

Abstract
The genome-scale model (GEM) of metabolism in the bacterium Escherichia coli K-12 has been in development for over a decade and is now in wide use. GEM-enabled studies of E. coli have been primarily focused on six applications: (1) metabolic engineering, (2) model-driven discovery, (3) prediction of cellular phenotypes, (4) analysis of biological network properties, (5) studies of evolutionary processes, and (6) models of interspecies interactions. In this review, we provide an overview of these applications along with a critical assessment of their successes and limitations, and a perspective on likely future developments in the field. Taken together, the studies performed over the past decade have established a genome-scale mechanistic understanding of genotype–phenotype relationships in E. coli metabolism that forms the basis for similar efforts for other microbial species. Future challenges include the expansion of GEMs by integrating additional cellular processes beyond metabolism, the identification of key constraints based on emerging data types, and the development of computational methods able to handle such large-scale network models with sufficient accuracy.
Keywords: constraint-based modeling, Escherichia coli , metabolic engineering, metabolism, network reconstruction
Introduction
Whole-genome sequencing along with decades worth of detailed biochemical and enzymatic data (e.g., bibliomic data) on microbial metabolism has led to the reconstruction of metabolic networks at the genome scale (so-called, GENREs or genome-scale reconstructions; Price et al, 2004; Feist et al, 2009; Henry et al, 2010; Thiele and Palsson, 2010). Integrating this information in a structured fashion has enabled its translation into computational models that can be used to calculate metabolic phenotypes (Palsson, 2009; Pfau et al, 2011; Lewis et al, 2012). In addition, other omics data types that have been generated can be interpreted in the context of a reconstruction and computational model to analyze cellular functions under specific conditions. Taken together, this information becomes a de facto knowledge base. Genome-scale models (GEMs) are a structured format of such a knowledge base that can be used to perform computational and quantitative queries to answer various questions about the capabilities of organisms and their likely phenotypic states (Palsson, 2006; Orth et al, 2010).
Escherichia coli is one of the most important model organisms in biology and its metabolic GEM has aided the development of microbial systems biology. The history of the metabolic network reconstruction process for E. coli, and the formulation and testing of its metabolic GEM now spans over a decade (Figure 1). One of the earliest studies to systematically analyze E. coli utilized a simplified constraint-based model of acetate overflow (Majewski and Domach, 1990). Subsequent pre-genome-scale studies expanded upon the constraint-based approach to include reactions involved in central carbohydrate metabolism, amino acid synthesis, and nucleotide synthesis to evaluate the biocatalyst production potential of E. coli (Varma et al, 1993; Varma and Palsson 1993) using flux balance analysis (FBA; Varma and Palsson, 1994a). The ability of FBA, in particular, and constraint-based modeling, in general, to quantitatively describe the metabolic physiology of E. coli observed experimentally (Varma and Palsson, 1994b) arguably solidified the value of systems level analysis in understanding microbial metabolism. The sequencing of the E. coli genome (Blattner et al, 1997), the advent of the lambda-red system for efficient genome manipulation (Datsenko and Wanner, 2000), and information readily available on annotated content of E. coli in databases and detailed biochemical reviews led to a steady increase in content of the E. coli GEM in the genomic era. Reconstruction efforts in the 2000s built off of successive versions, each adding new subsystems (e.g., fatty acid, alternate carbon metabolism, and cell wall synthesis, respectively) as the reconstructions strived to incorporate all of the existing content in literature and newly appearing data. Analysis of the rate at which new content was added to the latest metabolic GEM (Orth et al, 2011) indicates that mostly newly characterized content is now left to include in the reconstruction. Future expansion for the metabolic GEM is likely to come from characterizing promiscuity of known enzymes, and the addition of protein synthesis will open the door for more detailed examination of other cellular processes and integration with other omics data sets.
Figure 1.

History of the E. coli expression and metabolic reconstructions. Shown in the upper portion of the graph are 2 milestone efforts contributing to the reconstruction of the E. coli transcription and translation network, and shown in the bottom portion of the graph are 7 milestone efforts contributing to the reconstruction of the E. coli metabolic network. For each of the two reconstructions shown (Allen and Palsson, 2003; Thiele et al, 2009) in the upper graph, the number of included transcription units (blue diamonds), genes (green triangles) and components (purple squares) are displayed. For each of the seven reconstructions shown (Majewski and Domach, 1990; Varma and Palsson, 1993; Pramanik and Keasling, 1997; Edwards and Palsson, 2000; Reed et al, 2003; Feist et al, 2007; Orth et al, 2011) in the bottom graph, the number of included reactions (blue diamonds), genes (green triangles) and metabolites (purple squares) are displayed. Moreover, listed is noteworthy content expansion that each successive reconstruction provided over previous efforts. For example, Varma et al (1993), and Varma and Palsson (1993) included amino acid and nucleotide biosynthesis pathways in addition to the content that Majewski and Domach (1990) characterized. The start of the genomic era (Blattner et al, 1997) marked a significant increase in included components for successive iterations of the network reconstruction. The significant increase in the number of reactions in 2007 (Feist et al, 2007) was, in large part, due to the removal of many lumped reactions, which were often included for lipid and cell wall biosynthesis in earlier metabolic reconstructions. Thiele et al (2009) expanded the initial work of Allen and Palsson (2003) by increasing the scope of the transcription and translation network from a few example pathways to all known genes involved in protein synthesis (i.e., expression). Not included on the timeline is a metabolic reconstruction based upon Reed et al (2003), which was modified to include additional reactions from the KEGG (Kanehisa et al, 2008) database and incorporated into the MetaFluxNet software package (Lee et al, 2005).
In over a decade of model-driven development of systems biology for E. coli, over 200 peer-reviewed studies have appeared, as summarized in Figure 2 and Supplementary Figure S1, and as documented in greater detail in Supplementary Table S1. This review aims to determine the benefits and drawbacks of the uses of the E. coli GEM to date, what has been accomplished, what has been missed, and what is likely to lie ahead for this field in its next decade of development.
Figure 2.

The detailed usage of the E. coli metabolic GEM over time. The cumulative and new number of studies published per year separated according to (A) the metabolic reconstruction used (Edwards and Palsson, 2000; Reed et al, 2003; Feist et al, 2007; Orth et al, 2011), (B) in silico (i.e., strictly computational prediction) or combined in silico and in vivo (i.e., computational usage of the model and experimental validation or data generation guided by the model) and (C) the application category of the study. BD, model-driven discovery; BE, studies of evolutionary processes; II, interspecies interaction; ME, metabolic engineering; NA, analysis of network properties; PB, prediction of cellular phenotypes.
Categories of uses of GEMs
The E. coli GEM has been applied to answer different biological questions (Figure 3), most frequently in the categories of (1) metabolic engineering, (2) model-driven discovery, (3) prediction of cellular phenotypes, (4) analysis of biological network properties, (5) studies of evolutionary processes, and (6) models of interspecies interactions. In a previous review we described and categorized the uses of the E. coli GEM appearing in 64 papers published before 2007 (Feist and Palsson, 2008). Similarly, Oberhardt et al (2009) have reviewed applications enabled by GEMs for organisms other than E. coli through 2009, and specific reviews on GEM-enabled studies in plants (Sweetlove and Ratcliffe, 2011) and Saccharomyces cerevisiae (Osterlund et al, 2012) have recently appeared. However, the E. coli GEM remains the oldest and arguably the most extensively utilized GEM, and given the extensive uses of the E. coli GEM that have appeared since 2007, we now have a sufficient amount of studies to critically assess what GEMs can and cannot do by focusing on the E. coli GEM as a subject. For example, metabolic engineering and model-driven discovery are categories of uses in E. coli that have matured over the years into workflows that can be continuously repeated to tackle a diverse set of biological questions. In particular, strain design has matured from academic to industrial; thanks to the advent of sustainable processes that can be applied from one target compound to another. The development of these iterative workflows in systems biology is a theme that will be highlighted and discussed in greater detail as they appear in each category. In another example, recent studies modeling interspecies interactions signal an expansion of the field from single cell models to ecosystem level models. We have also highlighted a collection of noteworthy studies (Supplementary Figure S2) from the pool of uses that, we feel, demonstrate how a GEM can be well utilized to deduce biological complexities.
Figure 3.

Six categories of uses and number of studies for each use of the E. coli metabolic GEM. The original five categories defined in 2008 (Feist and Palsson, 2008) include (A) metabolic engineering, (B) model-driven discovery, (C) prediction of cellular phenotypes, (D) analysis of biological network properties and (E) studies of evolutionary processes. A new category has been added, (F) interspecies interaction. The addition of this category signifies a growing trend in the field to explore the interaction of the E. coli metabolic network with other organisms and across different environmental conditions. Specifically, studies have explored host/pathogen interactions (Jain and Srivastava, 2009), cocultures (Wintermute and Silver, 2010; Hanly and Henson, 2011; Tzamali et al, 2011), ecology (Klitgord and Segrè, 2010) and chemotaxis (Kugler et al, 2010). The number of studies in this category is expected to increase, as the interest in understanding the complexities of microbial interactions and ecosystems continues to grow. The complete lists of the studies for each category are included in Supplementary Table S1.
A detailed overview of the successes and limitations of the E. coli GEM implementation for the categories outlined above have been summarized in Table I and will be presented in detail in each section below. At the conclusion of each section, we will then assess how the limitations of GEMs can be overcome with further development and refinement. We also highlight systems biology workflows made possible by GEMs. Furthermore, we will attempt to identify the future challenges that need to be overcome to bring us closer to the ultimate goal of establishing a comprehensive and multi-scale mechanistic understanding of the genotype–phenotype relationship of microbial metabolism. It should be stressed that although this review is focused on the E. coli GEM, the findings documented here can be readily extended to metabolic modeling at the genome scale in general.
