

Abstract
The androgen receptor (AR) is the principal therapeutic target in prostate cancer. For the past 70 years, androgen deprivation therapy (ADT) has been the major therapeutic focus. However, some patients do not benefit, and those tumors that do initially respond to ADT eventually progress. One recently described mechanism of such an effect is growth and survival-promoting effects of the AR that are exerted independently of the AR ligands, testosterone and dihydrotestosterone. However, specific ligand-independent AR target genes that account for this effect were not well characterized. We show here that c-Myc, which is a key mediator of ligand-independent prostate cancer growth, is a key ligand-independent AR target gene. Using microarray analysis, we found that c-Myc and AR expression levels strongly correlated with each other in tumors from patients with castration-resistant prostate cancer (CRPC) progressing despite ADT. We confirmed that AR directly regulates c-Myc transcription in a ligand-independent manner, that AR and c-Myc suppression reduces ligand-independent prostate cancer cell growth, and that ectopic expression of c-Myc attenuates the anti-growth effects of AR suppression. Importantly, treatment with the bromodomain inhibitor JQ1 suppressed c-Myc function and suppressed ligand-independent prostate cancer cell survival. Our results define a new link between two critical proteins in prostate cancer – AR and c-Myc – and demonstrate the potential of AR and c-Myc-directed therapies to improve prostate cancer control.
Introduction
Prostate cancer is the most common cancer in men in the United States with 241,740 new cases anticipated this year [1]. Despite screening and early treatment, prostate cancer commonly recurs, and 28,170 men are predicted to die from prostate cancer this year [1]. Nearly all of these prostate cancer deaths are attributable to metastatic, castration-resistant prostate cancer (CRPC) that has progressed despite androgen deprivation therapy (ADT) – the most common treatment for patients with recurrent or advanced prostate cancer.
ADT works by lowering levels of the potent AR ligands testosterone and dihydrotestosterone (DHT) or interfering with binding of androgen ligands to the androgen receptor (AR) protein, the principal therapeutic target in prostate cancer [2]. Despite ADT, including novel and more potent treatments, all prostate cancers eventually progress [3], [4]. At progression, the AR is ubiquitously expressed [5], [6].
There are several possible explanations for AR-dependent mechanisms of progression despite the suppression or interference with androgen ligands. These include intratumoral androgen synthesis, the generation of constitutively active AR transcript variants, AR gene amplification, activating AR mutations, or activation of the AR by growth factors [7]–[16]. It is also now clear that the AR protein can promote the activation of AR ligand-independent pathways distinct from AR’s canonical ligand-activated pathways in CRPC [17]. However, critical downstream AR target genes of this type that account for AR dependent, ligand-independent prostate cancer cell survival have not been fully clarified. The study of such AR target genes and mechanisms by which AR regulates their expression will improve our understanding of castration-resistance and lead to the identification of key AR dependent proteins whose activity may control growth and survival of CRPC cells. Such targets and pathways would naturally become high priorities for drug development.
To understand genes that might account for that effect, we focused on c-Myc. This is because: 1) c-Myc overexpression promotes prostate cancer development [18]; 2) c-Myc is upregulated in androgen ligand-dependent prostate cancer and further upregulated in CRPC [19], [20]; and 3) prior reports have demonstrated that c-Myc, like AR, contributes to ligand-independent prostate cancer cell growth [21]. Our review of prior data that localized AR binding sites throughout the genome by chromatin immunoprecipitation (ChIP) showed that the AR localizes to an enhancer element of the c-Myc gene [17]. However, it was unclear if c-Myc was a direct AR target gene and whether androgen ligands were necessary for AR regulation of c-Myc expression.
We determined that c-Myc upregulation in human CRPC tumors correlates with AR upregulation, and we confirmed that c-Myc is a direct AR target gene using chromatin immunoprecipitation (ChIP) assays. c-Myc suppression achieves the same overall effects as AR suppression, and c-Myc overexpression attenuates the anti-growth effects of AR suppression. While AR promotes c-Myc expression, treatment with androgen ligands did not increase c-Myc expression. Thus, AR promotes the expression of c-Myc in a ligand-independent manner, and c-Myc is a key AR target gene.
Finally, we treated prostate cancer cells with the BET bromodomain inhibitor JQ1 that suppresses c-Myc expression [22], [23]. Treatment with JQ1 achieved the same overall effect as c-Myc RNAi and reduced prostate cancer cell survival in androgen ligand-depleted conditions.
Our studies clarify that c-Myc is a key androgen ligand-independent AR target gene that contributes to androgen ligand-independent but AR-dependent prostate cancer cell survival. Our results also demonstrate the potential of AR-directed therapies or c-Myc-directed therapies in prostate cancer as adjuncts to ADT.
Results
AR and c-Myc are Concordantly Expressed in Metastatic CRPC
Both AR and c-Myc are critical survival pathways in prostate cancer, and expression levels of both AR and c-Myc are commonly increased in human CRPC tumors progressing despite ADT [24], [25]. However, it was unknown whether overexpression of AR and c-Myc was linked with the other in human CRPC tumors. Therefore, we determined the expression levels of AR and c-Myc using gene expression microarrays in 140 human CRPC tumors versus 15 normal prostate samples. Next, we examined the association of AR upregulation and c-Myc upregulation in the human CRPC tumors. AR mRNA levels in CRPC samples were strongly associated with c-Myc mRNA levels (Pearson correlation = 0.3698, 95% Confidence Interval: 0.2172–0.5048, two-tailed p-value<0.0001) (Figure 1A). We also calculated the odds ratio for c-Myc and AR upregulation in these CRPC specimens. There was a statistically significant association with AR upregulation and c-Myc upregulation (OR = 3.528, 95% Confidence Interval: 1.347 to 9.240, p-value: 0.0108 by Fisher's Exact Test) (Figure 1B).
