

Abstract
In recent years, the role played by the stromal microenvironment has been given growing attention in order to achieve a full understanding of cancer initiation and progression. Because cancer is a tissue-based disease, the integrity of tissue architecture is a major constraint toward cancer growth. Indeed, a large contribution of the natural resistance to cancer stems from stromal microenvironment components, the dysregulation of which can facilitate cancer occurrence. For instance, recent experimental evidence has highlighted the involvement of stromal cells in ovarian carcinogenesis, as epitomized by ovarian xenografts obtained by a double KO of the murine Dicer and Pten genes. Likewise, we reported the role of an ancient extracellular RNase, called Ribonuclease T2 (RNASET2), within the ovarian stromal microenvironment. Indeed, hyperexpression of RNASET2 is able to control tumorigenesis by recruiting macrophages (mostly of the anticancer M1 subtype) at the tumor sites. We present biological data obtained by RNASET2 silencing in the poorly tumorigenetic and highly RNASET2-expressing human OVCAR3 cell line. RNASET2 knockdown was shown to stimulate in vivo tumor growth early after microinjection of OVCAR3 cells in nude mice. Moreover, we have investigated by molecular profiling the in vivo expression signature of human and mouse cell xenografts and disclosed the activation of pathways related to activation of the innate immune response and modulation of ECM components. Finally, we provide evidence for a role of RNASET2 in triggering an in vitro chemotactic response in macrophages. These results further highlight the critical role played by the microenvironment in RNASET2-mediated ovarian tumor suppression, which could eventually contribute to better clarify the pathogenesis of this disease.
Despite its relatively low incidence rate, ovarian cancer is an extremely lethal disease, which globally claims 125,000 lives per year, making it the seventh leading cause of cancer-related deaths among women (1). Although the 5-y survival rate for stage I ovarian cancer is >90%, stage I diagnoses are more often the exception than the rule. Indeed, most patients present with advanced-stage tumors, for which the 5-y survival rate is a dismal 30% (1). Contributing to such a dramatic outlook is the rather poor understanding of the biological basis of the disease, which appears as a highly heterogeneous group of pathological entities (2, 3). Traditionally, the anatomical site from which most cases of ovarian cancer were thought to arise has been the ovarian surface epithelium (2, 3). This long-held belief has recently been challenged by several observations pointing to fallopian tube structures as the prevailing sites where insurgence of ovarian neoplasias occurs (4). According to this new paradigm, the ovary would rather represent a sort of “metastatic” secondary site, the primary site being localized in the fallopian tube parenchyma (4). Supporting this paradigm is a recent finding from Dicer/Pten double KO mice, in which ovarian carcinogenesis is initiated from abnormal proliferation within the stromal compartment of the fallopian tube (5).
The tumor microenvironment has recently become a crucial player in the carcinogenetic processes (6–9). Indeed, stromal components such as fibroblasts, immune/vascular cells, and ECM are all known to be engaged in a complex and dynamic cross-talk with the neighboring epithelium, which can crucially contribute to tumor suppression and resistance (6–9). Indeed, interaction of parenchymal cells with stromal components is known to be essential for the maintenance of tissue architecture and function (6–9), and several studies have documented the role played by the stroma as a key determinant of epithelial proliferation, cell death, motility, polarity, and differentiation (6, 9). Moreover, stromal abnormalities have long been known to exert profound influences on tumor induction and progression (6–9). However, although the notion of a dominant role of the microenvironment in providing signaling cues to suppress or revert a malignant phenotype is consolidated (6–9), a detailed understanding of the types and specific roles of stromal and immune cells and of the ECM dynamics within it is still lacking.
Our research group has recently contributed to this issue by investigating the role of the human RNASET2 gene (10–15). The latter codes for an extracellular RNase expressed in several tissues, including the ovary and tubal structures, and is highly conserved among phyla from viruses to humans, implying a crucial, evolutionary conserved function (16). In an ovarian cancer xenograft model, we previously found RNASET2 to carry out a strong oncosuppressive activity associated with an in vivo recruitment of macrophages into the tumor mass (15). Here, we report RNASET2 gene silencing in an independent ovarian cancer cell model to provide further evidence for its role in microenvironmental-mediated tumor suppression in vivo.
Results
RNASET2 Silencing in OVCAR3 Ovarian Cancer Cells Is Associated with Increased in Vivo Tumorigenesis.
