

Abstract
Multidrug resistance proteins (MRPs) mediate the ATP-dependent efflux of structurally diverse compounds, including anticancer drugs and physiologic organic anions. Five classes of chalcogenopyrylium dyes (CGPs) were examined for their ability to modulate transport of [3H]estradiol glucuronide (E217βG; a prototypical MRP substrate) into MRP-enriched inside-out membrane vesicles. Additionally, some CGPs were tested in intact transfected cells using a calcein efflux assay. Sixteen of 34 CGPs inhibited MRP1-mediated E217βG uptake by >50% (IC50 values: 0.7–7.6 µM). Of 9 CGPs with IC50 values ≤2 µM, two belonged to class I, two to class III, and five to class V. When tested in the intact cells, only 4 of 16 CGPs (at 10 µM) inhibited MRP1-mediated calcein efflux by >50% (III-1, V-3, V-4, V-6), whereas a fifth (I-5) inhibited efflux by just 23%. These five CGPs also inhibited [3H]E217βG uptake by MRP4. In contrast, their effects on MRP2 varied, with two (V-4, V-6) inhibiting E217βG transport (IC50 values: 2.0 and 9.2 µM) and two (V-3, III-1) stimulating transport (>2-fold), whereas CGP I-5 had no effect. Strikingly, although V-3 and V-4 had opposite effects on MRP2 activity, they are structurally identical except for their chalcogen atom (Se versus Te). This study is the first to identify class V CGPs, with their distinctive methine or trimethine linkage between two disubstituted pyrylium moieties, as a particularly potent class of MRP modulators, and to show that, within this core structure, differences in the electronegativity associated with a chalcogen atom can be the sole determinant of whether a compound will stimulate or inhibit MRP2.
Introduction
ATP-binding cassette (ABC) transporters belong to a superfamily of mostly membrane proteins that mediate the ATP-dependent transmembrane transport of many structurally diverse xenobiotics and endogenous metabolites. Several human ABC transporters have been implicated in the drug resistance that is commonly observed in tumors refractory to chemotherapy. The most frequently implicated in clinical drug resistance are P-glycoprotein, multidrug resistance protein 1 (MRP1), and ABCG2 (Tamaki et al., 2011). Despite their shared function in tumor cell resistance, however, each of these transporters has its own distinct pharmacological and physiologic functions in normal cells (Leslie et al., 2005; Vlaming et al., 2009; Sharom, 2011; Slot et al., 2011). Thus, in addition to conferring drug resistance in tumor cells, the three ABC proteins also have an important influence on the absorption, distribution, and/or elimination of drugs and other xenobiotics from different tissues in the body.
MRP1 was discovered more than 15 years after P-glycoprotein, and thus it is not surprising that fewer MRP1-specific modulators have been identified (Cole et al., 1992; Chen et al., 2001; Wang et al., 2004; Boumendjel et al., 2005; Norman et al., 2005). Similar to P-glycoprotein, MRP1 can confer resistance to natural product drugs in tumor cells (Cole et al., 1994). It also plays a different but still protective role in normal tissues because it is expressed at the interface of various so-called pharmacological sanctuary sites, including the blood-cerebrospinal fluid barrier (Wijnholds et al., 2000; Kato et al., 2009). Unlike P-glycoprotein, however, MRP1 is an efficient transporter of numerous organic anions, many of which are glutathione or glucuronide conjugates of drug metabolites (Leslie et al., 2005; Slot et al., 2011). Estradiol glucuronide (E217βG) is a physiologic metabolite effluxed by human MRP1 (Jedlitschky et al., 1996).
MK-571, a cysteinyl leukotriene receptor antagonist, is the most extensively used experimental inhibitor of MRP1 but does not inhibit P-glycoprotein (Gekeler et al., 1995). Several compounds have been identified that modulate both transporters (Narasaki et al., 1997; Kimura et al., 2002; Toppmeyer et al., 2002; Pellicani et al., 2012) despite the fact that MRP1 and P-glycoprotein share <20% sequence identity. Consequently, chemical entities proposed as P-glycoprotein modulators are now often screened for their activity against MRP1 (Wesolowska, 2011; Ebert et al., 2012).
Of eight MRP1 homologs, two (MRP2 and MRP4) are known contributors to xenobiotic disposition and elimination, although neither is thought to play a significant role in resistance in human tumors (Imaoka et al., 2007; Nies and Keppler, 2007; Krishnamurthy et al., 2008; Russel et al., 2008; Slot et al., 2011). Although the substrate specificities of MRP1, MRP2, and MRP4 vary, all three can transport E217βG, and are inhibited by MK-571. Little is known about the effect of P-glycoprotein or MRP1 modulators on MRP2 and/or MRP4, and further, few (if any) inhibitors specific for the latter transporters have been identified. However, MRP2 and MRP4 transport can be modulated by an array of organic anions, some of which are therapeutically useful agents (Bakos et al., 2000; de Wolf et al., 2007; El-Sheikh et al., 2007). Given the contributions of MRP2 and MRP4 to the pharmacokinetic profiles of numerous therapeutic agents (and hence their efficacy and/or safety) (Russel et al., 2008; Lagas et al., 2009), such information is clinically relevant.
We recently compared the effects of a series of compounds containing different chalcogen atoms (O, S, Se, Te; International Union of Pure and Applied Chemistry group 16) in a pyrylium core with various 2-, 4-, and 6-position substituents on P-glycoprotein and MRP1 function (Ebert et al., 2012). These chalcogenopyrylium (CGP) derivatives are lipophilic cations that were designed based on the ability of related rhodamine-based photosensitizers to modulate P-glycoprotein (Sawada et al., 2008). Although both the rhodamines and CGPs incorporate chalcogen atoms at the 1 position of their trisubstituted pyrylium core, a key structural distinction is the flexibility of the CGP backbone versus the more rigid rhodamines. In our earlier study (Ebert et al., 2012), we examined four classes of CGPs (classes I–IV) and noted similarities and differences in their effects on MRP1 and P-glycoprotein. In the present study, we have identified a fifth structurally distinct class of CGPs characterized by the presence of two symmetrically disubstituted chalcogen-containing pyrylium moieties connected by a methine or trimethine linkage that modulates the vesicular transport activity of MRP1. We subsequently determined the relative potency of a subset of compounds comprising all five classes of CGPs as well as their ability to inhibit MRP1 in a dye efflux assay. Finally, the specificity of the five most potent MRP1 inhibitors was determined by measuring their effects on MRP2 and MRP4 transport.
Materials and Methods
Synthesis and Characterization of Chalcogenopyrylium Dyes.
