

Abstract
Alterations in expression of the imprinted gene PHLDA2 are linked to low birth weight in both humans and the mouse. However birth weight is a summary measure of fetal growth and provides little information on the growth rate of the fetus in early and late pregnancy. To examine the relation of PHLDA2 expression with rates of fetal growth and explore associations with the infant's body composition in early childhood, we measured PHLDA2 mRNA levels in the term placenta of 102 infants whose mothers were participating in the Southampton Women's Survey (SWS). Higher PHLDA2 expression was associated with a lower fetal femur growth velocity between 19 and 34 weeks gestation. In addition, higher placental PHLDA2 gene expression was associated with a lower child's bone mineral content at four years of age, measured using dual-energy X-ray absorptiometry. The results suggest that placental PHLDA2 may provide a biomarker for suboptimal skeletal growth in pregnancies uncomplicated by overt fetal growth restriction.
Keywords: PHLDA2, Fetal growth, Bone mineral content, Imprinted genes, Placenta
Highlights
► We examined placental PHLDA2 expression in 102 pregnancies. ► Identified negative correlation between placental PHLDA2 and fetal femur growth. ► Identified negative correlation between placental PHLDA2 and bone density at 4 years. ► Placental PHLDA2 may provide a biomarker for suboptimal skeletal growth.
Introduction
The most common cause of low birth weight (LBW) at term in Western societies is placental insufficiency [1]. LBW infants have not attained their growth potential (reviewed in [2]) but in addition birth weight is an important indicator of both short and long term health. LBW infants have a 10–20 fold increased risk of dying in the perinatal period [3] and are at increased risk of developing chronic diseases including type 2 diabetes, hypertension and heart disease in later life (discussed in [4,5]). Postnatal ‘catch up’ growth is observed in 70–90% of LBW infants and is generally complete by two years of age [6,7]. It is now widely accepted that human fetuses are able to respond to a limited supply of nutrients by changing their physiology and metabolism but this predisposes to chronic disease in later life [8]. There is now extensive data from animal models to support this hypothesis [9–14].
Elevated expression of the human PHLDA2 gene has been reported in the term placentas of LBW infants in two independent studies [15,16]. In a study of routine, ultrasound-dated pregnancies, higher placental PHLDA2 expression was also shown to correlate with lower birth weight [17]. PHLDA2 belongs to a family of imprinted genes expressed from only the maternally-inherited allele [18]. In humans, PHLDA2 is expressed primarily in the placenta in the villous cytotrophoblast until term [19]. Similarly, expression in the mouse is predominantly placental [18,20,21]. In mice PHLDA2 gene knockout results in placentomegaly with an expansion of the junctional zone but has no apparent consequence for fetal weight, fetal viability or adult health [22]. In contrast, mice genetically engineered to over-express PHLDA2 show a reduced placental weight and there is reduced fetal growth late in gestation, suggesting placental insufficiency [23,24]. Data from the mouse model suggest that excess expression of PHLDA2 in the placenta of human LBW infants is not merely a consequence of a dysfunctional placenta but contributes to the reduction in growth. Determining placental PHLDA2 expression may therefore help identify infants who have not reached their full growth potential. Distinguishing these infants may allow targeted interventions early in life to optimize adult health.
In this study, we examined PHLDA2 expression in placentas of 102 infants born to mothers participating in the Southampton Women's Survey for whom there is detailed information about fetal growth and placental weight at term [25]. In addition to measurements of fetal growth velocity, we correlated placental expression of PHLDA2 with the infant's anthropometry, bone mass and body composition at birth (measured by dual-energy X-ray absorptiometry (DXA)) and, where data were available, bone mass and body composition in early childhood. We found no significant relationship between PHLDA2 expression and birth weight in this cohort, but there were relationships between higher placental PHLDA2 expression and lower femur growth rate between 19 and 34 weeks of gestation and lower bone mineral content at 4 years.
Subjects and methods
Cohort
Details of the Southampton Women's Survey (SWS) have been published previously [25]. In a group of pregnancies the placenta was collected within 30 min of delivery. The weight of the placenta was measured after removing any obvious blood clots, cutting the umbilical cord flush with its insertion into the placenta, trimming away surrounding membranes and removing the amnion from the basal plate. To ensure that samples collected were representative of the placenta as a whole, 5 villous tissue samples were selected using a stratified random sampling method and stored at − 80 °C. For this study, we selected 102 placentas (from 300 collected in total) based on availability of neonatal DXA data.