Ovarian cancer-derived OVCAR3 cells were used to investigate the effects of RNASET2 knockdown. This cell line was chosen on two main basic premises. First, RNASET2 transcript and protein levels are significantly higher compared with other ovarian cancer cell lines (10). Second, OVCAR3 cells are reported to be poorly tumorigenic in vivo (17). RNASET2 silencing was carried out in OVCAR3 cells by stably expressing an siRNA targeting the RNASET2 mRNA, resulting in complete abrogation of its expression (Fig. 1A). Three independent RNASET2 knockdown clones and three control clones (expressing a scrambled shRNA) were then chosen for further analyses. The clones were injected s.c. into nude mice, and tumor growth was followed for approximately 40 d. As depicted in Fig. 1B, after a few days of in vivo growth, the three RNASET2-silenced clones showed an increased growth kinetics compared with control clones. By the end of the in vivo assay, the average tumor weight from RNASET2-silenced xenografts was also significantly increased compared with control xenografts (Fig. 1C). We thus concluded that RNASET2 silencing conferred to the poorly tumorigenic OVCAR3 cell line an increased in vivo growth rate. Such behavior is substantially congruent with what we have previously reported following overexpression of RNASET2 into the highly tumorigenic, low RNASET2-expressing Hey3Met2 ovarian cancer cell line, as, in that case, tumorigenesis was strongly inhibited by overexpression of this gene (15).
Fig. 1.

RNASET2 silencing enhances tumor growth in vivo. (A) Characterization of RNASET2-silenced OVCAR3 cells by Western blot analysis. (Upper) Intracellular lysates were probed with an anti-RNASET2 polyclonal antibody. (Lower) The same blot was probed with an anti–α-tubulin monoclonal antibody for normalization. (B) Three control and three RNASET2-silenced OVCAR3 clones were inoculated s.c. in nude mice as described in Materials and Methods. Tumor growth was monitored every 2 d until day 40. At least five mice were inoculated for each tested clone. Bars represent SD values (*P < 0.5 and **P < 0.01, significant difference in tumor size between RNASET2-silenced and control tumors, Student t test). (C) The mean weights of the collected tumors are represented (P = 0.004, two-tailed Student t test).
Morphological Characterization of RNASET2-Silenced OVCAR3 Cell Tumors Grown in Vivo.
To compare the morphological features of RNASET2-silenced and control xenografts, tumor samples were excised and processed for histological and immunohistochemical (IHC) analyses (Table S1). As depicted in Fig. 2, all tumors from RNASET2-silenced OVCAR3 cells were characterized by a diffuse pattern (Fig. 2 A and C). Tumor cells were large and polygonal, with prominent nuclei and remarkable nucleoli. Significantly, atypical mitotic figures were frequent, and the mitotic and proliferative indexes were high (Fig. 2E and Table S1). By contrast, tumors from control OVCAR3 cells showed a widespread nests/micronests pattern, with focal fibrous septa (Fig. 2 B and D). Coagulative necrosis and apoptosis in central areas of the tumors were abundantly present, whereas mitotic and proliferative indexes were decreased (Fig. 2F and Table S1). A more differentiated pattern, documented by increased cytokeratin 7 staining, was also observed in control samples compared with RNASET2-silenced tumors (Fig. S1). The occurrence of areas of widespread apoptosis in control tumors, that was already observed in histological sections, was also confirmed by IHC analysis (Fig. S1).
Fig. 2.

Histological and immunohistochemical analysis of tumor samples. Parental and RNASET2-silenced OVCAR3-derived tumor sections were stained with H&E (A–D). Microphotographs show a prevailing diffuse pattern and some solid nests in the central areas of RNASET2-silenced tumors, filled by large and polygonal cells with prominent nuclei and nucleoli (A and C). By contrast, control tumors show widespread nests/micronests pattern, with focal slender fibrous septa (B and D). Tumor sections were also analyzed by IHC for Ki-67 as a marker for cell proliferation (E and F) and for CD68 (G and H), inducible Nitric Oxide Synthase (iNOS-1) (I and J), and dectin-1 (K and L) to detect murine host inflammatory cells, the whole macrophage cell population, and M1- and M2-polarized macrophages, respectively. Photomicrographs shown are representative images of multiple fields examined in at least four sections derived from tumor excised in two to three independent animals for each experimental group.
A relevant histological finding concerned the presence of inflammatory infiltrates in xenografts from control OVCAR3 cells. To characterize the nature of such infiltrating cells, we carried out IHC assays for several inflammatory markers (data not shown). Among the chosen markers the most significant was CD68, whose pattern pointed to the presence of murine macrophages as the most representative cell population of the infiltrate, as shown in Fig. 2 G and H. To discriminate between macrophage subpopulations, we carried out an IHC assay for markers able to differentiate the M1 and M2 macrophage cell subpopulations. As shown in Fig. 2 I and J, a consistent depletion of inducible nitric oxide synthase-positive M1 macrophages was observed in RNASET2-silenced tumors compared with control xenografts. By contrast, infiltration by dectin-1–positive M2 macrophages, which was less pronounced in control xenografts compared with M1 macrophages, was only slightly decreased in RNASET2-silenced tumors (Fig. 2 K and L). In keeping with these results, a subsequent analysis of OVCAR3-derived xenograft RNA by real-time quantitative PCR (qPCR) technique using primer pairs specific for murine markers of M1/M2 polarization showed a decreased expression for two of three M1-specific markers in RNASET2-silenced xenografts. By contrast, no effect on gene expression was observed for two M2-specific markers, whereas expression of the third marked tested was up-regulated in RNASET2-silenced xenografts (Fig. S2).
