

Abstract
Background
Y chromosomes are responsible for the initiation of male development, male fertility, and other male-related functions in diverse species. However, Y genes are rarely characterized outside a few model species due to the arduous nature of studying the repeat-rich Y.
Results
The chromosome quotient (CQ) is a novel approach to systematically discover Y chromosome genes. In the CQ method, genomic DNA from males and females is sequenced independently and aligned to candidate reference sequences. The female to male ratio of the number of alignments to a reference sequence, a parameter called the chromosome quotient (CQ), is used to determine whether the sequence is Y-linked. Using the CQ method, we successfully identified known Y sequences from Homo sapiens and Drosophila melanogaster. The CQ method facilitated the discovery of Y chromosome sequences from the malaria mosquitoes Anopheles stephensi and An. gambiae. Comparisons to transcriptome sequence data with blastn led to the discovery of six Anopheles Y genes, three from each species. All six genes are expressed in the early embryo. Two of the three An. stephensi Y genes were recently acquired from the autosomes or the X. Although An. stephensi and An. gambiae belong to the same subgenus, we found no evidence of Y genes shared between the species.
Conclusions
The CQ method can reliably identify Y chromosome sequences using the ratio of alignments from male and female sequence data. The CQ method is widely applicable to species with fragmented genome assemblies produced from next-generation sequencing data. Analysis of the six Y genes characterized in this study indicates rapid Y chromosome evolution between An. stephensi and An. gambiae. The Anopheles Y genes discovered by the CQ method provide unique markers for population and phylogenetic analysis, and opportunities for novel mosquito control measures through the manipulation of sexual dimorphism and fertility.
Background
Originating independently in diverse taxa, the XX/XY sex determination system is a striking example of convergent evolution [1-5]. In many species with Y chromosomes, a Y-linked dominant male-determining gene is hypothesized to initiate male sexual differentiation [2,6,7]. However, Y-linked dominant male-determining genes have eluded characterization with only a few exceptions. In therian mammals the dominant male-determining factor, SRY, is one of the few Y-linked genes retained after the degeneration of the Y chromosome [8-11]. In the medaka fish Oryzias latipes the Y-linked gene DMY functions as the dominant male determining gene [7].
Numerous insect species determine sex through the XX/XY sex-determination system, although insect Y chromosomes likely originated independently multiple times from diverse origins [1-3,12]. A notable exception is that while Drosophila species have Y chromosomes, sex is not determined by a gene on the Y, but instead the X-to-autosome ratio [13]. Although no Y-linked sex-determining genes have been characterized in insects, Y-linked sex-determining genes likely exist in Anopheles mosquitoes, the housefly Musca domestica, the Mediterranean fruit fly Ceratitis capitata, and possibly many more species with Y chromosomes [3,14,15]. Most insect Y chromosomes remain entirely unexplored.
Over time, male-beneficial genes may accumulate on the Y chromosome by gene duplications [16]. For example, all known protein-coding genes on the D. melanogaster Y chromosome originated from autosome-to-Y duplications [16-21]. Due to their exclusive presence in males, Y chromosomes may also serve as a sanctuary for genes advantageous to males, and deleterious to females [22].
Y chromosome genes have eluded all but the most determined attempts at characterization due to the heterochromatic and repetitive nature of the Y [19,23]. Using bacterial artificial chromosomes (BACs) and an iterative mapping and sequencing strategy, the sequence of the euchromatic region of the Y chromosome was obtained for humans and two other primates [9,10]. The iterative mapping strategy used in primates has not been implemented to sequence other Y chromosomes due to the cost associated with the technique. In insects, the sequences of Y genes have only been characterized in a few Drosophila species. The Drosophila Y gene sequences were found by a strategy, developed by Carvalho and colleagues, based on the comparison of sequences not anchored to chromosomes to known protein sequences [17,19]. This method may be difficult to implement in species without a reference genome where most of the sequences are anchored to chromosomes, and it failed to identify any Y genes in the African malaria mosquito An. gambiae[24].
In this study, we introduce the chromosome quotient (CQ) method, a novel approach to systematically discover Y genes. The CQ method takes advantage of high-throughput genome and transcriptome sequencing data and does not require a reference genome where most of the sequences are anchored to chromosomes. We used the CQ method to find Y sequences from two important malaria mosquitoes, An. stephensi and An. gambiae, members of the same subgenus Cellia[25]. Comparison of the Y sequences to transcriptome sequence data led to the discovery and characterization of six Anopheles Y genes, three from each species. Evolutionary analysis strongly suggests rapid Y chromosome evolution in Anopheles mosquitoes.
Results
The chromosome quotient (CQ) method
We developed the CQ method based on the following observations. Y chromosomes are present only in males. Therefore, sequences unique to the Y should be present in male sequence data and absent from female sequence data. Using this principal, we developed a method to identify Y sequences by comparing separate male and female sequence data to a reference genome. Unique Y sequences can be identified because they are only present in the male sequence data. However, searching for sequences exclusive to the male sequence data yielded only a few short sequences because most Y sequences appear to be at least partially repetitive. We realized that if the reference genome was fragmented into smaller pieces by removing repeats, more sequences exclusive to the male sequence data could be identified.
Y chromosomes appear to be primarily composed of repetitive sequences that are not exclusive to the Y. Since most Y-repeats have closely-related sequences on the autosome or X, they may appear be present in both the female and male sequence data. Recent duplications of autosome or X sequences to the Y may also cause Y sequences to appear to be present in both the male and female sequence data. We reduced the interference from repetitive sequences by using very strict alignment criteria. We require zero mismatches over the entire length of the reads from the male and female sequence data. We increased the number of Y chromosome sequences identified by allowing a few alignments from the female sequence data, as long as there were many more alignments from the male sequence data.
To define the number of alignments allowed from female data we use the ratio of female to male alignments, a parameter we call the chromosome quotient (CQ). For a given sequence Si, CQ(Si) = F(Si)/M(Si), where F(Si) is the number of alignments from female sequence data to Si, and M(Si) is the number of alignments from male sequence data to Si. The method by which chromosome quotients are calculated is found in the additional files (Additional file 1: Figure S1).
The chromosome quotient allows for the differentiation of Y sequences from autosome and X sequences (Additional file 2: Figure S2). Males and females share the same complement of autosomes, so autosomal sequences are present in both male and female sequence data in roughly the same quantities. Therefore, autosomal sequences have chromosome quotients distributed around one. Females have two X chromosomes while males have only one, so X chromosome sequences are present in female sequence data roughly twice as frequently as in male sequence data. Therefore, X chromosome sequences have chromosome quotients distributed around two. Unique Y sequences are present only in male sequence data, and therefore have chromosome quotients of zero. Repetitive Y sequences are present in both the male and female sequence data and have chromosome quotients greater than zero but less than the chromosome quotients of autosomal sequences. We set a threshold of CQ = 0.3 to differentiate Y sequences from autosome and X sequences.
Using the CQ method, a sequence is classified as Y-linked if it has more than 3.33 times as many alignments from male data than from female data, and therefore a CQ less than 0.3. In case the coverage of the male and female sequence data differs, chromosome quotients are normalized to the median chromosome quotient of known autosomal sequences.
Y chromosome sequences have distinctive chromosome quotients
To validate that the chromosome quotients of Y sequences are distinctive from those of autosome and X sequences, the CQ method was tested in H. sapiens and D. melanogaster. Autosome, X, and Y sequences were downloaded for both H. sapiens and D. melanogaster. The repetitive sequences indicated by RepeatMasker were removed, fragmenting the genomes into many smaller pieces. Male and female sequence data was located for both species. The chromosome quotients of the fragmented autosome, X, and Y sequences were calculated using the separate male and female sequence data (Figure 1, Table 1).