Fetal ultrasonography
In 58 pregnancies, measures of fetal size and growth velocity were available from ultrasound scans performed by a research sonographer at 19 and 34 weeks gestation. Using a high resolution ultrasound system (Kretz Voluson 730), head circumference (HC) was obtained using an ellipse superimposed on a static scan image of the horizontal plane at the level of the thalamus and the cavum septi pellucidi [26]. Abdominal circumference (AC) was also similarly measured using a transverse section of the fetal abdomen at the level of the fetal stomach and where a short section of umbilical vein can be identified. Femur length (FL) was measured in longitudinal section by placing the linear calipers at the ends of the diaphysis, with the femur horizontally positioned in the scan plane [26]. Three measurements were made of each parameter and the mean used in the statistical analysis [27]. Precision of the measurements was assessed by replicate examinations in 50 pregnancies at both 19 and 34 weeks. The coefficient of variation for triplicate linear measurements was 0.6% at 19 weeks and 0.4% at 34 weeks [27]. For elliptical measurements the values were 4.4% at 19 and 3.2% at 34 weeks.
Postnatal measurements
The infant's gestational age at birth was calculated from the date of the mother's last menstrual period (LMP) and confirmed by ultrasonography, or adjusted by an early dating scan if the LMP was unsure. Shortly after delivery, research midwives recorded neonatal anthropometric measures (including birth weight, head, abdominal and mid-upper arm circumferences and crown–heel length).
A subset of 42 mothers and children were invited to visit the Osteoporosis Centre at Southampton General Hospital for assessment of bone mass when the child was 4 years old. At this visit written informed consent for the DXA scan was obtained from the mother or father. The child's height (using a Leicester height measurer) and weight (in underpants only, using calibrated digital scales (Seca Ltd.)) were measured. A whole body DXA scan was obtained, using a Hologic Discovery instrument (Hologic Inc., Bedford, MA, USA). To encourage compliance, a sheet with appropriate colored cartoons was laid on the couch first; to help reduce movement artifact, the children were shown a suitable DVD cartoon. The total radiation dose for the scans was 4.7 microsieverts for whole body measurement (pediatric scan mode). The manufacturer's coefficient of variation (CV) for the instrument was 0.75% for whole body bone mineral density, and the experimental CV when a spine phantom was repeatedly scanned in the same position 16 times was 0.68%.
RNA extraction and cDNA synthesis
For each placenta 5 snap frozen samples were pooled and powdered in a frozen tissue press. Total RNA was extracted from 30 mg powdered placental tissue using the RNeasy fibrous tissue RNA isolation mini kit (Qiagen, UK) according to the manufacturer's instructions. The integrity of total RNA was confirmed by visualization of ribosomal bands with ethidium bromide under ultra violet illumination by agarose gel electrophoresis, in 1 × TAE buffer.
Total RNA (0.2 μg) was reverse transcribed with 0.5 μg random hexamer primer, 200 units M-MLV reverse transcriptase, 25 units recombinant RNasin ribonuclease inhibitor and 0.5 mM each of dATP, dCTP, dGTP and dTTP in a final reaction volume of 25 μl in 1 × MMLV reaction buffer (Promega, Wisconsin, USA). All 102 samples were produced in one batch to reduce variation.
Probe and primer design
Oligonucleotide probes and primers were designed using the Roche ProbeFinder version 2.45 for human. Probes were supplied by Roche from the human universal probe library and primers were synthesized by Eurogentec (Seraing, Belgium). PHLDA2: Forward 5′-atcacttggccagtttgctt-3′, Reverse 5′-gactggatgagggtgtcctg-3′, probe #3. Control genes (YWHAZ, UBC and TOP1) were selected using the geNormTM human Housekeeping Gene Selection Kit (Primer Design Limited, Southampton UK).
Real-time PCR using a Roche light-cycler 480. For Roche universal probe library probes the cycle parameters were 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. For primer design Perfect Probes the cycle parameters were 95 °C for 10 min, followed by 40 cycles of 95 °C for 10 s and 60 °C and 72 °C for 15 s. Intra-assay CV's for each gene were 5–8%. Each of the 102 samples was run on the same plate in triplicate. All mRNA levels are presented relative to the geometric mean of the three control genes.
PHLDA2 expression levels were quantified by Real-time PCR (QPCR) against three reference genes: tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, zeta polypeptide (YWHAZ), ubiquitin C (UBC) and topoisomerase (TOP1) [28].
Statistics
Summary data are presented as mean (SD) or median (inter-quartile range) depending on whether or not the data were normally distributed. Variables not normally distributed were transformed logarithmically. To investigate associations between PHLDA2 expression and parental body composition, fetal growth rates and infants body composition, Pearson's and Spearman's correlation coefficients were calculated where appropriate. Differences in PHLDA2 expression levels between different categories of maternal lifestyle were tested by t-test or one-way analysis of variance. Neonatal anthropometric measurements were adjusted for sex and gestational age and neonatal DXA measurements were adjusted for sex, gestational age and age at DXA. As there was a question regarding sex differences in mRNA levels between male and female placentas all mRNA data were adjusted for the sex of the baby [29]. Within group Z-scores were generated for femur length and abdominal circumference at 19 and 34 weeks. Royston models were fitted to fetal measurements to create z-scores for size and conditional growth rates [30]. To investigate whether there were sex differences in the relationship between PHLDA2 expression and the variables sex was included in regression analyses as appropriate and where an interaction was found data were analyzed separately by sex. Data were analyzed using Stata version 11.0 (Statacorp, Texas, USA).
