

An in vitro familial amyotrophic lateral sclerosis (FALS) model was established from human ESCs overexpressing either a wild-type or a mutant SOD1 (G93A) gene, and the phenotypes and survival of the spinal motor neurons were evaluated. This model is expected to help unravel the disease mechanisms involved in the development of FALS and also lead to potential drug discoveries based on the prevention of neurodegeneration.
Keywords: Embryonic stem cells, Experimental models, Neuron, Astrocytes
Abstract
The generation of amyotrophic lateral sclerosis (ALS) disease models is an important subject for investigating disease mechanisms and pharmaceutical applications. In transgenic mice, expression of a mutant form of superoxide dismutase 1 (SOD1) can lead to the development of ALS that closely mimics the familial type of ALS (FALS). Although SOD1 mutant mice show phenotypes similar to FALS, dissimilar drug responses and size differences limit their usefulness to study the disease mechanism(s) and identify potential therapeutic compounds. Development of an in vitro model system for ALS is expected to help in obtaining novel insights into disease mechanisms and discovery of therapeutics. We report the establishment of an in vitro FALS model from human embryonic stem cells overexpressing either a wild-type (WT) or a mutant SOD1 (G93A) gene and the evaluation of the phenotypes and survival of the spinal motor neurons (sMNs), which are the neurons affected in ALS patients. The in vitro FALS model that we developed mimics the in vivo human ALS disease in terms of the following: (a) selective degeneration of sMNs expressing the G93A SOD1 but not those expressing the WT gene; (b) susceptibility of G93A SOD1-derived sMNs to form ubiquitinated inclusions; (c) astrocyte-derived factor(s) in the selective degeneration of G93A SOD1 sMNs; and (d) cell-autonomous, as well as non-cell-autonomous, dependent sMN degeneration. Thus, this model is expected to help unravel the disease mechanisms involved in the development of FALS and also lead to potential drug discoveries based on the prevention of neurodegeneration.
Introduction
Amyotrophic lateral sclerosis (ALS) is a late-onset degenerative motor neuron disease affecting both upper and lower motor neurons [1]. Degeneration of motor neurons progressively leading to muscle wasting and ultimately to death is the hallmark of ALS [2]. Familial ALS (FALS), which accounts for approximately 20% of all ALS cases, is characterized by studied mutations in genes such as the superoxide dismutase 1 (SOD1), TDP-43, FUS, and ANG genes [3–6]. Neurodegeneration observed in ALS is affected by various factors, including generation of free radicals [7], glutamate-induced excitotoxicity, formation of protein aggregates overwhelming the proteasomal system [8, 9], mitochondrial dysfunction [10], and axonal transport perturbation [11]. Mutations in SOD1 (the copper/zinc-binding superoxide dismutase gene) can lead to FALS [12]. Several different single-amino acid substitutions within the SOD1 gene lead to a toxic gain of function, and in transgenic mice, expression of some SOD1 mutants leads to the development of ALS, which closely mimics the human disease [13–16]. Both cell-autonomous and non-cell-autonomous processes contribute to the neurodegeneration that is observed in ALS models [16–20]. Formation of protein-ubiquitin aggregates, a reduction in proteasomal function, and enhanced cell death are some of the characteristic features of ALS motor neurons [15, 21]. However, the precise mechanism(s) by which mutated SOD1 induces motor neuron degeneration remains unclear. Understanding the disease mechanism(s) involved in the progression of ALS and targeting of pathways that affect the disease are important for drug discovery, as well as therapeutic purposes.