Figure 1.

Y chromosome sequences have distinctive chromosome quotients. The distribution of chromosome quotients calculated for autosome, and X sequences from H. sapiens, D. melanogaster, An. stephensi, and An. gambiae, the distribution of chromosome quotients for Y sequences from H. sapiens, and D. melanogaster, the distribution of chromosome quotients from predicted Y sequences from An. stephensi, and the distribution of chromosome quotients from known and predicted Y sequences from An. gambiae. The predicted Y chromosome sequences from An. stephensi originated from the contigs of the 454 genome assembly. The predicted Y chromosome sequences from An. gambiae originated from the UNKN sequences and our own Illumina assembly.
Table 1.
The number of sequences and the normalized CQs of the positive control data
| Data set | Autosomal sequences | Normalized median CQ | X sequences | Normalized median CQ | Y sequences | Normalized median CQ |
|---|---|---|---|---|---|---|
|
H. sapiens |
581,737 |
1.00 |
39,844 |
1.85 |
7,934 |
0.00 |
| D. melanogaster | 48,870 | 1.00 | 18,011 | 1.96 | 139 | 0.025 |
The number of sequences and the normalized median chromosome quotients (CQs) of the positive control data sets illustrate that autosomal sequences have chromosome quotients distributed around one, X sequences have chromosome quotients distributed around two, and Y sequences have chromosome quotients near zero. The distributions of chromosome quotients from different chromosomes are shown in Figure 1.
In both species, autosomal sequences have chromosome quotients distributed around one, X sequences have chromosome quotients distributed around two, and most Y sequences have chromosome quotients near zero. The median chromosome quotients of autosome and X sequences are significantly greater than the median chromosome quotients of Y sequences (Mann Whitney U Test p = 0 for X-to-Y and auto-to-Y comparisons in both species). Thus, Y sequences can be identified by their distinctive chromosome quotients. The interval of chromosome quotients (0.0, 0.3) captures more than 67 percent of the known Y sequences with a rate of false positives less than 2.5 percent in H. sapiens and D. melanogaster (Table 2). Therefore, we chose this threshold for the discovery of Y chromosome sequences in the mosquitoes An. stephensi and An. gambiae.
Table 2.
The false positive and false negative rates of the CQ method
| Data set | Total autosome and X sequences | Autosome or X sequences with CQ < 0.3 | False positive rate | Total Y sequences | Y sequences with CQ <0.3 | False negative rate |
|---|---|---|---|---|---|---|
|
H. sapiens |
621,581 |
135 |
2.44% |
7,934 |
5,408 |
31.8% |
| D. melanogaster | 66,881 | 2 | 1.85% | 139 | 106 | 23.7% |
The false positive rate is highly dependent on the CQ cut-off below which a sequence is classified as Y-linked. In this study, we used a CQ cut-off of 0.3 as it minimized false positives while keeping false negatives relatively low. There is flexibility in selecting the CQ cut-off according to the focus of the study and the scale of downstream verification that the investigators plan to perform.
Discovery of novel Y sequences in Anopheles mosquitoes
We used the CQ method to discover novel Y sequences in two malaria mosquitoes. In this study, we consider a sequence to be Y-linked if it has a chromosome quotient less than 0.3, meaning it has more than 3.33 times more alignments from male data than from female data.
We first searched for Y sequences in the Asian malaria mosquito An. stephensi, a species with a draft genome generated by 454 sequencing [GenBank: ALPR00000000]. We performed Illumina sequencing on male and female An. stephensi genomic DNA [SRA: SRP013838]. Chromosome quotients were first calculated for all known autosome and X contigs in the An. stephensi genome (Figure 1). All of the autosome and X sequences had chromosome quotients greater than 0.3. Chromosome quotients were then calculated for 113,570 contigs from the An. stephensi genome. We identified 317 candidate Y sequences with chromosome quotients less than 0.3, encompassing more than 130,000 bases (Figure 1). Five of these sequences were tested for male-specific amplification with PCR on male and virgin-female genomic DNA (Table 3). All five sequences amplified a product exclusively in male genomic DNA (Figure 2).
Table 3.
The chromosome quotients of sequences tested for Y-linkage with PCR
| Sequence name | Number of female alignments | Number of male alignments | Normalized chromosome quotient |
|---|---|---|---|
|
sYG1 |
1 |
553 |
0.002 |
|
sYG2 |
0 |
29 |
0.000 |
|
sYG3 |
1 |
158 |
0.005 |
|
gYG1 |
3 |
1631 |
0.002 |
|
gYG2 |
2 |
692 |
0.003 |
|
gYG3 |
19 |
340 |
0.056 |
|
An. stephensi Y sequence 1 |
0 |
211 |
0.000 |
|
An. stephensi Y sequence 2 |
1 |
305 |
0.003 |
|
An. stephensi Y sequence 3 |
4 |
148 |
0.023 |
|
An. stephensi Y sequence 4 |
10 |
335 |
0.026 |
|
An. stephensi Y sequence 5 |
2 |
61 |
0.029 |
| An. gambiae Y sequence 1 | 13 | 101 | 0.129 |
All of the sequences listed amplified a PCR product exclusively in male genomic DNA, and all have chromosome quotients less than 0.3. We made an exception for sYG2 as it only has 29 alignments from male data, and the cutoff was 30 (see Methods). Even at lower stringency, sYG2 still had no female alignments so we decided to include it. The chromosome quotients for gYG1 and gYG2 were calculated from the transcripts because of the long introns present in the genes.
Figure 2.
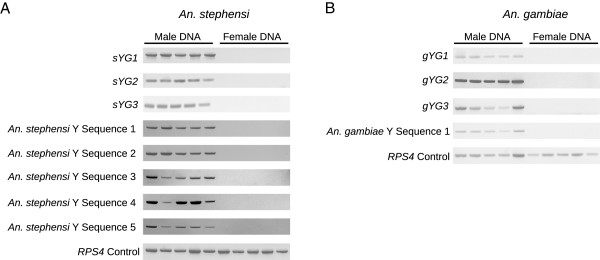
Male-specific PCR amplification of Y genes and intergenic Y sequences. (A) PCR performed with five male and five female genomic DNA samples shows male-specific amplification of three An. stephensi Y genes along with five An. stephensi intergenic Y sequences. A RPS4 ribosomal protein gene was amplified in both male and female genomic DNA confirming the integrity of the genomic DNA samples. (B) PCR performed with five male and five female genomic DNA samples shows male-specific amplification of the three An. gambiae Y genes, along with an intergenic An. gambiae Y sequence. A RPS4 ribosomal protein gene was amplified in both male and female genomic DNA confirming the integrity of the genomic DNA samples.
We then applied the CQ method to the genome of the African malaria mosquito, An. gambiae. We performed Illumina sequencing on male and female An. gambiae genomic DNA [SRA: SRP014730]. The An. gambiae AgamP3 genome assembly was downloaded from VectorBase. Repetitive sequences identified by RepeatMasker were removed, fragmenting the genome into many smaller pieces. Chromosome quotients were calculated for the fragmented autosome and X sequences from the AgamP3 genome assembly (Figure 1). The autosomal sequences had chromosome quotients distributed around one, and the X chromosome sequences had chromosome quotients distributed around two. Chromosome quotients were calculated for the fragmented Y chromosome sequences from the AgamP3 genome assembly. Of the 42 known An. gambiae Y sequences, 37 had chromosome quotients less than 0.3.
