

Abstract
Fluorescence Correlation Spectroscopy (FCS) is a frequently applied technique that allows for precise and sensitive analyses of molecular diffusion and interactions. However, the potential of FCS for in vitro or ex vivo studies has not been fully realized due in part to artifacts originating from autofluorescence (fluorescence of inherent components and fixative-induced fluorescence). Here, we propose the azadioxatriangulenium (ADOTA) dye as a solution to this problem. The lifetime of the ADOTA probe, about 19.4 ns, is much longer than most components of autofluorescence. Thus, it can be easily separated by time correlated single photon counting (TCSPC) methods. Here, we demonstrate the suppression of autofluorescence in FCS by using ADOTA labeled Hyaluronan macromolecules (HAs) with Rhodamine 123 added to simulate diffusing fluorescent background components. The emission spectrum and decay rate of Rhodamine 123 overlap with the usual sources of autofluorescence, and its diffusion behavior is well known. We show that the contributions from Rhodamine 123 can be eliminated by time-gating or by fluorescence lifetime correlation spectroscopy (FLCS). While the pairing of ADOTA and time-gating is an effective strategy for the removal of autofluorescence from fluorescence imaging, the loss of photons leads to erroneous concentration values with FCS. On the other hand, FLCS eliminates autofluorescence without such errors. We then show that both time gating and FLCS may be used successfully with ADOTA-labeled HA to detect the presence of hyaluronidase, the over-expression of which has been observed in many types of cancer.
Keywords: Fluorescence, Fluorescence Correlation Spectroscopy, Hyaluronan, Time Correlated Single Photon Counting
Introduction
Fluorescence Correlation Spectroscopy (FCS) is increasingly applied in the study of interact and mobility on the single molecule scale. The technique analyzes the fluctuations of fluorescent molecules as they diffuse through the detection volume of a confocal microscope. FCS has been widely employed in vitro to study molecular diffusion, which in turn provides information about the size of the molecule and/or the viscosity of the surrounding medium [1]. It has also been employed as a method for precise determination of the concentration of a particle of interest, a method that does not depend on the number of fluorescent probes attached to the particles [2]. FCS can also be combined with Förster Resonance Energy Transfer (FRET) to extract information about molecular interactions on the Angstrom (Å) scale, one molecule at a time, thereby avoiding the effects of averaging over millions of molecules, including those that were poorly labeled [3–6]. As the instrumentation involved in FCS is the same as that for confocal imaging, and the observation is performed from a single, diffraction-limited spot, the technique is ideally suited for in vitro studies involving heterogeneous mobility and concentration throughout a living cell. However, such studies are small in number, and one of the factors limiting the use of FCS is autofluorescence within cellular and tissue samples.
Autofluorescence from endogenous fluorophores is ubiquitous in biological samples and plagues fluorescence experiments. Even with advanced techniques and equipment, it is very hard to separate and eliminate autofluorescence, as the autofluorescent entities resemble commonly employed fluorescent probes. For example, the popular fluorescein and Rhodamine dyes are best used with a 470 nm, 488 nm, or 532 nm excitation sources, which unfortunately provide the most efficient excitation of flavins and flavoproteins [7, 8]. Furthermore, their emission spectra overlap, making spectral separation nearly impossible. Efforts to remove autofluorescence from fluorescence imaging have not been particularly successful, and most background suppression methods are completely unsuitable for measurements on the single molecule level. For example, the simplest solution is to overwhelm the background signal with heavy loading of the probe, but FCS requires very low concentrations of the probe (in the nM range) such that the fluorescence fluctuations do not average out [1]. Therefore, heavy loading of the probe to overcome autofluorescence is counterproductive. Single molecule experiments also require very high collection efficiencies, and thus chemical treatments [9–11] are generally unsuitable, as they reduce the signal from the probe as well as the background. Other methods of background suppression involve spectral unmixing of fluorescent species, but maybe the most frustrating problem aspect of autofluorescence is its variability between biological samples. Even if the emission spectrum of the probe is perfectly characterized, the variation in autofluorescence within and between samples makes it nearly impossible to characterize the autofluorescence spectrum well enough for these numerical methods [12, 13].