The study of FALS using animal models mimicking the human disease condition is very informative and useful for investigating certain aspects of ALS, but its usefulness is limited since the biology of disease, including its progression and drug responses, is different between rodents and humans. Thus, it is beneficial to establish human cell-based disease models that might help in unraveling the disease mechanisms. Human pluripotent stem cells, with their ability to regenerate indefinitely and differentiate into almost any cell lineage, offer attractive options for the development of disease models. We have recently reported an efficient differentiation protocol to derive spinal motor neurons (sMNs) from human and primate embryonic stem cells (ESCs) [22]. In this report we describe the development of a human ESC (hESC)-based ALS disease model, overexpressing the mutant G93A SOD1 gene, that mimics the in vivo disease in terms of sMN degeneration. Two different sMN degeneration mechanisms were observed in our ALS disease model: a cell-autonomous mechanism and a non-cell-autonomous mechanism that is astrocyte-dependent. We also show the formation of ubiquitin inclusions in mutant SOD1-expressing motor neurons that in part may be responsible for sMN death. Thus, this model is expected to help unravel disease mechanisms involved in the development of FALS and also lead to potential drug discoveries based on the prevention of neurodegeneration in ALS.
Materials and Methods
Culture of hESCs
The procedure for maintenance of KhES-1 hESCs was essentially the same as described previously [23]. Briefly, hESCs were cultured on mitomycin C-treated mouse embryonic fibroblasts in primate ES medium (ReproCELL, Tokyo, http://www.reprocell.com/en) supplemented with 5 ng/ml fibroblast growth factor 2 (Wako Chemical, Osaka, Japan, http://www.wako-chem.co.jp/english). The hESC line was used in conformity with the Guidelines for Derivation and Utilization of Human Embryonic Stem Cells of the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan.
Establishment of SOD1-Overexpressing hESC Lines
The expression vector pEF1/myc-HIS (Invitrogen, Carlsbad, CA, http://www.invitrogen.com) was digested by EcoRV and NotI and then ligated with wild-type (WT) or G93A SOD1 cDNA. The resulting SOD1 expression vectors were digested with PvuI and then introduced into KhES-1 hESCs by electroporation. G418-resistant clones were picked and the expression of endogenous and exogenous SOD1 was confirmed by reverse transcription-polymerase chain reaction (RT-PCR) using the following primers: SOD1 forward, GTCTGAGGCCCCTTAACTCA, and reverse, TCTGGCAAAATACAGGTCATTG for endogenous SOD1, and EF3UT forward, AGGTCACCCATTCGAACAAA, and reverse, ACAGTGGGAGTGGCACCTT for exogenous SOD1 expression. SOD enzyme activity was measured by SOD assay kit-WST (Dojindo Molecular Technologies Inc., Tokyo, http://www.dojindo.com). Undifferentiated hESC clones with high SOD enzyme activity were induced to neural differentiation with Noggin [24]. The expression of exogenous SOD1 in differentiated neural cells was confirmed by RT-PCR.
Motor Neuron and Astrocyte Differentiation of hESCs
Spinal motor neuron differentiation and enrichment protocols were described in an earlier report [22]. Briefly, neural stem cells from either hESC-derived neurospheres or Noggin-induced neural rosettes were dissociated using Accutase (Innovative Cell Technologies, San Diego, http://www.innovativecelltech.com). Dissociated cells were grown on poly-l-lysine-, laminin-, and fibronectin-coated culture dishes. The astrocyte differentiation method was a modified mouse primary astrocyte differentiation protocol [25]. Briefly, neurospheres of fewer than four passages or more than five passages were cultured in N2B27 medium with or without 80 ng/ml bone morphogenetic protein 4 (R&D Systems Inc., Minneapolis, http://www.rndsystems.com) and 80 ng/ml leukemia inhibitory factor (Millipore, Billerica, MA, http://www.millipore.com), respectively, for 7 days before neurospheres were dissociated. Typically, the efficiency of astrocyte differentiation was approximately 80%. All schematic procedures of neural differentiation are shown in supplemental online Figure 1.