Table 1. Strengths and limitations of the metabolic GEM applications.
| Application | What the model can do | What the model cannot do |
|---|---|---|
| Strengths of the E. coli GEM | Areas for future progress | |
| Metabolic engineering | Gene deletion (combinatorial) | Limited coverage of molecular biology |
| Gene addition | Predicting the effects of perturbations to regulatory elements | |
| Gene over- and under-expression | Predicting allosteric inhibition | |
| Rapidly test the systemic effects of heterologous pathway additions | There is no explicit representation of metabolite concentrations | |
| Design biomarkers/biosensors for characteristic function | Account for enzyme kinetics | |
| Determine media supplementation strategiesMap high-throughput data to identify bottlenecks | Cannot accurately predict the performance of nonnative genes/proteins in E. coli | |
| Design strains through evolution | ||
| Biological discovery | Predict growth on different carbon sources/media conditions | Predict the regulation of isozymes/parallel pathways |
| Guide the functional assignment of network gaps | Predict enzyme promiscuity | |
| Guide the discovery of previously uncharacterized gene product functions (graph theory analysis) | Predictive power is inherently limited, because the model is not complete in scope | |
| Guide the reannotations of incorrectly annotated genes | Predict the expression of genes | |
| Connect orphan metabolites to known reactions | Predict the functional state of proteins (e.g., posttranslational modification) | |
| Phenotypic behavior | Predict optimal cellular behavior | Differentiate between computed alternate optimal flux distributions of the cell a priori |
| Understand energetics and occurrence of suboptimal behavior | Explain the reasons for suboptimal performance a priori | |
| Infer impact of regulationProvide a context for which experimental data can be interpreted | Provide a framework for incorporating additional regulatory interactions that are currently under development | |
| Predict and understand absolute and conditional gene essentiality | ||
| Predict and understand shifts in growth conditions | ||
| Network analysis | Evaluate metabolic networks from a systems view through node and link dependencies, essentialities, overall network robustness | Does not always include the biological mechanisms behind the network connectionsFew predictions can be experimentally validated |
| Describe the complex interactions of the components of the metabolic network | ||
| Evaluate modularity of function | ||
| Evaluate regulation based on network structure | ||
| Bacterial evolution | Predict essential genes | Account for changes in regulatory elements |
| Predict the endpoint of evolution | Predict the time-course of evolution | |
| Understand the basis for epistatic interactions and mutational effects | Predict location of mutations in the genomePredict the effects of mutations in the genome | |
| Provide insights into evolutionary trajectories | Account for strain-specific genomic differences | |
| Interspecies interaction | Model the exchange of metabolites | Model interactions that affect metabolic regulation |
| Analyze high-throughput data from different strains | Inability to measure flux exchange in multi cell-type systems | |
| Determine the cost/benefit ratio for different types of commensalism | There are still too many unknowns to accurately build an interactions network | |
| Limited ability to define individual genetic content in large communities | ||
| Limited spatial knowledge in large communities |
Metabolic engineering
Current industrial processes rely upon nonrenewable resources that cannot sustain the growing world population indefinitely. The development of biosustainable processes that can convert renewable resources into commodity items is therefore of paramount socio-economic importance. Bacteria have recently emerged as a means by which to achieve bio sustainability (Lee et al, 2012). Through metabolic engineering, the native biochemical pathways of bacteria can be manipulated and optimized to more efficiently produce industrial and therapeutically relevant compounds. The E. coli GEM has guided metabolic engineers towards the production of an assortment of compounds, including organic acids, amino acids, and alcohols to name a few (see Supplementary Table S3 for a comprehensive list).
Contrary to random mutagenesis and screening, rational strain design uses the GEM to predict cellular phenotypes from a systems level using genomic, stoichiometric, kinetic, and regulatory knowledge to identify engineering strategies, which can then be implemented in vivo. These strategies include gene deletions (Fong et al, 2005), gene over- and underexpression (Fowler et al, 2009), mapping high-throughput data onto the network reconstruction to identify bottlenecks or competing pathways (Lee et al, 2007), and more recently, integration of nonnative pathways into standard microbial production hosts for production of compounds that are either not natively found in, or only synthesized in minute concentrations by the host (Jung et al, 2010; Xu et al, 2011; Yim et al, 2011). More advanced methods have even allowed for the identification of strategies that couple bacterial growth to target product overproduction (Burgard et al, 2003; Patil et al, 2005; Kim and Reed, 2010). A so-called ‘growth-coupled’ strain leads to a more robust strain that is less likely to lose the genetically engineered genotype, or be outcompeted by alternate bacterial phenotypes in a bioprocess environment. A cadre of algorithms, first appearing in 2003 (Burgard et al, 2003), with increasing biological detail (Kim and Reed, 2010) or alternative optimization methods (Patil et al, 2005) have been developed (Supplementary Table S2) and extensively reviewed (Oberhardt et al, 2009; Copeland et al, 2012).
Engineering strategies found using model-driven analysis can often be nonintuitive and highlight some of the most interesting recent findings using GEMs. For instance, researchers used the GEM to not only determine a gene that needed to be upregulated, but were able to tune the expression level of this gene after subsequent GEM analysis of a deleterious overexpression event (Lee et al, 2007). In another GEM-enabled study, the highest flavanone production was predicted and experimentally determined by strategically knocking out genes to not only increase the production of the redox carrier (NADPH) to drive the heterologous flavanone catalyst, but also to maintain the optimal redox potential of the cell (i.e., the ratio of NADPH to NADP+) (Fowler et al, 2009; Chemler et al, 2010). For a final example, researchers improved the production potential of the nonnative metabolite 1,4-butanediol in E. coli by over three orders of magnitude (Yim et al, 2011). The researchers were able to rewire the host cell to produce the compound via native and nonnative pathways by ensuring that the production of 1,4-butanediol was the only means by which the host cell could maintain a redox balance and grow anaerobically (Yim et al, 2011). These successes highlight the need to analyze genetic alterations at the systems level where one can not only predict the activation of pathways that compensate for lost functionalities following gene deletions, but also predict engineering strategies that couple cellular goals to target compound overproduction.
Engineering strategies derived from the E. coli GEM have also led to nonviable and suboptimal phenotypes. Even in the above studies (Fowler et al, 2009; Chemler et al, 2010; Yim et al, 2011), the authors had to carefully select which predicted knockout designs were constructed in vivo, due to, e.g., known isozymes that result in nonviability when deleted simultaneously. Hence, a strong understanding of metabolic biochemistry is a prerequisite for successful strain design. Many potential engineering strategies cannot be addressed with the current generation of GEMs as they do not account for translational regulation and detailed enzyme kinetics. For example, strains generated using random knockouts via transposon libraries and screening for lycopene overproduction identified gene deletion targets in regulatory elements (Alper et al, 2005) that cannot be predicted by the current GEM. Similarly, as GEM does not account for optimal codon usage, the in vivo performance of nonnative genes and proteins cannot be predicted.
The E. coli GEM has tilted the field of metabolic engineering towards advanced rational strain design by enabling researchers to explore a vast native and nonnative genetic space in designing strains for improved metabolite production. More complete biochemical information will greatly aid metabolic engineering by allowing for genome-scale reconstructions that account for cellular functions beyond those accounted for in the metabolic models (e.g., regulation, expression, and enzyme kinetics). This implies the need for greater experimental method development to deduce the details of expression, posttranscriptional and posttranslational modifications, and enzyme kinetics. Advancements in pathway finding procedures to identify heterologous pathways that are not native to the host, and the techniques to optimize the expression of nonnative enzymes will expand the type of compounds that can be overproduced at an industrial scale. Overall, the ability of the E. coli GEM to aid systems level analysis for rational strain design will only continue to improve the speed with which viable production strains can be designed and constructed.
Biological discovery
There are aspects of bacterial functions that remain uncharacterized. Even in E. coli, the most studied and best-known bacterium, 34% of the genes have an unknown function (Orth et al, 2011). In order to more efficiently expand our current understanding of cellular functions, an iterative workflow is needed that allows researchers to (1) account for what is known, (2) identify gaps in our knowledge, and (3) allow for the design of experiments to elucidate these gaps. The E. coli GEM has enabled the implementation of such a workflow to discover new features of microbial metabolism (Supplementary Table S4).
The function of uncharacterized open reading frames (ORFs) can be elucidated by comparing growth phenotypes from in silico model predictions of gene deletion mutants to in vivo experimental data (Box 1). Discrepancies between GEM predictions and experimental results can point to where current knowledge is missing or where there are functional discrepancies. This, in turn, allows one to systematically formulate testable hypotheses. For example, incorrect predictions made for talAB mutants grown on xylose led the authors to discover a novel pathway catalyzed by the gene products of pfkA and fbaA (Nakahigashi et al, 2009). Various algorithms have been implemented to aid researchers in this process by parsing the vast number of biochemical pathways of metabolism to reconcile in silico growth predictions with experimental data (Reed et al, 2006; Satish Kumar et al, 2007; Barua et al, 2010). These algorithms suggest network modifications (including assignment of enzymatic function to uncharacterized ORFs) that can then be confirmed by researchers in vivo (Reed et al, 2006). For example, one study used a combination of graph-theory-based and comparative genomic analyses to identify yneI (sad) as the gene responsible for the NAD+/NADP+-dependent succinate semialdehyde dehydrogenase, which the authors experimentally confirmed (Fuhrer et al, 2007).
