

Abstract
RNase E, an essential endoribonuclease of Escherichia coli, interacts through its C-terminal region with multiple other proteins to form a complex termed the RNA degradosome. To investigate the degradosome's proposed role as an RNA decay machine, we used DNA microarrays to globally assess alterations in the steady-state abundance and decay of 4,289 E. coli mRNAs at single-gene resolution in bacteria carrying mutations in the degradosome constituents RNase E, polynucleotide phosphorylase, RhlB helicase, and enolase. Our results show that the functions of all four of these proteins are necessary for normal mRNA turnover. We identified specific transcripts and functionally distinguishable transcript classes whose half-life and abundance were affected congruently by multiple degradosome proteins, affected differentially by mutations in degradosome constituents, or not detectably altered by degradosome mutations. Our results, which argue that decay of some E. coli mRNAs in vivo depends on the action of assembled degradosomes, whereas others are acted on by degradosome proteins functioning independently of the complex, imply the existence of structural features or biochemical factors that target specific classes of mRNAs for decay by degradosomes.
Keywords: RNase E, rhlB helicase, enolase, polynucleotide phosphorylase
The turnover of mRNA is a necessary component of normal genetic regulation within cells. In eubacteria, mRNA degradation results from the combined action of endo- and exoribonucleases. In Escherichia coli, the exoribonuclease polynucleotide phosphorylase (PNPase), the RhlB RNA helicase, and the glycolytic enzyme enolase assemble on the C-terminal region of the endoribonuclease RNase E as constituents of a protein complex termed the RNA degradosome (refs. 1–4; for recent reviews, see refs. 5 and 6). Two heat shock proteins, GroEL and DnaK (4), and polyphosphate kinase (Ppk) (7) also are associated with degradosomes in substoichiometric amounts. The N-terminal half of RNase E, which contains the catalytic domain of the enzyme (8), is not sufficient for degradosome formation (3, 9) but can associate the degradosome protein complex with the cytoplasmic membrane (11).
The E. coli ams/rne locus, which encodes RNase E, is required for bulk RNA turnover (12, 13) as well as for the processing of 9S rRNA (14, 15). Investigations of the decay of individual transcripts (16–19) and global investigations of mRNA abundance in rne mutants (20) have indicated a broadly important role for this enzyme. However, the RNase E region that provides a scaffold for degradosome formation is not essential for cell survival and growth (17, 21, 22), and truncated RNase E proteins lacking this domain are active in vivo as well as in vitro (8, 23).
Although the degradosome commonly has been viewed as an RNA decay “machine” (e.g., ref. 24), until recently (11) it was not known whether assembled degradosomes actually exist as such in living cells. Moreover, whether degradosome formation is a significant factor in mRNA decay in vivo has been controversial (21, 22). Studies by Ow et al. (22) using bacteria synthesizing truncated RNase E proteins concluded that degradosome assembly is dispensable for normal RNA decay, whereas Lopez et al. (21) found that similar RNase E truncation mutations retard both bulk mRNA decay and degradation of the lacZ and thrS transcripts.
There is evidence that individual degradosome constituents can functionally interact during decay of at least some RNAs. For example, PNPase and RNase E cooperate in the degradation of RNA I, an antisense regulator of replication of ColE1-type plasmids (25), and RhlB helicase can stimulate degradation of the malE–malF transcript by PNPase in vitro (1). Studies of reconstituted degradosomes in vitro (26, 27) have yielded analogous results. However, degradosome proteins can remain unattached to RNase E (2, 11); even in bacteria that have the ability to form the assembled degradosome complex, only 5–10% of cellular enolase and 10–20% of PNPase are estimated to be present in the complex (1, 11). Thus, mutations in degradosome constituents may affect the actions of unassociated constituents as well the actions of proteins in assembled degradosomes. Consistent with the possibility that degradosome components may operate in other formats, RhlB helicase has been shown to interact with PNPase independently of RNase E (28).
To better define the roles of RNase E, PNPase, RhlB helicase, and enolase in RNA metabolism both within assembled degradosome and as independently functioning enzymes, we undertook a DNA microarray-based, genome-scale investigation of the effects of mutations in these proteins on E. coli mRNA abundance and half-life; the methods used were similar to those employed to investigate these parameters in other contexts (20, 29). Here we report evidence that the functions of all four of the above degradosome proteins are needed for normal mRNA decay in E. coli and that assembled degradosome components work in concert to regulate transcripts of some E. coli metabolic pathways but not others. Our findings imply the existence of structural features or biochemical factors that distinguish among different classes of mRNAs targeted for degradation.
