

Abstract
To date, only a small number of commercial chemicals have been tested and documented as developmental neurotoxicants. Moreover, an increasing number of epidemiological, clinical and experimental studies suggest an association between toxicant or drug exposure during the perinatal period and the development of metabolic-related diseases and neurotoxicity later in life. The four speakers in this symposium presented their research results on different neurotoxic chemicals as they relate to the developmental origins of health and adult disease (DOHaD). Philippe Grandjean presented epidemiological data on children exposed to methylmercury and discussed the behavioral outcome measures as they relate to age and stage of brain development. Donald A. Fox presented data that low-to-moderate dose human equivalent gestational lead exposure produced late-onset obesity, and motor and coordination dysfunction only in male mice. Didima de Groot discussed the role of caloric restriction and/or high fat diets during gestation and/or postnatal development in mediating the metabolic and neurotoxic effects of developmental methylmercury exposure in rats. Merle G. Paule addressed the long-term changes in learning, motivation and short-term memory in aged Rhesus monkeys following 24 hour exposure to ketamine during early development. Overall, these presentations addressed fundamental issues in the emerging areas of lifetime neurotoxicity testing, differential vulnerable periods of exposure, nonmonotonic dose-response effects and neurotoxic risk assessment.
Keywords: methylmercury, lead, ketamine, DOHaD, behavior, obesity
Introduction
To date, only a small number of commercial chemicals have been tested and documented as developmental neurotoxicants. Moreover, an increasing number of epidemiological, clinical and experimental studies suggest an association between toxicant or drug exposure during the perinatal period and the development of metabolic-related diseases and neurotoxicity later in life. Compelling epidemiological, pharmacological and toxicological evidence shows that there are several vulnerable periods of growth and development during which environmental interactions with the immune system and genome increase susceptibility to diseases during aging [1-4]. These findings define the Developmental Origins of Health and Disease (DOHaD) paradigm. The gestational period is particularly sensitive to altered nutritional and hormonal homeostasis, chemical/toxicant exposures and increased proinflammatory cytokines [4-9].
Epidemiological Evidence of a Developmental Window of Susceptibility to Neurotoxicant Exposure: Age-Dependent Outcomes (Philippe Grandjean)
Among the small number of environmental chemicals that are known to cause developmental neurotoxicity, methylmercury (MeHg) has an interesting, if not dramatic history, which is still unfolding (Table 1). As MeHg usually originates from seafood, the prenatal exposure is generally much higher than early postnatal exposures. The most serious consequences occurred first in Minamata, Japan, in the 1950s. However, not until 2003 was international consensus achieved [10], that exposure limits for this neurotoxicant should aim at protecting the developing brain, rather than the nervous system in general, irrespective of age. During recent decades, discussions focused on several sources of uncertainty, which initially were thought capable of erroneously inflating the apparent neurotoxicity findings. These concerns were clarified.
One issue is the extent and the implications of imprecise exposure assessments in epidemiological studies. Some exposure biomarkers may have a relative imprecision as high as 50%, thereby substantially biasing the dose-effect relationships toward the null [11]. Along with my colleagues in the Faroe Islands and our international partners, we carried out prospective studies of birth cohorts to document the exposure-associated neurobehavioral deficits at different ages. Our first cohort was examined at 22 years of age. Through this research, we addressed several important concerns, including the significance of the age at outcome assessment, the sensitivity of tests to MeHg neurotoxicity, and possible differences in vulnerability. Our results supported the choice of early school age (i.e., 7 years) as an appropriate stage of development for neurobehavioral assessment. We found that for each doubling in MeHg exposure, the child’s brain development was delayed for 1-2 months of development: with some differences between domains. Subsequent studies have been in agreement with this initial finding and indicated that the deficits are likely to be permanent. This conclusion is also in agreement with a study on functional MR scanning during simple tasks, where subjects highly exposed to MeHg prenatally revealed abnormalities in brain areas activated [12]. As these changes were present at adolescence, it seems likely that developmental MeHg neurotoxicity is permanent. That does not mean that compensation may occur.
In structural equation models, Esben Budtz-Jørgensen compared the difference in performance between ages 7 and 14 years (unpublished results). The tendencies only approached statistical significance. However, it appeared that those subjects who performed well at age 7 years, no matter their mercury exposure level, improved more than the cohort average during the subsequent 7 years. The opposite was true for subjects with a poor performance at age 7. These findings suggest that the distribution of cognitive skills may not be moved to the left by a neurotoxic exposure, but it may rather be squeezed toward the left, thus preserving most of the children with initial high performance levels.
Another issue of concern was that known or unknown confounders could affect the mercury-associated effects seen in the Faroes and New Zealand. Perhaps, co-exposure to PCBs, another marine pollutant, could cause some of the neurotoxicity observed. However, adjustment of the Faroes data for PCB exposure did not eliminate the mercury effects [11]. Recent data from the Faroes support this conclusion. If these effects were explained by another pollutant (or other confounder, whether chemical, social or genetic), then this parameter should be more closely associated with the cord-blood Hg concentration than with the maternal hair-Hg to cause the greater effect estimates for the cord-blood level than for the hair level. The confounder also should be a better risk indicator when mothers with variable mercury exposure were excluded. It is difficult to imagine a confounder that would satisfy these requirements.
An additional concern involved so-called negative confounding, as MeHg exposure usually originates from fish and seafood that also contains essential nutrients [13]. Thus, the competing effects of mercury and nutrients must be ascertained so that properly adjusted measures could be generated for each component’s effects on the relevant outcomes and neither of them was underestimated. In the Faroes, adjustment for maternal fish intake during pregnancy only resulted in small increases in the calculated MeHg toxicity [14]. The limited effect was due to the poor correlation between fish intake and mercury exposure biomarkers. Information has now become available from the Seychelles [15], where mutual adjustment for fish intake and MeHg exposure revealed effects of both – in opposite directions – while neither had clear effects without the adjustment. In this case, one could conclude that the mercury exposure deprived the child of the benefits from the seafood nutrients.
Overall, these issues were crucial to the proper appreciation of the dose-response relationships and calculation of exposure limits. Taking into account imprecision and negative confounding would likely cut the lowest current exposure limit – the one used by the US Environmental Protection Agency – by 50% or more [16]. In addition to underestimating the developmental neurotoxicity, the increased risk incurred during fetal development was recognized with a delay. Although evidence was first published in 1952 that MeHg was a developmental neurotoxicant, international consensus and regulation only was reached 50 years later (Table 1). Thus, the evolution of insights into MeHg neurotoxicity demonstrates the challenges in documenting neurodevelopmental deficits due to prenatal neurotoxicant exposure [17].