The An. gambiae sequences that were unable to be anchored to chromosomes, known as UNKN in the AgamP3 assembly, were fragmented by removing the repeats indicated by RepeatMasker. Chromosome quotients were calculated for these UNKN sequences. From the UNKN sequences, we identified 16 novel candidate Y sequences, encompassing more than 9,000 bases (Figure 1).
We also tested the CQ method de novo by generating a genome assembly from male G3 strain Illumina sequence data using ABySS. Chromosome quotients were calculated for all the sequences in the Illumina assembly. From the Illumina assembly, we identified 17 novel candidate Y sequences encompassing more than 6,000 bases (Figure 1). All Y sequences reported in this study are provided in the additional files (Additional file 3).
Discovery and expression profile of six Anopheles Y genes
Comparison of the Y sequences to An. stephensi and An. gambiae RNA-sequencing data with blastn led to the identification of six novel Y genes, three from each species (Table 3). The An. stephensi and An. gambiae Y genes were given the names sYG (An. stephensi Y gene) and gYG (An. gambiae Y gene) respectively, followed by a number designating the order in which they were discovered. All six Y genes amplified a product exclusively in male genomic DNA (Figure 2). Sequencing of RT-PCR products (Figure 3) confirmed transcription of all six genes. cDNA sequences for genes with confirmed introns were used to determine the structure of these genes, and are provided in Additional file 3 (Figure 4). Interestingly, all six genes were expressed in the early embryo, 2–12 hours after egg deposition (Figure 3). None of the six genes were expressed in 0–1 hour embryos, when only maternal transcripts are present, or in the adult female where no Y chromosome is present [26,27].
Figure 3.

Expression profile of six Anopheles Y genes. (A) The expression profile of the three An. stephensi Y genes sYG1, sYG2, and sYG3. (B) The expression profile of the three An. gambiae Y genes gYG1, gYG2, and gYG3. For both species, a ribosomal protein gene (RPS4) was used as positive control. RT-PCR was performed with primer-sets that demonstrated male-specific amplification in PCR on genomic DNA (Additional file 1: Table S1). The expression profile was performed on time points spanning from 0–2 hour embryos to adults. Mixed larvae refers to mixed L1 to L4 instars; h, hour; d, day.
Figure 4.
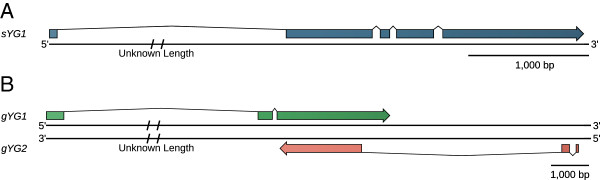
The structure of sYG1, gYG1, and gYG2 as determined by comparing cDNA and genome sequences. (A) The structure of sYG1. The first intron of sYG1 is likely very long as the start of the 5′ RACE aligns to a different scaffold in the draft An. stephensi genome. (B) gYG1 and gYG2 are overlapping genes that encode distinct RNAs from opposite strands.
The three An. stephensi Y genes include two recent duplications to the Y and a novel Y gene
sYG1 [GenBank: KC822952] shares 95 percent nucleotide identity with a gene mapped to chromosome 2R (Table 4, Figure 5). We call this autosomal paralog of sYG1, AsA-bbx (An. stephensi autosomal bobby sox). Both sYG1 and AsA-bbx are homologous to the D. melanogaster gene bobby sox (bbx) and to two closely linked An. gambiae genes, AGAP003896 and AGAP003897, which are located on the An. gambiae 2R chromosome arm. AsA-bbx is part of a large contig and the synteny of its neighboring genes is conserved among Anopheles and Aedes mosquitoes (Figure 5) suggesting that AsA-bbx is the ancestral copy, and sYG1 was formed from a recent duplication of AsA-bbx to the Y. Phylogenetic analysis suggests that this duplication of sYG1 occurred after the evolutionary divergence of An. stephensi and An. gambiae (Figure 5). 5′ and 3′ RACE was performed on sYG1 to characterize its full-length cDNA (Figure 4). Copy number determination using digital droplet PCR [28], designed to detect both sYG1 and AsA-bbx, indicates two autosomal AsA-bbx copies per diploid genome (a single copy per haploid genome) and six-to-nine copies of sYG1 in males (Figure 6). Chromosomal fluorescence in situ hybridization (FISH) with probes designed according to sYG1 hybridized to the An. stephensi Y on male mitotic chromosomes and on the 2R arm on female polytene chromosomes (Figure 5). Similar to the digital PCR analysis, this FISH result confirms that AsA-bbx is a single-copy autosomal gene located in subdivision 18A of the 2R arm, while sYG1 is a multi-copy gene on the Y.
Table 4.
Five of the six Y genes share high nucleotide identities to non-Y sequences
| Y gene 1 | Non-Y paralog | Identity (E-value) 2 |
|---|---|---|
|
sYG1 |
AsA-bbx3 |
95% (0) |
|
sYG2 |
None 4 |
Not applicable |
|
sYG3 |
An. stephensi AGAP0000485 |
94.8% (0) |
|
gYG1 |
3 L:34084227–34084914 6 |
94% (0) |
|
gYG2 |
3 L:34084227–34084914 7 |
94% (0) |
| gYG3 | 2 L: 5229785–5230624 8 | 89% (0) |
The most similar non-Y sequences to the Y genes in the An. stephensi and An. gambiae genomes. 1. Query sequences are the nucleotide sequences of the Y genes shown in Additional file 3. 2. blastn is used to obtain e-values. Percent identities reflect the entire sequence except for gYG1 and gYG2. 3. AsA-bbx is orthologous to An. gambiae AGAP003896/7. AsA-bbx is an autosomal paralog of sYG1. 4. sYG2 has no similar sequence in the rest of the An. stephensi genome except for a 55 bp repetitive region. 5. sYG3 is closely related to an annotated gene in An. stephensi, which is orthologous to An. gambiae AGAP000048. 6. gYG1 appears to be a composite of different fragments that share high similarities to different un-annotated non-Y sequences in An. gambiae. Some fragments are repeated in the genome. Shown in this table is a 688 base fragment that is 94 percent identical to sequences in chromosome 2R, 3 L as well as unmapped scaffolds. 7. gYG2 and gYG1 overlap, which is why the best non-Y match is identical between the two Y genes. gYG2 and gYG1 are transcribed from opposite strands. 8. gYG3 is most similar to an un-annotated 2 L sequence and an unmapped sequence in An. gambiae.
Figure 5.
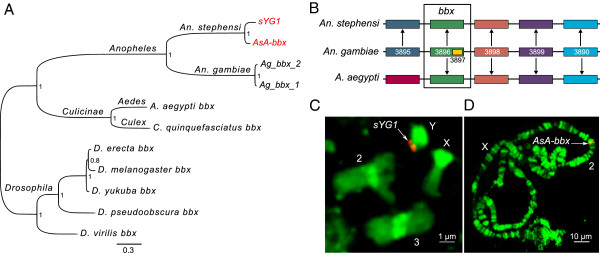
sYG1 is the result of a recent autosome-to-Y duplication. (A) Phylogenetic analysis of bobby sox (bbx) genes from different species. Alignments were produced by Clustalw. Phylogenetic inference was performed using MrBayes v3.1.2. The number near each node is the credibility score provided by MrBayes. Note that Ag_bbx_1 and Ag_bbx_2 are the two An. gambiae autosomal genes AGAP003986 and AGAP003987, respectively. (B) Synteny of the genes located around bobby sox is largely maintained in An. gambiae, An. stephensi, and Aedes aegypti. Genes indicated by the same color are orthologs. The An. gambiae gene names are preceded by AGAP00, the VectorBase gene designations. An. gambiae gene AGAP003897 is in an intron of AGAP003896. A. aegypti orthologs were assigned by VectorBase. An. stephensi orthologs were assigned by our own genome annotation. (C) FISH with a probe designed according to sYG1 hybridized to the An. stephensi Y chromosome. Mitotic chromosome slide preparations were prepared from imaginal discs of 4th instar larvae. Mitotic chromosome FISH was used because the An. stephensi Y chromosome does not polytenize in salivary glands and the ovarian polytene chromosomes do not contain a Y chromosome. (D) The autosomal copy of bobby sox in An. stephensi, AsA-bbx, was mapped to chromosome 2R with FISH on polytene chromosomes. Polytene chromosome slide preparations were obtained using ovaries of half gravid females. From these results we can deduce that AsA-bbx is the ancestral copy of bobby sox in An. stephensi, and that sYG1 is the result of a recent duplication from chromosome 2R to the Y.