Materials and Methods
Strains and Culture Conditions. The following E. coli strains were used: wild-type N3433 (lacZ, relA, spot1, thi1) (30); pnp mutant strain YHC012 (Tn5::pnp, lacZ, relA, spot1, thi1) (10); rhlB mutant SU02 (lacZ, relA, spot1, thi1, ΔrhlB) (present study); SH3208 [hisΔtrpE5(λ)] and mutant BZ453 (17) contains a truncated rne gene that encodes amino acids 1–602 of the Rne protein; wild-type strain K10 [garB10, fhuA22, ompF627( ), fadL701(
), fadL701( ), relA1, pit-10, spoT1, rrnB-2, mcrB1, creC510] (4); and mutant DF261 [garB10, fhuA 22, ompF627(
), relA1, pit-10, spoT1, rrnB-2, mcrB1, creC510] (4); and mutant DF261 [garB10, fhuA 22, ompF627( ), fadL701(
), fadL701( ), eno-2, relA1, pit-10, spoT1, rrnB-2, mcrB1, creC510] (31).
), eno-2, relA1, pit-10, spoT1, rrnB-2, mcrB1, creC510] (31).
Cultures were grown in M9 media supplemented with 0.2% tryptone/0.2% glycerol/1 mM MgSO4/0.0001% thiamine at 30°C in a shaking water bath. Media for strain DF261 and its isogenic wild-type strain were further supplemented with 40 mM succinate. Strains SH3208 and BZ453 were also studied in LB media. M9 plus 0.2% glucose agar plates, supplemented with tryptone, MgSO4, and thiamine as above, with and without 0.2% succinate were used to test strain DF261 for reversion. Eno mutants do not grow on glucose minimal media without succinate (31). The log-phase generation time for each strain was determined (Table 1).
Table 1. Median mRNA half-lives and generation times.
| Strain | Median half-life, min | Generation time, min |
|---|---|---|
| K10 | 2.1 ± 0.2 | 73 |
| DF261 (Eno-) | 2.8 ± 0.3 | 78 |
| N3433 | 3.7 ± 0.4 | 75 |
| SU02 (RhlB-) | 4.0 ± 1.2 | 75 |
| YHC012 (PNP-) | 4.0 ± 0.4 | 84 |
| SH3208 | 3.2 ± 0.3 | 81 |
| BZ453 (Rne truncation) | 6.0 ± 0.6 | 84 |
Generation times were determined for each mutant strain and its respective parental strain by following growth at 30°C in M9 media supplemented as described in Materials and Methods by following culture density at OD540. Strain K10 is the parental strain of DF261. N3433 is the parental strain of both SU02 and YHC012. SH3208 is the parental strain of BZ453. Median mRNA half-lives for each strain are expressed in minutes; indicated 95% confidence intervals were estimated as described in Materials and Methods.
Construction of an rhlB Mutant Strain Isogenic to N3433. Inactivation of the rhlB gene was accomplished by using procedures described in ref. 32, and PCR primers having extensions homologous to sequences flanking the rhlB ORF (underlined): RhlBKO-5′ [AAC TAA AGG GTT GAC TTT ATT TCA CCG GAT ACG CTT TGT GTA GGC TGG AGC TGC TTC (base pairs 3,963,309–3,963,282 of the E. coli genome; ref. 33)] and RhlBKO-3′ [TAC AGT TTG AAT GAT TTT GAG TAT GAC ATT TTT TAT CAT ATG AAT ATC CTC CTT AGT (base pairs 3,961,944–3,961,977)]. Verification of recombination into the rhlB locus was made by PCR using primer sequences homologous to rhlB flanking sequences (underlined): RhlB1-5′ CTC TCG AGG GCT GCA GGG AAT AGT AGC TGA TAT TC and RhlB430-3′ GGG GAT CCA AGC TTA TAC CTG AAC GAC GAC GAT T. These primers also contain extensions with restriction enzyme sites, indicated in boldface. The first confirmed recombinant strain in BW25113 was named SU01. The kanamycin (Km) resistance cassette within the disrupted rhlB locus was mobilized from strain SU01 and transferred to an N3433 background, creating strain SU02 by P1 (chloramphenicol-resistance) transduction as described (34).
Verification of Degradosome Protein Absence by Western Blotting. One milliliter of culture grown as described above was collected by centrifugation for Western blot analyses performed as described (4). Antibodies used were polyclonal anti-PNPase (1:8,000), anti-Eno (1:5,000), anti-RhlB (1:5,000), and monoclonal anti-RNase E (1:2,000) directed against the N-terminal region of RNase E. Chemiluminescence using an ECL kit (Amersham Pharmacia) was used for detection of degradosome protein bands.
Microarray Measurement of RNA Abundance. Relative mRNA abundance in pnp, eno, and rhlB mutants was measured for 4,289 genes by using parental strains grown under identical conditions as a reference. Late log-phase RNA samples (OD540 = 0.7) from an rne C-terminal deletion strain were similarly studied. RNA was isolated, and fluorescently labeled cDNA was synthesized as described (20). Hybridizations and data collection were performed according to ref. 20. Data were managed by using the Stanford Microarray Database (http://genome-www.stanford.edu/microarray) (35). Microarray spots with regression correlation of >0.4 and signal detectable at 300 fluorescence units above local background in >70% of arrays were used for analysis.
