

Abstract
JC and BK polyomaviruses were discovered over 40 years ago and have become increasingly prevalent causes of morbidity and mortality in a variety of distinct, immunocompromised patient cohorts. The recent discoveries of eight new members of the Polyomaviridae family that are capable of infecting humans suggest that there are more to be discovered and raise the possibility that they may play a more significant role in human disease than previously understood. In spite of this, there remains a dearth of specific therapeutic options for human polyomavirus infections and an incomplete understanding of the relationship between the virus and the host immune system. This review summarises the human polyomaviruses with particular emphasis on pathogenesis in those directly implicated in disease aetiology and the therapeutic options available for treatment in the immunocompromised host.
1. Introduction
Polyomaviruses (PyV) are small (diameter 40–50 nm), nonenveloped, circular, double-stranded DNA viruses of the family Polyomaviridae. To date, 32 PyV species have been described, ten of which have been reported to infect humans (HPyVs), although not all of those have yet been definitively linked with disease [1]. In recent years, there has been a significant increase in the study of HPyVs as eight novel species have been discovered since 2007: KIPyV [2], WUPyV [3], Merkel cell polyomavirus (MCV) [4], HPyV6 [5], HPyV7 [5], trichodysplasia spinulosa-associated PyV (TSV) [6], HPyV9 [7], and MW PyV/HPyV10 [8, 9]. Prior to this, most clinicians would have been familiar with JC PyV (JCV) and BK PyV (BKV), the first two HPyVs, which were discovered in 1971 in patients who were immunosuppressed: JCV was identified in brain tissue from a patient with progressive multifocal leukoencephalopathy (PML) [10] and BKV from the urine of a renal transplant patient [11]. Seroprevalence studies subsequently demonstrated that both JCV and BKV were far more prevalent in the general population than the incidence of the diseases that they caused (PML and BKV-associated nephropathy (BKVN), resp.) [12]. The increased incidence of JCV/PML in association with the HIV-1 pandemic and the emergence of BKV/BKVN in association with renal transplantation (and haemorrhagic cystitis in bone marrow transplant recipients) highlighted the importance of the host immune system in the control of these latent infections and the pathogenesis of these diseases [13, 14].
Until the early part of this century, the JCV/BKV pattern of disease has been the hallmark of HPyV infection: asymptomatic primary infection occurring almost universally in childhood, from which time, the virus remains latent in the human host; viral reactivation—as evidenced by the presence of viral DNA in urine or, less frequently, blood—occurring intermittently throughout life but rarely causing disease in the otherwise immunocompetent host; and occasional cases of PyV-associated disease in the profoundly immunosuppressed, susceptible host. Recent events and discoveries, however, suggest it may be time to reconsider this paradigm. There have been multiple reports of the development of PML as a side effect of the immunomodulatory therapies (monoclonal antibodies) natalizumab [15–17], rituximab [15, 18], efalizumab [15, 19], and infliximab [20] used to treat various chronic medical conditions. In addition, eight novel HPyVs have been discovered, at least one of which (MCV) does not appear to follow the traditional disease course described above for JCV and BKV [4]. Taken in conjunction, these findings provide an opportunity to reevaluate HPyVs and their role in human disease. This review will focus primarily on JCV, BKV, MCV, and TSV on account of their now well established disease associations. However, it will also discuss the clinical and epidemiological data that currently exist for HPyVs 3, 4, 6, 7, 9, and 10.
2. Classification
The family Polyomaviridae came into existence in 2000, when the International Committee on Taxonomy of Viruses formally split the genera of the Papovaviridae family—the polyomaviruses and papillomaviruses—to form two new families, Polyomaviridae and Papillomaviridae [21]. The name polyomavirus, meaning “many tumours” is derived from Greek, and based on the fact that the first polyomavirus isolated—murine polyomavirus—caused the formation of multiple tumour sites when inoculated into newborn mice [22]. Indeed, injection of BKV and JCV into rodents also leads to the formation of multiple tumours [13, 23]. However, until the discovery of MCV, there was no direct association between the HPyVs and tumour formation in humans. The ten known HPyVs, adult seroprevalence, clinical disease and risk groups are summarized in Table 1. The family Polyomaviridae now comprises two mammalian genera, Orthopolyomavirus (consisting of two separate lineages: I and II) and Wukipolyomavirus, an avian genus, Avipolyomavirus, and a fifth distinct group—yet to be named—of which HPyV10 is currently the only member [13], see Figure 1. Of note, the HPyVs do not form a distinct cluster: JCV and BKV are found in Orthopolyomavirus lineage I, with MCV, TSV, and HPyV9 in lineage II. The remaining human PyVs (excluding HPyV10) are in the Wukipolyomavirus genus [1].
Table 1.
The human polyomaviruses, associated disease and immunocompromised risk groups.
| Human polyomavirus | Adult seroprevalence | Clinical disease | Patient's risk groups |
|---|---|---|---|
| JCV | 50%–80% | Progressive multifocal leukoencephalopathy | HIV-infected, immunomodulatory therapies |
| BKV | ≥90% | BKV nephropathy, haemorrhagic cystitis, ureteral stenosis | Solid organ and HSCT transplant recipients |
| MCPyV | 60%–80% | Merkel cell carcinoma | >50 years of age, immune suppression |
| WUPyV | ≥69% | No strong association | Not defined |
| KIPyV | ≥55% | No strong association | Not defined |
| HPyV6 | ≥83% | No strong association | Not defined |
| HPyV7 | ≥64% | No strong association | Not defined |
| TSV | 70%–80% | Trichodysplasia spinulosa | Transplant recipients, immune suppression |
| HPyV9 | 34%–70% | No strong association | Not defined |
| MWPyV | Not defined | No strong association | Not defined |
Figure 1.
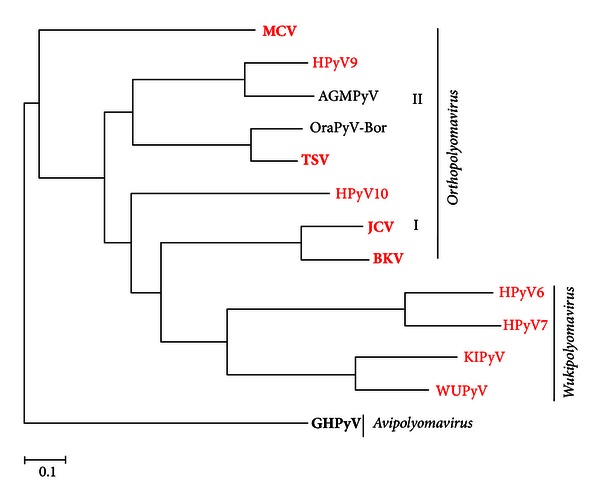
Phylogenetic relationships of the human polyomaviruses. Human polyomaviruses are presented in red with those associated with clinical disease in bold. The mammalian genera within the Polyomaviridae family: Orthopolyomavirus and Wukipolyomavirus and the single Avipolyomavirus genus member (employed as an outgroup) are indicated. Maximum likelihood phylogenetic analysis was performed on polyomaviral whole genome nucleotide sequences. Abbreviations and GenBank accession numbers employed as follows: AGMPyV, African green monkey polyomavirus (NC004763); BKV, BK polyomavirus (NC001538); goose haemorrhagic polyomavirus (GHPyV) genus (NC004800); HPyV6, human polyomavirus 6 (NC 004800); HPyV7, human polyomavirus 7 (HM011565); JCV, JC polyomavirus (NC001699); KIPyV, KI polyomavirus (NC 009238); MCV, The Merkel cell polyomavirus (HM011539); HPyV9, human polyomavirus 9 (HQ696595); MWHPyV, The Malawi polyomavirus (JX262162); TSV, trichodysplasia spinulosa-associated polyomavirus (GU989205); WUPyV, WU polyomavirus (NC009539).
The outer shell of the PyV capsid is constructed of 360 molecules of the major capsid protein VP1, organised into 72 pentamers, with each pentamer associated with a single copy of the minor structural protein VP2 or VP3. Only VP1 is exposed on the surface of the capsid and thus determines receptor specificity [23]. VP2 and VP3 are believed to play a role in stabilising the virus particle outside of the host cell, and—following alterations in the capsid structure that take place on cell entry—in traversing the intracellular interior [13, 23]. The genomic structure is highly related among the primate PyVs with a genome of around 5000 base pairs in length encoding six major viral proteins divided into three regions: the early coding region, the late coding region; and the noncoding control region (NCCR; see Figure 2). Each half of the PyV genome carries approximately half of the open reading frames, with replication proceeding in a bidirectional, temporally defined manner from the origin of replication (ORI) within the NCCR so that early and late transcribing regions are physically separated by the NCCR [23]. The PyV early proteins are translated from a series of alternative splicing events derived from a common mRNA precursor. The early coding region—transcribed before DNA replication begins—encodes large T antigen (TAg) and small t antigen (tAg). The BKV genome encodes three early proteins including the truncated tumour antigen (truncTAg) expressed from an alternatively spliced BKV early mRNA [24]. The late coding region—expressed after the onset of DNA replication—encodes the three viral structural proteins, VP1, VP2, and VP3, as well as the accessory agnoprotein. The PyV tumour antigens are multifunctional regulatory proteins that are essential for viral replication: in addition to driving the host cell towards the S phase of the cell cycle so that viral replication can occur; they also initiate viral DNA replication and they regulate transcription from the host and viral genomes. The agnoprotein appears to be multifunctional, with highly varied roles attributed to it, from viral transcription regulation to inhibition of host DNA repair to functioning as a viroporin [25–28].
Figure 2.

Schematic diagram illustrating the organisation of the dsDNA genome of BK virus. The open reading frames are represented by arrows with alternative splicing events highlighted by dashed lines. The origin of replication (ORI) within the noncoding control region (NCCR), from which transcription of early and late mRNAs proceeds, is indicated. The agnoprotein and truncated T antigen genes have not been described in all polyomaviruses.
3. JC Polyomavirus
3.1. Modes of Transmission and Epidemiology of JCV
The first case of demyelinating disease described with the term PML was found in a patient with chronic lymphocytic leukaemia and Hodgkin's lymphoma in 1958 [29], but accounts of potential cases can be traced as far back as 1930 [23, 30, 31]. A viral aetiology for PML was first proposed in 1959, based on observations of inclusion bodies in the nuclei of damaged oligodendrocytes [32]. However, it was not until 1971 that the causative agent was identified [10]. Padgett and colleagues isolated the virus from a mixed culture of glial cells and named it after the initials of the patient. The capacity of JCV to cause haemagglutination of human type O erythrocytes [33] facilitated seroprevalence studies, which demonstrated a worldwide distribution [34] and revealed that a large percentage of the population were asymptomatically infected before adulthood [35, 36]. Subsequently, more recent studies have confirmed these findings, with a reported prevalence for JCV of ~50%–80% in the general population [12, 37–39], although these rates vary among populations and age groups [40]. In addition, it has been shown that at any given time, approximately one-fifth of the population sheds JCV in urine [14]. Virus has also been detected in stool samples and is prevalent in sewage and rivers worldwide [41–45] raising the possibility of transmission through ingestion of nonsterile water. Full-length genome sequencing has identified seven JCV types, numbered 1–8 (type 5 was found to be a minor member of type 3), each with multiple subtypes [46]. The different types of JCV are associated with distinct human populations [46] and have been used to map population movements [47–50]. It has been hypothesised that type 6 is the original JCV type and that JCV coevolved with human populations, diverging as humans migrated out of Africa [51]. Types 1 and 4 are generally associated with Europeans, types 3 and 6 with Africans, type 2A with Asians, and 2D and 7C with Asians and South Asians. Types 2E, 8A, and 8B are found in Western Pacific populations with type 8A found only in Papua New Guinea [52–54]. Subtype 2B, found more often in Asians and Eurasians, has been associated with an increased risk of PML [46, 55]; type 4 has been associated with a lower disease risk [56].
In the majority of individuals, JCV infection is controlled by the healthy immune system [23], an interpretation supported by the epidemiology of PML. PML is an AIDS-defining illness, occurring in 3%–5% of HIV-infected individuals [14]. However, the rarity of PML prior to the AIDS pandemic—when it was associated primarily with B cell lymphoproliferative disorders [57, 58]—indicates that a reduction of CD4+ T-cells leads to a lack of immune control of JCV. In addition, non-HIV-related CD4+ T-cell reduction has also been associated with PML [59, 60]. Conversely, a cytotoxic T-cell response has been associated with greater control of JCV and longer PML survival rates [61, 62]. Furthermore, the use of highly active antiretroviral therapy (HAART) for the treatment of HIV has led to a reduced rate of PML in HIV-infected individuals despite having no demonstrable direct effect on JCV replication [23].
Indeed, both the EuroSIDA [63] and Swiss HIV cohort [64] studies have reported the clinical benefit of HAART on the incidence of PML, with reduced annual post-HAART rates of 0.6-0.7 per thousand. In immunocompromised individuals who are not infected with HIV, PML remains rare. In immunocompromised individuals who are not infected with HIV, PML remains rare. In a large population-based investigation, Amend and colleagues reported annual rates per 100 000 of 11.1 in chronic lymphocytic leukaemia, 10.8 in autoimmune vasculitis, 8.3 in non-Hodgkin's lymphoma, and 2.4 in systemic lupus erythematosus [63]. Studies in patients with rheumatoid arthritis have reported rates of 0.4 [66] to 1.0 [67] per 100,000, with the latter Swedish study also reporting a rate in the general population of 0.3. In individuals with multiple sclerosis (MS), however, the risk of PML has increased with the use of monoclonal antibodies, in particular natalizumab: the incidence of PML has risen from 0.09 per 1000 (for those who are anti-JCV negative) to 11.1 per 1000 for those who have received 24–48 months of natalizumab [68]. Similarly, in bone marrow transplant patients, the risk of PML appears to have surpassed that of HIV infected individuals, with both Amend et al. (35.4 per 100,000) [65] and Mateen and colleagues (1.24 per 1000) [69] reporting PML incidence rates in this group that are greater than that recorded in the EuroSIDA and Swiss HIV cohorts.
In spite of the fact that JC virus was identified as the aetiological agent of PML over 40 years ago, the definitive route of viral transmission and subsequent transport to the brain remain to be fully elucidated. The capacity of the virus to interact with B cells in the brain and replicate at low levels within B cells suggested a probable haematogenous route of CNS transmission [70–73]. Additional evidence that tonsillar stromal cells could be one of the initial sites of infection [57] led to the first working hypothesis that following primary infection—either via respiratory or oral acquisition—the virus is trafficked by infected lymphocytes from stromal or immune cells in the upper respiratory system to the bone marrow or kidneys, where it can persist for the life of the host. CD34+ haematopoietic stem cells harbor the virus in the bone marrow, and these cells migrate into the peripheral circulation and undergo differentiation to pre-B and mature B cells, augmenting JCV expansion [74]. Following immunosuppression, the virus mobilises from the bone marrow, and crosses the blood-brain barrier (BBB), with lytic infection commencing when the oligodendrocytes become infected [58].
The second working hypothesis for the pathogenesis of PML proposes that either the brain or the kidney may serve as a site of latency, indicating that JCV is already present in the brain at the time of the immune insult and that PML results from a loss of immune surveillance. In this model, JCV reaches the brain—possibly through B cells—during the viral dissemination that occurs following primary infection, reaching glial cells where it remains latent [23]. In support of this hypothesis, JCV DNA has been found in the brains of both healthy and immunocompromised patients without PML and other neurological disorders [75–78]. However, this pathway does not account for the very low incidence of PML in allograft recipients who are immunosuppressed for substantial periods of time for graft protection [23].
3.2. Pathogenesis of PML
Regardless of the site of viral latency or which of the above models is correct, the fundamental premise is that at least four events must occur before latent JCV can cause lytic infection of oligodendrocytes in the brain: (i) the host immune system must be compromised; (ii) the viral NCCR (discussed in more detail below) must acquire changes that increase viral transcription and replication in both B cells and glial cells; (iii) transcription factors that bind to the recombined NCCR sequence motifs must be present and/or upregulated in infected haematopoietic progenitor, B cells, and/or glial cells; (iv) free virus or virus in B cells must cross the BBB and be carried to the brain, where the virus is passed to oligodendrocytes and lytic infection takes place [23]. These events may occur in the bone marrow, in CD34+ lymphocyte precursors or B cells in the periphery, or in the brain. Significantly, in cases of PML, latent JCV DNA has been demonstrated in pathologic tissue from lymph, spleen, or bone marrow biopsies taken months to years before the patient developed neurological disease [79].
The PyV NCCR is the most variable portion of the viral genome, both within a single virus, as well as across genera of viruses [80–86]. It is thought to be the main determinant of cell type specificity, containing the origin of replication and numerous transcription factor binding sites [23]. In JCV infection, the NCCR varies greatly between isolates from PML patients. However, an “archetype” sequence (also known as CY) has been isolated from urine specimens from both PML patients and healthy individuals but is rarely found in PML lesions [23]. The NCCR from the original Mad-1 isolate of JCV contains an enhancer element that exists as a 98-bp direct tandem repeat and therefore contains duplicate TATA boxes, which can position mRNA start sites [87, 88] as well as multiple transcription factor binding sites [89]. The Mad-1 NCCR tandem repeat structure has been termed the “prototype” JCV NCCR sequence and is composed of three blocks of sequence, named “a,” “c,” and “e” with the TATA box found in “a.” Although Mad-1 was the first isolated NCCR sequence, many JCV isolates from PML patients do not possess the second TATA box, indicating it may not be essential for viral replication [90, 91]. The NCCR sequence of the “archetype” JCV is composed of a single copy of the 98-bp repeat of a-c-e, with 23-bp (“b”) and 66-bp (“d”) sequence blocks between “a,” “c,” and “e” to yield an a-b-c-d-e structure. However, archetype virus is rarely associated with PML [92]. Thus, the consistent isolation of tandem repeat-like NCCR sequences including the 98-bp tandem repeat in PML lesions strongly suggests this structure plays an important role in disease pathogenesis [82, 91, 93–95]. As a general rule, prototype and prototype-like sequences are generally found in PML tissue, while kidney-and urine-derived NCCR sequences are normally identical to archetype [23]. It has been proposed that all JCV isolates contain NCCRs that derive from the archetype sequence [92, 96, 97]; however, a mechanism for this derivation in the host has yet to be determined. Nonetheless, the prevailing disease pathogenesis model holds that the archetype-like sequences are transmitted from person-to-person and then undergo deletions and duplications within the infected host, leading to PML-like NCCR sequences which traffic to the brain. This “rearrangement” of the NCCR may take place in lymphoid cells, like B cells, since they possess the required enzymes for immunoglobulin gene rearrangement. Indeed, prototype-like sequences have been detected in lymphocytes from peripheral blood [57, 73, 98, 99] and the bone marrow [58, 94, 100]. Regardless of how the repeat NCCR variants are generated, this form of JCV is the pathogenic form that has repeatedly been isolated from PML. Compared with the archetype, this sequence contains significantly more transcription factor binding sites, which are essential to viral gene expression. Specifically, the archetype sequence does not contain Spi-B-binding sites, which are important for early viral gene expression [101], and possesses a reduced number of NF-1 binding sites, which are essential for fully activating viral transcription in the brain and cells of the lymphoid system. Spi-B is a transcription factor that binds to sequences in the JCV promoter/enhancer [74] and has been shown to be upregulated in B cells, glial cells, and haematopoietic progenitor cells in which JCV can replicate. The expression of Spi-B is also upregulated in patients with multiple sclerosis who are treated with the monoclonal antibody natalizumab (discussed below) [74]. NF-1 is a nuclear transcription factor and a cell-specific regulator of JCV promoter/enhancer activity. In humans, the NF-1 family of DNA-binding proteins is encoded by four discrete genes, one of which is NF-1 class X (NF-1X). NF-1X has also been shown to be upregulated in B cells, glial cells, and haematopoietic progenitor cells in which JCV can replicate [101–103]. These data suggest that changes in transcription factors can affect viral transcription during the maturation process of B cells.