Together, these results are totally in keeping with our previous data from the Hey3Met2 ovarian model (15), in which a strong infiltration of inflammatory cells (mostly M1 macrophages) was observed in RNASET2-overexpressing tumor xenografts (15). The results presented here thus provide further support to the notion that RNASET2-mediated tumor suppression is associated with in vivo recruitment of the M1 subclass of macrophages, which is known to have antitumorigenic properties (18).
Gene Expression Profile of RNASET2-Silenced OVCAR3 Cell Tumor Xenografts.
To investigate the molecular pathways underlying RNASET2-mediated tumor suppression in vivo, we next investigated the gene expression profile of the tumor xenografts. To evaluate the contribution of human cancer cells and murine host stroma, total RNA extracted from tumor xenografts was fluorescently labeled and hybridized to human and mouse whole-genome microarrays. By using a fold induction or repression greater than 1.5 and a P value lower than 0.01 as inclusion criteria, 104 human genes were found to be up-regulated and 189 were down-regulated in RNASET2-silenced xenografts (Dataset S1).When the murine host stromal transcriptional profile was investigated, 98 murine genes were shown to be up-regulated in RNASET2-silenced xenografts, compared with 272 down-regulated genes (Dataset S2).
Searches were then made for significantly enriched gene classes, as defined by Gene Ontology (GO) annotation and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis. As shown in Fig. S3A and Dataset S3, RNASET2-regulated human genes turned out to be significantly enriched in GO biological process classes mainly related to leukocyte activation (12 genes) and development (53 genes). Significantly, most human genes related to leukocytes activation turned out to be down-regulated in RNASET2-silenced OVCAR3 cells, suggesting a positive regulatory role for cells of the immune/inflammatory system by RNASET2. On the contrary, KEGG analysis displayed enrichment in pathways mainly related to immunity, cell adhesion, and cancer. To our surprise, GO and KEGG analysis did not show a significant enrichment in terms related to apoptosis. This was an unexpected finding, considering the increased apoptotic rate that we observed by IHC analysis in control OVCAR3 cell-derived xenografts (Fig. S1). However, a closer inspection of the microarray data showed a few apoptosis-related genes whose expression was indeed increased in control xenografts compared with RNASET2-silenced tumors. We thus confirmed by the more sensitive real-time qPCR technique that expression of five proapoptotic genes [Galanin (GAL), anterior pharynx defective 1 homolog B (APH1B), tumor necrosis factor receptor superfamily, member 25 (TNFRSF25), unc-13 homolog D (UNC13-D), and nuclear protein transcriptional regulator 1 (NUPR1)] and two antiapoptotic genes [adhesion molecule with Ig like domain 1 (AMIGO) and annexin A4 (ANXA4)] was down-regulated and up-regulated, respectively, in RNASET2-silenced xenografts. Thus, although not detected by microarray analysis, these genes might play a role in the decreased apoptotic rate observed in RNASET2-silenced xenografts.
When we turned to the host stromal transcriptional profile of the xenografts, a GO search returned gene classes related to cell adhesion and, most importantly, to immune response (Fig. S3B). The annotated murine genes in these two GO classes are listed in Dataset S4. Interestingly, similar results were obtained by KEGG analysis, with enrichment in pathways related to immunity and cell adhesion. As already reported for the analysis of the human cancer cell transcriptome, most murine genes related to immune system roles turned out to be down-regulated in RNASET2-silenced OVCAR3 cells, again suggesting a positive regulatory role for RNASET2 within the context of cells of the immune/inflammatory system. The significant enrichment of genes involved in immunological functions is in keeping with recent data pointing to a role for members of the T2 RNase gene family in the immune response (Discussion). Being aware of the sensitivity limits of the microarray hybridization technology, we aimed to further investigate whether RNASET2 might induce other immunologically competent genes. We thus compared by qPCR control and RNASET2-silenced tumor xenografts for expression levels of 70 genes belonging to the mouse chemokine gene family. Strikingly, 35 additional genes were shown to be significantly down-regulated in RNASET2-silenced tumors in addition to the 11 genes already detected by microarray analysis (Dataset S5). Among the 35 gene entries, we found several genes with an established role in monocyte/macrophage chemotaxis, such as chemokine (C-C motif) ligand (Ccl)-2, Ccl-7, chemokine (C-C motif) receptor (Ccr)-2, Ccr-6, Ccr-7, Cx3cl-1, and Cxcl-12, suggesting that RNASET2 might act in vivo to trigger a consistent chemoattractive response in cells of the innate immune system.