Analysis of Microarray Transcript Abundance Data. The “t score pattern-based rules” feature of the web-based gabriel software package (http://gabriel.stanford.edu) (36, 37), based on the statistical approach of Tusher et al. (38), was used to estimate false discovery rates (FDRs) in various transcript sets. Before analysis, missing values were estimated by using the K nearest neighbors method as implemented in the significance analysis for microarrays (SAM) package for genes with <50% missing data for each mutant–parental pair. FDRs at different t score thresholds were estimated by bootstrap analysis of a randomized data set, and thresholds yielding FDRs of 5% or less were applied to the experimental data we obtained. Determination of altered abundance for transcripts of previously documented operons was as described (20).
Determination of Half-Lives. Individual mRNA half-life determinations from microarray data were accomplished generally as described in ref. 29. After transcriptional arrest by rifampicin, mRNA samples were collected after 1.5-, 3-, 4.5-, and 6-min intervals. Analysis of samples harvested at both the first and last time points was carried out in duplicate for each time series. The average of the normalized fluorescence ratios from the duplicate hybridizations was used in subsequent calculations.
The array-determined half-lives of the sdhA, pntA, accC, and cysN transcripts were confirmed by quantitative two-step RT-PCR, as described in Supporting Text, which is published as supporting information on the PNAS web site.
Results
Construction and Verification of E. coli Strains Mutated in Specific Degradosomal Proteins. Previous work has identified E. coli mutant strains lacking PNPase or enolase (10, 31, 39). The absence of these enzymes in strains YHC012 and DF261, respectively, was verified by Western blotting (Fig. 1A). Expression of a truncated form of RNase E in strain BZ453 was similarly confirmed.
Fig. 1.

rhlB gene disruption and verification of degradosome component protein depletion in mutant strains. (A) Western blot-based verification of degradosome mutant protein expression. Shown are the amido-black-stained poly(vinylidene difluoride) (PVDF) membrane (Left) and corresponding Western blots (Right) conducted to verify mutant protein expression. PAGE and Western blotting were carried out as described in Materials and Methods. The target of antibodies used to probe each lane is indicated above the lanes corresponding to each parental/mutant strain pair. Deletion mutants YHC012 and SU02 failed to express PNPase and RhlB helicase (lanes 2 and 4, respectively). Strain DF261 contains a nonsense point mutation in the eno gene that also resulted in the absence of detectable protein expression (lane 6). Strain BZ453, carrying a chromosomal deletion of the C terminus of RNase E expressed an appropriately truncated RNase E protein (1–602 aa; lane 8). (B) A schematic diagram of the rhlB locus and flanking ORFs before and after deletion by homologous recombination. H1 and H2 indicate sequences complementary to the RhlBKO 5′ and 3′ primers. FRT is the Flp recombination target site. Arrowheads indicate the direction of ORFs. (C and D) PCR verification of rhlB disruption in strains SU01 and SU02. Agarose gels (2%) assessing PCR products are shown. Primer pairs rhlB and rhlBKO are as described in Materials and Methods; they amplify the rhlB locus, 1.2 kbp, and the 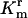 cassette, 1.6 kbp, respectively. M indicates lanes containing a 1-kbp DNA ladder. In lanes marked BW, N, and P, template DNA for PCR was from strains BW25113, N3433, and plasmid pKD4, respectively. In lanes marked 1 and 2, template DNA for PCR was from candidate recombinant colonies; in C these are derivatives of BW25113 after introduction of the rhlBKO PCR construct, and in D these are derivatives of N3433 after P1 transduction. For each candidate kanamycin-resistant colony, in contrast to the parental strains BW25113 and N3433, the rhlB gene-specific primer pair failed to produce a PCR product, whereas the rhlBKO primers resulted in a DNA fragment with an expected DNA size of a PCR fragment containing the kanamycin gene encoded in the plasmid pKD4.
cassette, 1.6 kbp, respectively. M indicates lanes containing a 1-kbp DNA ladder. In lanes marked BW, N, and P, template DNA for PCR was from strains BW25113, N3433, and plasmid pKD4, respectively. In lanes marked 1 and 2, template DNA for PCR was from candidate recombinant colonies; in C these are derivatives of BW25113 after introduction of the rhlBKO PCR construct, and in D these are derivatives of N3433 after P1 transduction. For each candidate kanamycin-resistant colony, in contrast to the parental strains BW25113 and N3433, the rhlB gene-specific primer pair failed to produce a PCR product, whereas the rhlBKO primers resulted in a DNA fragment with an expected DNA size of a PCR fragment containing the kanamycin gene encoded in the plasmid pKD4.