Low-Level Human Equivalent Gestational Lead Exposure Is A Risk Factor for Late-Onset Metabolic Syndrome and Neurodegeneration in Humans and Animals (Donald A. Fox)
The incidence of obesity in developed nations has reached epidemic proportions but common sense causes such as increased consumption of calorically-laden foods and decreased physical activity do not completely account for it [18,19]. The DOHaD Hypothesis states that fetal and neonatal reprogramming of metabolism significantly contributes to an increased risk of cardiovascular and neurodegenerative diseases as well as obesity and related metabolic disorders in adulthood [20-24]. Several synthetic organic and inorganic environmental toxicants appear capable of such reprogramming [18,19,25,26]. Heavy metals represent one such class of toxicant since fetal exposure to tin compounds or lead and cadmium produce significantly increased offspring weight on the day of birth, [27,28]. Epidemiological studies reveal persistent increases in body mass index (BMI) during childhood and subsequently young adulthood following moderate-level gestational lead exposure (GLE) and postnatal lead exposure. Similarly, the adverse effects of developmental lead exposure on cognitive, auditory and retinal function, visual-motor function, and motor function and coordination are well-documented in children and developing experimental animals [29-42]. However, the long-term effects of GLE or early postnatal lead exposure on these measures are mostly unexplored [8,43].
The experiments described herein were designed to investigate the long-term effects of low- (peak blood lead concentrations ([BPb])≤10 μg/dL), moderate- (peak [BPb]>10 and ≤25 μg/dL) and high-level (peak [BPb] >26 μg/dL) human equivalent gestational lead exposure on year-old C57BL/6 male and female mice. Gender differences were examined because early developmental lead exposure produces an increased risk for attention, visual motor and fine-motor deficits in males [44-47] and male and female animals exhibit differences in metal disposition and lead neurotoxicity [48,49]. C57BL/6 female mice were exposed to low (27 ppm), moderate (55 ppm) or high (109 ppm) lead containing drinking water throughout gestation and until postnatal day 10 (PN10). [BPb] in control, low-, moderate- and high-dose GLE mice was ≤1, ≤10, ~25 and ~40 μg/dL, respectively, on PN10 and by PN30 and at 12 months of age all were ≤1 μg/dL as described [8,42]. Water, food, and dams’ weight as well as offspring measures were monitored throughout pretreatment, mating and pregnancy, after delivery and until tissue collection at PN60 (adult). No significant treatment-related differences were observed [44]. Four major outcome measures were assessed: body weight, cumulative Wahman running wheel activity for five consecutive nights, cumulative spontaneous activity during 30 minutes in an Optovarimax behavioral monitor after 15 minutes of acclimation, motor coordination assessed on a Columbus rotarod after training as described [44].
GLE had no significant effect on male or female body weight at PN0, PN10, PN60 or PN180, although by PN60 males in all groups weighed significantly more than females [44]. In contrast, Figure 1A shows that year-old low-dose (+26%), moderate-dose (+21%) and high-dose (+13%) male GLE mice weighed significantly more than age-matched controls [8]. Moreover, GLE produced nonmonotonic dose-dependent responses since the alterations were consistently larger in the low-dose, than high-dose, GLE group. These responses are characteristic of inverted U-shaped dose-response curves often observed in lead neurotoxicity studies [50]. Interestingly, as described [8], there were no significant treatment-related effects of GLE on the body weight of 12 month-old female mice (Figure 1A). Thus, GLE produced late-onset obesity in male, but not female, mice. The gender selective effect is consistent with reports that boys have increased susceptibility to neurobehavioral alterations and cognitive deficits produced by low- to moderate-level GLE compared to age-matched girls [44,45,47].
Figure 1. Brain activity assessed with 18F-FDG microPET imaging of cerebellum.
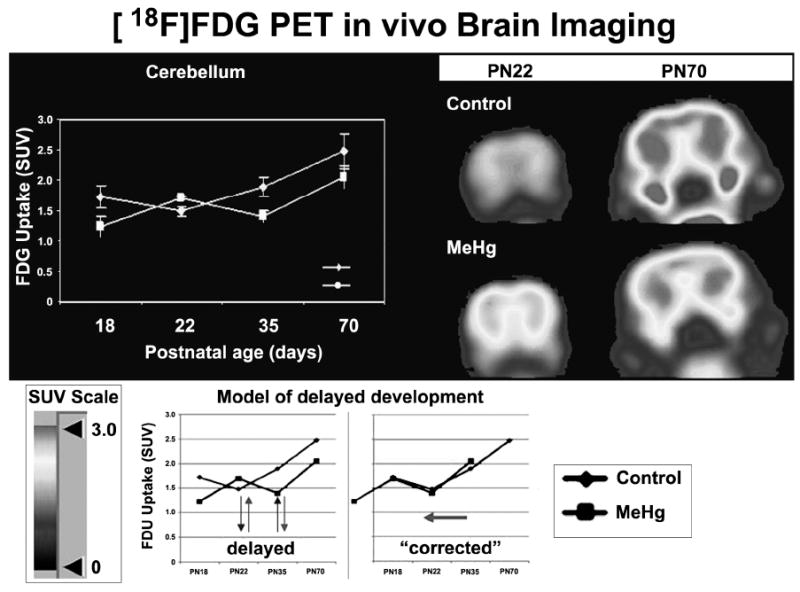
Brains of F1-rats prenatally exposed to MeHg (1.5 mg MeHg/kg body weight/day by gavage from gestational day 6 to postnatal day 10. Control rats received corn oil. The FDG uptake patterns in brain samples exposed to MeHg show a phase-shift compared to control samples, suggesting a delay in development and/or onset of brain activity. On PND 61 a persisting overall decrement in brain activity was observed. Abbreviations: 18F fluorindeoxyglucose; SUV, Standard Uptake Value; F1, first generation offspring; MeHg, methylmercury.
Since 12 month-old GLE males weighed more than age-matched control males (Figure 1A), the possibility that excess body weight made the GLE males less active was investigated. This hypothesis was not confirmed as there were no treatment-related differences in male running wheel activity [44]. However, 12 month-old male mice in the low-dose and high-dose GLE groups exhibited significantly less cumulative spontaneous activity than age-matched controls: -52% and -35%, respectively (Figure 1B: left panel). The low-dose GLE males were significantly less active than high-dose GLE males. In contrast, there were no treatment-related differences for female mice (Figure 1B: left panel).
