

Abstract
Lactoferrin is considered as a part of the innate immune system that plays a crucial role in preventing bacterial growth, mostly via an iron sequestration mechanism. Recent data show that bovine lactoferrin prevents late-onset sepsis in preterm very low birth weight neonates by serving as an iron chelator for some bacterial strains; thus, it is very important to control the iron saturation level during diet supplementation. An accurate estimation of lactoferrin iron saturation is essential not only because of its clinical applications but also for a wide range of biochemical experiments. A comprehensive method for the quantification of iron saturation in lactoferrin preparations was developed to obtain a calibration curve enabling the determination of iron saturation levels relying exclusively on the defined ratio of absorbances at 280 and 466 nm (A 280/466). To achieve this goal, selected techniques such as spectrophotometry, ELISA, and ICP-MS were combined. The ability to obtain samples of lactoferrin with determination of its iron content in a simple and fast way has been proven to be very useful. Furthermore, a similar approach could easily be implemented to facilitate the determination of iron saturation level for other metalloproteins in which metal binding results in the appearance of a distinct band in the visible part of the spectrum.
Electronic supplementary material
The online version of this article (doi:10.1007/s00216-013-6943-9) contains supplementary material, which is available to authorized users.
Keywords: Lactoferrin, Iron saturation, Absorption ratio, ICP-MS
Introduction
Lactoferrin (Lf), a protein being a member of the transferrin family, is highly abundant in colostrum and milk, but is also present in other extracellular excretions. This glycoprotein bears remarkable resemblance to transferrin—both proteins have a molecular weight of about 80 kDa and their structure comprises two lobes, each one capable of binding one ferric ion. The most important difference is lactoferrin possessing the ability to bind iron more tightly (binding constants of ∼1022–1024 and 1020–1021 M−1 for lactoferrin and transferrin, respectively, were reported) [1–3] and retain it at much lower pH values [4] than serotransferrin. Another lactoferrin-specific feature is a highly basic N-terminal region that allows interaction with negatively charged molecules such as lipopolysaccharide [5] or glycosaminoglycans [6]. Those two properties grant its ability to act as an antimicrobial agent and affecting immune response by binding to receptors on target cells.
Lactoferrin undergoes glycosylation in various species, but the number and location of glycosylation sites vary depending on the protein origin. Human lactoferrin has three potential N-glycosylation sites (Asn137, Asn478, and Asn623); cow, goat, and sheep lactoferrin has five (Asn233, Asn281, Asn368, Asn476, and Ans545) [7]. Folding of human lactoferrin does not depend on the number and location of bound glycan chains, and the role of glycosylation has not been elucidated so far. Several studies revealed that deglycosylation of human lactoferrin does not affect the binding of iron and other molecules, but decreases the proteolytic resistance of lactoferrin [8]. However, it has been reported that the glycosylation of camel lactoferrin influences the protein structure and iron binding mode within the cleft of the C-lobe. Supposedly, localization of the glycosylation site in the C-lobe inhibits the domain closer and weakens iron binding [9]. Lactoferrin is a basic protein with an isoelectric point of 8.7. To our knowledge, there are no data showing the influence of glycosylation or iron saturation on protein pI value. Basing on results reported for transferrin glycoforms (pentasialotransferrin, pI = 5.2; tetrasialotransferrin, pI = 5.4; trisialotransferrin, pI = 5.6; and disialotransferrin, pI = 5.7) [10], it should be expected that glycosylation should decrease the observed protein isoelectric point as a consequence of increasing the number of glycosylated arginine residues. Noticeable changes of the pI values should be anticipated upon saturation of latoferrin with iron (pI of 5.2 and 5.6 were reported for Fe2-Tf and Fe-Tf, respectively) [11]. The natural microheterogeneity of lactoferrin (different glycosylation status and iron saturation levels) makes lactoferrin isoforms difficult to separate.
Since nearly all living organisms need iron to live, iron scavengers—such as lactoferrin—might have a static effect on the growth of microorganisms in the gastrointestinal tract. The antimicrobial activity of lactoferrin has been frequently reported—its influence on bacterial, viral, protozoan, and fungal pathogen growth has been numerously reviewed [12–14]. However, very little attention has been paid to the iron saturation level of lactoferrin used in those studies. The iron content of lactoferrin preparations is an aspect somewhat neglected in the literature—only a few groups investigated the more accurate determination of the percentage saturation of lactoferrin with ferric ions and differentiation between the effects of the apo and holo forms on the studied pathogen species or target human cells. It has been shown that iron-free and iron-saturated lactoferrin affects human enterocytes in a different way [15, 16]. Furthermore, apolactoferrin has been described as more potent in reducing the growth of numerous bacterial strains [17–20].
Neither accurate determination of Lf iron content nor the preparation of apo- and hololactoferrin is a trivial issue. The simplest way of calculating the iron content in the preparation is based on spectrophotometric measurement. Total protein concentration can be determined using the absorption coefficient at 280 nm, while the concentration of the holo form can be estimated by employing molar absorption coefficient at 466 nm. The band in the visible part of spectrum with maximum approx. 470 nm is characteristic of iron-saturated lactoferrin. In the literature, several different absorption coefficients were described for both apo- and hololactoferrin; some of them are presented in electronic supplementary material (ESM) Table S1. However, the application of absorption coefficients to calculate the protein concentration is limited to the situation when only the apo or the holo form is present in solution. When the preparation is a mixture of both forms, the use of this method can lead to substantial deviations. To improve the accuracy of protein content determination, some groups applied ELISA [21], the method of Lowry [22], or the method of Dumas using a nitrogen–protein analyzer [23, 24]. Calculation of the iron content using the spectrophotometric method faces a problem related with the coexistence of diferric and monoferric lactoferrin forms. About 47–58 % of human serum transferrin is monoferric, while 16–52 % is in the apo form and differric transferrin accounts for 2–31 % depending on the origin of the blood samples [25]. To our knowledge, there are no analogous data reported for lactoferrin. Human lactoferrin was found to be saturated in 20 % [26], while bovine lactoferrin is saturated with iron in 15–19 % [27, 28]. The similarity of proteins of the lactoferrin family suggests that a significant percentage of lactoferrin may be in the monoferric form. The iron content of lactoferrin preparations can be accurately determined using inductively coupled plasma–mass spectrometry (ICP-MS) [29, 30] or optical emission spectrometry [31, 32] as well as atomic absorption spectrometry techniques. Recently, the commercially available iron/TIBC kit was also applied for the determination of iron saturation levels in apo- and holotransferrin, with no data for lactoferrin available yet [33].
