

Abstract
Brown adipose tissue (BAT) is a highly thermogenic organ that converts lipids and glucose into heat. Many of the metabolic and gene transcriptional hallmarks of BAT activation, namely increased lipolysis, uncoupling protein-1 (UCP1) mRNA, and glucose uptake, are regulated by the adrenergic second messenger, cAMP. Cyclic nucleotide phosphodiesterases (PDEs) catalyze the breakdown of cAMP, thereby regulating the magnitude and duration of this signaling molecule. In the absence of adrenergic stimulus, we found that it required a combination of a PDE3 and a PDE4 inhibitor to fully induce UCP1 mRNA and lipolysis in brown adipocytes, whereas neither PDE inhibitor alone had any substantial effect under basal conditions. Under submaximal β-adrenoceptor stimulation of brown adipocytes, a PDE3 inhibitor alone could potentiate induction of UCP1 mRNA, whereas a PDE4 inhibitor alone could augment lipolysis, indicating differential roles for each of these two PDEs. Neither induction of UCP1 nor lipolysis was altered by inhibition of PDE1, PDE2, or PDE8A. Finally, when injected into mice, the combination of PDE3 and PDE4 inhibitors stimulated glucose uptake in BAT under thermoneutral and fasted conditions, a response that was further potentiated by the global ablation of PDE8A. Taken together, these data reveal that multiple PDEs work in concert to regulate three of the important pathways leading to BAT activation, a finding that may provide an improved conceptual basis for the development of therapies for obesity-related diseases.
Introduction
Brown fat function and its relation to obesity and obesity-related diseases were widely studied in rodents throughout the 1980s and 1990s. However, a revival of interest in brown adipose tissue (BAT) has occurred because of the discovery that adult humans possess functionally active depots of BAT (Cypess et al., 2009; van Marken Lichtenbelt et al., 2009; Virtanen et al., 2009). Previously, it was shown that animals receiving a high-fat diet have increased expression of uncoupling protein-1 (UCP1) and increased thermogenesis because of higher sympathetic tone (Rothwell and Stock, 1979). It also is known that animals lacking functional BAT, because of ablation of UCP1, gain more weight than their wild-type counterparts under thermo-neutral conditions (Feldmann et al., 2009). In rodent models of obesity, administration of β3-adrenoceptor agonists not only leads to enhanced lipolysis in white adipocytes (Murphy et al., 1993), but also enhanced insulin sensitivity, glucose use, and triglyceride clearance (Yoshida et al., 1994; Liu et al., 1998; Bartelt et al., 2011). These latter effects are partially mediated by an increase in thermogenesis, whereby circulating fatty acids mobilized from white fat stores are converted into heat by activated brown fat (Weyer et al., 1999). Recently, increased fatty acid oxidation by brown fat has been shown in adult human males exposed to cold (Ouellet et al., 2012). It is this glucose and fatty acid clearing characteristic of brown fat that makes it such an attractive potential target for drug development to treat obesity-related diseases, such as diabetes.
Differentiated brown adipocytes are activated by norepinephrine, which increases cAMP production and stimulates cAMP-dependent protein kinase (PKA) (Janssens et al., 2008). Activation of PKA in turn leads to breakdown of triglyceride stores and increased expression and activation of UCP1 to generate heat (Fedorenko et al., 2012). Increased cAMP also results in increased glucose uptake and metabolism in several tissue types, including brown adipocytes but not in white adipocytes (Taylor et al., 1976; Marette and Bukowiecki, 1989; Jensen, 2007).
However, to date, an effective pharmacological strategy for combating obesity that selectively targets the cAMP-signaling system, particularly via the β3-adrenoceptor, in human brown adipose tissue has not been not been developed because of low expression of this receptor in humans (Barbe et al., 1996; Deng et al., 1996; Larsen et al., 2002; Ursino et al., 2009). Moreover, pharmacological strategies that avoid β-receptor activation will likely be required to avoid cardiovascular and other β-receptor–mediated adverse effects. This is of particular importance, because it has been speculated by several groups that any therapy aimed at increasing the amount of brown fat tissue by differentiation of adipocyte precursors into mature brown adipocytes may not be effective without also providing a means of increasing the activity of the new brown fat tissue (Sell et al., 2004; Gesta et al., 2007; Cannon and Nedergaard, 2009). It is therefore imperative to better understand the mechanisms that regulate the acute activation phases of heat production in vivo.
Although the cAMP dependency of brown fat metabolism has been extensively described, the field has only begun to address the roles played by the negative regulators of cyclic nucleotide signaling, the cyclic nucleotide phosphodiesterases (PDEs), that suppress or modulate each of the thermogenic processes described above. Previous studies have identified the expression of PDE2, PDE3, and PDE4 in rat BAT (Coudray et al., 1999), and at least one study suggested the presence of a PDE1 activity in differentiated brown adipocytes from NMRI mice (Bronnikov et al., 1999). However, which distinct PDE subtype regulates each of the various cAMP-dependent processes of brown fat activation has yet to be determined. Selective inhibitors to most of the PDE families have been developed, and inhibition of PDEs often leads to activation of cAMP-dependent physiologic processes, thereby making PDEs attractive pharmacological targets (Bender and Beavo, 2006).
In this study, we have determined which PDE subtypes are important regulators of three major cAMP-dependent pathways of BAT activation, namely UCP1 gene expression, lipolysis, and glucose uptake. We have found that the activity of both PDE3 and PDE4 can regulate basal UCP1 expression, lipolysis, and glucose uptake and that combining inhibitors to these PDEs synergistically stimulates each of these processes in BAT. We have also identified PDE8A as a potential regulator of BAT glucose uptake in vivo in conjunction with PDE3 and PDE4. Of most importance, these findings indicate that a single selective PDE inhibitor alone is not sufficient to substantially activate basal BAT function, but rather a combinatorial inhibitor approach may prove to be more effective.
Materials and Methods
Animals.
Wild-type C57/Bl6 mice were purchased from Taconic (Hudson, NY) or the Jackson Laboratory (Bar Harbor, MA). A description on the generation of the PDE8A knock-out (PDE8A−/−) mouse line is provided elsewhere (Vasta et al., 2006). For the experiments involving PDE8A−/− mice reported here, wild-type littermate mice aged 10–16 weeks were used as controls. The Institutional Animal Care and Use Committee of the University of Washington, in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals, approved all procedures.
Real-Time Polymerase Chain Reaction.
Mouse interscapular brown adipose tissue was excised, weighed, and disrupted using a rotor-stator tissue homogenizer. RNA was isolated using the RNeasy Lipid Tissue Mini kit (Qiagen, Valencia, CA). RNA was isolated from cells in culture by using either the QIAshredder system and RNeasy Mini kit (Qiagen) or the NucleoSpin RNA II kit (Macherey-Nagel, Bethlehem, PA). cDNA was then generated from 0.125–0.5 μg RNA with use of SuperScript III reverse transcriptase (Invitrogen, Grand Island, NY) and oligo-dT primers modified with a 5′-phosphate. Relative gene expression was determined by performing real-time polymerase chain reaction (PCR) on a MX3000P quantitative PCR system (Stratagene, La Jolla, CA) and analyzed using Mx-Pro software. Real-time PCR was run with iTaq SYBR Supermix (Bio-Rad Laboratories, Hercules, CA) with the following thermoprofile: 95°C for 15 seconds, followed by 60°C for 45 seconds for 40 cycles. Primer (IDT, Coralville, IA) sequences for the different PDE isoforms are provided in the supplement (Supplemental Table 1). All primer sets were determined to anneal on different exons.
Culturing and Differentiation of Immortalized Brown Adipocyte Precursors.
The immortalized brown adipocyte precursor cell line was a generous gift from Dr. Bruce Spiegelman’s laboratory (Dana Farber Cancer Institute, Boston, MA). The precursor isolation, immortalization procedure, and growth conditions have been described previously (Uldry et al., 2006). In brief, cells were routinely seeded at 25,000 cells/cm2 (day 0) in culture medium, composed of high-glucose Dulbecco’s modified Eagle’s medium (DMEM) (HyClone, Thermo Scientific, Waltham, MA) supplemented with 10% fetal bovine serum (HyClone, Thermo Scientific) and 50 U/50 μg penicillin/streptomycin (Gibco, Life Technologies, Grand Island, NY). The following day (day 1), the media was changed to induction medium, which was culture medium supplemented with 20 nM insulin, 1 nM triiodo-thyronine (Sigma-Aldrich, St. Louis, MO), 0.5 μM dexamethasone (Sigma-Aldrich), 0.5 mM isobutyl-1-methylxanthine (IBMX; Sigma-Aldrich), and 0.125 mM indomethacin (Sigma-Aldrich). On day 3, the media was changed to culture medium supplemented with 20 nM insulin and 1 nM triiodo-thyronine and subsequently changed every day thereafter. By day 6, the cells exhibited a fully differentiated phenotype with a large accumulation of multilocular fat droplets. All experiments were conducted on day 6 unless otherwise stated.
