxygen species, and microbial pathogens. Several signaling pathways have been implicated in the induction of autophagy, including AMPK, TorC1, and the class III phosphoinositide 3-kinase (PI3K). If AMPK were responsible for autophagic induction in Dictyostelium, differences would have been expected between the two mutants since only midA− and not ndufaf5− cells seem to have chronic activation of AMPK as determined by recovery experiments after AMPK knockdown. Of interest, genome-wide screening with a small interfering RNA library in human neuroblastoma cells showed that inhibition of several proteins could induce bulk autophagy in a manner dependent on PI3K (Lipinski et al., 2010). Some of these proteins, such as the complex IV subunit COX5A, were located in mitochondria, implicating directly an oxidative phosphorylation defect in autophagic induction (Lipinski et al., 2010). More work is needed to determine the signaling pathways and the mechanisms of bulk autophagy regulation in response to mitochondrial dysfunction.
In conclusion, we described the first model for Ndufaf5 deficiency in a simple eukaryote. The lack of Ndufaf5 or the expression of proteins carrying pathogenic mutations leads to a loss-of-function phenotype involving AMPK-independent phenotypes in growth and development. In addition, we described unexpected connections between CI deficiency and cellular homeostasis mechanisms such as autophagy that should be considered in future studies as new players in the cytopathology of the mitochondrial disease.
MATERIALS AND METHODS
Dictyostelium growth, transformation, and development
Dictyostelium AX4 cells were grown in HL5 medium (ForMedium, Hunstanton, United Kingdom) or in association with Klebsiella aerogenes (SM plates) as previously described (Sussman, 1987). Transfections were carried out as previously described (Pang et al., 1999). For synchronous development, Dictyostelium cells growing in HL5 medium were centrifuged, resuspended in PDF buffer (20 mM KCl, 9 mM K2HPO4, 13 mM KH2PO4, 1 mM CaCl2, 1 mM MgSO4, pH 6.4), and deposited onto nitrocellulose filters (Shaulsky and Loomis, 1993). Spore viability was measured as described before (Eichinger and Rivero, 2006).
Disruption of ndufaf5
For the disruption construct, N- and C-terminal fragments from Dictyostelium ndufaf5 gene were generated by PCR using Dictyostelium AX4 genomic DNA and sequentially cloned at either side of a blasticidin-resistance (BsR) cassette derived from pUCBsrBam vector (Adachi et al., 1994). The insertion of the BsR cassette by homologous recombination interrupted the gene at the coding nucleotide 206, which corresponds to the encoded amino acid 69, located 10 nucleotides upstream of the small intron of the gene. Because the C-terminal fragment begins at the encoded amino acid 85, a small deletion of 16 amino acids was created, which would also generate a frameshift. After transfection of wild-type AX4 cells and BsR selection, cells were plated on SM plates for clonal isolation and screened for homologous recombination into the target gene. To verify the absence of Ndufaf5 mRNA, RNA was isolated from wild-type and mutant strains (developed for 14 h) using TRIzol Reagent (Sigma-Aldrich, St. Louis, MO) and used in RT-PCRs with the oligonucleotides indicated in Supplemental Table S1.
ATP measurements and mitochondrial enzyme activities
For ATP measurements 5 × 106 wild-type or ndufaf5− cells grown in HL5 medium were washed and resuspended in water to a final volume of 1 ml. A 900-μl amount was mixed with 100 μl of trichloroacetic acid 10% in H2O and vortexed for 10 min. After centrifugation (10,000 × g, 5 min) the ATP content was measured in the supernatant using the ATP Determination Kittime-stable assay (Proteinkinase.de, Biaffin, Kassel, Germany). The luminescence was measured in a GloMax96 luminometer (Promega, Madison, WI). Respiratory enzyme activities and citrate synthase activity were measured as previously described (Tiranti et al., 1995; Carilla-Latorre et al., 2010; Rodenburg, 2011).
