

Abstract
Recent evidence suggests that specialized lipid mediators derived from polyunsaturated fatty acids control resolution of inflammation, but little is known about resolution pathways in vascular injury. We sought to determine the actions of D-series resolvin (RvD) on vascular smooth muscle cell (VSMC) phenotype and vascular injury. Human VSMCs were treated with RvD1 and RvD2, and phenotype was assessed by proliferation, migration, monocyte adhesion, superoxide production, and gene expression assays. A rabbit model of arterial angioplasty with local delivery of RvD2 (10 nM vs. vehicle control) was employed to examine effects on vascular injury in vivo. Local generation of proresolving lipid mediators (LC-MS/MS) and expression of RvD receptors in the vessel wall were assessed. RvD1 and RvD2 produced dose-dependent inhibition of VSMC proliferation, migration, monocyte adhesion, superoxide production, and proinflammatory gene expression (IC50≈0.1–1 nM). In balloon-injured rabbit arteries, cell proliferation (51%) and leukocyte recruitment (41%) were reduced at 3 d, and neointimal hyperplasia was attenuated (29%) at 28 d by RvD2. We demonstrate endogenous biosynthesis of proresolving lipid mediators and expression of receptors for RvD1 in the artery wall. RvDs broadly reduce VSMC responses and modulate vascular injury, suggesting that local activation of resolution mechanisms expedites vascular homeostasis.—Miyahara, T., Runge, S., Chatterjee, A., Chen, M., Mottola, G., Fitzgerald, J. M., Serhan, C. N., Conte, M. S. D-series resolvin attenuates vascular smooth muscle cell activation and neointimal hyperplasia following vascular injury.
Keywords: inflammation, intracellular signaling, resolvin
Atherosclerosis is now recognized as a chronic inflammatory disease of the vascular wall (1, 2). Clinical effectiveness of interventions commonly used to treat arterial occlusive diseases, whether surgical (bypass graft, endarterectomy) or catheter-based (angioplasty, stenting), remains limited by the development of recurrent vessel narrowing (restenosis), a manifestation of the local response to injury. Hundreds of thousands of such procedures are performed annually in the United States, with rates of failure that approach 50% within 5 yr depending on the circulatory bed (coronary or peripheral) and method applied (3–6). Thus, the public health effect of restenosis remains high despite improving endovascular and drug delivery technologies in the present era. Inflammatory cells are acutely recruited to sites of vascular damage, where they initiate processes of repair including clearance of cellular debris and recruitment of other local and blood-borne cells. In this milieu, resident vascular cells [endothelial cells (ECs) and vascular smooth muscle cell (VSMCs)] undergo phenotypic transformation and become activated to secrete proinflammatory cytokines [e.g., tumor necrosis factor α (TNF-α), interleukin-1β (IL-1β), and monocyte chemoattractant protein 1 (MCP-1)] and express cell adhesion molecules that promote leukocyte recruitment, migration, and differentiation. Leukocytes, particularly the monocytes/macrophages, elaborate cytokines and growth factors that potentiate VSMC activation in a paracrine fashion. The net result is the development of neointimal hyperplasia, a lesion comprising cells (predominantly VSMCs and myofibroblasts) and extracellular matrix, which, when excessive, leads to luminal renarrowing. Thus the magnitude and extent (both temporal and spatial) of the acute inflammatory response following vascular interventions are key determinants of the tissue repair/remodeling response and the ultimate clinical outcome. Current approaches to control this injury response include the local delivery of antiproliferative, antiinflammatory agents (e.g., paclitaxel, sirolimus) that have significant cytotoxicity and delay rather than accelerate tissue healing.
Recent evidence indicates that resolution of inflammation is an active, rather than a passive, process, orchestrated in part by specific proresolving lipid mediators (7–9). Four distinct families of proresolving mediators (termed lipoxins, resolvins, protectins, and maresins) are described that act via specific G-protein-coupled receptors (GPCRs) to down-regulate proinflammatory signals and enhance resolution. Eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) are ω-3 polyunsaturated fatty acids (PUFAs) that serve as local precursors for the E- and D-series resolvins (RvEs and RvDs), respectively. Resolvins were initially identified using unbiased lipidomics of resolving exudates in murine acute inflammation (9–11). RvDs are biosynthesized physiologically from the sequential oxygenation of DHA by lipoxygenases (8). Both RvEs and RvDs have beneficial actions in several animal models of inflammation, including sepsis (12), peritonitis (11, 13–17), colitis (18, 19), retinopathy (20), and periodontal disease (21, 22). A few studies have examined the potential antiatherosclerotic effects of proresolving mediators, including their direct effects on vascular cells and their interactions with leukocytes (23–25). Currently the role of proresolving mediators in the acute response to vascular injury is unknown. It is noteworthy that multiple clinical trials have examined the effects of ω-3 PUFA supplementation on coronary restenosis, with conflicting results. A recent meta-analysis (26) suggests there is considerable probability of a modest benefit on the prevention of restenosis but noted significant heterogeneity across trials in sample size, follow-up time, dosage and formulation of fish oils, and methods of end point ascertainment. Variability in the metabolism of PUFAs between individuals and their conversion to active lipid mediators (e.g., resolvins) at sites of vascular injury is also likely and as yet undefined.
Recently, we demonstrated that receptors for RvD1 (ALX/FPR2, also a receptor for lipoxin A4) and RvE1 (Chem R23) are expressed by human VSMCs (HVSMCs), and exposure to the ligands modulated VSMC migration responses to platelet-derived growth factor (PDGF) via PDGF receptor phosphorylation (24). Based on our earlier findings as well as emerging results from other experimental models of inflammatory disease (12, 19) we hypothesized that RvDs (RvD1 and RvD2) would counteract proinflammatory and growth factor signaling pathways that regulate VSMC phenotype and neointima formation. The present results support a potentially novel role for proresolving mediators in general, and RvDs specifically, in the therapeutic manipulation of vascular injury.
MATERIALS AND METHODS
Cell isolation and culture
Primary cultures of human greater saphenous vein VSMCs were isolated from saphenous vein discarded at the time of bypass operation in a University of California–San Francisco Institutional Review Board-approved protocol as described previously (27). VSMCs were maintained in Dulbecco's modified Eagle's medium (DMEM; low glucose; HyClone Laboratories, Logan, UT, USA) containing 10% FBS (Invitrogen Life Technologies, Grand Island, NY, USA) and used between passages 2 and 5.
Cell proliferation
Cell proliferation assay was performed as described previously (27). VSMCs were seeded onto 24-well plates at a density of 5000 cells/well, and then treated with RvD1 or RvD2 (0.01, 1, or 100 nM) in medium containing 10% FBS. Medium was replenished every 2 d. Alamar Blue (Invitrogen) assays were conducted every 48 h according to the manufacturer's protocol. Fluorescence measurements (excitation 506 nm, emission 590 nm) were made on medium aliquots; a standard curve was generated by correlating emission intensity with viable cell counts using trypan blue exclusion.
Transwell migration
VSMC migration was assayed using 8-μm-pore transwell inserts, as described previously (28). Cells were pretreated with RvD1 or RvD2 (0.01, 1, or 100 nM), or vehicle (0.1% ethanol) control for 30 min before the addition of PDGF-AB or PDGF-BB (50 ng/ml; Sigma-Aldrich, St. Louis, MO, USA) to the bottom wells. All antichemotactic compounds were present in both top and bottom wells for the full duration of chemotaxis experiments (6–9 h). In some experiments, pertussis toxin (PTX 100 ng/ml; Calbiochem EMD Chemicals, San Diego, CA, USA), anti-GPR32 (10 μg/ml; GeneTex, Irvine, CA, USA), or anti-FPR2/ALX (10 μg/ml; FN-1D6-A1, Genovac, Freiburg, Germany) neutralizing antibodies were added to the cells 15 min before the addition of RvDs. All treatment conditions were performed in triplicate wells.