PDE Activity Assay.
Differentiated brown adipocytes were washed three times with phophate-buffered saline, then lysed by briefly sonicating in ice cold 50 mM Tris, pH 7.4, 2 mM EDTA, 1 mM dithiothreitol, 1 mM sodium orthovanadate, 200 nM phenylmethylsulfonyl fluoride, and 1× protease inhibitor cocktail III (Calbiochem, EMD Millipore, Darmstadt, Germany). PDE activity was measured as described previously (Hansen and Beavo, 1982; Soderling et al., 1998). In brief, the activity assay was performed in 40 mM MOPS, pH 7.5, 15 mM magnesium acetate, 2 mM EGTA, and 0.2 mg/ml bovine serum albumin (BSA; Sigma-Aldrich) supplemented with ∼100,000 cpm 3H-cAMP or 3H-cGMP (PerkinElmer, Waltham, MA) in a final volume of 0.25 ml. The total substrate and inhibitor concentrations are indicated in the figure legends. The reaction time and amount of lysate were maintained so that less than 30% of the substrate was hydrolyzed.
Preparation of Freshly Isolated Primary Brown Adipocytes.
Primary brown adipocytes were isolated using a modified Rodbell method (Rodbell, 1964; Matthias et al., 2000). Routinely, the interscapular fat pads from 3–5 C57/Bl6 mice were pooled and initially placed in a 20-ml plastic scintillation vial (Research Products International, Mount Prospect, IL) containing filter-sterilized Krebs-Ringer HEPES Isolation Buffer (20 mM HEPES, 118.5 mM NaCl, 25.3 mM NaHCO3, 1.2 mM NaH2PO4, 1.2 mM MgSO4, 10 mM D-glucose, 10 mM D-fructose, 4% bovine serum albumin, pH 7.4) warmed to 37°C and equilibrated with 95% O2–5% CO2 bubbling through a diffuser. Whole tissue was pre-incubated in 3 ml Isolation Buffer supplemented with 2.0 mg/ml Collagenase (Sigma-Aldrich; C6885) shaking in a Dubnoff shaking water bath (Thermo Scientific) for 7 minutes shaking at 90 cycles/min. Every 2 minutes, the tissue was removed and gently vortexed for 10 seconds. At the end of the 7 minutes, the tissue was removed and rinsed with Isolation Buffer. The tissue was then finely minced with surgical blades (Bard Parker, Thermo Scientific; no. 371110) in a scissor fashion and placed back into a plastic scintillation vial containing 4 ml fresh Isolation Buffer containing collagenase. The mixture was gassed with 95%/5% O2/CO2 for 2 minutes bubbling through a 25-gauge syringe needle. The vial was then placed in a Dubnoff incubator shaker for 40 minutes shaking at 120 cycles/min and gently vortexed every 5 minutes for 10 seconds. The tissue fragments were then homogenized further by triturating gently with a plastic pasture pipette. The mixture was then filtered through a 70-μm nylon filter (BD Falcon 352350; BD Biosciences, San Jose, CA), and the filtrate was diluted with Isolation Buffer to 12 ml. Usually, it was necessary to gently press the remaining tiny tissue fragments through the filter with a gloved finger. The collected cells were centrifuged at 70g, and the infranatant below the floating brown adipocytes was removed through a tube with a peristaltic pump. The cells were then washed twice more by flotation with 6 ml 37°C Isolation Buffer that contained 4% Fatty Acid-Free BSA (Sigma-Aldrich) and gassed with 95%/5% O2/CO2, centrifuging after each wash at 70g. Finally, the cells were resuspended in 1ml FA-Free Isolation Buffer and counted on a hemacytometer. Cells containing multilocular fat droplets were identified as brown adipocytes. The yield usually ranged from 0.5 × 106 to 2 × 106 adipocytes per 3–5 fat pads.
Glycerol Release Assay.
Freshly isolated brown adipocytes were diluted to a final concentration of 75,000 cells/ml into 200 μl FA-Free Isolation Buffer containing pharmacological agents. The tubes were placed in heating blocks in a Dubnoff shaking water bath at 37°C and shaken at 90 cycles/min. The reaction was terminated by freezing the tubes in liquid nitrogen. Tubes were thawed in ice water and centrifuged at 16,000g at 4°C. The infranatant was removed, and the glycerol content was measured using the reagents listed in the Free Glycerol Determination Kit (Sigma-Aldrich). Background lipolysis in a given experiment was defined as glycerol released in the presence of 1 μM propranolol (Sigma-Aldrich) and was subtracted from the total.
Determination of cAMP Concentration by Enzyme Immunoassay.
Brown adipocyte precursors were grown and differentiated in 12-well plates and preincubated with PDE inhibitors in DMEM for 30 minutes, then isoproterenol in DMEM for 5 minutes. The media were removed, and the reaction was stopped with ice cold 99% ethanol-1% hydrochloric acid. The ethanol acid containing the cAMP was pipetted into Eppendorf tubes and dried in a speed vacuum (Savant, Thermo Scientific). The samples were then resuspended to 200 μl, and cAMP was measured according to the manual for the cAMP EIA Kit (American Qualex Molecular, San Clemente, CA). The cell monolayer was then resuspended in water, and protein content was assessed by BCA assay (Pierce, Rockford, IL).
Western Blotting.
Differentiated brown adipocytes were lysed in 50 mM Tris, pH 8.0, 0.5% Triton-X100, 150 mM NaCl, 1 mM EDTA, 1 mM dithiothreitol, 1 mM sodium orthovanadate, 1 mM sodium fluoride, 200 nM phenylmethylsulfonyl fluoride, and 1× protease inhibitor cocktail III (Calbiochem, EMD Millipore) and denatured in sample buffer (125 mM Tris, 20% glycerol, 2% SDS, 2% 2-mercaptoethanol) by boiling for 5 minutes. The samples were then separated by 10% SDS-PAGE at 0.75 mm thickness and transferred to polyvinylidene difluoride membranes for 1 hour at 60 V. Membranes were blocked overnight by shaking on an orbital shaker at 4°C in 5% BSA in Tris-buffered saline containing 0.05% Tween-20 (TBST). Membranes were probed for phospho-CREB (cAMP response element-binding protein) Ser-133 [1:1000 (v/v); 9198S; Cell Signaling Technology, Danvers, MA) or glyceraldehyde 3-phosphate dehydrogenase [1:20,000 (v/v); Fitzgerald Industries, Acton, MA) overnight with shaking on an orbital shaker at 4°C in 5% BSA in TBST. Membranes were then washed three times for 10 minutes with TBST at room temperature, rinsing with TBST between washes, then probed with secondary antibody conjugated to horseradish peroxidase [goat anti-rabbit 1:5000 (v/v), goat anti-mouse 1:20,000 (v/v); Bio-Rad Laboratories] in 5% BSA in TBST at room temperature for 1 hour. The washing steps were repeated as before, and membranes were developed with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific). Immunoreactivity was imaged using a Bio-Rad Molecular Imager ChemiDoc XRS+ with Image Laboratory Software, and band intensity was quantified using ImageJ. For the detection of PDE8A in whole mouse BAT, tissue was harvested from wild-type and PDE8A−/− mice and homogenized as described elsewhere (Nolan et al., 2004). The homogenates were centrifuged at 10,000g for 15 minutes, and the protein content of the infranatant was determined. Samples were denatured as described above. Thirty micrograms protein was separated by SDS-PAGE and transferred to polyvinylidene difluoride for 3 hours at 60 V, and membranes were blocked in 5% milk in TBST for 1 hour at room temperature. Membranes were probed for PDE8A [1:1200 (v/v); PDE8-121AP; FabGennix International, Frisco, TX] or UCP1 [1:1000 (v/v); C-17; Santa Cruz Biotechnology] in 5% milk in TBST overnight at 4°C. Membranes were washed three times for 10 minutes with TBST and then probed with secondary antibody conjugated to horseradish peroxidase [goat anti-rabbit 1:3000 (v/v) (Bio-Rad Laboratories), or rabbit anti-goat 1:3000 (Jackson Immunoresearch Laboratories, West Grove, PA)] for 1 hour at room temperature. The membranes were washed and developed as described above. Immunoreactivity was imaged using autoradiography film (Genesee Scientific, San Diego, CA).