We used PCR products and a one-step gene-inactivation procedure (32) to construct a strain deleted for the rhlB locus (Fig. 1B). Candidate kanamycin-resistant colonies were tested for rhlB deletion by PCR using primers specific for the rhlB gene and the rhlB knockout; typical results are shown in Fig. 1B. The verified gene deletion was moved to strain N3433 by P1 transduction to create strain SU02, as described in Materials and Methods, and the deletion was confirmed by PCR in strain SU02 (Fig. 1C). Absence of detectable RhlB protein in the deletion mutant SU02 was shown by Western blotting (Fig. 1 A). Whereas it has been believed that rhlB is an essential gene (1, 40), our results indicate that deletion of the rhlB locus of E. coli strain N3433 does not preclude bacterial viability or affect generation time under the culture conditions we used (Table 1). Very recent findings by Khemici and Carpousis (41) independently show that rhlB is not essential for E. coli survival.
Global Effects of Degradosome Protein Mutations on mRNA Decay. Comparison of median mRNA half-life between strains encoding Eno, RhlB, Pnp and Rne mutant proteins and their respective parental strains showed prolongation of the median half-life for all four mutants (Table 1 and Fig. 2). Although prolongation was of borderline statistical significance for the rhlB and pnp mutants, these results suggested that the function of all four of the degradosome components studied is required for normal mRNA turnover in vivo. Confirmation of this conclusion was provided by quantitative RT-PCR-based half-life determinations for the cysN, accC, pntA, and sdhA transcripts, which yielded data in good agreement with our microarray half-life determinations and confirmed by an independent method that mutation of any of the studied degradosome component proteins can alter mRNA half-life in E. coli (Table 2 and Fig. 3).
Fig. 2.

Histograms of mRNA half-life frequencies in mutant and parental strains. Each histogram shows the distribution of measured half-lives for Eno (A), Pnp (B), RhlB (C), and N-terminal Rne (D) mutants and their respective parental strains. Half-life ranges are indicated on the x axis in each histogram, and the fraction of transcripts in each range is indicated on the y axis. y axis values are expressed as a fraction of the total number of transcripts for which half-lives were determined. Source half-life data are shown in Table 3, which is published as supporting information on the PNAS web site.
Table 2. RT-PCR confirmation of microarray-determined half-lives.
| Microarray
|
RT-PCR
|
|||
|---|---|---|---|---|
| WT | Mutant | WT | Mutant | |
| Enolase cysN | 1.0 | 3.1 | 1.0 ± 0.1 | 2.0 ± 0.8 |
| RhIB accC | 1.6 ± 0.4 | 3.3 ± 0.4 | 2.5 ± 0.1 | 3.5 ± 1.0 |
| PNPase pntA | 2.9 ± 0.1 | 6.1 | 3.8 ± 0.1 | 5.8 ± 0.1 |
| Rne N-Ter truncation*sdhA | 2.5 | 6.6 | 2.0 ± 0.1 | 4.6 ± 0.8 |
By using DNA microarrays, prolongation of mRNA half-lives for the cysN, accC, pntA, and sdhA transcripts were observed in the enolase, RhIB, PNPase, and Rne C-terminal deletion strains, respectively. These results were confirmed by using quantitative RT-PCR to follow transcript decay as described in Materials and Methods.
In LB media
Fig. 3.

Quantitative RT-PCR determination of mRNA half-lives. The decay of the accC transcript in strain SU02 (rhlB-) was followed by using quantitative RT-PCR. (Lower) Raw abundance data for the 16S rRNA internal control are shown. (Upper) Normalized abundance data for the decay of the transcript are shown. Half-life for the transcript was determined from the slope of the log-transformed abundance versus time data as described in Materials and Methods.
During our microarray investigations of mRNA half-life, we observed different median mRNA half-lives among the parental strains N3433, K10, and SH3208, used as controls for comparison with degradosomal protein mutants (N3433, 3.7 ± 0.4 min; K10, 2.1 ± 0.2 min; SH3208, 3.2 ± 0.3 min). These strains, which all are “wild type” with respect to known pathways of mRNA degradation, were cultured at 30°C in M9 media having identical supplementation, except for the addition of 40 mM succinate for K10, as described in Materials and Methods; the generation times were found to be approximately equal (73, 75, and 81 min, respectively). Our observation suggests either that the addition of succinate during growth of strain K10 shortens median mRNA half-life, despite the absence of a detectable effect on cell growth, or that mutations thought to be “silent” with respect to mRNA decay can affect transcript degradation.
Earlier work (29, 42) has shown that mRNAs encoded by genes having related biological functions commonly have similar half-lives. This observation, which was made in strain MG1655 by both groups, was evident in our current studies in strains K10, N3433, and SH3208 (Table 4, which is published as supporting information on the PNAS web site). Moreover, the same specific correlations between gene function and mRNA half-life were seen for all of these strains.