The syntheses of 15 of the 22 CGPs examined in the present study have been described previously, and these compounds have been assigned to one of four structural classes designated CGP I–IV (Fig. 1) (Ebert et al., 2012). Five of 7 compounds (V-1, V-2, V-5, V-6, V-7) comprising a fifth class of structurally distinct CGPs (Fig. 2A) were selected from a library of chalcogenopyrylium methine and trimethine dyes, previously prepared by literature methods for evaluation as photosensitizers (Detty and Murray, 1982; Powers et al., 1989; Detty et al., 1990; Bellnier et al., 1999). Two additional class V CGPs (V-3, V-4) were synthesized as follows [using starting materials as described (Anderson and Stang, 1976; Leonard et al., 1999; Simard et al., 2000)]: 1) CGP V-3: (2,6-di-tert-butyl-4-((2,6-di-tert-butyl-4H-pyran-4-ylidene)methyl)selenopyrylium trifluoromethanesulfonate)-2,6-di-tert-butyl-4-methylpyrylium trifluoromethanesulfonate (0.077 g, 0.25 mmol) and 2,6-di-tert-butyl-4H-selenopyran-4-one (0.10 g, 0.25 mmol) in acetic anhydride (2 ml) were heated at 130°C for 2 hours. The reaction mixture was diluted with 2 ml of ether and chilled, precipitating a dark-purple product, which was collected by filtration and washed with ether. The crude product was dissolved in a minimal amount of acetonitrile, and was triturated with diethyl ether, precipitating 0.122 g (80%) of V-3 as a dark-purple solid [m.p. 147–148.5°C; 1H NMR (500 MHz, CD2Cl2) δ7.74 (br s, 2 H), 6.98 (br s, 2 H), 6.37 (s, 1 H), 1.535 (s, 18 H), 1.43 (s, 18 H); HRMS (EI) m/z 461.2308 (calculated for C27H41O80Se+: 461.2317); λmax (H2O) 550 nm (ε 9.5 × 104 M−1 cm−1); elemental analysis calculated for C27H41OSe•CF3SO3: C, 55.16; H, 6.78; found: C, 55.13; H, 6.67]. 2) CGP V-4: (2,6-di-tert-butyl-4-((2,6-di-tert-butyl-4H-pyran-4-ylidene)methyl)telluropyrylium hexafluorophosphate)-2,6-di-tert-butyl-4-methylpyrylium hexafluorophosphate (0.26 g, 0.75 mmol) and 2,6-di-tert-butyl-4H-telluropyran-4-one (0.307 g, 0.96 mmol) in acetic anhydride (0.5 ml) were heated at 130°C for 35 minutes. The product was dissolved in a minimal amount of acetonitrile and was triturated with diethyl ether, precipitating 0.42 g (86%) of V-4 as a dark-green solid [m.p. 149–153°C: 1H NMR (400 MHz, CD2Cl2) δ7.73 (s, 2 H), 6.96 (s, 2 H), 6.65 (s, 1 H), 1.48 (s, 18 H), 1.39 (s, 18 H); 13C NMR (300 MHz, CD3CN) δ177.40, 159.37, 156.16, 122.39, 110.98, 43.43, 38.16, 32.42, 28.01; HRMS (EI) m/z 511.226 (calculated for C27H41O130Te+: 511.2210); λmax (H2O) 587 nm (ε 9.2 × 104 M−1 cm−1); elemental analysis calculated for C27H41OTe∙PF6: C, 49.57; H, 6.32; found: C, 49.63; H, 6.36].
Fig. 1.

Chemical structures of class I–IV CGPs examined in this study. The structures of 10 CGPs belonging to four distinct chemical classes shown previously to inhibit MRP1 transport activity are illustrated. aPercentage inhibition of ATP-dependent [3H]E217βG uptake by MRP1-enriched inside-out membrane vesicles (tested at a single concentration of 30 μM except for CGP I-2, which was tested at 5 μM; data from Ebert et al., 2012).
Fig. 2.

Chemical structures and effect of class V CGPs on MRP1-mediated E217βG uptake into inside-out membrane vesicles. (A) Shown is the backbone chemical structure of the class V CGPs with the functional groups of the seven class V CGPs examined in this study. (B) [3H]E217βG uptake was measured in the presence of a single concentration of class V CGP (30 μM except for V-7, which was tested at 5 μM). Bars represent the means of values obtained in two independent experiments. Control: 1% DMSO (vehicle, solid bar).
The structures of the CGPs were verified using standard analytical methods (1H NMR, 13C NMR, mass spectrometry, UV-visible near infrared absorption spectroscopy, high-resolution mass spectrometry, and elemental analysis). The full chemical names and quantum fluorescence of the CGPs can be found in Supplemental Table 1. Purity of the CGPs was determined by high-performance liquid chromatography to be ≥94%. n-Octanol/water partition coefficients (log P values) were determined experimentally as before (Ebert et al., 2012). Stock solutions of CGPs in dimethylsulfoxide (DMSO) were prepared and kept at −20°C in the dark and diluted as needed.
Cell Culture.
The human embryonic kidney (HEK) cell line and the SV40-transformed HEK293T cell line were maintained in Dulbecco’s modified Eagle's medium (DMEM) supplemented with 4 mM l-glutamine and 7.5% fetal bovine serum (FBS). A stably transfected MRP1 overexpressing the HEK cell line was generated by standard procedures using a pcDNA3.1(−) expression vector containing the entire human MRP1 cDNA transcript and selection in G418 (Ito et al., 2001b; Ebert et al., 2012). The stable cell line was designated HEK-MRP1 and maintained in DMEM/7.5% FBS supplemented with 500 µg ml−1 G418. All cultures were grown at 37°C in 5% CO2/95% air. For membrane vesicle preparations, HEK-MRP1 cells were seeded onto 150-mm plates and collected at confluence.
Cells transiently expressing MRP2 and MRP4 were generated as follows. HEK293T cells were seeded at approximately 18 × 106 cells in 150-mm plates in DMEM/7.5% FBS. Twenty-four hours later, when cells were 90–95% confluent, pcDNA3.1(−) expression vectors containing either human MRP2 or human MRP4 cDNA (20 µg per plate) were transfected into the HEK293T cells using Lipofectamine 2000 (75 µl; Invitrogen, Carlsbad, CA) (Ito et al., 2001a; Hoque et al., 2009). After 6 hours at 37°C, the medium was replaced with fresh medium, and 48 hours later, cells were collected by centrifugation. Cell pellets were overlaid with homogenization buffer consisting of 250 mM sucrose/50 mM Tris pH 7.4/0.25 mM CaCl2 with protease inhibitors (Roche, Mississauga, ON, Canada), snap frozen in liquid nitrogen, and kept at −80°C until needed.
Preparation of Membrane Vesicles.
Membrane vesicles were prepared essentially as described by Loe et al. (1996). Cell pellets were thawed on ice, resuspended in homogenization buffer with protease inhibitors, and then disrupted by argon cavitation. After centrifugation at 1400g, the supernatant was retained, and the remaining pellet was resuspended in homogenization buffer with 0.5 mM EDTA. Following a second centrifugation at 1400g, the supernatants from the first and second centrifugations were combined and overlaid onto a cushion composed of 35% (w/v) sucrose/1 mM EDTA/50 mM Tris, pH 7.4. After centrifugation at 100,000g, the interface layer containing membranes was removed and placed in 25 mM sucrose/50 mM Tris, pH 7.4 buffer, and the membranes were collected by centrifugation again at 100,000 × g. The membranes were then washed with Tris-sucrose buffer (TSB; 250 mM sucrose, 50 mM Tris, pH 7.4) and centrifuged at 55,000g. The pellet was resuspended in TSB, and vesicles were prepared by passage through a 1-ml syringe with a 27-gauge needle. Vesicles were stored at −80°C until needed, and protein concentrations were determined using the Bradford method.
Immunoblotting for MRP Proteins.
The presence of MRP1, MRP2, and MRP4 in the membrane vesicles was confirmed by immunoblot analysis. Proteins were first resolved by electrophoresis on a 7% polyacrylamide gel and then electrotransferred onto polyvinylidene fluoride membranes (Pall Corporation, Ville St. Laurent, QC, Canada). After transfer, the blots were washed in Tris-buffered saline containing 0.1% Tween-20 and blocked in 4% (w/v) skim milk powder in Tris-buffered saline/Tween 20 for 1 hour. Blots were then incubated overnight at 4°C with the human MRP1-specific monoclonal antibody (MAb) QCRL-1 (diluted 1:10,000) (Cole laboratory), MRP2-specific MAb M2I-4 (diluted 1:5000; Alexis Laboratories, San Diego, CA), or MRP4-specific MAb M4I-10 (diluted 1:5000; Alexis Laboratories) (Hipfner et al., 1996; Létourneau et al., 2007; Hoque et al., 2009). After washing, MRP1 and MRP2 blots were incubated with horseradish peroxidase–conjugated goat antimouse antibody (Pierce Biotechnology, Rockford, IL), and MRP4 blots were incubated with horseradish peroxidase–conjugated goat antirat antibody (Chemicon) in blocking solution for 1–2 hours and then washed before incubating with chemiluminescence blotting substrate (PerkinElmer, Woodbridge, ON, Canada) and exposing the blot to film.