18F-Flurodeoxyglucose and PET Scanning.
The mice were fasted overnight before the day of imaging. On the day of imaging, the mice were housed in portable containers that were warmed by placing them on heating pads (Deltaphase Isothermal Pad; Braintree Scientific, Braintree, MA). Thirty minutes before administration of flurodeoxyglucose (FDG), mice received an intraperitoneal injection of a single PDE inhibitor, a combination of PDE inhibitors, or vehicle control. The injected mouse was then placed in a heated chamber. The chamber was warmed using heating pads and maintained at a temperature of 30–35°C. The mouse was awake in the chamber and allowed to move freely. After 30 minutes, the mouse was administered 200–300 µCi of FDG via retro-orbital injection. The amount of FDG administered (in microcuries) was approximately 10 times the weight of the mouse in grams. The mouse was placed in a warmed induction chamber and kept lightly sedated using 1–2% isoflurane anesthesia. After a 40-minute uptake period, the mouse was placed on the small animal PET scanner (Inveon Dedicated PET system; Siemens, Munich, Germany) and imaged for 20 minutes. During the PET imaging, the mouse was kept warm by a small heating pad on the table and maintained while anesthetized with 2–3% isoflurane. A transmission scan was also collected for attenuation correction. After imaging, the mouse was euthanized and the tissue was harvested. The data were reconstructed using software provided by the vendor. The three-dimensional ordered subsets expectation maximization– maximum a posterior (3D OSEM-MAP) method was used to reconstruct the images. A smoothing parameter to achieve 1.5 mm image resolution was used. The images were analyzed using Siemens’s ASIPRO analysis software. The system was calibrated to report image voxel values in radioactivity concentration (i.e., nCi/g). Regions of interest were drawn encompassing the two BAT regions in a coronal plane of the mouse, as shown in Supplemental Fig. 7. The maximum pixel values from the two regions of interest were averaged and used to determine the standardized uptake value (SUV) for FDG. The SUV is a semiquantitative metric used to express glucose uptake in tissue. It is calculated using the following formula:
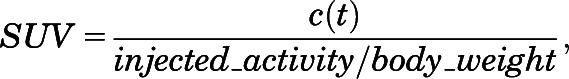 |
where c(t) was defined as the maximum activity concentration. Injected activity was corrected to the start of scanning. Body weight of the mouse was measured in grams. When using SUV as an uptake metric, it was important that all imaging be done at the same time after injection—in this case, 40 minutes.
Drugs.
SCH51866 [(+)-cis-5,6a,7,8,9,9a-hexahydro-2-[4-[trifluro-methyl] phenylmethyl]-5-methyl-cylopent[4,5]imidazo [2,1-b]purin-4(3H)one] (Schering-Plough/Merck, Whitehouse Station, NJ) is a selective PDE1 inhibitor that does not distinguish between PDE1 isoforms and was a gift from the manufacturer (structure published in Vemulapalli et al., 1996). BAY 60-7550 [2-(3,4-dimethoxybenzyl)-7-{(1R)-1-[(1R)-1-hydroxyethyl]-4-phenylbutyl}-5-methyllimidazo[5,1-f][1,2,4]triazin-4(3H)-one] (Cayman Chemicals, Ann Arbor, MI) is a PDE2 inhibitor. Cilostamide (Tocris, Bristol, UK) is a nonselective PDE3 inhibitor. Rolipram (Enzo Life Sciences, New York, NY) is a selective PDE4 inhibitor that does not distinguish between PDE4 isoforms. PF-04957325 (Pfizer, New York, NY) is a selective PDE8 inhibitor that will inhibit PDE8A and PDE8B with similar affinities and was a gift from the manufacturer (structure published in Vang et al., 2010). IBMX (Sigma-Aldrich) is a nonselective PDE inhibitor. H-89 (Tocris) is a protein kinase inhibitor with substantial selectivity for PKA. PDE and PKA inhibitors were dissolved in dimethyl-sulfoxide (Sigma-Aldrich) and stored at −20°C. 8-Bromoadenosine-3′,5′-cyclic monophosphorothioate (Rp-8-Br-cAMPS; BioLog, Bremen, Germany) is a competitive inhibitor to the cAMP-binding site on the regulatory subunit of PKA and was dissolved in water. 8-(4-Chlorophenylthio)-2'-O-methyladenosine-3′,5′-cyclic monophosphate (8-pCPT-2-O-Me-cAMP; BioLog) is an exchange protein activated by cAMP (Epac) agonist and was dissolved in water. 8-Bromoguanosine 3′,5′-cyclic monophosphate (8-Br-cGMP) is a protein kinase-G agonist and was dissolved in water. 10 mM isoproterenol (Sigma-Aldrich) was dissolved in 1 M ascorbic acid immediately before use in gene induction experiments. For lipolysis experiments, isoproterenol was dissolved in water immediately before use. For glucose uptake experiments, isoproterenol was dissolved in phosphate-buffered saline immediately before injection into mice.
Results
PDE Expression in Mouse Brown Adipocyte Models.
We first assessed the PDE mRNA expression profile in mouse interscapular brown fat tissue, freshly isolated primary adipocytes, and brown adipocytes differentiated from a brown adipocyte precursor cell line in vitro. Quantitative real-timePCR analysis revealed that the mRNAs for PDE1A, 2A, 3B, 4B, 4D, and 8A were the predominant ones expressed in whole brown fat tissue (Fig. 1A), primary brown adipocytes isolated from BAT tissue via collagenase digestion (Fig. 1B), and immortalized brown adipocyte precursors on day 6 after differentiation (Fig. 1C). Because it was thought that PDE8A expression is limited to a small number of tissues (Soderling et al., 1998), expression of PDE8A in mouse BAT was unexpected. We further verified the presence of PDE8A protein by Western blot analysis (Supplemental Fig. 1) and also found that both PDE8A RNA and protein was absent in the BAT of PDE8A−/− mice. (Fig. 1A; Supplemental Fig. 1). In addition, we did not detect any major compensatory changes in the expression of other PDEs in the BAT from PDE8A−/− mice, compared with wild-type littermates (Fig. 1A). These data indicate that the pattern of PDE expression from whole BAT tissue is largely representative of that found in isolated brown adipocytes and that these patterns are similar among all models of BAT used.
Fig. 1.

PDE mRNA expression profile in multiple models of BAT is similar. (A) RNA was extracted from whole mouse interscapular BAT from wild-type and PDE8A−/− littermates and then analyzed by real-time (RT)-PCR as described in Materials and Methods. Mean values ± S.E.M. are expressed relative to GAPDH (glyceraldehyde 3-phosphate dehydrogenase) mRNA (n = 3). The PDE gene name is abbreviated as a number and letter. (B) Isolated primary mouse brown adipocytes were pooled from 3–5 mice and analyzed by RT-PCR as described in Materials and Methods. Mean values ± S.E.M. are expressed normalized to PDE3B mRNA (n = 3). (C) RNA from immortalized brown adipocyte precursor cells on day 6 of differentiation was isolated and analyzed by RT-PCR as described in Materials and Methods. Mean values ± S.E.M. are expressed relative to 18S mRNA (n = 5–6).
Enzyme activity assays confirmed the functional presence of the major PDEs identified by the mRNA profile in immortalized brown adipocytes. The PDE1 family is stimulated by calcium-calmodulin, and PDE1A has an approximately 70-fold higher selectivity for cGMP, compared with cAMP, at low substrate levels. We therefore confirmed the presence of PDE1 activity by measuring cGMP hydrolysis in the presence and absence of calcium and calmodulin in the whole BAT extract. This hydrolytic activity was increased by 2.7-fold by calcium and was fully inhibited by 100 nM SCH51866, a relatively selective PDE1 inhibitor (IC50 for PDE1A, 10 nM) (Dunkern and Hatzelmann, 2007) (Fig. 2A).
Fig. 2.