C-Terminal Deletion of RNase E and Mutations in Other Degradosome Proteins Result in both Congruent and Noncongruent Changes in Transcript Abundance. While comparing mRNA half-lives in degradosome component mutants and their parental strains, we observed that mutation of individual degradosome components had noncongruent effects on certain transcripts but congruent effects on others; for example, whereas only the pnp mutation affected the half-lives of the cstA, cirA, fkpA, and ribA transcripts, the stability of the asnS, serA, gdhA, glgB, and thrS transcripts increased in both bacteria lacking the C-terminal end of RNase E, which is necessary for assembly of the degradosome complex, and those mutated in pnp.
We made parallel observations when we examined transcript abundance in bacteria containing the C-terminal Rne deletion or individual loss-of-function mutations in rhlB, eno, and pnp. Because the mutant and parental strains we used were otherwise isogenic and were grown under identical conditions, changes in transcript abundance associated with each degradosome component mutation were presumed to be primary or secondary effects of the mutation on mRNA decay and were examined as additional parameters of degradosome function. We hypothesized that disparate effects of these mutations on transcript abundance may indicate degradosome-independent actions of the components, whereas congruent effects are likely to reflect the action of the assembled degradosome machine working as a unit. Significant alterations were identified by gabriel (36, 37) as described in Materials and Methods. This analysis identified multiple sets of transcripts affected in common by the RNase E deletion mutation and a loss-of-function mutation in one, two, or three other degradosome component proteins. Within these sets, the distribution of encoded protein functional classes as assigned by ref. 43 was studied by using the Gene Ontology (GO) rules feature of gabriel (36, 37).
We identified 119 transcripts that showed increased abundance in the four degradosome component mutants studied. The identities of these transcripts and their respective relative abundances are shown graphically in Fig. 4; source data for this figure are reported in Table 5, which is published as supporting information on the PNAS web site. Functional classification of genes whose transcripts were overrepresented among those satisfying the t score criteria identified the classes “Macromolecule synthesis or modification” and “Global (P < 0.02, gabriel GO rule).” The pattern-based algorithm of gabriel also identified 166 genes that showed congruently lowered abundance in the enolase, PNPase, and Rnetruncation mutants (Fig. 4; source data reported in Table 5). Although increased mRNA stability can lead directly to increased transcript abundance, the decreased abundance we observed for these transcripts in the presence of degradosomal protein mutations is presumed to be a secondary effect. Included among this latter group of transcripts are members of the osmotically responsive RpoS-dependent regulon: treA, otsA, otsB, dps, osmB, osmE, and katE (44–46).
Fig. 4.

Comparative transcript abundances in degradosome mutants. The figure shows a false-color diagram of log-phase transcript abundance profiles in four degradosome mutants (scale as indicated). Genes included in the figure showed significant changes in abundance relative to parental strains in multiple degradosome mutants. (Upper) (red side bar) The diagram includes the 119 transcripts that demonstrated increased abundance in all four mutants studied: Eno, RhlB, Pnp, and Rne C-terminal deletion. (Lower) (green side bar) A display of the 166 transcripts that showed decreased abundance in strains carrying Eno, Pnp, and Rne C-terminal deletion mutations.
The abundance of the transcripts of the CysPUWAM, CysK, CysDNC, and CysJIH operons, which are involved in sulfate/thiosulfate uptake and its utilization in the conversion of serine to cysteine, increased an average of 2.9, 2.8, 3.4, and 3.7 times, respectively, in the pnp deletion and 7.6, 6.7, and 13.1 times, respectively, in the RNase E C-terminal deletion. Paralleling this increase was a decrease in the abundance of transcripts encoding enzymes in the glycolytic pathway in the pnp mutant, as has been observed (20) also in Δrne bacteria that overexpress just the N-terminal half of RNase E from a plasmid-encoded construct.
Transcript Abundance Alterations Specific to Individual Degradosome Protein Mutants. The effects of mutations in degradosome component proteins on some transcripts were disparate, suggesting that these transcripts decay independently of the degradosome complex. For example, the DF261 enolase mutation resulted in increased abundance of mRNAs encoded by regulons involved in the uptake and utilization of multiple carbon sources, including glycerol, fructose, maltose, galacticol, and mannose, whereas abundance of these same transcripts was decreased in the RNase E truncation mutant. Specifically, transcripts of the anaerobic glp regulon consisting of the glpTQ operon, involved in glycerol uptake, and the glpABC operon, involved in glycerol catabolism, were increased in abundance in the enolase mutant and decreased in the Rne truncation mutant, arguing that decay of these transcripts is not carried out coordinately by components of a degradosome “machine.” Similarly, divergent effects of mutations in different degradosome proteins were seen for transcripts encoding transport and utilization systems for fructose, maltose, galacticol, and mannose (i.e., the fruBKA, malKM/lamB, gatYZABCD, and manXYZ operons). Data are presented in Table 5.