Figure 1. AR and c-Myc levels are positively correlated in CRPC specimens.

A) Z-scores (to normal prostate specimens) of AR versus c-Myc mRNA expression across 140 human CRPC metastases. The Pearson correlation coefficient, linear regression, and F test for significantly non-zero slope were performed for each pair of genes. B) Fisher’s exact test and odds ratio on the contingency table analyzing the co-occurrence of tumors with AR or c-Myc z-scores greater than 2.
AR Suppression Reduces the Growth of AR Ligand-dependent and AR Ligand–independent Castration-resistant Prostate Cancer Cells
We suppressed expression of the AR with RNAi in prostate cancer cells grown in charcoal-stripped, androgen ligand-depleted serum. AR RNAi reduced cell growth of both androgen ligand-dependent LNCaP cells and their CRPC derivatives called LNCaP-abl (Figure 2A). Of note, both of these cells only express the full-length AR transcript. AR suppression with RNAi in the 22RV1 CRPC cell line that expresses both full-length AR and an AR transcript variant achieved the same effect (Figure 2A). In all cell lines, AR suppression reduced cell growth without inducing apoptosis (data not shown), suggesting a defect in proliferation. Thus, despite androgen ligand depletion, AR suppression further reduces prostate cancer cell growth.
Figure 2. AR and c-Myc promote ligand-independent prostate cancer cell growth.

A) LNCaP, abl and 22RV1 cells were transfected with 50 nM of non-targeted control (NTC) or AR RNAi oligonucleotides. Cells were switched to charcoal-stripped serum on the day of transfection. Cell growth was determined 5 days later for LNCaP and 6 days later for abl and CRPC 22RV1 with the trypan blue exclusion method. B) Immunoblot for AR expression. The lower bands in the AR immunoblot in 22RV1 cells reflect the presence of an AR transcript variant [7]. B) LNCaP, abl and 22RV1 cells were transfected with 50 nM of NTC or c-Myc RNAi oligonucleotides. Cells were switched to charcoal-stripped serum on the day of transfection. Cell growth was determined 5 days later for LNCaP cells and 6 days later for abl and 22RV1 with the trypan blue exclusion method. Immunoblot for c-Myc protein expression. C) LNCaP cells with stable overexpression of empty vector (EV) or c-Myc were generated. These cells were transfected with 50 nM of non-targeted control (NTC) or AR siRNA oligonucleotides. Cell growth was determined 6 days later with the trypan blue exclusion method. Immunoblot for AR and c-Myc protein expression. The higher bands on the c-Myc immunoblot in the c-Myc-overexpressing cells represent the ectopically-expressed c-Myc. *denotes p<0.05 compared to NTC.
c-Myc Suppression Recapitulates the Effect of AR Suppression, and c-Myc Overexpression Attenuates the Anti-tumor Activity of AR Suppression
To determine if c-Myc also influenced prostate cancer cell growth independent of androgen ligands, we suppressed c-Myc using RNAi. Like AR downregulation, c-Myc down-regulation suppressed ligand-independent growth of LNCaP, abl, and 22RV1 cells (Figure 2B). Further, we simultaneously suppressed AR and c-Myc with RNAi. Co-suppression of both proteins did not reduce cell growth more than suppression of either AR or c-Myc by itself (Figure S1).
We also demonstrated that c-Myc overexpression conferred ligand-independent growth to ligand-dependent LNCaP cells propagated long-term in castrate conditions, which is concordant with a prior report (Figure S2) [21]. Next, we suppressed AR with RNAi in LNCaP cells overexpressing empty vector or c-Myc and quantified cell growth. c-Myc overexpression was protective against the growth suppressive effects of AR RNAi (Figure 2C). This demonstrates that c-Myc at least partially contributes to AR’s effects on promoting ligand-independent prostate cancer cell survival.
AR but not Androgens Promote c-Myc Expression
The c-Myc oncogene is commonly upregulated in prostate cancer, and c-Myc upregulation promotes ligand-independent prostate cancer cell survival [21]. However, the dependency of c-Myc expression on AR had not been established. Accordingly, we examined previously published ChIP microarray data that localized AR throughout the genome of androgen ligand-dependent LNCaP cells and their Abl CRPC derivatives [17]. AR was reported to be bound to an enhancer element of the c-Myc gene in both of these cell lines. Therefore, we next used ChIP to confirm these results.
First, we grew cells in charcoal-stripped, androgen ligand-depleted serum and determined the effect of treatment with the androgen ligand R1881 on AR occupancy and histone acetylation, a mark of active transcription, at the c-Myc enhancer element using ChIP. We also measured the effects of R1881 treatment at the well-described, ligand-activated gene KLK3. AR and high levels of histone acetylation were present at the c-Myc enhancer even when cells were grown in ligand-depleted serum (Figure 3A). Further, the addition of R1881 to culture did not enhance AR occupancy or histone acetylation at c-Myc (Figure 3A). This contrasts with the effect of R1881 at the KLK3 gene enhancer- increased enrichment of AR and histone acetylation (Figure 3A).
Figure 3. c-Myc expression is not activated by androgen ligands.