Figure 6.
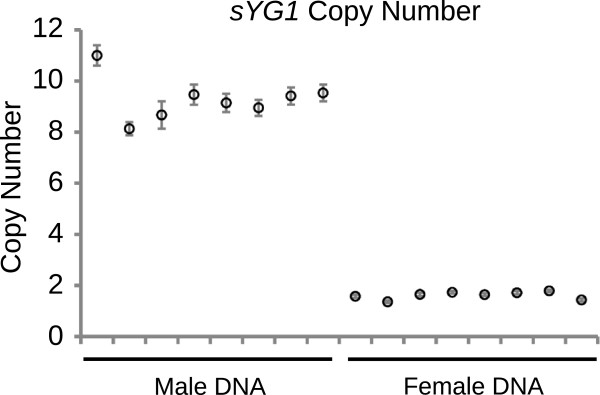
sYG1 is a multi-copy gene on the Y. Copy number determination using digital droplet PCR designed to detect both sYG1 and AsA-bbx indicates two autosomal AsA-bbx copies (seen in females) and six-to-nine copies of sYG1 in males (8 to 11 copies minus the two autosomal copies). Since we know that sYG1 hybridizes to the Y in FISH, we can infer that the extra copies of sYG1 in males is due to the fact that there are many copies of sYG1 on the Y. Digital PCR partitions a sample into as many as 20,000 individual droplets and uses TaqMan chemistry with fluorescently labelled probes to accurately determine the copy-number of genes. An autosomal reference gene (sequence provided in Additional file 3) was set as having two copies per diploid genome. The copy number of sYG1/AsA-bbx was determined by comparison to the autosomal reference. Error bars represent poisson error.
With the exception of a 55 base repetitive region, sYG2 [GenBank: KC822953] matched no other sequences in the An. stephensi genome using blastn or tblastx with an e-value threshold of 1e-2. sYG2 has no homolog in An. gambiae as indicated by blastn and tblastx searches of the PEST, M, and S genome assemblies as well as the trace files.
sYG3 [GenBank: KC840348] shares 94.8 percent nucleotide identity with an autosomal or X gene within the 355,000 base scaffold00149 of the An. stephensi genome. This gene is homologous to AGAP000048 on the An. gambiae X chromosome (Tables 4 and 5). AGAP000048 and its neighboring genes showed conserved synteny in An. gambiae, An. stephensi, and Culex quinquefasciatus, suggesting that the autosomal or X paralog of sYG3 is ancestral. Thus, we deduce sYG3 is also a recent duplication to the An. stephensi Y that occurred after the divergence of An. stephensi and An. gambiae.
Table 5.
Y gene homologs
| Y gene | Homolog1 | Maximum percent identity2 | E-value 2 | Homolog putative function |
|---|---|---|---|---|
|
sYG1 |
AGAP003896 |
35% |
1e-39 |
Bobby sox (bbx) HMG-box transcription factor |
|
sYG2 |
No homology found |
- |
- |
- |
|
sYG3 |
AGAP000048 |
37% |
2e-13 |
Adenomatous polyposis coli protein |
|
gYG1 |
AGAP005574 |
47% |
1e-16 |
Unknown |
|
AGAP011774 |
36% |
9e-08 |
Unknown |
|
|
AGAP0010793 |
89% 3 |
0.034 3 |
Unknown |
|
|
gYG2 |
AGAP0117344 |
38% 4 |
1e-5 4 |
Unknown |
| gYG3 | AGAP012527 | 33% | 2e-26 | General transcription factor II repeat domain |
The annotated homologs of the Y genes provide clues to putative functions of the Y genes we discovered. 1. Homologs were identified using blastx against the An. gambiae protein database (AgamP3.6 Gene Build) and the NCBI non-redundant protein database. Not surprisingly, the best matches are to An. gambiae sequences because the queries are from An. gambiae and An. stephensi. The An. gambiae genes AGAP003896 and AGAP000048 are orthologous to the autosomal or X paralogs of sYG1 and sYG3, respectively. The other homologs listed in column 2 are either distantly related or only share partial overlap. In cases where the Y genes match more than one related homolog, the homolog with the best e-value is shown. The three homologs shown for gYG1 are unrelated. 2. Maximum percent identities and e-values were obtained by blastx against the An. gambiae protein database (AgamP3.6 Gene Build). For Y genes that have confirmed introns (sYG1, gYG1, and gYG2), transcript sequences were used as the query. 3. The match to AGAP001079 is very short at the amino acid level although the nucleotide identity is 92 percent, and 98 percent in two longer fragments (e-value of 6×10-18). The sense strand of gYG1 is reverse-complementary to AGAP001079. 4. The sense strand of gYG2 is reverse-complementary to AGAP011734.
The three An. gambiae Y genes have closely related autosomal or X paralogs in the An. gambiae genome
gYG1 [GenBank: KC840350] and gYG2 [GenBank: KC845524] are two new genes found in the Y chromosome sequences of the AgamP3 genome assembly. Although they overlap on opposite strands, there are unique transcribed regions that allowed specific RT-PCR products to be amplified for each gene (Figures 3, 4). Both genes are highly transcribed from the early embryo until the adult male, including in the testes. Sequencing of RACE and RT-PCR products confirmed the organization of these two genes (Figure 4). Both genes have closely-related autosomal or X paralogs although these paralogs are not annotated (Table 4). Homologs of gYG1 and gYG2 were not identified in An. stephensi by blastn and tblastx searches of the genome assemblies as well as the 454 trace files, although a short repetitive region of gYG1 does have significant alignments to the An. stephensi genome from tblastx.
A portion of the gYG1 transcript shares more than 92 percent nucleotide identity with the AGAP001079 group of genes on both the X and autosomes, while the flanking regions are distantly but significantly related to AGAP005574 and AGAP011774, respectively. A portion of the gYG2 transcript shares greater than 90 percent nucleotide identity with a multi-copy non-coding sequence in the An. gambiae genome. Another part of gYG2 also contains a region that is homologous to AGAP011734 (Table 5). The gYG1 and gYG2 transcripts are reverse complementary to AGAP001079 and AGAP011734, respectively. This transcription orientation coupled with the fact that gYG1 and gYG2 are in opposite orientation may indicate functions related to double-stranded RNA formation, rather than coding for proteins. Because there are no known orthologs to AGAP001079, AGAP005574, and AGAP011774 we could not determine the ancestry of the gYG1 and gYG2 genes relative to their paralogs.
gYG3 [GenBank: KC840349] was discovered in an assembly of our own An. gambiae Illumina data. There are two similar sequences in the An. gambiae genome, both sharing 89 percent nucleotide identity with gYG3 (Table 4). One is on chromosome 2 L and the other is not mapped but its chromosome quotient suggests it is an autosomal or X sequence. Thus, gYG3 is again involved in a recent duplication, although we cannot assign the direction of the duplication. A blastx search shows that gYG3 is distantly related to AGAP012527 (OrthoDB Group EOG5VHHNX), which belongs to a large family of general transcription factor II-I repeat domain-containing proteins (Table 5).