Over the past decade, several immunomodulatory therapies, used for the treatment of autoimmune conditions, have been associated with cases of PML [15–20]. The known mechanism of action of each of these therapies has shed light on the host immune control of JCV. Natalizumab is a humanised monoclonal antibody for the treatment of relapsing-remitting multiple sclerosis (RRMS). The antibody binds the α4 chain of the α4/β1 and β7 integrin dimer also known as very late antigen-4 (VLA-4) [104]. VLA-4 mediates cell migration and infiltration in immune signaling, through binding its ligand—the vascular cell adhesion molecule (VCAM)—and facilitating the extravasation of leucocytes through endothelial cells to the sites of inflammation. In RRMS, the aim of the monoclonal antibody is to prevent leucocyte infiltration into the brain. However, natalizumab treatment also prevents developing B cells from attaching to a VCAM, forcing them to migrate from the bone marrow [74] and resulting in an increase in CD34+ progenitor cells in both the bone marrow and peripheral blood [105] and of factors involved in B cell differentiation, including Spi-B, in the peripheral blood [106]. Spi-B is also increased in CD34+ cells and B cells in natalizumab-treated patients. The risk of PML increases as treatment progresses, and the incidence of PML is estimated to be approximately 3.85 per 1000 patients treated with more than 24 infusions. Rituximab is an anti-CD20 humanised monoclonal antibody that fixes complement. Binding of CD20, an antigen expressed on B cells, results in downregulation of the B cell receptor and cytolytic apoptosis of CD20+ cells [107], resulting in depletion of CD20+ cells in the peripheral blood and cerebrospinal fluid (CSF) [14, 108]. In this setting, pre-B and B cells may be mobilised from the bone marrow and lymph nodes to replace CD20+ cells, leading to higher levels of CD34+ progenitors in the peripheral blood [14]. Efalizumab is a humanised monoclonal antibody against CD11a, a subunit of the leucocyte function-associated antigen type 1 (LFA-1), a T-lymphocyte adhesion molecule. LFA-1 binds intercellular adhesion molecule 1 (ICAM-1) which allows migration of T lymphocytes from circulation into sites of inflammation [109]. Efalizumab also downmodulates expression of VLA-4 resulting in T-cell hyporesponsiveness [110]. The drug was withdrawn from the market due to the occurrence of PML at an incidence of approximately 1 in 500. Infliximab is a humanized monoclonal antibody against tumour necrosis factor alpha (TNF-α) [111] that also induces apoptosis in TNF-α producing T-cells [112, 113]. The drug has been associated with an increase in infections or reactivation of latent infections [114], probably due to a blockage of TNF-α and T-cell reduction.
Finally, it should be noted that the rate of JCV disease in HIV-infected individuals remains significantly greater than in patients with other underlying causes of immunosuppression [23]. This is believed to be due to several factors: the duration and extent of immunosuppression, changes in cytokine secretion induced by HIV, viral interactions in coinfected cells and increased BBB permeability allowing for B cells infected by JCV to enter the brain [115]. Briefly, in HIV infection, the CD8+ T-cell response required to control JCV infection [116–120] is suboptimal because of the depletion in the CD4+ T-cells required to maintain that response [121]. In addition, HIV Tat protein has been shown to increase transcription from JCV [41, 122–128]; indeed, archetype JCV can replicate in cells expressing HIV Tat [29, 125, 129]. Furthermore, HIV infection of the brain causes upregulation of cytokines that attract lymphocytes [130] as well as an increase in cell adhesion molecules that may facilitate BBB crossing of JCV-infected cells. Finally, the astrocyte and neuronal damage caused by HIV proteins [131–135] lead to increased inflammation and further infiltration by JCV-infected lymphocytes, which may facilitate the onset of PML [23].
3.3. JCV-Associated Clinical Disease
The classic triad of PML consists of cognitive impairment, visual deficit and motor dysfunction [74], although symptoms and clinical presentation may vary based on the location and size of the lesion(s). Patients typically present with motor deficits, altered level of consciousness, ataxia, and visual symptoms [136, 137]. Seizures have been reported in PML, but this is believed to be due to the location of the lesions and does not herald a poorer prognosis [137]. Atypical (defined as non-PML) CNS presentations of JC infection have been described. JCV encephalopathy, indicating JC virus infection of the gray matter of the brain, has been reported in an HIV-negative woman with a history of lung cancer [138]; the extension of classic PML lesions into gray matter has also been described [139, 140]. JCV has also been implicated as a causative agent of meningitis in both immunocompromised and immunocompetent individuals [138]. Although not typically part of the routine screen for “viral meningitis” patients, one study has reported a prevalence of 1.5% for JCV in a mixed (immunocompetent and immunocompromised) cohort [141].
JCV-granule cell neuronopathy (JCV-GCN): while changes—enlarged and hyperchromatic nuclei [142]—in the granule cell layer of the cerebellum have been long recognized in PML it was unclear whether these cells were infected by JCV or the victims of collateral damage from the destruction of glial cells. However, in 2003, productive infection of granule cell neurons in the cerebellum was finally described, albeit in the presence of classic PML [143]. Subsequently, JCV was found in the brain of a patient with cerebellar atrophy in the absence of white matter PML lesions. JCV-GCN was proposed to be a novel syndrome distinct from PML and has since been reported in both HIV-positive and HIV-negative patients. Interestingly, the comparison of CSF-isolated virus and cerebellar virus NCCRs from a patient with AIDS showed differences in transcription factor binding-sites [144].
Magnetic resonance imaging (MRI) is the imaging modality of choice if a clinical diagnosis of PML is suspected, with lesions typically manifesting as high-signal intensity on T2-weighted and FLAIR sequences [23]. The lesions are usually multifocal, bilateral, and asymmetrical, involving the uncinate fibres, sparing the gray matter, and demonstrating a predilection for the posterior parts of the brain, although they may occur anywhere [145, 146]. The lesions may appear hypointense on T1-weighted images and do not enhance with the administration of gadolinium, as there is very little or no inflammation [147]. In the early stages of disease, the lesions are often subcortical, subsequently spreading to deep periventricular white matter [147]. Radiological findings alone are not sufficient to confirm a diagnosis of PML. Antibody testing is not currently of diagnostic significance after the onset of symptoms, although it may be used in risk stratification protocols for patients commencing immunomodulatory therapy [23]. The confirmatory test for suspected PML is the demonstration of JCV DNA in the CSF or brain by PCR. Detection of JCV DNA in blood is not of diagnostic significance as viraemia may be present in the absence of PML, and a percentage of PML patients are not viraemic [148]. The specificity of quantitative PCR can be optimised by targeting unique sequences within the JCV T antigen gene that are necessary for infection. In addition, the detection sensitivity of some assays can be as low as 10 copies/mL [149]. The prognostic significance of the magnitude of the viral load in the CSF has not been established [150]. Of note, other changes in the CSF in PML tend to be nonspecific, with a mild increase in protein, but a normal cell count and normal glucose. Interestingly, in the era of HAART and in those patients with MS in whom the immune system is relatively intact, the copy numbers of JCV can be quite low and difficult to detect [151, 152]. In this situation, brain biopsy may be indicated, as the MRI appearance is not pathognomonic for the disease. In brain tissue, JCV infection can be demonstrated by immunohistochemistry, in situ hybridization, or PCR analysis [153].
3.4. Association of JCV with Human Cancer
JC virus has the capacity to transform cells in culture and induce tumours of neural origin in animals, including rodents and non-human primates [154–156]. In human cancer, however, the data are less conclusive and conflicting reports of the presence of the JCV genome and the T antigen in tumours of both neural and nonneural origin exist. A comprehensive review of the available data in this controversial area is beyond the scope of this report. However, Del Valle and colleagues have recently performed such a review [157]. Although the authors ultimately conclude that JCV involvement in the genesis of neural tumours is a possibility that can neither be confirmed nor excluded at this time, they do highlight the intriguing fact that the cellular signaling pathways that have been identified as targets of JCV TAg in molecular experiments and in experiments with JCV early region transgenic mice are the same pathways that are observed to be dysregulated in human tumours that are immunopositive for TAg [157]. Ultimately, given the prevalence of JCV in the general population, large-scale epidemiological studies will be required to fully investigate the role—if any—of JCV in human cancers [23].
4. BK Polyomavirus
4.1. Modes of Transmission and Epidemiology of BKV
BK virus (BKV) was first isolated from a Sudanese renal transplant recipient (initials BK) with ureteral stenosis [11]. BKV acquisition is thought to occur subclinically early in childhood via the respiratory route, or accompanied by mild illness, such as tonsillitis, following contact with aerosols or fomites [158]. Seroconversion to BKV has been demonstrated in paired sera from children hospitalised with acute upper respiratory tract infection with multiple nonintegrated BKV genomes also detected in tonsillar tissue [159]. Evidence also exists to support other transmission modes for BKV, particularly, the faeco-urino-oral route and BKV seroconversion following organ transplantation, particularly in renal allograft recipients, has been established [44, 160–162]. BKV acquisition via semen, blood transfusion, and transplacental vertical transmission has also been put forward, with conflicting results in the latter case [163–168]. Population-based BKV seroprevalence studies indicate that 80%–90% of children are exposed and infected by ten years of age with a median age of 4-5 years [12, 39]. Waning of BKV immunity following the establishment of an infection has been suggested by decreases in antibody titres throughout life [12, 37]. This contrasts with serological correlates of JCV immunity, which remain stable and increase during life suggesting that differing transmission routes for each PyV and/or heterotypic immune responses to prior BKV exposure may afford some protection to infection from subsequent immunologic challenge with JCV [169].
There are four distinct serotypes and subtypes (genotypes) of BKV: I, II, III and IV with subtype I (the most prevalent) distributed worldwide, subtype IV in East Asia and Europe, and subtypes II and III rarely described [170]. BKV subtypes are routinely distinguished based on viral capsid protein VP1 nonsynonymous nucleotide polymorphisms and putative antigenic determinants of the BKV subtypes have been mapped within N-terminal residues 61–83 [171, 172]. Geographical separation of subgroups within BKV subtypes has been described with genetic studies showing subgroup Ia is most prevalent in Africans and the presumed ancestral subtype that coevolved with humans in an out of Africa dispersal, subgroup Ib1 significantly higher in Southeast Asians, Ib2 in Europeans and West Asians and Ic in Northeast Asians [170, 173, 174]. BKV subtype IV is particularly prevalent in East Asia [175], but has also been described in European populations [173, 176–179]. BKV subtype IV subgroups (IVa1, IVa2, IVc1, IVc2, IVb1 and IVb2) are found almost exclusively in Asia except IVc2 which occurs in Northeast Asia and Europe [180]. BKV subtyping has also provided insights into the mode of transmission. Second generation Japanese-Americans and Americans in California showed the European Ib2 lineage to predominate in both groups whereas Ic is most prevalent in Japan which suggests that transmission occurs outside the family [181]. There is no clear association with urinary excretion of a particular BKV subtype and human disease, and immunological status does not affect excretion of discrete BKV subtypes [13, 175].
4.2. BKV-Associated Clinical Disease
Following infection early in life, BKV remains latent in the tubular epithelium of the renal and urogenital tract [182]. Symptomatic reactivation of BKV in immunocompetent individuals is rare; however, the asymptomatic shedding of BKV in urine has been described in 7% of healthy adults without corresponding viraemia in paired plasma samples [183]. Three main clinical entities have been described associated with the BKV reactivation in the iatrogenically immunocompromised host: late-onset haemorrhagic cystitis, BKV nephropathy, and ureteral stenosis.
Haemorrhagic cystitis (HC) is characterised by haemorrhage of the bladder mucosa with painful micturation which ranges from microscopic haematuria to clot retention and renal failure. HC-associated reactivation of BKV is a frequently encountered condition in immunocompromised haematopoietic stem cell transplant (HSCT) recipients leading to significant morbidity and occasional mortality [184]. HC is either an early-onset, preengraftment event arising from chemotherapeutic agents, particularly metabolites of cyclophosphamide and/or irradiation or a viral-associated postengraftment, late-onset event; the majority of which are due to reactivation of BKV but may also arise from cytomegalovirus and adenoviruses. Late-onset BKV-associated HC occurs in 6%–29% of HSCT recipients and normally two months after transplant [185]. Numerous studies have identified an association between reactivation of BKV, with both viruria and/or viraemia, and late-onset HC and an overall lowering of patient survival [186, 187]. Other authors have seen no significant difference between BKV viruria in HC and non-HC groups and only correlated-disease progression with high-level reactivation in HSCT groups [188]. A case-control study evaluating the association of BKV viraemia with HC in HSCT recipients showed that plasma viral load of >104 copies/mL was detected in 63% of patients with HC and 57% of postengraftment BKV-HC cases compared with 5% of controls and importantly, BK viraemia occurred in 20 patients (67%) before clinical disease onset [189]. Saundh and colleagues have recently suggested that the monitoring of BKV viruria for early reactivation in the donor kidney may assist identifying patients at elevated risk of BKV-associated nephropathy (BKVN) [190].
BKVN develops in between 1%–10% of individuals who have undergone renal transplantation, generally within one year and up to 90% of these patients will lead to acute rejection [191]. Data from the United Network for Organ Sharing (UNOS; http://www.unos.org) show that graft loss attributable to BKVN was 7.5% (70/938) in 2009 and 5.7% (36/632) in 2010 [192].
Following the reactivation of latent BKV in the kidney, replication and lytic destruction of renal tubular epithelial cells occur resulting in tubular fluid accumulation in the interstitial compartment, characterised by an inflammatory interstitial nephropathy, associated with functional impairment due to tubular fibrosis and atrophy [193, 194]. No single risk factor has been definitively associated with BKVN in renal transplant recipients and the immunosuppressive regimen, and the intensity of immunosuppression appears to be the main factor resulting in BKV reactivation [13, 191]. With the triple immunosuppression and profound impairment of T-cell activation achieved by the increased usage of stronger calcineurin inhibitors such as tacrolimus, the use of antimetabolites like mycophenolate mofetil and anti-inflammatory corticosteroids has seen an increased incidence of BKVN [195]. A failure to mount or expand a cell-mediated immune response is further implicated in reactivation and replication of BKV, as interferon (IFN)-γ secreting BKV-specific T-cells were undetectable in renal transplant recipients who developed BKVN and correlated with higher levels of viraemia in BKV seropositive recipients [196, 197]. Strikingly, patients with BKVN treated by tapering of immunosuppression resulted in a reduction in plasma and urine viral loads, and the frequency of IFN-γ-secreting lymphocytes increased to the same level seen in healthy controls [196, 197]. In vitro investigations have also suggested that IFN-γ strongly inhibits replication/expression of BKV in primary human renal proximal tubule epithelial cells [198]. Taken together, the results suggest that cytokine and effector functions produced by cell-mediated immune responses are important in controlling viral reactivation and replication and clinical disease.
Humoral immunity is thought to be less important as BKV seropositive patients prior to transplantation are not protected from viral reactivation, replication, and BKVN [199]. Donor antibody levels are inversely proportional to viruria onset and directly proportional to viruria duration and peak urine viral load indicating donor origin for early BKV infection in renal transplant recipients [200]. Finally, viral-associated factors have been implicated in BKVN, and BKV NCCR and VP1 mutations have been described; however, it is unclear whether this may simply arise from a preexisting lack of immune control of viral replication which would naturally lead to higher viral sequence diversity [201, 202]. Other risk factors identified for the development of BKVN include mismatched HLA alleles, advanced age, male gender, white ethnicity, diabetes, recipient seronegativity and lack of HLA-C7 loci may also be associated with failure to control BKV replication [169, 193]. Interestingly, black renal transplant recipients had a lower risk of posttransplant BKV infection compared with white renal transplant recipients, independent of other confounding risk factors, suggesting that host factors that exist regulate viral latency and reactivation [203]. Genome wide association studies, such as have been conducted for hepatitis C virus to investigate ethnic differences in treatment responses, could conceivably be undertaken to potentially identify host genetic variation associated with poor prognosis [204].
Ureteral stenosis, necessitating percutaneous nephrostomy, has been associated with BKV viraemia when compared to aviraemic renal transplant recipients within one year of engraftment [205]. BKV-associated reversible upper urinary tract obstruction secondary to HC leading to ureteral stenosis has also been reported, though less frequently, in HSCT recipients [206].
Definitive diagnosis of BKVN requires a biopsy to be taken for histopathology to determine the severity of scarring, atrophy, interstitial fibrosis, and inflammation. However, as disease progresses following asymptomatic reactivation of latent virus in the kidney, monitoring for viruria and viraemia is undertaken by real-time PCR approaches—typically targeting the conserved T antigen gene—so a reduction in immunosuppression can be instigated early before extensive organ damage or allograft rejection can occur [207]. BKV-specific real-time PCR in plasma or sera are generally favoured over detection in urine as asymptomatic viruria is common and sustained viraemia is a better predictor for the development of BKVN [208]. Alternative approaches such as urinary cytology to detect renal tubular epithelial cells with intranuclear basophilic inclusion bodies on Papanicolaou staining (decoy cells) have low-positive predictive value in diagnosing BKVN compared to PCR-based approaches [208]. A cutoff of 104 viral copies per mL of serum or plasma is commonly employed, and this approach of viral monitoring and tapering of immunosuppression to prevent development of nephropathy and graft dysfunction has been previously shown to be effective in cost-benefit analysis [209].
A potentially major breakthrough in the prevention and treatment of BKVN was suggested by a recent longitudinal serological study of kidney transplant recipients which demonstrated that BKV subtype I and subtype IV are serologically distinct using sensitive new methodologies [210]. In particular, the authors relied on BKV reporter vectors (pseudovirions) to evaluate serotype-specific neutralising antibodies rather than more traditional recombinant virus-like particles (VLP) ELISAs which crucially detect both neutralising and nonneutralising antibodies in the latter case. Using these antibody-mediated neutralisation assays, 5% and 49% of kidney transplant recipients were BKV subtypes I and IV naïve respectively pre-transplant, and 100% of BK subtype I and 43% of BK subtype IV seronegative patients pretransplant seroconverted in a type-specific manner. A model is presented where BKVN can arise from a de novo infection arising from a BKV subtype IV-infected kidney leading to replication in immunococompromised patients without prior exposure to this rarer BK subtype. Interestingly, prior studies have reported higher seroprevalence of BKV subtype IV in patients with interstitial nephritis [211]. Pastrana and colleagues argue persuasively that induction of a neutralising antibody response to BK subtype IV, or all subtypes, by vaccination of kidney transplant patients immunological naïve for certain subtypes prior to transplantation may prevent replication and BKVN associated with virus present in the transplanted organ [210].
5. Merkel Cell Polyomavirus
5.1. Epidemiology of MCV
In contrast to the lack of evidence for a strong and unambiguous association of other PyVs with human cancers, particularly, JCV and BKV, reviewed in [212], the Merkel cell polyomavirus (MCV) since discovery in 2008 has been strongly implicated in cellular transformation in an highly aggressive primary cutaneous neuroendocrine skin neoplasm (associated with a poor prognosis) termed Merkel cell carcinoma (MCC) [213, 214]. MCV shares a similar epidemiological profile to other human PyVs with serosurveys indicating that the exposure and infection occur early in childhood or asymptomatically later in life and that adult MCV seroprevalence is 60%–80% [215–217]. The precise mode of MCV transmission is unclear but as MC polyomaviral DNA (and HPyV6 and HPyV7) is found predominantly on human skin and shed in encapsidated virions, acquisition is most likely by respiratory or cutaneous routes [5].
5.2. MCC and Immunity
Heath and colleagues defined the most prominent clinical features of MCC in the acronym: AEIOU (asymptomatic/lack of tenderness, expanding rapidly, immune suppression, older than 50 years, and ultraviolet-exposed site on a person with fair skin), where 89% of primary MCCs had ≥3 of these findings [218]. Prior to the discovery of MCV, a defect in cellular immunity, indicative of an infectious disease aetiology for MCC, was suggested by a strikingly higher incidence (>13-fold increased risk) in HIV-infected individuals with clinical AIDS compared to the general population [219]. Notably, chronic lymphocytic leukaemia (CLL) was also found to be >30-fold overrepresented in MCC patients [218].
In addition, in association with iatrogenic immunosuppression, transplantation and MCC cases were reported following liver, heart, bone marrow and particularly renal allografts [220]. Discontinuation of cyclosporine and azathioprine immunosuppressive therapy and temporary regression of MCC metastases has also been reported [221]. Cases of MCC have also been seen in patients with a diverse array of autoimmune disorders, including systemic lupus erythematosus, chronic sarcoidosis, myasthenia gravis and Behçet's disease, correlating with the increased usage of potent immunosuppressive agents in the treatment of these conditions, such as fludarabine and rituximab which induce profound lymphopenia [222–225]. Age-specific incidence data for primary MCC also indicate that this is a disease of the elderly (90% of patients being older than 50 years) which correlates with age-related waning immune surveillance and impaired immunity [218]. Interestingly, there is a male predominance of MCC with a ratio of 1.4 : 1 (58.5% male and 41.5% female), and, increasingly, gender-based differences in inflammatory responses to pathogens are being recognized [218, 226]. Titres of anti-MCV antibodies are elevated in MCC patients suggesting that a defect in immune surveillance leads to viral replication and viraemia before tumorigenensis [227]. Adoptive immunotherapies may therefore offer promise for the treatment of MCV-MCC in elderly patients and other groups with impaired immunity as spontaneous remission of MCC has been reported which is thought to occur by T-cell-mediated immune response and tumour cell apoptosis [228].