LNCaP, abl, and 22RV1 cells were grown in charcoal-stripped serum for 72 hours and then treated with 10 nM R1881 (or ethanol vehicle) for 4 hours. A) Chromatin immunoprecipitation was performed to determine the enrichment of AR and histone H3 acetylation (AcH3) at the c-Myc and KLK3 enhancer elements. B) QRT-PCR was performed to determine the mRNA levels of KLK3 and c-Myc relative to actin. C) Immunoblotting was performed to determine the protein levels of AR, c-Myc, and actin. *denotes p<0.05 compared to vehicle.
We next determined the effect of R1881 treatment on expression of c-Myc or KLK3. R1881 treatment increased KLK3 expression (Figure 3B). However, R1881 treatment did not increase c-Myc expression. Figure 3B,C).
Next, we treated prostate cancer cells with MDV3100, a potent, new androgen antagonist [26]. MDV3100 treatment suppressed expression of KLK3 but did not affect expression of c-Myc. (Figure S3). These results further support the notion that androgen ligands do not promote expression of c-Myc.
To determine if the AR was capable of regulating c-Myc in a ligand-independent manner, we used RNAi to suppress the expression of AR and measured c-Myc expression. RNAi-mediated suppression of AR reduced c-Myc mRNA and protein expression (Figure 4A, B). We performed ChIP assays and confirmed that AR RNAi reduced AR and histone acetylation from the c-Myc enhancer (Figure 4C). This was most significant in the 22RV1 cell line, although strong trends were also seen in LNCaP and Abl cells. Thus, c-Myc is a direct AR target gene, and AR RNAi suppresses c-Myc expression at least in part through depletion of AR and histone acetylation from the c-Myc enhancer. We also overexpressed AR in the M12 prostate cancer cell line that does not normally express AR. AR overexpression increased c-Myc mRNA and protein expression (Figure S4). This further supports the notion that AR activates c-Myc expression in a ligand-independent manner.
Figure 4. AR promotes ligand-independent expression of c-Myc.

LNCaP, abl and 22RV1 cells were transfected with 50 nM of non-targeted control (NTC) or AR RNAi oligonucleotides. Cells were then grown in charcoal-stripped serum for 96 hours. At the end of the treatment, cells were harvested to extract mRNA and protein. A) QRT-PCR was performed to determine the levels of c-Myc relative to actin. B) Immunoblotting was performed to determine the levels of AR, c-Myc and actin. C) Parallel treatments were performed and cells were cross-linked and processed for ChIP to determine AR and histone H3 acetylation (AcH3) enrichment at the c-Myc enhancer. *denotes p<0.05 compared to NTC.
AR Suppression Recapitulates the Effect of c-Myc Suppression
We next determined whether RNAi-mediated suppression of AR recapitulated the effect of RNAi-mediated suppression of c-Myc on expression of well-described c-Myc target genes (Figure 5) [27], [28]. Both c-Myc RNAi and AR RNAi reduced expression of the c-Myc-activated gene E2F1; conversely, c-Myc and AR RNAi both increased expression of the c-Myc-repressed gene CDKN1A (Figure 5). Recent reports demonstrate that mitotic genes, including KIF11, AURKB, and TPX2, are key c-Myc target genes [29]–[31]. AR RNAi recapitulated the effect of c-Myc RNAi and also reduced expression of these genes (Figure 5). This demonstrates that AR suppression disrupts c-Myc function and expression of well-established c-Myc target genes.
Figure 5. AR suppression recapitulates the effect of c-Myc suppression on c-Myc target gene expression.

LNCaP and abl cells were transfected with 50 nM of non-targeted control (NTC) and either A) AR or B) c-Myc RNAi oligonucleotides. Cells were switched to charcoal-stripped serum on the day of transfection and harvested 96 hours later. QRT-PCR was performed to determine the levels of the indicated c-Myc target genes relative to actin. *denotes p<0.05 compared to NTC.
The BET Bromodomain Inhibitor JQ1 Suppresses c-Myc Function and Reduces AR Ligand-independent Prostate Cancer Cell Survival
Our results demonstrate that c-Myc is an important AR target gene but that c-Myc’s expression is not activated by androgenic ligands. Currently, therapies to suppress AR expression are not yet available. However, recent work demonstrates that a BET bromodomain inhibitor called JQ1 suppresses c-Myc expression and c-Myc function because c-Myc is a bromodomain target gene [22], [23]. Therefore, we treated prostate cancer cells with JQ1. JQ1 treatment reduced mRNA and protein levels of c-Myc (Figure 6A,B) and suppressed c-Myc function as measured by c-Myc target gene expression (Figure 6B). Finally, like c-Myc RNAi, JQ1 treatment with nanomolar concentrations reduced ligand-independent prostate cancer cell survival (Figure 6C).
Figure 6. JQ1 treatment suppresses c-Myc expression and function and reduces ligand-independent prostate cancer cell survival.

LNCaP, abl, and 22RV1 cells were grown in charcoal-stripped serum and treated with vehicle, 50 nM, 250 nM or 500 nM JQ1 every 24 hours for 72 hours. A) Immunoblotting was performed to determine the protein levels of c-Myc. B) QRT-PCR was performed to determine the mRNA level of c-Myc and c-Myc targets genes KIF11, CDKN1A, TPX2, and AURKB relative to actin. *denotes p<0.05 compared to vehicle. C) Cell viability was determined at the end of treatment with the trypan blue exclusion method. p<0.01 for the 250 nM and 500 nM doses vs. vehicle in all three cell lines.