PDE enzymatic activities were confirmed in differentiated immortalized brown adipocytes. (A) PDE1 activity was detected by the addition of calcium/calmodulin to whole extract with use of 1 μM cGMP as substrate. The increase in activity was inhibited with 100 nM SCH51866 (n = 3–8). (B) PDE2 activity was detected by stimulating cAMP-hydrolyzing activity with cGMP. PDE3 and PDE4 were first inhibited with cilostamide and rolipram. cAMP-hydrolysis using 1 μM substrate was then stimulated by 1 μM cGMP. The increased activity was inhibited by the addition of 50 nM BAY 60-7550 (n = 3–4). (C) PDE3 activity was detected by the addition of 10 μM cilostamide with use of 1 μM cAMP as substrate. PDE4 activity was detected by the addition of 10 μM rolipram with use of 1 μM cAMP as substrate (n = 3–4). (D) PDE8 activity was defined as PDE activity that is insensitive to 100 μM IBMX and is inhibited by 10 nM PF-04957325 with 12 nM cAMP substrate (n = 3–6). Data are presented as mean percentage of total cyclic-nucleotide hydrolysis ± S.E.M., and statistical analysis in (D) was performed using paired Students t test: *P < 0.05 between IBMX alone and IBMX+PF.
PDE2 is a dual-substrate PDE, because it hydrolyzes cAMP and cGMP with approximately equal specificity. However, its cAMP-hydrolyzing activity is stimulated by cGMP. Therefore, to assay for PDE2 activity, we measured the hydrolysis of cAMP in the presence and absence of 1 μM cGMP. We included 10 μM cilostamide and 10 μM rolipram to these reactions to eliminate background PDE3 and PDE4 activity and to eliminate potential complications in the interpretation of the data, because PDE3s can be inhibited by cGMP. Under these conditions, the addition of 1 μM cGMP increased hydrolytic activity toward cAMP by 2.8-fold. This increased activity was completely inhibited by the addition of 50 nM of the PDE2 inhibitor, BAY 60-5770, a 10-fold excess over the reported IC50 for PDE2 (Bender and Beavo, 2006) (Fig. 2B).
PDE3 and PDE4 activities were detected by measuring the hydrolysis of 1 μM cAMP in the presence and absence of 200 nM cilostamide (PDE3 inhibitor) or 10 μM rolipram (PDE4 inhibitor), respectively. These inhibitor concentrations are 10-fold higher than their reported IC50 values for PDE3 and PDE4 (Bender and Beavo, 2006). Cilostamide inhibited total cAMP-hydrolyzing activity by approximately 60%, whereas rolipram inhibited total activity by approximately 40%. When combined together, approximately 88% of the total cAMP PDE activity was inhibited (Fig. 2C).
Finally, because the PDE8 family is insensitive to IBMX and has a Km for cAMP below 100 nM, PDE8 activity was detected by assay of cAMP hydrolytic activity at 0.012 μM substrate in the presence of 100 μM IBMX and the presence or absence of 10 nM PF-04957235. PF-04957235 decreased the IBMX-insensitive activity by 46% (P < 0.05), showing the presence of PDE8 activity in these extracts (Fig. 2D).
PDE3 and PDE4 Regulate Basal UCP1 and Peroxisome Proliferator-Activated Receptor-γ Coactivator mRNA Expression.
Because it is widely accepted that cAMP stimulates expression of UCP1 mRNA (Cannon and Nedergaard, 2004), we next tested which cAMP-dependent PDE or PDEs regulate the pool(s) of cAMP that control induction of UCP1 gene transcription in differentiated brown adipocytes. For these studies, an immortalized brown adipocyte precursor cell line was differentiated into mature brown fat cells in vitro. The differentiated adipocytes were then pretreated with selective inhibitors to PDE1 (SCH51866), PDE2 (BAY 60-7550), PDE3 (cilostamide), PDE4 (rolipram), or PDE8 (PF-04957235), either individually or in combination. Somewhat unexpectedly, when administered individually, none of the PDE inhibitors increased the basal expression of UCP1 mRNA. However, when administered together, 10 μM cilostamide and 10 μM rolipram resulted in an approximately 40-fold increase in basal UCP1 mRNA expression, similar to that seen with a high dose of isoproterenol (10 μM) (Supplemental Fig. 2A) or IBMX (200 μM) (Fig. 3A). This latter observation strongly suggests that inhibition of PDE3 and PDE4 together is sufficient to explain the stimulatory effect of the nonselective PDE inhibitor, IBMX, on induction of UCP1. The same magnitude of potentiation was observed when the concentration of cilostamide was reduced to 300 nM (Fig. 3B). The PDE8-selective inhibitor, PF-04957325 (200 nM), did not stimulate UCP1 mRNA expression when administered either alone or in combination with IBMX, cilostamide, or rolipram (Fig. 3C; Supplemental Fig. 2B). Similarly, PDE1 or PDE2 inhibitors administered either alone or in combination with PDE3 and PDE4 inhibitors did not stimulate expression of UCP1 mRNA, nor did they augment the effect of combined PDE3 and PDE4 inhibition (Supplemental Fig. 2C). In addition, we detected a 2.7-fold increase of UCP1 mRNA in the interscapular brown fat of fasted, warmed mice that were injected with the combination of cilostamide and rolipram (P < 0.05), where an injection of either drug alone had no significant effect, compared with an injection of vehicle (Fig. 3D). A similar pattern of stimulation was also observed in immortalized brown adipocytes for basal expression of peroxisome proliferator-activated receptor-γ coactivator mRNA (Supplemental Fig. 3), a transcription factor also regulated by cAMP that is important for UCP1 gene induction during BAT activation. Finally, PDE3 and PDE4 inhibitors administered individually did not stimulate basal cAMP accumulation, but when administered together, they increased cAMP by 9.6-fold (Fig. 4). Together, these results suggest that the combined inhibition of PDE3 and PDE4 removes an important physiologic suppressive effect of these two PDEs on basal cAMP signaling and BAT activation in the absence of β-adrenoceptor agonists.
Fig. 3.

PDE3 and PDE4 inhibitors increase Ucp1 and Pgc-1α mRNA expression in differentiated brown adipocytes. UCP1 mRNA expression was measured in differentiated brown adipocytes that were (A) pretreated with 10 μM cilostamide, 10 μM rolipram, both, or 200 μM IBMX for 30 minutes, then stimulated by vehicle or 1 nM isoproterenol for 4 hours (n = 3–13); (B) treated with 300 nM cilostamide or 10 μM cilostamide with or without 10 μM rolipram (n = 3–8); or (C) pretreated with either 200 nM PF-04957325, 200 μM IBMX, or both for 30 minutes, then stimulated by vehicle or 1 nM isoproterenol for 4 hours (n = 3). UCP1 mRNA was quantified relative to 18S mRNA with use of RT-PCR as described in Materials and Methods. (D) UCP1 mRNA expression was measured from the RNA extracted from the interscapular BAT pads harvested from mice undergoing PET scanning after the procedure was terminated (Materials and Methods). UCP1 mRNA was quantified relative to GAPDH (glyceraldehyde 3-phosphate dehydrogenase) mRNA with use of real-time (RT)-PCR as described in Materials and Methods. Data are presented as mean fold over vehicle control ± S.E.M., and statistical analyses were performed using one-way analysis of variance with Dunnet post hoc test: ***P < 0.001; *P < 0.05, versus vehicle in each group.
Fig. 4.

PDE3 and PDE4 inhibitors stimulate cAMP accumulation in differentiated brown adipocytes. Differentiated brown adipocytes were pretreated with PDE inhibitors for 30 minutes, and then stimulated with vehicle or 1 or 10 nM isoproterenol for 5 minutes. cAMP was measured as described in Materials and Methods. Data are presented as mean ± S.E.M. (n = 3–5), and statistical analyses were performed using one-way analysis of variance with Dunnett post hoc (***P < 0.001, versus vehicle control in each group) and by two-way analysis of variance with Bonferonni post hoc (#P < 0.05, versus vehicle control).
PDE3B Controls β-Adrenoceptor Stimulation of UCP1 mRNA Expression.
Our finding that both PDE3 and PDE4 together control basal UCP1 expression prompted us to determine whether inhibition of the same combination of PDEs regulates the increase in induction of UCP1 mRNA caused by β-adrenoceptor agonists. Typically, inhibition of the PDE or PDEs that regulate a given process will shift the dose-response curve of agonists for that process to the left or up and to the left. Because β-adrenoceptor activation is known to stimulate expression of UCP1 mRNA, we hypothesized that a PDE inhibitor or combination of inhibitors would potentiate a low dose of isoproterenol based on the dose-response relationship (Supplemental Fig. 2A). For these experiments, cells were pretreated with individual inhibitors to PDE1, PDE2, PDE3, PDE4, and PDE8, followed by 1 nM isoproterenol. We found that only pretreatment with the PDE3 inhibitor, cilostamide, dose-dependently potentiated the expression of UCP1 mRNA in response to this low dose of isoproterenol (P < 0.001 at 10 μM cilostamide). In contrast, a PDE4 inhibitor, rolipram, had no effect on isoproterenol-induced UCP1 expression (Fig. 3A). Of interest, cAMP accumulation did not follow the same pattern of potentiation (Fig. 4). At 1 nM isoproterenol, neither cilostamide nor rolipram potentiated global cAMP accumulation, even though UCP1 mRNA was substantially increased under these conditions. PDE1, PDE2, and PDE8 inhibitors also did not potentiate induction of UCP1 by isoproterenol (Fig. 3C; Supplemental Fig. 4).