The most convenient and easily accessible way of determining the iron content in a lactoferrin preparation, which is valuable not only for biomedical research but also for drug development sector, would be the usage of the ratio of absorbances at 280 and 466 nm. Such approach has been described several times in the literature. In the early 1980s, the value of 27–28 was proposed for the bovine lactoferrin fully saturated with iron [34], while quite recently, the value of 22 was suggested for fully saturated non-glycosylated human serum transferrin [35]. However, there is no information about using this parameter for lactoferrin only partially saturated with iron. To address the aforementioned problems, we have combined several techniques such as spectrophotometry, ELISA, and ICP-MS in order to prepare the calibration curve enabling determination of the iron saturation level of bovine lactoferrin relying solely on the A 280/466 ratio.
Materials and methods
Materials
Bovine lactoferrin was purchased from DMV International (estimated purity, 95 %; Veghel, the Netherlands) and the reference sample from Sigma-Aldrich Co. (≥85 % pure; St. Louis, MO, USA) and stored at 4 °C. Citric acid, acetic acid, and sodium chloride were analytical grade reagents supplied by POCH S.A. (Gliwice, Poland). Bovine β-lactoglobulin, lysozyme from chicken egg, ribonuclease A from bovine pancreas, bovine serum albumin, ferritin from equine spleen type I (saline solution), β-galactosidase from Escherichia coli, and other chemicals were of analytical grade and purchased from Sigma-Aldrich Co. Buffers and reagents in all experiments were prepared using Milli-Q water.
Electrophoresis: SDS-PAGE
The abundance of different protein species was examined using 10 % polyacrylamide gel electrophoresis under denaturing conditions [36]. Gels were stained with Coomassie Brilliant Blue and documented using GelDoc-It™ 310 Imaging System (Upland, CA, USA).
Preparation of bovine apo- and hololactoferrins
Lactoferrin supplied by the manufacturers was used without further purification. For apolactoferrin, preparation containing 50 mg/mL protein was dissolved in water and dialyzed extensively against 100 mM citrate buffer for 24 h, followed by dialysis against distilled water for 24 h [37]. Temperature (4 and 20 °C) and buffer pH (2.0–5.0) were modified in order to monitor their impact on iron desaturation. Iron saturation was calculated based on the A 280/A 466 ratio according to the calibration curve presented in “Results and discussion.” We define the iron saturation level of lactoferrin as the percentage of iron-binding sites occupied by ferric ions assuming that 2 mol of iron(III) ions is bound per 1 mol of protein. Thus, the given values refer to the percentage of differic lactoferrin. For some of these samples, the ICP-MS and ELISA tests were carried out and were used to prepare a calibration curve.
Hololactoferrin was prepared by the reaction of 50 mg/mL lactoferrin solution in 50 mM Tris–HCl, 150 mM NaCl (pH 7.4) with ferric nitrate salt in the presence of nitrilotriacetic acid (NTA) as well as different concentrations of sodium bicarbonate [24]. After incubation, excess iron was removed by dialysis against the same buffer solution without ferric salts for 24 h and against water for another 24 h. Various incubation times, temperatures, as well as ratios of Lf/Fe/NTA were employed to examine their effect on iron saturation efficiency; detailed conditions are depicted in the captions of figures.
Resaturation of apolactoferrin
The apolactoferrin prepared by dialyzing a sample of the original lactoferrin to citrate buffer (100 mM, pH 2.0, 3.0, and 4.0) at room temperature as described above was resaturated with iron ions in order to evaluate its chelation. Lf was incubated (1 h, room temperature) with ferric nitrate salt in the presence of nitrilotriacetic acid at a Lf/Fe/NTA ratio of 1:4:4.
FPLC procedures
The chromatographic system ÄKTA Purifier 10 (GE Healthcare, Life Sciences), with an injection loop of 50 μL, fraction collector FRAC-901, and UV/VIS (Monitor UV-900, GE Pharmacia) as well as conductivity (Monitor pH/C-900, GE Pharmacia) detectors were applied for protein separation.
Size exclusion chromatography
A Superdex 200 10/300 GL column (10 × 300-mm ID, 13-μm particle diameter; Tricorn, GE Healthcare Life Science) was equilibrated with phosphate buffer (50 mM, 150 mM NaCl, pH 8.4) at a flow rate of 0.5 mL/min and was kept at 20 °C. All buffers before use in fast protein liquid chromatography (FPLC) system were degassed and filtered through a 0.45-μm filter. Calibration of the column was achieved by using the following molecular mass markers: ribonuclease A (13.7 kDa), lysozyme (14.3 kDa), β-lactoglobulin (35.0 kDa), bovine serum albumin (66 kDa), ferritin (440 kDa), and β-galactosidase (465 kDa). The sample of lactoferrin (DMV International or Sigma-Aldrich) was diluted in the same buffer, filtered through a 0.22-μm membrane filter, and subsequently loaded into a 50-μl loop and injected into the column. After sample introduction, the protein was eluted with the aforementioned buffer. Detection wavelength was set at 280 nm.