MRP-Mediated Uptake of 3H-Labeled Organic Anions by Inside-Out Membrane Vesicles.
ATP-dependent uptake of [6,7-3H]E217βG (45 Ci/mmol−1; PerkinElmer) by MRP1-, MRP2-, and MRP4-enriched membrane vesicles was measured using a 96-well rapid filtration method, as previously described (Létourneau et al., 2007; Hoque et al., 2009). Reactions were carried out in duplicate in 96-well round-bottom plates in a final reaction volume of 30 µl in TSB. Stock solutions of CGPs were diluted as needed in TSB and then added to both the reaction mix [containing either AMP or ATP (4 mM), MgCl2 (10 mM), and E217βG/[3H]E217βG] and the membrane vesicle preparations. In some experiments, leukotriene C4 (LTC4)/[3H]LTC4 (146 Ci/mmol−1; PerkinElmer) was used instead of E217βG/[3H]E217βG. The reaction plate was allowed to acclimatize to 37°C (or 23°C for LTC4 transport experiments) for 3 minutes prior to initiating the uptake reaction by adding the reaction mix (24 µl) to the membrane vesicles (6 µl). For MRP1-mediated E217βG uptake, 2 µg of vesicle protein was incubated with reaction mix containing [3H]E217βG (400 nM, 20 nCi) for 3 minutes at 37°C. For MRP1-mediated LTC4 uptake, 2 µg of vesicle protein was incubated with reaction mix containing [3H]LTC4 (50 nM, 10 nCi) for 1 minute at 23°C. For MRP2-mediated uptake of E217βG, 6 µg of membrane vesicle protein was incubated with [3H]E217βG (400 nM, 40 nCi) for 4 minutes at 37°C. To measure MRP4-mediated E217βG uptake, 5 µg of vesicle protein was incubated with [3H]E217βG (1 µM, 60 nCi) for 10 minutes at 37°C.
[3H]E217βG (or [3H]LTC4) uptake was stopped by rapid dilution in ice-cold TSB followed by rapid filtration of the wells’ contents onto a Unifilter-96 GF/B filter plate using a 96-well Filtermate Harvester apparatus (Packard BioScience, Meriden, CT). Radioactivity was measured by liquid scintillation counting on a TopCount NXT Microplate Counter (PerkinElmer). To determine ATP-dependent uptake, uptake in the presence of AMP was subtracted from uptake in the presence of ATP and modulator, and was expressed as a percentage of [3H]E217βG (or [3H]LTC4) uptake in the absence of modulator (control). Curve-fitting and IC50 values were determined using GraphPad Prism 3.0 software (GraphPad, San Diego, CA). To determine if IC50 values were significantly different from one another, one-way analyses of variance with ad-hoc Dunnett’s tests or unpaired students t tests were performed using GraphPad Prism 3.0; P values <0.05 were considered statistically significant.
MRP1-Mediated Calcein Efflux from Intact HEK-MRP1 Cells.
The effect of the CGPs on MRP1-mediated calcein efflux from intact HEK-MRP1 cells was measured using the protocol of van Zanden et al. (2005) modified as follows. HEK-MRP1 cells were plated into 96-well clear-bottom, black-sided plates precoated with 0.01% poly-L-lysine (1 × 105 cells per well in 100 µl). After 1.5 hours at 37°C, stock solutions of CGPs in DMSO that had been diluted in OptiMem (GIBCO-Invitrogen, Burlington, ON, Canada) as needed were added to the plates in a volume of 50 µl such that their final concentration was 10 µM. As a vehicle control, 50 µl of TSB was added in place of the CGPs. Reactions were carried out in triplicate. After incubating the plates for 30 minutes at 37°C, 50 µl of calcein-AM (Cedarlane, Burlington, ON, Canada; 24 µM in OptiMem with 0.1% bovine serum albumin) was added to the wells to a final concentration of 6 µM. After 15 minutes at 37°C, the medium was aspirated and replaced with 200 µl of prewarmed OptiMem with 10% FBS. After 10 minutes at 37°C to allow calcein-AM hydrolysis and calcein efflux to occur, the plate was placed on ice and the medium replaced with 200 µl of ice-cold phosphate-buffered saline. Fluorescent calcein remaining in the cells was immediately measured on a Spectramax Gemini XS fluorimeter (Molecular Devices, Sunnyvale, CA; excitation wavelength 495 nm; emission wavelength 515 nm). All of the CGPs tested had little or no fluorescence (Supplemental Table 1), and thus had no impact on the data obtained with this assay. Efflux activity was calculated from the raw fluorescence values as follows. The calcein fluorescence [arbitrary units (AU)] remaining in the vehicle-treated control HEK-MRP1 cells (no CGP) was subtracted from the AU remaining in the HEK-MRP1 cells after incubation with a CGP, and then expressed relative to the maximal value of calcein efflux, which was calculated as the difference between the AU remaining in the HEK cells (no MRP1, no CGP) minus the AU remaining in the vehicle control HEK-MRP1 cells (no CGP). To determine if the effects of the CGPs on MRP1-mediated calcein efflux were different from one another, unpaired Student’s t tests were performed using GraphPad Prism 3.0. P values <0.05 were considered statistically significant.
Results
Class V CGPs Represent a Potent Class of Modulators of MRP1-Mediated [3H]E217βG Vesicular Transport.
In a previous study, a series of 32 CGPs were tested at a single concentration for their ability to modulate MRP1-mediated [3H]E217βG vesicular uptake activity. Ten of these compounds, belonging to four different classes of CGPs, were identified that inhibited MRP1-mediated [3H]E217βG uptake by >50% (70–96%) (Ebert et al., 2012) (Fig. 1). In the present study, we tested seven additional CGPs comprising a fifth class of CGPs (Fig. 2A) and found that six of these class V CGPs inhibited [3H]E217βG uptake by >80% when tested at a single concentration of 30 μM, or in the case of CGP V-7, 5 μM (Fig. 2B). The only exception was CGP V-2, which inhibited E217βG uptake by only 40% at 30 μM.
Concentration-dependent inhibition experiments were then carried out to determine the relative potencies of the 16 CGPs from the five classes of CGPs showing >50% inhibition of MRP1-mediated [3H]E217βG uptake. The concentration-response curves obtained exhibited a classic sigmoid shape, and representative graphs for CGPs I-5, III-1, V-3, V-4, and V-6 are presented in Fig. 3 (A–E). The IC50 values of the 16 CGPs ranged from 0.7 to 7.6 µM, and are summarized in Table 1. Nine of the 16 CGPs were quite potent, with IC50 values <2 µM. These included the two class III CGPs (III-1, III-2) which had comparable IC50 values of 1.6 and 2.0 µM, respectively, and two of the four class I CGPs (IC50 values of 1.8 µM for CGP I-3 and 0.7 µM for CGP I-4), with CGP I-4 being significantly (2-fold) more potent than CGP I-3 (P < 0.05). Five of six class V CGPs had comparable IC50 values <2 µM (0.9–1.4 µM), whereas the IC50 of CGP V-1 was 4 µM. On the other hand, the IC50 values for CGP I-2 and CGP I-5 were comparable at 3.9 and 2.5 µM, respectively (P > 0.05), and similarly, the two class II CGPs (CGP II-13, II-14) had IC50 values of 7.6 and 4.9 µM, respectively (P > 0.05). CGP IV-1 and CGP IV-3 had IC50 values of 5.3 and 3.3 µM, with CGP IV-3 being almost 2-fold more potent than CGP IV-1 (P < 0.05). Thus, of the nine CGPs that had IC50 values ≤2 µM, two belonged to class I (CGP I-3, I-4), two belonged to class III (CGP III-1, III-2), and five to class V (CGP V-3, V-4, V-5, V-6, V-7).