GFP expression, mitochondrial labeling, and confocal microscopy
Dictyostelium C20orf7 was amplified from genomic DNA and cloned into pGEM-T Easy vector (Promega). The fragment was subsequently cloned into pDV-CGFP-CTAP vector (GeneBank: EF028672), where the expression of the gene is under the control of the actin-15 promoter. Thereby, a C-terminal C20orf7-GFP fused protein was expressed in the indicated strains. For mitochondrial labeling 1 × 106 cells transfected with the aforementioned construct were incubated in HL5 (ForMedium) at 22ºC for 1 h with 500 nM MitoTracker Red CMXRos (Molecular Probes, Eugene, OR). After washing for 1 h with fresh HL5 medium, cells were deposited on a microscope coverslip and fixed with 3.7% formaldehyde in HL5 for 15 min. After washing with phosphate-buffered saline (PBS; 136 mM NaCl, 2.7 mM KCl, 10,1 mM Na2HPO4, 1.8 mM KH2PO4), they were mounted with Prolong (Molecular Probes) and analyzed by confocal microscopy. Confocal microscopy was performed using a Zeiss LSM 710 laser-scanning microscope (Zeiss, Jena, Germany) with a Plan-Apochromat 63×/1.40 objective. Software Zen 2009 and Photoshop CS (Adobe, San Jose, CA) were used to process all images.
Site-directed mutagenesis
Amino acid substitutions G86V, L165F, and L235P were performed by PCR using the oligonucleotides listed in Supplemental Table S1 containing the desired mutations. The template was ndufaf5 cloned in pGEM-T Easy (see earlier description). After the PCR the DNA was digested with DpnI enzyme, and after purification it was transformed into DH5α bacteria for cloning. The DNA was extracted by miniprep, and the mutations were confirmed by sequencing. The fragments were then cloned in pDV-CGFP-CTAP as previously described for the wild-type gene. The constructs were transformed into ndufaf5− cells, and the resulting stably transformed strains were analyzed for growth in liquid (HL5) and SM plates. Mitochondrial localization of the mutated proteins was analyzed by laser confocal microscopy as described.
Phototaxis
Qualitative and quantitative experiments for slug phototaxis were carried out with wild-type and ndufaf5− slugs as previously described (Darcy et al., 1994; Carilla-Latorre et al., 2010). AMPKas transfection and copy number quantification were done as before (Bokko et al., 2007).
Autophagy analyses
For analysis of autophagosomes, wild-type, ndufaf5−, and midA− cells were transfected with a construct expressing the autophagic marker GFP-Atg18 described previously (King et al., 2011). After hygromycin selection, cells were kept in HL5 medium and visualized by confocal microscopy as described. Z-stacks were selected to get maximum projections covering the whole cell. For starvation, cells grown in HL5 were washed two times and incubated for 1 h in PDF buffer and visualized as before. For mitophagy experiments, Atg18-GFP–transfected cells growing in HL5 medium were incubated for 1 h with MitoTracker Red CMX Ros and treated as before. For DAPI staining, cells were allowed to attach to a coverslip and fixed with 3% paraformaldehyde in PBS. After three washes with PBS, cells were stained with 1 μg/ml DAPI (Sigma-Aldrich) for 5 min and mounted for confocal microscopy as described.
Autophagic flux was measured as before (Calvo-Garrido et al., 2011). Briefly, GFP-Tkt-1–transfected cells growing in HL5 were washed and resuspended in fresh HL5 medium to a final concentration of 1 × 106 cells/ml. NH4Cl was added to a concentration ranging from 0 to 100 mM to slow down the autophagic kinetics, allowing degradation of GFP-Tkt-1 to be observed as previously published (Calvo-Garrido et al., 2011). After 2 h, NH4Cl was added again for an additional 2 h (4 h total incubation). Cells were collected by centrifugation and resuspended in RIPA buffer. The protein content was quantified using the Bradford assay (Bio-Rad, Hercules, CA). A 3.5-μg amount of protein was loaded onto a 12% SDS–PAGE gel. After blotting to a polyvinylidene fluoride membrane, GFP and GFP-Tkt-1 were detected with α-GFP antibody (Sigma-Aldrich).
Statistical analysis
Except where otherwise specified, results are shown as mean values with SD from at least three independent experiments. The significance of differences between groups was determined using the Student's t test. Phototaxis was analyzed using directional statistics based on the von Mises distribution as previously described (Annesley and Fisher, 2009b).
Bioinformatics analysis
Ndufaf5 protein sequences from humans D. discoideum and a representative group of homologues in α, β, and γ-proteobacteria (19 proteins), obtained previously using Blast, were aligned using ClustalW2 (Larkin et al., 2007) and displayed with Jalview (Waterhouse et al., 2009). The alignment was curated with Jalview, eliminating extended gaps and N- and C- terminal residues for the next-step analysis. A phylogenetic tree using distance matrix methods (neighbor joining) was generated with ClustalW2 (Larkin et al., 2007).