Due to the difficulty in spectral separation of autofluorescence and common probes, temporal separation of autofluorescence based on its rate of fluorescence decay could be advantageous. Unfortunately, there is a great deal of overlap in the decay rates of common organic dyes and autofluorescence, whose lifetimes range from ps up to 6 ns [14–16]. Thus we recently presented azadioxatriangulenium (ADOTA) dye [17, 18] as a promising solution to the problem of autofluorescence [19]. The fluorescence lifetime of ADOTA is much longer than the usual range of autofluorescence emission lifetimes, that is, it continues to fluoresce long after the autofluorescence has died out. The strategy, referred to as “time-gating,” entails ignoring all detected photons after each excitation pulse until the point where the background has decayed completely, and the probe continues to fluoresce. This method requires hardware equipped for time correlated single photon counting (TCSPC) with pulsed excitation sources, such that the fluorescence decay after each pulse can be analyzed. Unfortunately it also involves a loss of detected photons. It would seem that lanthanide-based probes, with lifetimes on the order of μs or ms [20], would be even more beneficial to the time gating procedure. However, lifetimes this long are associated with very low photon fluxes, making it very hard to collect adequate statistics. An adequate photon flux is important for imaging, but it is of dire importance for single molecule experiments, where collecting adequate statistics is the greatest challenge. FCS is especially difficult, because the photon count from each molecule is limited by its transit time through the detection volume. We have already demonstrated the use of ADOTA and its moderately long lifetime in the time-gated removal of autofluorescence from ocular tissue imaging [19].
Fluorescence lifetime correlation spectroscopy (FLCS) is an extension of FCS based on TCSPC that has been recently developed to separate the autocorrelation functions (ACFs) of multiple, distinct diffusing species in solution. This method assigns a weighting to each photon to base upon the probability of its origination from the species of interest, as determined by the elapsed time between the excitation and the photon’s collection, which depends upon the species’ respective lifetimes. The collection of weights as a function of delay time from excitation forms a continuous function, or filter. The benefit of FLCS is that photons are not excluded, and as a consequence, statistics are not seriously affected. Furthermore, since it is not necessary to wait for the short-lived species to die out before beginning detection, FLCS is able to form ACFs for separate species even when they have fairly similar lifetimes. However, FLCS alone does not solve the problem of autofluorescence as, once again, the heterogeneity of autofluorescence prevents one from characterizing its decay. Complicating things further, the lifetime of the probe can vary with its environment, the variation can be used as a means of identifying heterogeneity within a cell due to Förster Resonance energy transfer (FRET) or self-quenching. This phenomenon makes fluorescence lifetime imaging (FLIM) an active field of research. Therefore, a moderately long lifetime dye like ADOTA is needed [21], since its radiative decay rate is substantially different than typical autofluorescence. If, for example, the detected emission displays a two component exponential decay, 4 ns and 19 ns, one may say with near certainty that the 19 ns component is due solely to the probe with no influence from autofluorescent species.
As a model for our study, we used ADOTA and TCSPC separation techniques for the detection of hyaluronidase. The hyaluronidases (HA-ase) make up a family of enzymes that degrade Hyaluronan (HA), and its overexpression has been linked to several forms of cancer [22–25] as well as tumor activity [26, 25, 27]. Recently, we successfully demonstrated HA-ase detection by its digestion of fluorescein labeled HA as detected by cross-correlated FCS [28]. In this work we show that autofluorescence can be successfully removed from FCS experiments by the combination of TCSPC techniques (time-gating and FLCS) with the moderately long-lived dye ADOTA. We demonstrate these methods with the successful detection of hyaluronidase activity by time gated FCS and FLCS in the presence of simulated and severe autofluorescence. We use the dye Rhodamine 123 to simulate diffusing autofluorescence, as its fluorescent lifetime (4 ns) is in the typical range of the lifetime of autofluorescence. This system is an ideal test for these autofluorescence separation techniques, due to the great difference in size between HA and Rhodamine 123 molecules. The difference in size leads to a large, easily detectable difference in diffusion rates, by which one may confirm the removal of emission from the fast-moving Rhodamine 123. We first demonstrate the separation techniques on a mixture of HA labeled with ADOTA (HA-ADOTA) and Rhodamine 123, assessing the accuracy of each technique in determining diffusion speed and concentration. Then we show that an assay may be constructed for the detection of hyaluronidase in the presence of an overabundance of autofluorescence, simulated by Rhodamine 123.
Materials and Methods
Sodium hyaluronate from bacterial fermentation was obtained from Acros Organics (Thermo Fisher Scientific, NJ, USA). Dimethyl sulfoxide (DMSO), guanidine hydrochloride, acetaldehyde, cyclohexyl isocyanide, Sephadex G-75, and bovine testes hyaluronidase (EC 3.2.1.35, type 1-S, 451 U/mg) all were obtained from Sigma–Aldrich (Sigma–Aldrich, St. Louis, MO, USA). Dulbecco’s phosphate-buffered saline (PBS) was purchased from Invitrogen Life Technologies (Invitrogen Corporation, CA, USA) and was adjusted to pH 6.0 with 0.1 N HCl after reconstitution in deionized water (dH2O). Slide-A-Lyser dialysis cassettes (10,000 molecular weight cutoff) were purchased from Pierce Chemical (Thermo Fisher Scientific). All compounds, solvents and materials were used as received; water was used directly from a Millipore purification system.