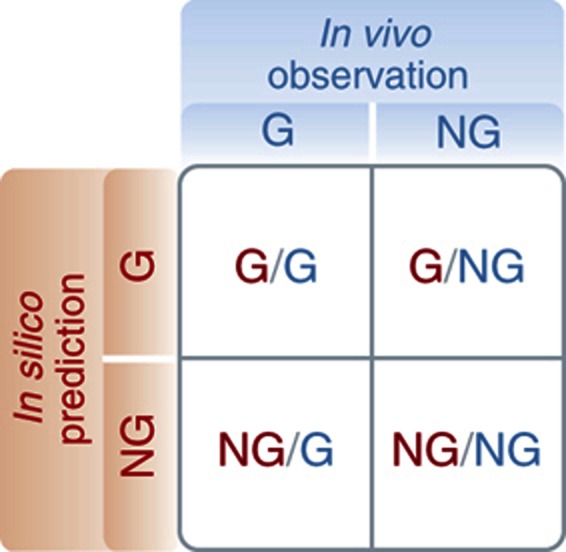
Growth phenotyping.
Growth phenotypes from single-gene deletion mutants based on in silico model predictions can be compared to in vivo experimental data to elucidate or confirm function of ORFs. The results of growth phenotyping studies can be classified into four categories:
·Growth/Growth (G/G): the model and experimental data show growth
·Growth/No Growth (G/NG): the model predicts growth, but the experimental data indicates no growth
·No Growth/Growth (NG/G): the model predicts no growth, but the experimental data indicates growth
·Growth/No Growth (NG/NG): the model and experimental data show no growth.
The G/NG case indicates that the model over-estimates the metabolic capabilities of the organism, while the NG/G case indicates that the model under-estimates the metabolic capabilities of the organism. Metabolic over-predictions are commonly caused by reactions that are absent in vivo, reactions that are down-regulated or inhibited under a specific environmental condition, or the biomass formulation is includes an erroneous metabolite. Metabolic under-predictions often represent knowledge gaps in that the model does not account for an unknown isozyme, parallel pathway, or some other functionality of the organism. G/G can be regarded as a consistency check and NG/NG can be regarded as a form of model validation.
 |
Box 1 Figure The four categories of growth phenotypes.
Although the GEM-enabled workflow (Covert et al, 2004; Joyce et al, 2006; Reed et al, 2006; Barua et al, 2010; Holm et al, 2010) has advanced our understanding of metabolism greatly, many aspects of bacterial metabolism are still waiting to be uncovered. One such aspect is the transcriptional regulation of metabolism. Researchers have attempted to integrate the transcriptional regulatory network (TRN) with the metabolic network to better understand and predict regulation. For example, the TRN was used to elucidate changes in expression of oxygen regulators between oxic and anoxic conditions (Covert et al, 2004). In another example, the TRN was used to confirm and refine the regulatory and functional assignment of various regulatory and metabolic genes, which included the novel finding that D-allose induces rpiR (Barua et al, 2010). However, the Boolean formulation of the TRN regulatory rules only allows one to model regulatory interactions as either on or off. Consequently, complex regulatory interactions involving a multitude of transcription factors, binding constants, and environmental dependencies along with posttranscriptional and posttranslational modifications that may account for in silico and in vivo discrepancies cannot be identified using the current GEMs.
Efforts are underway to better understand bacterial regulation. Large-scale, genome-wide screens to deduce the function of transcriptional regulators and the development of new formalisms to account for and integrate transcriptional regulation in the model are in progress (Cho et al, 2011). RNA sequencing-based technology will greatly assist researchers in elucidating the interactions of the transcriptional network by providing a richer data set than the existing methods (e.g., RNA microarrays and ChIP-chip; Cho et al, 2011). Other posttranscriptional and posttranslational modifications (these include, for instance, by small RNA at the transcript level or by phosphorylation, methylation, glycosylation, acetylation, or carboxylation, to name several, at the protein level) also contribute to the regulation of metabolic function in prokaryotes. Exemplary experimental efforts to better understand small RNA regulation of transcription (Wassarman and Saecker, 2006), and the conservation of phosphorylation in serine, tyrosine, and threonine metabolism (Macek et al, 2008) have demonstrated that the link between gene expression and metabolite profiles are far more complex than once thought. Continued experimental efforts and improved computational efforts to deduce and model the complexities of regulation will be needed to expand the scope of biological discoveries that the current model can assist with.
Phenotypic functions
For simple organisms, the physiology of bacteria is remarkably complex. The diverse set of biochemical pathways in bacteria have conferred a vast phenotypic potential that have enabled them to thrive in a plethora of environments ranging from volcanic vents on the bottom of the ocean, clouds, glaciers, and the human gut. In order to understand this phenotypic potential, researchers have turned to GEMs to interpret and predict cellular phenotypes. Constraint-based modeling with GEMs (Palsson, 2006) has allowed researchers to rapidly predict growth phenotypes in various genetic (Fong and Palsson, 2004) and environmental (Ibarra et al, 2002) conditions, explore different objectives of microbial metabolism (Schuetz et al, 2007; Ow et al, 2009) to examine the driving force behind cellular function, and better understand the suboptimal behavior of cells following perturbation (Segre et al, 2002; Link et al, 2010) and latent pathway activation (Nishikawa et al, 2008).
When phenotypic predictions are made using the GEM, one finds a large solution space of potential phenotypes that would allow the organism to survive in a given genetic and environmental background. Many of these solutions of metabolic network usage that would allow for survival may not be observed under physiological conditions. Consequently, researchers have formulated ways to confine the solution space to more accurately represent the experimentally observed phenotype of the cell for a given growth condition by incorporating constraints. Regulatory control of the metabolic genes provides a means to constrain the allowable solution space by specifying what genes are active in the metabolic network for a given environmental condition. Researchers have been able to show that while over 50% of the flux distribution is constrained by metabolism in a given environmental condition, an additional 20% can be attributed to the TRN (Shlomi et al, 2007). In addition, the TRN allows the organism to rapidly and efficiently adapt its metabolism to a wide range of environmental conditions by altering the expressed metabolic genes (Barrett et al, 2005; Samal and Jain, 2008). Spatial constraints in the form of molecular crowding (Beg et al, 2007; Vazquez et al, 2008), growth-associated metabolite dilution (Benyamini et al, 2010), membrane occupancy (Zhuang et al, 2011), super coiling of the DNA (Sonnenschein et al, 2011), and indirect protein–protein interactions to facilitate the organization of enzymes in the cytoplasm (Perez-Bercoff et al, 2011) have shown promise for increasing the predictive power of the E. coli GEMs. For instance, a mechanistic constraint on the available space on the cytoplasmic membrane was introduced to better explain respiro-fermentation physiology (Zhuang et al, 2011). Researchers have also shown that the physical structure of the DNA correlates more closely to the metabolic state than the regulatory network (Sonnenschein et al, 2011). Thermodynamic analysis provides another means to constrain the solution space by removing infeasible reaction loops that violate energy balance (Ederer and Gilles, 2007; Fleming et al, 2009), by refining reaction directionality, and by defining allowable flux ranges via calculation of the in vivo change of Gibbs free energy of reactions (Kümmel et al, 2006; Henry et al, 2007; Flamholz et al, 2012). Intimately tied to thermodynamic analysis is the integration of high-throughput metabolomics data. Several studies have incorporated metabolomics data into the E. coli GEM to better calculate the in vivo change in Gibbs free energy of reaction in order to better confine the feasible flux range of reactions and identify reactions that are potentially under allosteric regulation (Zamboni et al, 2008; Yizhak et al, 2010).
The metabolic model can be used as a scaffold onto which high-throughput data types, such as fluxomic (Herrgard et al, 2006; Choi et al, 2007; Chen et al, 2011), transcriptomic (Becker and Palsson, 2008; Portnoy et al, 2010), and proteomic data (Lewis et al, 2010), can be mapped to gain insight into context-specific phenotypes. Fluxomics data can be used to directly compare intracellular flux distribution as predicted by constraint-based models (Herrgard et al, 2006; Chen et al, 2011) using the GEM and can even be incorporated as additional constraints (Choi et al, 2007). Recent work has also demonstrated that fluxomic data can be integrated into a computational framework to explain suboptimal behavior as a trade-off between near optimal growth under one condition, and the ability to quickly adapt to a new growth condition (Schuetz et al, 2012). Transcriptomic data provide the experimentalist with a powerful means to decipher the phenotypic behavior of the cell due to its ability to qualitatively or quantitatively determine what genes are expressed by the cell under the given experimental conditions (Portnoy et al, 2010). A combination of transcriptomic and proteomic data have also allowed researchers to better understand the physiology of adapted strains and the mechanism for this adaption (Lewis et al, 2010).
Although the E. coli GEM has aided our understanding of cellular metabolism, it has limitations. For instance, one should be aware that alternate optimal flux distributions of the cell may confound the researcher’s ability to determine the true physiological state. This has been demonstrated when comparing fluxomic data to in silico predictions (Chen et al, 2011) and also in studies of adapted E. coli mutants (Fong and Palsson, 2004; Charusanti et al, 2010) where variations found in evolved replicates reflected the possible existence of multiple flux distributions that lead to equivalent growth phenotypes. The methods to predict cellular physiology present a user bias in the form of an objective function that must be validated for specific growth conditions (Schuetz et al, 2007). The suboptimal state of the cell can also be predicted, but the most utilized method (Segre et al, 2002) provides little insight into the biological driving force for suboptimal performance (Shlomi et al, 2005). It appears instead that a Pareto optimal solution of multiple (and at times) conflicting objectives can better explain the biological significance of suboptimal behavior than any one method (Schuetz et al, 2012). Incorporating thermodynamic constraints has been demonstrated to greatly reduce the solution space of metabolism. Unfortunately, the calculation of Gibbs free energy of reaction is hampered by the limited availability of experimentally determined standard Gibbs free energy of formation for a majority of the metabolites in the E. coli GEM. Therefore, the free energies of reaction can only be estimated (Jankowski et al, 2008).