Cell shape measurement
VSMCs were cultured in chamber slides in serum-free medium for 16 h. Cells were then pretreated with RvD1 or vehicle (0.1% ethanol) for 2 h, followed by the addition of PDGF-BB (50 ng/ml) for 1 h. Cells were washed twice in phosphate-buffered saline (PBS), permeabilized with 0.1% Triton-X, and then fixed in 3.7% formaldehyde, labeled with Alexa Fluor 568 phalloidin (Invitrogen), and mounted with DAPI containing mounting medium (Vectashield, Vector Laboratories, Burlingame, CA, USA). Cell area and length/width ratio were determined by outlining the cell dimensions and computing 2-dimensional area using ImageJ analysis software (U.S. National Institutes of Health, Bethesda, MD, USA). For each condition, dimensions were measured from 10 randomly selected cells, and all treatment conditions were performed in triplicate.
Cell viability assay
Cells were plated onto 24-well plates and treated with or without RvD1 or RvD2 (1, 10, and 100 nM) for 8 h. MTT viability assay was performed per manufacturer's instructions (TOX1 assay; Sigma-Aldrich).
Monocyte adhesion
A static monocyte adhesion assay was performed as described previously (29). U937 monocytes were labeled with 1 μM of calcein-AM (Invitrogen) for 30 min at 37°C in PBS. The cells were washed 3 times and resuspended in PBS without serum at a concentration of 1.0 × 106 cells/ml. VSMCs were grown to 100% confluence in 96-well dishes and treated with cytokines [10 ng/ml TNF-α (Sigma-Aldrich) or 1 nM IL-1β (R&D Systems, Minneapolis, MN, USA)] in the presence or absence of serial concentration of RvD1 or RvD2 (0.01–100 nM, Cayman Chemical, Ann Arbor, MI, USA) for 4 h at 37°C. Labeled monocytes (200 μl; 2.0×105 cells) were added to each well. After a 15-min incubation at 37°C, unbound cells were washed off with PBS 3 times. Fluorescence measurements (excitation 494 nm, emission 517 nm) of bound monocytes were made with a plate reader (Spectra Max M2; Molecular Devices, Sunnyvale, CA, USA).
Analysis of gene expression by reverse transcriptase-polymerase chain reaction (RT-PCR)
Cells were plated onto 6-well plates at 100% confluency in DMEM plus 10% FBS and allowed to attach overnight. Cells were then made quiescent by placing in serum-free medium for 48–72 h prior to start of experiments. Cells were pretreated with or without RvD1 or RvD2 at the indicated concentrations for 1 h, followed by the addition of cytokine (TNF-α, 10 ng/ml) and left incubated for 18 h. In a separate set of experiments to examine the role of G-protein receptors, VSMCs were exposed to TNF-α and RvD2 (10 nM), as described above, in the presence or absence of PTX (100 ng/ml). Total RNA from rabbit sample or cells was isolated with PureLink RNA Mini Kit (Ambion Life Technologies, Carlsbad, CA, USA) with RNase-free DNase treatment according to manufacturer's protocol. Total RNA (1 μg; from either VSMCs or rabbit tissue) was used to generate cDNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA) for subsequent RT-PCR reactions. SYBR Green-amplified DNA was detected by incorporation of SYBR Green (Qiagen, Germantown, MD, USA). Dissociation curve analyses were performed to confirm the specificity of the SYBR Green signal. Data were normalized to ≥2 reference genes. The primers used are listed in Table 1. Most primer pairs spanned across an intron. PCR parameters included an initial 10-min denaturation step at 95°C, followed by a cycling program of 95°C for 10 s, 60°C for 30 s for 40 cycles (CFX 96 Real Time System; Bio-Rad, Hercules, CA, USA).
Table 1.
Primers used in quantitative RT-PCR
| Primer | Forward sequence | Reverse sequence | Exon |
|---|---|---|---|
| Hu VCAM-1 | TGCAAGTCTACATATCACCCAAGAATA | GGTAGACCCTCGCTGGAACA | M |
| Hu ICAM-1 | GCCGGCCAGCTTATACACAA | TGGCCACGTCCAGTTTCC | M |
| Hu TNF-α | AGAGGGCCTGTACCTCATCTACTC | GGTTGACCTTGGTCTGGTAGGA | M |
| Hu IL-1β | CCCTAAACAGATGAAGTGCTCCTT | GGTGGTCGGAGATTCGTAGCT | M |
| Hu MCP-1 | CAGCAGCAAGTGTCCCAAAG | GAATCCTGAACCCACTTCTGCTT | M |
| Hu IL-6 | CCGGGAACGAAAGAGAAGCT | AGCAGCCCCAGGGAGAAG | M |
| Hu IL-1α | GAATCAGAAATCCTTCTATCATGTAAGC | ACTACCACCATGCTCTCCTTGAA | M |
| Hu HPRT | CAAGCTTGCTGGTGAAAAGGA | TGAAGTACTTATAGTCAAGGGCATATC | M |
| Hu UBC | ATTTGGGTCGCGGTTCTTG | TGCCTTGACATTCTCGATGGT | M |
| Rb VCAM-1 | GGTCTACATTTCACCCAAGAATACAG | ACTGGTAGACCCTCGCTGGAA | M |
| Rb ICAM-1 | AGACGCAGCTGAGCAAGGA | CACAGTCGGAAAAGCAGATGAG | G |
| Rb TNF-α | GGAAGAGCAGTCCCCAAACA | GGGCTAGAGGCTTGTCACTCA | M |
| Rb IL-1β | TGTACCTGTCCTGCGTGATGA | TCGTTTTTCCATCTTCTTCTTTGG | M |
| Rb MCP-1 | TGGGTCCAGGATGCCAT | AGTCGTGTGTTCTTGGGTTGTG | S |
| Rb IL-6 | ACGACCACGATCCACTTCATC | AAGGACACCCGCACTCCAT | S |
| Rb IL-1α | GAGTCGGCAAAGAAATCAAGATG | GCAGAGCTGTATTCCTCATTTTCA | G |
| Rb HPRT | GTGAAAAGGACCCCTCGAAGT | TCATTATAGTCAAGGGCATATCCTACA | M |
| Rb GAPDH | TCCCCGAGACACGATGGT | ACAACATCCACTTTGCCAGAGTT | S |
G, unknown exon; M, multiple exons; S, single exon.
Transcription factor assay
VSMCs were pretreated with or without RvD1 (10 nM) for 30 min, followed by addition of TNF-α (10 ng/ml) for 2 h, and nuclear extracts were prepared. NFκB and AP-1 activity were measured as the nuclear translocation and DNA binding of the p65 and c-Jun subunit in 2.5 μg nuclear extracts from cells, using a commercially available ELISA kit (Trans AM NFκB p65 and Trans AM c-Jun; Active Motif, Carlsbad, CA, USA).
Western blotting
Human VSMCs were lysed in buffer (20 mM Tris-Hcl, 137 mM NaCl, 10% glycerol, 1% Nonidet P-40) with protease inhibitors for 1 h at 4°C. After centrifugation (15,000 g for 20 min), the whole-cell extract (supernatant) was collected and was heated at 100°C in Laemlli buffer for 6 min. The lysate (50 μg) was then run on NuPAGE 10% Bis-Tris gel (Invitrogen) and transferred to a PVDF membrane that was probed with anti-GPR32 antibody (1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA, for rabbit artery; Novus Biologicals, Littleton, CO, USA, for human VSMCs), anti-FPR2/ALX (1:500; Santa Cruz Biotechnology) and anti-β-actin (1:5000; Sigma-Aldrich) using a QDot 625 Western blotting kit (Invitrogen).
Detection of superoxide production
Cells were seeded and grown on chamber slides at a density of 10,000 cells/chamber for 2 d, followed by treatment with 10 ng/ml TNF-α with or without RvD1 (10 and 100 nM) in serum-free medium for 4 h. After incubation, cells were incubated with dihydroethidium (DHE; 5 μM; Invitrogen) in PBS for 30 min at 37°C in a humidified chamber protected from light. DAPI nuclear counterstaining was utilized. For frozen sections, 30-μm-thick sections were incubated with DHE (10 μM) in PBS for 30 min in the same conditions described above. Fluorescence was detected with a TRITC filter allowing the detection of the DHE emission wavelength of 590–620 nm. The fluorescence lamp gain was standardized for all images and analyses. Fluorescence quantitation was performed using ImageJ analysis software. For VSMCs, the fluorescence intensity was determined for the DHE-stained control cells (no treatment control) from each experimental group, and then individual treated cells from the same group were normalized to the corresponding value. For frozen sections, the fluorescence intensity was determined for the DHE-stained control vessels (uninjured abdominal aorta) from each experimental group, and then individual treated vessels from the same group were normalized to the corresponding value.