Results
Descriptive statistics
In this study, PHLDA2 gene expression was examined in the placentas from 102 infants collected as part of the Southampton Women's Survey. All were singleton, term deliveries (37 weeks gestation or greater). 53 of the infants were male and 49 were female. Descriptive statistics are given in Table 1.
Table 1.
Pre-pregnant maternal and neonatal characteristics.
| Mean or median (SD or inter quartile range) | Number of subjects | |
|---|---|---|
| Maternal age (years) | 30.9 (3.9) | 102 |
| Maternal height (cm) | 162.3 (6.5) | 101 |
| Maternal body mass index (kg/cm2) | 25.2 (23.0–29.3) | 101 |
| Maternal mid upper arm circumference (cm) | 29.8 (27.2–32.7) | 101 |
| Maternal arm muscle area (cm2) | 36.7 (31.1–43.5) | 101 |
| Offspring's birth weight (g) | 3503 (453) | 102 |
| Placental weight (g) | 470 (96) | 101 |
| Birth weight/Placental weight ratio | 7.5 (6.8–8.4) | 101 |
| Neonatal abdominal circumference (cm) | 31.7 (1.8) | 102 |
| Neonatal crown heel length (cm) | 49.7 (1.8) | 102 |
| Neonatal mid upper arm circumference (cm) | 11.6 (1.0) | 102 |
Relationships between neonatal parameters and term placental expression of PHLDA2
Within this cohort of 102 infants, no association was found between the placental expression level of PHLDA2 and birth weight, placental weight or other neonatal anthropometric or body composition measurements at birth (Table 2).
Table 2.
Associations between placental PHLDA2 expression and neonatal parameters. N = 102.
|
PHLDA2 relative mRNA levels |
||
|---|---|---|
| r | P | |
| Birth weight | − 0.02 | 0.88 |
| Placental weight | 0.01 | 0.92 |
| Placental/birth weight ratio | 0.02 | 0.87 |
| Head circumference | − 0.03 | 0.78 |
| Abdominal circumference | 0.03 | 0.76 |
| Crown heel length | − 0.10 | 0.30 |
| Ponderal index | 0.09 | 0.36 |
| Mid upper arm circumference | 0.07 | 0.48 |
| Neonatal bone mineral content | 0.07 | 0.49 |
| Neonatal lean mass | − 0.03 | 0.79 |
| Neonatal fat mass | 0.07 | 0.46 |
Relationships between fetal growth parameters and term placental expression of PHLDA2
Longitudinal fetal ultrasound data was available at both 19 and 34 weeks for 58 fetuses within the cohort of 102 infants. There were no differences in the birth parameters between this subset of 58 pregnancies and the 43 pregnancies without full fetal scan data (data not shown). A lower 19–34 week femur length z-score change (linear growth velocity) was significantly associated with higher term placental PHLDA2 mRNA levels (Table 3, Fig. 1). Fetal femur length was not significantly related to placental PHLDA2 expression at 19 weeks of gestation but shorter fetal femur length at 34 weeks was associated with higher term placental expression of PHLDA2 (r = 0.35, P = 0.01) (Supplementary figure). This data suggests reduced femur growth velocity late in gestation in infants with a higher expression level of placental PHLDA2.
Table 3.
Associations between placental PHLDA2 expression and fetal parameters assessed by ultrasound. N = 59 at 19 weeks and 58 at 34 weeks. Significant correlations are highlighted in bold.
|
PHLDA2 relative mRNA levels |
||
|---|---|---|
| r | P | |
| 19 wk abdominal circumference z-score | 0.01 | 0.93 |
| 34 wk abdominal circumference z-score | 0.23 | 0.09 |
| 19–34 wk abdominal circumference growth velocity z-score | 0.28 | 0.04 |
| 19 wk Head circumference z-score | 0.15 | 0.27 |
| 34 wk head circumference z-score | 0.005 | 0.97 |
| 19–34 wk head circumference growth velocity z-score | − 0.05 | 0.71 |
| 19 wk femur length z-score | 0.13 | 0.34 |
| 34 wk femur length z-score | − 0.36 | 0.01 |
| 19–34 wk femur length growth velocity z-score | − 0.42 | 0.001 |
Fig. 1.