Discussion
Here we have shown that the CQ method can reliably identify Y sequences using the ratio of alignments from male and female sequence data. Using the CQ method we identified 350 Y chromosome sequences encompassing more than 145,000 bases of novel Y chromosome sequences from the malaria mosquitoes An. stephensi and An. gambiae.
It is also possible to identify Y sequences by coverage depth [25]. In male sequence data, Y sequences should have half the coverage of autosomal sequences. In mixed male and female sequence data, Y sequences should have one-quarter the coverage of autosomal sequences [29]. The CQ method does not directly rely on coverage depth to identify Y sequences. While Y sequences often do have low coverage depth, it is often not sufficient to classify a sequence as Y-linked solely by low coverage depth. Mis-assemblies of the reference genome, and allelic differences between the reference genome and the short-read sequence data could lead to low coverage depth causing errant classification of Y-linkage. Unmasked repetitive sequences may lead to high coverage depth, obfuscating many Y sequences. Instead, the CQ method uses the ratio of female to male alignments to a reference sequence. The CQ method takes into account both the lack of female alignments and the presence of male alignments when classifying sequences as Y-linked, reducing the overall rate of false positives and false negatives. Mis-assemblies and allelic variation can be detected by the lack of male reads aligned to a sequence. For a sequence to be classified as Y-linked with the chromosome quotient it must have many alignments from males, and zero or a few alignments from females.
Although the functions of the Y genes that we found are not yet clear, there are some tantalizing hints to their potential functions. sYG1 is homologous to an HMG-box transcription factor involved in a number of developmental functions. HMG-box transcription factors include the two key genes involved in male-determination in mammals [30,31]. sYG3 shares homology with a general transcription factor II repeat domain containing protein, indicating the possibility that it is a Y-linked transcription factor. Five of the Y genes are clearly expressed in the testes suggesting that they may perform male-specific roles. All six Y genes are expressed in the early embryo, which is the time when sexual differentiation begins [26,27].
Our research offers an opportunity to investigate Y chromosome evolution, which until now has been restricted to limited lineages. Primate Y chromosomes have been shown to undergo strict conservation following rapid gene loss [9]. In D. melanogaster, however, almost all the Y genes are the result of duplications to the Y. Here we have shown that two of the three An. stephensi Y genes were recently acquired from the autosomes or the X. All three An. gambiae Y genes have highly similar paralogs on the autosomes or X (Table 4), again consistent with recent duplication. Thus, gene acquisitions through duplications appear to be a major source of Y genes in Anopheles mosquitoes. This is intriguing given that unlike D. melanogaster, the Anopheles Y is thought to harbor a dominant male-determining factor that initiates sexual differentiation, similar to humans and several non-Drosophila flies [6].
So far, no conservation between the genes on the two Anopheles Y chromosomes has been found, although the two species belong to the same subgenus. It is possible that we simply missed the common Y genes that originated prior to the divergence between An. gambiae and An. stephensi. However, such shared Y genes are less likely to have highly similar autosomal or X paralogs compared to recent duplications, and they may even be unique to the Y. Y genes that are unique or significantly diverged from their autosomal or X paralogs are the easiest to find with the CQ method. To mitigate the effect of potential underrepresentation of Y genes in the Anopheles genome sequences, we compared the trace sequence files of both species and found no indications of shared Y genes. Thus, the evidence so far suggests that the An. gambiae and An. stephensi Y chromosomes are very different in their gene content. This conclusion is consistent with the observation that two of the three Y genes in An. stephensi were recently acquired after the divergence of the two mosquitoes. Further investigation of Y genes in additional Anopheles species will reveal whether the evolutionary scenarios demonstrated by the Drosophila Y chromosomes (10), namely recent Y gene acquisition and Y chromosome replacement, are widely applicable in species with a dominant male-determining factor on the Y.
Some of the Y genes described in this study have unique Y chromosome sequences. Because of their exclusive paternal inheritance and general lack of recombination, these Y sequences could serve as powerful molecular markers for the investigation of population structure and incipient speciation, which are often relevant to vectorial capacity [32]. Only female mosquitoes bite and transmit pathogens. Genetic strategies to control mosquito-borne diseases may include the release of sterile males (Sterile Insect Technique, SIT), the release of insects carrying a dominant lethal gene (RIDL), or the replacement of a pathogen-susceptible population with a pathogen-resistant one [33-37]. Under these scenarios, the release of males is either required or preferred. A better understanding of the possible functions of mosquito Y genes in sexual differentiation will facilitate the production of sterile males for improved SIT. It may also enable the production of highly competitive males to improve SIT and RIDL and to help spread pathogen resistance genes.
Conclusions
In this study, we have shown that Y sequences can be identified by their distinctive chromosome quotients, the ratio of female to male alignments. Although the genomes of many organisms with Y chromosomes have been sequenced, Y sequences have been characterized from only a small subset of these genomes. Y sequences are surely present in these genomes, but are probably fragmented into short contigs challenging the traditional experimental methods of finding Y sequences. The quality of the genome assembly is not a limiting factor in finding Y sequences, making the CQ method widely applicable to species with fragmented genome. Autosome or X sequences rarely have chromosome quotients less than 0.3, giving the CQ method a low false positive rate. The CQ method is able to identify the majority of known Y sequences from species we have analyzed. We have also shown that the CQ method can identify recent duplications to the Y that share more than 94 percent nucleotide identity with autosomal or X paralogs. Coupling the CQ method with transcriptome sequencing allowed us to identify six Y genes in An. stephensi and An. gambiae. Analysis of the six Y genes indicates rapid Y chromosome evolution between An. stephensi and An. gambiae. The Anopheles Y genes characterized in this study provide unique markers for population and phylogenetic analysis, and opportunities for novel mosquito control measures through the manipulation of sexual dimorphism and fertility. The identification of Y genes in more organisms will help us understand the evolution and functions of Y chromosomes.
Methods
The CQ method
The chromosome quotient is the normalized ratio of female to male alignments to a given reference sequence. For a given sequence Si, CQ(Si) = F(Si)/M(Si), where F(Si) is the number of alignments from female sequence data to Si, and M(Si) is the number of alignments from male sequence data to Si.
To calculate chromosome quotients we wrote a program we call CQ-calculate (Available from: http://sourceforge.net/projects/cqcalculate/files/CQ-calculate.pl/download). CQ-calculate was written in Perl and designed to rapidly calculate chromosome quotients. There are three inputs to CQ-calculate: reference sequences, male-specific sequence data, and female-specific sequence data. The reference sequences are required to be in FASTA format. The male and female sequence data can be in either FASTQ or FASTA format. Preferably, the male and female sequence data should be from either highly inbred populations, or from a pool of many individuals to adequately sample genetic variation. For the best results, the male and female sequence data should be from the same colony or population to minimize the risk of bacterial or viral contamination exclusive to either the male or female data. The male and female sequence data is aligned to the reference sequences using the ultrafast read aligner Bowtie [38]. CQ-calculate uses stringent alignment criteria requiring the entire read to align with zero mismatches. To account for differences in coverage between the male and female sequence data, the chromosome quotients of the reference sequences are normalized to the median chromosome quotient of known autosomal sequences.