Rabbit femoral artery balloon injury
All animal experiments were performed under a protocol approved by the University of California–San Francisco Animal Care Committee. New Zealand white rabbits (NZWRs; Western Oregon Rabbit Company, Philomath, OR, USA) weighing 3 to 5 kg and maintained on a normal chow diet (LabDiet Rabbit Diet High Fiber; Purina, St. Louis, MO, USA) were used in all experiments. Animals were anesthetized for surgical procedures with intramuscular injection of ketamine (25 mg/kg) and xylazine (5 mg/kg) and maintained with inhalation of isoflurane. The balloon injury model was made as described previously (30). Rabbits underwent bilateral external iliac and femoral artery injury with a 2F Fogarty balloon catheter (Applied Medical Resources Corp., Rancho Santa Margarita, CA, USA). After balloon injury, RvD2 (10 nM, Cayman Chemical) was infused to the luminal area of the right femoral artery, and control vehicle (normal saline with 0.1% ethanol) was infused to the left femoral artery and then left undisturbed for 20 min. For evaluation of acute effects, the rabbits were euthanized at 3 d after injury by intravenous overdose of sodium nembutal (n=6). The femoral arteries were harvested, and cryosection was made for immunohistochemistry, and total RNA from treated and control tissue was isolated. For evaluation of subacute responses regarding intimal hyperplasia, the rabbits were euthanized at 28 d after injury (n=8), and the tissue was perfusion fixed in 10% formalin and processed as described previously (27).
In a separate set of experiments, rabbits underwent unilateral iliac and femoral artery injury with a 2F Fogarty balloon catheter with a sham operation on the contralateral side. Rabbits were euthanized after 30 min or 3 d (n=2 for each time point). Tissues (aorta, iliac, and femoral arteries, spleen, lymph node) were collected and immediately snap-frozen for metabololipidomic profiling by liquid chromatography-dual mass spectrometry (LC-MS/MS) or Western blot, and cryosections were made for immunohistochemistry.
Sample extraction and mediator lipidomics
All samples for LC-MS/MS analysis were extracted with solid-phase extraction columns as described previously (31). Prior to sample extraction, 500 pg of deuterium-labeled internal standards d8-5S-hydroxyeicosatetraenoic acid (HETE), d4-leukotriene B4, (LTB4) d5-lipoxin A4 (LXA4) and d4-prostaglandin E2 (PGE2) were added to facilitate the quantification of sample recovery. Extracted samples were analyzed by a LC-UV-MS/MS system, QTrap 5500 (AB Sciex, Framingham, MA, USA) equipped with two Shimadzu LC-20AD pumps (Shimadzu Corp., Kyoto, Japan). An Agilent Eclipse Plus C18 column (100×4.6 mm×1.8 μm; Agilent Technologies, Santa Clara, CA, USA) was used with a gradient of methanol/water/acetic acid of 60:40:0.01 (v/v/v) to 100:0:0.01 at 0.4 ml/min flow rate. Identification was conducted using previously published criteria using a minimum of 6 diagnostic ions (31). Quantification was carried out based on the peak area of the multiple reaction monitoring transition and the linear calibration curve for each compound.
Immunofluorescent staining
All immunofluorescent staining was completed on 6-μm-thick frozen sections fixed by ice-cold acetone for 10 min. Anti-Ki67 (1:100, NCL-L-Ki67-MM1; Novocastra Laboratories, Newcastle on Tyne, UK) was utilized for cell proliferation detection. Anti-CD45 (1:200, L12/201; AbD Serotec, Oxford, UK) was utilized for leukocyte infiltration detection. GPR32 (1:25, S-13; Santa Cruz Biotechnology) and FRP2/ALX (1:25, C-17; Santa Cruz Biotechnology) were utilized for detection of each receptor. Goat anti-mouse IgG conjugated with Alexa Fluor 488 or 568 or donkey anti-goat IgG-conjugated Alexa Fluor 488 (1:100, respectively; Invitrogen) was used as secondary antibody. DAPI nuclear counterstaining was utilized on all the immunofluorescent staining. Photography was completed with a Olympus BX51 microscope (Olympus America, Center Valley, PA, USA) with an EXFO X-cite 120 system (EXFO Photonic Solutions, Mississauga, ON, Canada), Olympus DP70 digital microscope camera, and DPController software (Olympus). Six vessel zones (at ×200 view) were selected randomly on 4 coordinate axes of every stained rabbit arterial cross section. The proportion of Ki67 was calculated by absolute positive cell number divided by DAPI-positive nuclei. Leukocyte infiltration index was calculated as number of nuclei in the CD45 positive area divided by the total number of DAPI-positive nuclei.
Morphometry
Verhoeff-Van Gieson elastin staining was performed on perfusion-fixed rabbit femoral arterial cross sections to evaluate neointimal hyperplasia on 4 coordinate axes of every stained rabbit arterial cross section. Lumen circumference, internal elastic lamina, and external elastic lamina were delineated by hand, and planimetry was completed using ImageJ software.
Reendothelialization
Immunostaining was performed on perfusion-fixed rabbit femoral arterial cross sections to evaluate reendolelialization at 28 d after balloon injury. Sodium citrate buffer was used for antigen retrieval, monoclonal antibody against CD31 (1:30, JC70A; Dako Denmark A/S, Glostrup, Denmark) was used for primary antibody, and sections were counterstained with hematoxylin. The extent of reendotheliarization was scored in the following manner on 4 coordinate axes of every stained rabbit arterial cross section: score 1, 0–25%; score 2, 25–50%; score 3, 50–75%; score 4, 75–100%.
Statistical analysis
Data are shown as means ± se. Direct comparisons were made using unpaired or paired Student's t test, and multiple group comparisons were made using 1-way or 2-way ANOVA followed by Dunnett's or Bonferroni's post hoc tests where appropriate. In all cases, a level of P < 0.05 was considered significant.
RESULTS
RvD1 and RvD2 attenuate VSMC proliferation and migration
Following acutely denuding mechanical injury such as balloon angioplasty, exposed luminal VSMCs take on a proinflammatory, proliferative, and migratory phenotype that may be modeled using standard in vitro assays. Proliferation of VSMCs in standard growth medium (10% serum) was reduced in dose-dependent fashion by exposure to both RvD1 (Fig. 1A) and RvD2 (Supplemental Fig. S1A), with maximum effect (59% inhibition in RvD1 and 63% inhibition in RvD2) observed at 100 nM (P<0.01). The inhibitory concentration for 50% effect (IC50) appeared in the range 0.1–1 nM. MTT assays indicated that VSMCs exposed to either RvD1 or RvD2 (1–100 nM) for 8 h did not display any significant loss of cell viability, indicating a cytostatic rather than a cytotoxic effect (Supplemental Fig. S1B). The growth-inhibiting effects of RvD1 (10 nM) were markedly attenuated by anti-ALX antibody but not anti-GPR32 (Fig. 1B), suggesting relative receptor specificity for this effect at this dose.
Figure 1.