Two mutational events following a loss of immune surveillance appear critical to cancer development in MCC patients; firstly, MCV is clonally integrated in an apparently unbiased location in the tumour genomes, and, secondly, the TAg helicase domain associated with NCCR binding and thus viral (lytic) replication is abolished; however, all mutations downstream of the LXCXE Rb tumour suppressor-binding motif are retained [4, 229]. The current model for MCC development is that some form of immune compromise (either age-related, iatrogenic, inherited; or infectious disease-related immunodeficiency) leads to a failure of cell-mediated immune surveillance of MCV, and virus integration into the host genome with abrogation of replicative capability through TAg mutation leading to clonal expansion. The discovery of MCV has led to better diagnostics for MCC but also critically the identification of potential treatments and the more rational design of therapeutics, for example, the identification of small molecule inhibitors of the survivin oncoprotein which was found upregulated following MCV binding of the tumour suppressor Rb [230].
6. KI Polyomavirus
KI polyomavirus (KIPyV, named for the Karolinska Institute in which it was first identified) was discovered as part of a systemic “molecular screening” search for unknown viruses in clinical respiratory tract samples in 2007 [2]. Using their own previously described methodology [231], the authors screened cell-free supernatants of 20 randomly selected nasopharyngeal aspirates that were submitted to the Karolinska University Laboratory for the diagnosis of respiratory tract infections. Of sequence reads from 374 clones, 75 were categorised as viral sequences: of these, 69 matched human rhinovirus or enterovirus species, 5 closely matched respiratory syncytial virus, and one showed weak amino acid similarity to the VP1 protein of the simian PyV SV40. The complete consensus viral genome sequence of this clone was determined from the original patient sample, identified as a polyomavirus and demonstrated on phylogenetic analysis to be clearly separate from all other known polyomaviruses [2]. Molecular prevalence studies performed on several sample sets detected KIPyV in 6/637 (1%) nasopharyngeal aspirates and 1/192 (0.5%) faeces samples but in none of 150 urine, 192 whole blood, 96 leucocyte, or 33 serum samples. Of interest, five of six KIPyV positive samples had another respiratory virus detected by standard diagnostic techniques (three RSV, influenza, and human metapneumovirus), suggesting that KIPyV may not have been responsible for the symptoms prompting nasopharyngeal sampling.
Since its discovery, KIPyV DNA has been detected in respiratory specimens worldwide [232–237], suggesting widespread infection in humans. Indeed, Kean and colleagues have reported KIPyV seroprevalence rates of 55% in a population of healthy adult blood donors and paediatric blood samples. Of note, the seroprevalence in children 1–5 years of age (children <1 year were excluded to avoid the confounding effect of maternal antibody) was 44.6%, rising to 60.9% in 10–15 year olds, indicating that the primary exposure to the virus occurs in childhood [12]. In spite of KIPyVs widespread distribution, its pathogenicity or capacity to cause respiratory disease remains unconfirmed, as molecular prevalence studies in which control groups were included detected viral sequences at similar or higher frequencies in asymptomatic patients [236, 238]. In addition, the link between KIPyV and disease is complicated by the high rate of coinfection with other viruses [2, 232–234]. Nevertheless, the virus has been the sole pathogen identified in some cases [239]. KIPyV has been detected in blood from 4/130 (3.1%) healthy blood donors and in 2/62 (3.2%) HIV-infected individuals [240]. KIPyV has also been detected in the stool of patients with haematological disorders; however, the presence of other viruses capable of causing gastrointestinal disease makes causality difficult to establish. Nevertheless, an association between the presence of KIPyV and diarrhoea, compared to KIPyV-negative patients, has been reported [241]. The reported detection of KIPyV in the central nervous system [242] has not been confirmed, but the reason for the conflicting results is not known [243]. In order to determine if—like other HPyVs—KIPyV was more prevalent in immunocompromised patients, Kuypers and colleagues tested 2732 nasal washes during the first year after allogeneic HSCT from 222 patients [244]. After one year, the cumulative incidence estimate for KIPyV was 26%. Age <20 years and detection of a respiratory virus in the previous two weeks predicted KIPyV detection. Sputum production and wheezing were associated with detection of KIPyV in the past week. However, there was no association with acute graft versus host disease, CMV reactivation, neutropenia, lymphopenia, hospitalization, or death. The authors did not find a clear role for KIPyV (and WUPyV) as respiratory pathogens, and concluded that routine testing for these viruses in immunocompromised patients could not be recommended at this time [244].
7. WU Polyomavirus
WU polyomavirus (WUPyV, named for Washington University) was discovered in 2007 in a nasopharyngeal aspirate from a three-year-old child with pneumonia [3]. When standard respiratory virus PCR assays yielded no pathogen, total nucleic acid was randomly amplified, cloned, and shotgun sequenced. Of 384 reads obtained, six were classified as viral sequences that yielded three unique regions, each of which possessed limited homology to known PyV proteins. Subsequent analysis of the sequence data following the discovery of KIPyV revealed amino acid identities of 65% to 69% between WUPyV and KIPyV. Furthermore, an additional three of eight sequences that were previously unclassified demonstrated 58%–84% amino acid identity to KIPyV VP1 and VP2 proteins [3]. Molecular testing detected the novel virus in 37/1245 (3%) respiratory samples in 5/410 (1%) of upper respiratory specimens and in 1/480 (0.2%) bronchoalveolar lavage samples. Thirty-three of 37 positive specimens were from children under three years of age, and WUPyV was the sole virus detected in 12 patients with clinical evidence of respiratory tract infection. As with KIPyV, co-infections were common, with 25 of 37 samples yielding additional respiratory viruses, predominantly rhinovirus and human bocavirus. WUPyV DNA was not detected in any of 727 urine samples screened by PCR, the majority of which were obtained from renal transplant patients.
Since its discovery, WUPyV has also been detected in clinical samples worldwide [232, 234, 235, 237, 245–249]. Kean and colleagues reported WUPyV seroprevalence of 69% overall, from a baseline of 44.6% in 1–5 years olds, rising to 59.9% in 10–15 years olds and 70.9% in adults aged 50 years and older [12]. Again, this indicates the majority of human infection occurs in childhood. As for KIPyV, the pathogenicity of WUPyV given its detection in asymptomatic individuals remains to be resolved [243]. WUPyV has also been detected in blood [240], stool [241], and in unconfirmed reports in the CNS [242, 250], although, in contrast to KIPyV, the prevalence of WUPyV was higher in HIV-infected individuals (4.6%) than blood donors (0.8%) [240]. In allogeneic HSCT recipients, the cumulative annual incidence for WUPyV was 8%; as with KIPyV, age <20 years was predictive of detection [244]. Sputum production and wheezing were also associated with WUPyV detection in the preceding month. However, there was no association with acute graft versus host disease, CMV reactivation, neutropenia, lymphopenia, hospitalisation, or death. Routine testing of respiratory samples from immunocompromised individuals, as for KIPyV, is not recommended based on the currently available data [244].
8. Human Polyomavirus 6
Human polyomavirus 6 (HPyV6) was recovered in 2010 from the skin of healthy volunteers in a study designed to retrieve full-length wild-type MCV DNA from skin [5]. In addition to MCV, however, sequencing of the cloned rolling circle amplification (RCP) products also revealed the existence of two previously unknown PyVs, termed as HPyV6 and 7 (discussed below). Complete HPyV6 genomes were cloned from 5/35 individuals, with repeat sampling suggesting a chronic viral infection. Serology studies performed by Schowalter and colleagues on 95 samples from blood donors yielded an HPyV6 seroprevalence rate of 69%. These findings have been confirmed by others, with Nicol and colleagues reporting seroprevalence rates of 37.5% of 1–4 years olds, increasing to 61.8% in 15–19 years olds and 98.2% in those aged 80 years and older [215]. The increasing incidence with age suggests HPyV6 infection occurs throughout life. At present, there is no known disease association for HPyV6. However, viral DNA has been detected in faeces and nasopharyngeal swabs in transplant recipients [251]. The authors of the latter study note, however, that they cannot exclude contamination of these samples with virus shed from the skin.
9. Human Polyomavirus 7
Human polyomavirus 7 (HPyV7) was first identified on the skin of healthy volunteers enrolled in an MCV study as described above [5]. Complete HPyV7 genomes were cloned from 4/35 individuals, with repeat sampling suggesting chronic infection. The HPyV7 genome was 68% identical to HPyV6 at the nucleotide level. HPyV7 seroprevalence rates are lower than those of HPyV6. Schowalter's group reported HPyV7 seroprevalence rates of 35% in 95 adult blood donors, a finding confirmed by Nicol and colleagues, who reported rates of 10.4% in 1–4 years olds, increasing to 36% in 15–19 years olds, reaching 85.7% in individuals aged 80 years or more [215]. Again, the continued increase with age is indicative of infection occurring throughout life. HPyV7 has been detected in urine and nasopharyngeal swab samples from a liver transplant recipient [251]; however, there is no known disease association for HPyV7 at this time.
10. Trichodysplasia Spinulosa-Associated Polyomavirus
Trichodysplasia spinulosa (TS) is an extremely rare (<1/1000000 prevalence) folliculocentric skin disease that has been described in iatrogenically immunocompromised hosts, particularly solid organ transplant recipients, and also in individuals with nontransplant-associated haematological malignancies, particularly acute lymphocytic leukaemia, receiving chemotherapy [6, 252–262]. TS is characterised by the development of predominantly facial, 1–3 mm follicular papules and keratotic protrusions (spicules or spines) often with accompanying alopecia of the eyelashes and brows [252, 254–256, 258–262]. An infectious aetiology for TS had been suspected since the description of 40–45 nm viral particles with transmission electron microscopy by Haycox (1999) and coworkers within the nuclei of the abnormally matured follicular inner root sheath cells overproducing trichohyalin [254].
The TS-associated polyomavirus (TSV), the eighth described human PyV, was identified by rolling circle amplification using the bacteriophage φ29 DNA polymerase and template derived from the plucked spicules of a TS patient [6]. The TS patient had received a standard immunosuppressive regimen (tacrolimus, mycopheolate mofetil and methylprednisolone) for cardiac transplant 18 months prior to presentation to dermatologists. Interestingly, the patient was treated with rituximab one year posttransplant, with concomitant tapering of immunosuppressive treatment, following the development of an EBV-positive B cell lymphoma [6]. The 5232-bp circular dsDNA TSV genome formed a monophyletic clade with Bornean orangutan PyV and was more closely related genetically to MCV than to BKV or JCV [6]. The 100% TSV positivity and active infection characterised by high viral titres (>106 copies/cell) in skin lesions is strongly indicative of an aetiological relationship in disease pathogenesis [6, 263]. Age-specific seroprevalence studies in the human population have demonstrated that TSV is widespread in all age groups (41%–70% by age 10 and 70%–80% among adults) suggestive of primary exposure and the establishment of latency in early childhood with acquisition in adulthood a relatively rare event [215, 264, 265]. Furthermore, sensitive TSV-specific molecular assays failed to detect any active TSV infections in sera from a large elderly hospitalised population [266], and an age-specific decrease in anti-TSV antibody titres has also been observed [215]. Taken together, these findings suggest that TSV, like BKV, establishes a sub-clinical persistent infection early in childhood, that TSV does not replicate in adulthood in immunocompetent individuals and that progression from a latent to lytic cycle accompanies immunocompromise leading to active replication and associated disease.
11. Human Polyomavirus 9
Human polyomavirus 9 (HPyV9) was first discovered in the serum of a kidney transplant patient in 2011 [7]. Leendertz and colleagues screened 597 clinical samples collected from immunocompromised (renal transplant, HIV-infected, and PML) individuals, having previously identified more than 20 novel PyVs in non-human primates [267]. Phylogenetic analysis indicated that the HPyV9 genome was more similar to the genome of the African Green Monkey-derived lymphotropic polyomavirus (LPV) than to those of other PyVs. Interestingly, prior seroepidemiological studies had demonstrated that ≤30% of human sera had strong reactions to antigens derived from LPV [267, 268]. It appears that these findings can now be explained by cross-reactivity with HPyV9 [269, 270]. Nicol and colleagues reported HPyV9 seroprevalence rates of ~10% in clinical samples from children aged 1–7, rising to ~33% in healthy adult blood donors, the increasing prevalence with age suggesting that HPyV infection occurs throughout life. The same group later confirmed these findings in a larger patient cohort, with HPyV9 seroprevalence reaching 34% in 15–19 years olds and continuing to rise to 70% in subjects aged 80 years and older. Trusch and colleagues reported comparable seroprevalence rates in healthy children (13% in 2–5 years olds) and adolescents/young adults (35% in 11–20 years olds). Conversely, however, they also reported that HPyV9 seroprevalence peaked at 53% in 21–30 year olds, declining subsequently to 35% in subjects aged 60 years and older [270]. They also found a higher HPyV9 seroprevalence in renal and HSCT recipients, when compared with healthy controls. In contrast, liver transplant recipients and patients with neurological dysfunction demonstrated no such difference. HPyV9 DNA has also been detected in the urine of a child one week following liver transplant [251], and in respiratory samples from pregnant and nonpregnant females [271]. In contrast, HPyV9 DNA was not discovered in Japanese patients with CLL [272]. However, at present, there is no known disease association for HPyV9. Given that only five of the original 597 samples reported by Scuda and colleagues yielded positive HPyV9 DNA results by PCR (on repeat testing), and assuming an overall seroprevalence of 30%, it is probable that HPyV9—like other HPyVs—only causes disease in a small percentage of those infected (if at all).
12. MW Polyomavirus/Human Polyomavirus 10/MX Polyomavirus
Malawi polyomavirus (MWPyV) was identified by shotgun sequencing of DNA from virus-like particles isolated from a faeces sample collected from a healthy child from Malawi [9]. Siebrasse and colleagues subjected the purified DNA to 454 sequencing and identified six reads that aligned to tAg and VP1 proteins of known polyomaviruses. Phylogenetic analysis of the completed viral genome identified a novel polyomavirus that is highly divergent from other members of the Polyomaviridae family. Indeed, the different VP1 and VP2 tree topologies generated for MWPyV suggest it may be derived from an ancestral recombination event [9]. Molecular prevalence studies detected MWPyV in 12/514 (2.3%) stool samples from children presenting with diarrhoea. Interestingly, three of the positive samples were from a 5-year-old lung transplant recipient, taken over a period of four months, raising the possibility either of chronic infection or prolonged asymptomatic shedding: eight of the other nine patient samples were negative for all organisms tested, except MWPyV.
At the time the paper of Siebrasse's group was in press, another group was in the process of reporting a similar finding [8]. Buck and colleagues set out to identify unknown viruses in skin specimens taken from a patient with WHIM (warts, hypogammaglobulinemia, infection, and myelokathexis) syndrome, which is marked by an individual's relative inability to control human papillomavirus (HPV) infections [8]. Following rolling circle amplification, several cloned restriction fragments were homologous to various human and animal PyVs. Sequencing of the entire genome revealed what appeared to be a previously unknown human PyV, which the authors proposed to name HPyV10. However, subsequent comparative analysis of the nucleotide sequences of the MWPyV isolates demonstrated that they were from 95% to 99% identical to that of HPyV10, and as such constitutes two strains of the same viral species rather than two separate novel species.
Following these two reports, a third paper subsequently appeared reporting the discovery of a novel PyV in acute diarrhoeal samples from children [273]. Yu and colleagues employed an unbiased deep sequencing approach to identify a novel highly divergent HPyV in stool samples from children. The initial sequence was discovered in a sample from a two-year-old child with diarrhoea from Mexico, hence the proposed name MXPyV. The virus differed substantially from the other nine known PyVs, with amino acid sequence identities ranging from 13%–44%. Subsequent molecular prevalence studies performed by the group detected MXPyV in 12/96 (12.5%) stool samples from children in Mexico, in 18/546 (3.3%) stool samples in California, and in 4/96 (4.2%) in Chile [273]. However, no association between MXPyV and the presence of diarrhoeal symptoms was noted. MXPyV was also detected in 1/136 (0.74%) respiratory samples from hospitalised children with pneumonia in Mexico. MXPyV was not detected in any of 480 plasma and urine samples from renal (n = 283) or solid organ and bone marrow transplant (n = 193) recipients. When the complete sequence of MXPyV was compared with the novel MWPyV/HPyV10 above, it transpired that it was almost identical, sharing 99.8% or 99.7% identity, respectively. As such, the three viruses are different variants of the same species, and likely represent the first members of a new subclade of PyVs. Nevertheless, the seroprevalence of this virus in different human populations remains unknown, as does its capacity to cause disease.
13. Polyomavirus-Encoded MicroRNAs: Immune Evasion, Establishment of Persistent Infections, and Potential Therapeutic Applications
MicroRNAs (miRNAs) are small (~22 nucleotide), non-coding, posttranscriptional regulators of gene expression, initially identified in model organisms, and subsequently described in metazoans and viruses [274, 275]. Seminal work from the Sullivan Laboratory identified the existence of an SV40 virally-encoded miRNA in an antisense orientation to the TAg early gene target leading to an autoregulatory loop and downregulation of TAg mRNA and evasion from CD8+ cytotoxic T lymphocyte (CTL) immune surveillance [276]. These monkey SV40-encoded miRNAs were subsequently shown to be evolutionarily conserved in both sequence and function in the human pathogens BKV and JCV [277]. Intriguingly, these miRNAs are expressed in JCV PML brain tissue which suggests that the viral miRNAs could be therapeutic targets using appropriately delivered synthetic oligonucleotides, that is, anti-miRNAs, termed antagomirs [277, 278].
This was extended by elegant work which demonstrated that a viral miRNA identical in sequence between BKV and JCV was shown to target the stress-induced ligand ULBP3—a protein recognized by the natural killer (NK) cell receptor NKG2D [279]. BKV and JCV miRNA-mediated downregulation of ULBP3 decreased NKG2D-mediated killing of virus-infected cells by NK cells. Conversely, inhibition of the viral miRNA during infection leads to enhanced NK cell killing of infected cells implicating these virally-encoded miRNAs in the establishment and maintenance of lifelong persistence by compromising the host immune system [279]. MCV has also been shown to encode a miRNA from the late strand in antisense orientation to early viral transcripts in 50% of Merkel cell tumours and predicted to target host mRNAs leading to immune evasion [280, 281]. Moreover, in MCC, MCV-encoded microRNAs were identified that potentially regulate T- and B-cell receptor signalling hampering viral (tumour) immune recognition [280].
It is unclear whether detection of virally encoded miRNAs has diagnostic or prognostic clinical utility—in an analogous manner to the detection of BKV DNA in serum and plasma and VP1 mRNA in urine samples or JCV DNA in CSF—and longitudinal studies of immunocompromised renal-transplant recipients and patients receiving immunomodulatory agents are of interest to see whether PyV miRNA presence or absence in different compartments precede development of PyV-associated disease.
14. Antiviral Treatment
At present, there is no antiviral therapy specifically licensed for the treatment of either JCV or BKV infections. Despite anecdotal reports of response to various treatments in the literature, all controlled studies have failed to show any efficacy for the drugs tested against PML [23]. This includes cidofovir (CDV), cytosine arabinoside (Ara-C), and mefloquine. However, as DNA viruses, many of the available DNA polymerase inhibitors exhibit a level of in vitro activity against PyVs and have been used in the clinical setting [147]. To date, with the exception of TSV, there is little information of the benefit of antiviral therapy for the majority of the novel HPyVs, and the available evidence base is outlined in Table 2.
Table 2.