CQ-calculate can run on modest computers, and the only software requirements are: Linux, Perl, BioPerl, and bowtie. The time CQ-calculate takes to run is dependent on the genome size and coverage of the male and female sequence data. On a server running Ubuntu 12.04 with an Intel 3930 K six-core processor, it took approximately five minutes to calculate the chromosome quotients for all the sequences in the An. stephensi genome. For the much larger human genome, CQ-calculate took less than one hour. Memory requirements are dependent on the size of the reference genome but are typically minimal. CQ-calculate is easy to run, and is applicable to any heterogametic genome where separate male and female sequence data are available.
The CQ method uses the number of alignments from male and female sequence data to determine whether a sequence is Y-linked. The number of alignments affects the confidence with which a sequence can be classified as Y-linked. Thresholds were examined from one male alignment to 50 male alignments. Increasing the threshold for male alignments decreases the number of false positive results but also increases the number of false negatives (Additional file 4: Table S1). A threshold of 30 male alignments was chosen as it balances the number of false positives with the total number of sequences that can be analyzed. However, the threshold is flexible and can be increased for higher confidence in Y-linkage or decreased for a lower rate of false negatives.
We noticed that there are sequences with thousands of alignments from male and female data, but that still have many more male alignments than female alignments leading CQ-calculate to identify these sequences as Y-linked. Some of these sequences are known to be located on the autosomes or X, so we hypothesize that these are highly repetitive sequences with copies on the autosomes or X, but have many more copies on the Y. These highly repetitive sequences can be removed by setting a threshold for female alignments. In this study, we set a stringent threshold for female alignments of 30. Thus, to classify a sequence as Y-linked using the CQ method it must have a chromosome quotient less than 0.3, more than 30 alignments from male data, and less than 30 alignments from female data.
The coverage of the next-generation sequence data can affect the calculation of chromosome quotients. Since a threshold of alignments is required to classify a sequence as Y-linked, higher coverage male and female sequence data leads to the identification of more Y-sequences. We have tested a range of coverages and found that chromosome quotients can still be accurately calculated with as low as 5× coverage, but at this low coverage the number of Y sequences identified is reduced, and the false positive rate is increased. Above 10× coverage, many more Y sequences can be identified. With more than 20× coverage, many short Y sequences can be identified with a low rate of false positives.
The CQ method positive control
We tested the CQ method on the genomes of H. sapiens and D. melanogaster. Since repeats can obscure Y sequences, we downloaded the softmasked reference genomes of H. sapiens (hg19 assembly) and D. melanogaster (dm3 assembly) from the USCS Genome Bioinformatics website. In a genome that is softmasked, repetitive sequences identified by RepeatMasker are replaced with lowercase nucleotides. We removed these repeats, fragmenting the genomes into smaller pieces. Fragments shorter than 250 bases were removed to mitigate false positives. The number of sequences resulting from the fragmentation, and the N50 size of the fragments were calculated (Additional file 5: Table S2).
H. sapiens male and female specific next-generation sequence data from a single male and female were downloaded from the 1000 Genomes Project [HG00234 and HG00235] [39]. Male and female D. melanogaster next-generation sequence data for pooled five day old mated adults was downloaded from the NCBI Sequence Read Archive [SRA: SRP007888]. The coverage of the H. sapiens and D. melanogaster data was calculated (Additional file 6: Table S3). Chromosome quotients were calculated for all the fragments of the H. sapiens and D. melanogaster genomes and then normalized to the median chromosome quotient of the known autosomal sequences.
Y gene finding in the Asian malaria mosquito An. Stephensi
454 sequencing was performed on pools of approximately 30 adult An. stephensi from the Indian wild type strain. Newbler 2.6 was used to assemble the An. stephensi 454 sequence data into contigs and scaffolds. The contigs of this assembly are available from the NCBI [GenBank: ALPR00000000]. The number of contigs and N50 contig size were calculated (Additional file 5: Table S2). Pools of approximately 30 male and female An. stephensi of the Indian wild type strain were sequenced using the Illumina Genome Analyzer II [SRA: SRP013838] (Additional file 6: Table S3).
An. stephensi sequences have been anchored to chromosomes using chromosomal in situ hybridization [40]. The short probe sequences from FISH that hybridized to known autosome and X sequences were compared to the An. stephensi genome scaffolds using blastn requiring 95 percent identity. Autosome and X scaffolds were recovered. The scaffolds were then split into fragments by the ambiguous nucleotide N. Fragments shorter than 250 bases were removed. The chromosome quotients of these sequences were calculated.
Chromosome quotients were calculated for all the contigs from the An. stephensi genome using the male and female Illumina data mentioned above and normalized to the median chromosome quotient of autosomal sequences. Y genes were identified in the contigs with chromosome quotients less than 0.3 by comparison to transcriptome sequence data. Using blastn requiring 100 percent identity and an e-value less than 1×10-5 we compared the Y-linked contigs to transcriptome sequence data raw reads from An. stephensi Indian wild type strain. The time points compared were: 0–1 hour embryos, 2–4 hour embryos, 4–8 hour embryos, 8–12 hour embryos, mixed-instar larva, pupa, adult females, and adult males [SRA: SRP013839]. The number of reads for each time point was calculated (Additional file 6: Table S4).
We searched for evidence of the An. stephensi Y chromosome gene sYG2 in the An. gambiae genome using blastn and tblastx. Blastn searches with a word size seven and e-value threshold of 1×10-2 using sYG2 as the query against the An. gambiae PEST, M and S genome assemblies yielded no significant similarity. Additionally blastn searches with word size seven and e-value threshold of 1×10-2 using sYG2 as the query against the PEST, M, and S trace files revealed no significant alignments. Tblastx searches with an e-value threshold of 1×10-5 yielded no significant similarity to the An. gambiae genome.
Y gene finding in the African malaria mosquito An. gambiae
The AgamP3 genome assembly of the An. gambiae PEST strain was downloaded from VectorBase. The genome was divided into seven parts: the arms of the two autosomes, the X chromosome, fragments of the Y chromosome, and unmapped sequences referred to as UNKN. The repetitive sequences of the AgamP3 genome assembly were masked using RepeatMasker by VectorBase and indicated by lowercase nucleotides.
The repetitive sequences indicated by RepeatMasker were removed from the autosomes, X, Y, and UNKN sequences creating many smaller fragments. Sequences shorter than 250 bases were removed to mitigate false positives. The number and N50 size of the fragmented autosome, X, Y, and UNKN sequences were calculated (Additional file 5: Table S2). Illumina sequencing of male and virgin female An. gambiae was performed on pools of approximately 30 individuals from the G3 strain from the same colony [SRA: SRP014730] (Additional file 6: Table S3). Chromosome quotients were calculated for the fragmented An. gambiae autosomes, X, Y, and UNKN sequences using the Illumina sequence data mentioned above.
We were concerned that Sanger sequencing, which was used to sequence the An. gambiae genome, may be biased against the heterochromatic Y chromosome. We attempted to circumvent this bias by assembling our male G3 strain Illumina data and searching for Y sequences. We assembled the An. gambiae male Illumina sequence data using ABySS single-end assembly with the kmer setting 31 [41]. The number of contigs and the N50 contig size of the assembly were calculated (Additional file 5: Table S2). Since the N50 contig size was so short for this assembly, no further fragmentation was deemed necessary. Chromosome quotients were calculated for all the contigs in this assembly using the male and female An. gambiae Illumina data.
We searched for Y genes in the sequences with chromosome quotients less than 0.3 using the raw reads from An. gambiae transcriptome sequence data. Using blastn requiring 100 percent identity and an e-value less than 1×10-5 we compared the Y-linked contigs to transcriptome sequence data from adult male An. gambiae [SRA: SRP014756]. Using the same parameters, we also compared the UNKN sequences that we inferred to be Y-linked by their CQs to the same transcriptome data (Additional file 7: Table S4).