To investigate the long-term effects of GLE on inter-limb balance and motor coordination, rotarod behavior was assessed. The mean latency to fall from the rotarod decreased significantly in 12 month-old GLE males compared to age-matched controls: -61% and -28%, respectively (Figure 1B: right panel). In contrast, the mean latency to fall from the rotarod was not significantly different in 12 month-old female control and GLE mice (Figure 1B: right panel). These curvilinear results in 12 month-old male GLE mice are consistent with “negative” dose-response curves since high-dose GLE produced less deviation from control than did low-dose GLE [51]. Consistent with the negative dose-response curve, rotarod activity was unchanged in adult male rats exposed to 5- to 40-fold higher lead levels during gestation and lactation or lifetime [51,52].
The most compelling finding is that GLE is a delayed obesogen and that the weight gain was greater in low- and moderate-dose, than high-dose, GLE mice. These nonmonotonic obesogenic effects appear selective for GLE since low- or moderate-level postnatal lead exposure with similar [BPb] did not alter body weights in developing or 12 month-old mice [8], lifetime exposure to 5- to 20-fold higher lead levels did not affect body weight of 14 month-old male or female rats [53].
The rotarod results are reminiscent of the poorer fine motor control, visual motor function and postural balance found in children, adolescents and young adults with low-to-moderate developmental lead exposure [36,44-47]. Interestingly, there is an increased risk of neuromotor deficits in males [47]. Our results suggest that low-level GLE contributes to persistent neuromotor and balance deficits, and is a risk factor for injuries in older males.
In summary, these results demonstrate that GLE mice with peak [BPb] ≤10 μg/dL, the current low-level of concern [54], have permanent sex-specific motor abnormalities and late-onset obesity. The nonmonotonic dose-dependent responses reveal that low-level GLE produces the most adverse effects. These data raise complex issues for risk assessment and indicate that lifetime measures of dose-response toxicant exposure should be a component of the neurotoxic risk assessment process.
Maternal Diet Contributes to Metabolic Syndrome-Like Diseases Following Early Mercury Exposure: A Rat Model (Didima de Groot)
Animal studies have shown that early exposure to chemicals may reprogram (neuro)physiological set-points and thereby contribute to metabolic syndrome-like diseases later in life. Moreover, these studies suggest that maternal nutrition may enhance the effects of chemical exposure. Results of an epidemiological study on a cohort of individuals born in the north of Holland during or shortly after the period of extreme famine in the winter of 1944-1945 (the Dutch Famine model) show an increased incidence of obesity, diabetes, cardiovascular and neurodegenerative diseases (schizophrenia; stress sensitivity) in the adult life [55-64]. These higher incidences were associated with the highly reduced maternal food intake during pregnancy, which suddenly stopped and increased when the famine finished after the liberation of Holland. This sudden change in food intake may have altered preprogrammed set-points and increased sensitivity to chronic low levels of toxic substances, such as MeHg found in the environment.
To test the hypothesis that maternal diet enhances the vulnerability of the offspring exposed to toxic substances, we studied the effects of MeHg exposure in rats after prenatal in utero exposure (Study I) and after maternal diet manipulation followed by early postnatal MeHg exposure to the offspring (Study II).
Study I
F0-female rats were dosed daily by gavage with MeHg dissolved in corn oil (0, 0.1, 0.4, 0.7, 1.0, 1.5, 2.0 mg/kg body weight) between gestation day 6 and lactation day 10. The effects on F1-offspring were assessed for conventional guideline endpoints [Guidelines US-EPA OPPTS 870.6300]: bodyweight, clinical signs, developmental landmarks, neuropathology, and behavior. In addition, more advanced tests were explored to improve the sensitivity and relevance for regulatory toxicology testing. These were: 1) brain gene expression levels using Affymetrix Rat 230-2.0 chips (n = 68, 3-6 replicates/treatment), 2) cerebellar volume and neuron numbers using stereology, 3) metabolic brain activity with in vivo [18F] fluorindeoxyglucose (18F FDG) brain microPET, and 4) excitability of neuronal networks with electrically evoked hippocampal field potentials. In the field of regulatory toxicology, these advanced tests were introduced as novel indicators of the development of brain structure and function.
No effects were found for any of the conventional guideline endpoints of development (body weight and physical, sensory or sexual landmarks), behavior (motor activity, functional observational battery, active and passive avoidance, auditory startle response) and neuropathology (brain size and weight, microscopic slide reading and linear morphometry). In contrast the novel indicators of development showed that prenatal exposure to MeHg induced subtle and persistent effects, even long after cessation of exposure (Figure 2).
Figure 2. Percent control body weight, cumulative spontaneous activity and motor coordination in 12 month-old male and female mice following human equivalent gestational lead exposure.
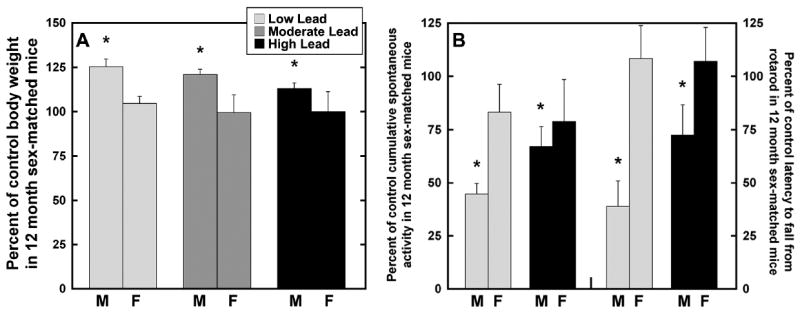
(A) Twelve month-old male mice in the low, moderate and high lead -dose groups weighed significantly more than age-matched controls: 26%, 21% and 13%, respectively. Gestational lead exposure produced nonmonotonic dose-dependent responses. There were no significant treatment-related differences in body weigh in female mice. (B) Twelve month-old male mice in the low and high lead -dose groups exhibited significantly less cumulative spontaneous activity than age-matched controls: -52% and -35%, respectively. The mean latency to fall from the rotarod significantly decreased in 12 month-old males in the low and high lead -dose groups compared to age-matched controls: -61% and -28%, respectively. Gestational lead exposure produced curvilinear dose-dependent responses. There were no significant treatment-related differences in cumulative spontaneous activity or mean latency to fall from the rotarod in female mice. *p<0.05 compared to sex-matched controls.