Supplementary Material
Acknowledgments
This work was supported by Grants BFU2009-09050 and BFU2012-32536 from the Spanish Ministerio de Ciencia e Innovación. Sequence data for Dictyostelium were obtained from the Genome Sequencing Centers of the University of Cologne, Cologne, Germany; the Department of Genome Analysis, Institute of Molecular Biotechnology, Jena, Germany; the Baylor College of Medicine, Houston, TX; and the Sanger Center in Hinxton, Cambridge, United Kingdom. We thank Diego Navarro for his assistance with confocal microscopy and Rosa Calvo for her help and advice during the course of this work.
Abbreviations used:
- AMPK
AMP-activated protein kinase
- CI
mitochondrial complex I or NADH:ubiquinone oxidoreductase
- NDUFAF
NADH dehydrogenase (ubiquinone) complex I assembly factor
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-11-0796) on March 27, 2013.
REFERENCES
- Adachi H, Hasebe T, Yoshinaga K, Ohta T, Sutoh K. Isolation of Dictyostelium discoideum cytokinesis mutants by restriction enzyme-mediated integration of the blasticidin S resistance marker. Biochem Biophys Res Commun. 1994;205:1808–1814. doi: 10.1006/bbrc.1994.2880. [DOI] [PubMed] [Google Scholar]
- Annesley SJ, Fisher PR. Dictyostelium discoideum—a model for many reasons. Mol Cell Biochem. 2009a;329:73–91. doi: 10.1007/s11010-009-0111-8. [DOI] [PubMed] [Google Scholar]
- Annesley SJ, Fisher PR. Dictyostelium slug phototaxis. Methods Mol Biol. 2009b;571:67–76. doi: 10.1007/978-1-60761-198-1_4. [DOI] [PubMed] [Google Scholar]
- Bokko PB, Francione L, Bandala-Sanchez E, Ahmed AU, Annesley SJ, Huang X, Khurana T, Kimmel AR, Fisher PR. Diverse cytopathologies in mitochondrial disease are caused by AMP-activated protein kinase signaling. Mol Biol Cell. 2007;18:1874–1886. doi: 10.1091/mbc.E06-09-0881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges HR, Fearnley IM, Hirst J. The subunit composition of mitochondrial NADH:ubiquinone oxidoreductase (complex I) from Pichia pastoris. Mol Cell Proteomics. 2010;9:2318–2326. doi: 10.1074/mcp.M110.001255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo SE, et al. High-throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex I deficiency. Nat Genet. 2010;42:851–858. doi: 10.1038/ng.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo-Garrido J, Carilla-Latorre S, Kubohara Y, Santos-Rodrigo N, Mesquita A, Soldati T, Golstein P, Escalante R. Autophagy in Dictyostelium: genes and pathways, cell death and infection. Autophagy. 2010;6:686–701. doi: 10.4161/auto.6.6.12513. [DOI] [PubMed] [Google Scholar]
- Calvo-Garrido J, Carilla-Latorre S, Mesquita A, Escalante R. A proteolytic cleavage assay to monitor autophagy in Dictyostelium discoideum. Autophagy. 2011;7:1063–1068. doi: 10.4161/auto.7.9.16629. [DOI] [PubMed] [Google Scholar]
- Carilla-Latorre S, et al. MidA is a putative methyltransferase that is required for mitochondrial complex I function. J Cell Sci. 2010;123:1674–1683. doi: 10.1242/jcs.066076. [DOI] [PubMed] [Google Scholar]
- Carroll J, Fearnley IM, Skehel JM, Runswick MJ, Shannon RJ, Hirst J, Walker JE. The post-translational modifications of the nuclear encoded subunits of complex I from bovine heart mitochondria. Mol Cell Proteomics. 2005;4:693–699. doi: 10.1074/mcp.M500014-MCP200. [DOI] [PubMed] [Google Scholar]
- Darcy PK, Wilczynska Z, Fisher PR. Genetic analysis of Dictyostelium slug phototaxis mutants. Genetics. 1994;137:977–985. doi: 10.1093/genetics/137.4.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efremov RG, Baradaran R, Sazanov LA. The architecture of respiratory complex I. Nature. 2010;465:441–445. doi: 10.1038/nature09066. [DOI] [PubMed] [Google Scholar]