Fig. 3.
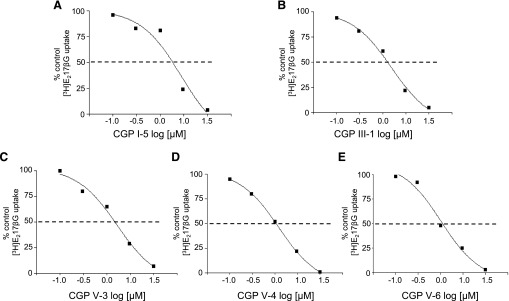
Concentration dependence of CGP-mediated inhibition of E217βG uptake by MRP1. Shown are representative concentration-response curves illustrating the effects of increasing concentrations of selected CGPs on MRP1-mediated uptake of [3H]E217βG into inside-out MRP1-enriched membrane vesicles. Each data point represents the mean of duplicate determinations. (A) CGP I-5; (B) CGP III-1; (C) CGP V-3; (D) CGP V-4; and (E) CGP V-6. Similar results were obtained in 2–3 additional independent experiments, and means (± S.D.) can be found in Table 1.
TABLE 1.
Relative inhibitory potencies of CGPs on MRP1-mediated E217βG uptake
| CGP | Chalcogen Atom | IC50a (E217βG Uptake) | Log Pb |
|---|---|---|---|
| μM | |||
| Class I | |||
| I-2 | S | 3.9 ± 1.9 (3) | 1.6 ± 0.1 |
| I-3 | S | 1.8 ± 0.1 (3) | 1.5 ± 0.2c |
| I-4 | Se | 0.7 ± 0.3 (3) | 1.6 ± 0.1c |
| I-5 | Te | 2.5 ± 0.5 (4) | 1.6 ± 0.1 |
| Class II | |||
| II-13 | S | 7.6 ± 3.1 (3) | 1.0 ± 0.1 |
| II-14 | Se | 4.9 ± 3.9 (3) | 1.1 ± 0.1 |
| Class III | |||
| III-1 | S | 1.6 ± 0.4 (3) | 2.1 ± 0.1 |
| III-2 | S | 2.0, 1.8 | 0.5 ± 0.1 |
| Class IV | |||
| IV-1 | Te | 5.3 ± 0.7 (3) | 1.4 ± 0.1 |
| IV-3 | Te | 3.3 ± 0.6 (3) | 1.1 ± 0.1 |
| Class V | |||
| V-1 | Te | 4.7, 3.2 | 2.5 ± 0.2 |
| V-3 | Se | 1.1 ± 0.4 (4) | 1.9 ± 0.1 |
| V-4 | Te | 1.2 ± 0.2 (3) | 1.9 ± 0.1 |
| V-5 | Te | 1.2, 0.9 | 1.8 ± 0.2 |
| V-6 | Se | 0.9 ± 0.1 (3) | 1.9 ± 0.1d |
| V-7 | Te | 1.4, 1.4 | 2.4 ± 0.1d |
The values shown represent the means ± S.D. (n) of IC50 values obtained in 2–4 independent experiments. When the compound was tested only twice, both values obtained are shown.
The values shown are experimental log P values that were derived as described in Materials and Methods.
From Sawada et al. (2008).
From Detty et al. (1990).
Modulation of MRP1-Mediated Calcein Efflux from Intact HEK-MRP1 Cells by CGPs.
To further examine the efficacy of the 16 CGPs identified as modulators of MRP1-mediated E217βG transport, a calcein efflux assay using intact cells (the HEK-MRP1 cell line and an untransfected HEK cell line as a control) was used. Of the 16 CGPs tested, only five inhibited MRP1-dependent efflux of calcein >25% at 10 µM (Fig. 4). Three of these were from class V (CGP V-3, V-4, V-6) and inhibited calcein efflux by 66% ± 19%, 46% ± 10%, and 59% ± 23%, respectively, whereas CGP III-1 inhibited efflux by 52% ± 9%. None of the four was significantly more effective than another (P > 0.05). On the other hand, CGP I-5 was significantly less effective than CGP V-3, V-4, V-6, and III-1 (P < 0.05) and inhibited calcein efflux by only 23% ± 4%. The complete structures of these five CGP modulators of MRP1-mediated calcein efflux from intact cells are shown in Supplemental Fig. 1.
Fig. 4.

Effect of CGPs on cellular efflux of calcein from HEK-MRP1 cells. The ability of the indicated CGPs (10 µM) to inhibit calcein efflux from HEK-MRP1 cells was measured (hatched bars) and compared with the relative difference in efflux from control HEK-MRP1 cells (ctrl). The bars represent the means (± S.D.) of 3–4 independent experiments, each performed in triplicate. *II-5 is a significantly less effective inhibitor of calcein efflux than CGP V-3, V-4, V-6, and III-1 (P < 0.05); however, there were no significant differences between CGP V-3, V-4, V-6, and III-1 (P > 0.05).
Class V CGPs V-3, V-4, and V-6 Are Also Potent Modulators of MRP1-Mediated [3H]LTC4 Vesicular Transport.
Because V-3, V-4, and V-6 were the most active class V CGPs in the calcein efflux assay as well as the [3H]E217βG transport assay, it was of interest to determine if these compounds inhibited the transport of LTC4, an MRP1 substrate with a binding site known to be pharmacologically distinct from the binding site for E217βG (Maeno et al., 2009). The IC50 values (LTC4 transport) obtained for V-3, V-4, and V-6 were 3.7 ± 0.6, 7.6 ± 1.5, and 1.3 ± 0.4, respectively.
CGPs I-5, III-1, and V-3, V-4, and V-6 Differentially Modulate MRP2-Mediated Vesicular Transport of [3H]E217βG.
To determine whether the five CGPs that were active in both MRP1 transport assays were specific inhibitors of MRP1, or could modulate the activity of other MRP transporters, membrane vesicles were prepared from transfected HEK293T cells expressing human MRP2 (as confirmed by immunoblotting) (Fig. 5A). The relative potencies of the five CGPs on MRP2-mediated [3H]E217βG uptake were then determined by concentration-response experiments in the presence of five different concentrations of CGP (to a maximum of 30 µM). CGP I-5 was ineffective over the entire concentration range tested (0.1–30 µM) (Fig. 5B), whereas the IC50 values for CGPs V-4 and V-6 were 2.0 (± 1.3) µM (n = 3) and 9.2 (± 2.0) µM (n = 3), respectively (Fig. 5, E and F). In contrast, CGPs III-1 and V-3 stimulated [3H]E217βG uptake activity to a maximum of 238% (± 31%) (n = 4) and 224% (± 44%) (n = 4) at approximately 30 µM (Fig. 5, C and D).
Fig. 5.

Concentration-dependent effects of CGPs on MRP2-mediated uptake of [3H]E217βG into inside-out membrane vesicles. (A) Immunoblot of membrane vesicle proteins prepared from MRP2-transfected and untransfected HEK293T (HEK) cells is shown. MAb M2I-4 was used to detect MRP2. (B–F) Effect of CGPs on MRP2-mediated [3H]E217βG uptake activity. Results shown are representative concentration response curves. Data points represent means of duplicate determinations. (B) CGP I-5; (C) CGP III-1; (D) CGP V-3; (E) CGP V-4; and (F) CGP V-6. Similar results were obtained in at least two additional independent experiments. Means (± S.D.) can be found in the text.
CGPs V-3, V-4, V-6, I-5, and III-1 Are Effective Modulators of MRP4-Mediated Vesicular Transport of [3H]E217βG at 10 µM but Not 1 µM.