Preparation of Hyaluronan-ADOTA Probe (HA-ADOTA)
The starting material for the preparation of the active ester of the AzaDiOxaTriaAngulenium (ADOTA-NHS) was prepared as described by Martin and Smith [29]. Further information regarding this preparation is given in [19].
HA was dissolved to 1.25 mg/mL in dH2O and then diluted 1:2 in DMSO. The ADOTA-NHS was dissolved in DMSO and added to the HA solution for a final ADOTA-NHS concentration of 50 μg/mL. Acetaldehyde and cyclohexyl isocyanide were added to 0.04% (vol/vol) and the reaction was allowed to proceed for 48 h at 25 °C. Afterwards, the solution was diluted 1:14 in ethanol:guanidine HCl (50 μL of 3 M guanidine HCl per 900 μL of 100% ethanol) and the HA-ADOTA allowed to precipitate overnight at −20 °C. The precipitate was then dissolved in 1 mL of dH2O followed by extensive dialysis against dH2O.
HA-ADOTA with simulated autofluorescence
The HA-ADOTA was diluted with PBS (pH = 6) to 0.17 nM, before a volume of dilute Rhodamine 123 was added to simulate autofluorescence. Rhodamine 123 is an organic dye whose emission at 560 nm and lifetime ~4ns both overlap with most common sources of autofluorescence [7, 8]. Rhodamine derivatives are commonly employed in FCS measurements and calibrations [30], and thus it provides a very controlled FCS contaminate by which these methods could be tested. The sample was prepared to resemble a case of extreme autofluorescence, or, alternatively, an extremely low concentration of the probe in the presence of autofluorescence. Thus, the concentration of the HA-ADOTA probe in this solution was 0.16 nM compared to the 10.34 nM concentration of Rhodamine 123. It should be noted that the molecular brightness of Rhodamine 123 is much higher than that of ADOTA. Then, 50 μL of the sample were dropped onto a coverslip, and the detection volume was raised 20 μm above the top surface of the coverslip into the sample using the backscattering image. The fluorescence signal was collected for 10 min at room temperature.
Hyaluronidase (HA-ase) Assay
An assay was created to detect the presence of hyaluronidase by the digestions of the HA-ADOTA substrate in the presence of (simulated) autofluorescence. The HA-ADOTA solution was diluted to an appropriate level with PBS (pH=6) and distributed into 300 μL aliquots. Then 30 μL of the Rhodamine 123 solution was added to each aliquot, followed by 20 μL of the hyaluronidase. The final concentrations of HA-ADOTA and Rh123 were 3nM and 2nM, respectively. With each measurement, the sample was incubated at room temperature for 30 min after the addition of hyaluronidase, and data was collected for 10 min at room temperature. For a control, the same procedure was followed, but 30 μL of PBS (pH=6) was added instead of the Rhodamine 123 solution to each aliquot. The same series of measurements with known levels of HA-ase were performed on the control solutions.
Equipment
Measurements were conducted on a Microtime 200 system from PicoQuant, GmbH (Berlin, Germany). The system was arranged as the shown in Figure 1. Excitation was provided by a 470 nm pulsed laser diode operating at 13 MHz. With the 19.4 ns lifetime of ADOTA, a low repetition rate was desired so that the dye could completely decay before the next pulse arrived. However, one must bear in mind that while data is being collected on the ps scale in relation to the excitation pulse, it is simultaneously being collected on the nanosecond scale in order to elucidate the diffusion behavior of the molecule. Diffusion is generally observed in the range of time lags from 100 ns to 1 s, and therefore a laser repetition rate of 10 MHz, or one pulse every 100 ns, will lead to artifacts in the correlation. The laser pulses were directed into the sample by a 60x 1.2 numerical aperture water immersion objective, part of an Olympus IX71 microscope. Samples were placed upon a No. 1 glass coverslip from Menzel-Gläser (Gerhard Menzel GmbH, Braunschweig, Germany). Scattered light was removed by a 488 nm long-pass filter, and the light passed through a 50 μm pinhole. Detection was made by a single hybrid photomultiplier assembly (PicoQuant). This hybrid detector records data free of afterpulsing, which makes a dual detector, cross-correlation setup unnecessary, and thus improves the signal to noise ratio. All data processing was performed by the SymPhoTime software, version 5.3.2, also from PicoQuant. Determining the size of the detection volume is of critical importance for quantitative FCS measurements, and its size is easily influenced by many factors: the objective and its correction collar setting, laser power, the coverslip, etc. Thus the volume was first calibrated by measuring Rhodamine 123, whose diffusion constant is well established [30].
Figure 1.