Antiviral therapies for human polyomavirus infection.
| Antiviral agent | Mechanism of action | HPyV |
In vitro
activity |
Clinical syndrome | Study design | Patient group | Clinical benefit | References |
|---|---|---|---|---|---|---|---|---|
| — | — | — | — | [274] | ||||
| Cytosine arabinoside (Ara-C) |
Nucleoside analogue (DNA polymerase inhibitor) |
JCV | Yes | PML | Case series/reports | HIV, non-HIV, dermatomyositis | Yes | [282–285] |
| PML | Case series/reports | HIV, non-HIV, dermatomyositis | No | [282, 286–289] | ||||
| PML | RCT | HIV | No | [290] | ||||
|
| ||||||||
| — | — | — | — | [274] | ||||
| PML | Pilot study | HIV | No | [281] | ||||
| JCV | No | PML | Multicohort analysis | HIV | No | [279] | ||
| PML | Multicentre Retrospective analysis | HIV | No | [278] | ||||
| Cidofovir (CDV) |
Nucleotide phosphonate analogue (DNA polymerase inhibitor) | PML | Case series | HIV | No | [280] | ||
| PML | Case reports | HIV, the Heerfordt syndrome, SLE | Yes | [275–277] | ||||
| — | — | — | — | [291] | ||||
| BKV | Yes | HC | Case series | HSCT | Yes | [292] | ||
| BKVN | Nonrandomised Controlled study (low-dose CDV) | Renal T'plant | Yes | [293] | ||||
| TSV | N/A | TS | Case report | Heart T'plant, CLL | Yes | [6, 294] | ||
|
| ||||||||
| JCV | Yes | — | — | — | — | [295, 296] | ||
| PML | Case report | Idiopathic CD4+ lymphocytopaenia | Yes | [297] | ||||
| CMX001 | Lipid conjugate of Cidofovir | |||||||
| BKV | Yes | — | — | — | — | [291] | ||
| BK Viruria | RCT | Renal T'plant | Pending | ClinicalTrials.gov-NCT00793598 | ||||
|
| ||||||||
| — | — | — | — | [298, 299] | ||||
| PML | Case report (mirtazapine) | Dermatomyositis | Yes | [285] | ||||
| Serotonin receptor 2A (5HT2AR) antagonists | Inhibits JCV receptor binding and cell entry | JCV | Yes | PML | Case report (mirtazapine) |
HIV | Yes | [300] |
| PML | Case report (chlorpromazine) |
CLL/HSCT | No | [301] | ||||
|
| ||||||||
| Mefloquine | Unknown: may directly inhibit large T antigen | JCV | Yes | — | — | — | — | [302] |
| PML | Case report | Sarcoidosis, AML/UBCT | Yes | [303, 304] | ||||
| PML | RCT | HIV | No | [23] | ||||
|
| ||||||||
| Leflunomide | Inhibits pyrimidine synthesis; inhibits tyrosine kinase | BKV | Yes | — | — | — | — | [305, 306] |
| BKVN | Case series | Renal T'plant | Yes | [307] | ||||
| BKVN | Case series | Renal T'plant | No | [308] | ||||
| BKVN | Case series | Renal T'plant | No | [309] | ||||
|
| ||||||||
| FK778 | Derived from the active metabolite of Leflunomide | BKVN | RCT | Renal T'plant | No | [310] | ||
|
| ||||||||
| Fluoroquinolones | Inhibit large T antigen helicase activity | BKV | Yes | — | — | — | — | [311, 312] |
| BK viraemia | Case series | Renal T'plant | Yes | [313] | ||||
| Renal T'plant | Yes (3 mo.) |
[314] | ||||||
| No (12 mo.) |
[314] | |||||||
| HC | Case series | HSCT | Yes | [315] | ||||
|
| ||||||||
| Mammalian target of rapamycin (mTOR) inhibitors |
Reduce translation and cell cycle progression |
BKV | Yes (sirolimus) | — | — | — | — | [316] |
| BKVN | Case series | Renal T'plant | Yes | [317] | ||||
| BKVN | Case report | Renal T'plant | No | [199] | ||||
| BK viraemia | Prospective Nonrandomised controlled study | Renal T'plant (Paediatric) | No | [318] | ||||
This table lists the available therapeutic options for human polyomavirus infections. KIPyV, WUPyV, HPyV6, 7, 9, and 10 have not been included as there are presently no definitive disease associations for these viruses. Abbreviations are defined in the accompanying text.
Cidofovir (CDV) is an acyclic nucleotide phosphonate analogue of deoxycytosine monophosphate licensed for the treatment of CMV retinitis, which has shown in vitro activity for non-human PyVs and BKV in cell culture, although a study employing JCV and a human neuroglial cell line showed no significant effect on replication [292]. Subsequent case reports in the literature suggested a possible clinical benefit from CDV in the treatment of PML [293, 295, 319]. However, larger case series and an analysis of six international cohorts of HIV-infected individuals affected by PML showed no significant impact of CDV on overall survival [291, 296, 297, 320]. It should be highlighted that all clinical studies to date have been retrospective or observational [147]. As such, to reach a definitive conclusion on the efficacy of CDV for PML, a randomised control trial (RCT) is needed. In contrast, CDV has improved clinical outcomes and decreased viruria and viraemia in BMT patients with BKV-associated HC [294]. In addition, low-dose CDV has been associated with enhanced graft survival in renal transplant patients with BKV interstitial nephritis [282]. However, the drug has limited treatment potential in renal transplant patients due to its toxicity and its limited oral bioavailability [283]. In this respect, of interest is the recent development of CMX001, an orally administered, bioavailable hexadecyloxypropyl lipid conjugate of CDV with reduced nephrotoxicity. CMX001 was found to reduce JCV replication by as much as 60% in human cell lines [284, 285]. It has also been successfully used in a patient with PML and idiopathic CD4+ lymphocytopenia [286]. While both in vitro studies reported decreasing cell viability with increasing concentrations of CMX001, suggesting toxicity remains an issue for this drug, Chimerix has recently reported that the drug is well tolerated in healthy volunteers at doses up to 2 mg/kg [287]. CMX001 is also able to inhibit BKV replication in human renal proximal tubule epithelial cells more rapidly and with a longer-lasting effect than CDV [288]. A double-blind randomised placebo-controlled trial of CMX001 in posttransplant subjects with BKV viruria has recently been completed and results are awaited (ClinicalTrials.gov-NCT00793598). Finally, topical CDV gel has been used to successfully treat TSV infection in a 15-year-old male heart transplant recipient and a 57-year-old woman with CLL [6, 289].
Ara-C is a nucleoside analogue that was effective in decreasing JCV replication in cultured human neuroglial cells [292]. In the clinical setting, reports in the literature have described positive [290, 298, 299, 321] and negative [300, 301, 322, 323] results. The only randomised clinical trial to date (ACTG 243) demonstrated that Ara-C administered either intravenously or intrathecally did not prolong survival for PML patients [302].
JCV enters host cells through binding of the virus to the primary receptor α2,6-linked sialic acid moieties and the secondary receptor serotonin receptor 2A (5HT2AR) [303]. Consequently, blocking access to this receptor—either through the use of antibodies or serotonin receptor antagonists—has been studied as a potential therapeutic approach for PML. To date, in vitro studies have demonstrated that ketanserin and ritanserin are effective at limiting JCV infection in human brain-derived cell culture [304]. In addition, newer antipsychotics such as risperidone and olanzapine have been reported to be up to ten times more potent in vitro at inhibiting JCV infection [305]. Furthermore, treatment of PML with mirtazapine alone and in combination has been associated with favourable outcomes [299, 306]. However, subsequent in vitro studies have shown that 5HT2AR is not essential for JCV infection of certain cells in the human brain [307, 308], and the only clinical report of a non-HIV PML case treated with chlorpromazine and CDV did not show any clinical benefit [309]. Consequently, the role of serotonin receptor antagonists in the treatment of PML remains to be determined.
Mefloquine is an antimalarial drug that has been shown to inhibit JCV replication in vitro. The viral target of the drug is unknown but may directly inhibit the PyV T antigen [310]. Initial case reports showed that mefloquine treatment of PML was successful in lowering the viral burden in the brain and improved clinical symptoms [311, 312]. However, a multicentre clinical trial of mefloquine in PML patients failed to demonstrate a reproducible reduction in JCV DNA in PML patients or reduced clinical progression of PML [23].
Leflunomide is a calcineurin inhibitor (immunosuppressant) licensed for the treatment of rheumatoid arthritis known to block dihydroorotate dehydrogenase, tyrosine kinase, and pyrimidine synthesis. In addition, it has been reported to have modest in vitro activity inhibiting BK viral replication [313, 314]. In clinical studies, leflunomide alone, in combination with CDV, decreased urine and plasma BK viral loads in renal transplant recipients, although drug monitoring is required to ensure adequate therapeutic levels [315]. However, not all studies have demonstrated a benefit [316, 317], and some argue that the addition of drug therapy in treatment of BKVN provides no benefit compared with the standard of care, that is, the reduction of immunosuppression alone [318].
The fluoroquinolones (FQ) are a class of antibiotics that have been used—in combination with immunosuppression reduction—in the treatment of BKVN and have been shown to have in vitro activity against BKV [324]. These antibiotics inhibit bacterial DNA replication by inhibiting type II topoisomerases and are thought to have activity against the viral helicase TAg [325]. In clinical studies, ciprofloxacin prophylaxis has been shown to reduce the rate of BK viraemia in renal transplant recipients [326, 327] and to reduce the incidence of severe HC in allogeneic HSCT [328]. However, the long-term effectiveness and optimal duration of FQ prophylaxis against BKV infection remain unknown [327].
Mammalian target of rapamycin (mTOR) inhibitors such as sirolimus reduce translation and cell cycle progression and reduce BKV replication in vitro [329]. An initial retrospective study, albeit with small numbers, suggested that renal allograft recipients on mTOR inhibitors cleared BK viruria and viraemia more quickly than those on other immunosuppressive agents [330]. However, other reports have not supported these findings [207, 331]. It may be possible that mTOR inhibitors are preferable to calcineurin inhibitors from the perspective of reducing the risk of BKV-associated disease, but at this point, there are no RCT data to suggest a benefit from mTOR inhibitors as antiviral treatments in this setting [13].
While there is no single reason for the lack of effective antiviral therapy for PyV infection, there is probably a number of challenges that explain the existing dearth. First, should the drug target the virus or aim to stimulate the host immune system? Second, if the virus is the target, which stage of the life cycle should be chosen: the primary infection, latency-site cell entry, or virus replication? Prevention of primary infection would only be achievable through large-scale population immunization, an approach that is not likely to be cost effective; while the inhibition of latency-site cell entry may be feasible (some virus receptors have been identified as mentioned above), the impact of such agents on preexisting latent virus is likely to be negligible. Thus, successful therapies will have to retard intracellular virus replication, requiring sufficient doses of active drug to be able to cross the BBB without causing significant toxicity, a significant problem for existing agents. Third, would a single specific antiviral agent be effective against both “prototype” and “archetype” JC virus, for example, or could such an agent drive the emergence of resistance? Fourth, given these unknowns, not to mention the relatively small number of suitable patients requiring antiviral treatment (and thus available for RCTs), is it reasonable to expect a single pharmaceutical company to take on the economic risk of developing such an agent, or will government input be required? Furthermore, if government input is required, would that constitute an appropriate allocation of limited resources in the current straitened times? If HIV-infected patients still constitute the population at greatest risk of PML for example, and the majority can be successfully treated with combination ART; perhaps, the more prudent approach to the management of PyV infected individuals is a risk reduction strategy, as evidenced by the prescreening of MS patients for JC antibodies before commencing natalizumab therapy. In summary, there are no currently available RCT data to recommend the use of specific antiviral therapy for the treatment of PyV infection, although the commencement of antiretroviral therapy is recommended in HIV-associated PML. For non-HIV PML and BKV disease in transplant recipients, withdrawal from, or reduction in, immunosuppression—insofar as is possible—is the present mainstay of treatment.
Acknowledgments
The authors would like to thank Dr. Jonathan Dean, Dr. Suzie Coughlan, and Ms. Ines Freitas for helpful comments and invaluable assistance during the preparation of this paper.
References
- 1.Feltkamp MC, Kazem S, van der Meijden E, et al. From Stockholm to Malawi: recent developments in studying human polyomaviruses. Journal of General Virology. 2013;94, part 3:482–496. doi: 10.1099/vir.0.048462-0. [DOI] [PubMed] [Google Scholar]
- 2.Allander T, Andreasson K, Gupta S, et al. Identification of a third human polyomavirus. Journal of Virology. 2007;81(8):4130–4136. doi: 10.1128/JVI.00028-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gaynor AM, Nissen MD, Whiley DM, et al. Identification of a novel polyomavirus from patients with acute respiratory tract infections. PLoS Pathogens. 2007;3(5, article e64) doi: 10.1371/journal.ppat.0030064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319(5866):1096–1100. doi: 10.1126/science.1152586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schowalter RM, Pastrana DV, Pumphrey KA, Moyer AL, Buck CB. Merkel cell polyomavirus and two previously unknown polyomaviruses are chronically shed from human skin. Cell Host and Microbe. 2010;7(6):509–515. doi: 10.1016/j.chom.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van der Meijden E, Janssens RW, Lauber C, Bouwes Bavinck JN, Gorbalenya AE, Feltkamp MC. Discovery of a new human polyomavirus associated with trichodysplasia spinulosa in an immunocompromized patient. PLoS Pathogens. 2010;6(7) doi: 10.1371/journal.ppat.1001024.e1001024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scuda N, Hofmann J, Calvignac-Spencer S, et al. A novel human polyomavirus closely related to the African green monkey-derived lymphotropic polyomavirus. Journal of Virology. 2011;85(9):4586–4590. doi: 10.1128/JVI.02602-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buck CB, Phan GQ, Raiji MT, et al. Complete genome sequence of a tenth human polyomavirus. Journal of Virology. 2012;86(19):p. 10887. doi: 10.1128/JVI.01690-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Siebrasse EA, Reyes A, Lim ES, et al. Identification of MW polyomavirus, a novel polyomavirus in human stool. Journal of Virology. 2012;86(19):10321–10326. doi: 10.1128/JVI.01210-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Padgett BL, Walker DL, ZuRhein GM, Eckroade RJ, Dessel BH. Cultivation of papova-like virus from human brain with progressive multifocal leucoencephalopathy. The Lancet. 1971;1(7712):1257–1260. doi: 10.1016/s0140-6736(71)91777-6. [DOI] [PubMed] [Google Scholar]
- 11.Gardner SD, Field AM, Coleman DV, Hulme B. New human papovavirus (B.K.) isolated from urine after renal transplantation. The Lancet. 1971;1(7712):1253–1257. doi: 10.1016/s0140-6736(71)91776-4. [DOI] [PubMed] [Google Scholar]
- 12.Kean JM, Rao S, Wang M, Garcea RL. Seroepidemiology of human polyomaviruses. PLoS Pathogens. 2009;5(3) doi: 10.1371/journal.ppat.1000363.e1000363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bennett SM, Broekema NM, Imperiale MJ. BK polyomavirus: emerging pathogen. Microbes and Infection. 2012;14(9):672–683. doi: 10.1016/j.micinf.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Major EO. Progressive multifocal leukoencephalopathy in patients on immunomodulatory therapies. Annual Review of Medicine. 2010;61:35–47. doi: 10.1146/annurev.med.080708.082655. [DOI] [PubMed] [Google Scholar]
- 15.Carson KR, Focosi D, Major EO, et al. Monoclonal antibody-associated progressive multifocal leucoencephalopathy in patients treated with rituximab, natalizumab, and efalizumab: a review from the Research on Adverse Drug Events and Reports (RADAR) Project. The Lancet Oncology. 2009;10(8):816–824. doi: 10.1016/S1470-2045(09)70161-5. [DOI] [PubMed] [Google Scholar]
- 16.Kleinschmidt-DeMasters BK, Tyler KL. Progressive multifocal leukoencephalopathy complicating treatment with natalizumab and interferon beta-1a for multiple sclerosis. The New England Journal of Medicine. 2005;353(4):369–374. doi: 10.1056/NEJMoa051782. [DOI] [PubMed] [Google Scholar]
- 17.Van Assche G, Van Ranst M, Sciot R, et al. Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn’s disease. The New England Journal of Medicine. 2005;353(4):362–368. doi: 10.1056/NEJMoa051586. [DOI] [PubMed] [Google Scholar]
- 18.Carson KR, Evens AM, Richey EA, et al. Progressive multifocal leukoencephalopathy after rituximab therapy in HIV-negative patients: a report of 57 cases from the Research on Adverse Drug Events and Reports project. Blood. 2009;113(20):4834–4840. doi: 10.1182/blood-2008-10-186999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sobell JM, Weinberg JM. Patient fatalities potentially associated with efalizumab use. Journal of Drugs in Dermatology. 2009;8(3):p. 215. [PubMed] [Google Scholar]
- 20.Kumar D, Bouldin TW, Berger RG. A case of progressive multifocal leukoencephalopathy in a patient treated with infliximab. Arthritis and Rheumatism. 2010;62(11):3191–3195. doi: 10.1002/art.27687. [DOI] [PubMed] [Google Scholar]
- 21.Delbue S, Comar M, Ferrante P. Review on the relationship between human polyomaviruses-associated tumors and host immune system. Clinical and Developmental Immunology. 2012;2012:10 pages. doi: 10.1155/2012/542092.542092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stewart SE, Eddy BE, Borgese N. Neoplasms in mice inoculated with a tumor agent carried in tissue. Journal of the National Cancer Institute. 1958;20(6):1223–1243. doi: 10.1093/jnci/20.6.1223. [DOI] [PubMed] [Google Scholar]
- 23.Ferenczy MW, Marshall LJ, Nelson CD, et al. Molecular biology, epidemiology, and pathogenesis of progressive multifocal leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clinical Microbiology Reviews. 2012;25(3):471–506. doi: 10.1128/CMR.05031-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abend JR, Joseph AE, Das D, Campbell-Cecen DB, Imperiale MJ. A truncated T antigen expressed from an alternatively spliced BK virus early mRNA. Journal of General Virology. 2009;90(5):1238–1245. doi: 10.1099/vir.0.009159-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Darbinyan A, Siddiqui KM, Slonina D, et al. Role of JC virus agnoprotein in DNA repair. Journal of Virology. 2004;78(16):8593–8600. doi: 10.1128/JVI.78.16.8593-8600.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khalili K, White MK, Sawa H, Nagashima K, Safak M. The agnoprotein of polyomaviruses: a multifunctional auxiliary protein. Journal of Cellular Physiology. 2005;204(1):1–7. doi: 10.1002/jcp.20266. [DOI] [PubMed] [Google Scholar]
- 27.Safak M, Khalili K. Physical and functional interaction between viral and cellular proteins modulate JCV gene transcription. Journal of NeuroVirology. 2001;7(4):288–292. doi: 10.1080/13550280152537111. [DOI] [PubMed] [Google Scholar]
- 28.Suzuki T, Orba Y, Okada Y, et al. The human polyoma JC virus agnoprotein acts as a viroporin. PLoS Pathogens. 2010;6(3) doi: 10.1371/journal.ppat.1000801.e1000801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Åström KE, Mancall EL, Richardson EP., Jr. Progressive multifocal leuko-encephalopathy: a HITHERTO unrecognized COMPLICATION o CHRONIC lymphatic leukÆmia AND hodgkin’s disease. Brain. 1958;81(1):93–111. doi: 10.1093/brain/81.1.93. [DOI] [PubMed] [Google Scholar]