Comparison of MeHg and control animals revealed reciprocal regulation of gene expression between postnatal day (PND) 21 and PND70 in cerebellum and cerebrum. Compared to controls, genes down-regulated by MeHg at PND21 are up-regulated at PND 70 while genes up-regulated at PND 21 are down-regulated at PND70. These reciprocal gene expression changes at PND21 and PND70 suggest a delay in development and/or onset of brain activity. The effects of MeHg detected by microarray gene expression reflect a large spectra of conventional endpoints including brain structure (e.g., neuron quantity, brain size and development), brain function (e.g., synaptic transmission, long-term depression) and behavior (e.g., learning, locomotion, startle response).
The volume of different brain regions was analysed in control and 1 mg MeHg dose group (n = 10 rats/group) and particularly male F1-rats show a small reduction both on PND22 and on PND62: The volume of the total rhombencephalon and medulla oblongata on PND62 is significantly reduced. On PND22, a significant loss of cerebellar granular neurons was found in female rats (-13.9%) and in male rats (-20.9%). Group means of -6.0% (females) and -7.5% (males) were observed on PND62.
Control rats and rats exposed to MeHg at dose 1.5 mg/kg (n = 4 rats/group) were analyzed at PND18, 22, 37 and 61. Similar to the results obtained by gene expression profiling, the FDG uptake patterns in brain samples exposed to MeHg show a phase-shift as compared to control samples, suggesting a delay in development and/or onset of brain activity (Figure 2). In addition, on PND61 there is a persisting overall decrement in brain activity, consistent with long-term effects of prenatal and perinatal exposure to MeHg on adult animals observed by microarray gene expression analyses.
Neuronal excitability was assessed by synaptic excitation analysis in the CA1 area of the hippocampus. F1-offspring of the control group and 1.5 mg MeHg dose group (n = 5 rats/group; 2 samples/rat) were analyzed at PND28 and PND68. A paired-pulse depression in the 28 days MeHg exposed group is shown (Figure 2), which could be the result of depletion of the neurotransmitter in the presynaptic terminal. An increased paired-pulse facilitation was discovered in the PND68 MeHg exposed group. This likely resulted from an increased neurotransmitter release. At this age the inhibitory cells are fully developed, therefore a MeHg-induced change in the excitatory-inhibitory network on the inhibitory cells is possible. The hippocampus is essential for learning and memory. These results imply that prenatal and perinatal exposure to MeHg changes functionality in adult animals.
Study II
F0-female rats were kept on control (CONT), high-caloric (HIGH) or deficient-caloric (DEF) diet from 6 weeks premating onwards. Pups were cross-fostered at birth obtaining 5 groups with respect to the diet during gestation (G) (cont, high, def) and lactation (L) (CONT, HIGH, DEF): Gcont/LCONT; Ghigh/LHIGH; Gdef/LDEF; Ghigh/LDEF, Gdef/LHIGH. The Gdef/LHIGH group can be considered equivalent to the “Dutch Famine model”. F1 offspring were injected daily (subQ) from PND2 to 21 with saline (control) or MeHg (3 mg/kg body weight). In addition to the conventional guideline endpoints, insulin levels, gastrointestinal and pancreatic hormone concentrations of F1 rats were measured. These were: 1) insulin levels on PND250 (glucose tolerance test), 2) gastrointestinal and pancreatic hormone concentrations (insulin, amylin, ghrelin, gastric inhibitory peptide (GIP), glucagon-like peptide-1 (GLP-1), peptide YY, pancreatic polypeptide (PP) measured 0, 15, 30, 60, and 90 min after ingestion (by gavage) of butter oil or olive oil on PND140 (fat test).
Specifically the reprogrammed Gdef/LHIGH group, the “Dutch Famine Rat model”, showed catch-up growth during lactation, persistently increased body weight at adulthood, and an increased response in the glucose tolerance test. This suggests an increased prevalence for obesity and diabetes. Moreover, an increased vulnerability to MeHg was observed in this group as demonstrated by neural impairment of the auditory startle response that was not observed produced by diet or MeHg exposure alone. Olive oil fed females Ghigh/LHIGH, Gdef/LHIGH, Gdef/LDEF, and Ghigh/LDEF had lower slopes for all tested hormones (except PP) as compared to control fed females. Amylin, ghrelin, and GLP-1 levels fluctuated in the Gdef/LHIGH group confirming an increased prevalence for obesity, diabetes, and neural impairment.
In summary, Study I showed that prenatal in utero exposure to MeHg induced subtle and persistent changes in brain structure and activity in the offspring when utilizing new advanced indicators of neurotoxicity (i.e., gene profiling, stereological analysis, MicroPET and neural excitability). No effects were found using conventional guideline endpoints. Study II showed that maternal food intake enhanced the vulnerability of the offspring for postnatal exposure to MeHg as well as increased the susceptibility for obesity, diabetes and neural impairment in later life. Three additional and important advantages of the new indicators, compared to the conventional toxicology guideline studies, is that they were more sensitive, more scientifically informative, and 50% fewer animals were required to produce statistically significant results.
Acute neonatal exposure to ketamine and long-lasting deficits in learning, motivation and concept formation in rhesus monkeys (Merle G. Paule)
Our increased ability to keep premature and compromised infants alive results in an ever-increasing population in our nation’s neonatal intensive care units. Part of this success lies in the increased number of complicated surgical and other interventions that occur in this already-at-risk population. Many of these are conducted under various forms of anesthesia and sedation. Concerns over the potential adverse effects of these kinds of drug exposures prompted the need for experimental studies to address this important issue.
Blockade of N-methyl-D-aspartate (NMDA) receptors by the anesthetic ketamine causes robust increases in apoptotic cell death during the rat brain growth spurt [65-69]. Excitatory amino acids (EEAs) play critical roles during development by regulating neuronal survival, axonal and dendritic structure, synaptogenesis and plasticity [70-72]. EEA receptors also play important roles in long-term potentiation [4], which is important for learning and memory processes [73-75]. The infant brain is thought to be more sensitive to NMDA receptor manipulation than the adult brain [74,76].
Studies in rats exposed to a single episode of an anesthetic cocktail typical of those used in children demonstrated subsequent deficits in learning behaviors when tested as young adults [77]. Thus, it was important to determine if similar phenomena occurred in nonhuman primates. Ketamine-induced increases in abnormal cell death occurred in the rhesus monkey [78] from the middle of the third trimester of pregnancy to postnatal days 5/6 (not seen by PND 35) [79,80]. Next we determined if there were associated functional consequences in primates, as demonstrated in rodents, with a goal toward making predictions about the effects of ketamine-induced general anesthesia during development on later cognitive function in humans. Several cognitive function tasks from the National Center for Toxicological Research (NCTR) Operant Test Battery (OTB) [81] were utilized in this regard. These included assessments of motivation, color discrimination, learning, and short-term memory as described by Paule et al. [82].