Advancements in methods to obtain and integrate high-quality omics data with the model will aid in overcoming many current limitations in accurately predicting phenotypic behavior. For example, genome-scale metabolomics is hampered by the biochemical diversity, range of physiological concentrations, and chemical liability of the species that comprise the intracellular metabolome. Consequently, multiple analytical platforms and well-tested analytical procedures (Buscher et al, 2009) are needed to accurately assay the full metabolome, which is costly, time-consuming, and technically challenging. The enzyme kinetic and thermodynamic information that can be obtained from metabolomics can directly improve the accuracy of the metabolic model, and can also be correlated with gene expression profiles (Jozefczuk et al, 2010) to assist in unraveling the dynamics between transcriptome and metabolome. Another promising area is the formulation of a genome–scale isotope mapping model (Ravikirthi et al, 2011) for implementation with metabolic flux analysis. An expansion of metabolic flux analysis to the genome–scale and the ability to determine the intracellular distribution of atoms beyond carbon will enhance our knowledge of in vivo flux states.
Biological network analysis
The metabolic reaction network is a highly complex, interwoven, and nonlinear system that responds to environmental and genetic perturbations. In order to elucidate and understand the relationship between the network structure and function, many researchers have turned to network analysis. This exercise is mathematical in nature. In network analysis, biochemical reactions are transformed into a unipartite or bipartite graph, where the nodes and links take the form of metabolites and enzymatic reactions. Once formulated as a graph, the network can be sampled and explored using a variety of minimally biased mathematical and algorithmic methods to arrive at biologically insightful conclusions. The following paragraphs will focus on the most recent advances in biological network analysis; the reader is referenced to Feist and Palsson (2008) and Oberhardt et al (2009) for less recent examples not covered in the main text.
Much progress has been made in the analysis of link and node essentiality, whereby the consequences of removing a link (i.e., the reaction) from the network is examined. As links have varying degrees of dependence upon one another, one must look to higher-order combinations of link removals to better understand the network properties of the E. coli GEMs. For example, synthetic lethals, which are defined as two genes whose independent deletion is not lethal, but simultaneous elimination is lethal, are often a consequence of network redundancy or parallel pathways (Ghim et al, 2005). The converse of synthetic lethals, synthetic rescues, which are defined as a gene pair where the deletion of one of the genes is lethal, but the simultaneous deletion of both genes is nonlethal, can be used to rescue a nonviable single-gene deletion phenotype by rewiring the network in such a way as to compensate for the deleterious effect of the initial genetic perturbation (Motter et al, 2008; Kim and Motter, 2009). To illustrate, it has been shown that the overexpression of udhA improves the growth of E. coli pgi knockout strains on glucose minimal media (Kim and Motter, 2009). Higher-order epistatic interactions have also been analyzed to predict nonintuitive combinations of lethal and auxotrophic-inducing/rescuing gene deletions (Suthers et al, 2009).
It is important to emphasize that although the E. coli metabolic network is analogous to other interaction networks (e.g., the internet) and can be interrogated using network theory (Almaas et al, 2004; Samal et al, 2011), the E. coli network is unique in that it is a biological network that describes a highly evolved function. Network analysis can easily be taken out of context or provide little insight if the function of the metabolic network is not taken into account. For example, graph theory can deduce the topological properties of the model without providing any information about the underlying biology. A prerequisite of biologically meaningful network analysis is a biologically functional random network, to which one can compare the properties of the E. coli GEM (Samal and Martin, 2011; Basler et al, 2012). However, the in vivo experimental validation of such comparisons (i.e., to a random network) is infeasible. In addition, many of the network analysis methods become computationally challenged for large biochemical networks. Examples include elementary mode (Schuster et al, 1999) and extreme pathway analyses (Schilling et al, 2000), which have been, for the most part, limited to small-scale networks due to the combinatorial explosion inherent to the methods (Klamt and Stelling, 2002; Yeung et al, 2007). It should be noted that numerical efforts have been made to scale pathway analysis to GEMs by calculating only a subset of elementary modes (Wessely et al, 2011). In addition, the E. coli metabolic network model is subject to iterative updates. Analyses obtained between older and newer models can lead to vastly different results. For instance, 81% of the coupling relations identified using flux coupling analysis changed between iJR904 and iAF1260 due to missing reactions in the older network model (Marashi and Bockmayr, 2011).
Network analysis using GEMs has largely been depicted as a strictly in silico undertaking. Recent progress, however, indicates that network analysis has practical applications. For example, the recent advances in elementary mode analysis can be readily extended to strain design (de Figueiredo et al, 2009) and nonnative pathway finding procedures (Larhlimi et al, 2012). In another example, progress has been made in applying network analysis of the E. coli metabolic GEM to discover novel drug targets (Plaimas et al, 2008; Kim et al, 2010; Shen et al, 2010). The E. coli GEM is particularly suitable for this application by enabling pharmaceutical researchers to analyze the complex interactions of the network as a whole, to elucidate target links and nodes that would allow for complete system collapse or would severely cripple the network if removed. A potential viable workflow for antimicrobial drug discovery was recently presented (Shen et al, 2010). The workflow invoked network analysis to identify novel antimicrobial targets combined with computational screening to identify inhibitory molecules against them followed by experimental validation (Shen et al, 2010). This GEM-aided workflow could reduce the expensive and time-consuming experimental methods in drug discovery (Shen et al, 2010), and is readily extendible to other bacterial species.
Bacterial evolution
The genomic content and phenotypic landscape of bacterial species are constantly adapting to meet the demands of the imposed environmental conditions. Adaption occurs via elimination of individual reactions by loss-of-function mutations, alterations in gene expression and enzyme capacity, alterations in enzyme kinetics, and through the addition of new reactions by horizontal gene transfer, gene duplication, and gain-of-function mutations. The E. coli GEM has been proven to be most useful in modeling microbial evolution through elimination and addition of new metabolic network content, and acting as a scaffold to aid in the understanding of bacterial evolution.
Computational frameworks using GEMs have been employed to simulate bacterial evolution through random gene deletions. These studies have shown that there appears to be a conserved reaction set that is similar for organisms with similar lifestyles (Pal et al, 2006), which reflects the common enzymatic machinery required to metabolize specific carbon sources. It has also been shown that although genes are lost at random, the order in which genes are lost follows a coordinated and consistent pattern—40% of which can be accounted for by the metabolic model when compared with available phylogenic data (Yizhak et al, 2011). The E. coli GEM also provides a context by which phylogeny data can be understood. Comparative genomics in the context of constraint-based modeling with the E. coli GEM has led researchers to assert that the dominant mechanism of bacterial evolution in E. coli appears to be horizontal gene transfer. Horizontal gene transfer is highly dependent upon the genomic content of the organism (Pal et al, 2005b; Notebaart et al, 2009) and involves genes that are mostly environment-specific and located at the periphery of the metabolic network (Pal et al, 2005a).
The E. coli GEM can act as a scaffold on which similar bacterial strains can be reconstructed, and their divergent evolutions understood. Because of the high standard of biochemical accuracy of the E. coli metabolic GEM (e.g., 97% of the included genetic content of the most recent E. coli GEM has been experimentally validated (Orth et al, 2011), many researchers have based the reconstruction of specific pathways or the entire organism on the reactions of the E. coli GEM (e.g., Salmonella typhimurium ; AbuOun et al, 2009). More recently, the pangenome of the species E. coli was reconstructed based on iAF1260, and was used to generate five strain-specific GEMs that include commensal strain K-12 W3110, two enterohemorrhagic O157:H7 strains EDL933 and Sakai, and two uropathogenic strains CFT073 and UTI89 (Baumler et al, 2011). The study found that pathogenic E. coli appear to be more adapted to growth under anaerobic conditions than commensal E. coli (Baumler et al, 2011). The use of the E. coli GEM to rapidly construct strain-specific models will continue to increase, particularly as the cost of genome sequencing of microbes continues to fall and the available number of sequenced and annotated strains continues to rise.
Although the metabolic model allows a vast region of genotypic space to be explored in order to model and understand bacterial evolution, the space is currently limited to metabolic genes. Changes in the regulation of metabolism during evolution are not accounted for in GEMs. Although horizontal gene transfer and gene loss can be modeled up to the resolution of a core metabolic gene set (Pal et al, 2006), and the predicted gene-loss order can be compared with evidence provided by comparative genomics (Yizhak et al, 2011), limitations remain in determining the precise genes and their exact loss, the location of mutations in the genome, and predicting their effect on the physiology of the organism. In addition, comparison of the evolutionary trajectories of different bacterial strains is hampered by the fact that strain-specific portions of the genomes remain largely uncharacterized.
The use of GEMs in modeling and understanding bacterial evolution will benefit from studies of adaptive laboratory evolution (ALE). ALE is an experimental procedure that introduces a selection pressure (e.g., fastest growth) in a controlled environmental setting that allows for a time-resolved depiction of changes in the organism’s genome that occur during the process of adaption (Conrad et al, 2011). These changes can then be reintroduced and their effect on the organism’s fitness studied (Herring et al, 2006; Conrad et al, 2009; Lee and Palsson, 2010; Applebee et al, 2011). The GEM provides a context for understanding these mutations by allowing the researcher to model the growth physiology of the adapted network.