RvD1 and RvD2 attenuate proliferation and migration responses of human VSMCs in vitro. A) HVSMC proliferation assay performed in normal growth medium (10% serum) as described in Materials and Methods. Dose-dependent inhibition of VSMC proliferation is shown for RvD1 (n=3). **P < 0.01 vs. control; 2-way ANOVA with Dunnett's post hoc test. B) Proliferation assay performed in 0.5 or 10% serum as noted, with addition of RvD1 (10 nM) and indicated antibodies to GPCRs (n=3). *P < 0.01, **P < 0.0001 vs. 10% FBS + RvD1 + IgG control; #P < 0.05, ##P < 0.0001 vs. 10% FBS + vehicle; 1-way ANOVA with Dunnett's post hoc test. C, D) VSMC migration response to PDGF-AB with RvD1/RvD2 cotreatment, using a transwell assay. Results are expressed as percentage change in migration from unstimulated control (no PDGF). PDGF-neutralizing antibody serves as positive control for inhibition. A dose-dependent inhibition of chemotaxis is demonstrated for both RvD1 and RvD2. Effects of RvD1 on chemotaxis are sensitive to anti-GPR32 blocking antibody, but not to anti-ALX (n=5). NS, not significant. *P < 0.05, **P < 0.01 vs. PDGF-AB; #P < 0.05 vs. PDGF-AB + RvD1; 1-way ANOVA with Dunnett's post hoc test. E, F) Cell shape changes assessed with phalloidin staining and quantitative cytometry. E) RvD1 induces cell shape changes, including an increase decrease of length/width ratio, and inhibits the typical cytoskeletal response to PDGF-AB (n=3). *P < 0.05, **P < 0.01 vs. PDGF-AB; #P < 0.05, ##P < 0.01 vs. vehicle control; unpaired t test. F) Cytoskeletal changes induced by RvD1 are abrogated by PTX and anti-GPR32, but not by anti-ALX (n=3). *P < 0.05, **P < 0.01 vs. isotype control; #P < 0.05 vs. negative control; 1-way ANOVA with Dunnett's post hoc test. Results are means ± se.
VSMC chemotaxis toward a PDGF gradient was significantly reduced by RvD1 and RvD2 in a dose- and GPCR-dependent fashion. Maximum percentage reduction in the cell migration assay was 38% in 100 nM RvD1 (P<0.01) and 41% in 100 nM RvD2 (P<0.01), and was similar for equivalent concentrations of PDGF-AB or PDGF-BB as the stimulus (Figs. 1C and 2 and Supplemental Data). The IC50 observed for this effect was ∼1 nM. We next examined the role of the known GPCRs for RvD1, ALX and GPR32, in mediating these actions. The effects of RvD1 on PDGF-AB- or PDGF-BB-induced chemotaxis were sensitive to inhibition by PTX and anti-GPR32 blocking antibody (Fig. 1D and Supplemental Fig. S2). However, the anti-ALX antibody had minimal effect on the antimigratory effects of RvD1 in these cells. Cell shape assays showed that RvD1 exposure caused rapid cytoskeletal changes in VSMCs, manifest as increased cell area and decreased length/width ratio consistent with decreased actin polymerization. Furthermore, RvD1 completely abrogated the spindle-like cytoskeletal changes induced by either PDGF-AB or PDGF-BB (Fig. 1E and Supplemental Fig. S3). These cytoskeletal effects were abrogated by both PTX and anti-GPR32, but not anti-ALX (Fig. 1F).
Figure 2.

Modulation of VSMC inflammatory responses by RvD1 and RvD2. A) Monocyte adhesion to VSMCs. HVSMCs were stimulated with TNF-α (10 ng/ml) for 4 h, in the presence or absence of RvD1 or RvD2 at the indicated doses. Labeled U937 monocytes were overlain, and cell adhesion assay was performed as described. Results are shown as relative percentage inhibition, expressed as a percentage of the maximal adhesion for the agonist (n=3). *P < 0.05, **P < 0.01; unpaired t test. B) RvD1 and RvD2 reduce cell adhesion molecule gene expression in VSMCs. VSMCs were treated with TNF-α (10 ng/ml) for 18 h in the presence or absence of RvD1 or RvD2 across a concentration range as shown. Expression of VCAM-1 and ICAM-1 was measured by quantitative RT-PCR (n=3). *P < 0.05, **P < 0.01; unpaired t test. C) Proinflammatory gene expression. VSMCs were stimulated with TNF-α as above in the presence or absence of RvD1 or RvD2 (0.1 nM or 10 nM), and qPCR was performed for multiple proinflammatory gene transcripts. Shown are significant reductions in the expression of TNF-α, IL-1β, MCP-1, IL-6, and IL-1α (n=3). *P < 0.05, **P < 0.01; unpaired t test. D) Effects of RvD2 on TNF-α stimulated gene expression in VSMCs are sensitive to PTX. VSMCs were exposed to TNF-α and RvD2 (10 nM) as described, in the presence or absence of PTX (100 ng/ml). RNA was harvested and analyzed for the expression of ICAM-1, IL-1β, and IL-1α by qPCR (n=3). *P < 0.05, **P < 0.01; 1-way ANOVA with Bonferroni's post hoc test). E) RvD1 modulates activity of transcription factors NFκB and AP-1 in TNF-α-stimulated VSMCs. VSMCs were treated with TNF-α (10 ng/ml) with or without RvD1 (10nM for 2 h), and nuclear extracts were prepared. Transcription factor activity was assessed as described in Materials and Methods (n=5). *P < 0.05; unpaired t test. Results are means ± se.
RvD1 and RvD2 reduce monocyte adhesion and proinflammatory gene expression in VSMCs
In a static cell adhesion assay, RvD1 and RvD2 produced a dose-dependent inhibition of U937 monocyte adhesion to cytokine (TNF-α, Fig. 2A; or IL-1β, Supplemental Fig, S4)-stimulated VSMCs. Peak inhibition of monocyte adhesion to TNF-α-stimulated VSMCs was observed at a dose of 10 nM, with a maximum percentage inhibition of 36% for RvD1, and 41% for RvD2; IC50 ∼ 1 nM. Effects seen were nearly identical for both inflammatory cytokines as stimuli.
Cytokine-stimulated gene expression in primary cultured VSMCs, including intercellular adhesion molecules and proinflammatory cytokines, was assessed using real-time quantitative RT-PCR. Both RvD1 and RvD2 produced a dose-dependent reduction of TNF-α-stimulated vascular cell adhesion molecule 1 (VCAM-1) and intercellular adhesion molecule 1 (ICAM-1) expression (Fig. 2B). In general, RvD2 was slightly more effective compared to RvD1. For VCAM-1, maximum percentage reduction was 69% in 100 nM RvD1 (P<0.01), and 76% in 100 nM RvD2 (P<0.01), and for ICAM-1 maximum percentage reduction was 63% in 100 nM RvD1 (P<0.01), and 70% in 100 nM RvD2 (P<0.01). There was a broad, significant reduction in the expression of multiple inflammatory cytokines, including TNF-α, IL-1β, MCP-1, IL-6, and IL-1α by both RvD1 and RvD2, with the greatest attenuation seen in IL-1β expression (Fig. 2C). To confirm the role of G-protein (Gα) receptors in RvD signaling in VSMCs, a separate set of experiments was conducted in the presence or absence of PTX, demonstrating abrogation of the inhibitory effects of RvD2 on ICAM-1, IL-1β, and IL-1α gene expression (Fig. 2D).
Given these broad observed effects on gene expression, we examined changes in the activity of key transcription factors NFκB and AP-1, which are known to regulate inflammatory pathways in VSMCs (Fig. 2E). Treatment with RvD1 (10 nM) significantly blunted TNF-α-stimulated NFκB and AP-1 activity compared to vehicle-treated controls (percentage inhibition: NFκB, 30%; AP-1, 40%; P<0.05).
Resolution pathways in vascular tissues: biosynthesis of proresolving mediators and expression of receptors
The biosynthesis of proresolving mediators has been extensively characterized in exudate models; however, the generation and accumulation of these mediators within the vessel wall is unknown. We performed metabololipidomic profiling of rabbit arteries using established LC-MS/MS protocols. A representative chromatographic profile from a balloon-injured (d 3) rabbit artery is shown in Fig. 3. We identified the presence of DHA-, EPA-, and arachidonic acid (AA)-derived precursors and downstream bioactive mediators within the injured vessel wall, including members of the RvD (RvD1, RvD5), maresin (MaR1), and lipoxin (LXB4) families of resolution agonists. The MS-MS spectra for RvD5, LXB4, and 17-hydroxydocosahexaenoic acid (HDHA), as examples of lipid mediators identified in the chromatographic profiles, are shown on the right of Fig. 3. The range of artery wall levels (picograms/100 mg tissue) measured for selected lipid mediators was RvD1 (0–374), RvD5 (0–64), MaR1 (0–81), LxB4 (0–270), 17-HDHA (192–635), 14-HDHA (60–178). There was a trend of increase in the global DHA metabolome in the injured as compared to uninjured arteries, though the sample size precludes statistical analysis. These studies demonstrate that endogenous pathways of proresolving mediator generation are present and biochemically active in the setting of vascular injury. It should be noted that the standard rabbit chow employed in these studies contains no fish sources and hence no appreciable DHA or EPA, with α-linolenic acid as the only dietary source of ω-3 PUFAs (0.24% total by weight; http://labdiet.com/pdf/5326.pdf).