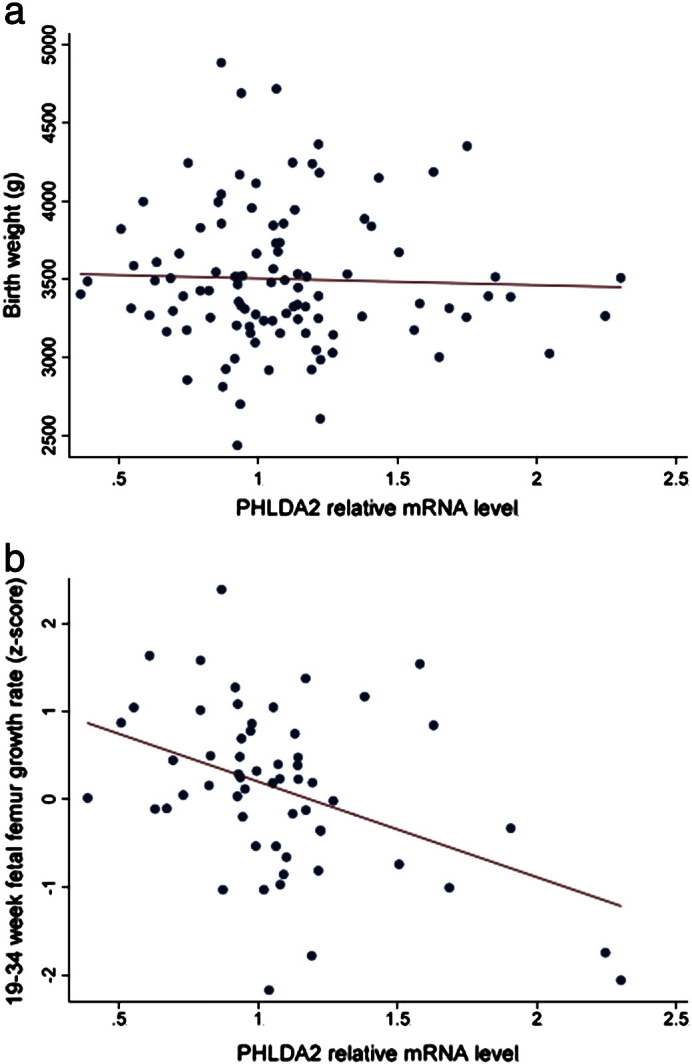
Placental PHLDA2 relative mRNA levels at birth compared to a) birth weight (r = − 0.02, P = 0.88) and b) 19–34 week fetal femur growth velocity (r = − 0.42, P = 0.001) (Royston femur length Z score) [30].
Supplementary figure.
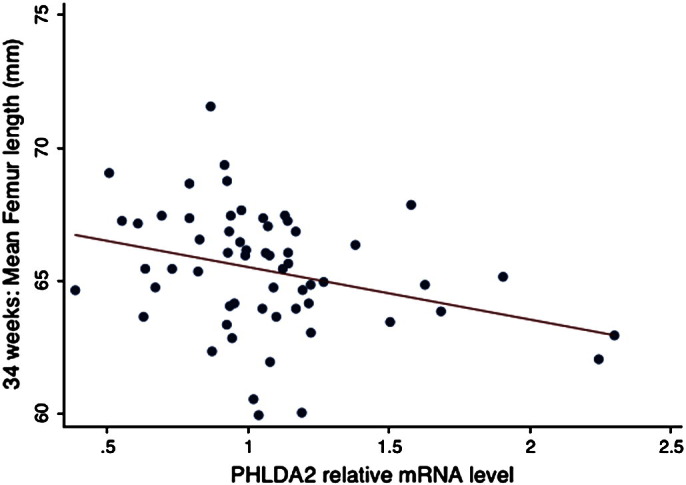
Scatter plot for absolute femur length at 34 weeks in mm. N = 58 subjects.
Placental expression of PHLDA2 was not related to fetal head circumference z-score at 19 weeks or 19–34 week fetal head circumference growth velocity (Table 3). Fetal abdominal circumference z-scores at 19 were not significantly related to placental expression of PHLDA2, but higher PHLDA2 expression was associated with a faster fetal abdominal circumference growth velocity between 19 and 34 weeks (Table 3).
Relationships between placental expression of PHLDA2 and childhood body composition at age 4 years
42 children in the study had DXA scans at age 4 years. There were no significant differences in the birth parameters collected for those who had DXA and those who did not (data not shown). In the 22 male and 20 female children followed to 4 years, there was an inverse relationship between placental PHLDA2 gene expression and bone mineral content, bone area and bone mineral density determined by DXA (Table 4). There were no significant correlations between either bone lean mass or fat mass and PHLDA2 expression when the data was analyzed by the sex of the infant or independently of sex (Table 4).
Table 4.