Task metrics included percent task completed, response rate and accuracy. Earlier studies [83] demonstrated that, in children, several metrics of these OTB tasks correlate positively with IQ scores, thus, their relevance. Earlier studies also demonstrated that, in general, the performance of well-trained monkeys is not different from that of children four to thirteen years of age, with the degree of similarity depending upon task and endpoint [84]. The translatability of monkey OTB data to humans is further supported by drug studies demonstrating that, where comparable data exist, psychotropic drug effects are the same or similar in both monkeys and humans [82].
Six PND5 or PND6 neonatal rhesus monkeys were exposed for 24 hours to ketamine-induced anesthesia (iv administration). Six controls were unexposed, but separated from their mothers for the same amount of time as the treated animals. Animals were reared by their natural mothers, without incident, until weaning at six months of age. At seven months of age, all animals began training in the NCTR OTB. For subjects to come to understand the rules governing particular OTB tasks, extensive training is required. During this training scores are assigned as animals demonstrate mastery of specifics. The OTB training scores of the ketamine-exposed animals closely tracked those of the controls until short-term memory task training began. This occurred about three months into training when the animals were approximately 10 months of age. This stage of training involves developing the concept of ‘matching’, whereby subjects are reinforced for ‘matching’ a previously shown ‘sample’ stimulus [83]. By the time ketamine-exposed animals mastered the memory task concept, they were three months behind the control animals. During mastery of learning task performance, the performance of controls and exposed animals was about the same for the first 10 months of training (~17 months of age), after which the performance of controls was significantly better than that of exposed animals. This disparity continued throughout the remainder of the observation period: the following two years [82]. The deficits manifest in the form of reduced response rate, accuracy and percent task completed. Similar persistent deficits were observed for response rates in both the motivation and color and position discrimination tasks [82].
These results provide proof of concept that a single episode of ketamine-induced general anesthesia during a sensitive period of brain growth can cause subsequent cognitive deficits in nonhuman primates. These effects are very long-term and possibly permanent. This observation is particularly troubling when one considers that they are seen in behaviors thought to reflect aspects of brain function related to intelligence in children.
Acknowledgments
Supported in part by Grants RO1 ES09797 (PG), RO1ES012482 (DAF), Core Grant EY07551 (DAF), NICHD (MGP), CDER/FDA (MGP), and NCTR/FDA (MGP). The authors also acknowledge the financial support of the local organizers. The findings and conclusions in the report by M.G. Paule are those of the author and do not necessarily represent the views of the FDA.
Footnotes
Conflicts of Interest
None of the authors has competing financial interests.
References
- 1.Cameron N, Demerath EW. Critical periods in human growth and their relationship to diseases of aging. Am J Phys Anthropol Suppl. 2002;35:159–184. doi: 10.1002/ajpa.10183. [DOI] [PubMed] [Google Scholar]
- 2.Gluckman PD, Hanson MA, Beedle AS. Early life events and their consequences for later disease: a life history and evolutionary perspective. Am J Hum Biol. 2007;19:1–19. doi: 10.1002/ajhb.20590. [DOI] [PubMed] [Google Scholar]
- 3.Heindell JJ. Animal models for probing the developmental basis of disease and dysfunction paradigm. Basic Clin Pharmacol Toxicol. 2008;102:76–81. doi: 10.1111/j.1742-7843.2007.00184.x. [DOI] [PubMed] [Google Scholar]
- 4.Bilbo SD, Schwarz JM. Early-life programming of later-life brain and behavior: a critical role for the immune system. Front Behav Neurosci. 2009;3:14. doi: 10.3389/neuro.08.014.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rice D, Barrone S., Jr Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ Health Perspect. 2000;108(Suppl 3):511–33. doi: 10.1289/ehp.00108s3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mendola P, Selevan SG, Gutter S, Rice D. Environmental factors associated with a spectrum of neurodevelopmental deficits. Ment Retard Dev Disabil Res Rev. 2002;8:188–97. doi: 10.1002/mrdd.10033. [DOI] [PubMed] [Google Scholar]
- 7.Taylor PD, Poston L. Developmental programming of obesity in mammals. Exp Physiol. 2007;92:287–98. doi: 10.1113/expphysiol.2005.032854. [DOI] [PubMed] [Google Scholar]
- 8.Leasure JL, Giddabasappa A, Chaney S, Johnson JE, Jr, Pothakos K, Lau YS, Fox DA. Low-level human equivalent gestational lead exposure produces sex-specific motor and coordination abnormalities and late-onset obesity in year-old mice. Environ Health Perspect. 2008;116:355–61. doi: 10.1289/ehp.10862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Newbold RR, Padilla-Banks E, Jefferson WN, Heindell JJ. Effects of endocrine disruptors on obesity. Int J Androl. 2008;31:201–8. doi: 10.1111/j.1365-2605.2007.00858.x. [DOI] [PubMed] [Google Scholar]
- 10.JECFA. Summary and conclusions; Sixty-first meeting of the Joint FAO/WHO Expert Committee on Food Additives; Rome. June 2003. [Google Scholar]
- 11.Grandjean P, Budtz-Jorgensen E, Keiding N, Weihe P. Underestimation of risk due to misclassification. Eur J Oncol. 2003;(Suppl 2):165–72. [Google Scholar]