Immunocytochemistry
Cells were fixed in freshly prepared 4% paraformaldehyde for 10 minutes at room temperature and were permeabilized by 0.5% Triton-X (Wako Chemical) for 5 minutes at room temperature. Primary antibodies used in this study included polyclonal antibodies against Nestin (1:200; Chemicon, Temecula, CA, http://www.chemicon.com), glial fibrillary acidic protein (GFAP) (1:400; DAKO, Glostrup, Denmark, http://www.dako.com), S100-β (1:200; Chemicon), and ubiquitin (1:200; Chemicon) or monoclonal antibodies against Oct4 (1:200; Santa Cruz Biotechnology Inc., Santa Cruz, CA, http://www.scbt.com) and MNR2 (HB9, 1:5; Developmental Studies Hybridoma Bank, Iowa City, IA, http://www.uiowa.edu/∼dshbwww). As secondary antibodies, goat anti-mouse immunoglobulin G Alexa Fluor 488 and 568 and goat anti-rabbit Alexa Fluor 488 and 568 (all at 1:1,000; Molecular Probes, Eugene, OR, http://probes.invitrogen.com) were used. Cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich, St. Louis, http://www.sigmaaldrich.com) for visualization of all nuclei. Image acquisition was performed using an Olympus IX71 epifluorescence inverted microscope with appropriate filter sets (Olympus, Tokyo, http://www.olympus-global.com) using single-channel acquisition on an Olympus DP70 charge-coupled device camera with DP software version 2.11.83.
Terminal Deoxynucleotidyl Transferase dUTP Nick-End Labeling Assay
Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) staining was performed using the In Situ Cell Death Detection Kit, Fluorescein (Roche Applied Science, Mannheim, Germany, https://www.roche-applied-science.com) following the manufacturer's instructions. Briefly, after staining with HB9 antibody and fluorescent-labeled secondary antibodies, the immunostained cells were treated with the reagents for the TUNEL assay at 37°C for 1 hour. Cells were washed three times with PBS and counterstained with DAPI.
Treatment of hESC-Derived sMNs with Conditioned Medium of hESC-Derived Astrocytes
hESC-derived astrocyte-conditioned medium (astrocyte-CM) was prepared by culturing hESC-derived astrocytes in N2B27 medium for 7 days. hESC-derived sMN culture was treated with astrocyte-CM and N2B27 medium (1:1 mixture) for 7 days. Apoptotic cell death in astrocyte-CM-treated sMNs was analyzed by HB9 immunostaining and TUNEL assay.
Results
Generation of SOD1-Overexpressing hESCs
To establish the cell lines with high SOD activity, the EF1α promoter-driven WT or G93A SOD1 expression vectors were integrated into KhES-1 hESC genome. After G418 selection, the expression of exogenous SOD1 in a total of 84 clones was confirmed by RT-PCR (data not shown). Clones with higher expression level were selected, and then SOD enzyme activity was measured. All SOD1-overexpressing hESCs showed enzyme activity that was at least three- to eightfold higher than that of the parental hESCs (Fig. 1A). Also, a positive correlation between the gene expression level and SOD enzyme activity has been verified (data not shown). Next, the expression of exogenous SOD1 was examined in differentiated neural cells because gene silencing easily occurred in differentiated cells even from undifferentiated hESCs with high expression level. Some of the clones were induced to undergo neural differentiation, and RT-PCR was carried out. Although exogenous SOD1 expression was suppressed in several clones, it was retained in some clones (Fig. 1B). Of them, two clones (105, expressing WT SOD1, and D06, expressing G93A SOD1) were selected for further study because the SOD1 expression levels were similar to each other in both undifferentiated and differentiated cells (Fig. 1A, 1B), although Southern analysis indicated that one and two copies of the SOD1 expression vector were integrated in the genome of clones 105 and D06, respectively (supplemental online Fig. 2).
Figure 1.
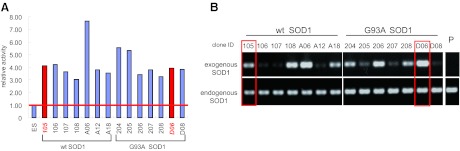
Establishment of SOD1-overexpressing human embryonic stem cell lines. (A): Relative SOD activity in undifferentiated human embryonic stem cells (hESCs) ectopically expressing wild-type SOD1 or G93A SOD1. (B): Reverse transcription-polymerase chain reaction analysis of exogenous and endogenous SOD1 expression in hESC-derived neurons. Red bars (A) and open rectangles (B) show clones selected for further analysis. Abbreviations: ID, identification number; P, parent hESC line KhES-1; SOD, superoxide dismutase; wt, wild-type.