Discussion
It is well-appreciated that the AR is a critical driver of prostate cancer cell survival and that AR accounts for progression to fatal CRPC despite treatment with ADT [24]. In many cases androgens persist intracellularly within CRPC tumors despite castrate serum levels of androgens [24], [32]. However, androgen ligand-independent but AR-dependent mechanisms that also promote survival of CRPC cells have been reported. These include activation of the AR by IL-6, AR gene amplification, and AR transcript variants that lack the androgen ligand binding domain [7]–[9], [15]. All of these mechanisms may contribute to prostate cancer progression despite ADT since none are directly targeted by ADT.
Recently, it was demonstrated that the AR protein promotes the expression of a gene program distinct from its canonical androgen ligand-directed targets in CRPC cells [17]. One such example is the AR target gene UBE2C that promotes ligand-independent prostate cancer proliferation [17]. Which of the other AR-induced gene products is critical for ligand-independent prostate cancer cell survival has been unclear. Our work demonstrates that the c-Myc oncogene is such a ligand-independent AR target gene.
c-Myc is commonly upregulated in prostate cancer, and c-Myc overexpression transforms normal prostatic epithelial cells in genetically engineered mouse models of prostate cancer and confers ligand-independent prostate cancer cell survival, but the dependency of c-Myc expression on the AR was unclear [18], [20], [21], [33]. We show here that AR and c-Myc are commonly upregulated in CRPC, and we confirmed that AR and c-Myc upregulation strongly correlated with each other in a large series of metastatic CRPC patient tumors (Figure 1).
We confirmed that AR suppression leads to loss of c-Myc expression in prostate cancer cell lines expressing full-length AR (LNCaP and Abl) and in another CRPC cell line 22RV1 that expresses both full-length AR and an AR transcript variant. Although we cannot exclude a role for the AR transcript variant in also promoting c-Myc expression, our results with AR RNAi in LNCaP and Abl and our results with AR overexpression in M12 cells demonstrate that full-length AR is capable of activating c-Myc expression (Figure 4, Figure S4).
Like AR RNAi, c-Myc RNAi reduced prostate cancer cell survival in androgen ligand-depleted conditions while co-suppression of AR and c-Myc was not more effective than suppression of either protein alone (Figure 2, Figure S1). c-Myc overexpression confers ligand-independent survival to prostate cancer cells (Figure S2), which matches a prior report [21]. We also showed that c-Myc overexpression attenuated the anti-tumor activity of AR suppression with RNAi (Figure 2C). Thus, c-Myc contributes to AR’s effects on promoting ligand-independent prostate cancer cell survival.
Despite the fact that AR promotes expression of c-Myc, treatment with androgen ligand did not increase c-Myc expression (Figure 3). Additionally, treatment with androgen ligands did not enhance AR occupancy at the c-Myc enhancer (Figure 3). This contrasts with the effects of androgen stimulation on expression of the KLK3 gene, a well-described androgen-activated gene, and AR occupancy at the KLK3 enhancer (Figure 3). To further confirm that AR promotes expression of c-Myc in a ligand-independent manner, we treated prostate cancer cells with the new, potent androgen antagonist MDV3100 [26]. Treatment with MDV3100 in a recent randomized, placebo-controlled phase III clinical trial improved overall survival of patients with CRPC [34]. However, in some patients there is no tumor response, and at progression the AR remains in the nucleus [3], [6], [34]. While MDV3100 treatment reduced expression of KLK3, MDV3100 treatment had no effect on c-Myc expression (Figure S3). These data further support the notion that AR promotes c-Myc expression in a ligand-independent manner. c-Myc is a critical factor in ligand-independent prostate cancer progression (Figure 2, Figure S2) [21]. Therefore, in the future, it will be important to measure c-Myc expression and function in CRPC patient tumors progressing despite more complete androgen interference with drugs such as MDV3100.
Because of the importance of c-Myc as a downstream contributor to AR’s effects on ligand-independent prostate cancer cell survival, we treated prostate cancer cells with the BET bromodomain inhibitor JQ1, a drug known to suppress expression of bromodomain target genes; foremost among which was c-Myc [22], [23]. In prior studies, JQ1 treatment in vitro and in vivo suppressed c-Myc expression and function and suppressed tumor growth without appreciable toxicity [22], [23].
We confirmed that JQ1 treatment of prostate cancer cells using nanomolar concentrations achieved the same overall effects as RNAi-mediated suppression of c-Myc – c-Myc mRNA and protein depletion, suppression of c-Myc function, and suppression of ligand-independent prostate cancer cell survival (Figure 6). In light of the involvement of c-Myc in critical physiological processes, targeting the c-Myc protein generally in multiple cell types through long-term administration could be undesirable [35], [36]. Clinical trials will be necessary to determine the safety of bromodomain inhibition. However, the results to date, including our own, suggest that this is a promising anti-tumor strategy for the treatment of CRPC (Figure 6) [22], [23].
Finally, that the AR controls expression of its target genes such as c-Myc in a tissue specific manner suggests that the ideal agent for suppression of c-Myc expression in prostate cancer cells specifically would target AR, itself. Indeed, our studies clarify that androgen ligand-independent but AR-dependent c-Myc gene upregulation is a mechanism by which the AR protein promotes ligand-independent survival of prostate cancer cells. Our studies support the worthiness of efforts to suppress AR’s ligand-independent function and expression of important ligand-independent AR target genes such as c-Myc (Figure 7). Drugs capable of suppressing AR expression are only now beginning to enter testing. These drugs include selective AR degraders and AR anti-sense oligonucleotides [37]–[40]. We await the results of these clinical studies to determine the safety, specificity, and efficacy of these agents in men with advanced prostate cancer.