We searched for the An. gambiae Y chromosome genes gYG1 and gYG2 in the An. stephensi genome using blastn and tblastx. Blastn searches with a word size of 7 and an e-value threshold of 1×10-5 using the sequence of gYG1 and gYG2 as a query yielded no similarity to the An. stephensi genome. Furthermore, using blastn we compared gYG1 and gYG2 to Illumina data from An. stephensi and we found no significant similarity using word size seven and an e-value threshold of 1×10-2. Tblastx searches with an e-value threshold of 1×10-5 only yielded significant alignments from a small repetitive part of gYG1 to the An. stephensi genome.
Molecular biology methods
Genomic DNA samples were isolated with Life Technologies DNAzol from male and virgin female mosquitoes from the Indian wild type strain of An. stephensi and the G3 strain of An. gambiae. Five male and female samples were prepared, each from five individuals. In the case of Y genes that have autosomal or X paralogs, primers for genomic DNA PCR were designed with the differences between the autosomal or X paralog and the Y sequence at the 3′ end of the primer. PCR was performed with Finnzymes Phire DNA Polymerase. RNA was isolated from embryos, larva, pupa, adult male, and adult female individuals with the Life Technologies mirVana RNA isolation kit using the total RNA isolation protocol. Complementary DNA was synthesized with Life Technologies SuperScript III RT. All primer sequences are available in the additional files (Additional file 8: Table S5). We used primer-sets that exhibited male-specific amplification to perform RT-PCR with the complementary DNA mentioned above as template with either Finnzymes Phire DNA Polymerase or TaKaRa rTaq. Rapid amplification of cDNA ends (RACE) was performed on sYG1, gYG2, and gYG3 using the SMARTer RACE cDNA Amplification Kit. The resulting sequences were assembled into full-length transcripts, and verified by sequencing complementary DNA. Digital PCR was performed with a QX100 Droplet Digital PCR System from Bio-Rad, on male and female genomic DNA with a probe that would hybridize to both sYG1 and AsA-bbx. A single copy autosomal gene, zeta DNA polymerase catalytic subunit, was used as the reference set as two copies per diploid genome. Chromosomal fluorescence in situ hybridization for sYG1 was performed on mitotic and polytene chromosomes using the method described in [42-44]. Phylogenetic analysis was performed with MrBayes [45]. The alignment and parameters used to infer the phylogeny are provided in the additional files (Additional file 9).
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
ABH conceived and implemented the CQ method and discovered the Y genes, performed PCR, and drafted the manuscript. YQ performed PCR and RACE. VT, MS, and IS performed FISH. IS and MS helped with manuscript revision. ZT initiated and designed the project, performed evolutionary analysis, and contributed to Y gene discovery and the writing of the manuscript. All authors read and approved the final manuscript.
Supplementary Material
The calculation of chromosome quotients.
Autosome, X, and Y sequences differ in the number of alignments from male and female sequence data.
Sequences referenced in the text including: the six Y genes, intergenic Y sequences tested for male-specific amplification, An. stephensi Y sequences, An. gambiae Y sequences, the autosomal homolog of sYG1, and the revised An. stephensi scaffold linking the autosomal homolog of sYG3 to a long scaffold.
Analysis of the male alignment threshold.
Statistics from the fragmented genome assemblies.
The coverage of the male and female sequence data used in the study.
The number of reads and read length from the RNA-seq data used in the study.
Sequences of the primers used in the study.
Alignment and parameters used to infer phylogeny.
Contributor Information
Andrew Brantley Hall, Email: brant@vt.edu.
Yumin Qi, Email: yuqi@vt.edu.
Vladimir Timoshevskiy, Email: timvl@vt.edu.
Maria V Sharakhova, Email: msharakh@vt.edu.
Igor V Sharakhov, Email: igor@vt.edu.
Zhijian Tu, Email: jaketu@vt.edu.
Acknowledgements
We thank Yann Jouvenot and Jack Regan at Bio-Rad for performing digital-droplet PCR. We thank Janet Webster for comments on the manuscript. We thank Xiaofang Jiang for help with statistics and figure preparation. We thank Randy Saunders for mosquito care. This work is supported by NIH Grants AI077680, AI105575 and AI094289, the Virginia Tech Experimental Station, and the Fralin Life Science Institute.
References
- Pease JB, Hahn MW. Sex chromosomes evolved from independent ancestral linkage groups in winged insects. Mol Biol Evol. 2012;29(6):1645–1653. doi: 10.1093/molbev/mss010. [DOI] [PubMed] [Google Scholar]
- Wilson MA, Makova KD. Genomic analyses of sex chromosome evolution. Annu Rev Genomics Hum Genet. 2009;10:333–354. doi: 10.1146/annurev-genom-082908-150105. [DOI] [PubMed] [Google Scholar]
- Baker RH, Sakai RK. Triploids and male determination in the mosquito, Anopheles culicifacies. J Hered. 1979;70(5):345–346. doi: 10.1093/oxfordjournals.jhered.a109271. [DOI] [PubMed] [Google Scholar]
- Charlesworth D. Plant sex determination and sex chromosomes. Heredity. 2002;88(2):94–101. doi: 10.1038/sj.hdy.6800016. [DOI] [PubMed] [Google Scholar]
- McLaren A. Sex determination in mammals. Trends Genet. 1988;4(6):153–157. doi: 10.1016/0168-9525(88)90020-0. [DOI] [PubMed] [Google Scholar]
- Marín I, Baker BS. The evolutionary dynamics of Sex determination. Science. 1998;281(5385):1990–1994. doi: 10.1126/science.281.5385.1990. [DOI] [PubMed] [Google Scholar]
- Matsuda M, Nagahama Y, Shinomiya A, Sato T, Matsuda C, Kobayashi T, Morrey CE, Shibata N, Asakawa S, Shimizu N. DMY is a Y-specific DM-domain gene required for male development in the medaka fish. Nature. 2002;417(6888):559–563. doi: 10.1038/nature751. [DOI] [PubMed] [Google Scholar]
- Charlesworth B, Charlesworth D. The degeneration of Y chromosomes. Philos Trans R Soc Lond B Biol Sci. 2000;355(1403):1563–1572. doi: 10.1098/rstb.2000.0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JF, Skaletsky H, Pyntikova T, Graves TA, van Daalen SKM, Minx PJ, Fulton RS, McGrath SD, Locke DP, Friedman C. Chimpanzee and human Y chromosomes are remarkably divergent in structure and gene content. Nature. 2010;463(7280):536–539. doi: 10.1038/nature08700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JF, Skaletsky H, Brown LG, Pyntikova T, Graves T, Fulton RS, Dugan S, Ding Y, Buhay CJ, Kremitzki C. Strict evolutionary conservation followed rapid gene loss on human and rhesus Y chromosomes. Nature. 2012;483(7387):82–86. doi: 10.1038/nature10843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallis MC, Waters PD, Graves JA. Sex determination in mammals–before and after the evolution of SRY. Cellular and molecular life sciences : CMLS. 2008;65(20):3182–3195. doi: 10.1007/s00018-008-8109-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koerich LB, Wang X, Clark AG, Carvalho AB. Low conservation of gene content in the Drosophila Y chromosome. Nature. 2008;456(7224):949–951. doi: 10.1038/nature07463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline TW, Meyer BJ. Vive la difference: males vs females in flies vs worms. Annu Rev Genet. 1996;30:637–702. doi: 10.1146/annurev.genet.30.1.637. [DOI] [PubMed] [Google Scholar]
