

Abstract
Dopamine acting through D2 receptors (D2Rs) controls lactotroph proliferation and prolactin (PRL) levels. Ablation of this receptor in mice results in lactotroph hyperplasia and prolactinomas in aged females. Alternative splicing of the Drd2 gene generates 2 independent isoforms, a long (D2L) and a short (D2S) isoform, which are present in all D2R-expressing cells. Here, we addressed the role of D2L and D2S on lactotroph physiology through the generation and analysis of D2S-null mice and their comparison with D2L-null animals. These mice represent a valuable tool with which to investigate dopamine-dependent isoform-specific signaling in the pituitary gland. We sought to assess the existence of a more prominent role of D2L or D2S in controlling PRL expression and lactotroph hyperplasia. Importantly, we found that D2L and D2S are specifically linked to independent transduction pathways in the pituitary. D2L-mediated signaling inhibits the AKT/protein kinase B kinase activity whereas D2S, in contrast, is required for the activation of the ERK 1/2 pathway. Under normal conditions, presence of only 1 of the 2 D2R isoforms in vivo prevents hyperprolactinemia, formation of lactotroph's hyperplasia, and tumorigenesis that is observed when both isoforms are deleted as in D2R−/− mice. However, the protective function of the single D2R isoforms is overridden when single isoform-knockout mice are challenged by chronic estrogen treatments as they show increased PRL production and lactotroph hyperplasia. Our study indicates that signaling from each of the D2R isoforms is sufficient to maintain lactotroph homeostasis in physiologic conditions; however, signaling from both is necessary in conditions simulating pathologic states.
Dopamine, a major neuromodulator of the central nervous system, is also critically involved in the regulation of prolactin (PRL) levels in the pituitary gland. The dopaminergic tone is indeed a powerful inhibitor of PRL synthesis and secretion and of lactotrophs' proliferation (1–3). Lactotrophs are remarkably plastic cells that undergo notable changes in size and number during the reproductive life of female mammals in response to hypothalamic and steroid hormones stimulation (1). Activation of dopamine D2 receptors (D2Rs) counterbalances these stimulations and maintains a normal homeostasis in these cells. Nevertheless, pituitary adenomas of lactotroph origin are quite frequent and represent one of the major causes of sterility in young women (4). Importantly, D2R-expressing adenomas regress in response to treatments with D2R-specific agonists (bromocriptine, cabergoline) (5); however, a percentage of these tumors become resistant to pharmacologic treatment due to decreased or absent D2R expression (6). We have previously shown that ablation of the D2R results in the development of lactotroph hyperplasia and prolactinomas, underlining the key role of D2R in the control of lactotroph physiology (3, 7). To date the molecular pathways by which D2R-mediated signaling regulates lactotrophs physiology, thus preventing the development of hyperplasia and tumors, are not completely elucidated. The identification of the pathways modulated by D2R-mediated signaling might lead to the development of alternative strategies for the treatment of D2R-resistant prolactinomas. In the pituitary, D2Rs have been reported to inhibit the cAMP pathway (8–10) and, in addition, to activate the ERK 1/2 pathway (11). In the brain, D2Rs have also been shown to couple to the AKT pathway in a cAMP-independent manner (12). Dysregulation of the ERK 1/2 and AKT pathways (13, 14) is possibly responsible for the altered proliferation and generation of lactotroph hyperplasia and tumors.
In addition to the multiple pathways involved in D2R-mediated signaling, the receptor in vivo is present in 2 molecularly distinct isoforms, long (D2L) and short (D2S), generated by alternative splicing of the Drd2 gene (15). The long isoform, D2L, differs from D2S, by the insertion of 29 amino acids in the region of the receptor that are critical for interacting with G proteins as well as with other proteins (the third intracellular loop), thus suggesting that each isoform might be coupled to distinct pathways (16, 17). Previous analyses of transgenic mice overexpressing either D2S or D2L in the lactotrophs were instrumental in showing the activation of the ERK 1/2 pathway by overexpression of D2S (11). However, these results were obtained in the presence of the endogenous receptor, leaving unanswered the question of the ability and specificity of each isoform on its own in modulating distinct transduction pathways in vivo. Furthermore, lack of D2R-specific ligands able to discriminate between D2L and D2S has prevented in vivo studies aimed at analyzing this question.
To overcome these issues, we have generated mice lacking exclusively the D2S isoform and have analyzed them in parallel with D2L-null mice previously developed in our laboratory (18). D2L−/− mice and the newly generated D2S−/− mice represent a unique tool to analyze, for the first time, the signal properties of each receptor in vivo. Molecular and cellular features of these mutant mice were studied to reveal how the absence of a single isoform would influence stimulation/inhibition of signaling pathways as well as on PRL synthesis and pituitary growth. Our results reveal a selective coupling of D2S and D2L receptors to the ERK 1/2 and AKT signaling pathways, respectively.
Strikingly, under physiologic conditions, signaling from only 1 isoform is sufficient to prevent lactotroph hyperplasia and tumor formation, which is instead observed in the absence of both isoforms in D2R−/− mice (3). In contrast, when D2L−/− or D2S−/− mice are challenged with chronic estrogen treatments they develop lactotroph hyperplasia, thus indicating that both isoforms are necessary for maintaining a normal homeostasis of these cells in pathologic conditions.
Materials and Methods
Animals
Female mice (4 and 8-mo-old) wild type (WT), D2R−/− (19), D2L−/− (18), and D2S−/− in an approximately 94% C57Bl6 background were obtained from mating of heterozygote mice from each line. Animals were kept in controlled conditions of light (12-h light, 12-h dark cycles), temperature (20–25°C), and humidity (45%–60%). Animals were fed standard laboratory chow and water ad libitum. All husbandry procedures and welfare policies were conducted according to the Guide for the Care and Use of Laboratory Animals, set forth by the Institute of Laboratory Animal Resources, Commission on Life Sciences, and National Research Council. All research was performed under protocols approved by the Institutional Animal Care and Use Committee (IACUC).
Generation of D2S−/− animals
The D2S−/− mouse line was generated at the Mouse Clinical Institute/Institut Clinique de la Souris (Illkirch, France) by a knock-in strategy aimed at preventing alternative splicing, thus allowing only the synthesis of the D2L isoform in vivo. The targeting vector was constructed as follows: a 3.6-kb fragment encompassing exon 3 and 4 of the Drd2 gene constituted the 5′-homology arm of the construct, whereas a 3.1-kb fragment encompassing exon 8 constituted the 3′-homology arm. These fragments were amplified by PCR from 129S2/SvPas cells DNA and subcloned into a vector containing the floxed neomycin resistance cassette. The cDNA fragment of the D2L isoform corresponding to exons 5–7 was then inserted between the 3.6-kb fragment followed by the floxed neomycin cassette and the 3.1-kb fragment. The construct was then linearized and electroporated in 129S2/SvPas mouse embryonic stem cells. After selection, targeted clones were identified by PCR using external primers and further confirmed by Southern blot with 5′- and 3′-external probes. Two positive embryonic stem clones were obtained (K191–327, K191–363) and injected into C57BL/6J blastocysts; male chimeras derived from these cells gave germ-line transmission; the neomycin cassette present in the targeted vector was then removed. The D2S−/− animals used in this article descend from clone K191–363.
Genotyping
PCR was performed on genomic DNA from tail biopsies of D2L−/− and D2S−/− mice. To genotype the D2L−/− line we used primers specific for the neomycin cassette located between exon 5 and exon 7, which replaces exon 6 of the Drd2 gene (Table 1). To genotype the D2S−/− line we used primers designed 362 bp upstream and 46 bp downstream of the lox P site (see Table 1 and Figure 1A). The D2R−/− line was genotyped by Southern blot analysis, as previously described (19).
Table 1.
Primers Used in RT-PCR and qRT-PCR
| Gene | Primers (5′ to 3′) |
|---|---|
| PRL | F:ctcaggccatcttggagaag |
| R:gaagtggggcagtcattgat | |
| GH | F:gctacagactctcggacctc |
| R:cggagcacagcattagaaaacag | |
| TSH | F:gggcaagcagcatccttttg |
| R:gtgtcatacaatacccagcacag | |
| D2R | Exon 2: |
| F:agtggccccactgccccaat | |
| R:tccagatagacgacccagggc | |
| Exon 5–7: | |
| F:ccttcatcgtcaccctgctgg | |
| R:ctccatttccagctcctgag | |
| GAPDH | F:aggtcggtgtgaacggatttg |
| R:tgtagaccatgtagttgaggtca | |
| Neomycin | F:ctcgacgttgtcactgaagc |
| R:cgtccagatcatcctgatcg | |
| Lox P site | F:ttgagatccagaccatgcccaatgg |
| R:agcccagacactgagtcaaactgc |
F, forward; R, reverse.
Figure 1.