Ion exchange chromatography
A MonoS 5/50 GL column (5 × 50-mm ID, 10-μm particle diameter; Tricorn, GE Healthcare, Life Science) was used for CIEX separation. Various conditions of the separation (different eluent compositions, pH, gradients, and flow rates) were applied to achieve the separation of three forms of lactoferrin: apo-, holo-, and monoferric. Finally, to obtain a homogenous Lf preparation, solutions of 50 mM potassium phosphate (pH 7.5), A, and 50 mM potassium phosphate, 1.5 M potassium chloride (pH 7.5), B, were used as eluents. The column was equilibrated at 1 mL/min with buffer A. Separation was monitored spectrophotometrically at 280 nm as well as by conductivity measurement, while the temperature of the column was kept at 21 °C. Lactoferrin solution (10 mg/mL; DMV International) was diluted in buffer A, filtered through a 0.22-μm membrane filter, and subsequently loaded into a 1-mL loop and injected into the column. After sample application, the column was washed with 5 column volumes at a flow rate of 2 mL/min with A. Lactoferrin adsorbed on the column was eluted with a step gradient—from 0 to 25 % B and from 25 to 75 % B at a flow rate of 1 mL/min—after which the column was washed with 5 column volumes with B. Fractions of 1 mL were collected during the chromatographic run. As a result of several purifications, no separation of peaks corresponding to the different iron-saturated lactoferrins was achieved. In order to discriminate the effect of glycosylation from those of iron saturation on protein separation, the fraction that was pooled from the central part of the major peak identified as a non-glycosylated form of Lf (Fig. 2) was lyophilized and modified to obtain both apo- and hololactoferrins, as described in “Preparation of bovine apo- and hololactoferrins.” The prepared non-glycosylated apo- and hololactoferrin samples were loaded separately into a 50-μl loop, injected into the MonoS column, and chromatographic separation was performed by utilizing the procedure described above.
Fig. 2.

Evaluation of the resolution properties of the MonoS 5/50 GL column used to separate bovine lactoferrin in a two-step purification. a MonoS chromatography of DMV International lactoferrin (the collected fraction highlighted in gray bar). b Fraction obtained from the first ion exchange separation was modified using the methods described in experimental section to get iron-saturated and iron-depleted lactoferrin forms. c Elution profiles of apo- and hololactoferrins from the MonoS column from the second separation: apo-Lf (black), holo-Lf (red)
A MonoQ 5/50 GL separation column was also used for AEX separation. Our attempts—involving usage of various conditions for the ion exchange chromatographic method—to isolate peaks corresponding to different iron-saturated lactoferrins were unsuccessful (results not shown).
Spectroscopic measurements
UV/VIS spectra were recorded using a Perkin Elmer Lambda 35 spectrophotometer, with photometric accuracy ±0.010 A (measured for potassium dichromate) and photometric reproducibility <0.001 A. Samples were appropriately diluted when necessary to obtain absorbance below 1. The obtained A 280/A 466 ratios were subsequently used to determine the iron saturation of lactoferrin preparations applying the calibration curve presented in “Results and discussion.” Measurements of circular dichroism (CD) spectra were conducted with a JASCO J-710 spectropolarimeter (JASCO, Tokyo, Japan). The range of measurement (250–187 nm) was selected after sample test scan. Measurements were carried out for lactoferrin purchased from DMV International and Sigma-Aldrich as well as the apo- and hololactoferrins obtained by purification (using cation exchange chromatography) and iron saturation/desaturation of commercially available products. All samples were prepared in 50 mM Tris–HCl buffer (pH 7.4).
Enzyme-linked immunosorbent assay
ELISA was conducted using bovine lactoferrin (bLf) ELISA kit (Alpha Diagnostic International, San Antonio, TX, USA). A standard curve was prepared with the supplied bLf standard with concentration ranging from 10 to 140 ng/mL. The tested samples were diluted 500,000 and 1,000,000 times in a sample diluent buffer and the test performed according to the manufacturer’s protocol. ELISA plates were read using Tecan Infinite 200 Reader plate at 450 nm to determine Lf concentration. Measurement at 630 nm was performed for background subtraction. Tests were performed for the samples with the lowest A 280/A 466 ratio as well as partially saturated lactoferrin samples, with the results used for calibration curve preparation.
Inductively coupled plasma–mass spectrometry
The total iron content in lactoferrin was measured by the application of ICP-MS. For ICP-MS measurements, lactoferrin samples with the lowest values of A 280/A 466 and partially saturated lactoferrin preparations were chosen. Prior to the determination of iron content, 100 μL of the samples was diluted with 6 mL of concentrated HNO3 and mineralized using Microwave Digestion System: Multiwave 3000 (Anton Paar) by employing 1,000-W power, 15-min ramp, followed by a 20-min hold with a rate of 0.4 bar s−1 up to 50 bar, at 213–230 °C. Iron concentration was determined in these samples with the ICP-MS technique performed with an ELAN DRCe Perkin Elmer spectrometer, employing the following conditions: plasma gas (Ar), 15 L min−1; Rf power, 1,270 W; nebulizer gas flow, 0.99 L min−1; auxiliary gas flow, 1.20 L min−1. The 57Fe isotope was used for the quantification of iron content in samples.
Results and discussion
Purity check
In order to verify the information provided by the manufacturers concerning the purity of lactoferrin preparations, two complementary techniques were exploited. First, denaturing electrophoresis in polyacrylamide gel was performed, yielding one distinct band of molecular weight approx. 80 kDa (Fig. 1a) for the DMV International and Sigma lactoferrin samples. Few lower-molecular-weight bands were also detected for both commercially available lactoferrin preparations. The presence of these bands was previously reported [38] and identified as degraded forms of lactoferrin. The purity of lactoferrin supplied by DMV International was densitometrically estimated as over 90 % and was higher than the purity of Lf preparation purchased from Sigma-Aldrich. The second approach involved the chromatographic separation of lactoferrin preparations on the gel filtration column. In order to overcome the problem of the unspecific binding of protein to the dextran resin observed when performing separation at pH ranging from 7.0 to 8.0, a higher value of pH 8.4 and increased salt concentration were used during the final size exclusion chromatography (SEC) separation. The main peak corresponding to the protein of molecular weight of approx. 80 kDa along with the minor peaks from smaller proteins were observed, which is consistent with electrophoretic analysis (Fig. 1b). Both approaches confirmed the high purity of DMV lactoferrin preparation. Therefore, DMV International’s preparation was used for further experiments; no additional purification procedures were performed in the following experiments.