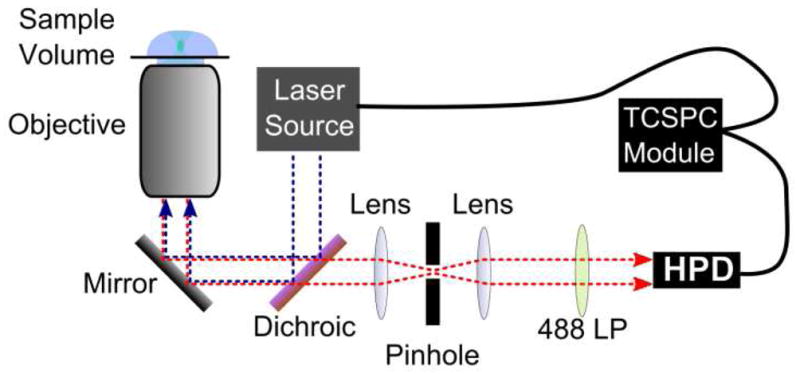
Setup for fluorescence correlation spectroscopy (FCS) and fluorescence lifetime correlation spectroscopy (FLCS). Excitation is provided by a 470 nm pulsed diode laser. It is reflected into a 60x 1.2 NA water immersion objective by a 49 wave pass filter. Detection was made with a hybrid photomultiplier detector assembly, and the signal was synchronized with the excitation by a time correlated single photon counting module.
Data Analysis
FCS analyses the fluctuation of emitted fluorescence over a period of time as the fluorescent source moves in and out of the microscope detection volume. FCS can only be successful if the number of fluorescent molecules flowing through the detection volume is small. Otherwise, molecules enter the detection volume as others exit, and the signal averages out to the mean. The upper limit on the concentrations available for study is determined by the size of the detection volume. It follows that, if the detection volume is precisely known, an unknown concentration may be easily obtained from FCS. For a detailed explanation of the autocorrelation performed upon the fluorescence fluctuation data, the fitting of the diffusion model to the autocorrelation data, and the calculation of concentration from this data, we refer the reader to the Electronic Supplementary Material.
The first method for the elimination of autofluorescence that we will employ is time gated detection. This involves discarding the photons that arrive soon after the excitation pulse, thereby eliminating a larger portion of the short-lived signal from autofluorescence than the long-lived probe. In the Electronic Supplementary Material, we present calculations showing that. With a 20 ns lifetime probe, 4 ns lifetime background, and a time gate that disregards all photons arriving within 30 ns of each excitation pulse, almost the entire background signal is eliminated, while 78% of the signal is eliminated.
FLCS is another method of separating fluorescent species based upon their radiative decay in the ps time range. The benefit of FLCS is that it does not exclude detected photons from the autocorrelation function. Rather, it assigns each photon a weight based upon the probability of it originating from the probe [31]. In the system presented here, we have a solution of HA macromolecules heavily labeled with the long-lived ADOTA (19.4 ns lifetime) combined with the comparatively short-lived Rhodamine 123 (~4 ns lifetime) to simulate autofluorescence. Figure 2 shows the fluorescence decay of ADOTA only (red) and Rhodamine 123 only (black) overlaid with the decay of the mixed solution (blue). It is clear that the decay of the mixed solution is a linear combination of the decay patterns from the two individual constituents of the mixture. First the decay of the mixed solution was fitted to an exponential decay model, and the following three components were determined: 1.4 ns, 3.8 ns, and 19.4 ns. The shortest component is largely due to the influence of Raman scattering, which is unavoidable in such experiments. The 3.8 ns component comes from Rhodamine 123, and the 19.4 ns component comes from ADOTA. The correct assignment of each component is critical for forming the FLCS filter, and it is here that we see the benefit of using ADOTA. The decay rate of autofluorescence can vary anywhere between the ps level and 6 ns [14–16]. Therefore it is very difficult to determine if a 4.0 ns component, for example, characterizes the probe, the autofluorescence, or a combination of the two. The restrictions to concentration on FCS make it impossible to simply overwhelm the autofluorescence by heavy loading with the probe. Once the lifetime component belonging to the probe is determined, the FLCS filter function can be constructed assigning weights to each photon. For a more detailed explanation of the FLCS filter construction, we refer the reader to [31]. The filter used in this experiment is displayed in the lower pane of Figure 2, which shows that the photons arriving soon after the excitation pulse are negatively weighted, as they most likely originated from Rhodamine 123. We excluded photons within the first 1 ns of the excitation pulse to exclude the strongest effects of Raman scattering.
Figure 2.
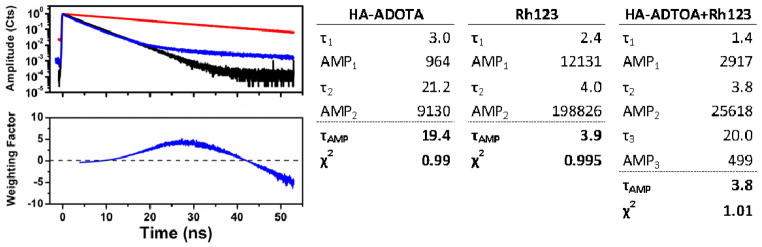
(top left) Decay curves corresponding to HA-ADOTA (red), Rhodamine 123 (black), and the combined solution of HA-ADOTA and Rhodamine 123 (blue). The table on the right displays the corresponding fitting parameters corresponding to each decay curve. (bottom left) the filter constructed from the long, 19.4 ns, component resulting from ADOTA.