Analysis of RNASET2 as Candidate Chemotactic Protein.
The results reported here earlier strongly suggest that RNASET2 might carry out its tumor-suppressive role by a direct recruitment of host macrophage cells into the tumor mass. We thus addressed whether a direct interaction of RNASET2 with cells of this lineage could take place.
To this end, we first investigated the occurrence of binding of recombinant RNASET2 to the cell surface of human macrophages. For this purpose, human monocyte-derived macrophages upon in vitro differentiation (i.e., M0) and M1/M2 polarized ones were used. After exposure to endotoxin-free recombinant RNASET2 protein purified from Pichia pastoris, a flow cytometric analysis showed significant and dose-dependent RNASET2 binding to the cell surface in all macrophage subpopulations tested (Fig. 3A).
Fig. 3.
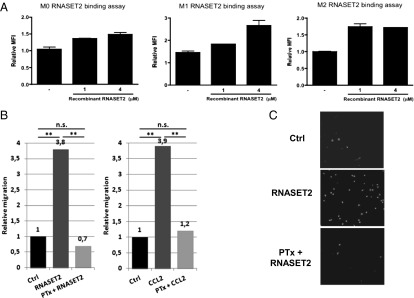
Binding of recombinant RNASET2 to the cell surface of macrophages and chemotactic activity on U937 cl.10 cell line. (A) Peripheral blood leucocyte (PBL)-derived human macrophages were detached from culture plates and starved in complete medium plus 1% FCS for 3 h at 37 °C. Binding assay was performed by adding recombinant RNASET2 for 3 h at 4 °C. Fc receptors were blocked by using 10% human serum for 15 min at 4 °C. Rabbit anti-human RNASET2 and normal rabbit IgG (as isotype control) were used to perform cell surface staining assay. Data are expressed as fold increase of mean fluorescence intensity (MFI) of anti-RNASET2 antibody-stained samples vs. isotype control-stained ones (error bars represent SEM). (B) U937 cl.10 cells were pretreated with 1 µg/mL PTx or left untreated. Human recombinant RNASET2 and CCL2 proteins were used as chemoattractants at the final concentrations of 8.172 µg/mL and 100 ng/mL, respectively. Statistical analysis was performed with the unilateral Student t test. (C) Fluorescence microscopy images of filters. Nuclei were stained with DAPI (magnification of 40×).
To investigate whether this binding could induce a cellular response, recombinant RNASET2 was used for in vitro chemotaxis assays on a human promyelocytic subclone showing low endogenous RNASET2 expression levels. Interestingly, we observed a significant RNASET2-mediated increase in the migration rate of these cells (Fig. 3 B and C). Importantly, such response was not affected by the expression system chosen for recombinant protein expression, as RNASET2 produced with baculovirus/insect cells and Pichia pastoris expression systems performed equally well in the chemotaxis assay. Moreover, the same chemotactic response was also observed when recombinant RNASET2 was applied to freshly prepared human primary monocytes isolated from buffy coats.
Taken together, these data support the possibility that the RNASET2 protein might directly evoke a chemotactic response in this cell lineage. This allowed us to hypothesize a receptor-mediated binding of the RNASET2 protein to the target cell surface. Intracellular signaling pathways in response to inflammatory chemokines are often triggered by the binding of signaling molecules to a cell-surface G protein-coupled receptor (19). Strikingly, when the in vitro migration assays on U937 cl.10 cells were carried out following pretreatment with pertussis toxin (PTx), a known inhibitor of G protein-coupled receptor function, cell migration in response to chemokine (C-C motif) ligand 2 (CCL-2) and recombinant RNASET2 protein was largely impaired (Fig. 3B). These results are consistent with the hypothesis of a receptor-mediated action of extracellular RNASET2.
Discussion
In the present study, we have extended some basic assumptions concerning the role of RNASET2 with regard to some of its pleiomorphic features, in particular those related to the control of cancer initiation. We have previously demonstrated that this RNase has the capacity to control ovarian tumorigenesis in vivo (15). Indeed, induced overexpression of RNASET2 was coupled with a strong reduction of ovarian tumorigenesis (15). The mechanism through which RNASET2 is acting was shown to take place in the microenvironment and consisted mainly of recruitment of M1-polarized host macrophages toward the site of tumor growth. Of note, this macrophage subpopulation is known to be associated with inhibition/suppression of cancer cells growth (19, 20). To gain more insights into the antineoplastic features of RNASET2, we reasoned that silencing its expression in ovarian cancer cell lines showing high endogenous levels of this protein would possibly be associated with the appearance of a cancer phenotype. Indeed, following their inoculation into nude mice, RNASET2-silenced OVCAR3 cells displayed completely different growth kinetics with respect to control OVCAR3 cells, with the appearance of conspicuous tumor masses just a few days after inoculation.