Fig. 1.

a SDS polyacrylamide gel electrophoresis of DMV International (left) and Sigma-Aldrich (right) bovine lactoferrin preparations. Lanes were loaded with 2.5, 5, and 10 μg of protein. Central lane was loaded with a molecular weight marker (two bands of 70 and 100 kDa denoted on the electropherogram). b SEC elution profile of DMV International lactoferrin obtained using the Superdex 200 10/300 GL column
The starting preparation of lactoferrin from DMV International contains approx. 10 % of holo-lactoferrin; therefore, the analytical cation exchange chromatography using MonoS was performed to separate apolactoferrin from its holo form according to the protocol described by Makino and Nishimura [39]. We observed that Lf was consistently eluted as one asymmetric peak. As a result of the first purification step, the major peak was observed at 0.63 M KCl and corresponds to a non-glycosylated form of Lf, whereas the small shoulder eluted at 0.53 M KCl contains protein with an N-linked glycan at the Asn281 position (Fig. 2a). Our results proved previous findings that N-linked glycosylation of bovine lactoferrin affects its elution from MonoS [40], but we have not been able to reproduce the observation of Makino and Nishimura [39] that apo- and iron-saturated lactoferrins are eluted at different positions from a MonoS column that allows their separation. To obtain homogeneous lactoferrin preparation, the MonoS elution fraction containing the non-glycosylated variant of lactoferrin was pooled, iron-saturated/desaturated, and subjected to re-chromatography (Fig. 2c).
MonoS chromatography of the non-glycosylated iron-saturated bovine lactoferrin (>65 %) showed no significant changes in the elution profile when compared with its apo form (<2 %), indicating that iron saturation does not affect bovine Lf binding and elution Lf from the MonoS column. During the second chromatographic step, homogenous apo- and hololactoferrins consistently eluted at 0.76 M KCl in a single symmetric peak. Our results are in line with earlier reports that apo- and iron-saturated human Lf bind equally well to anionic ligands and exhibit the same profile on MonoS [8, 41, 42]. It is possible that an extraordinary charge asymmetry encountered in lactoferrin protein (the N-terminus of this protein has strong cationic peptide regions) is the main driving force for electrostatic interactions. The degree of iron saturation, which probably alters lactoferrin pI and conformation, seems to be less important for ion exchange chromatography.
Method for the estimation of iron saturation in lactoferrin preparation
As mentioned before, employment of spectrophotometric measurements as a simple and rapid method for the estimation of iron content in the starting material as well as in the processed samples might not yield reliable and accurate results. This is mainly due to the high diversity of absorption coefficients for apo- and holo-Lf that can be found in the literature (see Table S1), which arise from the difficulty in the preparation of standards. We have shown that chromatographic separation between these two species is challenging and that no satisfactory results were obtained. Furthermore, most often, lactoferrin preparation is a mixture of various Lf species (apo, holo, monoferric N-domain, and monoferric C-domain forms), and using just one coefficient does not reflect their complexity. To overcome the problems associated with spectrophotometric measurements, a calibration curve for the estimation of iron content was prepared by the combination of two analytical methods. ELISA was employed for accurate and specific quantitation of Lf in the samples and ICP-MS served as a method for determining iron content. We still used UV/VIS spectrophotometry for the rapid and inexpensive screening of procedures leading to obtaining apo- and hololactoferrin preparations. Our goal was to show the relationship between the obtained ratios of A 280/A 466 (values that do not involve the application of molar absorption coefficients) and precise iron content in Lf samples determined using ELISA/ICP-MS tandem. The obtained data are presented in Fig. 3.
Fig. 3.

Correlation between the A 280/A 466 ratios and lactoferrin iron saturation calculated from ELISA and ICP-MS measurements
There is a nonlinear correlation between the measured absorbance ratios and iron saturation levels calculated from the values obtained with ELISA and ICP-MS measurements. This can be explained by the vast difference between the absorption coefficients for bands at 280 and 466 nm. The obtained data imply the power relationship shown in Eq. 1 for which the best fit was achieved (Fig. 3).
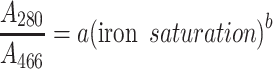 |
1 |
where 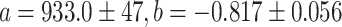
To our best knowledge, there have been no reports in the literature enabling the utilization of the A 280/A 466 ratio for the estimation of the iron content in lactoferrin preparation in a wide range of saturation. Based on the proposed fitting, the calculated value for 100 % hololactoferrin would be approx. 22, which is in very good agreement with the value of approx. 22 found for non-glycosylated human serum transferrin [35]. It must be noted that very small differences in the A 280/A 466 ratio for the highly saturated lactoferrin preparations give a large change in the iron saturation quantity; the inaccuracy of the calculated values can be as high as 10–15 %. The preparation of apolactoferrin with iron saturation below 2 % is characterized by the value of A 280/A 466 higher than approx. 530. According to the results presented in this work, a relatively large variation in the value of A 280/A 466 at very low iron saturation gives only a small contribution to the changes in iron content, while accuracy of the measurement of absorbance at 466 nm is decreasing. Thus, for the precise determination of iron content when saturation is below 2 %, the application of ELISA/ICP-MS tandem is recommended. Iron saturation for the bovine lactoferrin acquired from DMV International calculated using the presented calibration curve was found to be 7.72 ± 0.97 %, while the two independent measurements using ELISA and ICP-MS methods gave a value of 10.2 ± 0.2 %. Our results provide an effective method for a quick and simple estimation of the iron saturation levels in bovine lactoferrin preparations. However, if there is a need for a very accurate determination of iron concentration, only the application of ELISA/ICP-MS tandem can yield reliable results.