- Eichinger L, Rivero F, editors. Totowa, NJ: Humana Press; 2006. Dictyostelium discoideum Protocols. [Google Scholar]
- Escalante R. Dictyostelium as a model for human disease. Semin Cell Dev Biol. 2011;22:69. doi: 10.1016/j.semcdb.2010.12.001. [DOI] [PubMed] [Google Scholar]
- Escalante R, Vicente JJ. Dictyostelium discoideum: a model system for differentiation and patterning. Int J Dev Biol. 2000;44:819–835. [PubMed] [Google Scholar]
- Fassone E, et al. FOXRED1, encoding an FAD-dependent oxidoreductase complex-I-specific molecular chaperone, is mutated in infantile-onset mitochondrial encephalopathy. Hum Mol Genet. 2010;19:4837–4847. doi: 10.1093/hmg/ddq414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearnley IM, Carroll J, Walker JE. Proteomic analysis of the subunit composition of complex I (NADH:ubiquinone oxidoreductase) from bovine heart mitochondria. Methods Mol Biol. 2007;357:103–125. doi: 10.1385/1-59745-214-9:103. [DOI] [PubMed] [Google Scholar]
- Francione LM, Annesley SJ, Carilla-Latorre S, Escalante R, Fisher PR. The Dictyostelium model for mitochondrial disease. Semin Cell Dev Biol. 2011;22:120–130. doi: 10.1016/j.semcdb.2010.11.004. [DOI] [PubMed] [Google Scholar]
- Gabaldon T, Huynen MA. From endosymbiont to host-controlled organelle: the hijacking of mitochondrial protein synthesis and metabolism. PLoS Comput Biol. 2007;3:e219. doi: 10.1371/journal.pcbi.0030219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerards M, et al. Defective complex I assembly due to C20orf7 mutations as a new cause of Leigh syndrome. J Med Genet. 2009;47:507–512. doi: 10.1136/jmg.2009.067553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilson PR, Yu XC, Hereld D, Barth C, Savage A, Kiefel BR, Lay S, Fisher PR, Margolin W, Beech PL. Two Dictyostelium orthologs of the prokaryotic cell division protein TfsZ localize to mitochondria and are required for the maintenance of normal mitochondrial morphology. Eukaryot Cell. 2003;2:1315–1326. doi: 10.1128/EC.2.6.1315-1326.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haack TB, et al. Exome sequencing identifies ACAD9 mutations as a cause of complex I deficiency. Nat Genet. 2010;42:1131–1134. doi: 10.1038/ng.706. [DOI] [PubMed] [Google Scholar]
- Hansen JJ, et al. Hereditary spastic paraplegia SPG13 is associated with a mutation in the gene encoding the mitochondrial chaperonin Hsp60. Am J Hum Genet. 2002;70:1328–1332. doi: 10.1086/339935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heide H, et al. Complexome profiling identifies TMEM126B as a component of the mitochondrial complex I assembly complex. Cell Metab. 2012;16:538–549. doi: 10.1016/j.cmet.2012.08.009. [DOI] [PubMed] [Google Scholar]
- Hiltunen JK, Autio KJ, Schonauer MS, Kursu VA, Dieckmann CL, Kastaniotis AJ. Mitochondrial fatty acid synthesis and respiration. Biochim Biophys Acta. 2010;1797:1195–1202. doi: 10.1016/j.bbabio.2010.03.006. [DOI] [PubMed] [Google Scholar]
- Hiltunen JK, Schonauer MS, Autio KJ, Mittelmeier TM, Kastaniotis AJ, Dieckmann CL. Mitochondrial fatty acid synthesis type II: more than just fatty acids. J Biol Chem. 2009;284:9011–9015. doi: 10.1074/jbc.R800068200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst J. Why does mitochondrial complex I have so many subunits. Biochem J. 2011;437:e1–e3. doi: 10.1042/BJ20110918. [DOI] [PubMed] [Google Scholar]
- Hoefs SJ, et al. Baculovirus complementation restores a novel NDUFAF2 mutation causing complex I deficiency. Hum Mutat. 2009;30:E728–736. doi: 10.1002/humu.21037. [DOI] [PubMed] [Google Scholar]
- Janssen RJ, Nijtmans LG, van den Heuvel LP, Smeitink JA. Mitochondrial complex I: structure, function and pathology. J Inherit Metab Dis. 2006;29:499–515. doi: 10.1007/s10545-006-0362-4. [DOI] [PubMed] [Google Scholar]