Figure 7. AR and c-Myc are critical drivers of ligand-independent mechanisms of prostate cancer progression.
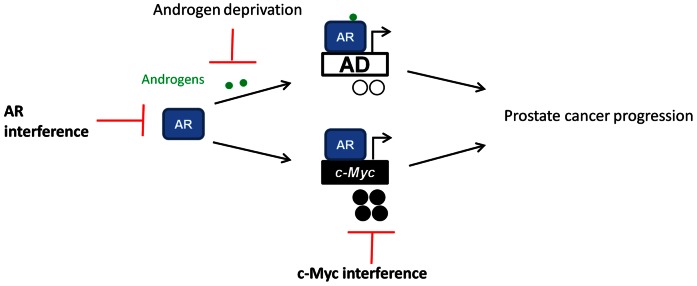
Currently, androgen deprivation therapies that interfere with androgen ligand activation of the AR are primarily used to treat this disease. These therapies suppress AR’s androgen ligand-dependent function and suppress expression of androgen ligand-dependent (AD) AR target genes. However, despite these treatments prostate cancer progression is inevitable. The AR also promotes the expression of androgen ligand-independent pathways such as c-Myc. The c-Myc gene is commonly upregulated in prostate cancer and contributes to androgen ligand-independent prostate cancer progression. This model strongly suggests that AR or c-Myc-directed therapies would complement current androgen deprivation strategies.
Methods
Cell Culture
LNCaP and 22RV1 cells were purchased from American Type Culture Collection (ATCC) and grown in 10% charcoal-stripped fetal bovine serum for all experiments. Abl cells, a CRPC derivative of LNCaP, were a kind gift from Zoran Culig, PhD and were grown in RPMI with 10% charcoal-stripped fetal bovine serum. M12 prostate cancer cells expressing empty vector or AR were a kind gift from Stephen R. Plymate, PhD, and grown as described previously [9].
For RNAi experiments, LNCaP and abl cells were transfected with RNAi oligonucleotides (AR: 5′-GACCUACCGAGGAGCUUUCUU-3′, and c-Myc: 5′-GAGCUAAAACGGAGCUUUUdTdT-3′) by using DharmaFECT 3 (Dharmacon) transfection reagent for a final concentration of 50 nM [41]. 22RV1 cells were transfected with siRNA oligonucleotides by using Lipofectamine2000 (Life Technologies) transfection reagent for a final concentration of 50 nM. Cells were harvested at indicated time points post-transfection.
R1881 (Sigma) was resuspended in 100% ethanol. MDV3100 was purchased (Selleckchem) and resuspended in DMSO. JQ1 was purchased from Sigma and resuspended in DMSO. Cell growth was determined at the end of treatment with the trypan blue exclusion method (Life Technologies) following manufacturers’ instructions. All cell culture experiments were performed with biological triplicate samples and confirmed in repeat experiments.
LNCaP cells with stable overexpression of c-Myc or empty vector were generated by transfecting LNCaP cells with pCDNA-DEST40-c-Myc (kindly provided by Rosalie Sears, PhD) or pCMV6-AN-His (Origene) and selecting with G418.
Colony-formation Assay
200,000 LNCaP cells with stable overexpression of empty vector (EV) or c-Myc were plated in 10 cm dishes. RPMI with 10% charcoal-stripped fetal bovine serum supplemented with 300 ug/ml G418 and 10 ug/ml bicalutamide treatment was added to the cells every other day for 14 days. Next, the cells were fixed with 4% formaldehyde and stained with syto60 (Invitrogen). Images were taken with Licor Odyssey imaging system.
Immunoblotting
Immunoblotting experiments were performed as described previously [42]. Final images were obtained with Licor Odyssey imaging system. Primary antibodies used were: AR (N-20, Santa Cruz); actin (Sigma) and c-Myc (Epitomics). Secondary antibodies were purchased from Licor.
QRT-PCR
RNA was extracted from cell pellets stored in RNAlater reagent (Life Technologies) using a MagMAX Total RNA Isolation kit (Life Technologies) or Trizol (Invitrogen) according to the manufacturer’s instructions. RNA concentration was determined using a NanoDrop ND-1000 spectrophotometer (NanoDrop). 250 ng–1 ug RNA (normalized for each experiment) was reverse-transcribed using a High-Capacity cDNA Reverse Transcription kit (Life Technologies) with random primers. Realtime (QRT)-PCR was performed using a 7500 Fast thermocycler (Life Technologies) with the following cycling program: 50°C for 2 min, 95°C for 10 min, 40 cycles of 95°C for 15 sec dissociation, 60°C for 1 min annealing/extension/read. 10 µL singleplex RTPCR reactions contained 1X Taqman universal standard mastermix, 1X Taqman hydrolysis probe, and 10 ng RNA-equivalent cDNA template. Human beta-actin (#4326315E) was used as an endogenous control. Primer information is included in Table S1. QRT-PCR for each biological replicate sample was performed with technical triplicates and analyzed with 7500 Software v2.0.5 and DataAssist Software v3.0 (Life Technologies).