- 30.Christensen E, Fog M. A case of Schilder’s disease in an adult with remarks to the etiology and pathogenesis. Acta Psychiatrica et Neurologica Scandinavica. 1955;30(1-2):141–154. doi: 10.1111/j.1600-0447.1955.tb06054.x. [DOI] [PubMed] [Google Scholar]
- 31.Richardson EP., Jr. Progressive multifocal leukoencephalopathy. The New England Journal of Medicine. 1961;265:815–823. doi: 10.1056/NEJM196110262651701. [DOI] [PubMed] [Google Scholar]
- 32.Cavanagh JB, Greenbaum D, Marshall AHE, Rubinstein LJ. Cerebral demyelination associated with disorders of the reticuloendothelial system. The Lancet. 1959;274(7102):524–529. doi: 10.1016/s0140-6736(59)91774-x. [DOI] [PubMed] [Google Scholar]
- 33.Osborn JE, Robertson SM, Padgett BL. Comparison of JC and BK human papovaviruses with Simian virus 40: restriction endonuclease digestion and gel electrophoresis of resultant fragments. Journal of Virology. 1974;13(3):614–622. doi: 10.1128/jvi.13.3.614-622.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brown P, Tsai T, Gajdusek DC. Seroepidemiology of human papovaviruses. Discovery of virgin populations and some unusual patterns of antibody prevalence among remote peoples of the world. American Journal of Epidemiology. 1975;102(4):331–340. doi: 10.1093/oxfordjournals.aje.a112169. [DOI] [PubMed] [Google Scholar]
- 35.Rziha HJ, Bornkamm GW, zur Hausen H. BK virus. I. Seroepidemiologic studies and serologic response to viral infection. Medical Microbiology and Immunology. 1978;165(2):73–81. doi: 10.1007/BF02122742. [DOI] [PubMed] [Google Scholar]
- 36.Walker DL, Padgett BL. The epidemiology of human polyomaviruses. Progress in Clinical and Biological Research. 1983;105:99–106. [PubMed] [Google Scholar]
- 37.Knowles WA, Pipkin P, Andrews N, et al. Population-based study of antibody to the human polyomaviruses BKV and JCV and the simian polyomavirus SV40. Journal of Medical Virology. 2003;71(1):115–123. doi: 10.1002/jmv.10450. [DOI] [PubMed] [Google Scholar]
- 38.Lundstig A, Dillner J. Serological diagnosis of human polyomavirus infection. Advances in Experimental Medicine and Biology. 2006;577:96–101. doi: 10.1007/0-387-32957-9_7. [DOI] [PubMed] [Google Scholar]
- 39.Stolt A, Sasnauskas K, Koskela P, Lehtinen M, Dillner J. Seroepidemiology of the human polyomaviruses. Journal of General Virology. 2003;84(6):1499–1504. doi: 10.1099/vir.0.18842-0. [DOI] [PubMed] [Google Scholar]
- 40.Major EO, Neel JV. The JC and BK human polyoma viruses appear to be recent introductions to some South American Indian tribes: there is no serological evidence of cross-reactivity with the simian polyoma virus SV40. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(26):15525–15530. doi: 10.1073/pnas.95.26.15525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ahmed W, Wan C, Goonetilleke A, Gardner T. Evaluating sewage-associated JCV and BKV polyomaviruses for sourcing human fecal pollution in a coastal river in Southeast Queensland, Australia. Journal of Environmental Quality. 2010;39(5):1743–1750. doi: 10.2134/jeq2010.0062. [DOI] [PubMed] [Google Scholar]
- 42.Hamza IA, Jurzik L, Stang A, Sure K, Überla K, Wilhelm M. Detection of human viruses in rivers of a densly-populated area in Germany using a virus adsorption elution method optimized for PCR analyses. Water Research. 2009;43(10):2657–2668. doi: 10.1016/j.watres.2009.03.020. [DOI] [PubMed] [Google Scholar]
- 43.McQuaig SM, Scott TM, Lukasik JO, Paul JH, Harwood VJ. Quantification of human polyomaviruses JC virus and BK Virus by TaqMan quantitative PCR and comparison to other water quality indicators in water and fecal samples. Applied and Environmental Microbiology. 2009;75(11):3379–3388. doi: 10.1128/AEM.02302-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vanchiere JA, Abudayyeh S, Copeland CM, Lu LB, Graham DY, Butel JS. Polyomavirus shedding in the stool of healthy adults. Journal of Clinical Microbiology. 2009;47(8):2388–2391. doi: 10.1128/JCM.02472-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vanchiere JA, Nicome RK, Greer JM, Demmler GJ, Butel JS. Frequent detection of polyomaviruses in stool samples from hospitalized children. Journal of Infectious Diseases. 2005;192(4):658–664. doi: 10.1086/432076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cubitt CL, Cui X, Agostini HT, et al. Predicted amino acid sequences for 100 JCV strains. Journal of NeuroVirology. 2001;7(4):339–344. doi: 10.1080/13550280152537201. [DOI] [PubMed] [Google Scholar]
- 47.Agostini HT, Yanagihara R, Davis V, Ryschkewitsch CF, Stoner GL. Asian genotypes of JC virus in native Americans and in a Pacific Island population: markers of viral evolution and human migration. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(26):14542–14546. doi: 10.1073/pnas.94.26.14542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ikegaya H, Zheng HY, Saukko PJ, et al. Genetic diversity of JC virus in the Saami and the Finns: implications for their population history. American Journal of Physical Anthropology. 2005;128(1):185–193. doi: 10.1002/ajpa.20189. [DOI] [PubMed] [Google Scholar]
- 49.Ryschkewitsch CF, Friedlaender JS, Mgone CS, et al. Human polyomavirus JC variants in Papua New Guinea and Guam reflect ancient population settlement and viral evolution. Microbes and Infection. 2000;2(9):987–996. doi: 10.1016/s1286-4579(00)01252-1. [DOI] [PubMed] [Google Scholar]
- 50.Stoner GL, Jobes DV, Fernandez Cobo M, Agostini HT, Chima SC, Ryschkewitsch CF. JC virus as a marker of human migration to the Americas. Microbes and Infection. 2000;2(15):1905–1911. doi: 10.1016/s1286-4579(00)01339-3. [DOI] [PubMed] [Google Scholar]
- 51.Pavesi A. African origin of polyomavirus JC and implications for prehistoric human migrations. Journal of Molecular Evolution. 2003;56(5):564–572. doi: 10.1007/s00239-002-2425-y. [DOI] [PubMed] [Google Scholar]
- 52.Cui X, Wang JC, Deckhut A, et al. Chinese strains (type 7) of JC virus are Afro-Asiatic in origin but are phylogenetically distinct from the Mongolian and Indian strains (type 2D) and the Korean and Japanese strains (type 2A) Journal of Molecular Evolution. 2004;58(5):568–583. doi: 10.1007/s00239-003-2579-2. [DOI] [PubMed] [Google Scholar]
- 53.Jobes DV, Friedlaender JS, Mgone CS, et al. New JC virus (JCV) genotypes from Papua New Guinea and Micronesia (type 8 and type 2E) and evolutionary analysis of 32 complete JCV genomes. Archives of Virology. 2001;146(11):2097–2113. doi: 10.1007/s007050170023. [DOI] [PubMed] [Google Scholar]
- 54.Yanagihara R, Nerurkar VR, Scheirich I, et al. JC virus genotypes in the western Pacific suggest Asian mainland relationships and virus association with early population movements. Human Biology. 2002;74(3):473–488. doi: 10.1353/hub.2002.0037. [DOI] [PubMed] [Google Scholar]
- 55.Agostini HT, Ryschkewitsch CF, Baumhefner RW, et al. Influence of JC virus coding region genotype on risk of multiple sclerosis and progressive multifocal leukoencephalopathy. Journal of NeuroVirology. 2000;6(supplement 2):S101–S108. [PubMed] [Google Scholar]
- 56.Dubois V, Moret H, Lafon ME, et al. JC virus genotypes in France: molecular epidemiology and potential significance for progressive multifocal leukoencephalopathy. Journal of Infectious Diseases. 2001;183(2):213–217. doi: 10.1086/317927. [DOI] [PubMed] [Google Scholar]
- 57.Monaco MCG, Atwood WJ, Gravell M, Tornatore CS, Major EO. JC virus infection of hematopoietic progenitor cells, primary B lymphocytes, and tonsillar stromal cells: Implications for viral latency. Journal of Virology. 1996;70(10):7004–7012. doi: 10.1128/jvi.70.10.7004-7012.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Houff SA, Major EO, Katz DA, et al. Involvement of JC virus-infected mononuclear cells from the bone marrow and spleen in the pathogenesis of progressive multifocal leukoencephalopathy. The New England Journal of Medicine. 1988;318(5):301–305. doi: 10.1056/NEJM198802043180507. [DOI] [PubMed] [Google Scholar]
- 59.Haider S, Nafziger D, Gutierrez JA, Brar I, Mateo N, Fogle J. Progressive multifocal leukoencephalopathy and idiopathic CD4+lymphocytopenia: a case report and review of reported cases. Clinical Infectious Diseases. 2000;31(4):E20–E22. doi: 10.1086/318120. [DOI] [PubMed] [Google Scholar]
- 60.Puri V, Chaudhry N, Gulati P, Patel N, Tatke M, Sinha S. Progressive multifocal leukoencephalopathy in a patient with idiopathic CD4+T lymphocytopenia. Neurology India. 2010;58(1):118–121. doi: 10.4103/0028-3886.60423. [DOI] [PubMed] [Google Scholar]
- 61.Du Pasquier RA, Kuroda MJ, Zheng Y, Jean-Jacques J, Letvin NL, Koralnik IJ. A prospective study demonstrates an association between JC virus-specific cytotoxic T lymphocytes and the early control of progressive multifocal leukoencephalopathy. Brain. 2004;127(9):1970–1978. doi: 10.1093/brain/awh215. [DOI] [PubMed] [Google Scholar]
- 62.Marzocchetti A, Tompkins T, Clifford DB, et al. Determinants of survival in progressive multifocal leukoencephalopathy. Neurology. 2009;73(19):1551–1558. doi: 10.1212/WNL.0b013e3181c0d4a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.D’Arminio Monforte A, Cinque P, Mocroft A, et al. Changing incidence of central nervous system diseases in the EuroSIDA cohort. Annals of Neurology. 2004;55(3):320–328. doi: 10.1002/ana.10827. [DOI] [PubMed] [Google Scholar]
- 64.Khanna N, Elzi L, Mueller NJ, et al. Incidence and outcome of progressive multifocal leukoencephalopathy over 20 years of the swiss HIV cohort study. Clinical Infectious Diseases. 2009;48(10):1459–1466. doi: 10.1086/598335. [DOI] [PubMed] [Google Scholar]
- 65.Amend KL, Turnbull B, Foskett N, Napalkov P, Kurth T, Seeger J. Incidence of progressive multifocal leukoencephalopathy in patients without HIV. Neurology. 2010;75(15):1326–1332. doi: 10.1212/WNL.0b013e3181f73600. [DOI] [PubMed] [Google Scholar]
- 66.Palazzo E, Yahia SA. Progressive multifocal leukoencephalopathy in autoimmune diseases. Joint Bone Spine. 2012;79(4):351–355. doi: 10.1016/j.jbspin.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 67.Arkema EV, van Vollenhoven RF, Askling J. Incidence of progressive multifocal leukoencephalopathy in patients with rheumatoid arthritis: a national population-based study. Annals of the Rheumatic Diseases. 2012;71(11):1865–1867. doi: 10.1136/annrheumdis-2012-201638. [DOI] [PubMed] [Google Scholar]
- 68.Bloomgren G, Richman S, Hotermans C, et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. The New England Journal of Medicine. 2012;366(20):1870–1880. doi: 10.1056/NEJMoa1107829. [DOI] [PubMed] [Google Scholar]
- 69.Mateen FJ, Muralidharan R, Carone M, et al. Progressive multifocal leukoencephalopathy in transplant recipients. Annals of Neurology. 2011;70(2):305–322. doi: 10.1002/ana.22408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Atwood WJ, Amemiya K, Traub R, Harms J, Major EO. Interaction of the human polyomavirus, JCV, with human B-lymphocytes. Virology. 1992;190(2):716–723. doi: 10.1016/0042-6822(92)90909-9. [DOI] [PubMed] [Google Scholar]
- 71.Major EO, Amemiya K, Elder G, Houff SA. Glial cells of the human developing brain and B cells of the immune system share a common DNA binding factor for recognition of the regulatory sequences of the human polyomavirus, JCV. Journal of Neuroscience Research. 1990;27(4):461–471. doi: 10.1002/jnr.490270405. [DOI] [PubMed] [Google Scholar]
- 72.Rieckmann P, Michel U, Kehrl JH. Regulation of JC virus expression in B lymphocytes. Journal of Virology. 1994;68(1):217–222. doi: 10.1128/jvi.68.1.217-222.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tornatore C, Berger JR, Houff SA, et al. Detection of JC virus DNA in peripheral lymphocytes from patients with and without progressive multifocal leukoencephalopathy. Annals of Neurology. 1992;31(4):454–462. doi: 10.1002/ana.410310426. [DOI] [PubMed] [Google Scholar]
- 74.Major EO. History and current concepts in the pathogenesis of PML. Cleveland Clinic Journal of Medicine. 2011;78, supplement 2:S3–S7. doi: 10.3949/ccjm.78.s2.02. [DOI] [PubMed] [Google Scholar]
- 75.Bayliss J, Karasoulos T, Bowden S, et al. Immunosuppression increases latent infection of brain by JC polyomavirus. Pathology. 2011;43(4):362–367. doi: 10.1097/PAT.0b013e3283463558. [DOI] [PubMed] [Google Scholar]
- 76.Delbue S, Branchetti E, Boldorini R, et al. Presence and expression of JCV early gene large T antigen in the brains of immunocompromised and immunocompetent individuals. Journal of Medical Virology. 2008;80(12):2147–2152. doi: 10.1002/jmv.21313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Perez-Liz G, Del Valle L, Gentilella A, Croul S, Khalili K. Detection of JC virus DNA fragments but not proteins in normal brain tissue. Annals of Neurology. 2008;64(4):379–387. doi: 10.1002/ana.21443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tan CS, Ellis LC, Wüthrich C, et al. JC virus latency in the brain and extraneural organs of patients with and without progressive multifocal leukoencephalopathy. Journal of Virology. 2010;84(18):9200–9209. doi: 10.1128/JVI.00609-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Monaco MCG, Jensen PN, Hou J, Durham LC, Major EO. Detection of JC virus DNA in human tonsil tissue: evidence for site of initial viral infection. Journal of Virology. 1998;72(12):9918–9923. doi: 10.1128/jvi.72.12.9918-9923.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fiers W, Contreras R, Haegeman G, et al. Complete nucleotide sequence of SV40 DNA. Nature. 1978;273(5658):113–120. doi: 10.1038/273113a0. [DOI] [PubMed] [Google Scholar]
- 81.Frisque RJ. Nucleotide sequence of the region encompassing the JC virus origin of DNA replication. Journal of Virology. 1983;46(1):170–176. doi: 10.1128/jvi.46.1.170-176.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Frisque RJ, Bream GL, Cannella MT. Human polyomavirus JC virus genome. Journal of Virology. 1984;51(2):458–469. doi: 10.1128/jvi.51.2.458-469.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Law MF, Martin JD, Takemoto KK, Howley PM. The colinear alignment of the genomes of papovaviruses JC, BK, and SV40. Virology. 1979;96(2):576–587. doi: 10.1016/0042-6822(79)90113-2. [DOI] [PubMed] [Google Scholar]
- 84.Reddy VB, Thimmappaya B, Dhar R. The genome of Simian virus 40. Science. 1978;200(4341):494–502. doi: 10.1126/science.205947. [DOI] [PubMed] [Google Scholar]
- 85.Seif I, Khoury G, Dhar R. The genome of human papovavirus BKV. Cell. 1979;18(4):963–977. doi: 10.1016/0092-8674(79)90209-5. [DOI] [PubMed] [Google Scholar]
- 86.Yang RCA, Wu R. Comparative study of papovavirus DNA: BKV(MM), BKV(WT) and SV40. Nucleic Acids Research. 1979;7(3):651–668. doi: 10.1093/nar/7.3.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ghosh PK, Lebowitz P, Frisque RJ, Gluzman Y. Identification of a promoter component involved in positioning the 5’ termini of simian virus 40 early mRNAs. Proceedings of the National Academy of Sciences of the United States of America. 1981;78(1):100–104. doi: 10.1073/pnas.78.1.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gluzman Y, Sambrook JF, Frisque RJ. Expression of early genes of origin-defective mutants of simian virus 40. Proceedings of the National Academy of Sciences of the United States of America. 1980;77(7):3898–3902. doi: 10.1073/pnas.77.7.3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Frisque RJ. Regulatory sequences and virus-cell interactions of JC virus. Progress in Clinical and Biological Research. 1983;105:41–59. [PubMed] [Google Scholar]
- 90.Lynch KJ, Frisque RJ. Identification of critical elements within the JC virus DNA replication origin. Journal of Virology. 1990;64(12):5812–5822. doi: 10.1128/jvi.64.12.5812-5822.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Martin JD, King DM, Slauch JM, et al. Differences in regulatory sequences of naturally occurring JC virus variants. Journal of Virology. 1985;53(1):306–311. doi: 10.1128/jvi.53.1.306-311.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yogo Y, Kitamura T, Sugimoto C, et al. Isolation of a possible archetypal JC virus DNA sequence from nonimmunocompromised individuals. Journal of Virology. 1990;64(6):3139–3143. doi: 10.1128/jvi.64.6.3139-3143.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jensen PN, Major EO. Viral variant nucleotide sequences help expose leukocytic positioning in the JC virus pathway to the CNS. Journal of Leukocyte Biology. 1999;65(4):428–438. doi: 10.1002/jlb.65.4.428. [DOI] [PubMed] [Google Scholar]
- 94.Marzocchetti A, Wuthrich C, Tan CS, et al. Rearrangement of the JC virus regulatory region sequence in the bone marrow of a patient with rheumatoid arthritis and progressive multifocal leukoencephalopathy. Journal of NeuroVirology. 2008;14(5):455–458. doi: 10.1080/13550280802356837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Vaz B, Cinque P, Pickhardt M, Weber T. Analysis of the transcriptional control region in progressive multifocal leukoencephalopathy. Journal of NeuroVirology. 2000;6(5):398–409. doi: 10.3109/13550280009018304. [DOI] [PubMed] [Google Scholar]
- 96.Flaegstad T, Sundsfjord A, Arthur RR, Pedersen M, Traavik T, Subramani S. Amplification and sequencing of the control regions of BK and JC virus from human urine by polymerase chain reaction. Virology. 1991;180(2):553–560. doi: 10.1016/0042-6822(91)90069-n. [DOI] [PubMed] [Google Scholar]
- 97.Iida T, Kitamura T, Guo J, et al. Origin of JC polyomavirus variants associated with progressive multifocal leukoencephalopathy. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(11):5062–5065. doi: 10.1073/pnas.90.11.5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dorries K, Vogel E, Gunther S, Czub S. Infection of human polyomaviruses JC and BK in peripheral blood leukocytes from immunocompetent individuals. Virology. 1994;198(2):59–70. doi: 10.1006/viro.1994.1008. [DOI] [PubMed] [Google Scholar]
- 99.Schneider EM, Dorries K. High frequency of polyomavirus infection in lymphoid cell preparations after allogeneic bone marrow transplantation. Transplantation Proceedings. 1993;25(1):1271–1273. [PubMed] [Google Scholar]
- 100.Tan CS, Dezube BJ, Bhargava P, et al. Detection of JC virus DNA and proteins in the bone marrow of HIV-positive and HIV-negative patients: implications for viral latency and neurotropic transformation. Journal of Infectious Diseases. 2009;199(6):881–888. doi: 10.1086/597117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Marshall LJ, Dunham L, Major EO. Transcription factor Spi-B binds unique sequences present in the tandem repeat promoter/enhancer of JC virus and supports viral activity. Journal of General Virology. 2010;91(12):3042–3052. doi: 10.1099/vir.0.023184-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Messam CA, Hou J, Gronostajski RM, Major EO. Lineage pathway of human brain progenitor cells identified by JC virus susceptability. Annals of Neurology. 2003;53(5):636–646. doi: 10.1002/ana.10523. [DOI] [PubMed] [Google Scholar]
- 103.Monaco MCG, Sabath BF, Durham LC, Major EO. JC virus multiplication in human hematopoietic progenitor cells requires the NF-1 class D transcription factor. Journal of Virology. 2001;75(20):9687–9695. doi: 10.1128/JVI.75.20.9687-9695.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Engelhardt B, Kappos L. Natalizumab: targeting α4-integrins in multiple sclerosis. Neurodegenerative Diseases. 2008;5(1):16–22. doi: 10.1159/000109933. [DOI] [PubMed] [Google Scholar]
- 105.Jing D, Oelschlaegel U, Ordemann R, et al. CD49d blockade by natalizumab in patients with multiple sclerosis affects steady-state hematopoiesis and mobilizes progenitors with a distinct phenotype and function. Bone Marrow Transplantation. 2010;45(10):1489–1496. doi: 10.1038/bmt.2009.381. [DOI] [PubMed] [Google Scholar]