Results and discussion
Removal of Autofluorescence from HA-ADOTA solution
A solution of HA-ADOTA was combined with a Rhodamine 123 solution to simulate the background from autofluorescent material, which could be found in biological material. The concentration of of HA-ADOTA in the final solution was 0.16 nM, and the concentration of Rhodamine 123 was 10.34 nM. Figure 3 shows that the autocorrelation constructed from the time trace of this mixture shows the contributions from both the fast moving dye and the slow moving HA-ADOTA, and it is clear that a one-component, pure diffusion fitting model is insufficient. A two component fitting model can adequately describe the system, but as we will show in the next section, it lacks the capability to separate the diffusion characteristics of the two species accurately. Besides, a two component diffusion fitting would not always be possible when autofluorescence is involved, because the diffusion characteristics are not likely to be homogenous. Thus a time-resolved solution was employed through time gating. Figure 3 shows the autocorrelation formed from data time-gated with progressively larger time delays from the excitation pulse. With larger time delays, the single species, pure diffusion model produces a better fit, but the statistics grow progressively worse as the time delay is increased—photons originating from HA-ADOTA as well as Rh123 are excluded. A 15 ns delay results in a 42% reduction of in the detection of photons from HA-ADOTA and a 78% reduction in detection of photons originating from Rhodamine 123. It is possible to retain more of the photons from HA-ADOTA because of its long lifetime. However, the signal from HA-ADOTA is still diminished by the time-gating, so it is desirable to use the shortest time delay in which the autofluorescence and background are eliminated. With only a 15 ns time delay, the diffusion constant calculated from the resulting one species diffusion model is equal to that of a pure HA-ADOTA solution, within the error of the fitting routine. However, the autocorrelation factor at time lag 0 ms, G(0), continues to grow larger with delays longer than 15 ns, which leads to lower apparent concentrations. G(0) reaches a maximum at 30 ns time delay, before it starts to decline, due simply to lack of counts. At this point, G(0) is still 37% lower than that measured from a pure HA-ADOTA solution of equal concentration without time gating, and this results in a 59% overestimate of HA-ADOTA concentration.
Figure 3.
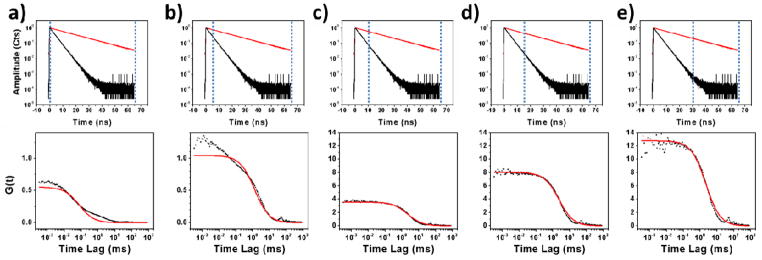
(top row) Decay curves of ADOTA (red) and Rhodamine 123 (black) with the time gating location represented by dotted blue lines. The time gate is located at (a) 0 ns, (b) 5 ns, (c) 10 ns, (d) 15 ns, and (e) 30 ns. One can see that the time gating eliminates much more of the signal from Rhodamine 123 than from ADOTA. (bottom row) Auto-correlation curves (black dots) formed from the HA-ADOTA solution mixed with Rhodamine 123 and a single component diffusion model fit to the data points (red). Each autocorrelation corresponds to the time gating shown above it. One can see from visual inspection (and an increase in χ2 from 63.0 to 6.0) that the data fitting improves significantly with the application of time-gating up to 10 ns. After 10 ns, the data fitting does not improve significantly, but G(0) continues to increase.
Next, FLCS was applied to the same data set in order to compare the two methods of filtering the background. The decay pattern was fitted to a three component pattern: two components assigned to scattering and Rhodamine 123 (1.4 ns and 3.8 ns), and one component assigned to ADOTA (20.0 ns). This assignment can be easily and confidently made due to the large difference in lifetime between ADOTA and the background constituents. A weighting function was then formed to correspond to the 20.0 ns component. The function, shown in Figure 2, negatively weights the photons arriving near in time to the excitation pulse, as these are likely to originate from the short-lived, background components of the fluorescence decay. The weighting function also positively weights the later-arriving photons likely to originate from ADOTA. As is seen in Figure 4, the autocorrelation formed using the FLCS method shows identical diffusion characteristics to that of pure HA-ADOTA. However, time gating left only 0.31 % of the originally detected photons to form the autocorrelation. In contrast, FLCS effectively leaves 6.95% of the original photons (after weighting) for the autocorrelation, thereby providing better statistics. By retaining more of the photons originating from the targeted dye molecule, FLCS produces more reliable concentration values. From Figure 5, we see that G(0) calculated using FLCS is much closer to that of the pure HA-ADOTA solution, differing by only 7% and resulting in a 7% underestimation of the concentration of HA-ADOTA. We must note that these methods were demonstrated successfully despite the fact that the concentration of Rh123 was 10 x higher than HA-ADOTA with high molecular brightness than HA-ADOTA as well.