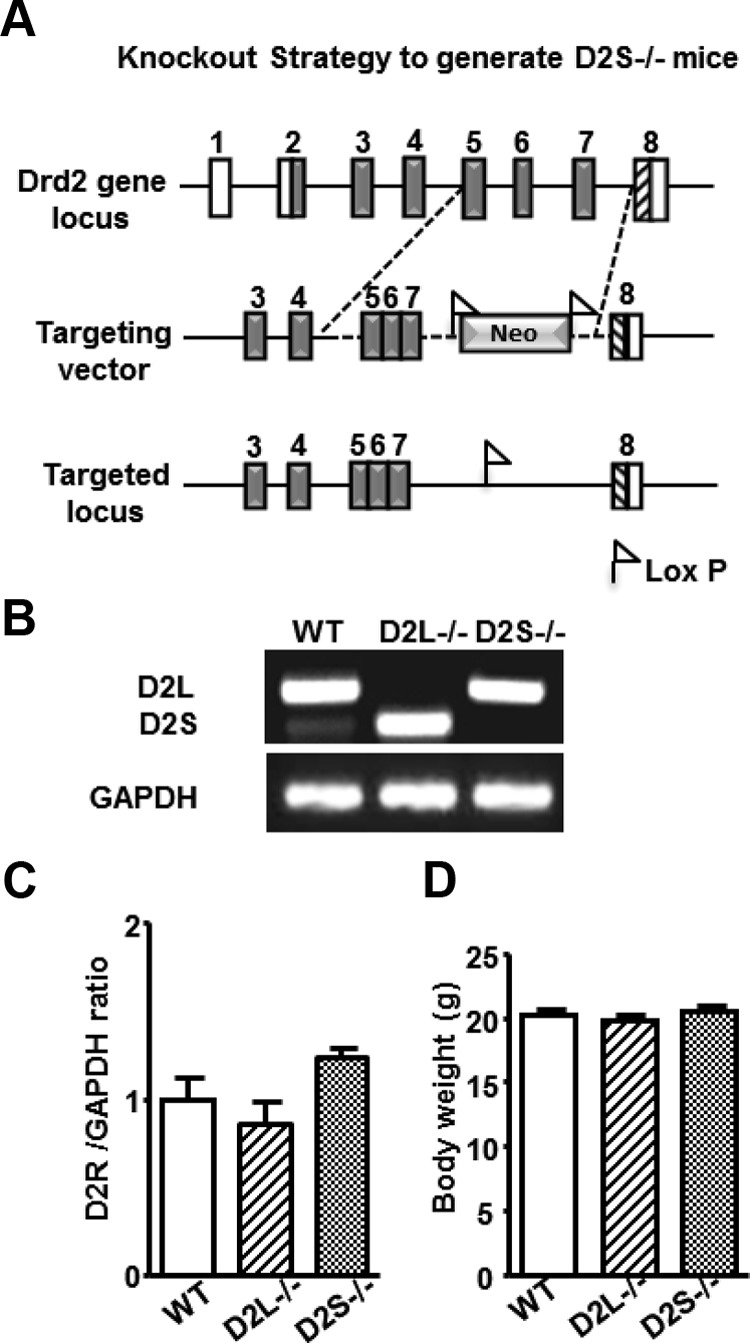
Characterization of the D2R Isoform's Specific Mutants. A, Representative diagram of the strategy used to generate D2S−/− mice. Rectangles represent Drd2 exons: in white are represented noncoding regions; gray represents the coding ones. Dashed lines indicate the genomic region that was manipulated to obtain the targeting vector including the substitution of the genomic regions between exon 5 and 7 with the corresponding region of the D2L cDNA and insertion of the floxed neomycin cassette. The neomycin cassette was then removed, as shown in the drawing of the targeted locus. B, RT-PCR of mRNA extracted from WT, D2L−/− and D2S−/− pituitaries. PCRs for the 3 genotypes were performed using primers specific for exon 5 and exon 7 (see Table 1) of the Drd2 gene. The upper band of 245 bp corresponds to D2L whereas the lower band of 158 bp corresponds to D2S. C, qRT-PCRs were performed to quantify the total amount of D2-specific mRNA in pituitaries of WT, D2L−/−, and D2S−/− mice, as indicated. Bars represent the ratio of expression levels between D2R-specific transcripts and GAPDH used as internal control. The ratio in WT was arbitrarily set as 1. qRT-PCRs were performed using primers specific for exon 2, which is common to both D2L and D2S and GAPDH (see Table 1). Data were calculated as ratio ± SEM using 4 animals per genotype and analyzed by 1-way ANOVA. D, Body weight of 10 week-old WT, D2L−/−, and D2S−/− mice, as indicated. Bars represent the mean ± SEM (n = 10); values were analyzed by 1-way ANOVA.
Haloperidol treatment
Four-month-old D2R−/−, D2L−/−, D2S−/−, and WT female littermates of each line were injected ip with haloperidol (2 mg/kg; Sigma, St. Louis, Missouri) or saline. Haloperidol was dissolved in a drop of glacial acetic acid and then brought up to volume in 0.9% NaCl; the solution was then adjusted to pH 6 with 5 M NaOH. Saline-injected mice received the vehicle used to dissolve haloperidol; 20 minutes after the injection animals were euthanized, and pituitary glands were dissected and rapidly frozen for Western blot analyses.
Estradiol treatment
Four-month-old D2R−/−, D2L−/−, D2S−/−, and WT female littermates for each line were injected sc with 25 mg/kg of 17β-estradiol (E2, Sigma) in 100 μL safflower oil (Sigma) or vehicle (VEH, safflower oil) once a week for 10 weeks. One week after the last injection, animals were humanely destroyed and pituitary glands were rapidly collected. Each gland was weighed and immediately frozen for protein or tissue analyses.
Western blot
Pituitary glands were rapidly collected and frozen in liquid nitrogen. Each pituitary was homogenized in 50 μL of lysis buffer (25 mM Tris, pH 6.8; 175 mM β-mercaptoethanol; 1% sodium dodecyl sulfate) containing protease inhibitor cocktail (Complete EDTA-free; Roche, Indianapolis, Indiana) and phosphatase inhibitor cocktail (PhosSTOP; Roche). Protein determination was made using the BCA kit (Thermo Scientific, Rockford, Illinois). Proteins (40 μg) were resolved in 10% sodium dodecyl sulfate-polyacrylamide gel and then transferred to polyvinyl difluoride membranes (Bio-Rad Laboratories, Hercules, California). Membranes were incubated for 1 hour in 5% nonfat dry milk-PBS-0.1% Tween 20 at room temperature and then overnight with the appropriate primary antibody in the same buffer at 4°C. The primary antibodies used were: antiphospho ERK 1/2 (1:1000, Cell Signaling Technology, Danvers, Massachusetts), anti-ERK 1/2 (1:1000, Cell Signaling Technology), antiphospho AKT Thr308 (1:200, Cell Signaling Technology), antiphospho AKT Ser473 (1:1000, Cell Signaling Technology), and anti-AKT (1:1000, Cell Signaling Technology). The next day, membranes were incubated for 1 hour at room temperature with the corresponding peroxidase-conjugated secondary antibody. Immunoreactivity was detected by enhanced chemiluminescence (Millipore Corp, Bedford, Massachusetts). Membranes were exposed to X-ray films and developed using an X-Ray developer. Quantifications were performed by NIH ImageJ (version 1.42q) software; the ratio between phosphorylated/total proteins in extracts from mutants was compared with that of WT values arbitrarily set to 1.
Hematoxylin-eosin staining
Cryostat sections (10 μm) were prepared from 4- and 8-month-old WT, D2R−/−, D2L−/−, and D2S −/− pituitaries. Sections were fixed in 4% PFA for 30 minutes, washed 3 times in PBS for 5 minutes and once in distilled water for 10 minutes. Tissue slides were dipped in hematoxylin for 30 seconds, immediately washed in tap water for 15 minutes, and then twice washed in distilled water for 10 minutes. Slides were then dipped for 30 seconds in eosin and washed in distilled water for 10 minutes. Afterward, tissue sections were dried and coverslips mounted in Permount (Fisher Scientific, Pittsburgh, Pennsylvania).
In situ hybridization
Cryostat sections (10 μm) were hybridized with 35S-CTP-labeled PRL, GH, and TSH probes prepared as previously described (19). Nuclei were stained with toluidine blue. Analyses were performed with a SP5 microscope (DMRE; Leica, Deerfield, Illinois).
RNA extraction, RT-PCR, and RT-qPCR
Total RNA was extracted from each pituitary gland using Trizol (Invitrogen, Carlsbad, California). After RNA quantification, 1 μg of total RNA was retro-transcribed using the iScript cDNA Synthesis kit (Bio-Rad Laboratories). Specific primers were used to detect mouse PRL, GH, TSH, and D2R (see Table 1). To normalize for RNA quantity, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as internal control. To assess for the ablation of the D2L or D2S isoforms at the mRNA level, we performed RT-PCR using 200 ng of cDNA as template in 25 μL PCR containing 4 mM MgCl2, 0.2 mM of each deoxynucleotide triphosphate, 0.8 μM of each primer, and 1.5 Taq DNA polymerase. The primers chosen were designed to recognize the presence or absence of exon 6. The forward primer was designed 101 bp upstream of exon 6 (in exon 5) and the reverse primer was designed 16 bp downstream from exon 6 (in exon 7) (see Table 1, Exon 5–7). PCRs were run as follows: after a first denaturation cycle at 94°C for 4 minutes, PCR was run for 40 cycles of 45 seconds at 94°C, 45 seconds at 60°C, 1 minute at 72°C, and a final extension period of 7 minutes at 72°C. Twelve microliters of each reaction were analyzed on 1% agarose gel.
To quantify the mRNA expression of PRL, GH, TSH, and the amount of D2R expressed in each mouse line, quantitative RT-PCR (qRT-PCR) was performed using the PTC-200 real time detection system (MJ Research, Watertown, Massachusetts). For a 20-μL reaction, 1 ng of cDNA template was mixed with the specific primers to a final concentration of 500 nM and 10 μl of iQ SYBR Green Supermix (Bio-Rad). The reactions were run as follows: after a first denaturation cycle at 95°C for 10 minutes, qRT-PCR was run for 40 cycles of 30 seconds at 95°C and 1 minute at 60°C.
Statistical analysis
One-way ANOVA was used to analyze the differences on pituitary and body weight, mRNA expression, and Western blot analyses between genotypes. Despite the fact that in all experiments WT mice of each line were compared to the respective mutant littermates, we observed that WT values did not statistically differ among lines and therefore they were pulled together and represented as 1 group in the figures. Student's t test was used to compare knockout values with WT.
Two-way ANOVA was used when animals received a treatment (ie, haloperidol or estradiol) followed by the appropriate post hoc test (Bonferroni's or Student's t test). At least 5 animals per group were analyzed in each experiment, and differences were considered with a P value < .05.
Results
Generation of D2S−/− mice
Previous studies have shown that absence of D2R in D2R−/− mice (3, 7) results in hyperplasia of the pituitary gland in young females and prolactinomas in aged ones (20). Transcription of the Drd2 gene leads to 2 cDNAs originated by a mechanism of alternative splicing generating two D2R isoforms, D2L and D2S (21). The role of each isoform in the regulation of lactotroph physiology and in the development of pituitary hyperplasia and tumors has never been assessed in vivo. To study the signaling specifically activated by D2L or D2S in lactotrophs, we sought to isolate and analyze pituitaries from D2L−/− and D2S−/− mice. We have previously reported the generation of D2L−/− mice (18). Here we report on the generation of mice in which the D2S isoform is selectively ablated. D2S−/− animals were generated by homologous recombination by substituting the genomic region encompassing exons 5- 6- 7 with the cDNA fragment corresponding to D2L (Figure 1A).