Interspecies interaction
There is a growing interest in better understanding host–pathogen interactions for the development of improved antimicrobials (Lebeis and Kalman, 2009), the use of microbes for environmental remediation (Singh et al, 2011), and for understanding and manipulating the microbiome of the human gut for improved health (Walter et al, 2011). Such applications would benefit greatly from a platform that would allow for the prediction and simulation of biological interactions. The E. coli GEM provides such a platform and has been successful in modeling the exchange of metabolites (Box 2) between different cell types (e.g., microbial species) and environmental conditions (Jain and Srivastava, 2009; Klitgord and Segrè, 2010; Wintermute and Silver, 2010).
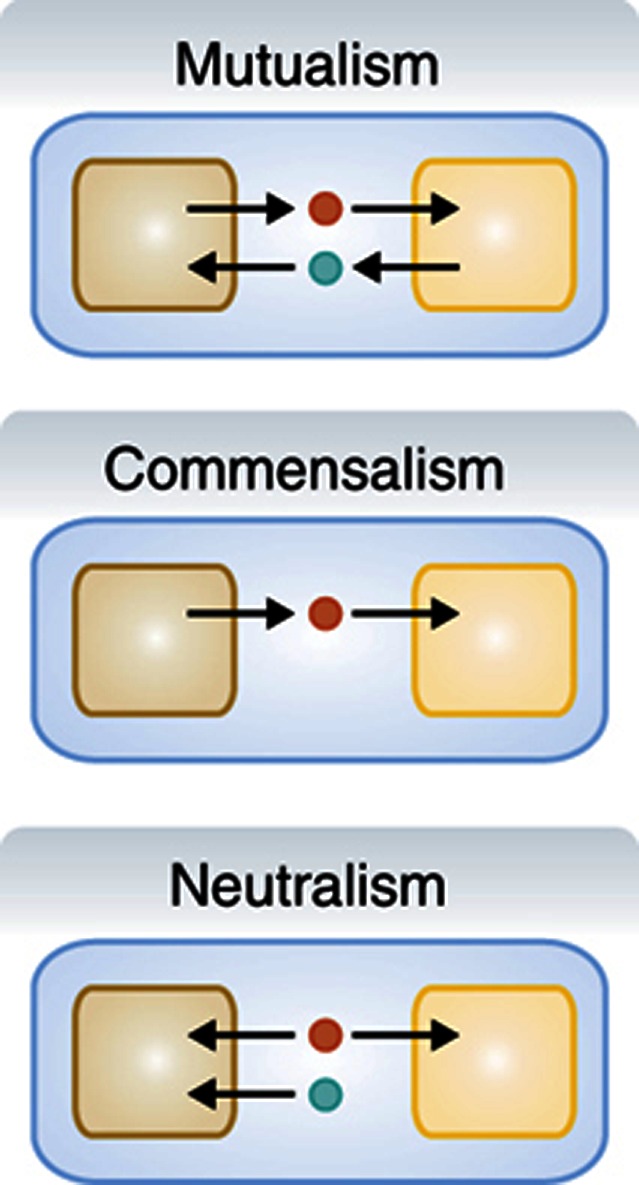
Microbial interactions .
A microbial interaction can be defined and modeled as an exchange of molecules between species in a given environment. There are three main types of interaction classes between microbial species:
Mutualism, also known as syntrophy or symbiosis, is where each organism produces an essential metabolite needed to support growth by the other organism.
Commensalism is where only one organism depends on the other for the production of an essential nutrient to support growth. A special case of commensalism, known as parasitism, is when the organism providing the essential nutrient comes at the cost of reduced fitness. Host/pathogen interactions are a type of parasitism.
Neutralism is where each organism can sustain growth in a given environment without the presence of the other organism. As each species are consuming the same resources, competition can arise between the organisms.
 |
Box 2 Figure Microbial interactions as defined by metabolite exchange.
Although interaction classes of interspecies interactions have been established, the mathematical formalism to model these interaction classes using GEMs has arisen only recently (Jain and Srivastava, 2009; Klitgord and Segrè, 2010; Wintermute and Silver, 2010; Hanly and Henson, 2011; Tzamali et al, 2011). Researchers have modeled host–pathogen interactions by directly incorporating pathogenic reactions into the stoichiometry of the host reaction network (e.g., to account for viral amino acid and nucleotide synthesis (Jain and Srivastava, 2009). Concatenated and joint stoichiometric models have been employed to study the exchange of metabolites between species (e.g., cocultures; Wintermute and Silver, 2010; Hanly and Henson, 2011) and the environment (Klitgord and Segrè, 2010). In contrast to multicellular stoichiometric models, which assume that the collection of microbes seeks to maximize the collective biomass, researchers have developed multicompetitor metabolic models to describe microbial communities, where each member seeks to maximize their own biomass (Tzamali et al, 2011). Together, these studies have found that the combined metabolisms of multiple species can utilize the capabilities of the environment better than a single species. Furthermore, although not every metabolic interaction is beneficial, metabolic interactions necessitate community formation.
Much work remains in the field of modeling interspecies interactions. For example, the measurement of metabolites exchanged between cells, needed to validate the accuracy of the in silico predictions, presents a strong technical challenge. In addition, the effect of biological interactions on regulatory elements is not yet accounted for. For instance, the extent to which other strains and the environment influence the regulation of metabolism (e.g., through quorum sensing) is unclear. Moreover, most of the large-community models do not differentiate the genetic content between individual species nor account for their spatial organization. Consideration of regulation, individual genetic content, and spatial organization will be needed to more accurately model and predict community-level biological processes (Zengler and Palsson, 2012).
The advent of single-cell sequencing technology (Tang et al, 2009) and other single-cell assays (Taniguchi et al, 2010) will benefit the study of interspecies interactions with GEMs. Such a ‘bottom-up’ approach would allow for the characterization of individual biological entities through, e.g., genomics and transcriptomics of individual species. From a ‘top-down’ approach, the total interaction of microbial communities can be characterized through genome-scale omic data. Genome sequencing and reconstruction of other bacterial species through manual curation or automatic reconstruction (e.g., model SEED; Henry et al, 2010), and the mapping of metabolite and reaction identifiers between reconstructions (e.g., RxnMet (Kumar et al, 2012)) will expand the number of species interactions that can be simulated together. On the basis of the genome-scale omic data that correspond with single cell data, developments in bioinformatics approaches that establish the relationships between individual biological entities and the growing number of reconstructions will allow researchers to piece together the contribution of each member on the biological community in order to generate complete community-level models.
In closing: what is likely for the future of GEMs
The number of applications focused on E. coli that have utilized the metabolic GEM have grown in size and scope (Feist and Palsson, 2008). This review distilled into six categories the ∼200 studies that have appeared in peer-reviewed manuscripts over the past 12 years. In each category, key examples and success stories were summarized and presented. In addition, we critically analyzed the current status of applications using the E. coli metabolic GEM to demonstrate what the model can and cannot do, and discussed the developments needed to overcome current limitations, as summarized in Table I. To help summarize the impact of GEM-aided analyses thus far, Supplementary Figure S2 highlights studies that have made a significant contribution to the E. coli GEM in particular, and our understanding of microbial metabolism, in general. Researchers are now able to complete the systems biology workflow and generate new biological knowledge with the help of the E. coli GEM (Figure 4). It should be emphasized that although this review has focused on the metabolic network, it is but one of several networks actively at work inside the cell (Feist et al, 2009).
Figure 4.

Iterative workflows. (A) A generic network reconstruction and model-driven systems biology workflow is a cyclic path that iterates between in silico predictions and in vivo observations. This general process has been followed in some of the more influential studies presented in this review. DNA sequencing and bibliomic data can be used to reconstruct and translate a biological system into a mathematical structure. Other omics data types that have been generated can be interpreted in the context of a reconstruction and computational model to analyze organism functions under specific conditions. This information becomes a de facto knowledge base that can be queried through a consortium of analytical methods. The aim of these methods is to hypothesize answers to complex biological questions that can often be nonintuitive or not readily apparent. Experiments can then be designed to test these predictions in order to either confirm GEM-derived explanations or move researchers one iteration closer to the answer. Studies that have successfully iterated through the E. coli GEM workflow that are presented as examples include (B) Reed et al (2006), (C) Shen et al (2010), (D) Yim et al (2011) and (E) Nakahigashi et al (2009).
The GEM of E. coli will continue to expand, as more cellular processes are mechanistically detailed and added to the organized GEM structure. The next significant increase in the applications of an E. coli GEM will likely come from mechanistically incorporating and integrating protein synthesis with metabolism. The integration of the transcriptional and translational machinery on the genome scale (Thiele et al, 2009, 2012) has now been completed. The operon structure that accounts for cellular regulation will follow protein synthesis as the next logical step of GEM expansion. The incorporation of DNA structure and transcription binding as a ready-to-compute biochemical network in a mathematical format would overcome the limitations presented by the current Boolean formulation of the TRN, and allow for complex regulatory interactions to be mechanistically modeled and predicted. It is conceivable that DNA synthesis, posttranslational modifications, and other cellular processes that involve biochemical interactions that can be described by a biochemical interaction network can also be incorporated into GEMs. In brief, what lies ahead for GEMs is the iterative expansion to include other cellular processes beyond metabolism, with the aid of omics data and the mathematical formalisms to model them (Figure 5). It is unclear which high-throughput data types and algorithms will be the major drivers for many of the applications enabled by GEMs with an expanded scope. However, it is clear that modeling with such expansive networks, whose components will carry activities across many orders of magnitude, will require greater computational accuracy and power given their size. Furthermore, the payoff for this increased complexity will be more accurate phenotype predictions after initial validation and gap filling is performed. GEM expansion will be a substantial but worthwhile endeavor that will unite many diverse aspects of microbiology and move the community closer to the ultimate goal of establishing a comprehensive mechanistic understanding of the genotype–phenotype relationship of microbes.