- Kagan RM, Clarke S. Widespread occurrence of three sequence motifs in diverse S-adenosylmethionine-dependent methyltransferases suggests a common structure for these enzymes. Arch Biochem Biophys. 1994;310:417–427. doi: 10.1006/abbi.1994.1187. [DOI] [PubMed] [Google Scholar]
- King JS, Veltman DM, Insall RH. The induction of autophagy by mechanical stress. Autophagy. 2011;7:1490–1499. doi: 10.4161/auto.7.12.17924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman WJ, Nijtmans LG, Dieteren CE, Roestenberg P, Valsecchi F, Smeitink JA, Willems PH. Mammalian mitochondrial complex I: biogenesis, regulation, and reactive oxygen species generation. Antioxid Redox Signal. 2010;12:1431–1470. doi: 10.1089/ars.2009.2743. [DOI] [PubMed] [Google Scholar]
- Larkin MA, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Lazarou M, Thorburn DR, Ryan MT, McKenzie M. Assembly of mitochondrial complex I and defects in disease. Biochim Biophys Acta. 2009;1793:78–88. doi: 10.1016/j.bbamcr.2008.04.015. [DOI] [PubMed] [Google Scholar]
- Lipinski MM, et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer's disease. Proc Natl Acad Sci USA. 2010;107:14164–14169. doi: 10.1073/pnas.1009485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, et al. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 2011;39:D225–D229. doi: 10.1093/nar/gkq1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie M, Ryan MT. Assembly factors of human mitochondrial complex I and their defects in disease. IUBMB Life. 2010;62:497–502. doi: 10.1002/iub.335. [DOI] [PubMed] [Google Scholar]
- McKenzie M, Tucker EJ, Compton AG, Lazarou M, George C, Thorburn DR, Ryan MT. Mutations in the gene encoding C8orf38 block complex I assembly by inhibiting production of the mitochondria-encoded subunit ND1. J Mol Biol. 2011;414:413–426. doi: 10.1016/j.jmb.2011.10.012. [DOI] [PubMed] [Google Scholar]
- Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mijaljica D, Prescott M, Devenish RJ. Mitophagy and mitoptosis in disease processes. Methods Mol Biol. 2010;648:93–106. doi: 10.1007/978-1-60761-756-3_6. [DOI] [PubMed] [Google Scholar]
- Niewmierzycka A, Clarke S. S-Adenosylmethionine-dependent methylation in Saccharomyces cerevisiae. Identification of a novel protein arginine methyltransferase. J Biol Chem. 1999;274:814–824. doi: 10.1074/jbc.274.2.814. [DOI] [PubMed] [Google Scholar]
- Nouws J, et al. Acyl-CoA dehydrogenase 9 is required for the biogenesis of oxidative phosphorylation complex I. Cell Metab. 2010;12:283–294. doi: 10.1016/j.cmet.2010.08.002. [DOI] [PubMed] [Google Scholar]
- Nouws J, Nijtmans LG, Smeitink JA, Vogel RO. Assembly factors as a new class of disease genes for mitochondrial complex I deficiency: cause, pathology and treatment options. Brain. 2012;135:12–22. doi: 10.1093/brain/awr261. [DOI] [PubMed] [Google Scholar]
- Ogilvie I, Kennaway NG, Shoubridge EA. A molecular chaperone for mitochondrial complex I assembly is mutated in a progressive encephalopathy. J Clin Invest. 2005;115:2784–2792. doi: 10.1172/JCI26020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliarini DJ, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang KM, Lynes MA, Knecht DA. Variables controlling the expression level of exogenous genes in Dictyostelium. Plasmid. 1999;41:187–197. doi: 10.1006/plas.1999.1391. [DOI] [PubMed] [Google Scholar]
- Penel S, Arigon AM, Dufayard JF, Sertier AS, Daubin V, Duret L, Gouy M, Perriere G. Databases of homologous gene families for comparative genomics. BMC Bioinformatics. 2009;10(Suppl 6):S3. doi: 10.1186/1471-2105-10-S6-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383–1435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- Rochet JC, Hay BA, Guo M. Molecular insights into Parkinson's disease. Prog Mol Biol Transl Sci. 2012;107:125–188. doi: 10.1016/B978-0-12-385883-2.00011-4. [DOI] [PubMed] [Google Scholar]