SOD1 Overexpression Does Not Affect Neural Differentiation of hESCs
Using the sMN differentiation protocol (supplemental online Fig. 1) [22], hESC-derived neural progenitor cells expressing either a WT SOD1 gene or a G93A mutant SOD1 gene were characterized. The numbers of WT and mutant SOD1-expressing hESCs were similar at the end of both the neural induction stage and the neural progenitor formation stage of neural differentiation (data not shown), suggesting that cell proliferation during differentiation was not different between transgenic cell lines. Then the expression of Nestin, a neural progenitor marker, was evaluated during neural differentiation. WT SOD1- or G93A SOD1-expressing cells stained strongly for Nestin, similar to parental hESCs, suggesting that overexpression of SOD1 does not affect neural progenitor formation (Fig. 2A). We then examined whether ectopic expression of SOD1 affects cell viability by measuring TUNEL-positive nuclei from WT and mutant SOD1-expressing cells that were differentiated into neural progenitors. Although more than 75% of the cells stained positively for Nestin, only a small fraction of cells were TUNEL-positive. Moreover, there were very few Nestin-TUNEL double-positive cells (Fig. 2B). This suggests that SOD1 expression does not significantly affect the cell viability of neural progenitors.
Figure 2.

Phenotypic analysis of SOD1-overexpressing human ESC-derived neural progenitor cells. (A): Immunocytochemical staining by Nestin (neural marker; green) and Oct-4 (ESC marker; red) of KhES-1, WT SOD1, and G93A SOD1 cells at the end of the neural progenitor formation stage. (B): Quantitation of TUNEL-positive, Nestin-positive, and double-positive cell ratios. Abbreviations: DAPI, 4′,6-diamidino-2-phenylindole; SOD, superoxide dismutase; TUNEL, terminal deoxynucleotidyl transferase dUTP nick-end labeling; WT, wild-type.
Mutant SOD1 Overexpression Leads to Spinal Motor Neuron-Specific Cell Death
Using a combination of all-trans retinoic acid and SAG, a Sonic hedgehog agonist (supplemental online Fig. 1), neural progenitors were terminally differentiated into sMNs. Approximately 20%–30% sMNs were typically obtained as HB9-positive postmitotic neurons [22]. Morphologically, G93A SOD1-expressing neurons looked much less healthy than their counterpart neurons expressing the WT SOD1 gene (Fig. 3A). Next, the viability of WT and mutant SOD1-expressing HB9-positive postmitotic neurons was evaluated. The sMN differentiation efficiency was not different between WT SOD1- and G93A SOD1-expressing hESCs (WT, 26.8 ± 1.8%; G98A, 24.9 ± 1.9%; Fig. 3). Expression of G93A SOD1 in sMNs significantly enhanced their apoptosis compared with WT SOD1-expressing neurons (Fig. 3B). Apoptotic cell death as assessed by TUNEL staining was significantly greater in G93A SOD1 neurons (Fig. 3C). Also, enhanced cell death in G93A SOD1-expressing cells was due to the specific death of sMN (Fig. 3D) but not other cell types, such as the astrocytes (glial fibrillary acidic protein-positive cells), which were unaffected by SOD1 expression (Fig. 3E). These results collectively indicate a selective degeneration and death of HB9-positive postmitotic sMNs, as is observed in in vivo mouse SOD1-ALS models [19].
Figure 3.