A closer examination of tumor sections revealed, among other distinctive features, a clear effect of RNASET2 silencing on the occurrence of stromal infiltrates. Indeed, comparative IHC analysis of the xenograft infiltrates revealed a substantial depletion of M1 macrophages in the RNASET2-silenced xenografts, suggesting that the microenvironment of control xenografts is consistently more tumor-inhibiting than the one in which RNASET2 is knocked down.
To provide molecular correlates of the RNASET2 induced biological responses, we looked at the in vivo gene expression profiles from xenograft samples. In this context, the results concerning the expression profile changes in the murine host stromal compartment are quite revealing. In general terms, our results substantiate the conclusions drawn from previous experimental data, which strongly suggest a role for RNASET2 in the innate immune response (16, 21, 22). In fact, members of the T2 RNase protein family were recently shown to modulate the mammalian innate immune system, as epitomized by the omega-1 protein secreted from Schistosoma mansoni, which is able to prime human and mouse dendritic cells for Th2 polarization of T cells (21, 22). Furthermore, a recent work from our group reported that the tumor suppressive role carried out by the RNASET2 gene in the Hey3Met2 ovarian cancer cell line model depends on the in vivo recruitment of cells from the macrophage lineage (15). Finally, a potential link between T2 ribonuclease-mediated modulation of the immune system and tumor suppression was recently established by a report showing that the Human T-lymphotropic virus 1 (HTLV-1) tax gene drives a strong down-regulation of the human RNASET2 gene (23). These experimental evidences, which collectively point to a role for RNASET2 in host immune response, are quite well recapitulated in the experimental data presented here. Indeed, not only is RNASET2 knock-down in OVCAR3 cells associated with increased tumor growth in vivo, but we could also detect the occurrence of a significant decrease of M1-polarized macrophage infiltration as a distinct feature of the RNASET2-silenced tumors. Within this frame, the in vivo mouse stromal gene expression profile provided a strong support to this hypothesis, as shown by the results of a GO search, which reported a significant increase in categories such as “immune response,” “defense response,” “innate immune response,” and “acute inflammatory response.” A confirmation of the role played by RNASET2 in modulating the innate immune response was also provided by a KEGG search, which showed a significant enrichment for the terms “chemokine signaling” (13 genes), “cytokine-cytokine receptor interaction” (12 genes), and “leukocyte transendothelial migration” (10 genes). A subsequent analysis by real-time PCR assay further showed that an increased number of mouse chemokine genes was actually affected in tumor xenografts following RNASET2 silencing. Finally, analysis of the in vivo expression profile for human genes in RNASET2-silenced xenografts reported the involvement of immune system-related pathways as well, such as “leukocyte activation” (12 genes) and “lymphocyte proliferation” (seven genes).
Taken together, these data strongly support the occurrence of an RNASET2-mediated tumor suppressive mechanism based on the establishment of cross-talk between cancer cells and cellular/structural components of the microenvironment, which results in an innate immune response against neoplastic cells. The basis of this presumed mechanism is being investigated by our group, and we started by demonstrating here that the RNASET2 protein actually displays chemotactic properties in vitro toward macrophage cells. Viewed from this perspective, these data allow us to hypothesize a role for RNASET2 similar to that of the alarmins (24), although we have to provide further experimental evidence for that. In this context, worth mentioning are our preliminary data indicating that RNASET2 protein expression levels are rather high in ovarian cancer cells from low-grade human ovarian tumors but progressively decrease in higher-grade/stage tumor samples. Concomitant with the immunological-based responses mediated by RNASET2, in this work, we also detected other mouse stromal gene expression profile changes that are mainly related to tissue structural/architectural features. Indeed, GO and KEGG analyses have highlighted several ECM component genes that are down-regulated in RNASET2-silenced ovarian xenografts. Several collagen genes and some matrix-remodeling enzymes were included in this list as well. Clearly, the observed down-regulation of these genes in the RNASET2-null xenografts is expected to affect the stromal-ECM compartment architectural features, which in turn would reorganize and impact the surrounding microenvironment. Further insights into RNASET2 stromal functions are also suggested by the observed down-regulation of several cell-adhesion molecules. Involvement in and modulation of cell-to-cell contacts seems therefore to be a prerogative of RNASET2 function. Worth mentioning are some cytoskeletal and cytoskeleton-associated genes that were also disclosed by GO analysis under the term “development” and whose expression turned out to be affected in RNASET2-silenced cells, suggesting that they could possibly impact the in vivo cell differentiation pattern of the tumor cells, as disclosed in our xenograft model. Thus, the gene expression profiles we have documented in vivo in our ovarian model system are basically in agreement with the apparent functional behavior endorsed by RNASET2 that has been proposed before now (16, 21–23). We emphasize its role in creating an environmental milieu that is basically tumor suppressive. The latter point is not irrelevant considering that some RNases are now in phase III clinical trials (25).