The five CGPs that were active in both MRP1 assays (I-5, III-1, V-3, V-4, V-6) were also tested for their ability to modulate [3H]E217βG uptake by MRP4. Following transfection of a human MRP4 cDNA expression vector into HEK293T cells, membrane vesicles were prepared and the presence of the transporter confirmed by immunoblotting (Fig. 6A). In contrast to MRP1 and MRP2, two immunoreactive bands were detected with the MRP4-specific MAb M4I-10. The reason for the two bands is currently unknown, but they have been observed previously and seem likely to be the result of variable MRP4 glycosylation (Hoque et al., 2009).
Fig. 6.

Effect of CGPs on MRP4-mediated uptake of [3H]E217βG into inside-out membrane vesicles. (A) Immunoblots of membrane vesicle proteins prepared from MRP4-transfected and untransfected HEK293T (HEK) cells are shown. MAb M4I-10 was used to detect MRP4. (B) Effect of selected CGPs on MRP4-mediated [3H]E217βG uptake activity. Bars represent the means of values obtained in two independent experiments. Open bars, 1 µM CGP; hatched bars, 10 µM CGP; solid bar, 1% DMSO (vehicle control). MV, membrane vesicles.
When tested at 1 µM, none of the five CGPs (I-5, III-1, V-3, V-4, or V-6) inhibited [3H]E217βG uptake by MRP4 by >20%. However, when tested at 10 µM, [3H]E217βG uptake was inhibited by >75% (77–86%) by all five CGPs (Fig. 6B).
Effect of CGP III-1 Analogs on MRP2- and MRP4-Mediated Vesicular Transport of [3H]E217βG.
In our previous study, five analogs of CGP III-1 (Supplemental Fig. 2) were examined for their ability to modulate MRP1-mediated [3H]E217βG uptake activity, but none were shown to be more efficacious than the parent compound (Ebert et al., 2012). In the present study, the five analogs were also tested for their effects on MRP2- and MRP4-mediated [3H]E217βG uptake (Supplemental Fig. 3). At 1 µM, CGP III-1-1, -2, and -3 had no effect on [3H]E217βG uptake by MRP2, whereas CGP III-1-4 and -5 stimulated uptake 1.5-fold (Supplemental Fig. 3A). When tested at 10 µM, four of the five CGP III-1 analogs stimulated [3H]E217βG uptake to varying degrees. The exception was CGP III-1-1, which still had no effect. Stimulation by CGP III-1-2 was weak (1.2-fold at 10 µM), whereas stimulation by CGP III-1-3 and III-1-5 was slightly greater (1.4-fold) at the same concentration. CGP III-1-4 appeared to be the most effective of the five analogs (1.7-fold stimulation) and was comparable to the parent CGP III-1 at 10 µM (1.8-fold stimulation). Taken together, the data indicate that, although some of the CGP III-1 analogs retained the ability to stimulate MRP2-mediated transport, the efficacy of only one of them approached that of parent CGP III-1.
The CGP III-1 analogs were also tested for their effects on MRP4-mediated [3H]E217βG uptake (Supplemental Fig. 3B). Little or no modulation was seen at 1 µM. However, at 10 µM, three of five CGP III-1 analogs (III-1-1, -3, and -4) inhibited [3H]E217βG uptake by MRP4 by approximately 60%. The other two (III-1-2 and -5) inhibited uptake by <20% at this concentration. Thus, none of the CGP III-1 analogs were as effective as the parent compound, which inhibited MRP4-mediated [3H]E217βG uptake by 77% at 10 µM. Overall, the CGP III-1 analogs showed a similar pattern of inhibition of [3H]E217βG uptake by MRP1 and MRP4, whereas for MRP2, the five analogs either had no effect or stimulated uptake, although to a lesser extent than the parent compound (Supplemental Fig. 3C).
Discussion
Over the past two decades, there has been widespread interest in the identification and development of modulators of MRP1, P-glycoprotein, and other drug transporters because of their influence on cellular accumulation of xenobiotics and hence efficacy and toxicity of antineoplastic and other therapeutic agents. Modulators have typically been identified by either testing drugs already in clinical practice (or derivatives thereof) or screening libraries of natural product or synthetic chemical entities. As rhodamine derivatives originally developed as photosensitizers and subsequently determined to be modulators of P-glycoprotein and/or MRP1, the CGPs are an example of the latter (Tombline et al., 2006; Sawada et al., 2008; Gannon et al., 2009; Ebert et al., 2012). In the present study, six of the seven molecules comprising a new class of CGPs (class V) tested initially at a single concentration inhibited MRP1-mediated E217βG uptake by >80%, thus identifying class V CGPs with their distinctive methine or trimethine linkage between two disubstituted pyrylium moieties as a particularly effective class of MRP1 modulators (Fig. 2).
When the 16 modulators that were identified in our initial screen and comprised all five classes of CGPs were further investigated, the IC50 values obtained for E217βG transport were all <10 µM (Table 1), demonstrating that, as a whole, this subset of CGPs is a potent group of MRP1 modulators. The majority of the most potent CGPs were the class V compounds described for the first time here. Thus, of nine CGPs with IC50 values (E217βG) <2 µM, five belonged to class V, two to class II, and two to class I.
Distinct from the class I–IV CGPs, class V compounds contain two CGP rings (either the same or different) linked by a methine or trimethine π–framework, and four identical hydrophobic groups at the 2 and 6 position of the two chalcogen-containing rings. The 2,6-tert-butyl substituents appear to impart particular potency on the class V CGPs, since CGP V-1 (which contains 2,6-phenyl substituents) is at least 3-fold less potent than the others. This difference may be related, at least in part, to differences in hydrophobicity, although the experimental log P values indicate that other factors are more important. Indeed, the length of the polymethine bridge connecting the two 2,6-disubstituted pyrylium moieties and the relative chalcogen atom electronegativity are known to impact the charge distribution (Detty et al., 1988; Calitree et al., 2007), and this is likely to influence interactions with MRP1. This may help explain the markedly different activities of the Se-containing CGP V-2 and CGP V-6, which are structurally identical except for their alkenyl bridges (1 carbon versus 3, respectively). On the other hand, the structures of the Te-containing CGP V-4 and CGP V-7 are identical except for the same 2-carbon difference in their alkenyl bridges, and yet their potencies as MRP1 modulators are the same. It may be that, for class V CGPs, charge distribution differences mediated by different chalcogen atoms can supercede charge distribution differences attributed to the linking bridge.
Of the active class I–IV CGPs, it is interesting to note that CGP I-3, I-4, and I-5 differ only in their chalcogen atom (S, Se, and Te, respectively). However, unlike class V CGPs, only one chalcogen atom mediates charge distribution. This suggests that Se confers optimal MRP1 modulating ability on this particular CGP structure, although the difference in potency relative to the S- and Te-containing analogs is modest. Thus, consistent with our previous study (Ebert et al., 2012), the identity of the chalcogen atom in these CGPs does not substantially influence their MRP1 inhibitory potency.
The vesicular transport assay used here is a convenient way to quickly and specifically identify chemical entities that interact with MRP1 (Slot et al., 2011; Ebert et al., 2012). However, because this assay measures substrate uptake into inside-out membrane vesicles, it does not take into account a potential modulator’s ability to cross the membrane of an intact cell, which is a prerequisite for activity in an intact organism. Thus, testing the CGPs in an intact cell system was an important next step in their evaluation. However, when the 16 CGPs shown to be inhibitors in the vesicular transport system were investigated for their ability to inhibit calcein efflux from MRP1-expressing cells, most showed little activity. Indeed, only five inhibited calcein efflux by >23%. Four of these (CGP V-3, V-4, V-6, and III-1) were significantly more effective than the fifth (CGP I-5) (Fig. 4). Thus, as observed in the vesicular transport assay, the class V CGPs show particular effectiveness against MRP1 in the cellular dye efflux assay. This may be related to the hydrophobicity of these compounds conferred by their four tert-butyl substituents, which would be expected to facilitate their uptake across the plasma membrane into the cell. These observations in intact cells support the conclusion that chemical structures composed of two 2,6-di-tert-butyl substituted chalcogenopyrylium moieties linked by a methine or trimethine bridge represent a particularly potent group of MRP1 modulators that have a strong potential to be active in vivo.