Figure 4.

Changes in diffusion rate with the suppression of background. The lines represent single component diffusion models fit to data collected from one experiment with combination HA-ADOTA and Rh 123 combination. The un-gated data (black) shows much faster diffusion, as Rh123 is more prominent. The apparent diffusion slows as Rh123 is suppressed with time gating at 5 ns (red), 55ns (magenta). The diffusion model resulting from the FLCS procedure (blue) is also shown along with that of a pure HA-ADOTA solution (dark gray).
Figure 5.
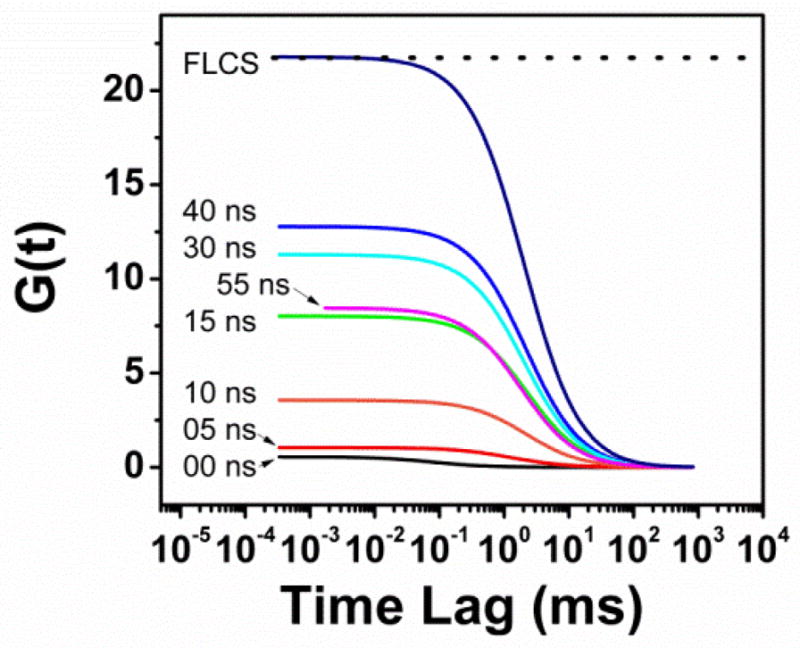
The effect of background suppression on G(0). As the effects from Rh123 are eliminated with time gating, G(0) increases until the lack of signal leads to erroneously low G(0) at 55 ns gating. It is seen that the FLCS method is much better for determining concentration, as it is nearly at the G(0) level for the pure HA-ADOTA solution (dotted line).
Suppression of Autofluorescence in HA-ase Assay
The above-mentioned method of autofluorescence suppression was employed in an assay for the detection of hyaluronidase activity. Various, known concentrations of hyaluronidase were added to HA-ADOTA solutions with Rh123 to simulate autofluorescence, and they were incubated for 30 min at RT. As the enzyme cleaved HA-ADOTA into many smaller molecules, the concentration of HA-ADOTA was monitored by FCS. As expected, FCS showed an increase in HA-ADOTA molecules with increasing hyaluronidase concentration, and a plot of the data is shown in Figure 6. The concentration is calculated by four methods: 1) a single component, pure diffusion model fit to the full data set, 2) a two component, pure diffusion model fit to the full data set, 3) a (one component) pure diffusion model fit to the autocorrelation formed after a 30 ns time-gate delay was applied, and 4) a (one component) pure diffusion model fit to the autocorrelation formed with an FLCS weighting function corresponding to 19.4 ns lifetime component originating from ADOTA. The concentrations of HA-ADOTA produced from these four methods are plotted alongside the control: a single component, pure diffusion model fit to the autocorrelation function collected from an HA-ADOTA solution of identical concentration cleaved by identical concentrations of hyaluronidase enzyme (no Rhodamine 123). From comparison with the control (no enzyme), it is quickly seen that the contributions to the signal from autofluorescence can drastically affect the concentration numbers calculated from simple, one component diffusion models. If the trend is assumed to be linear in this range of hyaluronidase concentration, the linear fit of each autofluorescence removal technique may be compared, and Table 1 shows the results. As expected, the one component and two component diffusion models exhibited such poor goodness of fit values that the information extracted is not meaningful. As before, the 30 ns gating method overestimated the HA-ADOTA concentration, while the FLCS method underestimated it, when compared to the one component diffusion model applied to the data from the solution of pure HA-ADOTA. However, the slopes of the linear fits for 30 ns gating and FLCS differed only by 3.2% and 2.2% respectively from that of pure HA-AD, thereby allowing a useful assay to be constructed after proper calibration.