- Rodenburg RJ. Biochemical diagnosis of mitochondrial disorders. J Inherit Metab Dis. 2011;34:283–292. doi: 10.1007/s10545-010-9081-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saada A, Edvardson S, Shaag A, Chung WK, Segel R, Miller C, Jalas C, Elpeleg O. Combined OXPHOS complex I and IV defect, due to mutated complex I assembly factor C20ORF7. J Inherit Metab Dis. 2012;35:125–131. doi: 10.1007/s10545-011-9348-y. [DOI] [PubMed] [Google Scholar]
- Saada A, et al. Mutations in NDUFAF3 (C3ORF60), encoding an NDUFAF4 (C6ORF66)-interacting complex I assembly protein, cause fatal neonatal mitochondrial disease. Am J Hum Genet. 2009;84:718–727. doi: 10.1016/j.ajhg.2009.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma LK, Lu J, Bai Y. Mitochondrial respiratory complex I: structure, function and implication in human diseases. Curr Med Chem. 2009;16:1266–1277. doi: 10.2174/092986709787846578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaulsky G, Loomis WF. Cell type regulation in response to expression of ricin-A in Dictyostelium. Dev. Biol. 1993;160:85–98. doi: 10.1006/dbio.1993.1288. [DOI] [PubMed] [Google Scholar]
- Sugiana C, et al. Mutation of C20orf7 disrupts complex I assembly and causes lethal neonatal mitochondrial disease. Am J Hum Genet. 2008;83:468–478. doi: 10.1016/j.ajhg.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussman M. Cultivation and synchronous morphogenesis of Dictyostelium under controlled experimental conditions. Methods Cell Biol. 1987;28:9–29. doi: 10.1016/s0091-679x(08)61635-0. [DOI] [PubMed] [Google Scholar]
- Tiranti V, et al. Maternally inherited hearing loss, ataxia and myoclonus associated with a novel point mutation in mitochondrial tRNASer(UCN) gene. Hum Mol Genet. 1995;4:1421–1427. doi: 10.1093/hmg/4.8.1421. [DOI] [PubMed] [Google Scholar]
- Torija P, Vicente JJ, Rodrigues TB, Robles A, Cerdan S, Sastre L, Calvo RM, Escalante R. Functional genomics in Dictyostelium: MidA, a new conserved protein, is required for mitochondrial function and development. J Cell Sci. 2006;119:1154–1164. doi: 10.1242/jcs.02819. [DOI] [PubMed] [Google Scholar]
- van Es S, Wessels D, Soll DR, Borleis J, Devreotes PN. Tortoise, a novel mitochondrial protein, is required for directional responses of Dictyostelium in chemotactic gradients. J Cell Biol. 2001;152:621–632. doi: 10.1083/jcb.152.3.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel RO, et al. Human mitochondrial complex I assembly is mediated by NDUFAF1. FEBS J. 2005;272:5317–5326. doi: 10.1111/j.1742-4658.2005.04928.x. [DOI] [PubMed] [Google Scholar]
- Vogel RO, et al. Cytosolic signaling protein Ecsit also localizes to mitochondria where it interacts with chaperone NDUFAF1 and functions in complex I assembly. Genes Dev. 2007;21:615–624. doi: 10.1101/gad.408407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. Mitochondrial DNA mutations in disease and aging. Environ Mol Mutagen. 2010;51:440–450. doi: 10.1002/em.20586. [DOI] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CC, MacCoss MJ, Howell KE, Yates JR., 3rd A method for the comprehensive proteomic analysis of membrane proteins. Nat Biotechnol. 2003;21:532–538. doi: 10.1038/nbt819. [DOI] [PubMed] [Google Scholar]
- Yang W, Hekimi S. Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans. Aging Cell. 2010;9:433–447. doi: 10.1111/j.1474-9726.2010.00571.x. [DOI] [PubMed] [Google Scholar]
- Zurita Rendon O, Shoubridge EA. Early complex I assembly defects result in rapid turnover of the ND1 subunit. Hum Mol Genet. 2012;21:3815–3824. doi: 10.1093/hmg/dds209. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.