Phenotypic analysis of SOD1-overexpressing human ESC (hESC)-derived spinal motor neurons. (A): Morphologically unhealthy cells were observed in spinal motor neurons (sMNs) derived from G93A SOD1-overexpressing hESCs, whereas WT SOD1 hESC-derived sMNs showed normal morphology. (B): Immunocytochemical staining of cells treated with or without sMN inducers (ATRA and SAG). Both TUNEL (green) and HB9 (red)-positive sMNs were observed in ATRA/SAG-treated G93A SOD1-overexpressing hESCs. (C): Total TUNEL-positive cell ratio. (D): HB9/TUNEL double-positive cell ratio. (E): GFAP/TUNEL double-positive cell ratio. These data indicated sMN-specific cell death in G93A SOD1 hESC-derived sMNs. Approximately 900 cells were used for calculating the ratio of immunopositive cells (TUNEL, HB9, and GFAP). Mean ± SEM; n = 9; Steel-Dwass test; ∗∗, p < .01. Abbreviations: ATRA, all-trans retinoic acid; DAPI, 4′,6-diamidino-2-phenylindole; GFAP, glial fibrillary acidic protein; K1, KhES-1; SOD, superoxide dismutase; TUNEL, terminal deoxynucleotidyl transferase dUTP nick-end labeling; WT, wild-type.
Mutant SOD1-Overexpressing hESC-Derived Astrocytes Secrete Factors That Are Toxic to sMNs
Recent reports indicated that factors secreted from astrocytes are involved in motor neuron death in SOD1-ALS models [17–20]. Therefore, to ascertain the involvement of astrocytes in cell death in our SOD1-ALS model, purified WT or G93A SOD1-expressing sMNs (up to 80% enriched) were exposed to CM obtained from WT or G93A SOD1-expressing astrocytes (approximately 80% differentiation efficiency) for 7 days (Fig. 4A). Cell death analysis was performed using the TUNEL assay (Fig. 4B). Astrocyte-CM did not affect the percentage of HB9-positive WT SOD1-expressing sMNs during CM treatment, although G93A SOD1 astrocyte-CM-treated G93A SOD1 sMNs were reduced slightly during 7 days of culture (Fig. 4C). However, apoptotic sMN death was detected in the population of WT SOD1-expressing sMNs, which were cultured in G93A SOD1 astrocyte-CM, much more than in the WT SOD1 sMN population treated with WT SOD1 astrocyte-CM (Fig. 4D, bar 1 vs. bar 3). This result suggests that hESC-derived astrocytes secrete factors toxic to sMNs. In addition, significantly more G93A SOD1-expressing sMNs died compared with WT SOD1 sMNs regardless of the source of astrocyte-CM, suggesting that mutant SOD1 expression in sMNs results in their cell death (Fig. 4D, bar 2 vs. bar 4). Together these results suggest the presence of two different mechanisms involved in reduced survival of sMNs: (a) an intrinsic sMN-dependent, cell-autonomous mechanism, and (b) an astrocyte-dependent (i.e., non-cell-autonomous) cell survival mechanism.
Figure 4.

G93A SOD1-overexpressing human ESC (hESC)-derived astrocyte supernatant-induced sMN death. (A): Experimental procedures to analyze the effect of astrocyte-CM on sMN cell death. (B): Immunocytochemical staining of astrocyte-CM-treated sMNs from either WT SOD1 or G93A SOD1-overexpressing hESCs. (C, D): Quantitative analysis of TUNEL-positive dead cells (C) and HB9/TUNEL-double-positive dead sMNs (D). Mean ± SEM; n = 9; Steel-Dwass test; ∗∗, p < .01. Abbreviations: AS CM, astrocyte-conditioned medium; CM, conditioned medium; DAPI, 4′,6-diamidino-2-phenylindole; DIV, days in vitro; sMN, spinal motor neuron; SOD, superoxide dismutase; TUNEL, terminal deoxynucleotidyl transferase dUTP nick-end labeling; WT, wild-type.