- 106.Lindberg RLP, Achtnichts L, Hoffmann F, Kuhle J, Kappos L. Natalizumab alters transcriptional expression profiles of blood cell subpopulations of multiple sclerosis patients. Journal of Neuroimmunology. 2008;194(1-2):153–164. doi: 10.1016/j.jneuroim.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 107.Reff ME, Carner K, Chambers KS, et al. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood. 1994;83(2):435–445. [PubMed] [Google Scholar]
- 108.McLaughlin P, Grillo-López AJ, Link BK, et al. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. Journal of Clinical Oncology. 1998;16(8):2825–2833. doi: 10.1200/JCO.1998.16.8.2825. [DOI] [PubMed] [Google Scholar]
- 109.Lebwohl M, Tyring SK, Hamilton TK, et al. A novel targeted T-cell modulator, efalizumab, for plaque psoriasis. The New England Journal of Medicine. 2003;349(21):2004–2013. doi: 10.1056/NEJMoa030002. [DOI] [PubMed] [Google Scholar]
- 110.Guttman-Yassky E, Vugmeyster Y, Lowes MA, et al. Blockade of CD11a by efalizumab in psoriasis patients induces a unique state of T-cell hyporesponsiveness. Journal of Investigative Dermatology. 2008;128(5):1182–1191. doi: 10.1038/jid.2008.4. [DOI] [PubMed] [Google Scholar]
- 111.Knight DM, Trinh H, Le J, et al. Construction and initial characterization of a mouse-human chimeric anti-TNF antibody. Molecular Immunology. 1993;30(16):1443–1453. doi: 10.1016/0161-5890(93)90106-l. [DOI] [PubMed] [Google Scholar]
- 112.Van den Brande JMH, Braat H, Van den Brink GR, et al. Infliximab but not etanercept induces apoptosis in lamina propria T-lymphocytes from patients with Crohn’s disease. Gastroenterology. 2003;124(7):1774–1785. doi: 10.1016/s0016-5085(03)00382-2. [DOI] [PubMed] [Google Scholar]
- 113.Van Den Brande JMH, Peppelenbosch MP, Van Deventer SJH. Treating Crohn’s disease by inducing T lymphocyte apoptosis. Annals of the New York Academy of Sciences. 2002;973:166–180. doi: 10.1111/j.1749-6632.2002.tb04628.x. [DOI] [PubMed] [Google Scholar]
- 114.Domm S, Cinatl J, Mrowietz U. The impact of treatment with tumour necrosis factor-α antagonists on the course of chronic viral infections: a review of the literature. British Journal of Dermatology. 2008;159(6):1217–1228. doi: 10.1111/j.1365-2133.2008.08851.x. [DOI] [PubMed] [Google Scholar]
- 115.Houff SA, Berger JR. The bone marrow, B cells, and JC virus. Journal of NeuroVirology. 2008;14(5):341–343. doi: 10.1080/13550280802348222. [DOI] [PubMed] [Google Scholar]
- 116.Du Pasquier RA, Clark KW, Smith PS, et al. JCV-specific cellular immune response correlates with a favorable clinical outcome in HIV-infected individuals with progressive multifocal leukoencephalopathy. Journal of NeuroVirology. 2001;7(4):318–322. doi: 10.1080/13550280152537175. [DOI] [PubMed] [Google Scholar]
- 117.Koralnik IJ, Du Pasquier RA, Letvin NL. JC virus-specific cytotoxic T lymphocytes in individuals with progressive multifocal leukoencephalopathy. Journal of Virology. 2001;75(7):3483–3487. doi: 10.1128/JVI.75.7.3483-3487.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lima MA, Bernal-Cano F, Clifford DB, Gandhi RT, Koralnik IJ. Clinical outcome of long-term survivors of progressive multifocal leukoencephalopathy. Journal of Neurology, Neurosurgery and Psychiatry. 2010;81(11):1288–1291. doi: 10.1136/jnnp.2009.179002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lima MA, Marzocchetti A, Autissier P, et al. Frequency and phenotype of JC virus-specific CD8+ T lymphocytes in the peripheral blood of patients with progressive multifocal leukoencephalopathy. Journal of Virology. 2007;81(7):3361–3368. doi: 10.1128/JVI.01809-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wüthrich C, Kesari S, Kim WK, et al. Characterization of lymphocytic infiltrates in progressive multifocal leukoencephalopathy: co-localization of CD8+T cells with JCV-infected glial cells. Journal of NeuroVirology. 2006;12(2):116–128. doi: 10.1080/13550280600716604. [DOI] [PubMed] [Google Scholar]
- 121.Matloubian M, Concepcion RJ, Ahmed R. CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. Journal of Virology. 1994;68(12):8056–8063. doi: 10.1128/jvi.68.12.8056-8063.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chowdhury M, Kundu M, Khalili K. GA/GC-rich sequence confers Tat responsiveness to human neurotropic virus promoter, JCV(L), in cells derived from central nervous system. Oncogene. 1993;8(4):887–892. [PubMed] [Google Scholar]
- 123.Chowdhury M, Taylor JP, Chang CF, Rappaport J, Khalili K. Evidence that a sequence similar to TAR is important for induction of the JC virus late promoter by human immunodeficiency virus type 1 Tat. Journal of Virology. 1992;66(12):7355–7361. doi: 10.1128/jvi.66.12.7355-7361.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Daniel DC, Kinoshita Y, Khan MA, et al. Internalization of exogenous human immunodeficiency virus-1 protein, Tat, by KG-1 oligodendroglioma cells followed by stimulation of DNA replication initiated at the JC virus origin. DNA and Cell Biology. 2004;23(12):858–867. doi: 10.1089/dna.2004.23.858. [DOI] [PubMed] [Google Scholar]
- 125.Nukuzuma S, Nakamichi K, Kameoka M, et al. Efficient propagation of progressive multifocal leukoencephalopathy-type JC virus in COS-7-derived cell lines stably expressing Tat protein of human immunodeficiency virus type 1. Microbiology and Immunology. 2010;54(12):758–762. doi: 10.1111/j.1348-0421.2010.00278.x. [DOI] [PubMed] [Google Scholar]
- 126.Remenick J, Radonovich MF, Brady JN. Human immunodeficiency virus Tat transactivation: induction of a tissue-specific enhancer in a nonpermissive cell line. Journal of Virology. 1991;65(10):5641–5646. doi: 10.1128/jvi.65.10.5641-5646.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Stettner MR, Nance JA, Wright CA, et al. SMAD proteins of oligodendroglial cells regulate transcription of JC virus early and late genes coordinately with the Tat protein of human immunodeficiency virus type 1. Journal of General Virology. 2009;90(8):2005–2014. doi: 10.1099/vir.0.011072-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Tada H, Rappaport J, Lashgari M, Amini S, Wong-Staal F, Khalili K. Trans-activation of the JC virus late promoter by the tat protein of type 1 human immunodeficiency virus in glial cells. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(9):3479–3483. doi: 10.1073/pnas.87.9.3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nukuzuma S, Kameoka M, Sugiura S, et al. Archetype JC virus efficiently propagates in kidney-derived cells stably expressing HIV-1 Tat. Microbiology and Immunology. 2009;53(11):621–628. doi: 10.1111/j.1348-0421.2009.00166.x. [DOI] [PubMed] [Google Scholar]
- 130.Berger JR, Chauhan A, Galey D, Nath A. Epidemiological evidence and molecular basis of interactions between HIV and JC virus. Journal of NeuroVirology. 2001;7(4):329–338. doi: 10.1080/13550280152537193. [DOI] [PubMed] [Google Scholar]
- 131.Churchill MJ, Wesselingh SL, Cowley D, et al. Extensive astrocyte infection is prominent in human immunodeficiency virus—associated dementia. Annals of Neurology. 2009;66(2):253–258. doi: 10.1002/ana.21697. [DOI] [PubMed] [Google Scholar]
- 132.Li W, Li G, Steiner J, Nath A. Role of Tat protein in HIV neuropathogenesis. Neurotoxicity Research. 2009;16(3):205–220. doi: 10.1007/s12640-009-9047-8. [DOI] [PubMed] [Google Scholar]
- 133.Masanetz S, Lehmann MH. HIV-1 Nef increases astrocyte sensitivity towards exogenous hydrogen peroxide. Virology Journal. 2011;8, article 35 doi: 10.1186/1743-422X-8-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tornatore C, Meyers K, Atwood W, Conant K, Major E. Temporal patterns of human immunodeficiency virus type 1 transcripts in human fetal astrocytes. Journal of Virology. 1994;68(1):93–102. doi: 10.1128/jvi.68.1.93-102.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Vincendeau M, Kramer S, Hadian K, et al. Control of HIV replication in astrocytes by a family of highly conserved host proteins with a common Rev-interacting domain (Risp) AIDS. 2010;24(16):2433–2442. doi: 10.1097/QAD.0b013e32833e8758. [DOI] [PubMed] [Google Scholar]
- 136.Berger JR. Progressive multifocal leukoencephalopathy and newer biological agents. Drug Safety. 2010;33(11):969–983. doi: 10.2165/11537510-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 137.Berger JR, Kaszovitz B, Post MJD, Dickinson G. Progressive multifocal leukoencephalopathy associated with human immunodeficiency virus infection: a review of the literature with a report of sixteen cases. Annals of Internal Medicine. 1987;107(1):78–87. doi: 10.7326/0003-4819-107-1-78. [DOI] [PubMed] [Google Scholar]
- 138.Tan CS, Koralnik IJ. Progressive multifocal leukoencephalopathy and other disorders caused by JC virus: clinical features and pathogenesis. The Lancet Neurology. 2010;9(4):425–437. doi: 10.1016/S1474-4422(10)70040-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Moll NM, Rietsch AM, Ransohoff AJ, et al. Cortical demyelination in PML and MS: similarities and differences. Neurology. 2008;70(5):336–343. doi: 10.1212/01.WNL.0000284601.54436.e4. [DOI] [PubMed] [Google Scholar]
- 140.Tallantyre EC, Paine SML, Sharp CP, Lowe JS, Gran B. Atypical progressive multifocal leukoencephalopathy associated with an unusual JC polyomavirus mutation. Archives of Neurology. 2009;66(8):1021–1024. doi: 10.1001/archneurol.2009.94. [DOI] [PubMed] [Google Scholar]
- 141.Behzad-Behbahani A, Klapper PE, Vallely PJ, Cleator GM, Bonington A. BKV-DNA and JCV-DNA in CSF of patients with suspected meningitis or encephalitis. Infection. 2003;31(6):374–378. doi: 10.1007/s15010-003-3078-5. [DOI] [PubMed] [Google Scholar]
- 142.Richardson EP, Jr., Webster HD. Progressive multifocal leukoencephalopathy: its pathological features. Progress in Clinical and Biological Research. 1983;105:191–203. [PubMed] [Google Scholar]
- 143.Du Pasquier RA, Corey S, Margolin DH, et al. Productive infection of cerebellar granule cell neurons by JC virus in an HIV+ individual. Neurology. 2003;61(6):775–782. doi: 10.1212/01.wnl.0000081306.86961.33. [DOI] [PubMed] [Google Scholar]
- 144.Roux D, Bouldouyre MA, Mercier-Delarue S, et al. JC virus variant associated with cerebellar atrophy in a patient with AIDS. Journal of Clinical Microbiology. 2011;49(6):2196–2199. doi: 10.1128/JCM.02057-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Shah R, Bag AK, Chapman PR, Curé JK. Imaging manifestations of progressive multifocal leukoencephalopathy. Clinical Radiology. 2010;65(6):431–439. doi: 10.1016/j.crad.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 146.Whiteman MLH, Post MJD, Berger JR, Tate LG, Bell MD, Limonte LP. Progressive multifocal leukoencephalopathy in 47 HIV-seropositive patients: neuroimaging with clinical and pathologic correlation. Radiology. 1993;187(1):233–240. doi: 10.1148/radiology.187.1.8451420. [DOI] [PubMed] [Google Scholar]
- 147.Tavazzi E, White MK, Khalili K. Progressive multifocal leukoencephalopathy: clinical and molecular aspects. Reviews in Medical Virology. 2012;22(1):18–32. doi: 10.1002/rmv.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Viscidi RP, Khanna N, Tan CS, et al. JC virus antibody and viremia as predictors of progressive multifocal leukoencephalopathy in human immunodeficiency virus-1-infected individuals. Clinical Infectious Diseases. 2011;53(7):711–715. doi: 10.1093/cid/cir507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ryschkewitsch C, Jensen P, Hou J, Fahle G, Fischer S, Major EO. Comparison of PCR-southern hybridization and quantitative real-time PCR for the detection of JC and BK viral nucleotide sequences in urine and cerebrospinal fluid. Journal of Virological Methods. 2004;121(2):217–221. doi: 10.1016/j.jviromet.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 150.Bossolasco S, Calori G, Moretti F, et al. Prognostic significance of JC virus DNA levels in cerebrospinal fluid of patients with HIV-associated progressive multifocal leukoencephalopathy. Clinical Infectious Diseases. 2005;40(5):738–744. doi: 10.1086/427698. [DOI] [PubMed] [Google Scholar]
- 151.Marzocchetti A, Di Giambenedetto S, Cingolani A, Ammassari A, Cauda R, De Luca A. Reduced rate of diagnostic positive detection of JC virus DNA in cerebrospinal fluid in cases of suspected progressive multifocal leukoencephalopathy in the era of potent antiretroviral therapy. Journal of Clinical Microbiology. 2005;43(8):4175–4177. doi: 10.1128/JCM.43.8.4175-4177.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Marzocchetti A, Sanguinetti M, Di Giambenedetto S, et al. Characterization of JC virus in cerebrospinal fluid from HIV-1 infected patients with progressive multifocal leukoencephalopathy: insights into viral pathogenesis and disease prognosis. Journal of NeuroVirology. 2007;13(4):338–346. doi: 10.1080/13550280701381324. [DOI] [PubMed] [Google Scholar]
- 153.Aksamit AJ, Sever JL, Major EO. Progressive multifocal leukoencephalopathy: JC virus detection by in situ hybridization compared with immunohistochemistry. Neurology. 1986;36(4):499–504. doi: 10.1212/wnl.36.4.499. [DOI] [PubMed] [Google Scholar]
- 154.London WT, Houff SA, Madden DL, et al. Brain tumors in owl monkeys inoculated with a human polyomavirus (JC virus) Science. 1978;201(4362):1246–1249. doi: 10.1126/science.211583. [DOI] [PubMed] [Google Scholar]
- 155.London WT, Houff SA, McKeever PE, et al. Viral-induced astrocytomas in squirrel monkeys. Progress in Clinical and Biological Research. 1983;105:227–237. [PubMed] [Google Scholar]
- 156.Ohsumi S, Ikehara I, Motoi M. Induction of undifferentiated brain tumors in rats by a human polyomavirus (JC virus) Japanese Journal of Cancer Research. 1985;76(6):429–431. [PubMed] [Google Scholar]
- 157.Del Valle L, White MK, Khalili K. Potential mechanisms of the human polyomavirus JC in neural oncogenesis. Journal of Neuropathology and Experimental Neurology. 2008;67(8):729–740. doi: 10.1097/NEN.0b013e318180e631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Goudsmit J, Baak ML, Slaterus KW, Van Der Noordaa J. Human papovavirus isolated from urine of a child with acute tonsillitis. British Medical Journal. 1981;283(6303):1363–1364. doi: 10.1136/bmj.283.6303.1363-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Goudsmit J, Wertheim-van Dillen P, van Strien A, van der Noordaa J. The role of BK virus in acute respiratory tract disease and the presence of BKV DNA in tonsils. Journal of Medical Virology. 1982;10(2):91–99. doi: 10.1002/jmv.1890100203. [DOI] [PubMed] [Google Scholar]
- 160.Andrews CA, Shah KV, Daniel RW, Hirsch MS, Rubin RH. A serological investigation of BK virus and JC virus infections in recipients of renal allografts. Journal of Infectious Diseases. 1988;158(1):176–181. doi: 10.1093/infdis/158.1.176. [DOI] [PubMed] [Google Scholar]
- 161.Bofill-Mas S, Formiga-Cruz M, Clemente-Casares P, Calafell F, Girones R. Potential transmission of human polyomaviruses through the gastrointestinal tract after exposure to virions or viral DNA. Journal of Virology. 2001;75(21):10290–10299. doi: 10.1128/JVI.75.21.10290-10299.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Bofill-Mas S, Pina S, Girones R. Documenting the epidemiologic patterns of polyomaviruses in human populations by studying their presence in urban sewage. Applied and Environmental Microbiology. 2000;66(1):238–245. doi: 10.1128/aem.66.1.238-245.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Boldorini R, Allegrini S, Miglio U, et al. BK virus sequences in specimens from aborted fetuses. Journal of Medical Virology. 2010;82(12):2127–2132. doi: 10.1002/jmv.21923. [DOI] [PubMed] [Google Scholar]
- 164.Boldorini R, Allegrini S, Miglio U, et al. Serological evidence of vertical transmission of JC and BK Polyomaviruses in humans. Journal of General Virology. 2011;92(5):1044–1050. doi: 10.1099/vir.0.028571-0. [DOI] [PubMed] [Google Scholar]
- 165.Brown DWG, Gardner SD, Gibson PE, Field AM. BK virus specific IgM responses in cord sera, young children and healthy adults detected by RIA. Archives of Virology. 1984;82(3-4):149–160. doi: 10.1007/BF01311159. [DOI] [PubMed] [Google Scholar]
- 166.Dolei A, Pietropaolo V, Gomes E, et al. Polyomavirus persistence in lymphocytes: prevalence in lymphocytes from blood donors and healthy personnel of a blood transfusion centre. Journal of General Virology. 2000;81(8):1967–1973. doi: 10.1099/0022-1317-81-8-1967. [DOI] [PubMed] [Google Scholar]
- 167.Pietropaolo V, Di Taranto C, Degener AM, et al. Transplacental transmission of human polyomavirus BK. Journal of Medical Virology. 1998;56(4):372–376. doi: 10.1002/(sici)1096-9071(199812)56:4<372::aid-jmv14>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 168.Zambrano A, Kalantari M, Simoneau A, Jensen JL, Villarreal LP. Detection of human polyomaviruses and papillomaviruses in prostatictissue reveals the prostate as a habitat for multipleviral infections. Prostate. 2002;53(4):263–276. doi: 10.1002/pros.10157. [DOI] [PubMed] [Google Scholar]
- 169.Jiang M, Abend JR, Johnson SF, Imperiale MJ. The role of polyomaviruses in human disease. Virology. 2009;384(2):266–273. doi: 10.1016/j.virol.2008.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Yogo Y, Sugimoto C, Zhong S, Homma Y. Evolution of the BK polyomavirus: epidemiological, anthropological and clinical implications. Reviews in Medical Virology. 2009;19(4):185–199. doi: 10.1002/rmv.613. [DOI] [PubMed] [Google Scholar]
- 171.Jin L, Gibson PE, Booth JC, Clewley JP. Genomic typing of BK virus in clinical specimens by direct sequencing of polymerase chain reaction products. Journal of Medical Virology. 1993;41(1):11–17. doi: 10.1002/jmv.1890410104. [DOI] [PubMed] [Google Scholar]
- 172.Jin L, Gibson PE, Knowles WA, Clewley JP. BK virus antigenic variants: sequence analysis within the capsid VP1 epitope. Journal of Medical Virology. 1993;39(1):50–56. doi: 10.1002/jmv.1890390110. [DOI] [PubMed] [Google Scholar]
- 173.Ikegaya H, Saukko PJ, Tertti R, et al. Identification of a genomic subgroup of BK polyomavirus spread in European populations. Journal of General Virology. 2006;87(11):3201–3208. doi: 10.1099/vir.0.82266-0. [DOI] [PubMed] [Google Scholar]
- 174.Zheng HY, Nishimoto Y, Chen Q, et al. Relationships between BK virus lineages and human populations. Microbes and Infection. 2007;9(2):204–213. doi: 10.1016/j.micinf.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 175.Chen Q, Zheng HY, Zhong S, et al. Subtype IV of the BK polyomavirus is prevalent in East Asia. Archives of Virology. 2006;151(12):2419–2429. doi: 10.1007/s00705-006-0814-z. [DOI] [PubMed] [Google Scholar]
- 176.Carr MJ, McCormack GP, Mutton KJ, Crowley B. Unique BK virus non-coding control region (NCCR) variants in hematopoietic stem cell transplant recepients with and without hemorrhagic cystitis. Journal of Medical Virology. 2006;78(4):485–493. doi: 10.1002/jmv.20566. [DOI] [PubMed] [Google Scholar]
- 177.Di Taranto C, Pietropaolo V, Orsi GB, Jin L, Sinibaldi L, Degener AM. Detection of BK polyomavirus genotypes in healthy and HIV-positive children. European Journal of Epidemiology. 1997;13(6):653–657. doi: 10.1023/a:1007371320999. [DOI] [PubMed] [Google Scholar]
- 178.Jin L, Pietropaolo V, Booth JC, Ward KH, Brown DW. Prevalence and distribution of BK virus subtypes in healthy people and immunocompromised patients detected by PCR-restriction enzyme analysis. Clinical and Diagnostic Virology. 1995;3(3):285–295. doi: 10.1016/s0928-0197(94)00044-1. [DOI] [PubMed] [Google Scholar]
- 179.Krumbholz A, Zell R, Egerer R, et al. Prevalence of BK virus subtype I in Germany. Journal of Medical Virology. 2006;78(12):1588–1598. doi: 10.1002/jmv.20743. [DOI] [PubMed] [Google Scholar]
- 180.Nishimoto Y, Takasaka T, Hasegawa M, et al. Evolution of BK virus based on complete genome data. Journal of Molecular Evolution. 2006;63(3):341–352. doi: 10.1007/s00239-005-0092-5. [DOI] [PubMed] [Google Scholar]