Figure 6.
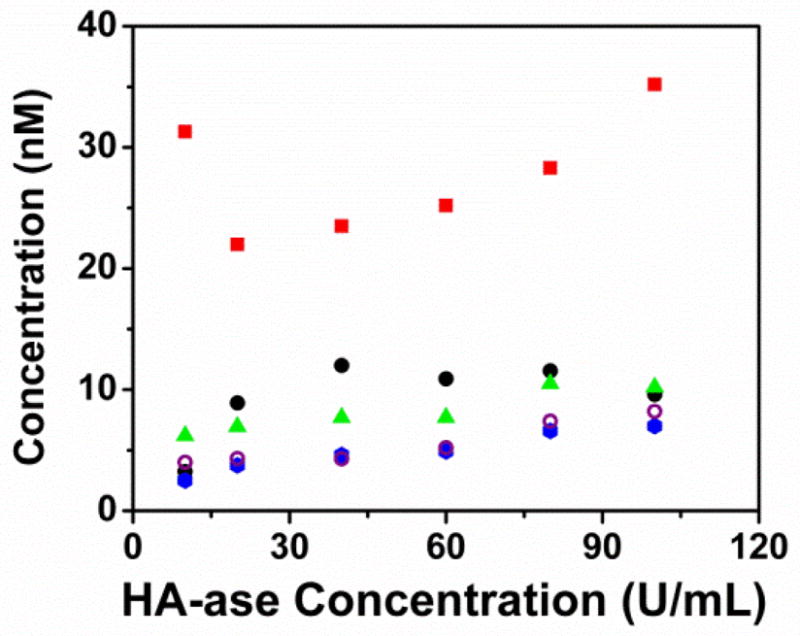
Concentration values collected during the HA-ase assay with the background provided by Rh123. At each HA-ase concentration, the concentration was extracted 4 ways: (red) a single species diffusion model fitted to un-gated data, (black) a two species diffusion model fitted to un-gated data, (green triangle) a single species diffusion model fitted to time gated data at 30 ns, (blue circle) a single species diffusion model fitted to data filtered by the FLCS method, and finally (open purple circles) a single species diffusion model fitted to data collected from an assay performed in the absence of Rh123. To preserve clarity, the linear regressions are omitted from this graph.
Table 1.
Fitting parameters from the linear regression performed on the 5 data sets shown in Figure 6.
| One Component | Two Component | 30 ns Gating | FLCS | HA-ADOTA One Component | |
|---|---|---|---|---|---|
| R-squared | 0.169 | 0.063 | 0.848 | 0.941 | 0.871 |
| Intercept | 6.603 +/−2.284 | 23.862 +/−3.778 | 5.776 +/−0.532 | 2.432 +/−0.323 | 3.054 +/−0.502 |
| Slope | 0.053 +/− 0.038 | 0.072 +/− 0.062 | 0.047 +/- 0.009 | 0.048 +/−0.005 | 0.049 +/−0.008 |
| % diff | 9.9% | 48.0% | 3.2% | 2.2% |
Conclusion
The combination of filtering on the ps scale and the employment of the moderately long-lived ADOTA dye provide a successful strategy to separate and eliminate unwanted autofluorescence during in vitro FCS experiments. The 19.4 ns lifetime of ADOTA dye is long enough that the signal of the probe can be easily distinguished from the wide variety of autofluorescence sources whose lifetimes vary from 0–5 ns, and it is still bright enough to employ in single molecule experiments like FCS. Rhodamine 123 was used to simulate diffusing autofluorescence sources, because its lifetime is 4 ns, and it is often used in FCS experiments. The time gating procedure quickly resolved the diffusion behavior of HA-ADOTA, even when eliminating only the first 15 ns of photons from each excitation cycle. However, much greater time gates were used to better determine the concentration of HA molecules, and the time gating method never fully resolved the true concentration—the reduction in photon statistics causes a systematic overestimation of concentration. Nonetheless, a successful assay can be constructed from this, as the true concentration of the HA substrate is not needed to calculate the concentration of hyaluronidase. The FLCS technique was able to successfully resolve the diffusion behavior and concentration, as this technique is not as detrimental to photon statistics as time gating. Though FLCS was more accurate in concentration values, it produced an identical trend to those of time gating and autofluorescence free HA-ADOTA in the hyaluronidase assay.
Supplementary Material
Acknowledgments
This work was supported by NIH grants R01EB12003 (Z.G.) and 1RO1HL090786-01A2 (J.B.) and the Danish Council for Independent Research, Technology and Production Sciences grant 10-093546 (T.J.S.). The authors would like to thank Dr. Badri Maliwal for helping with the preparation of ADOTA-NHS.