Figure 3.

Local biosynthesis of lipid mediators in vascular injury. Rabbits underwent unilateral iliofemoral artery balloon injury. Vessels were harvested 3 d after injury for biochemical and histological assays. Metabololipidomics of arterial injury. Representative chromatographic profiles from LC-MS/MS obtained from injured artery showing the presence of proresolving lipid mediators and precursors, including RvD5, MaR1, LXB4, and DHA- and EPA-derived monohydroxy acids. MS-MS spectra are on right.
We next asked whether GPCRs capable of mediating RvD1 effects are expressed within the vascular tissues. We have previously reported the presence of ALX expression in primary cultured human VSMCs (24). We confirmed expression of GPR32 in cultured human VSMCs by Western blot (Fig. 4A). Western blot analysis indicated that both GPR32 and ALX receptors are present in injured and uninjured rabbit arteries (Fig. 4B). Immunohistochemical analysis showed that GPR32 is strongly expressed in rabbit vessels (Fig. 4C–F), both in the endothelium and in the underlying medial VSMCs. Following balloon denudation, GPR32 expression appears increased in the medial layer (Fig. 4E). Although Western blot results of rabbit artery lysates were unequivocal, immunohistochemical staining for ALX in rabbit arteries was weaker and diffuse, and we are therefore unable to comment on its specific localization within the vessel wall.
Figure 4.
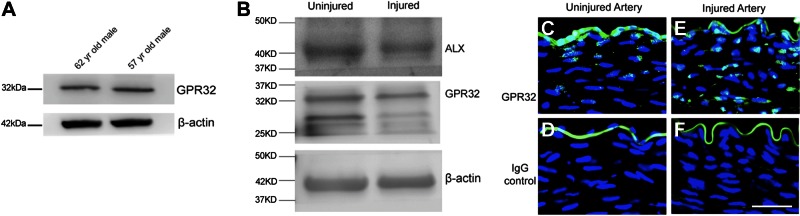
Expression of RvD1 receptors in VSMCs and rabbit artery tissues. A) Identification of GPR32 in human VSMCs. Western blot analysis (50 μg of total cell lysate) of primary cultured VSMCs from 2 donors using anti-GPR32 antibody and anti-β-actin antibodies. A single band is identified of the appropriate size for each protein. B) Identification of GPR32 and ALX in rabbit artery. Western blot analysis (50 μg of total artery lysate) using anti-GPR32 antibody, anti-ALX antibody, and anti-β-actin antibodies. A single band is identified for ALX; GPR32 blot shows band at appropriate size and smaller bands of unknown significance. C–F) Representative GPR32 immunostaining of uninjured (C, D) and injured (E, F) arteries. GPR32 receptor is expressed both in injured and uninjured vessels. Scale bar = 100 μm.
RvD2 modulates the acute vascular injury response
Following balloon angioplasty, rabbit femoral arteries were immediately exposed to RvD2 (10 nM) vs. vehicle by brief (20 min) intraluminal incubation. Short-term effects on the local tissue response were evaluated 3 d after injury (n=6). Cell proliferation (Ki-67 staining index) was decreased in RvD2-treated vessels compared to control (percentage inhibition 51%, P<0.01, paired t test; Fig. 5A–C). CD45 staining showed that leukocyte infiltration after injury was also inhibited by RvD2 (percentage inhibition 41%, P<0.05, paired t test; Fig. 5D–F). Inflammatory gene expression in the treated arteries was evaluated by quantitative PCR (qPCR), demonstrating significant reductions in TNF-α, MCP-1, and IL-1α expression in the RvD2-treated arteries vs. vehicle-treated controls (Fig. 5G).
Figure 5.

RvD2 treatment attenuates the acute response to vascular injury, in vivo. Rabbits (n=6) underwent bilateral femoral artery angioplasty, with immediate local delivery of RvD2 (10 nM) or vehicle control by intraluminal incubation as described. Vessels were harvested 3 d after injury for histological assays. A–C) RvD2 reduces the early proliferative response to angioplasty. Representative Ki-67 immunostaining of vehicle-treated (A) and RvD2-treated (B) arteries, with summary of quantitative analysis (C). D–F) RvD2 treatment attenuates early leukocyte recruitment to the injured vessel. Representative CD45-stained sections of vehicle-treated (D) and RvD2-treated (E) arteries, with summary of quantitation (F). G) RvD2 treatment modulates early inflammatory gene expression in the acutely injured artery in vivo. Shown are fold expression changes of VCAM-1, ICAM-1, TNF-α, IL-1β, MCP-1, IL-6, and IL-1α normalized to uninjured artery. Scale bars = 100 μm. Results are means ± se. *P < 0.05, **P < 0.01; paired t test.
RvD1 and RvD2 modulate VSMC superoxide production
Local generation of reactive oxygen species (ROS) such as superoxide potentiates inflammation and cell activation signaling in the vessel wall. ROS may be generated by recruited leukocytes or by resident vascular cells, including VSMCs. In vitro, RvD1 treatment significantly reduced TNF-α-induced superoxide production in VSMCs (Fig. 6A–D, I). In vivo, DHE staining demonstrated a significant reduction of injury-induced ROS production in RvD2-treated rabbit arteries 3 d after balloon injury (n=6, percentage inhibition 26%, P<0.05, paired t test; Fig. 6E–H, J).
Figure 6.

RvD treatment modulates superoxide production by VSMCs in vitro and in vivo. A–D, I) RvD1 treatment reduces TNF-α-induced superoxide production in cultured HVSMCs. A–D) Representative merged images of DHE staining, counterstained with DAPI of untreated VSMCs (A), positive control (TNF-α 10 ng/ml for 4 h; B), TNF-α with 10nM RvD1 (C), and TNF-α with 100nM RvD1 (D). I) Quantitative comparison of DHE staining intensity (n=3). *P < 0.05; 1-way ANOVA with Dunnett's post hoc test). E–H, J) RvD2 treatment reduces oxidative stress in the acutely injured rabbit artery (3 d postangioplasty). E–H) Representative images of DHE staining of an uninjured aorta (E), balloon injured and untreated iliac artery (F), vehicle-treated femoral artery (G), and RvD2-treated femoral artery (H). J) Quantitative comparison of staining intensity (n=6). *P < 0.05; paired t test. Scale bars = 200 μm (A–D); 100 μm (E–H). Results are means ± se.
RvD2 attenuates neointimal hyperplasia in balloon-injured rabbit arteries
The effects of locally delivered RvD2 treatment on neointimal hyperplasia were evaluated in the rabbit femoral artery 28 d after injury (n=8). Both neointimal area (0.27±0.03 vs. 0.38±0.04 mm2, P=0.002, paired t test) and neointima/media ratio (0.77±0.06 vs. 1.07±0.09 mm2, P=0.007, paired t test) were significantly reduced by RvD2 treatment (Fig. 7).
Figure 7.