A prominent feature of the transcript profile observed uniquely in response to the loss of RhlB helicase function was decreased abundance, relative to the N3433 parental strain, of transcripts encoding proteins involved in iron uptake and known to be regulated by the Fur repressor in an iron-dependent manner (47, 48). Data showing the highly specific effect of the helicase mutation on these mRNAs (i.e., transcripts of the entCEBA, fepB, cirA, fes/entF/fepE, exbBD, tonB, fecABCDE, fecIR, and nrdHIEF operons) are presented in Table 5. The unique effect, among degradosome components, of the RhlB helicase on operons regulated by the Fur repressor protein suggests a role for this protein in decay of the fur transcript, or alternatively in posttranscriptional regulation of genes targeted by Fur. The data favor the latter possibility because levels of the fur transcript were unchanged in the helicase mutant. One such Fur target is the small regulatory RNA ryhB, which along with its targets has recently been reported to be degraded by RNase E (49, 50). Consistent with a possibly important role for rhlB in modulating the actions of the Fur repressor protein is our finding that transcripts of the nuoABCEFGHIJKLMN operon, which previously have been speculated to be subject to Fur regulation, were increased in abundance in the rhlB mutant (Table 5). Additionally, transcripts of the sdhCDAB (succinate dehydrogenase) operon, which are inversely correlated with expression of ryhB (49), were all increased in abundance by the rhlB mutation. Interestingly, transcripts encoding three other RNA helicases belonging to the DEAD box family, of which RhlB is a member (i.e., the DeaD, RhlE, and SrmB helicases), also showed increased abundance in the rhlB mutant.
Our investigations of the effects of degradosome mutations on mRNA abundance also identified a set of transcripts that showed no significant change in abundance in any of the degradosome mutant strains we studied, suggesting that their steady-state level is independent of the actions of RNase E and other degradosomal proteins. Approximately 11% of transcripts were in this category, showing abundance that varied <0.5 SD (0.3 log2 units or 1.2 times) in all four mutants studied.
Discussion
Notwithstanding the scientific interest that the discovery of a complex containing proteins implicated in mRNA decay in E. coli has generated, a biological role for the assembled degradosome complex as an RNA decay machine and the importance of individual degradosome proteins in controlling the degradation and cellular level of mRNAs have been uncertain. The work reported here indicates that the actions of RNase E, PNPase, enolase, and RhlB helicase are required for normal RNA turnover in E. coli, establishing a role for each of these degradosome components in mRNA decay. Our results further show that the effects of mutations in degradosome components vary with individual messages; they provide evidence that the assembled degradosome complex mediates the decay of some transcripts, whereas other transcripts are likely to be degraded independently of the complex, possibly by recently identified assembled subsets of degradosome enzymes (28). Collectively, our findings imply the existence of structural features or biochemical factors that distinguish cellular categories of mRNAs marked for degradation. Previous results indicate that transcripts encoding genes having related functions show similar rates of decay (29, 42). The experiments reported here suggest that the function of the protein encoded by a transcript is also a determinant of whether degradation of the transcript is carried out by assembled degradosomes.
The observation that PNPase deletion and loss of the RNase E scaffold region have similar effects on transcripts of the glycolytic and cysteine biosynthesis pathways is consistent with the notion that the combined actions of the two degradosome proteins modulate expression of genes in these pathways. Previous studies show that glycolytic pathway transcripts are less abundant in cells expressing the N terminus of RNase E versus the full-length enzyme and that RNase E-mediated decay of the glucose-specific permease transcript, ptsG, is regulated by glycolytic flux (20, 51). Interestingly, one of the glycolytic pathway transcripts affected by deletion of either the C-terminal scaffold region of RNase E or deletion of the pnp gene encodes the degradosome component enolase, which itself has global effects on mRNA turnover. The mechanisms underlying both the congruent effects of RNase E and pnp mutations on glycolytic pathway mRNAs and the mechanism for the effects of enolase on mRNA turnover are unknown.
Deletion of rhlB, but not the loss of degradosome complex assembly secondary to RNase E truncation, was associated with decreased abundance of transcripts in the Fur regulon and increased abundance of transcripts encoding indirect targets of Fur (e.g., transcripts of the sdh and nuo operons). Although these findings phenotypically resemble the effects of activation of the Fur repressor by a level of high iron in the culture media (49, 52), the mutant and wild-type strains we studied were grown under identical conditions. Thus, rhlB mutant cells may be perturbed in their ability to accurately sense the iron level and/or may accumulate iron to a greater extent than wild-type bacteria. Potentially, loss of the rhlB helicase from cells may alter iron sensing by stabilizing helices within the regulatory RNA, ryhB, whose degradation by RNase E is modulated by the small RNA-binding protein Hfq (50) because the predicted effects of such stabilization on Fur regulon transcripts parallel those we have observed.