- 181.Yogo Y, Zhong S, Suzuki M, Shibuya A, Kitamura T. Occurrence of the European subgroup of subtype I BK polyomavirus in Japanese-Americans suggests transmission outside the family. Journal of Virology. 2007;81(23):13254–13258. doi: 10.1128/JVI.01018-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wiseman AC. Polyomavirus nephropathy: a current perspective and clinical considerations. American Journal of Kidney Diseases. 2009;54(1):131–142. doi: 10.1053/j.ajkd.2009.01.271. [DOI] [PubMed] [Google Scholar]
- 183.Egli A, Infanti L, Dumoulin A, et al. Prevalence of polyomavirus BK and JC infection and replication in 400 healthy blood donors. Journal of Infectious Diseases. 2009;199(6):837–846. doi: 10.1086/597126. [DOI] [PubMed] [Google Scholar]
- 184.O’Donnell PH, Swanson K, Josephson MA, et al. BK virus infection is associated with hematuria and renal impairment in recipients of allogeneic hematopoetic stem cell transplants. Biology of Blood and Marrow Transplantation. 2009;15(9):1038.e1–1048.e1. doi: 10.1016/j.bbmt.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Wong ASY, Chan KH, Cheng VCC, Yuen KY, Kwong YL, Leung AYH. Relationship of pretransplantation polyoma BK virus serologic findings and BK viral reactivation after hematopoietic stem cell transplantation. Clinical Infectious Diseases. 2007;44(6):830–837. doi: 10.1086/511863. [DOI] [PubMed] [Google Scholar]
- 186.Bogdanovic G, Priftakis P, Giraud G, et al. Association between a high BK virus load in urine samples of patients with graft-versus-host disease and development of hemorrhagic cystitis after hematopoietic stem cell transplantation. Journal of Clinical Microbiology. 2004;42(11):5394–5396. doi: 10.1128/JCM.42.11.5394-5396.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Cesaro S, Facchin C, Tridello G, et al. A prospective study of BK-virus-associated haemorrhagic cystitis in paediatric patients undergoing allogeneic haematopoietic stem cell transplantation. Bone Marrow Transplantation. 2008;41(4):363–370. doi: 10.1038/sj.bmt.1705909. [DOI] [PubMed] [Google Scholar]
- 188.Tanaka K, Hori T, Hatakeyama N, et al. Quantification of BK polyoma viruria in Japanese children and adults with hemorrhagic cystitis complicating stem cell transplantation. Journal of Medical Virology. 2008;80(12):2108–2112. doi: 10.1002/jmv.21328. [DOI] [PubMed] [Google Scholar]
- 189.Erard V, Kim HW, Corey L, et al. BK DNA viral load in plasma: Evidence for an association with hemorrhagic cystitis in allogeneic hematopoietic cell transplant recipients. Blood. 2005;106(3):1130–1132. doi: 10.1182/blood-2004-12-4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Saundh BK, Baker R, Harris M, et al. Early BK polyomavirus (BKV) reactivation in donor kidney is a risk factor for development of BKV-associated nephropathy. Journal of Infectious Diseases. 2013;207(1):137–141. doi: 10.1093/infdis/jis642. [DOI] [PubMed] [Google Scholar]
- 191.van Aalderen MC, Heutinck KM, Huisman C, et al. BK virus infection in transplant recipients: clinical manifestations, treatment options and the immune response. Netherlands Journal of Medicine. 2012;70(4):172–183. [PubMed] [Google Scholar]
- 192.Kuypers DR. Management of polyomavirus-associated nephropathy in renal transplant recipients. Nature Reviews Nephrology. 2012;8(7):390–402. doi: 10.1038/nrneph.2012.64. [DOI] [PubMed] [Google Scholar]
- 193.Boothpur R, Brennan DC. Human polyoma viruses and disease with emphasis on clinical BK and JC. Journal of Clinical Virology. 2010;47(4):306–312. doi: 10.1016/j.jcv.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Nickeleit V, Hirsch HH, Binet IF, et al. Polyomavirus infection of renal allograft recipients: from latent infection to manifest disease. Journal of the American Society of Nephrology. 1999;10(5):1080–1089. doi: 10.1681/ASN.V1051080. [DOI] [PubMed] [Google Scholar]
- 195.Mengel M, Marwedel M, Radermacher J, et al. Incidence of polyomavirus-nephropathy in renal allografts: influence of modern immunosuppressive drugs. Nephrology Dialysis Transplantation. 2003;18(6):1190–1196. doi: 10.1093/ndt/gfg072. [DOI] [PubMed] [Google Scholar]
- 196.Comoli P, Azzi A, Maccario R, et al. Polyomavirus BK-specific immunity after kidney transplantation. Transplantation. 2004;78(8):1229–1232. doi: 10.1097/01.tp.0000137932.44791.d3. [DOI] [PubMed] [Google Scholar]
- 197.Comoli P, Binggeli S, Ginevri F, Hirsch HH. Polyomavirus-associated nephropathy: update on BK virus-specific immunity. Transplant Infectious Disease. 2006;8(2):86–94. doi: 10.1111/j.1399-3062.2006.00167.x. [DOI] [PubMed] [Google Scholar]
- 198.Abend JR, Low JA, Imperiale MJ. Inhibitory effect of gamma interferon on BK virus gene expression and replication. Journal of Virology. 2007;81(1):272–279. doi: 10.1128/JVI.01571-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Hirsch HH, Knowles W, Dickenmann M, et al. Prospective study of polyomavirus type BK replication and nephropathy in renal-transplant recipients. The New England Journal of Medicine. 2002;347(7):488–496. doi: 10.1056/NEJMoa020439. [DOI] [PubMed] [Google Scholar]
- 200.Bohl DL, Storch GA, Ryschkewitsch C, et al. Donor origin of BK virus in renal transplantation and role of HLA C7 in susceptibility to sustained BK viremia. American Journal of Transplantation. 2005;5(9):2213–2221. doi: 10.1111/j.1600-6143.2005.01000.x. [DOI] [PubMed] [Google Scholar]
- 201.Luo C, Hirsch HH, Kant J, et al. VP-1 quasispecies in human infection with polyomavirus BK. Journal of Medical Virology. 2012;84(1):152–161. doi: 10.1002/jmv.22147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Olsen GH, Hirsch HH, Rinaldo CH. Functional analysis of polyomavirus BK non-coding control region quasispecies from kidney transplant recipients. Journal of Medical Virology. 2009;81(11):1959–1967. doi: 10.1002/jmv.21605. [DOI] [PubMed] [Google Scholar]
- 203.Sood P, Senanayake S, Sujeet K, et al. Lower prevalence of BK virus infection in African American renal transplant recipients: a prospective study. Transplantation. 2012;93(3):291–296. doi: 10.1097/TP.0b013e31823ec05a. [DOI] [PubMed] [Google Scholar]
- 204.Thomas DL, Thio CL, Martin MP, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature. 2009;461(7265):798–801. doi: 10.1038/nature08463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Geddes CC, Gunson R, Mazonakis E, et al. BK viremia surveillance after kidney transplant: single-center experience during a change from cyclosporine-to lower-dose tacrolimus-based primary immunosuppression regimen. Transplant Infectious Disease. 2011;13(2):109–116. doi: 10.1111/j.1399-3062.2010.00566.x. [DOI] [PubMed] [Google Scholar]
- 206.Khan H, Oberoi S, Mahvash A, et al. Reversible ureteral obstruction due to polyomavirus infection after percutaneous nephrostomy catheter placement. Biology of Blood and Marrow Transplantation. 2011;17(10):1551–1555. doi: 10.1016/j.bbmt.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Hirsch HH, Mohaupt M, Klimkait T, Limaye AP, Jerome KR. Prospective monitoring of BK virus load after discontinuing sirolimus treatment in a renal transplant patient with BK virus nephropathy. Journal of Infectious Diseases. 2001;184(11):1494–1496. doi: 10.1086/324425. [DOI] [PubMed] [Google Scholar]
- 208.Viscount HB, Eid AJ, Espy MJ, et al. Polyomavirus polymerase chain reaction as a surrogate marker of polyomavirus-associated nephropathy. Transplantation. 2007;84(3):340–345. doi: 10.1097/01.tp.0000275205.41078.51. [DOI] [PubMed] [Google Scholar]
- 209.Smith F, Panek R, Kiberd BA. Screening to prevent polyoma virus nephropathy in kidney transplantation: a cost analysis: brief communication. American Journal of Transplantation. 2009;9(9):2177–2179. doi: 10.1111/j.1600-6143.2009.02729.x. [DOI] [PubMed] [Google Scholar]
- 210.Pastrana DV, Brennan DC, Cuburu N, et al. Neutralization serotyping of BK polyomavirus infection in kidney transplant recipients. PLOS Pathogens. 2012;8(4) doi: 10.1371/journal.ppat.1002650.e1002650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Randhawa PS, Khaleel-Ur-Rehman K, Swalsky PA, et al. DNA sequencing of viral capsid protein VP-1 region in patients with BK virus interstitial nephritis. Transplantation. 2002;73(7):1090–1094. doi: 10.1097/00007890-200204150-00013. [DOI] [PubMed] [Google Scholar]
- 212.Abend JR, Jiang M, Imperiale MJ. BK virus and human cancer: innocent until proven guilty. Seminars in Cancer Biology. 2009;19(4):252–260. doi: 10.1016/j.semcancer.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Arora R, Chang Y, Moore PS. MCV and Merkel cell carcinoma: a molecular success story. Current Opinion in Virology. 2012;2(4):489–498. doi: 10.1016/j.coviro.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Spurgeon ME, Lambert PF. Merkel cell polyomavirus: a newly discovered human virus with oncogenic potential. Virology. 2013;435(1):118–130. doi: 10.1016/j.virol.2012.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Nicol JT, Robinot R, Carpentier A, et al. Age-specific seroprevalence of Merkel cell polyomavirus, human polyomaviruses 6, 7 and 9 and Trichodysplasia Spinulosa-associated polyomavirus. Clinical and Vaccine Immunology. 2013;20(3):363–368. doi: 10.1128/CVI.00438-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Tolstov YL, Pastrana DV, Feng H, et al. Human Merkel cell polyomavirus infection II. MCV is a common human infection that can be detected by conformational capsid epitope immunoassays. International Journal of Cancer. 2009;125(6):1250–1256. doi: 10.1002/ijc.24509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Tolstov YL, Knauer A, Chen JG, et al. Asymptomatic primary merkel cell polyomavirus infection among adults. Emerging Infectious Diseases. 2011;17(8):1371–1380. doi: 10.3201/eid1708.110079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Heath M, Jaimes N, Lemos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. Journal of the American Academy of Dermatology. 2008;58(3):375–381. doi: 10.1016/j.jaad.2007.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Engels EA, Frisch M, Goedert JJ, Biggar RJ, Miller RW. Merkel cell carcinoma and HIV infection. The Lancet. 2002;359(9305):497–498. doi: 10.1016/S0140-6736(02)07668-7. [DOI] [PubMed] [Google Scholar]
- 220.Agelli M, Clegg LX, Becker JC, et al. The etiology and epidemiology of merkel cell carcinoma. Current Problems in Cancer. 2010;34(1):14–37. doi: 10.1016/j.currproblcancer.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 221.Muirhead R, Ritchie DM. Partial regression of Merkel cell carcinoma in response to withdrawal of azathioprine in an immunosuppression-induced case of metastatic Merkel cell carcinoma. Clinical Oncology. 2007;19(1):p. 96. doi: 10.1016/j.clon.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 222.Gianfreda M, Caiffi S, De Franceschi T, et al. Merkel cell carcinoma of the skin in a patient with Myasthenia gravis. Minerva Medica. 2002;(3):219–222. [PubMed] [Google Scholar]
- 223.McLoone NM, McKenna K, Edgar D, Walsh M, Bingham A. Merkel cell carcinoma in a patient with chronic sarcoidosis. Clinical and Experimental Dermatology. 2005;30(5):580–582. doi: 10.1111/j.1365-2230.2005.01800.x. [DOI] [PubMed] [Google Scholar]
- 224.Nemoto I, Sato-Matsumura KC, Fujita Y, et al. Leukaemic dissemination of Merkel cell carcinoma in a patient with systemic lupus erythematosus. Clinical and Experimental Dermatology. 2008;33(3):270–272. doi: 10.1111/j.1365-2230.2007.02618.x. [DOI] [PubMed] [Google Scholar]
- 225.Satolli F, Venturi C, Vescovi V, Morrone P, De Panfilis G. Merkel-cell carcinoma in Behçet’s disease. Acta Dermato-Venereologica. 2005;85(1):p. 79. doi: 10.1080/00015550410021664. [DOI] [PubMed] [Google Scholar]
- 226.Casimir GJA, Duchateau J. Gender differences in inflammatory processes could explain poorer prognosis for males. Journal of Clinical Microbiology. 2011;49(1):478–479. doi: 10.1128/JCM.02096-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Pastrana DV, Tolstov YL, Becker JC, Moore PS, Chang Y, Buck CB. Quantitation of human seroresponsiveness to Merkel cell polyomavirus. PLoS Pathogens. 2009;5(9) doi: 10.1371/journal.ppat.1000578.e1000578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Wooff JC, Trites JR, Walsh NMG, Bullock MJ. Complete spontaneous regression of metastatic merkel cell carcinoma: a case report and review of the literature. American Journal of Dermatopathology. 2010;32(6):614–617. doi: 10.1097/DAD.0b013e3181cd3158. [DOI] [PubMed] [Google Scholar]
- 229.Shuda M, Feng H, Kwun HJ, et al. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(42):16272–16277. doi: 10.1073/pnas.0806526105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Arora R, Shuda M, Guastafierro A, et al. Survivin is a therapeutic target in Merkel cell carcinoma. Science Translational Medicine. 2012;4(133) doi: 10.1126/scitranslmed.3003713.133ra56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Allander T, Tammi MT, Eriksson M, Bjerkner A, Tiveljung-Lindell A, Andersson B. Cloning of a human parvovirus by molecular screening of respiratory tract samples. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(36):12891–12896. doi: 10.1073/pnas.0504666102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Abedi Kiasari B, Vallely PJ, Corless CE, Al-Hammadi M, Klapper PE. Age-related pattern of KI and WU polyomavirus infection. Journal of Clinical Virology. 2008;43(1):123–125. doi: 10.1016/j.jcv.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Babakir-Mina M, Ciccozzi M, Dimonte S, et al. Identification of the novel KI polyomavirus in the respiratory tract of an Italian patient. Journal of Medical Virology. 2008;80(11):2012–2014. doi: 10.1002/jmv.21303. [DOI] [PubMed] [Google Scholar]
- 234.Bialasiewicz S, Whiley DM, Lambert SB, et al. Presence of the newly discovered human polyomaviruses KI and WU in Australian patients with acute respiratory tract infection. Journal of Clinical Virology. 2008;41(2):63–68. doi: 10.1016/j.jcv.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Foulongne V, Brieu N, Jeziorski E, Chatain A, Rodière M, Segondy M. KI and WU polyomaviruses in children, France. Emerging Infectious Diseases. 2008;14(3):523–525. doi: 10.3201/eid1403.071206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Norja P, Ubillos I, Templeton K, Simmonds P. No evidence for an association between infections with WU and KI polyomaviruses and respiratory disease. Journal of Clinical Virology. 2007;40(4):307–311. doi: 10.1016/j.jcv.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Payungporn S, Chieochansin T, Thongmee C, Samransamruajkit R, Theamboolers A, Poovorawan Y. Prevalence and molecular characterization of WU/KI polyomaviruses isolated from pediatric patients with respiratory disease in Thailand. Virus Research. 2008;135(2):230–236. doi: 10.1016/j.virusres.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.van de Pol AC, Wolfs TFW, Jansen NJG, Kimpen JLL, Van Loon AM, Rossen JWA. Human bocavirus and KI/WU polyomaviruses in pediatric intensive care patients. Emerging Infectious Diseases. 2009;15(3):454–457. doi: 10.3201/eid1503.081203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Mourez T, Bergeron A, Ribaud P, et al. Polyomaviruses KI and WU in immunocompromised patients with respiratory disease. Emerging Infectious Diseases. 2009;15(1):107–109. doi: 10.3201/1501.080758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Babakir-Mina M, Ciccozzi M, Farchi F, et al. KI and WU polyomaviruses and CD4+ cell counts in HIV-1-infected patients, Italy. Emerging Infectious Diseases. 2010;16(9):1482–1485. doi: 10.3201/eid1609.100211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Babakir-Mina M, Ciccozzi M, Alteri C, et al. Excretion of the novel polyomaviruses KI and WU in the stool of patients with hematological disorders. Journal of Medical Virology. 2009;81(9):1668–1673. doi: 10.1002/jmv.21559. [DOI] [PubMed] [Google Scholar]
- 242.Barzon L, Squarzon L, Militello V, et al. WU and KI polyomaviruses in the brains of hiv-positive patients with and without progressive multifocal leukoencephalopathy. Journal of Infectious Diseases. 2009;200(11):1755–1758. doi: 10.1086/648095. [DOI] [PubMed] [Google Scholar]
- 243.Babakir-Mina M, Ciccozzi M, Perno CF, et al. The novel KI, WU, MC polyomaviruses: possible human pathogens? New Microbiologica. 2011;34(1):1–8. [PubMed] [Google Scholar]
- 244.Kuypers J, Campbell AP, Guthrie KA, et al. WU and KI polyomaviruses in respiratory samples from allogeneic hematopoietic cell transplant recipients. Emerging Infectious Diseases. 2012;18(10):1580–1588. doi: 10.3201/eid1810.120477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Abed Y, Wang D, Boivin G. WU polyomavirus in children, Canada. Emerging Infectious Diseases. 2007;13(12):1939–1941. doi: 10.3201/eid1312.070909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Tae HH, Chung JY, Ja WK, Sang WK, Hwang ES. WU polyomavirus in children with acute lower respiratory tract infections, South Korea. Emerging Infectious Diseases. 2007;13(11):1766–1768. doi: 10.3201/eid1311.070872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Le BM, Demertzis LM, Wu G, et al. Clinical and epidemiologic characterization of WU polyomavirus infection, St. Louis, Missouri. Emerging Infectious Diseases. 2007;13(12):1936–1938. doi: 10.3201/eid1312.070977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Lin F, Zheng M, Li H, et al. WU polyomavirus in children with acute lower respiratory tract infections, China. Journal of Clinical Virology. 2008;42(1):94–102. doi: 10.1016/j.jcv.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Neske F, Blessing K, Ullrich F, Pröttel A, Kreth HW, Weissbrich B. WU polyomavirus infection in children, Germany. Emerging Infectious Diseases. 2008;14(4):680–681. doi: 10.3201/eid0104.071325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Barzon L, Squarzon L, Pacenti M, Scotton PG, Palú G. Detection of WU polyomavirus in cerebrospinal fluid specimen from a patient with AIDS and suspected progressive multifocal leukoencephalopathy. Journal of Infectious Diseases. 2009;200(2):314–315. doi: 10.1086/599842. [DOI] [PubMed] [Google Scholar]
- 251.Siebrasse EA, Bauer I, Holtz LR, et al. Human polyomaviruses in children undergoing transplantation, United States, 2008–2010. Emerging Infectious Diseases. 2012;18(10):1676–1679. doi: 10.3201/eid1810.120359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Brimhall CL, Malone JC. Viral-associated trichodysplasia spinulosa in a renal transplant patient. Archives of Dermatology. 2012;148(7):863–864. doi: 10.1001/archdermatol.2012.29. [DOI] [PubMed] [Google Scholar]
- 253.Chastain MA, Millikan LE. Pilomatrix dysplasia in an immunosuppressed patient. Journal of the American Academy of Dermatology. 2000;43(1):118–122. doi: 10.1067/mjd.2000.100967. [DOI] [PubMed] [Google Scholar]
- 254.Haycox CL, Kim S, Fleckman P, et al. Trichodysplasia spinulosa—a newly described folliculocentric viral infection in an immunocompromised host. Journal of Investigative Dermatology Symposium Proceedings. 1999;4(3):268–271. doi: 10.1038/sj.jidsp.5640227. [DOI] [PubMed] [Google Scholar]
- 255.Heaphy MR, Jr., Shamma HN, Hickmann M, White MJ. Cyclosporine-induced folliculodystrophy. Journal of the American Academy of Dermatology. 2004;50(2):310–315. doi: 10.1016/s0190-9622(03)00774-6. [DOI] [PubMed] [Google Scholar]
- 256.Lee JSS, Frederiksen P, Kossard S. Progressive trichodysplasia spinulosa in a patient with chronic lymphocytic leukaemia in remission. Australasian Journal of Dermatology. 2008;49(1):57–60. doi: 10.1111/j.1440-0960.2007.00422.x. [DOI] [PubMed] [Google Scholar]