Associations between placental PHLDA2 expression and body composition assessed by DXA scan at age four years. N = 42. Significant correlations are highlighted in bold.
|
PHLDA2 relative mRNA levels |
||
|---|---|---|
| r | P | |
| Bone mineral content | − 0.38 | 0.01 |
| Bone area | − 0.392 | 0.010 |
| Bone mineral density | − 0.332 | 0.032 |
| Volumetric bone mineral density | − 0.147 | 0.353 |
| Lean mass | − 0.23 | 0.15 |
| Fat mass | − 0.20 | 0.21 |
| 4 year height | − 0.122 | 0.369 |
| 4 year weight (kg) | − 0.043 | 0.805 |
There were no significant interactions between PHLDA2 mRNA levels and sex with any fetal, neonatal or postnatal outcomes.
Parental parameters
Term placental PHLDA2 mRNA levels were not associated with maternal parity primiparous vs multiparous (values are mean (SD), 1.1 (0.4) vs 1.0 (0.4), P = 0.21), smoking (non-smoking 1.1 (0.3) vs smoking 1.2 (0.6), P = 0.18) or social class (social class I/II1.0 (0.3), IIIN/M1.2 (0.4), IV/V1.0 (0.3) P = 0.30) but levels were higher in mothers who, at recruitment to the study, reported that they undertook strenuous exercise compared to those who did not (1.2 (0.4) vs 1.0 (0.3). P = 0.02).
Term placental PHLDA2 mRNA levels were not associated with mother's own birth weight (r = 0.08, P = 0.47), height (r = − 0.05, P = 0.60), BMI (r = − 0.16, P = 0.11) or arm muscle area (r = − 0.10, P = 0.33). There were no significant interactions between term placenta PHLDA2 mRNA levels and sex for any maternal anthropometric outcomes. A lower paternal birth weight was associated with higher term placental PHLDA2 mRNA levels (R = − 0.35, P = 0.02).
Discussion
Our first key finding was the negative correlation between linear femur growth rate between 19 and 34 weeks of gestation and PHLDA2 expression in the term placenta in both male and female infants. A second finding was the negative correlation between bone mineral content at age 4 years and PHLDA2 expression in the term placenta.
Two independent studies have reported increased placental PHLDA2 expression at term in growth restricted infants [15,16]. In a third study, Apostolidou et al., reported a negative correlation between birth weight and placental PHLDA2 in 200 routine pregnancies [17]. We did not identify a significant negative trend between PHLDA2 expression in our 102 routine pregnancies which may suggest differences in the populations under study, differences in the way in which these measurements were taken or differences in the methodologies, for instance in our study we normalized to the geometric mean of three housekeeping genes which had been shown to be stably expressed in human placenta [28]. However, despite the lack of correlation with birth weight, we did identify a statistically significant negative correlation between PHLDA2 expression and linear growth rate in the 58 infants who underwent ultrasound scanning at 19 and 34 weeks. There was also a weak negative correlation between placental PHLDA2 and crown heel length at birth and also height at age 4, both of which might be anticipated to have a relationship with pre term femoral length. While these measurements did not reach significance, DXA data obtained at 4 years did reveal an inverse correlation between placental PHLDA2 expression and bone mineral content [31]. These data suggest that PHLDA2 may act to restrict skeletal growth and this restricted growth in utero has post natal consequences for skeletal integrity. As with birth weights, the lack of significant negative correlation of PHLDA2 expression with crown heel length at birth and height at age 4 might be explained by the imprecision of measurements taken on a single occasion over that obtained from scan data. It may well be that with a larger cohort these negative trends will achieve significance.
We have previously reported a direct causative effect between high PHLDA2 and late onset growth restriction in an animal model, which we attributed to placental insufficiency [23,24]. Data from an animal model employing bilateral uterine vessel ligation to mimic placental insufficiency suggests a link between suboptimal fetal growth and postnatal bone density [32]. PHLDA2 may act specifically to limit the transport of factors required for skeletal growth, for example by limiting calcium transport. Alternatively, PHLDA2 may indirectly affect calcium and bone metabolism through its role in regulating the placental hormones [24] involved in driving the maternal adaptations to pregnancy, which include increased maternal bone turnover. A third possibility is that PHLDA2 acts intrinsically to limit bone growth. PHLDA2 is expressed in human chondrocytes [33] and high expression of PHLDA2 has recently been reported in hypertrophic mouse chondrocytes, as compared to proliferative/resting chondrocytes [34]. Animal models will play an important role in distinguishing between an intrinsic or extrinsic mechanism for limiting skeletal growth. Whatever the mechanism turns out to be, we have identified a potential role for PHLDA2 in restricting early skeletal growth, which has post natal consequences for skeletal integrity.