- 12.White RF, Palumbo CL, Yurgelun-Todd DA, Heaton KJ, Weihe P, Debes F, et al. Functional MRI approach to developmental methylmercury and polychlorinated biphenyl neurotoxicity. Neurotoxicology. 2001 doi: 10.1016/j.neuro.2011.04.001. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi AL, Cordier S, Weihe P, Grandjean P. Negative confounding in the evaluation of toxicity: the case of methylmercury in fish and seafood. Crit Rev Toxicol. 2008;38:877–93. doi: 10.1080/10408440802273164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Budtz-Jorgensen E, Grandjean P, Weihe P. Separation of risks and benefits of seafood intake. Environ Health Perspect. 2007;115:323–7. doi: 10.1289/ehp.9738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stokes-Riner A, Thurston SW, Myers GJ, Duffy EM, Wallace J, Bonham M, et al. A longitudinal analysis of prenatal exposure to methylmercury and fatty acids in the Seychelles. Neurotoxicol Teratol. 2011;33:325–8. doi: 10.1016/j.ntt.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Budtz-Jorgensen E, Keiding N, Grandjean P. Effects of exposure imprecision on estimation of the benchmark dose. Risk Anal. 2004;24:1689–96. doi: 10.1111/j.0272-4332.2004.00560.x. [DOI] [PubMed] [Google Scholar]
- 17.Grandjean P, Landrigan PJ. Developmental neurotoxicity of industrial chemicals. Lancet. 2006;368:2167–78. doi: 10.1016/S0140-6736(06)69665-7. [DOI] [PubMed] [Google Scholar]
- 18.Baillie-Hamilton PF. Chemical toxins: a hypothesis to explain the global obesity epidemic. J Altern Complement Med. 2002;8:185–92. doi: 10.1089/107555302317371479. [DOI] [PubMed] [Google Scholar]
- 19.Heindell JJ. Role of exposure to environmental chemicals in the developmental basis of disease and dysfunction. Reprod Toxicol. 2007;23:257–9. doi: 10.1016/j.reprotox.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 20.Barker DJ, Eriksson JG, Forsén T, Osmond C. Fetal origins of adult disease: strength of effects and biological basis. Int J Epidemiol. 2002;31:1235–9. doi: 10.1093/ije/31.6.1235. [DOI] [PubMed] [Google Scholar]
- 21.Weiss B, Clarkson TW, Simon W. Silent latency periods in methylmercury poisoning and in neurodegenerative disease. Environ Health Perspect. 2002;110(Suppl 5):851–4. doi: 10.1289/ehp.02110s5851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gluckman PD, Hanson MA. Developmental origins of disease paradigm: a mechanistic and evolutionary perspective. Pediatr Res. 2004;56:311–7. doi: 10.1203/01.PDR.0000135998.08025.FB. [DOI] [PubMed] [Google Scholar]
- 23.Langley-Evans SC. Developmental programming of health and disease. Proc Nutr Soc. 2006;65:97–105. doi: 10.1079/pns2005478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taylor PD, Poston L. Developmental programming of obesity in mammals. Exp Physiol. 2007;92:287–98. doi: 10.1113/expphysiol.2005.032854. [DOI] [PubMed] [Google Scholar]
- 25.Newland MC, Rasmussen EB. Aging unmasks adverse effects of gestational exposure to methylmercury in rats. Neurotoxicol Teratol. 2000;22:819–28. doi: 10.1016/s0892-0362(00)00107-0. [DOI] [PubMed] [Google Scholar]
- 26.Landrigan PJ, Sonawane B, Butler RN, Trasande L, Callan R, Droller D. Early environmental origins of neurodegenerative disease in later life. Environ Health Perspect. 2005;113:1230–3. doi: 10.1289/ehp.7571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Antonio MT, Corpos I, Leret ML. Neurochemical changes in newborn’s brain after gestational cadmium and lead exposure. Tox Lett. 1999;104:1–9. doi: 10.1016/s0378-4274(98)00125-8. [DOI] [PubMed] [Google Scholar]
- 28.Grün F, Blumberg B. Environmental obesogens: organotins and endocrine disruption via nuclear receptor signaling. Endocrinology. 2006;147:S50–5. doi: 10.1210/en.2005-1129. [DOI] [PubMed] [Google Scholar]
- 29.Altmann L, Sveinsson K, Krämer U, Weishoff-Houben M, Turfeld M, Winneke G, Wiegand H. Visual functions in 6-year-old children in relation to lead and mercury levels. Neurotoxicol Teratol. 1998;20:9–17. doi: 10.1016/s0892-0362(97)00070-6. [DOI] [PubMed] [Google Scholar]
- 30.Lilienthal H, Lenaerts C, Winneke G, Hennekes R. Alteration of the visual evoked potential and the electroretinogram in lead-treated monkeys. Neurotoxicol Teratol. 1988;10:417–22. doi: 10.1016/0892-0362(88)90002-5. [DOI] [PubMed] [Google Scholar]
- 31.Lilienthal H, Kohler K, Turfeld M, Winneke G. Persistent increases in scotopic B.- wave amplitudes after lead exposure in monkeys. Exp Eye Res. 1994;59:203–9. doi: 10.1006/exer.1994.1098. [DOI] [PubMed] [Google Scholar]
- 32.Fox DA, Campbell ML, Blocker YS. Functional alterations and apoptotic cell death in the retina following developmental or adult lead exposure. Neurotoxicology. 1997;18:645–64. [PubMed] [Google Scholar]
- 33.Fox DA, Kala SV, Hamilton WR, Johnson JE, O’Callaghan JP. Low-level human equivalent gestational lead exposure produces supernormal scotopic electroretinograms, increased retinal neurogenesis and decreased dopamine utilization in rats. Environ Hlth Perspect. 2008;116:618–25. doi: 10.1289/ehp.11268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fox DA, Hamilton RW, Johnson JE, Xiao W, Chaney S, Mukherjee S, Miller DB, O’Callaghan JP. Gestational lead exposure selectively decreases retinal dopamine amacrine cells and dopamine content in adult mice. Toxicol Appl Pharm. 2011;256:258–67. doi: 10.1016/j.taap.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Osman K, Pawlas K, Schütz A, Gazdzik M, Sokal JA, Vahter M. Lead exposure and hearing effects in children in Katowice, Poland. Eviron Res. 1999;80:1–8. doi: 10.1006/enrs.1998.3886. [DOI] [PubMed] [Google Scholar]
- 36.Wasserman GA, Musabegovic A, Liu X, Kline J, Factor-Litvak P, Graziano JH. Lead exposure and motor functioning in 4(1/2)-year-old children: the Yugoslavia prospective study. J Pediatr. 2000;137:555–61. doi: 10.1067/mpd.2000.109111. [DOI] [PubMed] [Google Scholar]