- Schmidt R, Hediger M, Roth S, Nothiger R, Dubendorfer A. The Y-chromosomal and autosomal male-determining M factors of Musca domestica are equivalent. Genetics. 1997;147(1):271–280. doi: 10.1093/genetics/147.1.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willhoeft U, Franz G. Identification of the sex-determining region of the Ceratitis capitata Y chromosome by deletion mapping. Genetics. 1996;144(2):737–745. doi: 10.1093/genetics/144.2.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho AB. Origin and evolution of the Drosophila Y chromosome. Curr Opin Genet Dev. 2002;12(6):664–668. doi: 10.1016/S0959-437X(02)00356-8. [DOI] [PubMed] [Google Scholar]
- Bernardo Carvalho A, Koerich LB, Clark AG. Origin and evolution of Y chromosomes: Drosophila tales. Trends in genetics : TIG. 2009;25(6):270–277. doi: 10.1016/j.tig.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho AB, Lazzaro BP, Clark AG. Y chromosomal fertility factors kl-2 and kl-3 of Drosophila melanogaster encode dynein heavy chain polypeptides. Proc Natl Acad Sci. 2000;97(24):13239–13244. doi: 10.1073/pnas.230438397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho AB, Dobo BA, Vibranovski MD, Clark AG. Identification of five new genes on the Y chromosome of Drosophila melanogaster. Proc Natl Acad Sci. 2001;98(23):13225–13230. doi: 10.1073/pnas.231484998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos B, Araripe LO, Hartl DL. Polymorphic Y chromosomes harbor cryptic variation with manifold functional consequences. Science. 2008;319(5859):91–93. doi: 10.1126/science.1148861. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Bachtrog D. Sex-specific adaptation drives early Sex chromosome evolution in drosophila. Science. 2012;337(6092):341–345. doi: 10.1126/science.1225385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roldan ER, Gomendio M. The Y chromosome as a battle ground for sexual selection. Trends Ecol Evol. 1999;14(2):58–62. doi: 10.1016/S0169-5347(98)01567-5. [DOI] [PubMed] [Google Scholar]
- Krzywinski J, Nusskern DR, Kern MK, Besansky NJ. Isolation and characterization of Y chromosome sequences from the African malaria mosquito Anopheles gambiae. Genetics. 2004;166(3):1291–1302. doi: 10.1534/genetics.166.3.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski J, Chrystal M, Besansky N. Gene finding on the Y: fruitful strategy in drosophila does not deliver in anopheles. Genetica. 2006;126(3):369–375. doi: 10.1007/s10709-005-1985-3. [DOI] [PubMed] [Google Scholar]
- Coluzzi M, Cancrini G, Di Deco M. The polytene chromosomes of Anopheles superpictus and relationships with Anopheles stephensi. Parassitologia. 1970;12(2/3):101–112. [Google Scholar]
- Salz H, Erickson JW. Sex determination in Drosophila: the view from the top. Fly. 2010;4(1):60–70. doi: 10.4161/fly.4.1.11277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadros W, Lipshitz HD. The maternal-to-zygotic transition: a play in two acts. Development (Cambridge, England) 2009;136(18):3033–3042. doi: 10.1242/dev.033183. [DOI] [PubMed] [Google Scholar]
- Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, Bright IJ, Lucero MY, Hiddessen AL, Legler TC. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011;83(22):8604–8610. doi: 10.1021/ac202028g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N, Bellott DW, Page DC, Clark AG. Identification of avian W-linked contigs by short-read sequencing. BMC Genomics. 2012;13:183. doi: 10.1186/1471-2164-13-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashimada K, Koopman P. Sry: the master switch in mammalian sex determination. Development. 2010;137(23):3921–3930. doi: 10.1242/dev.048983. [DOI] [PubMed] [Google Scholar]
- Kozhukhar VG. SRY and SOX9: the main genetic factors of mammalian sex determination. Tsitologiia. 2012;54(5):390–404. [PubMed] [Google Scholar]
- Riehle MM, Guelbeogo WM, Gneme A, Eiglmeier K, Holm I, Bischoff E, Garnier T, Snyder GM, Li X, Markianos K. A cryptic subgroup of anopheles gambiae is highly susceptible to human malaria parasites. Science. 2011;331(6017):596–598. doi: 10.1126/science.1196759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins FH, Paskewitz SM. Malaria: current and future prospects for control. Annu Rev Entomol. 1995;40(1):195–219. doi: 10.1146/annurev.en.40.010195.001211. [DOI] [PubMed] [Google Scholar]
- Benedict MQ, Robinson AS. The first releases of transgenic mosquitoes: an argument for the sterile insect technique. Trends Parasitol. 2003;19(8):349–355. doi: 10.1016/S1471-4922(03)00144-2. [DOI] [PubMed] [Google Scholar]
- Bargielowski I, Alphey L, Koella JC. Cost of mating and insemination capacity of a genetically modified mosquito aedes aegypti OX513A compared to its wild type counterpart. PLoS One. 2011;6(10):e26086. doi: 10.1371/journal.pone.0026086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Valdez MR W, Nimmo D, Betz J, Gong H-F, James AA, Alphey L, Black WC. Genetic elimination of dengue vector mosquitoes. Proc Natl Acad Sci. 2011;108(12):4772–4775. doi: 10.1073/pnas.1019295108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deredec A, Godfray HC, Burt A. Requirements for effective malaria control with homing endonuclease genes. A ETAProc Natl Acad Sci USA. 2011;108(43):E874–E880. doi: 10.1073/pnas.1110717108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B. Aligning short sequencing reads with Bowtie. Curr Protoc Bioinformatics. 2010;11:11.7. doi: 10.1002/0471250953.bi1107s32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharakhova MV, Xia A, Tu Z, Shouche YS, Unger MF, Sharakhov IV. A physical map for an Asian malaria mosquito, Anopheles stephensi. AmJTrop Med Hyg. 2010;83(5):1023–1027. doi: 10.4269/ajtmh.2010.10-0366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009;19(6):1117–1123. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George P, Sharakhova MV, Sharakhov IV. High-throughput physical mapping of chromosomes using automated in situ hybridization. Journal of visualized experiments : JoVE. 2012. [DOI] [PMC free article] [PubMed]
- Sharakhova MV, Xia A, McAlister SI, Sharakhov IV. A standard cytogenetic photomap for the mosquito Anopheles stephensi (Diptera: Culicidae): application for physical mapping. J Med Entomol. 2006;43(5):861–866. doi: 10.1603/0022-2585(2006)43[861:ASCPFT]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Timoshevskiy VA, Sharma A, Sharakhov IV, Sharakhova MV. Fluorescent in situ hybridization on mitotic chromosomes of mosquitoes. Journal of visualized experiments : JoVE. 2012. [DOI] [PMC free article] [PubMed]
- Ronquist F, Huelsenbeck JP. MrBayes 3: bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19(12):1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The calculation of chromosome quotients.
Autosome, X, and Y sequences differ in the number of alignments from male and female sequence data.
Sequences referenced in the text including: the six Y genes, intergenic Y sequences tested for male-specific amplification, An. stephensi Y sequences, An. gambiae Y sequences, the autosomal homolog of sYG1, and the revised An. stephensi scaffold linking the autosomal homolog of sYG3 to a long scaffold.
Analysis of the male alignment threshold.
Statistics from the fragmented genome assemblies.
The coverage of the male and female sequence data used in the study.
The number of reads and read length from the RNA-seq data used in the study.
Sequences of the primers used in the study.
Alignment and parameters used to infer phylogeny.