Figure 5.

The future of the E. coli GEM. The most widely used E. coli reconstruction accounts only for metabolism (the ‘M’ matrix) (Feist et al, 2009). However, efforts are currently underway to integrate the operon structure that determines cellular regulation (the ‘O’ matrix), the transcriptional and translational machinery allowing for the expression of proteins (the ‘E’ matrix; Thiele et al, 2009) and other cellular processes (e.g., DNA replication, posttranslational modifications, etc.) with metabolism. The integration of these cellular processes, supported by high-throughput data types, into a single mathematical model, will allow researchers to more accurately compute complex phenotypes, and will guide the discovery of unknown aspects of cellular functions beyond that of just metabolism.
Supplementary Material
Acknowledgments
This work was funded by the NIH R01 GM057089 and The Novo Nordisk Foundation. We would like to thank Marc Abrams, Joshua Lerman, and Edward O’Brien for valuable input when composing the manuscript.
Footnotes
The authors declare that they have no conflict of interest.
References
- AbuOun M, Suthers PF, Jones GI, Carter BR, Saunders MP, Maranas CD, Woodward MJ, Anjum MF (2009) Genome scale reconstruction of a salmonella metabolic model. J Biol Chem 284: 29480–29488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen TE, Palsson BØ (2003) Sequence-based analysis of metabolic demands for protein synthesis in prokaryotes. J Theor Biol 220: 1–18 [DOI] [PubMed] [Google Scholar]
- Almaas E, Kovacs B, Vicsek T, Oltvai ZN, Barabasi AL (2004) Global organization of metabolic fluxes in the bacterium Escherichia coli. Nature 427: 839–843 [DOI] [PubMed] [Google Scholar]
- Alper H, Miyaoku K, Stephanopoulos G (2005) Construction of lycopene-overproducing E. coli strains by combining systematic and combinatorial gene knockout targets. Nat Biotechnol 23: 612–616 [DOI] [PubMed] [Google Scholar]
- Applebee MK, Joyce AR, Conrad TM, Pettigrew DW, Palsson BØ (2011) Functional and metabolic effects of adaptive glycerol kinase (GLPK) mutants in Escherichia coli. J Biol Chem 286: 23150–23159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett CL, Herring CD, Reed JL, Palsson BØ (2005) The global transcriptional regulatory network for metabolism in Escherichia coli attains few dominant functional states. Proc Natl Acad Sci USA 102: 19103–19108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barua D, Kim J, Reed JL (2010) An automated phenotype-driven approach (GeneForce) for refining metabolic and regulatory models. PLoS Comput Biol 6: e1000970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basler G, Grimbs S, Ebenhoh O, Selbig J, Nikoloski Z (2012) Evolutionary significance of metabolic network properties. J R Soc Interface 9: 1168–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumler D, Peplinski R, Reed J, Glasner J, Perna N (2011) The evolution of metabolic networks of E. coli. BMC Syst Biol 5: 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker SA, Palsson BØ (2008) Context-specific metabolic networks are consistent with experiments. PLoS Comput Biol 4: e1000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beg QK, Vazquez A, Ernst J, de Menezes MA, Bar-Joseph Z, Barabasi AL, Oltvai ZN (2007) Intracellular crowding defines the mode and sequence of substrate uptake by Escherichia coli and constrains its metabolic activity. Proc Natl Acad Sci USA 104: 12663–12668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benyamini T, Folger O, Ruppin E, Shlomi T (2010) Flux balance analysis accounting for metabolite dilution. Genome Biol 11: R43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattner FR, Plunkett G 3rd, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y (1997) The complete genome sequence of Escherichia coli K-12. Science 277: 1453–1474 [DOI] [PubMed] [Google Scholar]
- Burgard AP, Pharkya P, Maranas CD (2003) Optknock: a bilevel programming framework for identifying gene knockout strategies for microbial strain optimization. Biotechnol Bioeng 84: 647–657 [DOI] [PubMed] [Google Scholar]
- Buscher JM, Czernik D, Ewald JC, Sauer U, Zamboni N (2009) Cross-platform comparison of methods for quantitative metabolomics of primary metabolism. Anal Chem 81: 2135–2143 [DOI] [PubMed] [Google Scholar]
- Charusanti P, Conrad TM, Knight EM, Venkataraman K, Fong NL, Xie B, Gao Y, Palsson BØ (2010) Genetic basis of growth adaptation of Escherichia coli after deletion of pgi, a major metabolic gene. PLoS Genet 6: e1001186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemler JA, Fowler ZL, McHugh KP, Koffas MAG (2010) Improving NADPH availability for natural product biosynthesis in Escherichia coli by metabolic engineering. Metab Eng 12: 96–104 [DOI] [PubMed] [Google Scholar]
- Chen X, Alonso AP, Allen DK, Reed JL, Shachar-Hill Y (2011) Synergy between 13C-metabolic flux analysis and flux balance analysis for understanding metabolic adaption to anaerobiosis in E. coli. Metab Eng 13: 38–48 [DOI] [PubMed] [Google Scholar]
- Cho B-K, Palsson B, Zengler K (2011) Deciphering the regulatory codes in bacterial genomes. Biotechnol J 6: 1052–1063 [DOI] [PubMed] [Google Scholar]
- Choi HS, Kim TY, Lee DY, Lee SY (2007) Incorporating metabolic flux ratios into constraint-based flux analysis by using artificial metabolites and converging ratio determinants. J Biotechnol 129: 696–705 [DOI] [PubMed] [Google Scholar]
- Conrad TM, Joyce AR, Applebee MK, Barrett CL, Xie B, Gao Y, Palsson BØ (2009) Whole-genome resequencing of Escherichia coli K-12 MG1655 undergoing short-term laboratory evolution in lactate minimal media reveals flexible selection of adaptive mutations. Genome Biol 10: R118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad TM, Lewis NE, Palsson BØ (2011) Microbial laboratory evolution in the era of genome-scale science. Mol Syst Biol 7: 509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland WB, Bartley BA, Chandran D, Galdzicki M, Kim KH, Sleight SC, Maranas CD, Sauro HM (2012) Computational tools for metabolic engineering. Metab Eng 14: 270–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covert MW, Knight EM, Reed JL, Herrgard MJ, Palsson BØ (2004) Integrating high-throughput and computational data elucidates bacterial networks. Nature 429: 92–96 [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97: 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Figueiredo LF, Podhorski A, Rubio A, Kaleta C, Beasley JE, Schuster S, Planes FJ (2009) Computing the shortest elementary flux modes in genome-scale metabolic networks. Bioinformatics 25: 3158–3165 [DOI] [PubMed] [Google Scholar]
- Ederer M, Gilles ED (2007) Thermodynamically feasible kinetic models of reaction networks. Biophys J 92: 1846–1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards JS, Palsson BØ (2000) The Escherichia coli MG1655 in silico metabolic genotype: Its definition, characteristics, and capabilities. Proc Natl Acad Sci 97: 5528–5533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feist AM, Henry CS, Reed JL, Krummenacker M, Joyce AR, Karp PD, Broadbelt LJ, Hatzimanikatis V, Palsson BØ (2007) A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol Syst Biol 3: 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feist AM, Herrgard MJ, Thiele I, Reed JL, Palsson BØ (2009) Reconstruction of biochemical networks in microorganisms. Nat Rev Microbiol 7: 129–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feist AM, Palsson BØ (2008) The growing scope of applications of genome-scale metabolic reconstructions using Escherichia coli. Nat Biotechnol 26: 659–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flamholz A, Noor E, Bar-Even A, Milo R (2012) eQuilibrator--the biochemical thermodynamics calculator. Nucleic Acids Res 40: D770–D775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming RMT, Thiele I, Nasheuer HP (2009) Quantitative assignment of reaction directionality in constraint-based models of metabolism: application to Escherichia coli. Biophys Chem 145: 47–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong SS, Burgard AP, Herring CD, Knight EM, Blattner FR, Maranas CD, Palsson BØ (2005) In silico design and adaptive evolution of Escherichia coli for production of lactic acid. Biotechnol Bioeng 91: 643–648 [DOI] [PubMed] [Google Scholar]
- Fong SS, Palsson BØ (2004) Metabolic gene-deletion strains of Escherichia coli evolve to computationally predicted growth phenotypes. Nat Genet 36: 1056–1058 [DOI] [PubMed] [Google Scholar]
- Fowler ZL, Gikandi WW, Koffas MAG (2009) Increased malonyl coenzyme A biosynthesis by tuning the Escherichia coli metabolic network and its application to flavanone production. Appl Environ Microbiol 75: 5831–5839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrer T, Chen L, Sauer U, Vitkup D (2007) Computational prediction and experimental verification of the gene encoding the NAD+/NADP+-dependent succinate semialdehyde dehydrogenase in Escherichia coli. J Bacteriol 189: 8073–8078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghim CM, Goh KI, Kahng B (2005) Lethality and synthetic lethality in the genome-wide metabolic network of Escherichia coli. J Theor Biol 237: 401–411 [DOI] [PubMed] [Google Scholar]
- Hanly TJ, Henson MA (2011) Dynamic flux balance modeling of microbial co-cultures for efficient batch fermentation of glucose and xylose mixtures. Biotechnol Bioeng 108: 376–385 [DOI] [PubMed] [Google Scholar]