Apolactoferrin preparation
In acidic solutions (pH below 5), lactoferrin eagerly adopts more flexible and open conformation, which facilitates the release of ferric ion [7, 43]. Following the literature recommendation, we used citrate as an efficient mediator of iron transport from transferrins [37]. The efficiency of iron removal from Lf in citrate buffer was tested in a pH range of 2–5 (Fig. 4). Iron removal was carried out at room temperature (approx. 20 °C) since it was found that this process is slightly more efficient than at 4 °C. It is very likely that the protein structure becomes more flexible with temperature, which increases the probability of iron release. The optimal pH value for iron desaturation (obtaining <2 % of differic lactoferrin) was ≤3.5. Previous publications show similar results, which indicate that between pH 2 and 3, iron release from lactoferrin reaches a plateau [31, 44]. It has been shown that apolactoferrin, upon dialysis at pH 4, exhibits iron saturation of 2–2.5 % [24, 31].
Fig. 4.

Effect of citrate buffer pH during dialysis at room temperature on lactoferrin iron saturation. Data were obtained through the measurement of absorbance ratio at 280 and 466 nm and converted to lactoferrin iron saturation using the calibration curve described in previous section
Once retaining the ability of efficient ferric ion binding by the preparation of apolactoferrin, which is crucial, the apo-Lf sample was saturated with iron according to the procedure described in “Materials and methods.” We have observed an effective resaturation with ferric ions for the protein sample obtained at different pH values (Fig. 5), which supports the assumption that apolactoferrin preparation does not affect protein function—lactoferrin was proven to still be capable of binding iron. Moreover, the CD spectra of the original lactoferrin preparation and both apo- (pH 4) and holo-Lf do not differ (ESM Fig. S1). Therefore, procedures leading to obtaining those species of lactoferrin do not interrupt the secondary structure of the protein and, most probably, do not influence its functions.
Fig. 5.

Comparison of iron saturation of the original lactoferrin preparation (Lf DMV International) and hololactoferrins (holo-Lf) prepared by resaturation with ferric ions of Lf DMV International as well as apo-Lf obtained at various pH values
It seems surprising that effective resaturation occurs also for apolactoferrin preparations obtained at low pH 2.0 and 3.0. It was shown that at these pH values, apo-Lf undergoes thermal denaturation at room temperature [31], and one can expect a substantially lower iron binding. However, we observed a lower protein concentration for these conditions, which suggests that, indeed, a part of the protein molecules is denatured, but saturation with iron is only marginally influenced. Therefore, the use of pH 4 provides a compromise between preserving lactoferrin in a non-denatured state and productive iron release from the protein.
Hololactoferrin preparation
To obtain iron-saturated lactoferrin, reaction of Lf with ferric salts in neutral pH buffer (50 mM Tris–HCl, 150 mM NaCl, pH 7.4) was utilized to provide optimal conditions for an effective iron uptake. An iron chelator—nitrilotriacetic acid (NTA) [45]—was employed to prevent unspecific binding of ferric ions to protein molecules. NTA forms the 2:1 coordination complex with iron(III), Fe(NTA)2, with a pK ML2 of approx. 8 [46]. Therefore, it is a much weaker chelator than the lactoferrin binding pocket, however strong enough to shield the protein against iron binding in a nonspecific manner. A few different ratios of protein/iron/NTA were used, yielding comparable results measured by the percentage of iron saturation of Lf (Fig. 6a). Only when the excess of NTA was present in the solution was a noticeable decrease in the iron saturation of Lf observed. Nevertheless, lactoferrin affinity for iron is strong enough to sequester iron from the Fe(NTA)2 complex. For all following procedures, the protein/iron/NTA ratio of 1:4:4 was utilized. Similar conditions were also applied by other researchers [8, 24]. The binding of iron by lactoferrin involves the coordination of a synergistic carbonate anion that is coordinated in a bidentate manner [47]. Previous studies have proved that other ions such as citrate or oxalate can also substitute carbonate ion [48]. Our studies have shown that in the absence of the external added sodium bicarbonate as well as in its presence in the range 0.1–1.0 M, the extent of iron saturation is very similar (results not shown). Even without the addition of sodium bicarbonate, CO3 2− can still be employed as a synergistic ion due to the hydration of CO2 dissolved in aqueous solution. The engagement of nitrilotriacetate in coordination of iron in the lactoferrin pocket is unlikely, as shown by other studies [49].
Fig. 6.

Analysis of lactoferrin iron saturation under different conditions. a Influence of the Lf/Fe/NTA ratio on the iron saturation process carried out overnight. b Effect of saturation period on the overall efficiency of this process. Lf/Fe/NTA was kept as 1:4:4
Another factor examined was the time required for the full saturation of the iron binding pockets of lactoferrin. The change in the color of native lactoferrin was observed immediately upon the addition of a mixture of ferric salt and NTA. The undergoing reaction was examined employing stopped-flow spectroscopic measurements. All obtained results point to a fast and effective process of Lf iron saturation, reaching its plateau after approx. 10 s (ESM Fig. S2). This finding is in good agreement with previously reported data, which suggests that most of the iron uptake by lactoferrin occurs within a few seconds, followed by a very slow reaction giving only small changes in absorption at 466 nm [50]. In order to obtain a fully saturated protein, prolonged saturation periods were tested. However, no significant difference was observed as a result of the application of longer timescales (Fig. 6b). These observations confirm that lactoferrin acts as a thermodynamically and kinetically efficient chelator for iron(III) ion.