As a last point, we would like to comment on a recent finding suggesting the fallopian tube as a potential primary site of origin of high-grade serous ovarian carcinomas (5). More specifically, Kin et al. were recently able to demonstrate that the onset of the carcinogenetic process takes place in the stromal compartment of the fallopian tube in conditional Dicer/Pten double-knockdown mice (5). Intriguingly, these structures correspond to sites of strong RNASET2 expression, suggesting that this extracellular RNase might be, among other players, a good candidate for initiating and sustaining ovarian tumorigenesis.
In summary, we have investigated the putative mechanisms through which an extracellular RNase is involved in the control of ovarian tumorigenesis. The pleiomorphic functions of RNASET2 and the pathways that have been uncovered in the present study will likely contribute to shed some light on the dominant antitumoral role exhibited by the stromal/microenvironmental architectural determinants in ovarian cancer pathogenesis (6–9). Furthermore, interindividual genetic variation within all the molecular components comprising the networks where RNASET2 is likely to act might be exploited to better define the genetic basis of the cancer-resistance phenotype (7).
Materials and Methods
RNASET2 Knockdown in OVCAR3 Cells.
RNASET2 silencing was carried out by means of a lentiviral infection vector expressing an shRNA targeting nucleotides 814 to 832 within the RNASET2 mRNA (Genbank accession no. NM_003730.4). Oligonucleotide sequences for RNAi were generated with the pSicoOligomaker 1.5 software and subsequently aligned to the human genome sequence (UCSC Genome Browser) to test for specificity. Control scrambled sequences were designed having the same length and GC content as the test sequence, but with no significant match to the database. The sequences of the RNASET2-targeting and scrambled sequences are available on request. Sense and antisense oligonucleotides representing these sequences were annealed and cloned into HpaI/XhoI-digested pSicoR vector (provided by F. Nicassio, Fondazione Istituto FIRC di Oncologia Molecolare, Milan, Italy). The recombinant shRNA-expressing vectors were stably transfected into OVCAR3 cells with Lipofectamine 2000 (Invitrogen). The efficiency of RNASET2 knockdown was evaluated by Western blot analysis as described previously (14).
In Vivo Xenograft Studies.
Male nude mice 6 wk of age were challenged s.c. in the left flank with 2 × 106 OVCAR3 cells in 0.15 mL saline solution. Animals were monitored twice per week for weight and tumor growth, and killed at day 39. Separate experiments were performed with three different clones of both control and RNASET2-silenced cells (n ≥ 5 animals per clone). Animals were maintained in a pathogen-free facility and treated in accordance with European Union guidelines under the approval of the ethical committee of Ospedale San Raffaele (Institutional Animal Care and Use Committee protocol 418). Tumors were frozen for RNA extraction and microarray studies. Statistical significance of data was calculated by Student t test.
Histological and IHC Assays.
A total of nine formalin-fixed and paraffin-embedded tissue specimens from murine tumors were cut at 3 μm thickness and stained with H&E for routine histopathological analysis. The following parameters were evaluated by a senior pathologist (C.R.): type of growth, cell morphology, stroma infiltration, necrosis, apoptosis, and mitotic index. For IHC assays, formalin-fixed and paraffin-embedded additional sections were used. Sections were mounted on slides, deparaffinized and immersed in 10 mM sodium citrate, pH 6, and microwaved for antigen retrieval. Antibodies raised against the following markers were used: Ki-67 (Dako), CD68 (Dako), CD45RO (Dako), cytokeratin 7 (Ventana), anti-cleaved caspase-3 (Cell Signaling Technology), inducible nitric oxide synthase (Transduction Lab), and dectin-1 (Bioorbyt). Reactions were visualized by using an ABC immunoperoxidase system (Vector Labs) or UltraVision (Thermo Scientific) with DAB (Sigma) as chromogenic substrate. The extent of immunoreactivity in the samples was assessed by a senior pathologist considering the proportion of positive cells.
Gene Expression Profiling.
For microarray hybridization, RNA from tumor samples or cultured cells was purified and quantified by UV spectrophotometry. RNA quality was assessed with a model 2100 Bioanalyzer (Agilent). Cy3-labeled cRNA was generated from 500 ng input total RNA. For every sample, 1.65 µg labeled cRNA was hybridized to the SurePrint G3 Human or Mouse Gene Expression 8 × 60K Microarray (Agilent). The fluorescence intensities on scanned images were extracted and preprocessed by Agilent Feature Extraction Software (version 10.5.1.1). Quality control and array normalization was performed in the R statistical environment by using the Agi4 × 44PreProcess package downloaded from the Bioconductor Web site (www.bioconductor.org). The quantile normalization and default filtering steps were based on those described in the Agi4×44 PreProcess reference manual.