- Henry CS, Broadbelt LJ, Hatzimanikatis V (2007) Thermodynamics-based metabolic flux analysis. Biophys J 92: 1792–1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry CS, DeJongh M, Best AA, Frybarger PM, Linsay B, Stevens RL (2010) High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat Biotechnol 28: 977–982 [DOI] [PubMed] [Google Scholar]
- Herrgard MJ, Fong SS, Palsson BØ (2006) Identification of genome-scale metabolic network models using experimentally measured flux profiles. PLoS Comput Biol 2: e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring CD, Raghunathan A, Honisch C, Patel T, Applebee MK, Joyce AR, Albert TJ, Blattner FR, van den Boom D, Cantor CR, Palsson BØ (2006) Comparative genome sequencing of Escherichia coli allows observation of bacterial evolution on a laboratory timescale. Nat Genet 38: 1406–1412 [DOI] [PubMed] [Google Scholar]
- Holm AK, Blank LM, Oldiges M, Schmid A, Solem C, Jensen PR, Vemuri GN (2010) Metabolic and transcriptional response to cofactor perturbations in Escherichia coli. J Biol Chem 285: 17498–17506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarra RU, Edwards JS, Palsson BØ (2002) Escherichia coli K-12 undergoes adaptive evolution to achieve in silico predicted optimal growth. Nature 420: 186–189 [DOI] [PubMed] [Google Scholar]
- Jain R, Srivastava R (2009) Metabolic investigation of host/pathogen interaction using MS2-infected Escherichia coli. BMC Syst Biol 3: 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowski MD, Henry CS, Broadbelt LJ, Hatzimanikatis V (2008) Group contribution method for thermodynamic analysis of complex metabolic networks. Biophys J 95: 1487–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce AR, Reed JL, White A, Edwards R, Osterman A, Baba T, Mori H, Lesely SA, Palsson BØ, Agarwalla S (2006) Experimental and computational assessment of conditionally essential genes in Escherichia coli. J Bacteriol 188: 8259–8271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jozefczuk S, Klie S, Catchpole G, Szymanski J, Cuadros-Inostroza A, Steinhauser D, Selbig J, Willmitzer L (2010) Metabolomic and transcriptomic stress response of Escherichia coli. Mol Syst Biol 6: 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung YK, Kim TY, Park SJ, Lee SY (2010) Metabolic engineering of Escherichia coli for the production of polylactic acid and its copolymers. Biotechnol Bioeng 105: 161–171 [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, Yamanishi Y (2008) KEGG for linking genomes to life and the environment. Nucleic Acids Res 36: D480–D484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D-H, Motter AE (2009) Slave nodes and the controllability of metabolic networks. N J Phys 11: 113047 [Google Scholar]
- Kim J, Reed JL (2010) OptORF: Optimal metabolic and regulatory perturbations for metabolic engineering of microbial strains. BMC Syst Biol 4: 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TY, Kim HU, Lee SY (2010) Metabolite-centric approaches for the discovery of antibacterials using genome-scale metabolic networks. Metab Eng 12: 105–111 [DOI] [PubMed] [Google Scholar]
- Klamt S, Stelling J (2002) Combinatorial complexity of pathway analysis in metabolic networks. Mol Biol Rep 29: 233–236 [DOI] [PubMed] [Google Scholar]
- Klitgord N, Segrè D (2010) Environments that induce synthetic microbial ecosystems. PLoS Comput Biol 6: e1001002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugler H, Larjo A, Harel D (2010) Biocharts: a visual formalism for complex biological systems. J R Soc Interface 7: 1015–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Suthers PF, Maranas CD (2012) MetRxn: a knowledgebase of metabolites and reactions spanning metabolic models and databases. BMC Bioinform 13: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kümmel A, Panke S, Heinemann M (2006) Systematic assignment of thermodynamic constraints in metabolic network models. BMC Bioinform 7: 512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larhlimi A, Basler G, Grimbs S, Selbig J, Nikoloski Z (2012) Stoichiometric capacitance reveals the theoretical capabilities of metabolic networks. Bioinformatics 28: i502–i508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebeis SL, Kalman D (2009) Aligning antimicrobial drug discovery with complex and redundant host-pathogen interactions. Cell Host Microbe 5: 114–122 [DOI] [PubMed] [Google Scholar]
- Lee DH, Palsson BØ (2010) Adaptive evolution of Escherichia coli K-12 MG1655 during growth on a nonnative carbon source, L-1,2-propanediol. Appl Environ Microb 76: 4158–4168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Na D, Park JM, Lee J, Choi S, Lee SY (2012) Systems metabolic engineering of microorganisms for natural and non-natural chemicals. Nat Chem Biol 8: 536–546 [DOI] [PubMed] [Google Scholar]
- Lee KH, Park JH, Kim TY, Kim HU, Lee SY (2007) Systems metabolic engineering of Escherichia coli for L-threonine production. Mol Syst Biol 3: 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY, Woo HM, Lee DY, Choi HS, Kim TY, Yun H (2005) Systems-level analysis of genome-scale in silico metabolic models using MetaFluxNet. Biotechnol Bioproc E 10: 425–431 [Google Scholar]
- Lewis NE, Hixson KK, Conrad TM, Lerman JA, Charusanti P, Polpitiya AD, Adkins JN, Schramm G, Purvine SO, Lopez-Ferrer D, Weitz KK, Eils R, Konig R, Smith RD, Palsson BØ (2010) Omic data from evolved E. coli are consistent with computed optimal growth from genome-scale models. Mol Syst Biol 6: 390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis NE, Nagarajan H, Palsson BØ (2012) Constraining the metabolic genotype-phenotype relationship using a phylogeny of in silico methods. Nat Rev Microbiol 10: 291–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link H, Anselment B, Weuster-Botz D (2010) Rapid media transition: an experimental approach for steady state analysis of metabolic pathways. Biotechnol Progr 26: 1–10 [DOI] [PubMed] [Google Scholar]
- Macek B, Gnad F, Soufi B, Kumar C, Olsen JV, Mijakovic I, Mann M (2008) Phosphoproteome analysis of E. coli reveals evolutionary conservation of bacterial Ser/Thr/Tyr phosphorylation. Mol Cell Proteomics 7: 299–307 [DOI] [PubMed] [Google Scholar]
- Majewski RA, Domach MM (1990) Simple constrained-optimization view of acetate overflow in E. coli. Biotechnol Bioeng 35: 732–738 [DOI] [PubMed] [Google Scholar]
- Marashi S-A, Bockmayr A (2011) Flux coupling analysis of metabolic networks is sensitive to missing reactions. Biosystems 103: 57–66 [DOI] [PubMed] [Google Scholar]
- Motter AE, Gulbahce N, Almaas E, Barabasi AL (2008) Predicting synthetic rescues in metabolic networks. Mol Syst Biol 4: 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahigashi K, Toya Y, Ishii N, Soga T, Hasegawa M, Watanabe H, Takai Y, Honma M, Mori H, Tomita M (2009) Systematic phenome analysis of Escherichia coli multiple-knockout mutants reveals hidden reactions in central carbon metabolism. Mol Syst Biol 5: 306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa T, Gulbahce N, Motter AE (2008) Spontaneous reaction silencing in metabolic optimization. PLoS Comput Biol 4: e1000236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notebaart R, Kensche P, Huynen M, Dutilh B (2009) Asymmetric relationships between proteins shape genome evolution. Genome Biol 10: R19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberhardt MA, Palsson BØ, Papin JA (2009) Applications of genome-scale metabolic reconstructions. Mol Syst Biol 5: 320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth JD, Conrad TM, Na J, Lerman JA, Nam H, Feist AM, Palsson BØ (2011) A comprehensive genome-scale reconstruction of Escherichia coli metabolism--2011. Mol Syst Biol 7: 535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth JD, Thiele I, Palsson BØ (2010) What is flux balance analysis? Nat Biotechnol 28: 245–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterlund T, Nookaew I, Nielsen J (2012) Fifteen years of large scale metabolic modeling of yeast: developments and impacts. Biotechnol Adv 30: 979–988 [DOI] [PubMed] [Google Scholar]
- Ow DS-W, Lee D-Y, Yap MG-S, Oh SK-W (2009) Identification of cellular objective for elucidating the physiological state of plasmid-bearing Escherichia coli using genome-scale in silico analysis. Biotechnol Progr 25: 61–67 [DOI] [PubMed] [Google Scholar]
- Pal C, Papp B, Lercher MJ (2005a) Adaptive evolution of bacterial metabolic networks by horizontal gene transfer. Nat Genet 37: 1372–1375 [DOI] [PubMed] [Google Scholar]
- Pal C, Papp B, Lercher MJ (2005b) Horizontal gene transfer depends on gene content of the host. Bioinformatics 21: Suppl 2ii222–ii223 [DOI] [PubMed] [Google Scholar]
- Pal C, Papp B, Lercher MJ, Csermely P, Oliver SG, Hurst LD (2006) Chance and necessity in the evolution of minimal metabolic networks. Nature 440: 667–670 [DOI] [PubMed] [Google Scholar]
- Palsson B (2006) Systems Biology: Properties of Reconstructed Networks New York: Cambridge University Press, [Google Scholar]
- Palsson B (2009) Metabolic systems biology. FEBS Lett 583: 3900–3904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil KR, Rocha I, Forster J, Nielsen J (2005) Evolutionary programming as a platform for in silico metabolic engineering. BMC Bioinform 6: 308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Bercoff A, McLysaght A, Conant GC (2011) Patterns of indirect protein interactions suggest a spatial organization to metabolism. Mol Biosyst 7: 3056–3064 [DOI] [PubMed] [Google Scholar]
- Pfau T, Christian N, Ebenhoh O (2011) Systems approaches to modelling pathways and networks. Brief Funct Genomics 10: 266–279 [DOI] [PubMed] [Google Scholar]