Expression of D2L and D2S mRNAs in knockout mice was analyzed and compared with that of WT mice. In pituitaries from WT mice, the D2R isoforms are expressed in a 4:1 ratio (D2L:D2S) in favor of D2L; indeed RT-PCRs showed the presence of both a D2L (245 bp)- and a D2S (158 bp)-specific fragment, which respected the known ratio (10) (Figure 1B). Conversely, pituitary mRNAs from the D2L−/− mice showed the exclusive presence of the 158-bp fragment corresponding to D2S (Figure 1B); conversely, D2S−/− mice only showed the 245-bp fragment, which corresponds to the D2L isoform (Figure 1B). To quantify the expression of each isoform in D2L−/− and D2S−/− mice, we performed qRT-PCR using oligonucleotides corresponding to exon 2 (a region common to both isoforms). These analyses indicated that the total amount of mRNAs corresponding to the D2R gene in WT vs D2L−/− or D2S−/− pituitaries did not differ (Figure 1C). These results are in support of previous observations (18), indicating that genetic deletion of exon 6 (D2L−/−) or its replacement by a fragment of D2L cDNA (D2S−/−) does not affect transcription of the Drd2 gene. Thus, D2L−/− and D2S−/− pituitaries express the same amount of D2 transcripts although in each of these mutants only a single isoform (either D2L or D2S) is expressed (Figure 1C). These results also indicate that the absence of intron sequences located between exon 5 and 7 does not affect the total amount of D2R-specific transcripts. Indeed, when membrane extracts from pituitaries from WT, D2L−/−, and D2S−/− mice were analyzed by Western blot using a monoclonal antibodies directed against D2R (22, 23), the intensity of the band corresponding to D2Rs was similar in pituitary extracts of each genotype (Supplemental Figure 1 published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org). No D2R-specific bands were observed in extracts from D2R−/− pituitaries.
D2L−/− and D2S−/− mice have a normal life span that does not differ from that of WT littermates; moreover, these mice grow and reproduce normally. Body weight of animals of the WT and mutant genotypes also did not differ (Figure 1D).
Intracellular pathways
Signaling from D2Rs has been reported to affect multiple transduction pathways. To date, it is still not known whether both D2R isoforms contribute to maintenance of a normal lactotroph physiology or whether D2R isoforms are coupled to common or independent signaling pathways in the pituitary. Studies performed in a lactotroph-derived cell line (MMQ cells) using a D2R-specific agonist showed a sustained activation of ERK 1/2, which led to a concomitant reduction of the cell growth rate (11). In vivo studies using transgenic mice overexpressing the D2S isoform in lactotrophs showed that this isoform is critical for this effect (11); conversely overexpression of D2L in mice pituitaries did not appear to affect the ERK 1/2 pathway (11). A tight correlation between activation of ERK 1/2 and lactotroph proliferation was further supported by in vivo analyses on D2R−/− mice, which revealed a reduced level of ERK 1/2 phosphorylation in the pituitary as compared with WT littermates and the development of lactotroph hyperplasia and tumors (11).
More recently, D2R-mediated signaling has been shown to also regulate the AKT pathway in the striatum in a cAMP-independent fashion (24). In particular, D2L activation has been shown to be responsible for inhibiting phosphorylation of the Thr308 residue in striatal neurons (25). The generation of D2R isoform-specific knockout mice offers the unique possibility to assess which isoform is coupled to specific signaling in the pituitary gland. Thus, we analyzed and compared ERK 1/2 and AKT phosphorylation levels in pituitary extracts from 4-month-old mice lacking either isoforms of the receptors (D2R−/−), or only D2S (D2S−/−) but expressing D2L, or lacking D2L (D2L −/−) but expressing D2S and compared it with the levels obtained by analyses of WT pituitaries. These analyses were performed using antibodies that specifically recognize phosphorylated ERK1 and ERK2 and phosphorylated AKT on Thr308.
In agreement with a stimulatory effect of D2R-mediated signaling on ERK 1/2 activation (11) in the pituitary, the basal level of ERK 1/2 phosphorylation is 37 ± 2% (n = 5) lower in D2R−/− pituitary extracts as compared with WT levels (Figure 2A). Interestingly, as in D2R−/−, we observed a similar reduction (41 ± 9%, n = 5) of ERK 1/2 phosphorylation levels in pituitary extracts from age-matched D2S−/− mice vs WT littermates (Figure 2A). Conversely, analyses of D2L−/− pituitaries showed an 80 ± 16% increase of ERK 1/2 phosphorylation levels compared with pituitary extracts from WT littermates (Figure 2A). The increase of ERK 1/2 phosphorylation observed in D2L−/− extracts is likely to be dependent upon a higher expression of D2S in D2L−/− mice as compared with WT animals. Indeed, whereas in WT mice D2S mRNA accounts for only 20% of the total D2R mRNA, deletion of exon 6 in D2L−/− mice allows for 100% expression of this isoform (Figure 1B) (18). These results clearly establish a specific effect of D2S-mediated signaling on the ERK 1/2 pathway, further supporting previous studies performed in transgenic mice overexpressing D2S (11).
Figure 2.
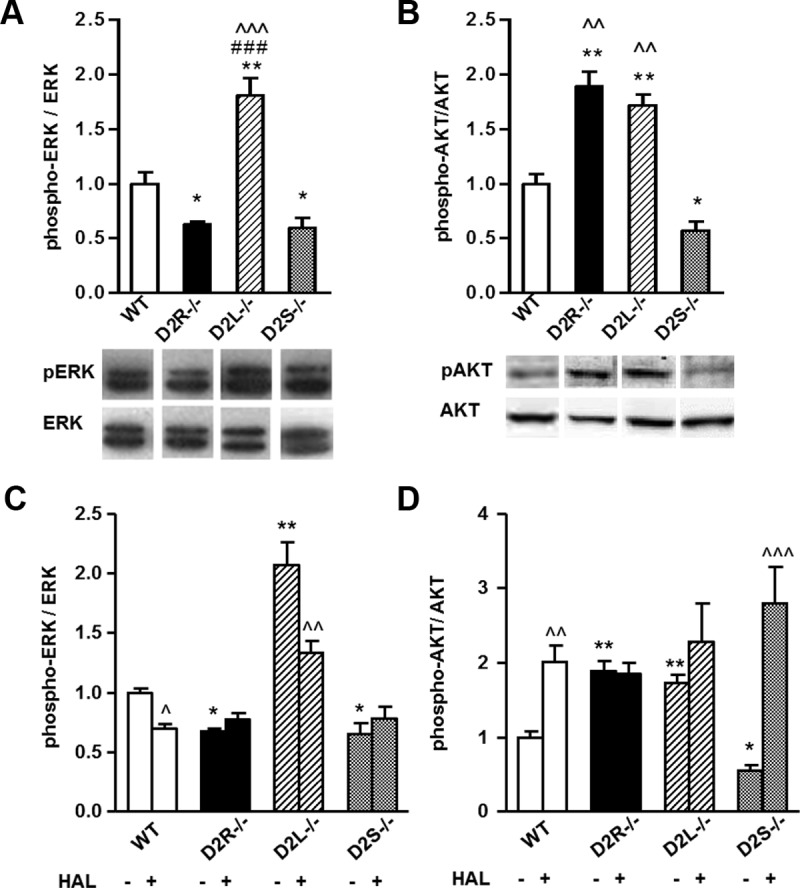
ERK 1/2 and AKT Phosphorylation Profiles in Pituitaries of 4-Month-Old Mice. Western blot analyses of proteins from pituitary extracts of 4-month-old mice. A, Quantifications of Western blots shown as phospho-ERK 1/2:total ERK 1/2 ratios normalized to the WT values, set as 1 (upper panel). Representative Western blots using phospho-ERK and total ERK antibodies (lower panel). B, Quantifications of Western blots shown as phospho-AKT:total AKT ratios normalized to WT values, set as 1 (upper panel). Representative Western blots using phospho-AKT and total AKT antibodies (lower panel). C and D, Western blot analyses of proteins from pituitary extracts of 4-month-old mice treated with haloperidol (HAL, ip injection, 2 mg/kg) or saline (0.9% NaCl) for 20 minutes. C, Bars represent the ratio between phospho-ERK:ERK, normalized to WT saline-treated values, set as 1. D, Same as in panel C for phospho-AKT:AKT ratios normalized to WT saline-treated values, set as 1. At least 5 animals per genotype were analyzed by 1-way ANOVA followed by Bonferroni (A, Genotype: F(3, 20)=20.94; B, Genotype: F(3, 22)=36.09) or 2-way ANOVA followed by Bonferroni (C, Genotype × treatment: F(3, 38) = 5.440; D, Genotype × treatment: F(3, 42) =5.965). A and B, * P < .05 vs WT extracts; ** P < .01 vs WT extracts, ^^ P < .01 vs D2S−/−, ^^^ P < .001 vs D2S−/−, ### P < .001 vs D2R−/−. C and D, * P < .05; ** P < .01 vs saline-treated WT extracts; ^ P < .05; ^^ P < .01; and ^^^ P < .001 vs the respective genotype saline treated.
Next, we analyzed AKT phosphorylation level on Thr308 in pituitary extracts from D2R−/−, D2L−/−, D2S−/−, and their respective WT controls (25). Importantly, these analyses showed that either the total loss of D2R-dependent signaling (D2R−/−) or of the D2L isoform in D2L−/− pituitaries resulted in 89 ± 13% (D2R−/−, n = 5) or 71 ± 10% (D2L−/−, n = 6) increase of AKT phosphorylation on Thr308 as compared with WT pituitary extracts (Figure 2C). These data are in agreement with results obtained in the striatum (25) and indicate that D2L-mediated signaling in the pituitary gland is linked, as in the brain, to inhibition of this pathway. Interestingly, AKT phosphorylation levels on Thr308 in pituitary extracts from 4 month-old D2S−/− animals was significantly lower (43 ± 7% decrease) than that found in WT extracts (Figure 2B); this result indicates that deletion of exon 6 in these mice and the consequent translation of all Drd2 transcripts into D2L further potentiates the inhibitory D2L-mediated signaling on AKT activity.