Mutant SOD1 Induces Abnormal Ubiquitin Inclusion Body Formation in sMN
Ubiquitinated and SOD1-immunopositive inclusions are the neuropathological features of SOD1-related FALS [26, 27]. Therefore, we examined whether ubiquitin inclusion bodies are formed in sMNs derived from the hESC-based ALS model developed in this study. Both ubiquitin and HB9-positive cells were detected in the sMNs derived from parental KhES-1 (K1) and WT SOD1- or SOD1 G93A-expressing hESCs (Fig. 5A). A smooth staining pattern of ubiquitin was detected in sMNs from K1 and WT SOD1, whereas both smooth and rough staining was observed in sMNs expressing G93A SOD1 (Fig. 5A). These abnormal rough staining patterns were counted as abnormal ubiquitin inclusions (Fig. 5B). Although there was no difference in the efficiency of sMN differentiation among K1, WT SOD1, and G93A SOD1, only G93A SOD1 induced the formation of abnormal ubiquitin inclusions in approximately half of the HB9-positive sMNs (Fig. 5B), indicating that this abnormal ubiquitin staining is a unique phenotype of mutant SOD1-expressing sMNs and might be equivalent to ubiquitinated inclusions in sMNs of ALS patients.
Figure 5.

Ubiquitin staining of SOD1 human ESC-derived sMNs. (A): Immunocytochemical staining of ubiquitin (green) and HB9 (red). An abnormal staining pattern was observed in G93A SOD1-expressing sMNs. Arrowheads show rough staining of ubiquitin or HB9, which might be abnormal ubiquitin inclusions. (B): Quantitative analysis of cells with ubiquitin inclusions (green bars), HB9-positive sMNs (purple bars), and sMNs with ubiquitin inclusions (yellow bars). Mean ± SEM; n = 9; Steel-Dwass test; ∗∗, p < .01. Abbreviations: sMN, spinal motor neuron; SOD, superoxide dismutase; Ub, ubiquitin; WT, wild-type.
Discussion
In this study, we developed an in vitro FALS model to investigate the ALS disease mechanism and perform drug discovery research. For performing these investigations using a cellular model, an adequate number of sMNs, the main target cells for degeneration in ALS, need to be generated. We recently reported a differentiation protocol that efficiently generates functional sMNs from human and primate ESCs [22]. Using a modification of the above protocol, hESCs expressing either an exogenous WT or G93A SOD1 gene were efficiently differentiated into sMNs and astrocytes. Thus, our differentiation protocol results in the generation of large numbers of sMNs and astrocytes.
A specific death/degeneration of sMNs was observed in the in vitro FALS model that we developed in this study. Interestingly, the expression of either the WT or mutant SOD1 gene did not affect the differentiation process, morphological characteristics, or cell viability until the formation of postmitotic sMNs, suggesting that a toxic gain of function of mutant SOD1 might affect only the terminally differentiated cells derived from hESCs. Moreover, sMN degeneration or death occurred both in the absence and in the presence of astrocytes in the culture, implicating cell-autonomous dependent and non-cell-autonomous dependent sMN death, respectively. Similar results showing cell-autonomous sMN death were recently reported in hESC-derived sMNs, wherein liposome transfection of mutant SOD1 expression vectors resulted in sMN cell death [28]. On the other hand, non-cell-autonomous sMN killing by astrocytes was reported to be important in ALS pathology. Soluble astrocyte-derived factors such as reactive oxygen species and prostaglandin D2 were proposed to mediate the non-cell-autonomous killing of sMNs [18–20]. We also observed that sMNs derived from either WT or G93A SOD1-expressing hESCs were susceptible to death when cultured in conditioned medium derived from mutant SOD1-expressing astrocytes. This result pointed out that hESC-derived astrocytes secreted one or more soluble factors involved in sMN cell death in our ALS model. Earlier reports describing the interaction of hESC-derived sMNs with astrocytes used primary glial cells derived from transgenic mice expressing the mutant SOD1 [18] or primary human astrocytes transfected with viral vectors expressing a mutant SOD1 [20]. Hence, ours is the first study wherein both sMNs and astrocytes are derived from the same hESCs stably expressing either the WT or the G93A SOD1 gene, thus enabling an accurate study of both cell-autonomous and the non-cell-autonomous dependent pathology in ALS.