In contrast to the negative relationship with fetal femur growth, PHLDA2 was positively associated with change in abdominal circumference from 19 to 34 weeks. There is much evidence to suggest that different fetal compartments may be differentially regulated and thus growth may not be concordant across these measurements. Femur length may be taken as a proxy for linear growth of the skeleton (crown rump or crown heel length are not measurable by ultrasound in late pregnancy); in contrast abdominal circumference is a composite measure of liver size and thickness of subcutaneous adipose tissue, potentially involving hormones such as IGF-1 and leptin [35,36]. There is no reason to suppose therefore, that femur length and abdominal circumference will relate in the same direction to a single regulator; indeed, we have previously demonstrated differences in relationships between postnatal skeletal indices and femur length compared with abdominal circumference growth in utero [31]. These results support the notion that birth weight is a relatively crude surrogate for fetal developmental and that a more detailed measurement of individual markers of fetal growth may give a more accurate assessment of the regulation of development in utero.
A key question is what drives deregulated expression of PHLDA2? In rodent models PHLDA2 responds to suboptimal in utero environments. Specifically, increased placental expression of PHLDA2 has been reported in response to hypoxia during pregnancy, decreased food consumption and maternal alcohol consumption [37,38]. In this study, we noted that PHLDA2 expression was higher in mothers who reported that they undertook strenuous exercise. A more extensive study will be critical in determining the relevance of this observation. Lower paternal birth weight was also associated with higher term placental PHLDA2 mRNA levels. PHLDA2 is imprinted and it is the paternally-inherited copy that is silenced. There is currently no evidence for full loss of imprinting of PHLDA2 in low birth weight pregnancies [15,16] but increased expression could occur as a consequence of the failure of the paternal genome to fully silence PHLDA2. In which case, exploring the relationship between both maternal and paternal lifestyles will be important.
Conclusion
In summary, higher expression of the placental growth regulator, PHLDA2, was associated with lower fetal femur growth velocity between 19 and 34 weeks gestation in fetuses who are within a normal birth weight range at birth. This suggests that the correct dosage of PHLDA2 may be critical for optimal skeletal growth in the third trimester of pregnancy. Alterations in bone mineral content suggest that high placental PHLDA2 may have long-term consequences for bone health. Different early life growth trajectories influence adult health and the identification of infants who have experienced sub-optimal growth using a molecular marker rather than by birth weight alone may be helpful in determining where to apply interventional strategies to improve long-term health.
The following are the supplementary materials related to this article.
Acknowledgments
We thank the families who took part in the Southampton Women's Survey (SWS) and the SWS research staff. This work was supported by the Medical Research Council, University of Southampton, the British Heart Foundation (MH), the Food Standards Agency (contract NO5049), the National Institute for Health Research (KMG) and Cardiff University (RMJ).
The author contributions: RMJ, RML and MAH designed and instigated the study of PHLDA2 in the Southampton Women's Survey placentas. CC, HMI, KG, NCH, SMR designed and/or implemented aspects of the Southampton Women's Survey within which the tissues were collected and pregnancy and postnatal measurements were made. RML and JKC collected the tissues and undertook the PCR analysis of gene expression. PAM undertook fetal ultrasound data. GN, SRC and HMI undertook the statistical analysis. All authors were involved in the preparation of the manuscript and approving the final version. RMJ takes responsibility for the integrity of the data analysis.
Edited by: Bjorn Olsen
Footnotes
Conflict of interest: The authors have no financial interests, direct or indirect, that might be construed as affecting the conduct or reporting of the work they have submitted.
Contributor Information
R.M. Lewis, Email: Rohan.Lewis@soton.ac.uk.
J.K. Cleal, Email: j.k.cleal@soton.ac.uk.
G. Ntani, Email: gn@mrc.soton.ac.uk.
S.R. Crozier, Email: src@mrc.soton.ac.uk.
P.A. Mahon, Email: p.mahon@soton.ac.uk.
S.M. Robinson, Email: smr@mrc.soton.ac.uk.
N.C. Harvey, Email: nch@mrc.soton.ac.uk.
C. Cooper, Email: cc@mrc.soton.ac.uk.
H.M. Inskip, Email: h.m.inskip@mrc.soton.ac.uk.
K.M. Godfrey, Email: kmg@mrc.soton.ac.uk.
M.A. Hanson, Email: m.hanson@soton.ac.uk.
R.M. John, Email: JohnRM@cf.ac.uk.