Chromatin Immunoprecipitation (ChIP)
5 µg of anti-AR antibody (N-20, Santa Cruz), anti-acetylated histone H3 (AcH3) antibody (Millipore) or normal rabbit (IgG) antibody (Millipore) were added to sheared, formaldehyde cross-linked chromatin derived from 1 to 2×106 cells to immunoprecipitate DNA overnight at 4°C. 1% of chromatin was removed prior to immunoprecipitation as input. Immune complexes were collected with protein A magnetic beads (Dynabeads, Life Technologies). After extensive washing, immune complexes were released, cross-links were reversed, and DNA was purified with mini-elute PCR purification kit (Qiagen) and eluted with 60 µl EB. Realtime QPCR was performed as described above using 1X SYBR GreenER mastermix (Life Technologies), 750 ng of each primer, and 2 µl of the immunoprecipitated DNA or 2 µl of the 1% input in a 20 µL reaction. Immunoprecipitated DNA was calculated as “fold enrichment over IgG” with ddCt method. Primer information is included in Table S1.
Statistical Analysis
Data are expressed as standard deviation of the mean. All PCR results and cell count results represent one single experiment performed in triplicate. p-values were calculated from one single experiment with two-tailed unpaired student’s T-test in Excel (Microsoft) or Study Results 1.0 software. Each experiment was confirmed in two to three separate experiments.
AR ChIP-microarray Data Mining
The AR ChIP-microarray data was downloaded from http://research4.dfci.harvard.edu/brownlab/datasets/index.php?dir=Wang_AR_Data/for both abl and LNCaP cells. We filtered the data requiring that each peak have a FDR <5% and be within 50 Kb of either the 5′ or 3′ end of a RefSeq gene. Using these criteria, we found overlapping peaks with proximity to the c-Myc locus at chr8∶128844491–128845592 (∼21.6 Kb from c-Myc) in abl cells and chr8∶128844572–128845592 (∼21.7 Kb from c-Myc) in LNCaP cells.
AR and c-Myc Expression in Human Castration-Resistant Prostate Cancer Tumors
Agilent 44 K whole human genome expression oligonucleotide microarrays (Agilent Technologies, Inc.) were used to profile 140 human castration-resistant prostate cancer soft tissue metastases from 55 patients. Tissue samples were collected from autopsies performed at the University of Washington Medical Center under the rapid autopsy program with Institutional Review Board approval as described previously [43]. The tumor samples were laser-capture microdissected, and total RNA was isolated and amplified as described previously [44]. Probe labeling and hybridization were performed following the Agilent suggested protocols, and fluorescent array images were collected using the Agilent DNA microarray scanner G2565BA. Expression ratios were normalized using the R software Bioconductor snm package and combined with expression profiles from 15 normal prostate specimens to create z-scores, which represent the number of standard deviations away from the mean of expression in the normal prostate group [45], [46]. GraphPad Prism v4.03 software was used to analyze the correlation of expression and strength of association between genes. A Pearson correlation coefficient, linear regression, and F test for significantly non-zero slope were performed for each pair of genes as well as a Fisher’s exact test and odds ratio on the contingency table analyzing the co-occurrence of tumors with AR or c-Myc z-scores greater than 2.
Supporting Information
Co-suppression of AR and c-Myc does not lead to greater anti-tumor activity than suppression of either protein by itself. A) LNCaP cells were transfected with 50 nM of NTC, AR, c-Myc, or both AR and c-Myc RNAi oligonucleotides. Cells were switched to charcoal-stripped serum on the day of transfection. Cell growth was determined 5 days later with the trypan blue exclusion method. *denotes p<0.01 compared to NTC. B) Immunoblotting was performed to determine the levels of AR, c-Myc, and actin.
(TIF)
c-Myc over expression promotes ligand-independent prostate cancer growth. A) The same number of LNCaP cells with stable overexpression of empty vector or c-Myc were grown in 10% charcoal-stripped fetal bovine serum. Cell number was determined 1, 4, and 7 days after plating with the trypan blue exclusion method. Cell growth was calculated compared to day 1. p<0.01 for both time points. B) The same number of LNCaP cells with stable overexpression of empty vector or c-Myc was plated. Cells were grown in charcoal-stripped serum supplemented with bicalutamide for 14 days. Colony formation was determined.
(TIF)
MDV3100 treatment does not reduce c-Myc expression. LNCaP, abl, and 22RV1 cells were grown in androgen-replete serum and treated with 10 µM MDV3100 or vehicle for 24 hours. QRT-PCR was performed to determine the mRNA levels of KLK3 and c-Myc relative to actin. *denotes p<0.001 compared to vehicle.
(TIF)
AR overexpression promotes c-Myc upregulation. AR or empty vector was stably overexpressed in M12 prostate cancer cells that do not express endogenous AR [9]. RNA and protein were harvested. A) QRT-PCR was performed to determine the level of c-Myc relative to actin. B) Immunoblotting was performed to determine the levels of AR, c-Myc and actin. *denotes p<0.003 compared to empty vector.
(TIF)
Primers used for ChIP-PCR and QRTPCR.