ABC proteins show little sequence conservation in their membrane, spanning domains which are largely responsible for determining each transporter’s distinct substrate profile. Even among the MRPs, the sequence conservation of their membrane spanning domains (MSDs) is rather limited, although they do share the common ability, at least in vitro, to transport various organic anions, a property that distinguishes them from P-glycoprotein, which transports only hydrophobic or neutral compounds. However, each of the MRPs has its own substrate profile, and although there are instances of substrate (and modulator) overlap, this overlap does not necessarily correlate well with sequence similarity among the proteins (Zelcer et al., 2001). For example, MRP1 and MRP2 are good transporters of the proinflammatory cysteinyl leukotriene C4, whereas MRP3 and MRP4 are not. Further, the potent glutathione-dependent tricyclic isoxazole inhibitor of MRP1, LY475776, does not interact with MRP2, MRP3, 4 or 5 (Mao et al., 2002; Dantzig et al., 2004; Norman et al., 2005).
In this study, we took advantage of the shared ability of MRP1, MRP2, and MRP4 to transport E217βG to examine the specificity of the five most potent CGP inhibitors of MRP1. Despite the fact that MRP1 and MRP4 share relatively few common substrates (and relatively less sequence conservation) than MRP1 and MRP2, all five CGPs (I-5, III-1, V-3, V-4, and V-6) were good inhibitors of E217βG uptake by MRP4, causing >75% inhibition at 10 µM (Fig. 6). In contrast, these five CGPs had some unanticipated effects on E217βG uptake by MRP2 (Fig. 5) that were quite distinct from their effects on MRP1 and MRP4. First, CGP I-5 had no effect on MRP2-mediated E217βG uptake even at 30 µM, a concentration that inhibited transport by MRP1 and MRP4 by >80% (Fig. 5B). Thus, since MRP1 and MRP4 are significantly less related to one another than MRP1 and MRP2, CGP I-5 is an example of a modulator whose activity against these three MRPs does not correlate with their relative sequence conservation.
The second distinctive effect of the five CGPs on MRP2 was that, whereas two of them (V-4, V-6) inhibited transport, two others (V-3, III-1) stimulated transport (>2-fold). Previous studies have identified several organic anions that inhibit one MRP transporter while stimulating another, leading to the conclusion that some MRPs contain at least two interacting ligand binding sites (Bakos et al., 2000; Zelcer et al., 2003; Wittgen et al., 2011). For example, both sulfinpyrazone and probenicid inhibit MRP1 and MRP4 but stimulate MRP2 (Bakos et al., 2000; Ito et al., 2001a; Smeets et al., 2004; Huisman et al., 2005). Consequently, the opposite effects observed for CGP V-4 and V-6 on MRP1 (inhibitory) versus MRP2 (stimulatory) are in themselves not surprising. On the other hand, the present observations are quite remarkable when it is noted that CGP V-3 and V-4 have completely opposite effects on MRP2 activity, and yet are structurally identical except for their chalcogen atom (Se versus Te) (Fig. 5, D versus E). This is the first time (to our knowledge) that the ability to stimulate versus inhibit MRP2 transport can be attributed to not only a single atom, but in particular, to a chalcogen atom. The significant difference in Se and Te electronegativity (Pauling scale 2.55 versus 2.10), which changes the inductive character of the carbon-chalcogen bond as well as the distribution of electron density in the π framework linking the two pyrylium moieties, appears to be sufficient, at least in class V compounds, to change dramatically the interactions with MRP2.
In conclusion, we have shown that CGPs are effective modulators not only of MRP1 but also its homologs MRP2 and MRP4. We have further identified class V CGPs with their distinctive linkage between two disubstituted chalcogenopyrylium moieties as a particularly effective class of MRP modulators in both membrane vesicle and intact cell assays, suggesting they are likely to be effective in vivo. Finally, we have shown that within the class V core structure, differences in the electronegativity associated with a chalcogen atom can be the sole determinant of whether a compound will stimulate or inhibit MRP2.
Supplementary Material
Acknowledgments
The authors thank Maureen Hobbs for assistance in the preparation of the figures and the manuscript.
Abbreviations
- ABC
ATP-binding cassette
- AU
arbitrary units
- CGP
chalcogenopyrylium
- DMEM
Dulbecco’s modified Eagle’s medium
- DMSO
dimethylsulfoxide
- E217βG
estradiol glucuronide
- FBS
fetal bovine serum
- HEK
human embryonic kidney
- LTC4
leukotriene C4
- MAb
monoclonal antibody
- MRP
multidrug resistance protein
- TSB
Tris-sucrose buffer
Authorship Contributions
Participated in research design: Myette, Conseil, Cole.
Conducted experiments: Myette, Conseil, Detty, Ebert, Wetzel.
Contributed new reagents or analytic tools: Conseil, Detty, Ebert, Wetzel.
Performed data analysis: Myette, Conseil, Cole, Detty, Ebert, Wetzel.
Wrote or contributed to the writing of the manuscript: Myette, Conseil, Detty, Cole.
Footnotes
This work was supported by a grant from the Canadian Institutes of Health Research [MOP 10519]; the Canada Research Chair program (S.P.C.C.); and the National Institutes of Health [Grant GM-94367] (to M.R.D.).