References
- 1.Henriksen T., Clausen T. The fetal origins hypothesis: placental insufficiency and inheritance versus maternal malnutrition in well-nourished populations. Acta Obstet Gynecol Scand. 2002;81:112–114. doi: 10.1034/j.1600-0412.2002.810204.x. [DOI] [PubMed] [Google Scholar]
- 2.Brodsky D., Christou H. Current concepts in intrauterine growth restriction. J Intensive Care Med. 2004;19:307–319. doi: 10.1177/0885066604269663. [DOI] [PubMed] [Google Scholar]
- 3.Monk D., Moore G.E. Intrauterine growth restriction — genetic causes and consequences. Semin Fetal Neonatal Med. 2004;9:371–378. doi: 10.1016/j.siny.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 4.Barker D.J. The developmental origins of well-being. Philos Trans R Soc Lond B Biol Sci. 2004;359:1359–1366. doi: 10.1098/rstb.2004.1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanson M.A. Cambridge University Press; 2006. Developmental origins of health and disease. [Google Scholar]
- 6.Ong K.K. Catch-up growth in small for gestational age babies: good or bad? Curr Opin Endocrinol Diabetes Obes. 2007;14:30–34. doi: 10.1097/MED.0b013e328013da6c. [DOI] [PubMed] [Google Scholar]
- 7.Hokken-Koelega A.C., De Ridder M.A., Lemmen R.J., Den Hartog H., De Muinck Keizer-Schrama S.M., Drop S.L. Children born small for gestational age: do they catch up? Pediatr Res. 1995;38:267–271. doi: 10.1203/00006450-199508000-00022. [DOI] [PubMed] [Google Scholar]
- 8.Barker D.J. The fetal and infant origins of adult disease. BMJ. 1990;301:1111. doi: 10.1136/bmj.301.6761.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gluckman P.D., Hanson M.A. The developmental origins of the metabolic syndrome. Trends Endocrinol Metab. 2004;15:183–187. doi: 10.1016/j.tem.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 10.Cottrell E.C., Ozanne S.E. Developmental programming of energy balance and the metabolic syndrome. Proc Nutr Soc. 2007;66:198–206. doi: 10.1017/S0029665107005447. [DOI] [PubMed] [Google Scholar]
- 11.Vuguin P.M. Animal models for small for gestational age and fetal programming of adult disease. Horm Res. 2007;68:113–123. doi: 10.1159/000100545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nathanielsz P.W. Animal models that elucidate basic principles of the developmental origins of adult diseases. ILAR J. 2006;47:73–82. doi: 10.1093/ilar.47.1.73. [DOI] [PubMed] [Google Scholar]
- 13.Armitage J.A., Khan I.Y., Taylor P.D., Nathanielsz P.W., Poston L. Developmental programming of the metabolic syndrome by maternal nutritional imbalance: how strong is the evidence from experimental models in mammals? J Physiol. 2004;561:355–377. doi: 10.1113/jphysiol.2004.072009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin-Gronert M.S., Ozanne S.E. Experimental IUGR and later diabetes. J Intern Med. 2007;261:437–452. doi: 10.1111/j.1365-2796.2007.01800.x. [DOI] [PubMed] [Google Scholar]
- 15.McMinn J., Wei M., Schupf N., Cusmai J., Johnson E.B., Smith A.C. Unbalanced placental expression of imprinted genes in human intrauterine growth restriction. Placenta. 2006;27:540–549. doi: 10.1016/j.placenta.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 16.Diplas A.I., Lambertini L., Lee M.J., Sperling R., Lee Y.L., Wetmur J. Differential expression of imprinted genes in normal and IUGR human placentas. Epigenetics. 2009;4:235–240. doi: 10.4161/epi.9019. [DOI] [PubMed] [Google Scholar]
- 17.Apostolidou S., Abu-Amero S., O'Donoghue K., Frost J., Olafsdottir O., Chavele K.M. Elevated placental expression of the imprinted PHLDA2 gene is associated with low birth weight. J Mol Med. 2007;85:379–387. doi: 10.1007/s00109-006-0131-8. [DOI] [PubMed] [Google Scholar]
- 18.Qian N., Frank D., O'Keefe D., Dao D., Zhao L., Yuan L. The IPL gene on chromosome 11p15.5 is imprinted in humans and mice and is similar to TDAG51, implicated in Fas expression and apoptosis. Hum Mol Genet. 1997;6:2021–2029. doi: 10.1093/hmg/6.12.2021. [DOI] [PubMed] [Google Scholar]
- 19.Saxena A., Morozov P., Frank D., Musalo R., Lemmon M.A., Skolnik E.Y. Phosphoinositide binding by the pleckstrin homology domains of Ipl and Tih1. J Biol Chem. 2002;277:49935–49944. doi: 10.1074/jbc.M206497200. [DOI] [PubMed] [Google Scholar]
- 20.Frank D., Mendelsohn C.L., Ciccone E., Svensson K., Ohlsson R., Tycko B. A novel pleckstrin homology-related gene family defined by Ipl/Tssc3, TDAG51, and Tih1: tissue-specific expression, chromosomal location, and parental imprinting. Mamm Genome. 1999;10:1150–1159. doi: 10.1007/s003359901182. [DOI] [PubMed] [Google Scholar]