The phosphorylation levels of AKT at Ser473 were not affected in any of the mutants tested as compared with WT extracts (data not shown).
Taken together these results clearly show that D2L and D2S in the pituitary gland regulate two different pathways: D2L-mediated signaling down-regulates the AKT pathway whereas D2S-mediated signaling is positively coupled to stimulation of the ERK 1/2 pathway. Interestingly, we noticed that absence of both isoforms in D2R−/− mice results in an 89% increase of AKT phosphorylation and in a 37% decrease of ERK 1/2 phosphorylation levels. Conversely, in D2L−/− and D2S−/− pituitary extracts we observed that the phosphorylation levels of these kinases were either both up-regulated as in D2L−/− pituitaries or both down-regulated as in D2S−/− pituitaries in comparison with levels observed in WT littermate pituitary extracts. The effects of D2S and D2L ablation on the ERK 1/2 and AKT pathways in the pituitary gland were also observed in male mice (Supplemental Figure 2), thus excluding a potential interference of estrous cycle in the observed alterations of these pathways.
To provide an in vivo demonstration that phosphorylation of ERK 1/2 and AKT is specifically dependent upon activation of D2Rs, we pharmacologically blocked D2R-dependent signaling using the D2-specific antagonist, haloperidol, in all mutants and WT littermates. Twenty minutes after an acute injection of haloperidol, animals of WT, D2R−/−, D2L−/−, and D2S−/− genotype were humanely destroyed and the pituitary glands were rapidly extracted and analyzed.
Importantly, and in line with our results, haloperidol treatment decreased ERK 1/2 phosphorylation levels only in WT and D2L −/− pituitaries but had no effect in D2S−/− mice, confirming that the D2S isoform is responsible in vivo for the activation of the ERK 1/2 pathway (Figure 2C). Similarly, blocking D2R-mediated signaling by haloperidol in WT and D2S−/− mice, but not in D2L−/− animals, resulted in increased AKT phosphorylation at Thr308 (Figure 2D), demonstrating that the D2L isoform in vivo is responsible for the inhibition of this pathway. Finally, haloperidol treatment, as expected, did not affect the phosphorylation levels of either kinases in D2R−/− extracts, due to the absence of both D2R isoforms.
Next, we performed analyses on pituitaries of 8-month-old mice. This is a time at which absence of D2R signaling in D2R−/− mice leads to the development of large lactotroph hyperplasia and prolactinomas (3). Remarkably, the ERK 1/2 and AKT phosphorylation profiles remained constant in both 4- and 8-month-old mice (Figure 3). ERK 1/2 phosphorylation increased by 74 ± 7% (n = 5) in D2L−/− and decreased by 59 ± 2% (n = 6) in D2S−/− mice (Figure 3A). Conversely the phosphorylation of AKT was found to be increased by 84 ± 2.5% (n = 5) in D2L−/− and decreased by 75 ± 3% (n = 5) in D2S−/− compared with WT animals (Figure 3B). It is also interesting to note that the only significant differences between young and old animals were observed in WT extracts in which the phosphorylation levels of both kinases were increased in older pituitary extracts with respects to the younger ones (data not shown). Interestingly, in D2R−/− mice whereas ERK 1/2 phosphorylation was equally down-regulated in 4 and 8-month-old mice, the level of AKT phosphorylation was lower in 8- as compared with 4-month-old mice. The mechanism underlying AKT reduction in the older pituitary is presently unknown; however, with aging the lactotroph of D2R−/− female pituitaries transition from hyperplasia to prolactinomas (3, 20), which might lead to the intervention of other factors that can modify AKT as well as other signaling pathways in fully transformed lactotrophs.
Figure 3.
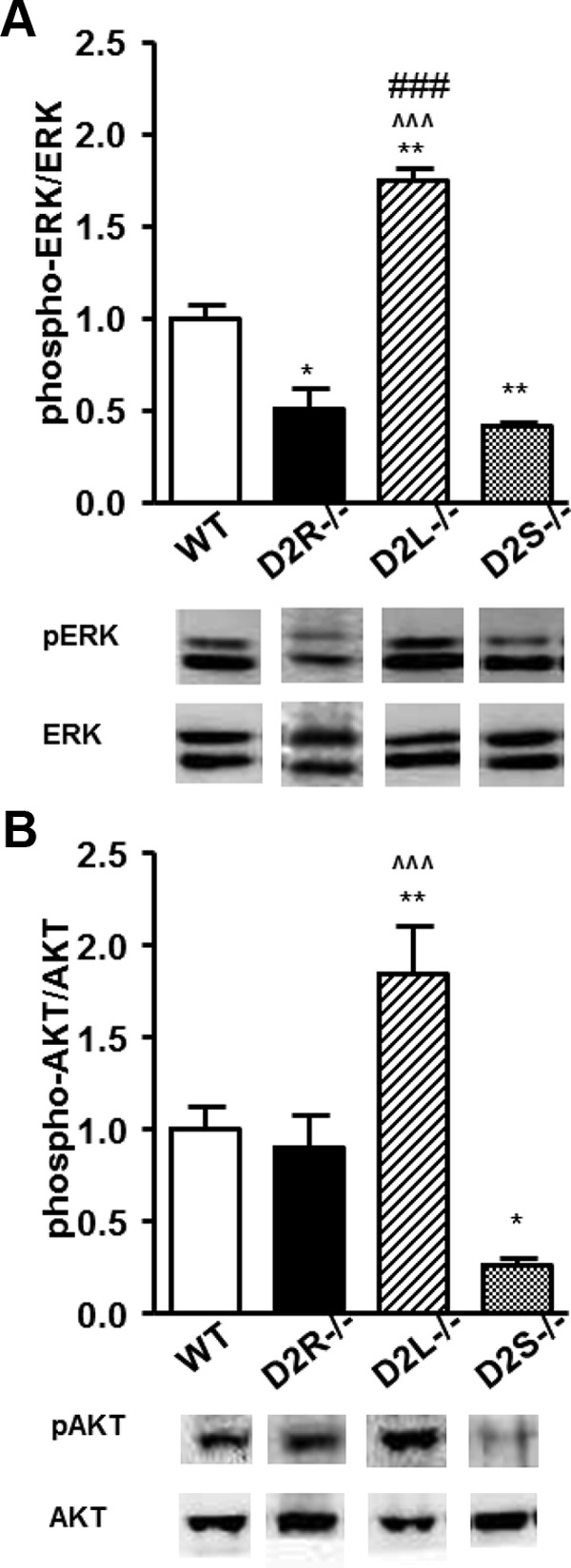
ERK 1/2 and AKT Phosphorylation Profiles in Pituitaries of 8-Month-Old Mice. Western blot analyses of pituitary extracts from 8-month-old WT, D2R−/−, D2L−/−, and D2S−/− mice using antibodies specific for total ERK, phospho-ERK, AKT, and phospho-AKT. Ratios between phospho-ERK/ERK (A) or phospho-AKT/AKT (B) were normalized to WT values set as 1. At least 5 animals per genotype were analyzed by 1-way ANOVA followed by Bonferroni (A, Genotype: F(3, 20)=53.60; B, Genotype: F(4, 20)=19.63). * P < .05 vs WT animals; ** P < .01 vs WT animals; ^^^ P < .01 vs D2S−/−; ### P < .001 vs D2R−/−.
The presence of only one D2R isoform prevents pituitary hyperplasia
The process of hyperplasia in D2R−/− precedes tumor formation and appears in 4-month-old females as observed by histologic examination (3). Thus, we analyzed the pituitary gland of WT and mutant females at this age and compared them with histological sections obtained from D2R−/− pituitaries. Interestingly, a comparative analysis of the weight of the pituitary glands of animals of each genotype (Figure 4A) revealed that only D2R−/− mice develop hyperplasia at 4 months of age (Figure 4, A and B), as previously reported (3). Conversely, the size and weight of the pituitaries from D2L−/− or D2S−/− mice did not differ from that of WT controls. Similarly, macroscopic observations of pituitary gland sections from mice of all the four genotypes stained by hematoxylin and eosin confirmed these results at the histologic level (Figure 4B).
Figure 4.
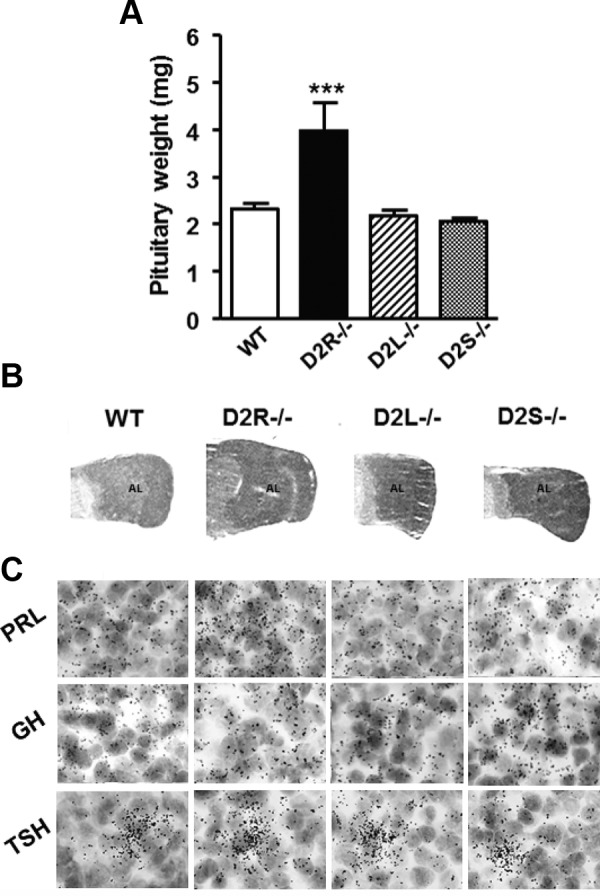
Pituitaries (4-mo-old) from D2L−/− and D2S−/− Mice Do Not Differ from the WT. A, Pituitary from 4-month-old females of the different genotypes, as indicated, were weighed and analyzed. Data represent the mean ± SEM of 7–10 pituitaries per group, analyzed by 1-way ANOVA (Genotype: F(3, 36)= 12.62). *** P < .01 vs WT, D2L−/−, and D2S−/−. B, Hematoxylin-eosin staining of pituitary sections from female animals of the different genotypes. These analyses showed a similar weight and histologic appearance between pituitaries of D2L−/−, D2S−/−, and WT mice. Conversely, the pituitaries of D2R−/− already at this age appear larger both in weight and histologic appearance. AL, anterior lobe. C, Expression of PRL, GH, and TSH mRNAs analyzed by in situ hybridization in pituitaries of WT and D2 mutants, as indicated. Magnification, ×100.