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
References
- Anderson AG, Stang PJ. (1976) A convenient two-step synthesis of 2,6-di-tert-butyl-4-methylpyridine, a sterically hindered non-nucleophilic base. J Org Chem 18:3034–3036 [Google Scholar]
- Bakos E, Evers R, Sinkó E, Váradi A, Borst P, Sarkadi B. (2000) Interactions of the human multidrug resistance proteins MRP1 and MRP2 with organic anions. Mol Pharmacol 57:760–768 [DOI] [PubMed] [Google Scholar]
- Bellnier DA, Young DN, Detty MR, Camacho SH, Oseroff AR. (1999) pH-dependent chalcogenopyrylium dyes as potential sensitizers for photodynamic therapy: selective retention in tumors by exploiting pH differences between tumor and normal tissue. Photochem Photobiol 70:630–636 [PubMed] [Google Scholar]
- Boumendjel A, Baubichon-Cortay H, Trompier D, Perrotton T, Di Pietro A. (2005) Anticancer multidrug resistance mediated by MRP1: recent advances in the discovery of reversal agents. Med Res Rev 25:453–472 [DOI] [PubMed] [Google Scholar]
- Calitree B, Donnelly DJ, Holt JJ, Gannon MK, Nygren CL, Sukumaran DK, Autschbach J, Detty MR. (2007) Tellurium analogues of rosamine and rhodamine dyes: synthesis, structure, 125Te NMR, and heteroatom contributions to excitation. Organometallics 26:6248–6257 [Google Scholar]
- Chen ZS, Aoki S, Komatsu M, Ueda K, Sumizawa T, Furukawa T, Okumura H, Ren XQ, Belinsky MG, Lee K, et al. (2001) Reversal of drug resistance mediated by multidrug resistance protein (MRP) 1 by dual effects of agosterol A on MRP1 function. Int J Cancer 93:107–113 [DOI] [PubMed] [Google Scholar]
- Cole SPC, Bhardwaj G, Gerlach JH, Mackie JE, Grant CE, Almquist KC, Stewart AJ, Kurz EU, Duncan AMV, Deeley RG. (1992) Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science 258:1650–1654 [DOI] [PubMed] [Google Scholar]
- Cole SPC, Sparks KE, Fraser K, Loe DW, Grant CE, Wilson GM, Deeley RG. (1994) Pharmacological characterization of multidrug resistant MRP-transfected human tumor cells. Cancer Res 54:5902–5910 [PubMed] [Google Scholar]
- Dantzig AH, Shepard RL, Pratt SE, Tabas LB, Lander PA, Ma L, Paul DC, Williams DC, Peng SB, Slapak CA, et al. (2004) Evaluation of the binding of the tricyclic isoxazole photoaffinity label LY475776 to multidrug resistance associated protein 1 (MRP1) orthologs and several ATP- binding cassette (ABC) drug transporters. Biochem Pharmacol 67:1111–1121 [DOI] [PubMed] [Google Scholar]
- Detty MR, McKelvey JM, Luss HR. (1988) Telluropyrylium dyes. 2. The electron-donating properties of the chalcogen atoms to the chalcogenopyrylium nuclei and their radical dications, neutral radicals, and anions. Organometallics 7:1131–1147 [Google Scholar]
- Detty MR, Merkel PB, Hilf R, Gibson SL, Powers SK. (1990) Chalcogenapyrylium dyes as photochemotherapeutic agents. 2. Tumor uptake, mitochondrial targeting, and singlet-oxygen-induced inhibition of cytochrome c oxidase. J Med Chem 33:1108–1116 [DOI] [PubMed] [Google Scholar]
- Detty MR, Murray BJ. (1982) Telluropyrylium dyes. 1. 2,6-Diphenyltelluropyrylium dyes. J Org Chem 47:5235–5239 [Google Scholar]
- de Wolf CJ, Yamaguchi H, van der Heijden I, Wielinga PR, Hundscheid SL, Ono N, Scheffer GL, de Haas M, Schuetz JD, Wijnholds J, et al. (2007) cGMP transport by vesicles from human and mouse erythrocytes. FEBS J 274:439–450 [DOI] [PubMed] [Google Scholar]
- Ebert SP, Wetzel B, Myette RL, Conseil G, Cole SPC, Sawada GA, Loo TW, Bartlett MC, Clarke DM, Detty MR. (2012) Chalcogenopyrylium compounds as modulators of the ATP-binding cassette transporters P-glycoprotein (P-gp/ABCB1) and multidrug resistance protein 1 (MRP1/ABCC1). J Med Chem 55:4683–4699 [DOI] [PubMed] [Google Scholar]
- El-Sheikh AA, van den Heuvel JJ, Koenderink JB, Russel FG. (2007) Interaction of nonsteroidal anti-inflammatory drugs with multidrug resistance protein (MRP) 2/ABCC2- and MRP4/ABCC4-mediated methotrexate transport. J Pharmacol Exp Ther 320:229–235 [DOI] [PubMed] [Google Scholar]
- Gannon MK, 2nd, Holt JJ, Bennett SM, Wetzel BR, Loo TW, Bartlett MC, Clarke DM, Sawada GA, Higgins JW, Tombline G, et al. (2009) Rhodamine inhibitors of P-glycoprotein: an amide/thioamide “switch” for ATPase activity. J Med Chem 52:3328–3341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gekeler V, Ise W, Sanders KH, Ulrich WR, Beck J. (1995) The leukotriene LTD4 receptor antagonist MK571 specifically modulates MRP associated multidrug resistance. Biochem Biophys Res Commun 208:345–352 [DOI] [PubMed] [Google Scholar]
- Hipfner DR, Almquist KC, Stride BD, Deeley RG, Cole SPC. (1996) Location of a protease-hypersensitive region in the multidrug resistance protein (MRP) by mapping of the epitope of MRP-specific monoclonal antibody QCRL-1. Cancer Res 56:3307–3314 [PubMed] [Google Scholar]
- Hoque MT, Conseil G, Cole SPC. (2009) Involvement of NHERF1 in apical membrane localization of MRP4 in polarized kidney cells. Biochem Biophys Res Commun 379:60–64 [DOI] [PubMed] [Google Scholar]
- Huisman MT, Chhatta AA, van Tellingen O, Beijnen JH, Schinkel AH. (2005) MRP2 (ABCC2) transports taxanes and confers paclitaxel resistance and both processes are stimulated by probenecid. Int J Cancer 116:824–829 [DOI] [PubMed] [Google Scholar]
- Imaoka T, Kusuhara H, Adachi M, Schuetz JD, Takeuchi K, Sugiyama Y. (2007) Functional involvement of multidrug resistance-associated protein 4 (MRP4/ABCC4) in the renal elimination of the antiviral drugs adefovir and tenofovir. Mol Pharmacol 71:619–627 [DOI] [PubMed] [Google Scholar]
- Ito K, Oleschuk CJ, Westlake C, Vasa MZ, Deeley RG, Cole SPC. (2001a) Mutation of Trp1254 in the multispecific organic anion transporter, multidrug resistance protein 2 (MRP2) (ABCC2), alters substrate specificity and results in loss of methotrexate transport activity. J Biol Chem 276:38108–38114 [DOI] [PubMed] [Google Scholar]
- Ito K, Olsen SL, Qiu W, Deeley RG, Cole SPC. (2001b) Mutation of a single conserved tryptophan in multidrug resistance protein 1 (MRP1/ABCC1) results in loss of drug resistance and selective loss of organic anion transport. J Biol Chem 276:15616–15624 [DOI] [PubMed] [Google Scholar]
- Jedlitschky G, Leier I, Buchholz U, Barnouin K, Kurz G, Keppler D. (1996) Transport of glutathione, glucuronate, and sulfate conjugates by the MRP gene-encoded conjugate export pump. Cancer Res 56:988–994 [PubMed] [Google Scholar]
- Kato S, Ito K, Kato Y, Wakayama T, Kubo Y, Iseki S, Tsuji A. (2009) Involvement of multidrug resistance-associated protein 1 in intestinal toxicity of methotrexate. Pharm Res 26:1467–1476 [DOI] [PubMed] [Google Scholar]
- Kimura Y, Aoki J, Kohno M, Ooka H, Tsuruo T, Nakanishi O. (2002) P-glycoprotein inhibition by the multidrug resistance-reversing agent MS-209 enhances bioavailability and antitumor efficacy of orally administered paclitaxel. Cancer Chemother Pharmacol 49:322–328 [DOI] [PubMed] [Google Scholar]
- Krishnamurthy P, Schwab M, Takenaka K, Nachagari D, Morgan J, Leslie M, Du W, Boyd K, Cheok M, Nakauchi H, et al. (2008) Transporter-mediated protection against thiopurine-induced hematopoietic toxicity. Cancer Res 68:4983–4989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagas JS, Vlaming ML, Schinkel AH. (2009) Pharmacokinetic assessment of multiple ATP-binding cassette transporters: the power of combination knockout mice. Mol Interv 9:136–145 [DOI] [PubMed] [Google Scholar]