Supplementary Material
Acknowledgments
Surveswaran Siddharthan assisted in the construction of strain SU02 and Kevin Pan and Richard Lin advised and assisted in the use of gabriel. We thank Dr. Barry Wanner for strain BW25113 and plasmids pKD4 and pKD46, Dr. Hans Bremer for strain HB191, Dr. Michael Cashel for anti-RhlB antibody, the Escherichia coli Genetic Stock Center at Yale University for strains K10 and DF261, and K. McDowall and M. Dreyfus for helpful comments on the manuscript. These studies were supported by National Institutes of Health Grant GM 54158 (to S.N.C.) and by National Science Council (Republic of China) Frontier Sciences Research Grant NSC90-91-2321-B-001 (to S.L.-C.). J.A.B. received support from National Institutes of Health Training Grant GM 07790.
Abbreviation: PNPase, polynucleotide phosphorylase.
References
- 1.Py, B., Higgins, C. F., Krisch, H. M. & Carpousis, A. J. (1996) Nature 381, 169-172. [DOI] [PubMed] [Google Scholar]
- 2.Carpousis, A. J., Van Houwe, G., Ehretsmann, C. & Krisch, H. M. (1994) Cell 76, 889-900. [DOI] [PubMed] [Google Scholar]
- 3.Kaberdin, V. R., Miczak, A., Jakobsen, J. S., Lin-Chao, S., McDowall, K. J. & von Gabain, A. (1998) Proc. Natl. Acad. Sci. USA 95, 11637-11642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miczak, A., Kaberdin, V. R., Wei, C. L. & Lin-Chao, S. (1996) Proc. Natl. Acad. Sci. USA 93, 3865-3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coburn, G. A. & Mackie, G. A. (1999) Prog. Nucleic Acid Res. Mol. Biol. 62, 55-108. [DOI] [PubMed] [Google Scholar]
- 6.Carpousis, A. J. (2002) Biochem. Soc. Trans. 30, 150-155. [PubMed] [Google Scholar]
- 7.Blum, E., Py, B., Carpousis, A. J. & Higgins, C. F. (1997) Mol. Microbiol. 26, 387-398. [DOI] [PubMed] [Google Scholar]
- 8.McDowall, K. J. & Cohen, S. N. (1996) J. Mol. Biol. 255, 349-355. [DOI] [PubMed] [Google Scholar]
- 9.Vanzo, N. F., Li, Y. S., Py, B., Blum, E., Higgins, C. F., Raynal, L. C., Krisch, H. M. & Carpousis, A. J. (1998) Genes Dev. 12, 2770-2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaberdin, V. R., Chao, Y. H. & Lin-Chao, S. (1996) J. Biol. Chem. 271, 13103-13109. [DOI] [PubMed] [Google Scholar]
- 11.Liou, G. G., Jane, W. N., Cohen, S. N., Lin, N. S. & Lin-Chao, S. (2001) Proc. Natl. Acad. Sci. USA 98, 63-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuwano, M., Ono, M., Endo, H., Hori, K., Nakamura, K., Hirota, Y. & Ohnishi, Y. (1977) Mol. Gen. Genet. 154, 279-285. [DOI] [PubMed] [Google Scholar]
- 13.Ono, M. & Kuwano, M. (1979) J. Mol. Biol. 129, 343-357. [DOI] [PubMed] [Google Scholar]
- 14.Apirion, D. & Lassar, A. B. (1978) J. Biol. Chem. 253, 1738-1742. [PubMed] [Google Scholar]
- 15.Misra, T. K. & Apirion, D. (1979) J. Biol. Chem. 254, 11154-11159. [PubMed] [Google Scholar]
- 16.Goodrich, A. F. & Steege, D. A. (1999) RNA 5, 972-985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kido, M., Yamanaka, K., Mitani, T., Niki, H., Ogura, T. & Hiraga, S. (1996) J. Bacteriol. 178, 3917-3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kokoska, R. J. & Steege, D. A. (1998) J. Bacteriol. 180, 3245-3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mudd, E. A., Carpousis, A. J. & Krisch, H. M. (1990) Genes Dev. 4, 873-881. [DOI] [PubMed] [Google Scholar]
- 20.Lee, K., Bernstein, J. A. & Cohen, S. N. (2002) Mol. Microbiol. 43, 1445-1456. [DOI] [PubMed] [Google Scholar]
- 21.Lopez, P. J., Marchand, I., Joyce, S. A. & Dreyfus, M. (1999) Mol. Microbiol. 33, 188-199. [DOI] [PubMed] [Google Scholar]
- 22.Ow, M. C., Liu, Q. & Kushner, S. R. (2000) Mol. Microbiol. 38, 854-866. [DOI] [PubMed] [Google Scholar]
- 23.Taraseviciene, L., Bjork, G. R. & Uhlin, B. E. (1995) J. Biol. Chem. 270, 26391-26398. [DOI] [PubMed] [Google Scholar]
- 24.Carpousis, A. J., Vanzo, N. F. & Raynal, L. C. (1999) Trends Genet. 15, 24-28. [DOI] [PubMed] [Google Scholar]