Absence of hyperplasia in 4-month-old mice, however, did not exclude the possibility that this process could be delayed in animals lacking only 1 of the 2 D2R isoforms. Thus, we repeated the analysis in 8-month-old mice. Remarkably, also at this time point the weight and histologic appearance of the pituitaries from D2L−/− and D2S−/− animals did not differ from that of WT animals (Figure 5), whereas a considerable hyperplasia was present in the pituitary of D2R−/− mice in support of previous results (3). Altogether, these data indicate that the presence of only 1 of the D2R isoforms is sufficient to maintain the control of lactotroph proliferation. Thus, either D2L or D2S are each individually able to prevent the pathologic growth observed in the absence of both isoforms-mediated D2R signaling.
Figure 5.
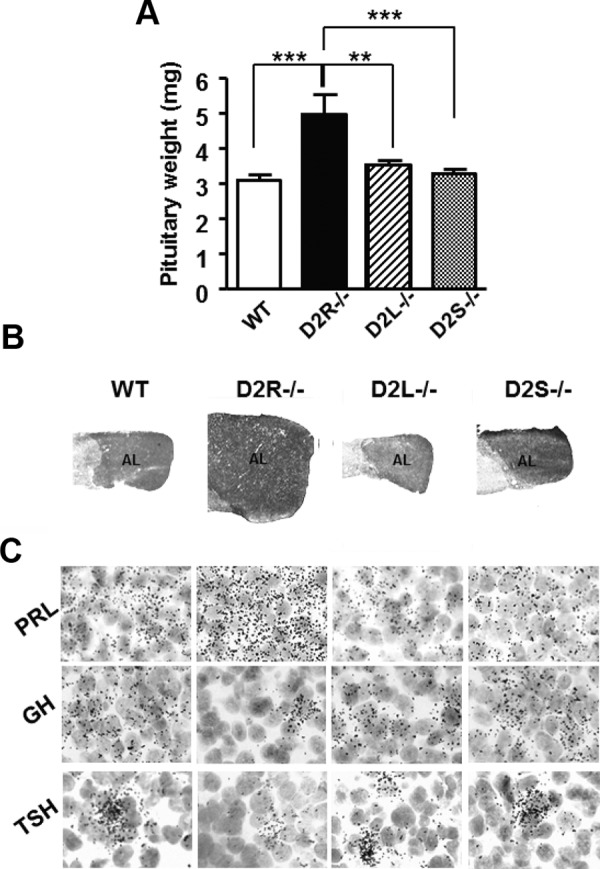
Absence of Hyperplasia in 8-Month-Old Pituitaries from[b] D2L−/− and D2S−/− Mice. A, Quantification of pituitary weight from 8-month-old animals of the different genotypes, as indicated. Data represent the mean ± SEM of 5–7 female pituitaries per group, analyzed by 1-way ANOVA followed by Bonferroni (Genotype: F(3, 32)=11.63). ***, P < .001 vs WT and D2S−/−; ** P < .01 vs D2L−/−. B, Hematoxylin-eosin staining of pituitary sections from females of the indicated genotypes. As previously observed at 4 months, also at 8 months the pituitaries of D2L−/− and D2S−/− mice do not differ from WT, whereas a clear difference is observed between D2R−/− pituitaries and all the other genotypes. AL, anterior lobe. C, Expression of PRL, GH, and TSH mRNAs analyzed by in situ hybridization in pituitaries of WT and D2 mutants, as indicated. Magnification, ×100.
Normal levels of pituitary hormones in D2L- and D2S-null animals
In the anterior pituitary, D2Rs are major regulators of PRL production; indeed, their activation inhibits the synthesis and release of PRL, a lactogenic hormone (1). Lactotrophs are the final cell product of a lineage that also originates thyrotrophs, producers of TSH, and somatotrophs, the cells that synthesize GH (26). Hence, we analyzed the expression level for these hormones in pituitaries from D2L and D2S mutants and compared these with analyses of D2R−/− and WT mice. The mRNA levels of PRL, GH, and TSH were measured by qRT-PCR on total mRNA (Figure 6) and by in situ hybridization (Figure 4C) on pituitary sections from 4-month-old animals of each genotype. In agreement with previous findings (3), D2R−/− pituitaries show a strong increase of PRL mRNA expression (Figures 4C and 6). Interestingly, PRL expression levels from D2L−/− and D2S−/− mice did not differ from WT mice (Figures 4C and 6A). At this age point, there were no differences in GH and TSH mRNA levels among genotypes (Figures 4C and 6A). These experiments were also repeated in 8-month-old mice and results similar to those obtained at 4 months for WT, D2L−/−, and D2S−/− mice were obtained (Figures 5C and 6B). Analyses of 8-month-old D2R−/− pituitary's RNAs showed increased PRL expression and reduction of GH and TSH mRNA expression (Figures 5C and 6B), as previously described (3). PRL inhibition in D2L−/− and D2S−/− pituitaries also indicates that both D2 isoforms are equally capable of inhibiting the cAMP pathway. Indeed, haloperidol treatment of WT, D2L−/−, and D2S−/− mice similarly induced an increase of cAMP response element binding protein phosphorylation on Ser133 in all genotypes (Supplemental Figure 3). In addition, levels of phosphorylated cAMP response element binding protein in the D2 mutants were similar to those in saline-treated pituitaries from WT mice, further supporting equal ability of only 1 of the D2 isoform to control cAMP levels. Altogether, these results suggest that only the simultaneous ablation of both D2R isoforms affects PRL mRNA levels, whereas the presence of either D2L or D2S is sufficient for regulating the inhibition of PRL synthesis.
Figure 6.
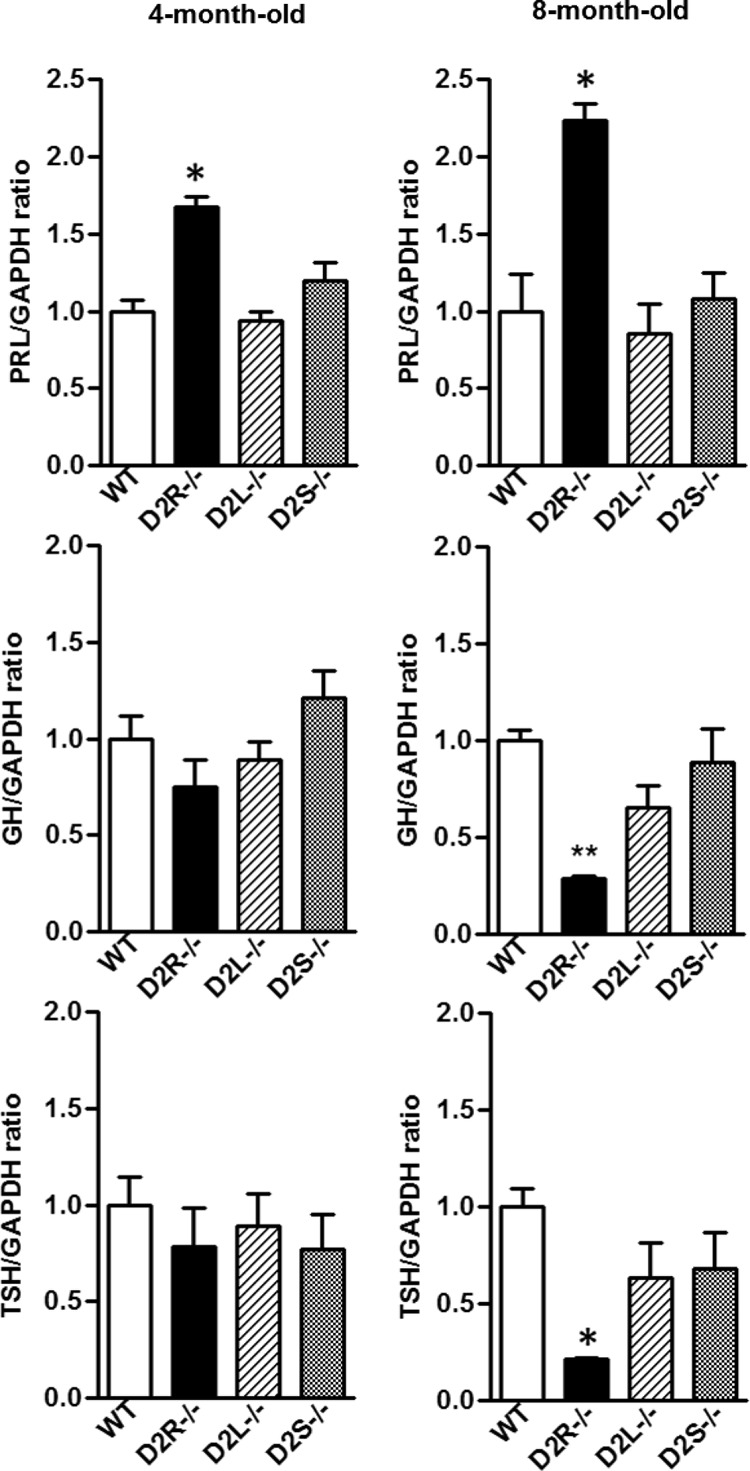
mRNA Expression Profiles for PRL, GH, and TSH in D2L−/− and D2S−/− Mice Do Not Differ from WT. Quantification of PRL, GH, and TSH mRNA expression levels from pituitaries of 4- (left panels) and 8-month-old female (right panels) of WT, D2R−/−, D2L−/−, and D2S−/− mice. qRT-PCRs were performed using primers specific for PRL, GH, TSH, and GAPDH. Data are represented as ratio between PRL:GAPDH, GH:GAPDH, and TSH:GAPDH and normalized to WT values, set as 1. At least 5 animals per genotype were used and results analyzed by 1-way ANOVA. * P < .05 D2R−/− vs WT, D2L−/−, and D2S−/−.