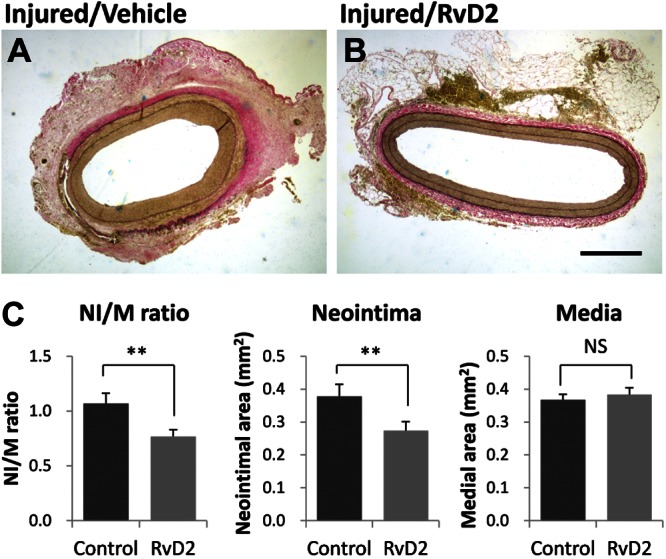
RvD2 treatment inhibits neointimal hyperplasia postangioplasty. Angioplasty and local treatment of bilateral rabbit (n=8) femoral arteries with RvD2 vs. vehicle control was performed as described, and vessels were explanted at 28 d by perfusion-fixation. A, B) Histomorphometric analysis was performed on elastin-stained sections of vehicle-treated (A) vs. RvD2-treated (10 nM, B) arteries. Scale bar = 500 μm. C) Summary of morphometric results. Neointima/media (NI/M) ratio and neointimal area were significantly reduced in RvD2-treated vessels. Results are means ± se. NS, not significant. **P <0.01; paired t test.
Reendothelialization in balloon-injured rabbit arteries
Reendothelialization was evaluated by immunostaining for CD31 in the rabbit femoral artery at 28 d after injury (n=8, 4 sections examined for each paired vessel). Since there are few branches in this injured segment, endothelialization primarily occurs from the proximal or distal ends of the treated artery. We found that reendothelialization was incomplete in all of the injured arteries examined, with an average of 25–50% CD31-positive coverage observed. No significant difference was observed between vehicle-treated and RvD2-treated vessels in the extent of reendothelialization (data not shown).
DISCUSSION
The present results document potent actions of the proresolving lipid mediators RvD1 and RvD2 on VSMC phenotypic responses that are characteristic in vascular injury. Dose-dependent inhibition of proliferation, migration, monocyte adhesion, superoxide production, inducible adhesion molecule, and inflammatory cytokine expression was observed, without measurable cytotoxicity. In a rabbit model of balloon arterial injury, local treatment with RvD2 significantly reduced proliferation, leukocyte infiltration, and oxidative stress at 3 d, and neointimal hyperplasia was significantly attenuated at 28 d. Furthermore we demonstrate that endogenous proresolving lipid mediators are available in the vessel wall, and vascular cells express the two known receptors for RvD1. Thus, collectively, these studies suggest a potential therapeutic opportunity for proresolution mediators in accelerating healing following acute vascular injury.
Accumulating evidence indicates that acute inflammation does not resolve in a passive fashion as previously assumed, but is actively terminated by a homeostatic process governed by specific lipid-derived mediators initiated by lipoxygenases (7–9). Much of the experimental evidence in this field has involved animal models of sterile inflammation, such as asthma (32–34), peritonitis (11, 13–17), and periodontitis (21, 22). The existence of a potential “resolution-deficit” is now considered a plausible mechanism for chronic and subacute inflammatory diseases, including atherosclerosis. There have been few studies examining the biological effects of resolvins on vascular cells, and fewer involving in vivo models of vascular disease. A recent study showed that 12/15-lipoxygenase expression protects mice against atherosclerosis via its role in the local biosynthesis of lipid mediators, including LXA4, RvD1, and protectin D1 (PD1) (23). These mediators exert potent agonist actions on macrophages and vascular ECs that can control the magnitude of the local inflammatory response. In contrast to early atherogenesis, the acute response to vascular injury emphasizes the central role of the VSMCs; hence, both the biological and the clinical/translational implications of the present work are distinct. We have previously shown that both lipoxins (LXA4) and resolvins (RvD1 and RvE1) have direct actions on VSMCs (24). The present study demonstrates a broad profile of activity in VSMCs, with direct relevance to the vascular injury response. While the molecular pathways of signal transduction remain to be fully elucidated, these findings have potential implications for the development of therapeutics specifically targeting resolution pathways to accelerate vascular healing and reduce scarring.
One of the salient findings of this study is the reduction in VSMC proinflammatory gene expression and adhesiveness to monocytes by RvDs. While prior studies have examined the direct actions of RvDs on monocytes/macrophages (35, 36), we demonstrate significant down-regulation of cell adhesion molecules on VSMCs known to mediate leukocyte adhesion. Furthermore, transcription factors regulating inflammatory pathways in VSMCs, including both NFκB and AP-1, were significantly down-regulated by RvD1. Previous studies showed that RvD1 reduced VCAM-1 expression in the choroid-retinal ECs (37), and protectin D1 (PD1) down-regulated the expression of VCAM-1 and MCP-1 in human aortic ECs (23). Exogenous DHA reduces expression of EC adhesion molecules, such as VCAM-1 and ICAM-1, thus regulating leukocyte:endothelial interactions (38). Vascular ECs modulate atherosclerosis development and progression via the expression of adhesion molecules that regulate the recruitment of leukocytes to the lesion. However, VSMCs also have an important role in the development of atherosclerotic lesions in humans, and are of particular relevance to the local response following acutely denuding mechanical injuries, such as angioplasty. In this setting, the transformation of resident and normally quiescent VSMCs to a proadhesive, proinflammatory cell is considered to be a critical early step in the pathogenesis of neointimal hyperplasia. The temporal and spatial extent of this phenotypic switch is also intimately linked with the magnitude of the response.
In addition to a reduction in inflammatory activation, we found that VSMC proliferation and migration were significantly reduced by RvD1 and RvD2. Previous studies have shown that LXA4 regulates PDGF-induced human renal mesangial cell proliferation and profibrotic change (39–42). A recent study also reported that RvE1 and D1 inhibit interstitial fibrosis in the obstructed kidney via inhibition of local fibroblast proliferation (43). Our previous study showed that both aspirin-triggered lipoxin (ATL) and RvE1 counterregulated PDGF-stimulated migration of human saphenous vein SMCs and decreased phosphorylation of the PDGF receptor β, thus changing the VSMC phenotype (24). We speculate that RvDs may directly counterregulate cytokine and growth factor signaling pathways in VSMCs that control proliferation and chemotaxis; however, further studies are needed to clarify the molecular mechanisms involved in these cellular responses.
The rabbit model of balloon arterial injury is an established model of neointimal hyperplasia, with known limitations in comparison to clinical disease. Important among these is the absence of an underlying atherosclerotic substrate. The local delivery approach employed in these experiments, i.e., a single, brief intraluminal exposure to drug, is a simplistic one with transient bioavailability. Further development of local vascular therapy with proresolution lipid mediators will require dedicated pharmacokinetics studies and likely more sophisticated delivery methods. Future preclinical studies are planned to address this. Nonetheless, using this strategy we observed attenuation of injury-induced cell proliferation, leukocyte infiltration, inflammatory gene expression, and superoxide production by RvD2 in the acute phase, which translated into a significant reduction of neointimal hyperplasia at 28 d after injury. As it is widely acknowledged that the processes of monocyte recruitment, VSMC proliferation, and VSMC migration have important roles in clinical restenosis, these effects of RvDs are considered very likely to contribute to the in vivo response observed and are of translational relevance.
A previous study reported that RvD1 reduced oxidative stress-induced peritonitis in mice (16). A more recent study showed that RvD2 decreased leukocyte:endothelial interactions in vivo by increased endothelial-dependent nitric oxide production and direct modulation of leukocyte adhesion receptor expression (12). In microbial sepsis initiated by cecal ligation and puncture (CLP), RvD2 sharply decreased both local and systemic bacterial burden; excessive cytokine production, including TNF-α, IL-1β, and IL-6; and neutrophil recruitment, while increasing peritoneal mononuclear cells and macrophage phagocytosis (12). Our study demonstrated that RvDs reduce inflammatory gene expression and superoxide production in VSMCs, in response to both exogenous cytokine and mechanical injury. Taken together, RvD1 and RvD2 demonstrate potent and broad effects to reduce vascular cell activation to a range of inflammatory stimuli that overlap acute and chronic injury.