- Plaimas K, Mallm JP, Oswald M, Svara F, Sourjik V, Eils R, Konig R (2008) Machine learning based analyses on metabolic networks supports high-throughput knockout screens. BMC Syst Biol 2: 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portnoy VA, Scott DA, Lewis NE, Tarasova Y, Osterman AL, Palsson BØ (2010) Deletion of genes encoding cytochrome oxidases and quinol monooxygenase blocks the aerobic-anaerobic shift in Escherichia coli K-12 MG1655. Appl Environ Microbiol 76: 6529–6540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pramanik J, Keasling JD (1997) Stoichiometric model of Escherichia coli metabolism: incorporation of growth-rate dependent biomass composition and mechanistic energy requirements. Biotechnol Bioeng 56: 398–421 [DOI] [PubMed] [Google Scholar]
- Price ND, Reed JL, Palsson BØ (2004) Genome-scale models of microbial cells: evaluating the consequences of constraints. Nat Rev Microbiol 2: 886–897 [DOI] [PubMed] [Google Scholar]
- Ravikirthi P, Suthers PF, Maranas CD (2011) Construction of an E. Coli genome-scale atom mapping model for MFA calculations. Biotechnol Bioeng 108: 1372–1382 [DOI] [PubMed] [Google Scholar]
- Reed J, Vo T, Schilling C, Palsson B (2003) An expanded genome-scale model of Escherichia coli K-12 (iJR904 GSM/GPR). Genome Biol 4: R54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JL, Patel TR, Chen KH, Joyce AR, Applebee MK, Herring CD, Bui OT, Knight EM, Fong SS, Palsson BØ (2006) Systems approach to refining genome annotation. Proc Natl Acad Sci USA 103: 17480–17484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samal A, Jain S (2008) The regulatory network of E. coli metabolism as a Boolean dynamical system exhibits both homeostasis and flexibility of response. BMC Syst Biol 2: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samal A, Martin OC (2011) Randomizing genome-scale metabolic networks. Plos One 6: e22295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samal A, Wagner A, Martin OC (2011) Environmental versatility promotes modularity in genome-scale metabolic networks. BMC Syst Biol 5: 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satish Kumar V, Dasika M, Maranas C (2007) Optimization based automated curation of metabolic reconstructions. BMC Bioinform 8: 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling CH, Letscher D, Palsson BØ (2000) Theory for the systemic definition of metabolic pathways and their use in interpreting metabolic function from a pathway-oriented perspective. J Theor Biol 203: 229–248 [DOI] [PubMed] [Google Scholar]
- Schuetz R, Kuepfer L, Sauer U (2007) Systematic evaluation of objective functions for predicting intracellular fluxes in Escherichia coli. Mol Syst Biol 3: 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuetz R, Zamboni N, Zampieri M, Heinemann M, Sauer U (2012) Multidimensional optimality of microbial metabolism. Science 336: 601–604 [DOI] [PubMed] [Google Scholar]
- Schuster S, Dandekar T, Fell DA (1999) Detection of elementary flux modes in biochemical networks: a promising tool for pathway analysis and metabolic engineering. Trends Biotechnol 17: 53–60 [DOI] [PubMed] [Google Scholar]
- Segre D, Vitkup D, Church GM (2002) Analysis of optimality in natural and perturbed metabolic networks. Proc Natl Acad Sci USA 99: 15112–15117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Liu J, Estiu G, Isin B, Ahn YY, Lee DS, Barabasi AL, Kapatral V, Wiest O, Oltvai ZN (2010) Blueprint for antimicrobial hit discovery targeting metabolic networks. Proc Natl Acad Sci USA 107: 1082–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlomi T, Berkman O, Ruppin E (2005) Regulatory on/off minimization of metabolic flux changes after genetic perturbations. Proc Natl Acad Sci USA 102: 7695–7700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlomi T, Eisenberg Y, Sharan R, Ruppin E (2007) A genome-scale computational study of the interplay between transcriptional regulation and metabolism. Mol Syst Biol 3: 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh JS, Abhilash PC, Singh HB, Singh RP, Singh DP (2011) Genetically engineered bacteria: An emerging tool for environmental remediation and future research perspectives. Gene 480: 1–9 [DOI] [PubMed] [Google Scholar]
- Sonnenschein N, Geertz M, Muskhelishvili G, Hutt M-T (2011) Analog regulation of metabolic demand. BMC Syst Biol 5: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suthers PF, Zomorrodi A, Maranas CD (2009) Genome-scale gene/reaction essentiality and synthetic lethality analysis. Mol Syst Biol 5: 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweetlove LJ, Ratcliffe RG (2011) Flux-balance modeling of plant metabolism. Front Plant Sci 2: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, Wang X, Bodeau J, Tuch BB, Siddiqui A, Lao K, Surani MA (2009) mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods 6: 377–382 [DOI] [PubMed] [Google Scholar]
- Taniguchi Y, Choi PJ, Li GW, Chen H, Babu M, Hearn J, Emili A, Xie XS (2010) Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science 329: 533–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele I, Fleming RM, Que R, Bordbar A, Diep D, Palsson BØ (2012) Multiscale modeling of metabolism and macromolecular synthesis in E. coli and its application to the evolution of codon usage. PLoS ONE 7: e45635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele I, Jamshidi N, Fleming RM, Palsson BØ (2009) Genome-scale reconstruction of Escherichia coli’s transcriptional and translational machinery: a knowledge base, its mathematical formulation, and its functional characterization. PLoS Comput Biol 5: e1000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele I, Palsson BØ (2010) A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat Protoc 5: 93–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzamali E, Poirazi P, Tollis IG, Reczko M (2011) A computational exploration of bacterial metabolic diversity identifying metabolic interactions and growth-efficient strain communities. BMC Syst Biol 5: 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma A, Boesch BW, Palsson BØ (1993) Biochemical production capabilities of Escherichia coli. Biotechnol Bioeng 42: 59–73 [DOI] [PubMed] [Google Scholar]
- Varma A, Palsson BØ (1993) Metabolic capabilities of Escherichia coli II. Optimal growth patterns. J Theor Biol 165: 503–522 [DOI] [PubMed] [Google Scholar]
- Varma A, Palsson BØ (1994a) Metabolic flux balancing: basic concepts, scientific and practical use. Nat Biotechnol 12: 994–998 [Google Scholar]
- Varma A, Palsson BØ (1994b) Stoichiometric flux balance models quantitatively predict growth and metabolic by-product secretion in wild-type Escherichia-Coli W3110. Appl Environ Microb 60: 3724–3731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez A, Beg QK, Demenezes MA, Ernst J, Bar-Joseph Z, Barabasi AL, Boros LG, Oltvai ZN (2008) Impact of the solvent capacity constraint on E. coli metabolism. BMC Syst Biol 2: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter J, Britton RA, Roos S (2011) Host-microbial symbiosis in the vertebrate gastrointestinal tract and the Lactobacillus reuteri paradigm. Proc Natl Acad Sci USA 108: Suppl 14645–4652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassarman KM, Saecker RM (2006) Synthesis-mediated release of a small RNA inhibitor of RNA polymerase. Science 314: 1601–1603 [DOI] [PubMed] [Google Scholar]
- Wessely F, Bartl M, Guthke R, Li P, Schuster S, Kaleta C (2011) Optimal regulatory strategies for metabolic pathways in Escherichia coli depending on protein costs. Mol Syst Biol 7: 515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wintermute EH, Silver PA (2010) Emergent cooperation in microbial metabolism. Mol Syst Biol 6: 407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Ranganathan S, Fowler ZL, Maranas CD, Koffas MA (2011) Genome-scale metabolic network modeling results in minimal interventions that cooperatively force carbon flux towards malonyl-CoA. Metab Eng 13: 578–587 [DOI] [PubMed] [Google Scholar]
- Yeung M, Thiele I, Palsson BØ (2007) Estimation of the number of extreme pathways for metabolic networks. BMC Bioinform 8: 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim H, Haselbeck R, Niu W, Pujol-Baxley C, Burgard A, Boldt J, Khandurina J, Trawick JD, Osterhout RE, Stephen R, Estadilla J, Teisan S, Schreyer HB, Andrae S, Yang TH, Lee SY, Burk MJ, Van Dien S (2011) Metabolic engineering of Escherichia coli for direct production of 1,4-butanediol. Nat Chem Biol 7: 445–452 [DOI] [PubMed] [Google Scholar]
- Yizhak K, Benyamini T, Liebermeister W, Ruppin E, Shlomi T (2010) Integrating quantitative proteomics and metabolomics with a genome-scale metabolic network model. Bioinformatics 26: i255–i260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhak K, Tuller T, Papp B, Ruppin E (2011) Metabolic modeling of endosymbiont genome reduction on a temporal scale. Mol Syst Biol 7: 479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamboni N, Kummel A, Heinemann M (2008) anNET: a tool for network-embedded thermodynamic analysis of quantitative metabolome data. BMC Bioinform 9: 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengler K, Palsson BØ (2012) A road map for the development of community systems (CoSy) biology. Nat Rev Microbiol 10: 366–372 [DOI] [PubMed] [Google Scholar]
- Zhuang K, Vemuri GN, Mahadevan R (2011) Economics of membrane occupancy and respiro-fermentation. Mol Syst Biol 7: 500. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.