Estradiol treatment induces pituitary enlargement of greater degree in D2R mutants
E2 is one of the most important factors in the regulation of lactotroph physiology and PRL secretion (27) (28). E2 treatments in vivo induce the expression and release of pituitary growth factors such as TGF-α, TGFβ3, fibroblast growth factor-β, and IGF-I (29). These factors, by acting in a paracrine and autocrine manner, induce the proliferation and differentation of lactotrophs (30, 31). Chronic E2 administration in females induces pituitary hyperplasia and, in some cases, prolactinomas (27).
To assess whether the presence of only 1 of the D2R isoforms might be sufficient to maintain PRL expression and lactotroph growth to WT levels as observed under normal conditions, we compared the effects of a chronic estrogen treatment in D2L−/−, D2S−/− females, and WT littermates. To do so, 4-month-old mice were administered E2 (25 mg/kg sc in safflower oil) once a week for a period of 10 weeks. Animals were then humanely destroyed and their pituitaries were weighed and analyzed. Interestingly, the weight and histologic analyses of pituitaries of treated animals showed an E2-induced significant enlargement of the gland in all animals as compared with vehicle-treated mice of each genotype (Figure 7, A and C). It is noteworthy that the weight and size of D2L−/− and D2S−/− pituitaries were larger than that of treated WT littermate pituitaries (Figure 7, A and C; P < .05; n = 5). Nevertheless, the weight and size of E2-treated D2R−/− pituitaries significantly exceeded that of D2L−/−, D2S−/−, and WT mice (Figure 7, A and C). The increased size of the pituitaries of D2R mutants was due to lactotroph hyperplasia as assessed by PRL in situ hybridization experiments (Figure 7D).
Figure 7.
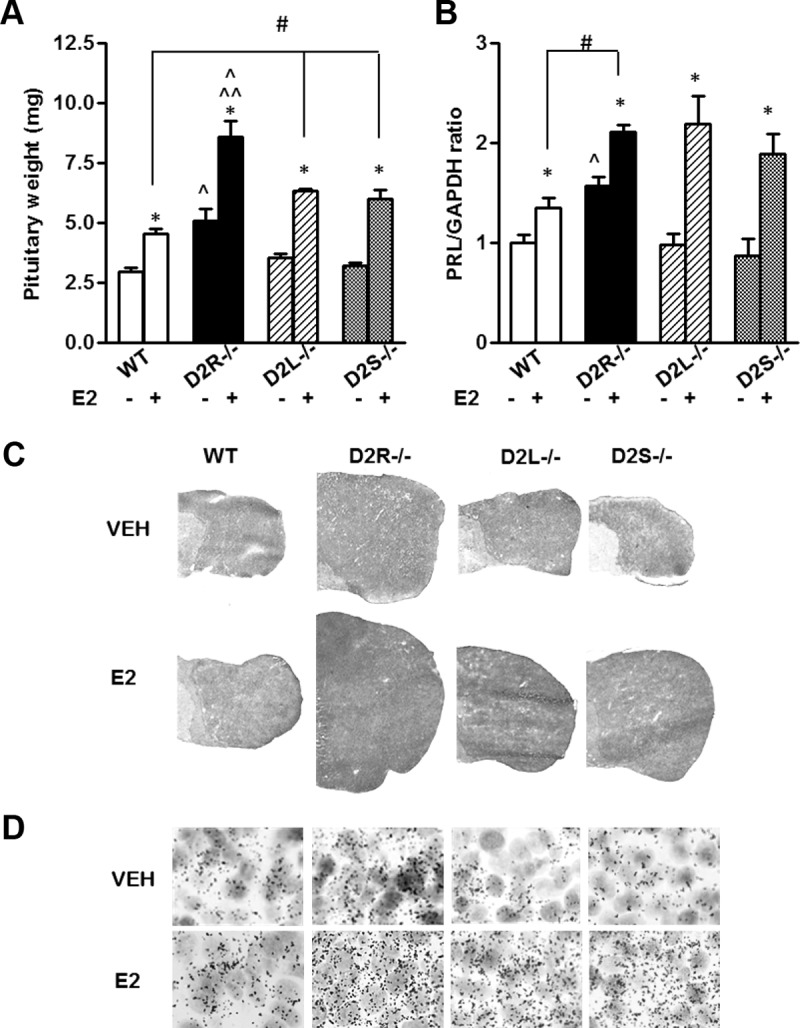
Chronic E2 Treatment Induces Hyperplasia in D2R Mutants. Animals (4 mo of age) were treated with E2 (sc injection, 0.5 mg/100 μL of safflower oil) or VEH (vehicle, safflower oil) for 10 weeks. At least 5 animals per genotype were used, and results were analyzed by 2-way ANOVA to compare the treatment and the genotype followed by the appropriate post hoc test. A, Bars represent pituitary weight among genotypes. Genotype × treatment: F(3, 42) =3.057. * P < .05 vs respective control injected with VEH; ○, P < .05 vs D2L−/− and D2S−/− with the same treatment; ^^ P < .01 vs WT with the same treatment; #, P < .05 vs WT with the same treatment. B, Quantification of PRL mRNA by qRT-PCR was performed using specific primers for PRL. Data are represented as fold change ± SEM of PRL mRNA/GAPDH. Main effect of treatment: F(1, 36)=27.09. Main effect of Genotype: F(3, 36) = 4.72. Student's t test: * P < .05 vs respective control injected with VEH; ^ P < .05 vs the other genotypes with the same treatment; # P < .05 vs WT with the same treatment. C, Hematoxylin-eosin staining of pituitary cryostat sections of animals of the indicated genotypes treated either with vehicle (VEH) or estrogens (E2). D, PRL mRNA expression analyzed by in situ hybridization in pituitaries of WT and D2 mutants treated either with vehicle (VEH) or estrogens (E2), as indicated. Magnification, ×100.
Lactotroph hyperplasia was further confirmed by the analysis of PRL mRNA levels in mice of each genotype with or without E2 treatment, because a strong increase in PRL expression was observed in all genotypes. However, the E2 treatment elicited a much stronger effect on PRL expression in all D2R mutants as compared with WT (Figure 7B). These results indicate that, although under normal conditions the presence of only 1 isoform is sufficient to maintain normal PRL expression and lactotroph proliferative rate, under E2 challenge this is no longer the case. Thus, a chronic E2 treatment bypasses the inhibitory signaling generated by the presence of either D2L or D2S and leads into higher PRL expression and pituitary hyperplasia. This indicates that an integral pathway of D2R-mediated signaling is necessary to maintain a normal physiologic response of lactotrophs to pathologic conditions.
Next, we analyzed phosphorylation of ERK 1/2 and AKT after estrogen treatment to compare them with their levels under normal conditions. Interestingly, E2 treatment appears to uncouple both isoforms from their respective signaling pathways. Indeed, in D2L−/− mice we observed a significant decrease of ERK 1/2 phosphorylation as compared with vehicle-treated control mice of the same genotype (Figure 8A). ERK 1/2 phosphorylation levels were also decreased significantly in pituitaries of WT-treated mice vs vehicle-treated controls; ERK 1/2 phosphorylation, in contrast, was not affected by estrogen treatment in D2S−/− and D2R−/− mice as compared with vehicle-treated animals of the same genotype. In D2S−/− mice we observed an increase of AKT phosphorylation at Thr308 to values that no longer differed from that of WT pituitaries (Figure 8B). Conversely, AKT phosphorylation at Thr308 did not differ between untreated and treated mice of both WT and D2L−/− genotype. Thus, estrogens appear to interfere with the signaling of either isoforms of the D2R.
Figure 8.
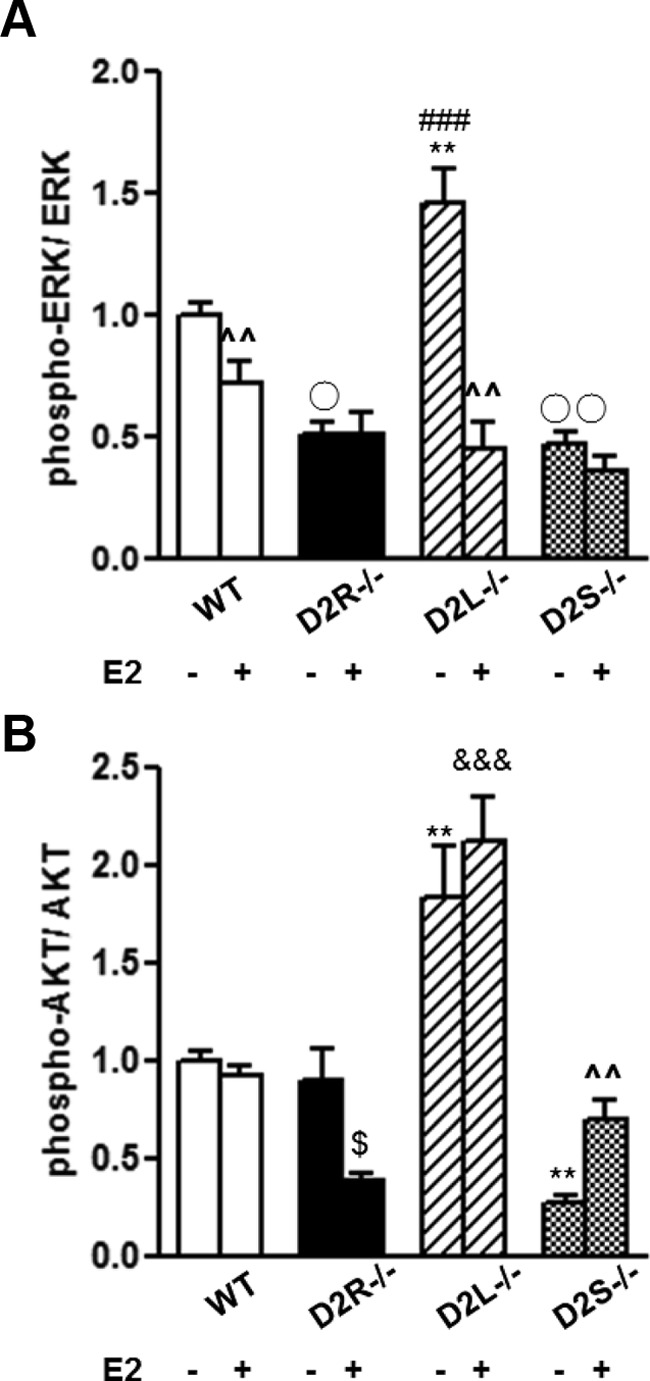
E2 Treatment on the Phosphorylation Prolife of ERK 1/2 and AKT. A, Quantification of phospho-ERK:ERK ratio in the pituitaries from WT, D2R−/−, D2L−/−, and D2S−/− animals treated with E2 for 10 weeks. Data represent the ratio between phospho-ERK and the total ERK normalized to the ratio of WT animals. B, Quantification of phospho-AKT/AKT as in panel A. At least 7 animals per genotype were analyzed by 2-way ANOVA followed by the appropriate post hoc test. A, Genotype × treatment: F(3, 49)=6.23. B, Genotype × treatment: F(3, 31)=4.83. A and B, ^^ P < .01 vs the same genotype without treatment; ** P < .01 vs the other genotypes without treatment; ○ P < .05, or ○○ P < .01 vs WT without treatment; $ P < .05 vs the same genotype without treatment; &&& P < .001 vs WT, D2R−/−, and D2S−/− with treatment.