Known receptors for RvD1 are members of the GPCR family, ALX (FPR2) and GPR32 (35). At present, specific receptors for RvD2 are yet to be identified. A common pattern emerging with resolvin signaling is the requirement for specific GPCRs, with often two distinct receptors capable of ligand binding and transduction. Recent studies on the effects of RvD1 on polymorphonuclear leukocyte adhesion have shown differential dose-response profiles for ALX and GPR32, as well as increased surface expression of these receptors in human neutrophils following proinflammatory stimuli (25). Interestingly, our data suggests that the effects of RvD1 on VSMC proliferation and migration may be differentially mediated by these two receptors. Moreover, Western blot and immunohistochemical analyses indicate that both GPR32 and ALX receptors are expressed in VSMCs, but GPR32 appears more strongly expressed in rabbit arteries. Further investigations are needed to more completely characterize the relative roles of GPR32 and ALX in VSMC signaling under various conditions, and the relationship between RvD receptor engagement and phenotypic responses of VSMCs to the ligands.
In summary, the present study demonstrates that RvDs broadly inhibit VSMC activation responses and modulate the vascular injury response in vivo, leading to a reduction in early inflammation and subsequent neointimal hyperplasia. These results suggest that antiinflammatory/proresolving lipid mediators, such as RvD1 or RvD2, may provide novel therapeutic strategies for the management of vascular diseases.
Supplementary Material
Acknowledgments
The authors thank Dr. Rong Wang for critical review of the manuscript.
This work was supported in part by funding from the Foundation for Accelerated Vascular Research (Vascular Cures), and U.S. National Institutes of Health grants 1P01GM095467 (C.N.S.), and F32 HL 104851-2 (S.R.).
C.N.S. is an inventor on patents (resolvins) assigned to Brigham and Women's Hospital (BWH) and licensed to Resolvyx Pharmaceuticals (Cambridge, MA, USA). C.N.S. is a scientific founder of Resolvyx Pharmaceuticals and owns equity in the company. C.N.S.'s interests were reviewed and are managed by BWH and Partners HealthCare in accordance with their conflict of interest policies.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- cDNA
- complementary DNA
- DHA
- docosahexaenoic acid
- DHE
- dihydroethidium
- DMEM
- Dulbecco's modified Eagle's medium
- EC
- endothelial cell
- EPA
- eicosapentaenoic acid
- GPCR
- G-protein-coupled receptor
- HDHA
- hydroxydocosahexaenoic acid
- HVSMC
- human vascular smooth muscle cell
- IC50
- inhibitory concentration for 50% effect
- ICAM-1
- intercellular adhesion molecule 1
- IL-1
- interleukin-1
- IL-1α
- interleukin-1α
- IL-1β
- interleukin-1β
- IL-6
- interleukin-6
- LC-MS/MS
- liquid chromatography-dual mass spectrometry
- LXA4
- d5-lipoxin A4
- LXB4
- lipoxin B4
- MaR
- maresin
- MCP-1
- monocyte chemoattractant protein 1
- PBS
- phosphate-buffered saline
- PD1
- protectin D1
- PDGF
- platelet-derived growth factor
- PTX
- pertussis toxin
- PUFA
- polyunsaturated fatty acid
- qPCR
- quantitative polymerase chain reaction
- RT-PCR
- reverse transcriptase-polymerase chain reaction
- ROS
- reactive oxygen species
- RvD
- D-series resolvin
- RvD1
- resolvin D1
- RvD2
- resolvin D2
- RvE
- E-series resolvin
- RvE1
- resolvin E1
- TNF-α
- tumor necrosis factor α
- VCAM-1
- vascular cell adhesion molecule 1
- VSMC
- vascular smooth muscle cell
REFERENCES
- 1. Hansson G. K. (2005) Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 352, 1685–1695 [DOI] [PubMed] [Google Scholar]
- 2. Libby P. (2002) Inflammation in atherosclerosis. Nature 420, 868–874 [DOI] [PubMed] [Google Scholar]
- 3. Conte M. S., Bandyk D. F., Clowes A. W., Moneta G. L., Seely L., Lorenz T. J., Namini H., Hamdan A. D., Roddy S. P., Belkin M., Berceli S. A., DeMasi R. J., Samson R. H., Berman S. S. (2006) Results of PREVENT III: a multicenter, randomized trial of edifoligide for the prevention of vein graft failure in lower extremity bypass surgery. J. Vasc. Surg. 43, 742–751; discussion 751 [DOI] [PubMed] [Google Scholar]
- 4. Norgren L., Hiatt W. R., Dormandy J. A., Nehler M. R., Harris K. A., Fowkes F. G. (2007) Inter-society consensus for the management of peripheral arterial disease (TASC II). J. Vasc. Surg. 45(Suppl. S), S5–S67 [DOI] [PubMed] [Google Scholar]
- 5. Rocha-Singh K. J., Jaff M. R., Crabtree T. R., Bloch D. A., Ansel G. (2007) Performance goals and endpoint assessments for clinical trials of femoropopliteal bare nitinol stents in patients with symptomatic peripheral arterial disease. Catheter Cardiovasc. Interv. 69, 910–919 [DOI] [PubMed] [Google Scholar]
- 6. Schillinger M., Sabeti S., Dick P., Amighi J., Mlekusch W., Schlager O., Loewe C., Cejna M., Lammer J., Minar E. (2007) Sustained benefit at 2 years of primary femoropopliteal stenting compared with balloon angioplasty with optional stenting. Circulation 115, 2745–2749 [DOI] [PubMed] [Google Scholar]
- 7. Serhan C. N., Savill J. (2005) Resolution of inflammation: the beginning programs the end. Nat. Immunol. 6, 1191–1197 [DOI] [PubMed] [Google Scholar]
- 8. Serhan C. N. (2007) Resolution phase of inflammation: novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu. Rev. Immunol. 25, 101–137 [DOI] [PubMed] [Google Scholar]
- 9. Serhan C. N., Chiang N., Van Dyke T. E. (2008) Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 8, 349–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Serhan C. N., Hong S., Gronert K., Colgan S. P., Devchand P. R., Mirick G., Moussignac R. L. (2002) Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med. 196, 1025–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Serhan C. N., Clish C. B., Brannon J., Colgan S. P., Chiang N., Gronert K. (2000) Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J. Exp. Med. 192, 1197–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Spite M., Norling L. V., Summers L., Yang R., Cooper D., Petasis N. A., Flower R. J., Perretti M., Serhan C. N. (2009) Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature 461, 1287–1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Arita M., Bianchini F., Aliberti J., Sher A., Chiang N., Hong S., Yang R., Petasis N. A., Serhan C. N. (2005) Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J. Exp. Med. 201, 713–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bannenberg G. L., Chiang N., Ariel A., Arita M., Tjonahen E., Gotlinger K. H., Hong S., Serhan C. N. (2005) Molecular circuits of resolution: formation and actions of resolvins and protectins. J. Immunol. 174, 4345–4355 [DOI] [PubMed] [Google Scholar]
- 15. Hong S., Gronert K., Devchand P. R., Moussignac R. L., Serhan C. N. (2003) Novel docosatrienes and 17S-resolvins generated from docosahexaenoic acid in murine brain, human blood, and glial cells: autacoids in anti-inflammation. J. Biol. Chem. 278, 14677–14687 [DOI] [PubMed] [Google Scholar]
- 16. Spite M., Summers L., Porter T. F., Srivastava S., Bhatnagar A., Serhan C. N. (2009) Resolvin D1 controls inflammation initiated by glutathione-lipid conjugates formed during oxidative stress. Br. J. Pharmacol. 158, 1062–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sun Y. P., Oh S. F., Uddin J., Yang R., Gotlinger K., Campbell E., Colgan S. P., Petasis N. A., Serhan C. N. (2007) Resolvin D1 and its aspirin-triggered 17R epimer: stereochemical assignments, anti-inflammatory properties, and enzymatic inactivation. J. Biol. Chem. 282, 9323–9334 [DOI] [PubMed] [Google Scholar]