- Leonard K, Nelen M, Raghu M, Detty MR. (1999) Chalcogenopyranones from disodium chalcogenide additions to 1,4-pentadiyn-3-ones. The role of enol ethers as intermediates. J Heterocycl Chem 36:707–717 [Google Scholar]
- Leslie EM, Deeley RG, Cole SPC. (2005) Multidrug resistance proteins: role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol Appl Pharmacol 204:216–237 [DOI] [PubMed] [Google Scholar]
- Létourneau IJ, Slot AJ, Deeley RG, Cole SPC. (2007) Mutational analysis of a highly conserved proline residue in MRP1, MRP2, and MRP3 reveals a partially conserved function. Drug Metab Dispos 35:1372–1379 [DOI] [PubMed] [Google Scholar]
- Loe DW, Almquist KC, Deeley RG, Cole SPC. (1996) Multidrug resistance protein (MRP)-mediated transport of leukotriene C4 and chemotherapeutic agents in membrane vesicles. Demonstration of glutathione-dependent vincristine transport. J Biol Chem 271:9675–9682 [DOI] [PubMed] [Google Scholar]
- Maeno K, Nakajima A, Conseil G, Rothnie A, Deeley RG, Cole SP. (2009) Molecular basis for reduced estrone sulfate transport and altered modulator sensitivity of transmembrane helix (TM) 6 and TM17 mutants of multidrug resistance protein 1 (ABCC1). Drug Metab Dispos 37:1411–1420 [DOI] [PubMed] [Google Scholar]
- Mao Q, Qiu W, Weigl KE, Lander PA, Tabas LB, Shepard RL, Dantzig AH, Deeley RG, Cole SPC. (2002) GSH-dependent photolabeling of multidrug resistance protein MRP1 (ABCC1) by [125I]LY475776. Evidence of a major binding site in the COOH-proximal membrane spanning domain. J Biol Chem 277:28690–28699 [DOI] [PubMed] [Google Scholar]
- Narasaki F, Oka M, Fukuda M, Nakano R, Ikeda K, Takatani H, Terashi K, Soda H, Yano O, Nakamura T, et al. (1997) A novel quinoline derivative, MS-209, overcomes drug resistance of human lung cancer cells expressing the multidrug resistance-associated protein (MRP) gene. Cancer Chemother Pharmacol 40:425–432 [DOI] [PubMed] [Google Scholar]
- Nies AT, Keppler D. (2007) The apical conjugate efflux pump ABCC2 (MRP2). Pflugers Arch 453:643–659 [DOI] [PubMed] [Google Scholar]
- Norman BH, Lander PA, Gruber JM, Kroin JS, Cohen JD, Jungheim LN, Starling JJ, Law KL, Self TD, Tabas LB, et al. (2005) Cyclohexyl-linked tricyclic isoxazoles are potent and selective modulators of the multidrug resistance protein (MRP1). Bioorg Med Chem Lett 15:5526–5530 [DOI] [PubMed] [Google Scholar]
- Pellicani RZ, Stefanachi A, Niso M, Carotti A, Leonetti F, Nicolotti O, Perrone R, Berardi F, Cellamare S, Colabufo NA. (2012) Potent galloyl-based selective modulators targeting multidrug resistance associated protein 1 and P-glycoprotein. J Med Chem 55:424–436 [DOI] [PubMed] [Google Scholar]
- Powers SK, Walstad DL, Brown JT, Detty MR, Watkins PJ. (1989) Photosensitization of human glioma cells by chalcogenapyrylium dyes. J Neurooncol 7:179–188 [DOI] [PubMed] [Google Scholar]
- Russel FGM, Koenderink JB, Masereeuw R. (2008) Multidrug resistance protein 4 (MRP4/ABCC4): a versatile efflux transporter for drugs and signalling molecules. Trends Pharmacol Sci 29:200–207 [DOI] [PubMed] [Google Scholar]
- Sawada GA, Raub TJ, William Higgins J, Brennan NK, Moore TM, Tombline G, Detty MR. (2008) Chalcogenopyrylium dyes as inhibitors/modulators of P-glycoprotein in multidrug-resistant cells. Bioorg Med Chem 16:9745–9756 [DOI] [PubMed] [Google Scholar]
- Sharom FJ. (2011) The P-glycoprotein multidrug transporter. Essays Biochem 50:161–178 [DOI] [PubMed] [Google Scholar]
- Simard TP, Yu JH, Zebrowski-Young JM, Haley NF, Detty MR. (2000) Soluble, infrared-absorbing croconate dyes from 2,6-di-tert-butyl-4-methylchalcogenopyrylium salts. J Org Chem 65:2236–2238 [DOI] [PubMed] [Google Scholar]
- Slot AJ, Molinski SV, Cole SPC. (2011) Mammalian multidrug-resistance proteins (MRPs). Essays Biochem 50:179–207 [DOI] [PubMed] [Google Scholar]
- Smeets PHE, van Aubel RA, Wouterse AC, van den Heuvel JJMW, Russel FGM. (2004) Contribution of multidrug resistance protein 2 (MRP2/ABCC2) to the renal excretion of p-aminohippurate (PAH) and identification of MRP4 (ABCC4) as a novel PAH transporter. J Am Soc Nephrol 15:2828–2835 [DOI] [PubMed] [Google Scholar]
- Tamaki A, Ierano C, Szakacs G, Robey RW, Bates SE. (2011) The controversial role of ABC transporters in clinical oncology. Essays Biochem 50:209–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tombline G, Donnelly DJ, Holt JJ, You Y, Ye M, Gannon MK, Nygren CL, Detty MR. (2006) Stimulation of P-glycoprotein ATPase by analogues of tetramethylrosamine: coupling of drug binding at the “R” site to the ATP hydrolysis transition state. Biochemistry 45:8034–8047 [DOI] [PubMed] [Google Scholar]
- Toppmeyer D, Seidman AD, Pollak M, Russell C, Tkaczuk K, Verma S, Overmoyer B, Garg V, Ette E, Harding MW, et al. (2002) Safety and efficacy of the multidrug resistance inhibitor Incel (biricodar; VX-710) in combination with paclitaxel for advanced breast cancer refractory to paclitaxel. Clin Cancer Res 8:670–678 [PubMed] [Google Scholar]
- van Zanden JJ, Wortelboer HM, Bijlsma S, Punt A, Usta M, Bladeren PJ, Rietjens IM, Cnubben NH. (2005) Quantitative structure activity relationship studies on the flavonoid mediated inhibition of multidrug resistance proteins 1 and 2. Biochem Pharmacol 69:699–708 [DOI] [PubMed] [Google Scholar]
- Vlaming ML, Lagas JS, Schinkel AH. (2009) Physiological and pharmacological roles of ABCG2 (BCRP): recent findings in Abcg2 knockout mice. Adv Drug Deliv Rev 61:14–25 [DOI] [PubMed] [Google Scholar]
- Wang S, Wan NC, Harrison J, Miller W, Chuckowree I, Sohal S, Hancox TC, Baker S, Folkes A, Wilson F, et al. (2004) Design and synthesis of new templates derived from pyrrolopyrimidine as selective multidrug-resistance-associated protein inhibitors in multidrug resistance. J Med Chem 47:1339–1350 [DOI] [PubMed] [Google Scholar]
- Wesołowska O. (2011) Interaction of phenothiazines, stilbenes and flavonoids with multidrug resistance-associated transporters, P-glycoprotein and MRP1. Acta Biochim Pol 58:433–448 [PubMed] [Google Scholar]
- Wijnholds J, deLange EC, Scheffer GL, van den Berg DJ, Mol CA, van der Valk M, Schinkel AH, Scheper RJ, Breimer DD, Borst P. (2000) Multidrug resistance protein 1 protects the choroid plexus epithelium and contributes to the blood-cerebrospinal fluid barrier. J Clin Invest 105:279–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittgen HGM, van den Heuvel JJ, van den Broek PHH, Dinter-Heidorn H, Koenderink JB, Russel FGM. (2011) Cannabinoid type 1 receptor antagonists modulate transport activity of multidrug resistance-associated proteins MRP1, MRP2, MRP3, and MRP4. Drug Metab Dispos 39:1294–1302 [DOI] [PubMed] [Google Scholar]
- Zelcer N, Huisman MT, Reid G, Wielinga P, Breedveld P, Kuil A, Knipscheer P, Schellens JH, Schinkel AH, Borst P. (2003) Evidence for two interacting ligand binding sites in human multidrug resistance protein 2 (ATP binding cassette C2). J Biol Chem 278:23538–23544 [DOI] [PubMed] [Google Scholar]
- Zelcer N, Saeki T, Reid G, Beijnen JH, Borst P. (2001) Characterization of drug transport by the human multidrug resistance protein 3 (ABCC3). J Biol Chem 276:46400–46407 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.