To detect expression differences among different cell populations a moderated t test was applied. Moderated t statistics were generated by Limma Bioconductor package, and modulated genes were chosen as those with a fold change greater than 1.5 and a P value lower than 0.01.
These genes were analyzed by using the WebGestalt Web site (http://bioinfo.vanderbilt.edu/webgestalt/) to identify GO category and KEGG pathways that are significantly overrepresented, using default settings (the top 10 pathways with the most significant P values found by a hypergeometric statistical test were identified). For GO analysis, the significance level for the adjusted P value was set at 0.01 and the number of minimum genes for a category was set at 2. For KEGG analysis, the top 10 results are shown, and the minimum genes for a category was set at 2. Microarray data were deposited in the National Center for Biotechnology Information Gene Expression Omnibus repository and are available under accession no. GSE43029.
Gene Expression Assays by Real-Time qPCR.
Total RNA was extracted from tumor samples with TriFast reagent (Euroclone) and reverse transcribed with random examers. Primers for real-time qPCR were selected within the same region of the mRNA used to design the probes for microarray experiments. Real time RT-PCR reactions were performed on ABI PRISM 7000 system (Applied Biosystems) in triplicate in a 25 μL volume containing target cDNA, 40 nM primers, 12.5 μL of Power SYBR Green Master Mix, and water. Samples were denatured at 95 °C for 15 s and annealed/extended at 60 °C for 1 min, for 40 cycles. Fluorescent signals generated during PCR amplification were monitored and analyzed with ABI PRISM 7000 SDS software (Applied Biosystems). Comparison of the amount of each gene transcript among different samples was made by using tubulin or GAPDH as reference. The amount of target RNA, normalized to the endogenous reference gene, was calculated by means of the difference in threshold cycle parameter (ΔΔCt). Statistical analysis was done by Student t test. All primer sequences are available on request.
In Vitro Assays.
Macrophages were obtained from buffy coat-derived human monocytes upon macrophage colony-stimulating factor (100 ng/mL) stimulation. LPS (100 ng/mL) plus IFN-γ (20 ng/mL) or IL-4 (20 ng/mL) was used for M1 and M2 differentiation, respectively. Macrophages were starved in complete medium plus 1% FCS for 2 h at 37 °C before the binding assay, which was performed by adding recombinant RNASET2 for 3 h at 4 °C.
Fc receptors were blocked by using 10% (vol/vol) human serum. Rabbit anti-human RNASET2 and normal rabbit IgG (as isotype control) were used to perform cell surface staining assay. Data were expressed as fold increase of mean fluorescence intensity of RNASET2-stained samples vs. isotype control-stained ones.
The U937 cl.10 cell line (provided by R. Accolla) is an HIV-permissive subclone derived from the human histiocytic lymphoma-derived U937. Cells were grown in RPMI/10% (vol/vol) FBS with 1% L-glutamine. The in vitro migration rate of U937 cl.10 cells was assessed in blind-well chambers (NeuroProbe) with 3-µm-pore polycarbonate filters separating the upper and lower chambers. Cells were resuspended in serum-free RPMI-1640 medium and incubated for 24 h at 37 °C, 5% CO2. PTx-pretreated and control U937 cl.10 cells were resuspended in serum-free medium at a final density of 6 × 105 cells/mL and placed in the upper compartment. Chemoattractant agents (8,172 µg/mL RNASET2; 100 ng/mL CCL2) were diluted in serum-free medium and added to the lower compartment. When reported, pretreatment with 1 µg/mL PTx (CalBiochem) was performed for 90 min at 37 °C, 5% CO2. After incubation, cells were washed twice in PBS solution and processed for the chemotaxis assay. Chambers were incubated for 21 h at 37 °C, 5% CO2, to allow migration to occur. After incubation, nonmigrated cells were scraped and filters were fixed, stained with H&E (Diff-Quik staining protocol; Medion Diagnostics), and mounted on slides (7.5 µg/mL DAPI in Vectashield mounting medium). DAPI-stained nuclei were counted, and relative migration values were calculated. As negative controls, the only protein storage buffers diluted in serum-free RPMI 1640 medium were used. Statistical analysis was performed by using two-sided Student t test for unpaired data.
Supplementary Material
Acknowledgments
This work was supported by Federico Ghidoni memorial funds (to F.A. and R.T.) and a grant from Fondazione Cariplo (to R.T.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE43029).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1222079110/-/DCSupplemental.