(DOCX)
Funding Statement
This publication was made possible with support from the Oregon Clinical and Translational Research Institute, grant number KL2 RR024141 from the National Center for Research Resources and the National Center for Advancing Translational Sciences of the National Institutes of Health (JA). This work was also supported by the Pacific Northwest Prostate Cancer SPORE/National Cancer Institute (P50CA097186) (JA, PN, IC), the Department of Defense (PC093509) (PSN), a Flight Attendant Medical Research Institute Young Clinical Scientist Award (JA), a Wayne D. Kuni & Joan E. Kuni Foundation Kuni Scholar Award (JA), and a Prostate Cancer Foundation Young Investigator Award (JA). With special thanks to Platt Electric, Bruce Burns, and The Burns Family Fund of the Oregon Community Foundation for their philanthropic support of this work. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Siegel R, Naishadham D, Jemal A (2012) Cancer statistics, 2012. CA Cancer J Clin 62: 10–29. [DOI] [PubMed] [Google Scholar]
- 2. Taplin ME (2007) Drug insight: role of the androgen receptor in the development and progression of prostate cancer. Nat Clin Pract Oncol 4: 236–244. [DOI] [PubMed] [Google Scholar]
- 3. Scher HI, Beer TM, Higano C, Anand A, Taplin M-E, et al. (2010) Antitumour Activity of MDV3100 in a Phase 1–2 Study of Castration-Resistant Prostate Cancer. Lancet 375: 1437–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, et al. (2011) Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med 364: 1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Efstathiou E, Titus M, Tsavachidou D, Tzelepi V, Wen S, et al. (2012) Effects of abiraterone acetate on androgen signaling in castrate-resistant prostate cancer in bone. J Clin Oncol 30: 637–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Efstathiou E, Titus MA, Tsavachidou D, Hoang A, Karlou M, Wen S, et al.. (2011) MDV3100 effects on androgen receptor (AR) signaling and bone marrow testosterone concentration modulation: A preliminary report. J Clin Oncol 29: abstr 4501.
- 7. Guo Z, Yang X, Sun F, Jiang R, Linn DE, et al. (2009) A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res 69: 2305–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, et al. (2009) Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res 69: 16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, et al. (2010) Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest 120: 2715–2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, et al. (2011) Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 19: 575–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mulholland DJ, Tran LM, Li Y, Cai H, Morim A, et al. (2011) Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell 19: 792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gottlieb B, Beitel LK, Nadarajah A, Paliouras M, Trifiro M (2012) The androgen receptor gene mutations database: 2012 update. Hum Mutat 33: 887–894. [DOI] [PubMed] [Google Scholar]
- 13. Linja MJ, Savinainen KJ, Saramaki OR, Tammela TL, Vessella RL, et al. (2001) Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer Res 61: 3550–3555. [PubMed] [Google Scholar]
- 14. Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinanen R, et al. (1995) In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet 9: 401–406. [DOI] [PubMed] [Google Scholar]
- 15. Ueda T, Bruchovsky N, Sadar MD (2002) Activation of the androgen receptor N-terminal domain by interleukin-6 via MAPK and STAT3 signal transduction pathways. J Biol Chem 277: 7076–7085. [DOI] [PubMed] [Google Scholar]
- 16. Chang KH, Li R, Papari-Zareei M, Watumull L, Zhao YD, et al. (2011) Dihydrotestosterone synthesis bypasses testosterone to drive castration-resistant prostate cancer. Proc Natl Acad Sci U S A 108: 13728–13733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang Q, Li W, Zhang Y, Yuan X, Xu K, et al. (2009) Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 138: 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ellwood-Yen K, Graeber TG, Wongvipat J, Iruela-Arispe ML, Zhang J, et al. (2003) Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell 4: 223–238. [DOI] [PubMed] [Google Scholar]
- 19. Hawksworth D, Ravindranath L, Chen Y, Furusato B, Sesterhenn IA, et al. (2010) Overexpression of C-MYC oncogene in prostate cancer predicts biochemical recurrence. Prostate Cancer Prostatic Dis 13: 311–315. [DOI] [PubMed] [Google Scholar]
- 20. Jenkins RB, Qian J, Lieber MM, Bostwick DG (1997) Detection of c-myc oncogene amplification and chromosomal anomalies in metastatic prostatic carcinoma by fluorescence in situ hybridization. Cancer Res 57: 524–531. [PubMed] [Google Scholar]
- 21. Bernard D, Pourtier-Manzanedo A, Gil J, Beach DH (2003) Myc confers androgen-independent prostate cancer cell growth. J Clin Invest 112: 1724–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, et al. (2011) BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146: 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, et al. (2011) Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A 108: 16669–16674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, et al. (2004) Molecular determinants of resistance to antiandrogen therapy. Nat Med 10: 33–39. [DOI] [PubMed] [Google Scholar]
- 25. Tomlins SA, Mehra R, Rhodes DR, Cao X, Wang L, et al. (2007) Integrative molecular concept modeling of prostate cancer progression. Nat Genet 39: 41–51. [DOI] [PubMed] [Google Scholar]