Despite changing the saturation conditions, the lowest ratio for A 280/466 was 26, which corresponds to approx. 80 % of differic lactoferrin form. In the literature, an absorbance ratio between 27 and 29 was accepted as a fully saturated lactoferrin [51]. However, our findings point out that a ratio of 22 must be reached in order to obtain 100 % saturation, in agreement with other studies performed for transferrin [35].
Conclusions
We proposed in this article the simple, rapid, and high-throughput method for obtaining a high-quality, substantially pure, structurally intact, and biologically functional samples of lactoferrin with its iron content determined. The suggested protocol can be employed for further studies aimed at verification of the biological/medicinal properties of the apo and holo forms of lactoferrin. Apolactoferrin has been attributed with multifunctional properties, including its potential to act as a promising antibacterial agent that could efficiently deprive bacteria of iron and inhibit their growth. However, it seems essential to retain its ability to bind ferric ions with high affinity—use of pH 4 seems a fine compromise between preserving lactoferrin function and obtaining its apo form with iron saturation levels of approx. 2 %. Nevertheless, whether apolactoferrin prepared in such a manner is truly an iron-depleted form for bacterial pathogens is an interesting question requiring further investigation.
Electronic supplementary material
Below is the link to the electronic supplementary material.
(PDF 123 kb)
Acknowledgments
This research was carried out with equipment purchased with financial support from the European Regional Development Fund within the framework of the Polish Innovation Economy Operational Program (contract no. POIG.0 2.01.00-12-0 23/08). G.M. acknowledges the financial support from the project Interdisciplinary PhD Studies “Molecular sciences for medicine” (co-financed by the European Social Fund within the Human Capital Operational Programme). Halina Mrowiec is kindly acknowledged for performing the ICP-MS measurements.
Footnotes
Grzegorz Majka and Klaudyna Śpiewak contributed equally to this work.
Contributor Information
Magdalena Strus, Phone: +48-12-6336033, Email: magdalena.strus@uj.edu.pl.
Małgorzata Brindell, Phone: +48-12-6632221, FAX: +48-12-6340515, Email: brindell@chemia.uj.edu.pl.
References
- 1.Aisen P, Leibman A. Lactoferrin and transferrin: a comparative study. Biochim Biophys Acta—Protein Struct. 1972;257(2):314–323. doi: 10.1016/0005-2795(72)90283-8. [DOI] [PubMed] [Google Scholar]
- 2.Baker EN (1994) Structure and reactivity of transferrins. In: Sykes AG (ed) Advances in inorganic chemistry, vol. 41. Academic, San Diego, pp 389–463
- 3.Aisen P, Leibman A, Zweier J. Stoichiometric and site characteristics of the binding of iron to human transferrin. J Biol Chem. 1978;253(6):1930–1937. [PubMed] [Google Scholar]
- 4.Mazurier J, Spik G. Comparative study of the iron-binding properties of human transferrins: I. Complete and sequential iron saturation and desaturation of the lactotransferrin. Biochim Biophys Acta—Gen Subjects. 1980;629(2):399–408. doi: 10.1016/0304-4165(80)90112-9. [DOI] [PubMed] [Google Scholar]
- 5.Appelmelk BJ, An YQ, Geerts M, Thijs BG, de Boer HA, MacLaren DM, de Graaff J, Nuijens JH. Lactoferrin is a lipid A-binding protein. Infect Immun. 1994;62(6):2628–2632. doi: 10.1128/iai.62.6.2628-2632.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.El Yazidi-Belkoura I, Legrand D, Nuijens J, Slomianny M-C, van Berkel P, Spik G. The binding of lactoferrin to glycosaminoglycans on enterocyte-like HT29-18-C1 cells is mediated through basic residues located in the N-terminus. Biochim Biophys Acta—Gen Subjects. 2001;1568(3):197–204. doi: 10.1016/S0304-4165(01)00222-7. [DOI] [PubMed] [Google Scholar]
- 7.Baker EN, Baker HM. A structural framework for understanding the multifunctional character of lactoferrin. Biochimie. 2009;91(1):3–10. doi: 10.1016/j.biochi.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 8.van Berkel PH, Geerts ME, van Veen HA, Kooiman PM, Pieper FR, de Boer HA, Nuijens JH. Glycosylated and unglycosylated human lactoferrins both bind iron and show identical affinities towards human lysozyme and bacterial lipopolysaccharide, but differ in their susceptibilities towards tryptic proteolysis. Biochem J. 1995;312(1):107–114. doi: 10.1042/bj3120107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khan JA, Kumar P, Paramasivam M, Yadav RS, Sahani MS, Sharma S, Srinivasan A, Singh TP. Camel lactoferrin, a transferrin-cum-lactoferrin: crystal structure of camel apolactoferrin at 2.6 Å resolution and structural basis of its dual role. J Mol Biol. 2001;309(3):751–761. doi: 10.1006/jmbi.2001.4692. [DOI] [PubMed] [Google Scholar]
- 10.de Jong G, van Eijk HG. Microheterogeneity of human serum transferrin: a biological phenomenon studied by isoelectric focusing in immobilized pH gradients. Electrophoresis. 1988;9(9):589–598. doi: 10.1002/elps.1150090921. [DOI] [PubMed] [Google Scholar]