- 21.Dunwoodie S.L., Beddington R.S. The expression of the imprinted gene Ipl is restricted to extra-embryonic tissues and embryonic lateral mesoderm during early mouse development. Int J Dev Biol. 2002;46:459–466. [PubMed] [Google Scholar]
- 22.Frank D., Fortino W., Clark L., Musalo R., Wang W., Saxena A. Placental overgrowth in mice lacking the imprinted gene Ipl. Proc Natl Acad Sci U S A. 2002;99:7490–7495. doi: 10.1073/pnas.122039999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salas M., John R., Saxena A., Barton S., Frank D., Fitzpatrick G. Placental growth retardation due to loss of imprinting of PHLDA2. Mech Dev. 2004;121:1199–1210. doi: 10.1016/j.mod.2004.05.017. [DOI] [PubMed] [Google Scholar]
- 24.Tunster S.J., Tycko B., John R.M. The imprinted PHLDA2 gene regulates extraembryonic energy stores. Mol Cell Biol. 2010;30:295–306. doi: 10.1128/MCB.00662-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Inskip H.M., Godfrey K.M., Robinson S.M., Law C.M., Barker D.J., Cooper C. Cohort profile: the Southampton Women's Survey. Int J Epidemiol. 2006;35:42–48. doi: 10.1093/ije/dyi202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Loughna P., Chitty L., Evans T., Chudleigh T. Fetal size and dating: charts recommended for clinical obstetric practice. Ultrasound. 2009;17:161–167. [Google Scholar]
- 27.Mahon P.A., Cooper C., Crozier S.R., Godfrey K.M. The use of 3D ultrasound to investigate fetal bone development. Norsk Epidemiologi. 2009;19:45–52. [Google Scholar]
- 28.Cleal J.K., Day P., Hanson M.A., Lewis R.M. Measurement of housekeeping genes in human placenta. Placenta. 2009;30:1002–1003. doi: 10.1016/j.placenta.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 29.Cleal J.K., Day P.L., Hanson M.A., Lewis R.M. Sex differences in the mRNA levels of housekeeping genes in human placenta. Placenta. 2010;31:556–557. doi: 10.1016/j.placenta.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 30.Royston P. Calculation of unconditional and conditional reference intervals for foetal size and growth from longitudinal measurements. Stat Med. 1995;14:1417–1436. doi: 10.1002/sim.4780141303. [DOI] [PubMed] [Google Scholar]
- 31.Harvey N., Mahon P., Robinson S., Nisbet C., Javaid M., Crozier S. Different indices of fetal growth predict bone size and volumetric density at 4 years old. J Bone Miner Res. 2010;25:920–927. doi: 10.1359/jbmr.091022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Romano T., Wark J.D., Owens J.A., Wlodek M.E. Prenatal growth restriction and postnatal growth restriction followed by accelerated growth independently program reduced bone growth and strength. Bone. 2009;45:132–141. doi: 10.1016/j.bone.2009.03.661. [DOI] [PubMed] [Google Scholar]
- 33.Andreas K., Lubke C., Haupl T., Dehne T., Morawietz L., Ringe J. Key regulatory molecules of cartilage destruction in rheumatoid arthritis: an in vitro study. Arthritis Res Ther. 2008;10:R9. doi: 10.1186/ar2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ulici V., James C.G., Hoenselaar K.D., Beier F. Regulation of gene expression by PI3K in mouse growth plate chondrocytes. PLoS One. 2010;5:e8866. doi: 10.1371/journal.pone.0008866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu J.L., Yakar S., LeRoith D. Conditional knockout of mouse insulin-like growth factor-1 gene using the Cre/loxP system. Proc Soc Exp Biol Med. 2000;223:344–351. doi: 10.1046/j.1525-1373.2000.22349.x. [DOI] [PubMed] [Google Scholar]
- 36.Yuen B.S., Owens P.C., Muhlhausler B.S., Roberts C.T., Symonds M.E., Keisler D.H. Leptin alters the structural and functional characteristics of adipose tissue before birth. FASEB J. 2003;17:1102–1104. doi: 10.1096/fj.02-0756fje. [DOI] [PubMed] [Google Scholar]
- 37.Shukla P.K., Sittig L.J., Ullmann T.M., Redei E.E. Candidate placental biomarkers for intrauterine alcohol exposure. Alcohol Clin Exp Res. 2011;35:559–565. doi: 10.1111/j.1530-0277.2010.01373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tomlinson T.M., Garbow J.R., Anderson J.R., Engelbach J.A., Nelson D.M., Sadovsky Y. Magnetic resonance imaging of hypoxic injury to the murine placenta. Am J Physiol Regul Integr Comp Physiol. 2011;298:R312–R319. doi: 10.1152/ajpregu.00425.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]