Effects of PDE3 and PDE4 Inhibition Are PKA Dependent.
Two major pathways for cAMP-mediated mechanisms for control of transcription have been described: the Epac pathway and the cAMP-dependent PKA pathway. We therefore wanted to test by which of these molecular pathway(s) PDE inhibition caused stimulation of UCP1 mRNA expression. We pretreated differentiated brown adipocytes with PDE3 and PDE4 inhibitors along with increasing doses of H-89, a known PKA inhibitor. Cilostamide and rolipram alone had minor effects on PKA-substrate phosphorylation and CREB phosphorylation (1.8- and 2.5-fold, respectively). However, when combined, they produced a 9.6-fold stimulation of CREB phosphorylation, compared with the control. H-89 was able to fully reduce expression of UCP1 mRNA and CREB phosphorylation at a dose of 25 μM (Fig. 5, A–C). Similarly, although 1 nM isoproterenol alone increased CREB-phosphorylation by 3.6-fold (P < 0.01), combining isoproterenol with cilostamide further increased CREB phosphorylation to 5.9-fold (P < 0.001). The difference between isoproterenol alone and isoproterenol with cilostamide was statistically significant (P < 0.05). Furthermore, H-89 (10 μM) was sufficient to inhibit the stimulatory effect of cilostamide on isoproterenol-stimulated UCP1 induction and on CREB phosphorylation (Fig. 5, D–E). We also saw a significant inhibition of UCP1 induction when cells were pretreated with a more selective PKA inhibitor (1mM Rp-8-Br-cAMPS) that binds to a different site on PKA than H-89 (Supplemental Fig. 5, A and B). A selective agonist for Epac, 8-pCPT-2-O-Me-cAMP, did not stimulate UCP1 mRNA (unpublished data). Because it has been shown that H-89 can inhibit protein kinase-G (Ki = 0.5 μM) with a Ki that is only 10-fold higher than the Ki for PKA (Ki = 0.05 μM) in vitro (Hidaka and Kobayashi, 1992), we also tested whether 8-Br-cGMP, an agonist to cGMP-dependent protein kinase, could stimulate accumulation of UCP1 mRNA; we observed no appreciable change after application of this inhibitor (unpublished data). Together, these data strongly suggest that the stimulatory effect of combined PDE3 and PDE4 inhibition on UCP1 mRNA accumulation is largely through the canonical PKA-dependent signaling pathway.
Fig. 5.

The potentiating effect of PDE inhibitors on Ucp1 mRNA and CREB phosphorylation is PKA dependent. (A) UCP1 mRNA expression was measured in differentiated brown adipocytes that were pretreated with H-89 for 1 hour, then 10 μM cilostamide and 10 μM rolipram for 4.5 hours. RNA was isolated, and UCP1 mRNA was quantified relative to 18S mRNA with use of real-time (RT)-PCR as described in Materials and Methods. Values were then normalized to the cilostamide and rolipram treatment in the absence of H-89 control. (B) CREB phosphorylation after 40 minutes of stimulation by 10 μM cilostamide and 10 μM rolipram in differentiated brown adipocytes. (C) Quantification of Western blot in (B). (D) UCP1 mRNA expression in differentiated brown adipocytes that were pretreated with H-89 at the indicated doses for 1 hour, followed by PDE inhibitors added 30 minutes before addition of 1 nM isoproterenol. RNA was isolated, and UCP1 mRNA was quantified relative to 18S mRNA with use of RT-PCR as described in Materials and Methods. Values were then normalized to the cilostamide and isoproterenol treatment in the absence of H-89 control. (E) CREB phosphorylation after 10 minutes of isoproterenol stimulation after a 30-minute preincubation with 10 μM cilostamide in differentiated brown adipocytes. (F) Quantification of Western blot in (E). Data are presented as mean ± S.E.M. (n = 3–7), and statistical analyses were performed by one-way analysis of variance with Dunnett post hoc: ***P < 0.001; **P < 0.01, versus vehicle control in each group; #P < 0.05; ###P < 0.001, versus 1 nM isoproterenol plus 10 μM cilostamide without H-89.
PDE3 and PDE4 in Combination Regulate Lipolysis in Primary Brown Adipocytes.
Noradrenergic regulation of BAT function also requires stimulation of lipolysis to provide both the energy for establishing the mitochondrial proton gradient and the allosteric activation of UCP1 by free fatty acids. Therefore, we were interested in determining which PDEs were important for cAMP-dependent regulation of lipolysis in brown fat. Freshly isolated primary brown adipocytes were pretreated with 10 μM cilostamide, 10 μM rolipram, or both and then treated with vehicle or isoproterenol for 1 hour. Lipolysis was measured as glycerol released into the media. We found that cilostamide alone did not significantly alter basal lipolysis or lipolysis stimulated by 10 nM isoproterenol. Conversely, rolipram alone significantly potentiated both basal and isoproterenol-stimulated glycerol release by 7.8-fold and 2.8-fold, respectively, over the vehicle control (Fig. 6A). This suggested that PDE4, but not PDE3, was a primary regulator of the pool of cAMP that regulates lipolysis in primary brown adipocytes. However, when both inhibitors were combined, there was an even larger 30–40-fold increase in lipolysis that is of the same magnitude as that seen with the nonselective PDE inhibitor IBMX (Fig. 6A). Of interest, despite substantial expression of PDE8A in brown fat, the PDE8 inhibitor did not augment lipolysis in primary brown adipocytes, either alone or in combination with IBMX, cilostamide, and/or rolipram (Fig. 6B; Supplemental Fig. 6, A–C). These data indicate synergistic roles for PDE3 and PDE4, but not for PDE8A, on lipolysis in brown adipocytes.
Fig. 6.

PDE3 and PDE4 inhibitors, but not the PDE8 inhibitor, increased glycerol production from isolated primary mouse brown adipocytes. Primary brown adipocytes were isolated from mouse interscapular brown fat pads as described in Materials and Methods. (A) Primary brown adipocytes were pretreated with 10 μM cilostamide, 10 μM rolipram, both, or 50 μM IBMX for 20 minutes and then stimulated with vehicle or 10 nM isoproterenol for 1 hour. (B) Primary brown adipocytes were pretreated with 1 μM PF-04957325 and/or 50 μM IBMX for 20 minutes and then stimulated with vehicle or 10 nM isoproterenol for 1 hour. Glycerol was measured as described in Materials and Methods. Data are expressed as mean mg glycerol/105 cells ± S.E.M. (n = 3–5), and statistical analyses were performed by one-way analysis of variance. ***P < 0.001, for Cil+Rol and IBMX versus vehicle control in each group; **P < 0.01 for IBMX and PF+IBMX versus vehicle; *P < 0.05 for Cil or Rol versus vehicle; and a two-tailed t test #P < 0.05, for PDE inhibitors versus vehicle in the presence of 10 nM isoproterenol.
Inhibition of PDE3 and PDE4 Stimulate In Vivo Glucose Uptake in BAT.