Neuronal cell death and the formation of inclusion bodies, which are ubiquitinated protein aggregates, are two major hallmarks of ALS. In this study, cell death and abnormal ubiquitin staining were observed frequently and selectively in hESC-derived sMNs expressing G93A SOD1. These cellular phenotypes might be equivalent to the sMN cell death and ubiquitinated inclusions observed in ALS patients. Therefore, these phenotypes detected in the hESC-derived FALS model might be useful for drug screening and discovery.
Conclusion
We have developed a hESC-based ALS disease model that mimics some aspects of in vivo ALS disease. This model is very useful to study the complex interactions between sMNs and surrounding astrocytes along with investigating the autonomous sMN degeneration due to the expression of mutant SOD1. A reduction in the formation of ubiquitin inclusions and the prevention of sMN death are two potential targets to study the disease mechanism(s) and develop small molecules that modulate these processes as potential therapeutic drugs.
Acknowledgments
This research was supported in part by a grant from the New Energy and Industrial Technology Development Organization (P05010, to N.N.) and in part by a Grant-in-Aid for Scientific Research of Japan Society for the Promotion of Science (23500446, to K.A.). The Institute for Integrated Cell-Material Sciences is supported by World Premier International Research Center Initiative, MEXT, Japan.
Author Contributions
T.W.: conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing; S.K.G.: collection and/or assembly of data, data analysis and interpretation, manuscript writing; N.T.: collection and/or assembly of data; H.I. and R.T.: provision of study material or patients; N.N.: conception and design, final approval of manuscript; K.A.: conception and design, manuscript writing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicate no potential conflicts of interest.
References
- 1.Ozdinler PH, Benn S, Yamamoto TH, et al. Corticospinal motor neurons and related subcerebral projection neurons undergo early and specific neurodegeneration in hSOD1G93A transgenic ALS mice. J Neurosci. 2011;31:4166–4177. doi: 10.1523/JNEUROSCI.4184-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kiernan MC, Vucic S, Cheah BC, et al. Amyotrophic lateral sclerosis. Lancet. 2011;377:942–955. doi: 10.1016/S0140-6736(10)61156-7. [DOI] [PubMed] [Google Scholar]
- 3.Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 4.Vance C, Rogelj B, Hortobágyi T, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wegorzewska I, Bell S, Cairns NJ, et al. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci USA. 2009;106:18809–18814. doi: 10.1073/pnas.0908767106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greenway MJ, Alexander MD, Ennis S, et al. A novel candidate region for ALS on chromosome 14q11.2. Neurology. 2004;63:1936–1938. doi: 10.1212/01.wnl.0000144344.39103.f6. [DOI] [PubMed] [Google Scholar]
- 7.Bruijn LI, Beal MF, Becher MW, et al. Elevated free nitrotyrosine levels, but not protein-bound nitrotyrosine or hydroxyl radicals, throughout amyotrophic lateral sclerosis (ALS)-like disease implicate tyrosine nitration as an aberrant in vivo property of one familial ALS-linked superoxide dismutase 1 mutant. Proc Natl Acad Sci USA. 1997;94:7606–7611. doi: 10.1073/pnas.94.14.7606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bruijn LI, Houseweart MK, Kato S, et al. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998;281:1851–1854. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- 9.Zetterström P, Stewart HG, Bergemalm D, et al. Soluble misfolded subfractions of mutant superoxide dismutase-1s are enriched in spinal cords throughout life in murine ALS models. Proc Natl Acad Sci USA. 2007;104:14157–14162. doi: 10.1073/pnas.0700477104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kong J, Xu Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci. 1998;18:3241–3250. doi: 10.1523/JNEUROSCI.18-09-03241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Williamson TL, Cleveland DW. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci. 1999;2:50–56. doi: 10.1038/4553. [DOI] [PubMed] [Google Scholar]