- 26. Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, et al. (2009) Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 324: 787–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Claassen GF, Hann SR (2000) A role for transcriptional repression of p21CIP1 by c-Myc in overcoming transforming growth factor beta -induced cell-cycle arrest. Proc Natl Acad Sci U S A 97: 9498–9503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fernandez PC, Frank SR, Wang L, Schroeder M, Liu S, et al. (2003) Genomic targets of the human c-Myc protein. Genes Dev 17: 1115–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kessler JD, Kahle KT, Sun T, Meerbrey KL, Schlabach MR, et al. (2012) A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science 335: 348–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. den Hollander J, Rimpi S, Doherty JR, Rudelius M, Buck A, et al. (2010) Aurora kinases A and B are up-regulated by Myc and are essential for maintenance of the malignant state. Blood 116: 1498–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lal A, Navarro F, Maher CA, Maliszewski LE, Yan N, et al. (2009) miR-24 Inhibits cell proliferation by targeting E2F2, MYC, and other cell-cycle genes via binding to “seedless” 3'UTR microRNA recognition elements. Mol Cell 35: 610–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, et al. (2008) Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res 68: 4447–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hawksworth D, Ravindranath L, Chen Y, Furusato B, Sesterhenn IA, et al. (2010) Overexpression of C-MYC oncogene in prostate cancer predicts biochemical recurrence. Prostate Cancer Prostatic Dis 13: 311–315. [DOI] [PubMed] [Google Scholar]
- 34.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, et al.. (2012) Increased Survival with Enzalutamide in Prostate Cancer after Chemotherapy. N Engl J Med. [DOI] [PubMed]
- 35. Muncan V, Sansom OJ, Tertoolen L, Phesse TJ, Begthel H, et al. (2006) Rapid loss of intestinal crypts upon conditional deletion of the Wnt/Tcf-4 target gene c-Myc. Mol Cell Biol 26: 8418–8426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, et al. (2008) Modelling Myc inhibition as a cancer therapy. Nature 455: 679–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Snoek R, Cheng H, Margiotti K, Wafa LA, Wong CA, et al. (2009) In vivo knockdown of the androgen receptor results in growth inhibition and regression of well-established, castration-resistant prostate tumors. Clin Cancer Res 15: 39–47. [DOI] [PubMed] [Google Scholar]
- 38. Zhang Y, Castaneda S, Dumble M, Wang M, Mileski M, et al. (2011) Reduced expression of the androgen receptor by third generation of antisense shows antitumor activity in models of prostate cancer. Mol Cancer Ther 10: 2309–2319. [DOI] [PubMed] [Google Scholar]
- 39. Bradbury RH, Hales NJ, Rabow AA, Walker GE, Acton DG, et al. (2011) Small-molecule androgen receptor downregulators as an approach to treatment of advanced prostate cancer. Bioorg Med Chem Lett 21: 5442–5445. [DOI] [PubMed] [Google Scholar]
- 40.Loddick S, Bradbury R, Broadbent N, Campbell H, Gaughan L, et al.. (2012) Preclinical profile of AZD3514: A small molecule-targeting androgen receptor function with a novel mechanism of action and the potential to treat castration-resistant prostate cancer. Cancer Res 72: abstr 3848.
- 41. Haag P, Bektic J, Bartsch G, Klocker H, Eder IE (2005) Androgen receptor down regulation by small interference RNA induces cell growth inhibition in androgen sensitive as well as in androgen independent prostate cancer cells. J Steroid Biochem Mol Biol 96: 251–258. [DOI] [PubMed] [Google Scholar]
- 42. Gibbs A, Schwartzman J, Deng V, Alumkal J (2009) Sulforaphane destabilizes the androgen receptor in prostate cancer cells by inactivating histone deacetylase 6. Proc Natl Acad Sci U S A 106: 16663–16668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Morrissey C, True LD, Roudier MP, Coleman IM, Hawley S, et al. (2008) Differential expression of angiogenesis associated genes in prostate cancer bone, liver and lymph node metastases. Clin Exp Metastasis 25: 377–388. [DOI] [PubMed] [Google Scholar]
- 44. True L, Coleman I, Hawley S, Huang CY, Gifford D, et al. (2006) A molecular correlate to the Gleason grading system for prostate adenocarcinoma. Proc Natl Acad Sci U S A 103: 10991–10996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mecham BH, Nelson PS, Storey JD (2010) Supervised normalization of microarrays. Bioinformatics 26: 1308–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Page ST, Lin DW, Mostaghel EA, Marck BT, Wright JL, et al. (2011) Dihydrotestosterone administration does not increase intraprostatic androgen concentrations or alter prostate androgen action in healthy men: a randomized-controlled trial. J Clin Endocrinol Metab 96: 430–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Co-suppression of AR and c-Myc does not lead to greater anti-tumor activity than suppression of either protein by itself. A) LNCaP cells were transfected with 50 nM of NTC, AR, c-Myc, or both AR and c-Myc RNAi oligonucleotides. Cells were switched to charcoal-stripped serum on the day of transfection. Cell growth was determined 5 days later with the trypan blue exclusion method. *denotes p<0.01 compared to NTC. B) Immunoblotting was performed to determine the levels of AR, c-Myc, and actin.
(TIF)
c-Myc over expression promotes ligand-independent prostate cancer growth. A) The same number of LNCaP cells with stable overexpression of empty vector or c-Myc were grown in 10% charcoal-stripped fetal bovine serum. Cell number was determined 1, 4, and 7 days after plating with the trypan blue exclusion method. Cell growth was calculated compared to day 1. p<0.01 for both time points. B) The same number of LNCaP cells with stable overexpression of empty vector or c-Myc was plated. Cells were grown in charcoal-stripped serum supplemented with bicalutamide for 14 days. Colony formation was determined.
(TIF)
MDV3100 treatment does not reduce c-Myc expression. LNCaP, abl, and 22RV1 cells were grown in androgen-replete serum and treated with 10 µM MDV3100 or vehicle for 24 hours. QRT-PCR was performed to determine the mRNA levels of KLK3 and c-Myc relative to actin. *denotes p<0.001 compared to vehicle.
(TIF)
AR overexpression promotes c-Myc upregulation. AR or empty vector was stably overexpressed in M12 prostate cancer cells that do not express endogenous AR [9]. RNA and protein were harvested. A) QRT-PCR was performed to determine the level of c-Myc relative to actin. B) Immunoblotting was performed to determine the levels of AR, c-Myc and actin. *denotes p<0.003 compared to empty vector.
(TIF)
Primers used for ChIP-PCR and QRTPCR.
(DOCX)