Interestingly, in D2R−/− pituitaries we observed no difference in ERK 1/2 phosphorylation levels upon estrogen treatment, in support of a D2R-specific effect on this pathway. In contrast, AKT phosphorylation was reduced by estrogen in these mice pituitaries, further suggesting, as in 8-month-old mice, the intervention of other molecules presently nonidentified, in the control of AKT signaling in the absence of D2R in aging and under estrogen challenge.
Discussion
The dopamine D2R is a critical modulator of lactotroph physiology. A remarkable illustration of this notion is provided by D2R-null female mice that develop pituitary hyperplasia and prolactinomas as they age (3, 7, 20, 40). Also in humans D2R signaling governs lactotroph functions, and indeed clinical therapies based on the use of D2R agonists, such as bromocriptine and cabergoline, are currently used to treat PRL-producing adenomas, which are frequent in young women. Nevertheless, a proportion of these tumors become resistant to these treatments likely due to major impairments of the D2R-mediated signaling (5). Alternative therapies for dopamine-resistant tumors should potentially aim at identifying downstream targets of D2R activity. D2R signaling is classically linked to the inhibition of the cAMP pathway (32); however, more recent studies have shown that activation of these receptors is key in the regulation of other signaling pathways, which play a critical role in cell growth and differentiation such as the ERK 1/2 (11) and AKT pathways (25).
D2L and D2S isoforms appear to share mostly similar pharmacologic and signaling properties when transfected in heterologous cells (15, 16, 33, 34). Interestingly, studies performed in the striatum using D2L−/− mice suggested that D2L and D2S might serve different functions (18), at least in this brain area. In agreement with a differential function and signaling of these isoforms in vivo, we had previously shown that overexpression of D2S, but not D2L, in the lactotrophs of transgenic mice resulted in an increased stimulation of the ERK 1/2 pathway (11). D2L, on the other hand, has been shown to activate a cAMP-independent signaling system in the striatum, which results in the inhibition of the AKT activity (12). To date it remained unexplored whether this signaling pathway operates also in the pituitary gland.
The present study provides the first in vivo comparative analysis of D2L- and D2S-mediated signaling in the pituitary gland in the absence of the other isoform. For this, we used D2L−/− mice (18) together with the newly generated D2S−/− mice; these animals allowed the analysis of isoform-specific D2R-mediated signaling and its effect on lactotroph growth and PRL expression. Mice were analyzed under physiologically normal conditions or after pharmacologic and hormonal challenge.
Importantly, we discovered that D2L and D2S activate different signaling pathways in the pituitary gland. In line with an inhibitory effect of D2R stimulation on the AKT pathway, absence of D2L results in an increase of the phosphorylation level of AKT on Thr308 with respect to WT, as previously reported in the brain (25). Remarkably, in D2L−/− animals ERK 1/2 phosphorylation is also increased, as compared with WT levels. The increase of ERK 1/2 phosphorylation with respect to WT pituitaries is likely due to the conversion of all Drd2 gene transcripts into D2S in these mice, leading to a concomitantly stronger activation by D2S of the ERK 1/2 pathway (11). In support of these data, analyses of D2S−/− mice showed a decreased ERK 1/2 phosphorylation levels with respect to pituitaries from WT littermates. Extracts from D2S−/− mice also showed a significantly stronger inhibition of the AKT phosphorylation on Thr308 with respect to WT mice. The stronger inhibition of the AKT pathway in D2S−/− pituitary is very likely dependent on the higher expression of D2L receptors in these mutants as compared with WT due to the conversion of all Drd2 transcripts into D2L. The effects of D2S and D2L ablation on the ERK 1/2 and AKT pathways remained constant with age, as shown by the analyses performed in 4-month-old vs 8-month-old female mice. Thus, signaling from the D2R isoforms in the pituitary gland shows a great level of selectivity in the coupling to specific signaling pathways. These effects are specific as shown by treatment of D2L−/− and D2S−/− mice with the D2R antagonist haloperidol that abolished the D2R isoform-dependent modulations on both pathways. It should be noted that results obtained in vivo in D2L−/− and D2S−/− mice on the dopamine-mediated modulation of the ERK and AKT pathways differ, in part, from those obtained in studies performed in rat GH cells ex vivo (35–42). We believe that these discrepancies are linked to the different model systems used in our studies and in other research. For instance, GH cells are of somatolactotroph origin and derived from a rat pituitary tumor; our analyses have been performed on pituitaries of D2 isoform knockout mice, which do not develop tumors. These differences might account for these discrepancies and suggest different outcomes of D2-mediated signaling in normal and transformed cells.
Importantly, analyses of extracts from D2R−/− mouse pituitaries summarize the effect of the deletion of each isoform. Indeed, in these mice, we observed a down-regulation of the ERK 1/2 phosphorylation and an up-regulation of the AKT on Thr308. Interestingly, whereas D2R−/− mice develop pituitary hyperplasia and prolactinomas as they age, D2L−/− and D2S−/− mice do not.
It is thus conceivable that the simultaneous ablation of both isoforms resulting in opposite outcomes on the ERK 1/2 (inhibitory) and AKT (stimulatory) signaling is responsible for the aberrant lactotroph proliferation observed in D2R−/− mice. Indeed, whereas in D2L−/− mice a concomitant up-regulation of both pathways is observed, in D2S−/− mice, these pathways are both down-regulated. One interesting hypothesis is that, under normal conditions, signaling from either D2L or D2S exerts a balance of the ERK 1/2 and AKT pathways able to compensate for the loss of a normal dopamine-mediated signaling. In line with these results, when PRL mRNA expression was analyzed, we did not obtain statistically different results between WT, D2L−/−, and D2S−/− mice. Similarly, we did not observe pituitary hyperplasia either in 4- or in 8-month-old D2L−/− and D2S−/− mice as compared with WT mice, whereas a clear hyperplasia was readily visible in D2R−/− mice.
It could be argued that the increase of D2L or D2S expression in each mutant, due to the conversion of Drd2 transcripts in only one of the isoform in the absence of the other, might be protective toward an aberrant proliferation of lactotrophs. To address this point we crossed D2S−/− or D2L−/− mice with D2R−/− mice in order to lower the expression of each isoform; however, also in this case the progenies from these matings did not show sign of lactotroph hyperplasia at all ages tested (data not shown).
We then treated D2L−/− and D2S−/− mice with estrogens in order to analyze lactotroph responses in these mutants after challenge. Estrogens induce lactotroph proliferation and PRL expression, and its effect is normally counterbalanced by dopamine (1). As previously discussed, under normal conditions PRL expression is not affected by loss of either D2L or D2S signaling as compared with WT littermates. However, after challenge with estrogens, we observed that E2 differentially affects pituitary growth and PRL expression in D2R mutants in comparison with untreated mutants or to WT-treated or untreated mice. Interestingly, we observed that in response to chronic estrogens, absence of one or both isoforms leads to a similar increase of PRL expression as compared with WT mice. In addition, estrogens treatment induced a hyperplasia of the anterior pituitary in both D2L−/− and D2S−/− mice in a manner similar to that obtained in D2R−/− mice. Nevertheless, D2L−/− and D2S−/− pituitaries did not reach the size of D2R−/− pituitaries, and the histologic examination did not show the presence of neoplasia. These results indicate that the presence of both isoforms is necessary to prevent hyperplasia and increased PRL levels upon a sustained estrogen challenge. Thereby we conclude that the presence of only 1 isoform of the D2R is sufficient to preserve lactotroph functions in physiologic conditions, but not in conditions simulating a pathologic state as modeled by the chronic estrogen treatment. These results also suggest that microadenomas in humans might be linked to simultaneous impairments of D2L or D2S and of hormone levels, whereas dopamine agonist-resistant tumors might result from a total loss of D2R-mediated signaling.
Supplementary Material
Acknowledgments
This work was supported by funds from the University of California Cancer Research Coordinating Committee (UC CRCC-43674) and National Institutes of Health (NIDA DA024689 and DA033554).
Disclosure Summary: The authors have nothing to declare.
Footnotes
- D2L
- long isoform of D2
- D2S
- short isoform of D2
- D2R
- D2 receptor
- E2
- 17β-estradiol
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- PRL
- prolactin
- qRT-PCR
- quantitative RT-PCR
- WT
- wild type.