- 12.Rosen DR, Siddique T, Patterson D, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 13.Nagai M, Aoki M, Miyoshi I, et al. Rats expressing human cytosolic copper-zinc superoxide dismutase transgenes with amyotrophic lateral sclerosis: Associated mutations develop motor neuron disease. J Neurosci. 2001;21:9246–9254. doi: 10.1523/JNEUROSCI.21-23-09246.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gurney ME, Pu H, Chiu AY, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 15.Cheroni C, Marino M, Tortarolo M, et al. Functional alterations of the ubiquitin-proteasome system in motor neurons of a mouse model of familial amyotrophic lateral sclerosis. Hum Mol Genet. 2009;18:82–96. doi: 10.1093/hmg/ddn319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamanaka K, Boillee S, Roberts EA, et al. Mutant SOD1 in cell types other than motor neurons and oligodendrocytes accelerates onset of disease in ALS mice. Proc Natl Acad Sci USA. 2008;105:7594–7599. doi: 10.1073/pnas.0802556105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Di Giorgio FP, Carrasco MA, Siao MC, et al. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat Neurosci. 2007;10:608–614. doi: 10.1038/nn1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Di Giorgio FP, Boulting GL, Bobrowicz S, et al. Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell. 2008;3:637–648. doi: 10.1016/j.stem.2008.09.017. [DOI] [PubMed] [Google Scholar]
- 19.Nagai M, Re DB, Nagata T, et al. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10:615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marchetto MC, Muotri AR, Mu Y, et al. Non-cell-autonomous effect of human SOD1 G37R astrocytes on motor neurons derived from human embryonic stem cells. Cell Stem Cell. 2008;3:649–657. doi: 10.1016/j.stem.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 21.Urushitani M, Kurisu J, Tsukita K, et al. Proteasomal inhibition by misfolded mutant superoxide dismutase 1 induces selective motor neuron death in familial amyotrophic lateral sclerosis. J Neurochem. 2002;83:1030–1042. doi: 10.1046/j.1471-4159.2002.01211.x. [DOI] [PubMed] [Google Scholar]
- 22.Wada T, Honda M, Minami I, et al. Highly efficient differentiation and enrichment of spinal motor neurons derived from human and monkey embryonic stem cells. PLoS One. 2009;4:e6722. doi: 10.1371/journal.pone.0006722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suemori H, Yasuchika K, Hasegawa K, et al. Efficient establishment of human embryonic stem cell lines and long-term maintenance with stable karyotype by enzymatic bulk passage. Biochem Biophys Res Commun. 2006;345:926–932. doi: 10.1016/j.bbrc.2006.04.135. [DOI] [PubMed] [Google Scholar]
- 24.Sakurai K, Shimoji M, Tahimic CG, et al. Efficient integration of transgenes into a defined locus in human embryonic stem cells. Nucleic Acids Res. 2010;38:e96. doi: 10.1093/nar/gkp1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Namihira M, Nakashima K, Taga T. Developmental stage dependent regulation of DNA methylation and chromatin modification in a immature astrocyte specific gene promoter. FEBS Lett. 2004;572:184–188. doi: 10.1016/j.febslet.2004.07.029. [DOI] [PubMed] [Google Scholar]
- 26.Tu PH, Raju P, Robinson KA, et al. Transgenic mice carrying a human mutant superoxide dismutase transgene develop neuronal cytoskeletal pathology resembling human amyotrophic lateral sclerosis lesions. Proc Natl Acad Sci USA. 1996;93:3155–3160. doi: 10.1073/pnas.93.7.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kato S, Horiuchi S, Liu J, et al. Advanced glycation endproduct-modified superoxide dismutase-1 (SOD1)-positive inclusions are common to familial amyotrophic lateral sclerosis patients with SOD1 gene mutations and transgenic mice expressing human SOD1 with a G85R mutation. Acta Neuropathol. 2000;100:490–505. doi: 10.1007/s004010000226. [DOI] [PubMed] [Google Scholar]
- 28.Karumbayaram S, Kelly TK, Paucar AA, et al. Human embryonic stem cell-derived motor neurons expressing SOD1 mutants exhibit typical signs of motor neuron degeneration linked to ALS. Dis Model Mech. 2009;2:189–195. doi: 10.1242/dmm.002113. [DOI] [PMC free article] [PubMed] [Google Scholar]