- 257.Matthews MR, Wang RC, Reddick RL, Saldivar VA, Browning JC. Viral-associated trichodysplasia spinulosa: a case with electron microscopic and molecular detection of the trichodysplasia spinulosa-associated human polyomavirus. Journal of Cutaneous Pathology. 2011;38(5):420–431. doi: 10.1111/j.1600-0560.2010.01664.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Osswald SS, Kulick KB, Tomaszewski MM, Sperling LC. Viral-associated trichodysplasia in a patient with lymphoma: a case report and review. Journal of Cutaneous Pathology. 2007;34(9):721–725. doi: 10.1111/j.1600-0560.2006.00693.x. [DOI] [PubMed] [Google Scholar]
- 259.Sadler GM, Halbert AR, Smith N, Rogers M. Trichodysplasia spinulosa associated with chemotherapy for acute lymphocytic leukaemia. Australasian Journal of Dermatology. 2007;48(2):110–114. doi: 10.1111/j.1440-0960.2007.00348.x. [DOI] [PubMed] [Google Scholar]
- 260.Schwieger-Briel A, Balma-Mena A, Ngan B, Dipchand A, Pope E. Trichodysplasia spinulosa—a rare complication in immunosuppressed patients. Pediatric Dermatology. 2010;27(5):509–513. doi: 10.1111/j.1525-1470.2010.01278.x. [DOI] [PubMed] [Google Scholar]
- 261.Sperling LC, Tomaszewski MM, Thomas DA. Viral-associated trichodysplasia in patients who are immunocompromised. Journal of the American Academy of Dermatology. 2004;50(2):318–322. doi: 10.1016/s0190-9622(03)01490-7. [DOI] [PubMed] [Google Scholar]
- 262.Wyatt AJ, Sachs DL, Shia J, Delgado R, Busam KJ. Virus-associated trichodysplasia spinulosa. American Journal of Surgical Pathology. 2005;29(2):241–246. doi: 10.1097/01.pas.0000149691.83086.dc. [DOI] [PubMed] [Google Scholar]
- 263.Kazem S, van der Meijden E, Kooijman S, et al. Trichodysplasia spinulosa is characterized by active polyomavirus infection. Journal of Clinical Virology. 2012;53(3):225–230. doi: 10.1016/j.jcv.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 264.van der Meijden E, Kazem S, Burgers MM, et al. Seroprevalence of trichodysplasia spinulosa-associated polyomavirus. Emerging Infectious Diseases. 2011;17(8):1355–1363. doi: 10.3201/eid1708.110114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Chen T, Mattila PS, Jartti T, et al. Seroepidemiology of the newly found trichodysplasia spinulosa-associated polyomavirus. Journal of Infectious Diseases. 2011;204(10):1523–1526. doi: 10.1093/infdis/jir614. [DOI] [PubMed] [Google Scholar]
- 266.Sadeghi M, Aronen M, Chen T, et al. Merkel cell polyomavirus and trichodysplasia spinulosa-associated polyomavirus DNAs and antibodies in blood among the elderly. BMC Infectious Diseases. 2012;12, article 383 doi: 10.1186/1471-2334-12-383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Leendertz FH, Scuda N, Cameron KN, et al. African great apes are naturally infected with polyomaviruses closely related to Merkel cell polyomavirus. Journal of Virology. 2011;85(2):916–924. doi: 10.1128/JVI.01585-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Takemoto KK, Furuno A, Kato K, Yoshike K. Biological and biochemical studies of African green monkey lymphotropic papovavirus. Journal of Virology. 1982;42(2):502–509. doi: 10.1128/jvi.42.2.502-509.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Nicol JT, Touze A, Robinot R, et al. Seroprevalence and cross-reactivity of human polyomavirus 9. Emerging Infectious Diseases. 2012;18(8):1329–1332. doi: 10.3201/eid1808.111625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Trusch F, Klein M, Finsterbusch T, et al. Seroprevalence of human polyomavirus 9 and cross-reactivity to African green monkey-derived lymphotropic polyomavirus. Journal of General Virology. 2012;934, part 4:698–705. doi: 10.1099/vir.0.039156-0. [DOI] [PubMed] [Google Scholar]
- 271.Csoma E, Sapy T, Meszaros B, et al. Novel human polyomaviruses in pregnancy: higher prevalence of BKPyV, but no WUPyV, KIPyV and HPyV9. Journal of Clinical Virology. 2012;55(3):262–265. doi: 10.1016/j.jcv.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 272.Imajoh M, Hashida Y, Taniguchi A, et al. Novel human polyomaviruses, Merkel cell polyomavirus and human polyomavirus 9, in Japanese chronic lymphocytic leukemia cases. Journal of Hematology & Oncology. 2012;5, article 25 doi: 10.1186/1756-8722-5-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Yu G, Greninger AL, Isa P, et al. Discovery of a novel polyomavirus in acute diarrheal samples from children. PLoS One. 2012;7(11) doi: 10.1371/journal.pone.0049449.e49449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Bauman Y, Mandelboim O. MicroRNA based immunoevasion mechanism of human polyomaviruses. RNA Biology. 2011;8(4):591–594. doi: 10.4161/rna.8.4.15587. [DOI] [PubMed] [Google Scholar]
- 275.Boss IW, Renne R. Viral miRNAs and immune evasion. Biochimica et Biophysica Acta. 2011;1809(11-12):708–714. doi: 10.1016/j.bbagrm.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Sullivan CS, Grundhoff AT, Tevethia S, Pipas JM, Ganem D. SV40-encoded microRNAs regulate viral gene expression and reduce susceptibility to cytotoxic T cells. Nature. 2005;435(7042):682–686. doi: 10.1038/nature03576. [DOI] [PubMed] [Google Scholar]
- 277.Seo GJ, Fink LHL, O’Hara B, Atwood WJ, Sullivan CS. Evolutionarily conserved function of a viral microRNA. Journal of Virology. 2008;82(20):9823–9828. doi: 10.1128/JVI.01144-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Osman A. MicroRNAs in health and disease—basic science and clinical applications. Clinical Laboratory. 2012;58(5-6):393–402. [PubMed] [Google Scholar]
- 279.Bauman Y, Nachmani D, Vitenshtein A, et al. An identical miRNA of the human JC and BK polyoma viruses targets the stress-induced ligand ULBP3 to escape immune elimination. Cell Host and Microbe. 2011;9(2):93–102. doi: 10.1016/j.chom.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 280.Lee S, Paulson KG, Murchison EP, et al. Identification and validation of a novel mature microRNA encoded by the Merkel cell polyomavirus in human Merkel cell carcinomas. Journal of Clinical Virology. 2011;52(3):272–275. doi: 10.1016/j.jcv.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Seo GJ, Chen CJ, Sullivan CS. Merkel cell polyomavirus encodes a microRNA with the ability to autoregulate viral gene expression. Virology. 2009;383(2):183–187. doi: 10.1016/j.virol.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 282.Kuypers DRJ, Bammens B, Claes K, Evenepoel P, Lerut E, Vanrenterghem Y. A single-centre study of adjuvant cidofovir therapy for BK virus interstitial nephritis (BKVIN) in renal allograft recipients. Journal of Antimicrobial Chemotherapy. 2009;63(2):417–419. doi: 10.1093/jac/dkn495. [DOI] [PubMed] [Google Scholar]
- 283.Safrin S, Cherrington J, Jaffe HS. Clinical uses of cidofovir. Reviews in Medical Virology. 1997;7(3):145–156. doi: 10.1002/(sici)1099-1654(199709)7:3<145::aid-rmv196>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 284.Gosert R, Rinaldo CH, Wernli M, Major EO, Hirsch HH. CMX001 (1-O-hexadecyloxypropyl-cidofovir) inhibits polyomavirus jc replication in human brain progenitor-derived astrocytes. Antimicrobial Agents and Chemotherapy. 2011;55(5):2129–2136. doi: 10.1128/AAC.00046-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Jiang ZG, Cohen J, Marshall LJ, Major EO. Hexadecyloxypropyl-cidofovir (CMX001) suppresses JC virus replication in human fetal brain SVG cell cultures. Antimicrobial Agents and Chemotherapy. 2010;54(11):4723–4732. doi: 10.1128/AAC.00837-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Patel A, Patel J, Ikwuagwu J. A case of progressive multifocal leukoencephalopathy and idiopathic CD4+ lymphocytopenia. Journal of Antimicrobial Chemotherapy. 2010;65(12):2697–2698. doi: 10.1093/jac/dkq359. [DOI] [PubMed] [Google Scholar]
- 287.Painter W, Robertson A, Trost LC, et al. First pharmacokinetic and safety study in humans of the novel lipid antiviral conjugate CMX001, a broad-spectrum oral drug active against double-stranded DNA viruses. Antimicrobial Agents and Chemotherapy. 2012;56(5):2726–2734. doi: 10.1128/AAC.05983-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Rinaldo CH, Gosert R, Bernhoff E, Finstad S, Hirsch HH. 1-O-hexadecyloxypropyl cidofovir (CMX001) effectively inhibits polyomavirus BK replication in primary human renal tubular epithelial cells. Antimicrobial Agents and Chemotherapy. 2010;54(11):4714–4722. doi: 10.1128/AAC.00974-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Wanat KA, Holler PD, Dentchev T, et al. Viral-associated trichodysplasia: characterization of a novel polyomavirus infection with therapeutic insights. Archives of Dermatology. 2012;148(2):219–223. doi: 10.1001/archdermatol.2011.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Aksamit AJ. Treatment of non-AIDS progressive multifocal leukoencephalopathy with cytosine arabinoside. Journal of NeuroVirology. 2001;7(4):386–390. doi: 10.1080/13550280152537292. [DOI] [PubMed] [Google Scholar]
- 291.Marra CM, Rajicic N, Barker DE, et al. A pilot study of cidofovir for progressive multifocal leukoencephalopathy in AIDS. AIDS. 2002;16(13):1791–1797. doi: 10.1097/00002030-200209060-00012. [DOI] [PubMed] [Google Scholar]
- 292.Hou J, Major EO. The efficacy of nucleoside analogs against JC virus multiplication in a persistently infected human fetal brain cell line. Journal of NeuroVirology. 1998;4(4):451–456. doi: 10.3109/13550289809114545. [DOI] [PubMed] [Google Scholar]
- 293.Razonable RR, Aksamit AJ, Wright AJ, Wilson JW. Cidofovir treatment of progressive multifocal leukoencephalopathy in a patient receiving highly active antiretroviral therapy. Mayo Clinic Proceedings. 2001;76(11):1171–1175. doi: 10.4065/76.11.1171. [DOI] [PubMed] [Google Scholar]
- 294.Savona MR, Newton D, Frame D, Levine JE, Mineishi S, Kaul DR. Low-dose cidofovir treatment of BK virus-associated hemorrhagic cystitis in recipients of hematopoietic stem cell transplant. Bone Marrow Transplantation. 2007;39(12):783–787. doi: 10.1038/sj.bmt.1705678. [DOI] [PubMed] [Google Scholar]
- 295.Yagi T, Hattori H, Ohira M, et al. Progressive multifocal leukoencephalopathy developed in incomplete Heerfordt syndrome, a rare manifestation of sarcoidosis, without steroid therapy responding to cidofovir. Clinical Neurology and Neurosurgery. 2010;112(2):153–156. doi: 10.1016/j.clineuro.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 296.Berenguer J, Miralles P, Arrizabalaga J, et al. Clinical course and prognostic factors of progressive multifocal leukoencephalopathy in patients treated with highly active antiretroviral therapy. Clinical Infectious Diseases. 2003;36(8):1047–1052. doi: 10.1086/374048. [DOI] [PubMed] [Google Scholar]
- 297.De Luca A, Ammassari A, Pezzotti P, et al. Cidofovir in addition to antiretroviral treatment is not effective for AIDS-associated progressive multifocal leukoencephalopathy: a multicohort analysis. AIDS. 2008;22(14):1759–1767. doi: 10.1097/QAD.0b013e32830a5043. [DOI] [PubMed] [Google Scholar]
- 298.Portegies P, Algra PR, Hollak CEM, et al. Response to cytarabine in progressive multifocal leucoencephalopathy in AIDS. The Lancet. 1991;337(8742):680–681. doi: 10.1016/0140-6736(91)92504-u. [DOI] [PubMed] [Google Scholar]
- 299.Vulliemoz S, Lurati-Ruiz F, Borruat FX, et al. Favourable outcome of progressive multifocal leucoencephalopathy in two patients with dermatomyositis. Journal of Neurology, Neurosurgery and Psychiatry. 2006;77(9):1079–1082. doi: 10.1136/jnnp.2006.092353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Antinori A, De Luca A, Ammassari A, et al. Failure of cytarabine and increased JC virus-DNA burden in the cerebrospinal fluid of patients with AIDS-related progressive multifocal leucoencephalopathy. AIDS. 1994;8(7):1022–1024. [PubMed] [Google Scholar]
- 301.Tubridy N, Wells C, Lewis D, Schon F. Unsuccessful treatment with cidofovir and cytarabine in progressive multifocal leukoencephalopathy associated with dermatomyositis. Journal of the Royal Society of Medicine. 2000;93(7):374–375. doi: 10.1177/014107680009300710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Hall CD, Dafni U, Simpson D, et al. Failure of cytarabine in progressive multifocal leukoencephalopathy associated with human immunodeficiency virus infection. The New England Journal of Medicine. 1998;338(19):1345–1351. doi: 10.1056/NEJM199805073381903. [DOI] [PubMed] [Google Scholar]
- 303.Elphick GF, Querbes W, Jordan JA, et al. The human polyomavirus, JCV, uses serotonin receptors to infect cells. Science. 2004;306(5700):1380–1383. doi: 10.1126/science.1103492. [DOI] [PubMed] [Google Scholar]
- 304.Nukuzuma S, Nakamichi K, Nukuzuma C, Takegami T. Inhibitory effect of serotonin antagonists on JC virus propagation in a carrier culture of human neuroblastoma cells. Microbiology and Immunology. 2009;53(9):496–501. doi: 10.1111/j.1348-0421.2009.00156.x. [DOI] [PubMed] [Google Scholar]
- 305.Altschuler EL, Kast RE. The atypical antipsychotic agents zisprasidone, risperdone and olanzapine as treatment for and prophylaxis against progressive multifocal leukoencephalopathy. Medical Hypotheses. 2005;65(3):585–586. doi: 10.1016/j.mehy.2005.01.037. [DOI] [PubMed] [Google Scholar]
- 306.Lanzafame M, Ferrari S, Lattuada E, et al. Mirtazapine in an HIV-1 infected patient with progressive multifocal leukoencephalopathy. Infezioni in Medicina. 2009;17(1):35–37. [PubMed] [Google Scholar]
- 307.Chapagain ML, Sumibcay L, Gurjav U, Kaufusi P, Kast RE, Nerurkar VR. Serotonin receptor 2A blocker (risperidone) has no effect on human polyomavirus JC infection of primary human fetal glial cells. Journal of NeuroVirology. 2008;14(5):448–454. doi: 10.1080/13550280802235916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Monaco MC, Maric D, Bandeian A, et al. Progenitor-derived Oligodendrocyte culture system from human fetal brain. Journal of Visualized Experiments. 2012;(70) doi: 10.3791/4274.e4274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Pöhlmann C, Hochauf K, Röllig C, et al. Chlorpromazine combined with cidofovir for treatment of a patient suffering from progressive multifocal leukoencephalopathy. Intervirology. 2007;50(6):412–417. doi: 10.1159/000112916. [DOI] [PubMed] [Google Scholar]
- 310.Brickelmaier M, Lugovskoy A, Kartikeyan R, et al. Identification and characterization of mefloquine efficacy against JC virus in vitro. Antimicrobial Agents and Chemotherapy. 2009;53(5):1840–1849. doi: 10.1128/AAC.01614-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Gofton TE, Al-Khotani A, O’Farrell B, Ang LC, McLachlan RS. Mefloquine in the treatment of progressive multifocal leukoencephalopathy. Journal of Neurology, Neurosurgery and Psychiatry. 2011;82(4):452–455. doi: 10.1136/jnnp.2009.190652. [DOI] [PubMed] [Google Scholar]
- 312.Kishida S, Tanaka K. Mefloquine treatment in a patient suffering from progressive multifocal leukoencephalopathy after umbilical cord blood transplant. Internal Medicine. 2010;49(22):2509–2513. doi: 10.2169/internalmedicine.49.3227. [DOI] [PubMed] [Google Scholar]
- 313.Bernhoff E, Tylden GD, Kjerpeseth LJ, Gutteberg TJ, Hirsch HH, Rinaldo CH. Leflunomide inhibition of BK virus replication in renal tubular epithelial cells. Journal of Virology. 2010;84(4):2150–2156. doi: 10.1128/JVI.01737-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Farasati NA, Shapiro R, Vats A, Randhawa P. Effect of Leflunomide and Cidofovir on replication of BK virus in an in vitro culture system. Transplantation. 2005;79(1):116–118. doi: 10.1097/01.tp.0000149338.97084.5f. [DOI] [PubMed] [Google Scholar]
- 315.Josephson MA, Gillen D, Javaid B, et al. Treatment of renal allograft polyoma BK virus infection with leflunomide. Transplantation. 2006;81(5):704–710. doi: 10.1097/01.tp.0000181149.76113.50. [DOI] [PubMed] [Google Scholar]
- 316.Faguer S, Hirsch HH, Kamar N, et al. Leflunomide treatment for polyomavirus BK-associated nephropathy after kidney transplantation. Transplant International. 2007;20(11):962–969. doi: 10.1111/j.1432-2277.2007.00523.x. [DOI] [PubMed] [Google Scholar]
- 317.Leca N, Muczynski KA, Jefferson JA, et al. Higher levels of leflunomide are associated with hemolysis and are not superior to lower levels for BK virus clearance in renal transplant patients. Clinical Journal of the American Society of Nephrology. 2008;3(3):829–835. doi: 10.2215/CJN.03930907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Guasch A, Roy-Chaudhury P, Woodle ES, Fitzsimmons W, Holman J, First MR. Assessment of efficacy and safety of FK778 in comparison with standard care in renal transplant recipients with untreated BK nephropathy. Transplantation. 2010;90(8):891–897. doi: 10.1097/TP.0b013e3181f2c94b. [DOI] [PubMed] [Google Scholar]
- 319.Reilmann R, Imai T, Ringelstein EB, et al. Remission of progressive multifocal leucoencephalopathy in SLE after treatment with cidofovir: a 4 year follow up. Journal of Neurology, Neurosurgery and Psychiatry. 2005;76(9):1304–1305. doi: 10.1136/jnnp.2004.057588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Kraemer C, Evers S, Nolting T, Arendt G, Husstedt IW. Cidofovir in combination with HAART and survival in AIDS-associated progressive multifocal leukoencephalopathy. Journal of Neurology. 2008;255(4):526–531. doi: 10.1007/s00415-008-0731-z. [DOI] [PubMed] [Google Scholar]
- 321.Nicoli F, Chave B, Peragut JC, Gastaut JL. Efficacy of cytarabine in progressive multifocal leucoencephalopathy in AIDS. The Lancet. 1992;339(8788):p. 306. doi: 10.1016/0140-6736(92)91376-j. [DOI] [PubMed] [Google Scholar]
- 322.Guarino M, D’Alessandro R, Rinaldi R, et al. Progressive multifocal leucoencephalopathy in AIDS: treatment with cytosine arabinoside. AIDS. 1995;9(7):819–820. [PubMed] [Google Scholar]
- 323.Moreno S, Miralles P, Díaz MD, et al. Cytarabine therapy for progressive multifocal leukoencephalopathy in patients with AIDS. Clinical Infectious Diseases. 1996;23(5):1066–1068. doi: 10.1093/clinids/23.5.1066. [DOI] [PubMed] [Google Scholar]
- 324.Portolani M, Pietrosemoli P, Cermelli C, et al. Suppression of BK virus replication and cytopathic effect by inhibitors of prokaryotic DNA gyrase. Antiviral Research. 1988;9(3):205–218. doi: 10.1016/0166-3542(88)90004-6. [DOI] [PubMed] [Google Scholar]
- 325.Ali SH, Chandraker A, DeCaprio JA. Inhibition of Simian virus 40 large T antigen helicase activity by fluoroquinolones. Antiviral Therapy. 2007;12(1):1–6. [PubMed] [Google Scholar]
- 326.Gabardi S, Waikar SS, Martin S, et al. Evaluation of fluoroquinolones for the prevention of BK viremia after renal transplantation. Clinical Journal of the American Society of Nephrology. 2010;5(7):1298–1304. doi: 10.2215/CJN.08261109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Wojciechowski D, Chanda R, Chandran S, et al. Ciprofloxacin prophylaxis in kidney transplant recipients reduces BK virus infection at 3 months but not at 1 Year. Transplantation. 2012;94(11):1117–1123. doi: 10.1097/TP.0b013e31826ec74e. [DOI] [PubMed] [Google Scholar]
- 328.Miller AN, Glode A, Hogan KR, et al. Efficacy and safety of ciprofloxacin for prophylaxis of polyomavirus BK virus-associated hemorrhagic cystitis in allogeneic hematopoietic stem cell transplantation recipients. Biology of Blood and Marrow Transplantation. 2011;17(8):1176–1181. doi: 10.1016/j.bbmt.2010.12.700. [DOI] [PubMed] [Google Scholar]
- 329.Liacini A, Seamone ME, Muruve DA, Tibbles LA. Anti-BK virus mechanisms of sirolimus and leflunomide alone and in combination: toward a new therapy for BK virus infection. Transplantation. 2010;90(12):1450–1457. doi: 10.1097/TP.0b013e3182007be2. [DOI] [PubMed] [Google Scholar]
- 330.Sanchez Fructuoso AI, Calvo N, Perez-Flores I, et al. Mammalian target of rapamycin signal inhibitors could play a role in the treatment of BK polyomavirus nephritis in renal allograft recipients. Transplant Infectious Disease. 2011;13(6):584–591. doi: 10.1111/j.1399-3062.2011.00649.x. [DOI] [PubMed] [Google Scholar]
- 331.Hymes LC, Warshaw BL. Five-year experience using sirolimus-based, calcineurin inhibitor-free immunosuppression in pediatric renal transplantation. Pediatric Transplantation. 2011;15(4):437–441. doi: 10.1111/j.1399-3046.2011.01477.x. [DOI] [PubMed] [Google Scholar]