Glucose uptake in brown adipocytes can be regulated by cAMP-dependent signaling (Marette and Bukowiecki, 1989). We therefore hypothesized that selective inhibition of the appropriate PDE(s) would potentiate glucose uptake in BAT. To test this, we treated fasted, warmed, wild-type, and PDE8A−/− littermates with 3 mg/kg cilostamide, 3 mg/kg rolipram, a combination of both, or the vehicle control, followed by an injection of 18F- FDG. The mice were then imaged using a PET scanner. Basal glucose uptake by BAT from wild-type and PDE8A−/− mice was very low (Fig. 7, A and B). Moreover, neither PDE inhibitor alone induced a significant increase in BAT glucose uptake activity, compared with the vehicle control in the wild-type mice. On the other hand, cilostamide treatment alone produced a small, but significant, increase in glucose uptake, compared with the vehicle control in the PDE8A−/− mice (2.7-fold; P < 0.05), although the maximum glucose uptake in response to cilostamide was not significantly different between the two genotypes (Fig. 7C). We then tested the effect of a combination of PDE3 and PDE4 inhibitors on glucose uptake in BAT. Treatment with a combination of both cilostamide and rolipram resulted in a large increase in BAT glucose uptake in both the wild-type and PDE8A−/− mice. Of surprise, the maximum response to both PDE inhibitors in the PDE8A−/− mice was consistently 1.8-fold higher than what was observed in the wild-type littermate controls (P < 0.01). Conversely, when injected with isoproterenol, there was no difference in BAT glucose uptake between wild-type and PDE8A−/− mice. Taken together, these data suggest that each of these three PDEs can suppress basal cAMP-dependent glucose uptake in BAT and that all three need to be inhibited to allow full stimulation in the absence of agonist. However, under conditions of β-adrenoceptor stimulation, this attenuation of glucose uptake by the PDEs can be overridden, presumably by the high adenylyl cyclase activity resulting from adrenergic stimulation.
Fig. 7.

PDE3 and PDE4 inhibitors stimulate 18F- FDG uptake in the interscapular BAT in vivo. (A) Wild-type and PDE8A−/− littermate mice were injected with 3mg/kg of cilostamide, rolipram, or both. FDG was injected retro-orbitally, and mice were imaged using positron emission tomography as described in Materials and Methods. (B) Quantification of BAT region of interest (ROI) expressed as SUV=Max count in ROI/injected dose/weight. (C) Wild-type and PDE8A−/− littermates were injected with 0.5 mg/kg isoproterenol or vehicle, and FDG uptake was quantified similarly. Data are presented as mean SUV ± S.E.M. (n = 3–4), and statistical analyses were performed by one-way analysis of variance with Dunnett post hoc (*P < 0.05; **P < 0.01; and ***P < 0.001 versus vehicle control in each group) and by two-way analysis of variance with Bonferonni post hoc (###P < 0.001 and n.s. for wild-type versus PDE8A−/− treated with cilostamide and rolipram). n.s., not significant.
Discussion
Although many brown fat–directed studies have focused on brown adipocyte differentiation or the recruitment of adipocyte precursors into more brown-like adipocytes, it also has been shown that simply having more brown fat is insufficient to influence whole animal metabolism if the tissue remains in a nonactivated state (Sell et al., 2004). Therefore, selective pharmacological stimulation of this nonstimulated state of BAT could provide an effective new treatment paradigm for obesity and obesity-related diseases in humans. In early studies, nonselective PDE inhibitors were shown to potentiate the thermogenic response of cold-exposed rats (Wang and Anholt, 1982). However, nonselective inhibitors of PDEs, such as theophylline, have a very narrow therapeutic index and substantial toxicity (Beavo et al., 2007). Studies with nonselective PDE inhibitors also cannot identify which specific PDEs are most important to BAT thermogenesis. The experiments described in this article were designed to test the hypothesis that inhibition of the specific PDEs that regulate BAT activation might provide a conceptual basis for the design of new PDE-related therapies for the treatment of obesity-related diseases.
In this study, we addressed the question of which subtype(s) of PDE regulate lipolysis, glucose uptake, and mRNA expression of UCP1. We identified PDE3 and PDE4 (likely PDE3B and PDE4B/D) as the major regulators of these major BAT processes both in vitro and in vivo (Figs. 2, 3, 6, and 7). Under basal conditions, the effects of the individual PDE3 and PDE4 inhibitors were synergistic in nature, because individual inhibitors to PDE3 and PDE4 had little or no effect on their own but, when combined, would produce a large, synergistic response. In fact, the same magnitude of potentiation on UCP1 expression was observed over a wide range of cilostamide doses, suggesting a truly synergistic relationship between rolipram and cilostamide (Fig. 3B). The only conditions in which a single selective PDE3 or PDE4 inhibitor was sufficient to potentiate UCP1 induction or lipolysis, respectively, were those in which an adrenoceptor agonist was also present (Figs. 3 and 6). Finally, we also observed that PDE8A may play a role in the regulation of basal BAT glucose uptake, but this global knockout effect was only observed when PDE3 and PDE4 were also inhibited (Fig. 7).
We also found that PDE1, PDE2, or PDE8 inhibitors had virtually no effect on UCP1 mRNA either alone or in any combination with either PDE3 or PDE4 inhibitors (Fig. 3C; Supplemental Fig. 2B), despite the fact that we can detect PDE1, PDE2, and PDE8 mRNAs and enzyme activities (Figs. 1 and 2, A and B) in extracts from these differentiated brown adipocytes. Therefore, inhibition of both PDE3 and PDE4 together is necessary to fully activate basal UCP1 induction and lipolysis and is sufficient to fully account for the IBMX response. We observed what appears to be a greater, though not statistically significant, effect of IBMX on cAMP levels, compared with the cilostamide-rolipram combination at 10 nM isoproterenol. This may suggest that PDE1 and PDE2 have some role under high cAMP levels, which is consistent with what has been shown previously in several other tissue types (Bronnikov et al., 1999; Beavo et al., 2007). Therefore, different PDEs can regulate different processes in BAT, but not all PDEs regulate all processes.
We next investigated which phosphodiesterase subtypes regulate adrenoceptor-mediated lipolysis and induction of UCP1 mRNA transcription. We found that PDE3 inhibition potentiated induction of UCP1 mRNA in response to 1 nM isoproterenol, whereas PDE1, PDE2, PDE4, and PDE8 inhibition did not (Fig. 3A; Supplemental Fig. 2C). This observation suggests that PDE3, but not PDE4, regulates a local pool of cAMP that is generated by the stimulation of β-adrenoceptors and leads to full induction of UCP1 mRNA. Because we did not observe changes in global cAMP accumulation at any of the various concentrations of isoproterenol and PDE inhibitors (Fig. 4), it is likely that the cAMP enzyme immunoassay method is not sensitive enough to detect small, local changes in cAMP concentration. However, under similar conditions, we were able to observe a potentiating effect on the PKA phosphorylation of CREB (Fig. 5, B and C) when isoproterenol and cilostamide were combined. This supports the model indicated by many investigators of compartmentalized cAMP signaling being critically regulated by specific PDEs—in this case, PDE3. In accordance with this finding, we observed that PDE4 inhibition potentiated lipolysis in primary brown adipocytes in response to 10 nM isoproterenol, whereas PDE3B and PDE8A inhibition did not (Fig. 6). Therefore, in contrast to a major role for PDE3 regulation of UCP1 induction in differentiated brown adipocytes, PDE4 seems to be the major regulator of lipolysis in brown adipocytes under adrenergic stimulation. These results suggest that PDE3 and PDE4 are, at least in part, in distinct compartments that are important for receptor-mediated signals, but not necessarily for basal adenylyl cyclase tone.
The similarities that we observed across two different brown adipocyte models with respect to PDE3 and PDE4 dual inhibition resulting in a stimulation of multiple BAT processes prompted us to test the significance of the paradigm in vivo. It has been shown that glucose uptake in brown adipocytes is a cAMP-dependent phenomenon and that IBMX can stimulate glucose uptake in isolated brown adipocytes (Marette and Bukowiecki, 1989). However, no study to date has determined which PDE subtype(s) regulate(s) this effect. If the PDE inhibitors stimulated basal BAT glucose uptake in the same pattern as they did for UCP1 and lipolysis, we expected injections of PDE3 or PDE4 inhibitors alone to have little, if any, effect on glucose uptake, but to have a substantial synergistic effect on glucose uptake when coinjected. Indeed, this is exactly what we observed (Fig. 7, A and B), indicating that PDE3 and PDE4, possibly working together, regulate/suppress basal glucose uptake into the BAT of wild-type mice in vivo.