- 11.Hovanessian AG, Awdeh ZL. Gel isoelectric focusing of human-serum transferrin. Eur J Biochem/FEBS. 1976;68(2):333–338. doi: 10.1111/j.1432-1033.1976.tb10819.x. [DOI] [PubMed] [Google Scholar]
- 12.Jenssen H, Hancock REW. Antimicrobial properties of lactoferrin. Biochimie. 2009;91(1):19–29. doi: 10.1016/j.biochi.2008.05.015. [DOI] [PubMed] [Google Scholar]
- 13.Yen C-C, Shen C-J, Hsu W-H, Chang Y-H, Lin H-T, Chen H-L, Chen C-M. Lactoferrin: an iron-binding antimicrobial protein against Escherichia coli infection. Biometals. 2011;24(4):585–594. doi: 10.1007/s10534-011-9423-8. [DOI] [PubMed] [Google Scholar]
- 14.Berlutti F, Pantanella F, Natalizi T, Frioni A, Paesano R, Polimeni A, Valenti P. Antiviral properties of lactoferrin—a natural immunity molecule. Molecules. 2011;16(8):6992–7018. doi: 10.3390/molecules16086992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang R, Lopez V, Kelleher SL, Lönnerdal B. Apo- and holo-lactoferrin are both internalized by lactoferrin receptor via clathrin-mediated endocytosis but differentially affect ERK-signaling and cell proliferation in Caco-2 cells. J Cell Physiol. 2011;226(11):3022–3031. doi: 10.1002/jcp.22650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang R, Lönnerdal B. Apo-and holo-lactoferrin stimulate proliferation of mouse crypt cells but through different cellular signaling pathways. Int J Biochem Cell B. 2012;44(1):91–100. doi: 10.1016/j.biocel.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 17.Arnold RR, Brewer M, Gauthier JJ. Bactericidal activity of human lactoferrin: sensitivity of a variety of microorganisms. Infect Immun. 1980;28(3):893–898. doi: 10.1128/iai.28.3.893-898.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arnold RR, Russell JE, Champion WJ, Brewer M, Gauthier JJ. Bactericidal activity of human lactoferrin: differentiation from the stasis of iron deprivation. Infect Immun. 1982;35(3):792–799. doi: 10.1128/iai.35.3.792-799.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ellison RT, Giehl TJ, LaForce FM. Damage of the outer membrane of enteric Gram-negative bacteria by lactoferrin and transferrin. Infect Immun. 1988;56(11):2774–2781. doi: 10.1128/iai.56.11.2774-2781.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crosa JH. Genetics and molecular biology of siderophore-mediated iron transport in bacteria. Microbiol Rev. 1989;53(4):517–530. doi: 10.1128/mr.53.4.517-530.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sui Q, Roginski H, Williams RPW, Wooster TJ, Versteeg C, Wan J. Effect of the ionic strength of pulsed electric field treatment medium on the physicochemical and structural characteristics of lactoferrin. J Agric Food Chem. 2010;58(22):11725–11731. doi: 10.1021/jf102171u. [DOI] [PubMed] [Google Scholar]
- 22.Soo Nam M, K-I S, Kumura H, Lee KK, Yu DY. Characterization of Korean native goat lactoferrin. Comp Biochem Physiol Part B: Biochem Mol B. 1999;123(2):201–208. doi: 10.1016/S0305-0491(99)00060-7. [DOI] [PubMed] [Google Scholar]
- 23.Brisson G, Britten M, Pouliot Y. Effect of iron saturation on the recovery of lactoferrin in rennet whey coming from heat-treated skim milk. J Dairy Sci. 2007;90(6):2655–2664. doi: 10.3168/jds.2006-725. [DOI] [PubMed] [Google Scholar]
- 24.Brisson G, Britten M, Pouliot Y. Heat-induced aggregation of bovine lactoferrin at neutral pH: effect of iron saturation. Int Dairy J. 2007;17(6):617–624. doi: 10.1016/j.idairyj.2006.09.002. [DOI] [Google Scholar]
- 25.Leibman A, Aisen P. Distribution of iron between the binding sites of transferrin in serum: methods and results in normal human subjects. Blood. 1979;53(6):1058–1065. [PubMed] [Google Scholar]
- 26.Lima MF, Kierszenbaum F. Lactoferrin effects of phagocytic cell function. II. The presence of iron is required for the lactoferrin molecule to stimulate intracellular killing by macrophages but not to enhance the uptake of particles and microorganisms. J Immunol. 1987;139(5):1647–1651. [PubMed] [Google Scholar]
- 27.Indyk HE, Filonzi EL. Determination of lactoferrin in bovine milk, colostrum and infant formulas by optical biosensor analysis. Int Dairy J/Int Dairy F. 2005;15(5):429–438. doi: 10.1016/j.idairyj.2004.09.003. [DOI] [Google Scholar]
- 28.Steijns JM, van Hooijdonk AC. Occurrence, structure, biochemical properties and technological characteristics of lactoferrin. Br J Nutr. 2000;84(1):S11–S17. doi: 10.1017/s0007114500002191. [DOI] [PubMed] [Google Scholar]
- 29.Volden J, Jørgensen CE, Rukke E-O, Egelandsdal B. Oxidative properties of lactoferrins of different iron-saturation in an emulsion consisting of metmyoglobin and cod liver oil. Food Chem. 2012;132(3):1236–1243. doi: 10.1016/j.foodchem.2011.11.092. [DOI] [PubMed] [Google Scholar]
- 30.Harada K, Kuniyasu A, Nakayama H, Nakayama M, Matsunaga T, Uji Y, Sugiuchi H, Okabe H. Separation of human serum transferrins with different iron-binding states by high-performance liquid chromatography using a pyridinium polymer column. J Chromatogr B. 2002;767(1):45–51. doi: 10.1016/S0378-4347(01)00529-1. [DOI] [PubMed] [Google Scholar]