References
- 1. Ben-Jonathan N, Hnasko R. Dopamine as a prolactin (PRL) inhibitor. Endocr Rev. 2001;22:724–763 [DOI] [PubMed] [Google Scholar]
- 2. Radl DB, Ferraris J, Boti V, Seilicovich A, Sarkar DK, Pisera D. Dopamine-induced apoptosis of lactotropes is mediated by the short isoform of D2 receptor. PloS One. 2011;6:e18097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Saiardi A, Bozzi Y, Baik JH, Borrelli E. Antiproliferative role of dopamine: loss of D2 receptors causes hormonal dysfunction and pituitary hyperplasia. Neuron. 1997;19:115–126 [DOI] [PubMed] [Google Scholar]
- 4. Molitch ME. Prolactinoma in pregnancy. Best Pract Res Clin Endocrinol Metab. 2011;25:885–896 [DOI] [PubMed] [Google Scholar]
- 5. Colao A, Savastano S. Medical treatment of prolactinomas. Nat Rev Endocrinol. 2011;7:267–278 [DOI] [PubMed] [Google Scholar]
- 6. Gillam MP, Molitch ME, Lombardi G, Colao A. Advances in the treatment of prolactinomas. Endocr Rev. 2006;27:485–534 [DOI] [PubMed] [Google Scholar]
- 7. Kelly MA, Rubinstein M, Asa SL, et al. Pituitary lactotroph hyperplasia and chronic hyperprolactinemia in dopamine D2 receptor-deficient mice. Neuron. 1997;19:103–113 [DOI] [PubMed] [Google Scholar]
- 8. Borgundvaag B, George SR. Dopamine inhibition of anterior pituitary adenylate cyclase is mediated through the high-affinity state of the D2 receptor. Life Sci. 1985;37:379–386 [DOI] [PubMed] [Google Scholar]
- 9. Ishida M, Mitsui T, Yamakawa K, et al. Involvement of cAMP response element-binding protein in the regulation of cell proliferation and the prolactin promoter of lactotrophs in primary culture. Am J Physiol Endocrinol Metab. 2007;293:E1529–E1537 [DOI] [PubMed] [Google Scholar]
- 10. Montmayeur JP, Borrelli E. Transcription mediated by a cAMP-responsive promoter element is reduced upon activation of dopamine D2 receptors. Proc Natl Acad Sci USA. 1991;88:3135–3139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Iaccarino C, Samad TA, Mathis C, Kercret H, Picetti R, Borrelli E. Control of lactotrop proliferation by dopamine: essential role of signaling through D2 receptors and ERKs. Proc Natl Acad Sci USA. 2002;99:14530–14535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Beaulieu JM, Gainetdinov RR, Caron MG. The Akt-GSK-3 signaling cascade in the actions of dopamine. Trends Pharmacol Sci. 2007;28:166–172 [DOI] [PubMed] [Google Scholar]
- 13. Cakir M, Grossman AB. Targeting MAPK (Ras/ERK) and PI3K/Akt pathways in pituitary tumorigenesis. Expert Opin Ther Targets. 2009;13:1121–1134 [DOI] [PubMed] [Google Scholar]
- 14. Dworakowska D, Wlodek E, Leontiou CA, et al. Activation of RAF/MEK/ERK and PI3K/AKT/mTOR pathways in pituitary adenomas and their effects on downstream effectors. Endocr Relat Cancer. 2009;16:1329–1338 [DOI] [PubMed] [Google Scholar]
- 15. Montmayeur JP, Bausero P, Amlaiky N, Maroteaux L, Hen R, Borrelli E. Differential expression of the mouse D2 dopamine receptor isoforms. FEBS Lett. 1991;278:239–243 [DOI] [PubMed] [Google Scholar]
- 16. Montmayeur JP, Guiramand J, Borrelli E. Preferential coupling between dopamine D2 receptors and G-proteins. Mol Endocrinol. 1993;7:161–170 [DOI] [PubMed] [Google Scholar]
- 17. Guiramand J, Montmayeur JP, Ceraline J, Bhatia M, Borrelli E. Alternative splicing of the dopamine D2 receptor directs specificity of coupling to G-proteins. J Biol Chem. 1995;270:7354–7358 [DOI] [PubMed] [Google Scholar]
- 18. Usiello A, Baik JH, Rougé-Pont F, et al. Distinct functions of the two isoforms of dopamine D2 receptors. Nature. 2000;408:199–203 [DOI] [PubMed] [Google Scholar]
- 19. Baik JH, Picetti R, Saiardi A, et al. Parkinsonian-like locomotor impairment in mice lacking dopamine D2 receptors. Nature. 1995;377:424–428 [DOI] [PubMed] [Google Scholar]
- 20. Asa SL, Kelly MA, Grandy DK, Low MJ. Pituitary lactotroph adenomas develop after prolonged lactotroph hyperplasia in dopamine D2 receptor-deficient mice. Endocrinology. 1999;140:5348–5355 [DOI] [PubMed] [Google Scholar]
- 21. Picetti R, Saiardi A, Abdel Samad T, Bozzi Y, Baik JH, Borrelli E. Dopamine D2 receptors in signal transduction and behavior. Crit Rev Neurobiol. 1997;11:121–142 [DOI] [PubMed] [Google Scholar]
- 22. Bertolino A, Fazio L, Di Giorgio A, et al. Genetically determined interaction between the dopamine transporter and the D2 receptor on prefronto-striatal activity and volume in humans. J Neurosci. 2009;29:1224–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Doi M, Yujnovsky I, Hirayama J, et al. Impaired light masking in dopamine D2 receptor-null mice. Nat Neurosci. 2006;9:732–734 [DOI] [PubMed] [Google Scholar]
- 24. Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. An Akt/β-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell. 2005;122:261–273 [DOI] [PubMed] [Google Scholar]
- 25. Beaulieu JM, Tirotta E, Sotnikova TD, et al. Regulation of Akt signaling by D2 and D3 dopamine receptors in vivo. J Neurosci. 2007;27:881–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Borrelli E, Heyman RA, Arias C, Sawchenko PE, Evans RM. Transgenic mice with inducible dwarfism. Nature. 1989;339:538–541 [DOI] [PubMed] [Google Scholar]
- 27. Sarkar DK. Genesis of prolactinomas: studies using estrogen-treated animals. Front Horm Res. 2006;35:32–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Toledano Y, Zonis S, Ren SG, Wawrowsky K, Chesnokova V, Melmed S. Estradiol partially recapitulates murine pituitary cell cycle response to pregnancy. Endocrinology. 2012;153:5011–5022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Takahashi S. Intrapituitary regulatory system of proliferation of mammotrophs in the pituitary gland. Zool Sci. 2004;21:601–611 [DOI] [PubMed] [Google Scholar]
- 30. Hentges S, Boyadjieva N, Sarkar DK. Transforming growth factor-β3 stimulates lactotrope cell growth by increasing basic fibroblast growth factor from folliculo-stellate cells. Endocrinology. 2000;141:859–867 [DOI] [PubMed] [Google Scholar]
- 31. Oomizu S, Takeuchi S, Takahashi S. Stimulatory effect of insulin-like growth factor I on proliferation of mouse pituitary cells in serum-free culture. J Endocrinol. 1998;157:53–62 [DOI] [PubMed] [Google Scholar]
- 32. Albert PR. G protein preferences for dopamine D2 inhibition of prolactin secretion and DNA synthesis in GH4 pituitary cells. Mol Endocrinol. 2002;16:1903–1911 [DOI] [PubMed] [Google Scholar]
- 33. Senogles SE. The D2 dopamine receptor isoforms signal through distinct Giα proteins to inhibit adenylyl cyclase. A study with site-directed mutant Giα proteins. J Biol Chem. 1994;269:23120–23127 [PubMed] [Google Scholar]
- 34. Liu YF, Civelli O, Grandy DK, Albert PR. Differential sensitivity of the short and long human dopamine D2 receptor subtypes to protein kinase C. J Neurochem. 1992;59:2311–2317 [DOI] [PubMed] [Google Scholar]
- 35. Conrad KE, Oberwetter JM, Vaillancourt R, Johnson GL, Gutierrez-Hartmann A. Identification of the functional components of the Ras signaling pathway regulating pituitary cell-specific gene expression. Mol Cell Biol. 1994;14:1553–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Romano D, Magalon K, Ciampini A, Talet C, Enjalbert A, Gerard C. Differential involvement of the Ras and Rap1 small GTPases in vasoactive intestinal and pituitary adenylyl cyclase activating polypeptides control of the prolactin gene. J Biol Chem. 2003;278:51386–51394 [DOI] [PubMed] [Google Scholar]
- 37. Romano D, Magalon K, Pertuit M, et al. Conditional overexpression of the wild-type Gsα as the gsp oncogene initiates chronic extracellularly regulated kinase 1/2 activation and hormone hypersecretion in pituitary cell lines. Endocrinology. 2007;148:2973–2983 [DOI] [PubMed] [Google Scholar]
- 38. Banihashemi B, Albert PR. Dopamine-D2S receptor inhibition of calcium influx, adenylyl cyclase, and mitogen-activated protein kinase in pituitary cells: distinct Gα and Gβγ requirements. Mol Endocrinol. 2002;16:2393–2404 [DOI] [PubMed] [Google Scholar]
- 39. Liu JC, Baker RE, Chow W, Sun CK, Elsholtz HP. Epigenetic mechanisms in the dopamine D2 receptor-dependent inhibition of the prolactin gene. Mol Endocrinol. 2005;19:1904–1917 [DOI] [PubMed] [Google Scholar]
- 40. Liu JC, Baker RE, Sun C, Sundmark VC, Elsholtz HP. Activation of Go-coupled dopamine D2 receptors inhibits ERK1/ERK2 in pituitary cells. A key step in the transcriptional suppression of the prolactin gene. J Biol Chem. 2002;277:35819–35825 [DOI] [PubMed] [Google Scholar]
- 41. Kanasaki H, Fukunaga K, Takahashi K, Miyazaki K, Miyamoto E. Involvement of p38 mitogen-activated protein kinase activation in bromocriptine-induced apoptosis in rat pituitary GH3 cells. Biol Reprod. 2000;62:1486–1494 [DOI] [PubMed] [Google Scholar]
- 42. Kievit P, Lauten JD, Maurer RA. Analysis of the role of the mitogen-activated protein kinase in mediating cyclic-adenosine 3′,5′-monophosphate effects on prolactin promoter activity. Mol Endocrinol. 2001;15:614–624 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.