- 25.Xu, F. & Cohen, S. N. (1995) Nature 374, 180-183. [DOI] [PubMed] [Google Scholar]
- 26.Coburn, G. A. & Mackie, G. A. (1998) J. Mol. Biol. 279, 1061-1074. [DOI] [PubMed] [Google Scholar]
- 27.Coburn, G. A., Miao, X., Briant, D. J. & Mackie, G. A. (1999) Genes Dev. 13, 2594-2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liou, G. G., Chang, H. Y., Lin, C. S. & Lin-Chao, S. (2002) J. Biol. Chem. 277, 41157-41162. [DOI] [PubMed] [Google Scholar]
- 29.Bernstein, J. A., Khodursky, A. B., Lin, P. H., Lin-Chao, S. & Cohen, S. N. (2002) Proc. Natl. Acad. Sci. USA 99, 9697-9702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goldblum, K. & Apirion, D. (1981) J. Bacteriol. 146, 128-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hillman, J. D. & Fraenkel, D. G. (1975) J. Bacteriol. 122, 1175-1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Datsenko, K. A. & Wanner, B. L. (2000) Proc. Natl. Acad. Sci. USA 97, 6640-6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blattner, F. R., Plunkett, G., III, Bloch, C. A., Perna, N. T., Burland, V., Riley, M., Collado-Vides, J., Glasner, J. D., Rode, C. K., Mayhew, G. F., et al. (1997) Science 277, 1453-1474. [DOI] [PubMed] [Google Scholar]
- 34.Miller, J. H. (1972) Experiments in Molecular Genetics (Cold Spring Harbor Lab. Press, Plainview, NY).
- 35.Gollub, J., Ball, C. A., Binkley, G., Demeter, J., Finkelstein, D. B., Hebert, J. M., Hernandez-Boussard, T., Jin, H., Kaloper, M., Matese, J. C., et al. (2003) Nucleic Acids Res. 31, 94-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pan, K. H., Lih, C. J. & Cohen, S. N. (2002) Proc. Natl. Acad. Sci. USA 99, 2118-2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang, H., Pan, K. H. & Cohen, S. N. (2003) Proc. Natl. Acad. Sci. USA 100, 3251-3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tusher, V. G., Tibishirani, R. & Chu, G. (2001) Proc. Natl. Acad. Sci. USA 98, 5116-5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Irani, M. & Maitra, P. K. (1974) Biochem. Biophys. Res. Commun. 56, 127-133. [DOI] [PubMed] [Google Scholar]
- 40.Kalman, M., Murphy, H. & Cashel, M. (1991) New Biol. 3, 886-895. [PubMed] [Google Scholar]
- 41.Khemici, V. & Carpousis, A. J. (2004) Mol. Microbiol. 51, 777-790. [DOI] [PubMed] [Google Scholar]
- 42.Selinger, D. W., Saxena, R. M., Cheung, K. J., Church, G. M. & Rosenow, C. (2003) Genome Res. 2, 216-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Riley, M. (1997) Nucleic Acids Res. 25, 51-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheung, K. J., Badarinarayana, V., Selinger, D. W., Janse, D. & Church, G. M. (2003) Genome Res. 13, 206-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hengge-Aronis, R., Klein, W., Lange, R., Rimmele, M. & Boos, W. (1991) J. Bacteriol. 173, 7918-7924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hengge-Aronis, R., Lange, R., Henneberg, N. & Fischer, D. (1993) J. Bacteriol. 175, 259-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Escolar, L., Perez-Martin, J. & de Lorenzo, V. (1998) J. Bacteriol. 180, 2579-2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Escolar, L., Perez-Martin, J. & de Lorenzo, V. (1999) J. Bacteriol. 181, 6223-6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Masse, E. & Gottesman, S. (2002) Proc. Natl. Acad. Sci. USA 99, 4620-4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moll, I., Afonyushkin, T., Vytvytska, O., Kaberdin, V. R. & Blasi, U. (2003) RNA 9, 1308-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kimata, K., Tanaka, Y., Inada, T. & Aiba, H. (2001) EMBO J. 20, 3587-3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McHugh, J. P., Rodriguez-Quinones, F., Abdul-Tehrani, H., Svistunenko, D. A., Poole, R. K., Cooper, C. E. & Andrews, S. C. (2003) J. Biol. Chem. 278, 29478-29486. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.