- 31.Sreedhara A, Flengsrud R, Prakash V, Krowarsch D, Langsrud T, Kaul P, Devold TG, Vegarud GE. A comparison of effects of pH on the thermal stability and conformation of caprine and bovine lactoferrin. Int Dairy J. 2010;20(7):487–494. doi: 10.1016/j.idairyj.2010.02.003. [DOI] [Google Scholar]
- 32.Sreedhara A, Flengsrud R, Langsrud T, Kaul P, Prakash V, Vegarud G. Structural characteristic, pH and thermal stabilities of apo and holo forms of caprine and bovine lactoferrins. Biometals. 2010;23(6):1159–1170. doi: 10.1007/s10534-010-9366-5. [DOI] [PubMed] [Google Scholar]
- 33.Kansal RG, Aziz RK, Kotb M. Modulation of expression of superantigens by human transferrin and lactoferrin: a novel mechanism in host–Streptococcus interactions. J Infect Dis. 2005;191(12):2121–2129. doi: 10.1086/430386. [DOI] [PubMed] [Google Scholar]
- 34.Brown EM, Parry RM. Spectroscopic study of bovine lactoferrin. Biochemistry (Mosc) 1974;13(22):4560–4565. doi: 10.1021/bi00719a014. [DOI] [PubMed] [Google Scholar]
- 35.Mason AB, Halbrooks PJ, Larouche JR, Briggs SK, Moffett ML, Ramsey JE, Connolly SA, Smith VC, MacGillivray RTA. Expression, purification, and characterization of authentic monoferric and apo-human serum transferrins. Protein Expr Purif. 2004;36(2):318–326. doi: 10.1016/j.pep.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 36.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227(5259):680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 37.Wakabayashi H, Yamauchi K, Kobayashi T, Yaeshima T, Iwatsuki K, Yoshie H. Inhibitory effects of lactoferrin on growth and biofilm formation of Porphyromonas gingivalis and Prevotella intermedia. Antimicrob Agents Chemother. 2009;53(8):3308–3316. doi: 10.1128/AAC.01688-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nuijens JH, van Berkel PH, Schanbacher FL. Structure and biological actions of lactoferrin. J Mammary Gland Biol Neoplasia. 1996;1(3):285–295. doi: 10.1007/BF02018081. [DOI] [PubMed] [Google Scholar]
- 39.Makino Y, Nishimura S. High-performance liquid chromatographic separation of human apolactoferrin and monoferric and diferric lactoferrins. J Chromatogr B. 1992;579(2):346–349. doi: 10.1016/0378-4347(92)80402-C. [DOI] [PubMed] [Google Scholar]
- 40.Anderson BF, Baker HM, Norris GE, Rice DW, Baker EN. Structure of human lactoferrin: crystallographic structure analysis and refinement at 2.8 Å resolution. J Mol Biol. 1989;209(4):711–734. doi: 10.1016/0022-2836(89)90602-5. [DOI] [PubMed] [Google Scholar]
- 41.van Berkel PH, Geerts ME, van Veen HA, Mericskay M, de Boer HA, Nuijens JH. N-terminal stretch Arg2, Arg3, Arg4 and Arg5 of human lactoferrin is essential for binding to heparin, bacterial lipopolysaccharide, human lysozyme and DNA. Biochem J. 1997;328(1):145–151. doi: 10.1042/bj3280145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Veen HA, Geerts MEJ, van Berkel PHC, Nuijens JH. Analytical cation-exchange chromatography to assess the identity, purity, and N-terminal integrity of human lactoferrin. Anal Biochem. 2002;309(1):60–66. doi: 10.1016/S0003-2697(02)00273-7. [DOI] [PubMed] [Google Scholar]
- 43.Grossmann JG, Neu M, Pantos E, Schwab FJ, Evans RW, Townes-Andrews E, Lindley PF, Appel H, Thies W-G, Hasnain SS. X-ray solution scattering reveals conformational changes upon iron uptake in lactoferrin, serum and ovo-transferrins. J Mol Biol. 1992;225(3):811–819. doi: 10.1016/0022-2836(92)90402-6. [DOI] [PubMed] [Google Scholar]
- 44.Stowell KM, Rado TA, Funk WD, Tweedie JW. Expression of cloned human lactoferrin in baby-hamster kidney cells. Biochem J. 1991;276(2):349–355. doi: 10.1042/bj2760349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bates GW, Billups C, Saltman P. The kinetics and mechanism of iron(III) exchange between chelates and transferrin. J Biol Chem. 1967;242(12):2810–2815. [PubMed] [Google Scholar]
- 46.Motekaitis RJ, Martell AE. The iron(III) and iron(II) complexes of nitrilotriacetic acid. J Coord Chem. 1994;31(1):67–78. doi: 10.1080/00958979408022546. [DOI] [Google Scholar]
- 47.Saponja JA, Vogel HJ. Quadrupolar central transition (QCT) and 13C NMR competition studies of metal ion binding to ovotransferrin. Can J Chem. 2011;89(7):779–788. doi: 10.1139/v11-019. [DOI] [Google Scholar]
- 48.Baker HM, Anderson BF, Brodie AM, Shongwe MS, Smith CA, Baker EN. Anion binding by transferrins: importance of second-shell effects revealed by the crystal structure of oxalate-substituted diferric lactoferrin. Biochemistry (Mosc) 1996;35(28):9007–9013. doi: 10.1021/bi960288y. [DOI] [PubMed] [Google Scholar]
- 49.Abdallah FB, El Hage Chahine J-M. Transferrins: iron release from lactoferrin. J Mol Biol. 2000;303(2):255–266. doi: 10.1006/jmbi.2000.4101. [DOI] [PubMed] [Google Scholar]
- 50.Pakdaman R, Chahine J-MEH. A mechanism for iron uptake by transferrin. Eur J Biochem. 1996;236(3):922–931. doi: 10.1111/j.1432-1033.1996.00922.x. [DOI] [PubMed] [Google Scholar]
- 51.Salmon V, Legrand D, Georges B, Slomianny M-C, Coddeville B, Spik G. Characterization of human lactoferrin produced in the baculovirus expression system. Protein Expr Purif. 1997;9(2):203–210. doi: 10.1006/prep.1996.0687. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 123 kb)