- 37.Rothenberg SJ, Poblano A, Schnaas L. Brainstem auditory evoked response at five years and prenatal and postnatal blood lead. Neurotoxicol Teratol. 2000;22:503–10. doi: 10.1016/s0892-0362(00)00079-9. [DOI] [PubMed] [Google Scholar]
- 38.Rothenberg SJ, Schnaas L, Salgado-Valladares M, Casanueva E, Geller AM, Hudnell HK, Fox DA. Increased ERG a- and b-wave amplitudes in 7- to 10-year-old children resulting from prenatal lead exposure. Invest Ophthalmol Vis Sci. 2002;43:2036–44. [PubMed] [Google Scholar]
- 39.Canfield RL, Henderson CR, Jr, Cory-Slechta DA, Cox C, Jusko TA, Lanphear BP. Intellectual impairment in children with blood lead concentrations below 10 microg per deciliter. N Engl J Med. 2003;348:1517–26. doi: 10.1056/NEJMoa022848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.He L, Perkins GA, Poblenz AT, Harris JB, Hung M, Ellisman MH, Fox DA. Bcl-xL overexpression blocks bax-mediated mitochondrial contact site formation and apoptosis in rod photoreceptors of lead-exposed mice. Proc Natl Acad Sci USA. 2003;100:1022–7. doi: 10.1073/pnas.0333594100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nagpal AG, Brodie SE. Supranormal electroretinogram in a 10-year-old girl with lead toxicity. Doc Ophthalmol. 2009;118:163–6. doi: 10.1007/s10633-008-9144-7. [DOI] [PubMed] [Google Scholar]
- 42.Giddabasappa A, Hamilton WR, Chaney S, Xiao W, Johnson J, Mukherjee, Fox DA. Low-level gestational lead exposure increases retinal progenitor cell proliferation and rod photoreceptor and bipolar cell neurogenesis in mice. Environ Health Perspect. 2011;119:71–7. doi: 10.1289/ehp.1002524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fox DA, Rubinstein SD. Age-related changes in retinal sensitivity, rhodopsin content and rod outer segment length in hooded rats following low-level lead exposure during development. Exp Eye Res. 1989;48:237–49. doi: 10.1016/s0014-4835(89)80073-9. [DOI] [PubMed] [Google Scholar]
- 44.Bhattacharya A, Shukla R, Bornschein RL, Dietrich KN, Keith R. Lead effects on postural balance of children. Environ Health Perspect. 1990;89:35–42. doi: 10.1289/ehp.908935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bhattacharya A, Shukla R, Dietrich KN, Bornschein RL. Effect of early lead exposure on the maturation of children’s postural balance: a longitudinal study. Neurotoxicol Teratol. 2006;28:376–385. doi: 10.1016/j.ntt.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 46.Baghurst PA, McMichael AJ, Wigg NR, Vimpani GV, Robertson EF, Roberts RJ, et al. Environmental exposure to lead and children’s intelligence at the age of seven years. The Port Pirie Cohort Study. N Engl J Med. 1992;327:1279–1284. doi: 10.1056/NEJM199210293271805. [DOI] [PubMed] [Google Scholar]
- 47.Ris MD, Dietrich KN, Succop PA, Berger OG, Bornschein RL. Early exposure to lead and neuropsychological outcome in adolescence. J Int Neuropsychol Soc. 2004;10:261–270. doi: 10.1017/S1355617704102154. [DOI] [PubMed] [Google Scholar]
- 48.Cory-Slechta DA, Virgolini MB, Thiruchelvam M, Weston DD, Bauter MR. Maternal stress modulates the effects of developmental lead exposure. Environ Health Perspect. 2004;112:717–730. doi: 10.1289/ehp.6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vahter M, Akesson A, Liden C, Ceccatelli S, Berglund M. Gender differences in the disposition and toxicity of metals. Environ Res. 2007;104:85–95. doi: 10.1016/j.envres.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 50.Davis JM, Svendsgaard DJ. U-shaped dose-response curves: their occurrence and implications for risk assessment. J Toxicol Environ Health. 1990;30:71–83. doi: 10.1080/15287399009531412. [DOI] [PubMed] [Google Scholar]
- 51.Ma T, Chen HH, Ho IK. Effects of chronic lead exposure on neurobehavioral function and dopaminergic neurotransmitter receptors in rats. Toxicol Lett. 1999;105:111–121. doi: 10.1016/s0378-4274(98)00388-9. [DOI] [PubMed] [Google Scholar]
- 52.Moreira EG, Vassilieff I, Vassilieff VS. Developmental lead exposure: behavioral alterations in the short and long term. Neurotoxicol Teratol. 2001;23:489–495. doi: 10.1016/s0892-0362(01)00159-3. [DOI] [PubMed] [Google Scholar]
- 53.Verlangieri AJ. Prenatal and postnatal chronic lead intoxication and running wheel activity in the rat. Pharmacol Biochem Behav. 1979;11:95–98. doi: 10.1016/0091-3057(79)90303-4. [DOI] [PubMed] [Google Scholar]
- 54.CDC. Preventing lead poisoning in young children: a statement by the Centers for Disease Control and Prevention. US Department of Health and Human Services, Public Health Service 1991 [Google Scholar]
- 55.Susser ES, Lin SP. Schizophrenia after prenatal exposure to the Dutch Hunger Winter of 1944-1945. Arch Gen Psychiatry. 1992;49:983–988. doi: 10.1001/archpsyc.1992.01820120071010. [DOI] [PubMed] [Google Scholar]
- 56.Lumey LH, Stein AD. In utero exposure to famine and subsequent fertility: The Dutch famine birth cohort study. Am J Public Hlth. 1997;87:1962–1966. doi: 10.2105/ajph.87.12.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hoek HW, Brown AS, Susser E. The Dutch Famine and schizophrenia spectrum disorders. Soc Psychiatry Psychiatr Epidemiol. 1998;33:373–379. doi: 10.1007/s001270050068. [DOI] [PubMed] [Google Scholar]
- 58.Roseboom TJ, van der Meulen JHP, Osmond C, Barker DJP, Ravelli ACJ, Bleker OP. Plasma lipid profiles in adults after prenatal exposure to the Dutch famine. Am J Clin Nutr. 2000;72:1101–1106. doi: 10.1093/ajcn/72.5.1101. [DOI] [PubMed] [Google Scholar]
- 59.Roseboom TJ, van der Meulen JHP, Osmond C, Barker DJP, Ravelli ACJ, Schroeder-Tanka JM, et al. Coronary heart disease after prenatal exposure to the Dutch Famine, 1944–45. Heart. 2000;84:595–598. doi: 10.1136/heart.84.6.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Roseboom TJ, van der Meulen JHP, Ravelli ACJ, Osmond C, Barker DJP, Bleker OP. Effects of prenatal exposure to the Dutch famine on adult disease in later life: an overview. Mol Cell Endocrinol. 2001;185:93–98. doi: 10.1016/s0303-7207(01)00721-3. [DOI] [PubMed] [Google Scholar]