References
- 1.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58(2):71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Auersperg N, Maines-Bandiera SL, Dyck HG, Kruk PA. Characterization of cultured human ovarian surface epithelial cells: Phenotypic plasticity and premalignant changes. Lab Invest. 1994;71(4):510–518. [PubMed] [Google Scholar]
- 3.Feeley KM, Wells M. Precursor lesions of ovarian epithelial malignancy. Histopathology. 2001;38(2):87–95. doi: 10.1046/j.1365-2559.2001.01042.x. [DOI] [PubMed] [Google Scholar]
- 4.Kurman RJ, Shih IeM. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer—shifting the paradigm. Hum Pathol. 2011;42(7):918–931. doi: 10.1016/j.humpath.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim J, et al. High-grade serous ovarian cancer arises from fallopian tube in a mouse model. Proc Natl Acad Sci USA. 2012;109(10):3921–3926. doi: 10.1073/pnas.1117135109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bissell MJ, Hines WC. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med. 2011;17(3):320–329. doi: 10.1038/nm.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klein G. Toward a genetics of cancer resistance. Proc Natl Acad Sci USA. 2009;106(3):859–863. doi: 10.1073/pnas.0811616106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klein G, Imreh S, Zabarovsky ER. Why do we not all die of cancer at an early age? Adv Cancer Res. 2007;98:1–16. doi: 10.1016/S0065-230X(06)98001-4. [DOI] [PubMed] [Google Scholar]
- 9.Liotta LA, Kohn EC. The microenvironment of the tumour-host interface. Nature. 2001;411(6835):375–379. doi: 10.1038/35077241. [DOI] [PubMed] [Google Scholar]
- 10.Acquati F, et al. Cloning and characterization of a senescence inducing and class II tumor suppressor gene in ovarian carcinoma at chromosome region 6q27. Oncogene. 2001;20(8):980–988. doi: 10.1038/sj.onc.1204178. [DOI] [PubMed] [Google Scholar]
- 11.Acquati F, et al. Tumor and metastasis suppression by the human RNASET2 gene. Int J Oncol. 2005;26(5):1159–1168. [PubMed] [Google Scholar]
- 12.Monti L, et al. RNASET2 as a tumor antagonizing gene in a melanoma cancer model. Oncol Res. 2008;17(2):69–74. doi: 10.3727/096504008784523658. [DOI] [PubMed] [Google Scholar]
- 13.Acquati F, et al. Molecular signature induced by RNASET2, a tumor antagonizing gene, in ovarian cancer cells. Oncotarget. 2011;2(6):477–484. doi: 10.18632/oncotarget.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patel S, et al. RNASET2—an autoantigen in anaplastic large cell lymphoma identified by protein array analysis. J Proteomics. 2012;75(17):5279–5292. doi: 10.1016/j.jprot.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 15.Acquati F, et al. Microenvironmental control of malignancy exerted by RNASET2, a widely conserved extracellular RNase. Proc Natl Acad Sci USA. 2011;108(3):1104–1109. doi: 10.1073/pnas.1013746108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luhtala N, Parker R. T2 Family ribonucleases: Ancient enzymes with diverse roles. Trends Biochem Sci. 2010;35(5):253–259. doi: 10.1016/j.tibs.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shaw TJ, Senterman MK, Dawson K, Crane CA, Vanderhyden BC. Characterization of intraperitoneal, orthotopic, and metastatic xenograft models of human ovarian cancer. Mol Ther. 2004;10(6):1032–1042. doi: 10.1016/j.ymthe.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 18.Sica A, et al. Macrophage polarization in tumour progression. Semin Cancer Biol. 2008;18(5):349–355. doi: 10.1016/j.semcancer.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 19.Murphy PM. The molecular biology of leukocyte chemoattractant receptors. Annu Rev Immunol. 1994;12:593–633. doi: 10.1146/annurev.iy.12.040194.003113. [DOI] [PubMed] [Google Scholar]
- 20.Verreck FAW, et al. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proc Natl Acad Sci USA. 2004;101(13):4560–4565. doi: 10.1073/pnas.0400983101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Everts B, et al. Omega-1, a glycoprotein secreted by Schistosoma mansoni eggs, drives Th2 responses. J Exp Med. 2009;206(8):1673–1680. doi: 10.1084/jem.20082460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steinfelder S, et al. The major component in schistosome eggs responsible for conditioning dendritic cells for Th2 polarization is a T2 ribonuclease (omega-1) J Exp Med. 2009;206(8):1681–1690. doi: 10.1084/jem.20082462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Polakowski N, Han H, Lemasson I. Direct inhibition of RNAse T2 expression by the HTLV-1 viral protein Tax. Viruses. 2011;3(8):1485–1500. doi: 10.3390/v3081485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chan JK, et al. Alarmins: Awaiting a clinical response. J Clin Invest. 2012;122(8):2711–2719. doi: 10.1172/JCI62423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Makarov AA, Kolchinsky A, Ilinskaya ON. Binase and other microbial RNases as potential anticancer agents. Bioessays. 2008;30(8):781–790. doi: 10.1002/bies.20789. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.