The pathway(s) and mechanism(s) by which cAMP stimulates glucose uptake in BAT are controversial. It has been suggested that there may be an indirect response of the cell to the increased ATP demand because of the uncoupling process stimulated by lipolysis in rat brown adipocytes (Marette and Bukowiecki, 1991). Because PDE3 and PDE4 inhibition stimulates lipolysis to the same order of magnitude as IBMX in freshly isolated primary adipocytes, our data suggest that the injection of these isozyme selective inhibitors into mice might synergistically stimulate glucose uptake possibly via free fatty acid stimulation of UCP1. Furthermore, one study indicated that there was an increase in glucose transporters to the surface of primary isolated mouse brown adipocytes stimulated by cAMP-elevating agents (including IBMX) (Omatsu-Kanbe and Kitasato, 1992), and another study indicated that the cAMP effect was largely on the affinity of GLUT-1 receptors, and not on translocation, in differentiated adipocyte precursors isolated from the stromal fraction of mouse BAT (Shimizu et al., 1998). Despite the lack of a potentiating effect of the PDE8-selective inhibitor on either UCP1 mRNA expression or lipolysis (Figs. 3 and 6), we found that glucose uptake in response to PDE3-PDE4 dual inhibition was 1.8-fold higher in BAT from PDE8A−/− mice than BAT from their wild-type littermates (Fig. 7, A and B), whereas isoproterenol had no differential effect between the two genotypes (Fig. 7C). These results suggest that there may be at least two cAMP-dependent pathways that converge on glucose uptake: one mediated by lipolysis and uncoupling via PDE3 and PDE4 and a second mediated more chronically by the global ablation of PDE8A. Additional studies are needed to elucidate this mechanism, because the global knockout of PDE8A could indirectly affect brown fat activation via some other cell type.
cAMP-dependent glucose uptake is a major point of distinction between BAT and WAT, and therapeutically targeting BAT’s insulin-independent glucose uptake might prove to be beneficial in an insulin-resistant condition. This is further suggested by the observation that activated human BAT can take up and metabolize circulating fatty acids and glucose (Ouellet et al., 2012) and that obese persons can exhibit reduced thermogenesis and low responsiveness to noradrenaline (Jung et al., 1979). In addition, because human fat tissues of any type express a negligible number of β3-adrenoceptors, selective activation of the tissue with β3-adrenoceptor agonist approaches have failed in clinical trials. Therefore, alternative approaches that bypass receptor-mediated activation may be useful in the future.
A synergistic relationship between PDE3 and PDE4 has been described previously in other tissues, such as rat vascular smooth muscle cells (Palmer et al., 1998) and even in the regulation of lipolysis in white adipocyte models from mice (Snyder et al., 2005). In addition, dual PDE3/PDE4 inhibitors have been tested clinically for the treatment of chronic obstructive pulmonary disease with limited success. However, these drugs were fairly well tolerated in early-phase clinical trials with low incidence of adverse effects that are traditionally associated with effective doses of PDE4 inhibitors alone (Banner and Press, 2009). To our knowledge, the effects of these dual inhibitors on BAT function have not been determined.
In summary, understanding the regulation of the basal state of cAMP/PKA in BAT may prove to be critically important in the design of future therapies for disease states. In this case, it appears that there are multiple BAT PDEs that suppress tissue activation in the absence of agonists that drive cAMP production. Removing this inhibition with the correct combination of PDE inhibitors is sufficient to fully activate these processes. It is now well known that identifying small molecules that target only subsets of PDEs is possible from a medicinal chemistry point of view, and examples of single molecules that selectively inhibit both PDE3 and PDE4 have been described (reviewed in Bender and Beavo, 2006). However, it remains to be seen whether such an approach, perhaps involving a single molecule that inhibits multiple PDEs, might be efficacious in a clinical setting.
Supplementary Material
Acknowledgments
The authors thank Dr. Bruce Spiegelman for providing the immortalized brown adipocyte cell line; Pfizer, Inc., for providing the PDE8 inhibitor PF-04957325; Shering-Plough for providing the PDE1 inhibitor SCH51866; Dr. Masami Shimizu-Albergine, Dr. Sergei Rybalkin, Dr. Stan McKnight, and Claude Beltejar for critical reading of the manuscript; and Barbara Lewellen and Gregory Garwin for technical assistance in conducting PET experiments.
Abbreviations
- BAT
brown adipose tissue
- BAY 60-7550
2-(3,4-dimethoxybenzyl)-7-{(1R)-1-[(1R)-1-hydroxyethyl]-4-phenylbutyl}-5-methyllimidazo[5,1-f][1,2,4]triazin-4(3H)-one
- BSA
bovine serum albumin
- CREB
cAMP response element-binding protein
- DMEM
Dulbecco’s modified Eagle’s medium
- Epac
exchange protein activated by cAMP
- FDG
flurodeoxyglucose
- IBMX
isobutyl-1-methylxanthine
- PCR
polymerase chain reaction
- PDE
phosphodiesterase
- PKA
protein kinase
- SCH51866
(+)-cis-5,6a,7,8,9,9a-hexahydro-2-[4-[trifluro-methyl] phenylmethyl]-5-methyl-cylopent[4,5]imidazo [2,1-b]purin-4(3H)one
- SUV
standardized uptake value
- TBST
Tris-buffered saline containing 0.05% Tween-20
- UCP1
uncoupling protein-1
Authorship Contributions
Participated in research design: Kraynik, Miyaoka, Beavo.
Conducted experiments: Kraynik, Miyaoka.
Performed data analysis: Kraynik, Miyaoka, Beavo.
Wrote or contributed to the writing of the manuscript: Kraynik, Miyaoka, Beavo.
Footnotes
This work was supported by the University of Washington Department of Medicinal Chemistry, Pharmacological Sciences Training [Grant GM08392]; the UW Diabetes Research Center Samuel and Althea Stroum Endowed Diabetes Fellowship [P30 DK017047]; and the National Institutes of Health [Grants GM083926 and P30 CA15704].
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Banner KH, Press NJ. (2009) Dual PDE3/4 inhibitors as therapeutic agents for chronic obstructive pulmonary disease. Br J Pharmacol 157:892–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbe P, Millet L, Galitzky J, Lafontan M, Berlan M. (1996) In situ assessment of the role of the beta 1-, beta 2- and beta 3-adrenoceptors in the control of lipolysis and nutritive blood flow in human subcutaneous adipose tissue. Br J Pharmacol 117:907–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartelt A, Bruns OT, Reimer R, Hohenberg H, Ittrich H, Peldschus K, Kaul MG, Tromsdorf UI, Weller H, Waurisch C, et al. (2011) Brown adipose tissue activity controls triglyceride clearance. Nat Med 17:200–205 [DOI] [PubMed] [Google Scholar]
- Beavo J, Francis SH, Houslay MD. (2007) Cyclic nucleotide phosphodiesterases in health and disease, CRC Press/Taylor & Francis, Boca Raton [Google Scholar]
- Bender AT, Beavo JA. (2006) Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev 58:488–520 [DOI] [PubMed] [Google Scholar]
- Bronnikov GE, Zhang SJ, Cannon B, Nedergaard J. (1999) A dual component analysis explains the distinctive kinetics of cAMP accumulation in brown adipocytes. J Biol Chem 274:37770–37780 [DOI] [PubMed] [Google Scholar]
- Cannon B, Nedergaard J. (2004) Brown adipose tissue: function and physiological significance. Physiol Rev 84:277–359 [DOI] [PubMed] [Google Scholar]
- Cannon B, Nedergaard J. (2009) Thermogenesis challenges the adipostat hypothesis for body-weight control. Proc Nutr Soc 68:401–407 [DOI] [PubMed] [Google Scholar]
- Coudray C, Charon C, Komas N, Mory G, Diot-Dupuy F, Manganiello V, Ferre P, Bazin R. (1999) Evidence for the presence of several phosphodiesterase isoforms in brown adipose tissue of Zucker rats: modulation of PDE2 by the fa gene expression. FEBS Lett 456:207–210 [DOI] [PubMed] [Google Scholar]
- Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, Kuo FC, Palmer EL, Tseng YH, Doria A, et al. (2009) Identification and importance of brown adipose tissue in adult humans. N Engl J Med 360:1509–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng C, Paoloni-Giacobino A, Kuehne F, Boss O, Revelli JP, Moinat M, Cawthorne MA, Muzzin P, Giacobino JP. (1996) Respective degree of expression of beta 1-, beta 2- and beta 3-adrenoceptors in human brown and white adipose tissues. Br J Pharmacol 118:929–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunkern TR, Hatzelmann A. (2007) Characterization of inhibitors of phosphodiesterase 1C on a human cellular system. FEBS J 274:4812–4824 [DOI] [PubMed] [Google Scholar]
- Fedorenko A, Lishko PV, Kirichok Y. (2012) Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell 151:400–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann HM, Golozoubova V, Cannon B, Nedergaard J. (2009) UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab 9:203–209 [DOI] [PubMed] [Google Scholar]
- Gesta S, Tseng YH, Kahn CR. (2007) Developmental origin of fat: tracking obesity to its source. Cell 131:242–256 [DOI] [PubMed] [Google Scholar]
- Hansen RS, Beavo JA. (1982) Purification of two calcium/calmodulin-dependent forms of cyclic nucleotide phosphodiesterase by using conformation-specific monoclonal antibody chromatography. Proc Natl Acad Sci USA 79:2788–2792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidaka H, Kobayashi R. (1992) Pharmacology of protein kinase inhibitors. Annu Rev Pharmacol Toxicol 32:377–397 [DOI] [PubMed] [Google Scholar]
- Janssens K, Boussemaere M, Wagner S, Kopka K, Denef C. (2008) Beta1-adrenoceptors in rat anterior pituitary may be constitutively active. Inverse agonism of CGP 20712A on basal 3′,5′-cyclic adenosine 5′-monophosphate levels. Endocrinology 149:2391–2402 [DOI] [PubMed] [Google Scholar]
- Jensen J. (2007) More PKA independent beta-adrenergic signalling via cAMP: is Rap1-mediated glucose uptake in vascular smooth cells physiologically important? Br J Pharmacol 151:423–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung RT, Shetty PS, James WP, Barrand MA, Callingham BA. (1979) Reduced thermogenesis in obesity. Nature 279:322–323 [DOI] [PubMed] [Google Scholar]
- Larsen TM, Toubro S, van Baak MA, Gottesdiener KM, Larson P, Saris WH, Astrup A. (2002) Effect of a 28-d treatment with L-796568, a novel beta(3)-adrenergic receptor agonist, on energy expenditure and body composition in obese men. Am J Clin Nutr 76:780–788 [DOI] [PubMed] [Google Scholar]
- Liu X, Pérusse F, Bukowiecki LJ. (1998) Mechanisms of the antidiabetic effects of the beta 3-adrenergic agonist CL-316243 in obese Zucker-ZDF rats. Am J Physiol 274:R1212–R1219 [DOI] [PubMed] [Google Scholar]
- Marette A, Bukowiecki LJ. (1989) Stimulation of glucose transport by insulin and norepinephrine in isolated rat brown adipocytes. Am J Physiol 257:C714–C721 [DOI] [PubMed] [Google Scholar]
- Marette A, Bukowiecki LJ. (1991) Noradrenaline stimulates glucose transport in brown adipocytes by activating thermoenesis. Evidence that fatty acid activation of mitochondrial respiration enhances glucose transport. Biochem J 277:119–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthias A, Ohlson KB, Fredriksson JM, Jacobsson A, Nedergaard J, Cannon B. (2000) Thermogenic responses in brown fat cells are fully UCP1-dependent. UCP2 or UCP3 do not substitute for UCP1 in adrenergically or fatty scid-induced thermogenesis. J Biol Chem 275:25073–25081 [DOI] [PubMed] [Google Scholar]
- Murphy GJ, Kirkham DM, Cawthorne MA, Young P. (1993) Correlation of beta 3-adrenoceptor-induced activation of cyclic AMP-dependent protein kinase with activation of lipolysis in rat white adipocytes. Biochem Pharmacol 46:575–581 [DOI] [PubMed] [Google Scholar]
- Nolan MA, Sikorski MA, McKnight GS. (2004) The role of uncoupling protein 1 in the metabolism and adiposity of RII beta-protein kinase A-deficient mice. Mol Endocrinol 18:2302–2311 [DOI] [PubMed] [Google Scholar]
- Omatsu-Kanbe M, Kitasato H. (1992) Insulin and noradrenaline independently stimulate the translocation of glucose transporters from intracellular stores to the plasma membrane in mouse brown adipocytes. FEBS Lett 314:246–250 [DOI] [PubMed] [Google Scholar]
- Ouellet V, Labbé SM, Blondin DP, Phoenix S, Guérin B, Haman F, Turcotte EE, Richard D, Carpentier AC. (2012) Brown adipose tissue oxidative metabolism contributes to energy expenditure during acute cold exposure in humans. J Clin Invest 122:545–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer D, Tsoi K, Maurice DH. (1998) Synergistic inhibition of vascular smooth muscle cell migration by phosphodiesterase 3 and phosphodiesterase 4 inhibitors. Circ Res 82:852–861 [DOI] [PubMed] [Google Scholar]
- Rodbell M. (1964) Metabolism of Isolated Fat Cells. I. Effects of Hormones on Glucose Metabolism and Lipolysis. J Biol Chem 239:375–380 [PubMed] [Google Scholar]
- Rothwell NJ, Stock MJ. (1979) A role for brown adipose tissue in diet-induced thermogenesis. Nature 281:31–35 [DOI] [PubMed] [Google Scholar]
- Sell H, Berger JP, Samson P, Castriota G, Lalonde J, Deshaies Y, Richard D. (2004) Peroxisome proliferator-activated receptor gamma agonism increases the capacity for sympathetically mediated thermogenesis in lean and ob/ob mice. Endocrinology 145:3925–3934 [DOI] [PubMed] [Google Scholar]
- Shimizu Y, Satoh S, Yano H, Minokoshi Y, Cushman SW, Shimazu T. (1998) Effects of noradrenaline on the cell-surface glucose transporters in cultured brown adipocytes: novel mechanism for selective activation of GLUT1 glucose transporters. Biochem J 330:397–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder PB, Esselstyn JM, Loughney K, Wolda SL, Florio VA. (2005) The role of cyclic nucleotide phosphodiesterases in the regulation of adipocyte lipolysis. J Lipid Res 46:494–503 [DOI] [PubMed] [Google Scholar]
- Soderling SH, Bayuga SJ, Beavo JA. (1998) Cloning and characterization of a cAMP-specific cyclic nucleotide phosphodiesterase. Proc Natl Acad Sci USA 95:8991–8996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor WM, Mak ML, Halperin ML. (1976) Effect of 3′:5′-cyclic AMP on glucose transport in rat adipocytes. Proc Natl Acad Sci USA 73:4359–4363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uldry M, Yang W, St-Pierre J, Lin J, Seale P, Spiegelman BM. (2006) Complementary action of the PGC-1 coactivators in mitochondrial biogenesis and brown fat differentiation. Cell Metab 3:333–341 [DOI] [PubMed] [Google Scholar]
- Ursino MG, Vasina V, Raschi E, Crema F, De Ponti F. (2009) The beta3-adrenoceptor as a therapeutic target: current perspectives. Pharmacol Res 59:221–234 [DOI] [PubMed] [Google Scholar]
- van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JM, Kemerink GJ, Bouvy ND, Schrauwen P, Teule GJ. (2009) Cold-activated brown adipose tissue in healthy men. N Engl J Med 360:1500–1508 [DOI] [PubMed] [Google Scholar]
- Vang AG, Ben-Sasson SZ, Dong H, Kream B, DeNinno MP, Claffey MM, Housley W, Clark RB, Epstein PM, Brocke S. (2010) PDE8 regulates rapid Teff cell adhesion and proliferation independent of ICER. PLoS ONE 5:e12011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasta V, Shimizu-Albergine M, Beavo JA. (2006) Modulation of Leydig cell function by cyclic nucleotide phosphodiesterase 8A. Proc Natl Acad Sci USA 103:19925–19930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemulapalli S, Watkins RW, Chintala M, Davis H, Ahn HS, Fawzi A, Tulshian D, Chiu P, Chatterjee M, Lin CC, et al. (1996) Antiplatelet and antiproliferative effects of SCH 51866, a novel type 1 and type 5 phosphodiesterase inhibitor. J Cardiovasc Pharmacol 28:862–869 [DOI] [PubMed] [Google Scholar]
- Virtanen KA, Lidell ME, Orava J, Heglind M, Westergren R, Niemi T, Taittonen M, Laine J, Savisto NJ, Enerbäck S, et al. (2009) Functional brown adipose tissue in healthy adults. N Engl J Med 360:1518–1525 [DOI] [PubMed] [Google Scholar]
- Wang LC, Anholt EC. (1982) Elicitation of supramaximal thermogenesis by aminophylline in the rat. J Appl Physiol 53:16–20 [DOI] [PubMed] [Google Scholar]
- Weyer C, Gautier JF, Danforth E., Jr (1999) Development of beta 3-adrenoceptor agonists for the treatment of obesity and diabetes—an update. Diabetes Metab 25:11–21 [PubMed] [Google Scholar]
- Yoshida T, Sakane N, Wakabayashi Y, Umekawa T, Kondo M. (1994) Anti-obesity and anti-diabetic effects of CL 316,243, a highly specific beta 3-adrenoceptor agonist, in yellow KK mice. Life Sci 54:491–498 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.