- 18. Arita M., Yoshida M., Hong S., Tjonahen E., Glickman J. N., Petasis N. A., Blumberg R. S., Serhan C. N. (2005) Resolvin E1, an endogenous lipid mediator derived from omega-3 eicosapentaenoic acid, protects against 2,4,6-trinitrobenzene sulfonic acid-induced colitis. Proc. Natl. Acad. Sci. U. S. A. 102, 7671–7676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bento A. F., Claudino R. F., Dutra R. C., Marcon R., Calixto J. B. (2011) Omega-3 fatty acid-derived mediators 17(R)-hydroxy docosahexaenoic acid, aspirin-triggered resolvin D1 and resolvin D2 prevent experimental colitis in mice. J. Immunol. 187, 1957–1969 [DOI] [PubMed] [Google Scholar]
- 20. Connor K. M., SanGiovanni J. P., Lofqvist C., Aderman C. M., Chen J., Higuchi A., Hong S., Pravda E. A., Majchrzak S., Carper D., Hellstrom A., Kang J. X., Chew E. Y., Salem N., Jr., Serhan C. N., Smith L. E. (2007) Increased dietary intake of omega-3-polyunsaturated fatty acids reduces pathological retinal angiogenesis. Nat. Med. 13, 868–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hasturk H., Kantarci A., Goguet-Surmenian E., Blackwood A., Andry C., Serhan C. N., Van Dyke T. E. (2007) Resolvin E1 regulates inflammation at the cellular and tissue level and restores tissue homeostasis in vivo. J. Immunol. 179, 7021–7029 [DOI] [PubMed] [Google Scholar]
- 22. Hasturk H., Kantarci A., Ohira T., Arita M., Ebrahimi N., Chiang N., Petasis N. A., Levy B. D., Serhan C. N., Van Dyke T. E. (2006) RvE1 protects from local inflammation and osteoclast- mediated bone destruction in periodontitis. FASEB J. 20, 401–403 [DOI] [PubMed] [Google Scholar]
- 23. Merched A. J., Ko K., Gotlinger K. H., Serhan C. N., Chan L. (2008) Atherosclerosis: evidence for impairment of resolution of vascular inflammation governed by specific lipid mediators. FASEB J. 22, 3595–3606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ho K. J., Spite M., Owens C. D., Lancero H., Kroemer A. H., Pande R., Creager M. A., Serhan C. N., Conte M. S. (2010) Aspirin-triggered lipoxin and resolvin E1 modulate vascular smooth muscle phenotype and correlate with peripheral atherosclerosis. Am. J. Pathol. 177, 2116–2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Norling L. V., Dalli J., Flower R. J., Serhan C. N., Perretti M. (2012) Resolvin D1 limits polymorphonuclear leukocytes recruitment to inflammatory loci: receptor-dependent actions. Arterioscler. Thromb. Vasc. Biol. 32, 1970–1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Filion K. B., El Khoury F., Bielinski M., Schiller I., Dendukuri N., Brophy J. M. (2010) Omega-3 fatty acids in high-risk cardiovascular patients: a meta-analysis of randomized controlled trials. BMC Cardiovasc. Disord. 10, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang G. J., Sui X. X., Simosa H. F., Jain M. K., Altieri D. C., Conte M. S. (2005) Regulation of vein graft hyperplasia by survivin, an inhibitor of apoptosis protein. Arterioscler. Thromb. Vasc. Biol. 25, 2081–2087 [DOI] [PubMed] [Google Scholar]
- 28. Ho K. J., Owens C. D., Longo T., Sui X. X., Ifantides C., Conte M. S. (2008) C-reactive protein and vein graft disease: evidence for a direct effect on smooth muscle cell phenotype via modulation of PDGF receptor-beta. Am. J. Physiol. Heart Circ. Physiol. 295, H1132–H1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Patricia M. K., Kim J. A., Harper C. M., Shih P. T., Berliner J. A., Natarajan R., Nadler J. L., Hedrick C. C. (1999) Lipoxygenase products increase monocyte adhesion to human aortic endothelial cells. Arterioscler. Thromb. Vasc. Biol. 19, 2615–2622 [DOI] [PubMed] [Google Scholar]
- 30. Conte M. S., Birinyi L. K., Miyata T., Fallon J. T., Gold H. K., Whittemore A. D., Mulligan R. C. (1994) Efficient repopulation of denuded rabbit arteries with autologous genetically modified endothelial cells. Circulation 89, 2161–2169 [DOI] [PubMed] [Google Scholar]
- 31. Dalli J., Serhan C. N. (2012) Specific lipid mediator signatures of human phagocytes: microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood 120, e60–e72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Aoki H., Hisada T., Ishizuka T., Utsugi M., Kawata T., Shimizu Y., Okajima F., Dobashi K., Mori M. (2008) Resolvin E1 dampens airway inflammation and hyperresponsiveness in a murine model of asthma. Biochem. Biophys. Res. Commun. 367, 509–515 [DOI] [PubMed] [Google Scholar]
- 33. Aoki H., Hisada T., Ishizuka T., Utsugi M., Ono A., Koga Y., Sunaga N., Nakakura T., Okajima F., Dobashi K., Mori M. (2010) Protective effect of resolvin E1 on the development of asthmatic airway inflammation. Biochem. Biophys. Res. Commun. 400, 128–133 [DOI] [PubMed] [Google Scholar]
- 34. Haworth O., Cernadas M., Yang R., Serhan C. N., Levy B. D. (2008) Resolvin E1 regulates interleukin 23, interferon-gamma and lipoxin A4 to promote the resolution of allergic airway inflammation. Nat. Immunol. 9, 873–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Krishnamoorthy S., Recchiuti A., Chiang N., Yacoubian S., Lee C. H., Yang R., Petasis N. A., Serhan C. N. (2010) Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc. Natl. Acad. Sci. U. S. A. 107, 1660–1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Titos E., Rius B., Gonzalez-Periz A., Lopez-Vicario C., Moran-Salvador E., Martinez-Clemente M., Arroyo V., Claria J. (2011) Resolvin D1 and its precursor docosahexaenoic acid promote resolution of adipose tissue inflammation by eliciting macrophage polarization toward an M2-like phenotype. J. Immunol. 187, 5408–5418 [DOI] [PubMed] [Google Scholar]
- 37. Tian H., Lu Y., Sherwood A. M., Hongqian D., Hong S. (2009) Resolvins E1 and D1 in choroid-retinal endothelial cells and leukocytes: biosynthesis and mechanisms of anti-inflammatory actions. Invest. Ophthalmol. Vis. Sci. 50, 3613–3620 [DOI] [PubMed] [Google Scholar]
- 38. De Caterina R., Cybulsky M. A., Clinton S. K., Gimbrone M. A., Jr., Libby P. (1995) Omega-3 fatty acids and endothelial leukocyte adhesion molecules. Prostaglandins Leukot. Essent. Fatty Acids 52, 191–195 [DOI] [PubMed] [Google Scholar]
- 39. McMahon B., Mitchell D., Shattock R., Martin F., Brady H. R., Godson C. (2002) Lipoxin, leukotriene, and PDGF receptors cross-talk to regulate mesangial cell proliferation. FASEB J. 16, 1817–1819 [DOI] [PubMed] [Google Scholar]
- 40. McMahon B., Stenson C., McPhillips F., Fanning A., Brady H. R., Godson C. (2000) Lipoxin A4 antagonizes the mitogenic effects of leukotriene D4 in human renal mesangial cells. Differential activation of MAP kinases through distinct receptors. J. Biol. Chem. 275, 27566–27575 [DOI] [PubMed] [Google Scholar]
- 41. Mitchell D., Rodgers K., Hanly J., McMahon B., Brady H. R., Martin F., Godson C. (2004) Lipoxins inhibit Akt/PKB activation and cell cycle progression in human mesangial cells. Am. J. Pathol. 164, 937–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rodgers K., McMahon B., Mitchell D., Sadlier D., Godson C. (2005) Lipoxin A4 modifies platelet-derived growth factor-induced pro-fibrotic gene expression in human renal mesangial cells. Am. J. Pathol. 167, 683–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Qu X., Zhang X., Yao J., Song J., Nikolic-Paterson D. J., Li J. (2012) Resolvins E1 and D1 inhibit interstitial fibrosis in the obstructed kidney via inhibition of local fibroblast proliferation. [E-pub ahead of print] J. Pathol. doi: 10.1002/path.4050 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.