- 61.Elias SG, Peeters PHM, Grobbee DE, van Noord PAH. Breast cancer risk after caloric restriction during the 1944–1945 Dutch Famine. J Natl Cancer Inst. 2004;96:539–546. doi: 10.1093/jnci/djh087. [DOI] [PubMed] [Google Scholar]
- 62.Painter RC, Roseboom TJ, Bleker OP. Prenatal exposure to the Dutch Famine and disease in later life: An overview. Reprod Toxicol. 2005;20:345–352. doi: 10.1016/j.reprotox.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 63.Roseboom T, de Rooij S, Painter R. The Dutch Famine and its long-term consequences for adult health. Early Human Develop. 2006;82:485–491. doi: 10.1016/j.earlhumdev.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 64.De Rooij SR. PhD thesis. University of Amsterdam; the Netherlands: 2007. Metabolic consequences of prenatal exposure to the Dutch Famine. Printed by: Buijten & Schipperheijn, Amsterdam. [Google Scholar]
- 65.Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vockler J, Dikranian K, et al. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;83:70–4. doi: 10.1126/science.283.5398.70. [DOI] [PubMed] [Google Scholar]
- 66.Hayashi H, Dikkes P, Soriano S. Repeated administration of ketamine may lead to neuronal degeneration in the developing rat brain. Paediatr Anaesth. 2002;12:770–4. doi: 10.1046/j.1460-9592.2002.00883.x. [DOI] [PubMed] [Google Scholar]
- 67.Scallet AC, Schmued LC, Slikker W, Jr, Grunberg N, Faustino PJ, Davis H, et al. Developmental neurotoxicity of ketamine: morphometric confirmation, exposure parameters, and multiple fluorescent labeling of apoptotic neurons. Toxicol Sci. 2004;81:364–70. doi: 10.1093/toxsci/kfh224. [DOI] [PubMed] [Google Scholar]
- 68.Zou X, Patterson TA, Sadovova N, Twaddle NC, Doerge DR, Zhang X, et al. Potential neurotoxicity of ketamine in the developing rat brain. Toxicol Sci. 2009;108:149–58. doi: 10.1093/toxsci/kfn270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shi Q, Guo L, Patterson TA, Dial D, Li Q, Sadovova N, et al. Gene expression profiling in the developing rat brain exposed to ketamine. Neuroscience. 2010;166:852–63. doi: 10.1016/j.neuroscience.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McDonald JW, Johnston MV. Physiological and pathophysiological roles of excitatory amino acids during central nervous system development. Brain Res Brain Res Rev. 1990;15:41–70. doi: 10.1016/0165-0173(90)90011-c. [DOI] [PubMed] [Google Scholar]
- 71.Komuro H, Rakic P. Modulation of neuronal migration by NMDA receptors. Science. 1993;260:95–7. doi: 10.1126/science.8096653. [DOI] [PubMed] [Google Scholar]
- 72.Reiprich P, Kilb W, Luhmann HJ. Neonatal NMDA receptor blockade disturbs neuronal migration in rat somatosensory cortex in vivo. Cereb Cortex. 2005;15:349–58. doi: 10.1093/cercor/bhh137. [DOI] [PubMed] [Google Scholar]
- 73.Tomita H, Shibata Y, Sakurai T, Okada Y. Involvement of a protein kinase C-dependent process in long-term potentiation formation in guinea pig superior colliculus slices. Brain Res. 1990;536:146–52. doi: 10.1016/0006-8993(90)90019-8. [DOI] [PubMed] [Google Scholar]
- 74.D’Souza SW, McConnell SE, Slater P, Barson AJ. N-methyl-D-aspartate binding sites in neonatal and adult brain. Lancet. 1992;339:1240. doi: 10.1016/0140-6736(92)91188-e. [DOI] [PubMed] [Google Scholar]
- 75.Huang EP, Stevens CF. The matter of mind: molecular control of memory. Essays Biochem. 1998;3:165–78. doi: 10.1042/bse0330165. [DOI] [PubMed] [Google Scholar]
- 76.Bittigau P, Sifringer M, Pohl D, Stadthaus D, Ishimaru M, Shimizu H, et al. Apoptotic neurodegeneration following trauma is markedly enhanced in the immature brain. Ann Neurol. 1999;5:724–35. doi: 10.1002/1531-8249(199906)45:6<724::aid-ana6>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 77.Jevtovic-Todorovic V, Hartman RE, Izumi Y, Benshoff ND, Dikranian K, Zorumski CF, et al. Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J Neurosci. 2003;23:876–82. doi: 10.1523/JNEUROSCI.23-03-00876.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Slikker W, Jr, Zou X, Hotchkiss CE, Divine RL, Sadovova N, Twaddle NC, et al. Ketamine-induced neuronal cell death in the perinatal rhesus monkey. Toxicol Sci. 2007;98:145–58. doi: 10.1093/toxsci/kfm084. [DOI] [PubMed] [Google Scholar]
- 79.Brambrink AM, Back SA, Avidan MS, Creeley CE, Olney JW. Ketamine and isoflurane anesthesia triggers neuronal and glial apoptosis in the neonatal Macaque. Proc Am Soc Anesth. 2010 Abs #A375. [Google Scholar]
- 80.Zou X, Patterson TA, Divine RL, Sadovova N, Zhang X, Hanig JP, et al. Prolonged exposure to ketamine increases neurodegeneration in the developing monkey brain. Int J Dev Neurosci. 2009;27:727–31. doi: 10.1016/j.ijdevneu.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 81.Paule MG. Validation of a behavioral test battery for monkeys. In: Buccafusco JJ, editor. Methods of Behavioral Analysis in Neuroscience. Boca Raton: CRC Press LLC; 2001. pp. 281–94. [Google Scholar]
- 82.Paule MG, Li M, Allen RR, Liu F, Zou X, Hotchkiss C, et al. Ketamine anesthesia during the first week of life can cause long-lasting cognitive deficits in rhesus monkeys. Neurotoxicol Teratol. 2011;33:220–30. doi: 10.1016/j.ntt.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Paule MG, Chelonis JJ, Buffalo EA, Blake DJ, Casey PH. Operant test battery performance in children: correlation with IQ. Neurotoxicol Teratol. 1999;21:223–30. doi: 10.1016/s0892-0362(98)00045-2. [DOI] [PubMed] [Google Scholar]
- 84.Paule MG, Forrester TM, Maher MA, Cranmer JM, Allen RR. Monkey versus human performance in the NCTR Operant Test Battery. Neurotoxicol Teratol. 1990;12:503–7. doi: 10.1016/0892-0362(90)90014-4. [DOI] [PubMed] [Google Scholar]