References
- 1.Lakowicz JR. Principles of Fluorescence Spectroscopy. Kluwer Academic/Plenum; 1999. [Google Scholar]
- 2.Weidemann T, Wachsmuth M, Knoch TA, Müller G, Waldeck W, Langowski J. Counting Nucleosomes in Living Cells with a Combination of Fluorescence Correlation Spectroscopy and Confocal Imaging. Journal of Molecular Biology. 2003;334:229–240. doi: 10.1016/j.jmb.2003.08.063. [DOI] [PubMed] [Google Scholar]
- 3.Fore S, Yuen Y, Hesselink L, Huser T. Pulsed-Interleaved Excitation FRET Measurements on Single Duplex DNA Molecules Inside C-Shaped Nanoapertures. Nano Lett. 2007;7:1749–1756. doi: 10.1021/nl070822v. [DOI] [PubMed] [Google Scholar]
- 4.Rarbach M, Kettling U, Koltermann A, Eigen M. Dual-Color Fluorescence Cross-Correlation Spectroscopy for Monitoring the Kinetics of Enzyme-Catalyzed Reactions. Methods. 2001;24:104–116. doi: 10.1006/meth.2001.1172. [DOI] [PubMed] [Google Scholar]
- 5.Kettling U, Koltermann A, Schwille P, Eigen M. Real-time enzyme kinetics monitored by dual-color fluorescence cross-correlation spectroscopy. PNAS. 1998;95:1416–1420. doi: 10.1073/pnas.95.4.1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koltermann A, Kettling U, Bieschke J, Winkler T, Eigen M. Rapid assay processing by integration of dual-color fluorescence cross-correlation spectroscopy: High throughput screening for enzyme activity. PNAS. 1998;95:1421–1426. doi: 10.1073/pnas.95.4.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vetrova EV, Kudryasheva NS, Visser AJWG, Van Hoek A. Characteristics of endogenous flavin fluorescence of Photobacterium leiognathi luciferase and Vibrio fischeri NAD(P)H:FMN-oxidoreductase. Luminescence. 2005;20:205–209. doi: 10.1002/bio.815. [DOI] [PubMed] [Google Scholar]
- 8.Visser AJWG, Ghisla S, Massey V, Müller F, Veeger C. Fluorescence Properties of Reduced Flavins and Flavoproteins. European Journal of Biochemistry. 1979;101:13–21. doi: 10.1111/j.1432-1033.1979.tb04210.x. [DOI] [PubMed] [Google Scholar]
- 9.Cowen T, Haven AJ, Burnstock G. Pontamine sky blue: A counterstain for background autofluorescence in fluorescence and immunofluorescence histochemistry. Histochemistry. 1985;82:205–208. doi: 10.1007/BF00501396. [DOI] [PubMed] [Google Scholar]
- 10.Schnell SA, Staines WA, Wessendorf MW. Reduction of Lipofuscin-like Autofluorescence in Fluorescently Labeled Tissue. J Histochem Cytochem. 1999;47:719–730. doi: 10.1177/002215549904700601. [DOI] [PubMed] [Google Scholar]
- 11.Clancy B, Cauller L. Reduction of background autofluorescence in brain sections following immersion in sodium borohydride. Journal of Neuroscience Methods. 1998;83:97–102. doi: 10.1016/s0165-0270(98)00066-1. [DOI] [PubMed] [Google Scholar]
- 12.Steinkamp JA, Stewart CC. Dual-laser, differential fluorescence correction method for reducing cellular background autofluorescence. Cytometry. 1986;7:566–574. doi: 10.1002/cyto.990070611. [DOI] [PubMed] [Google Scholar]
- 13.Van de Lest CH, Versteeg EM, Veerkamp JH, Van Kuppevelt TH. Elimination of autofluorescence in immunofluorescence microscopy with digital image processing. J Histochem Cytochem. 1995;43:727–730. doi: 10.1177/43.7.7608528. [DOI] [PubMed] [Google Scholar]
- 14.Schweitzer D, Gaillard ER, Dillon J, Mullins RF, Russell S, Hoffmann B, Peters S, Hammer M, Biskup C. Time-Resolved Autofluorescence Imaging of Human Donor Retina Tissue from Donors with Significant Extramacular Drusen. IOVS. 2012;53:3376–3386. doi: 10.1167/iovs.11-8970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Becker W. Fluorescence lifetime imaging – techniques and applications. Journal of Microscopy. 2012;247:119–136. doi: 10.1111/j.1365-2818.2012.03618.x. [DOI] [PubMed] [Google Scholar]
- 16.Schneckenburger H, Wagner M, Weber P, Strauss WSL, Sailer R. Autofluorescence Lifetime Imaging of Cultivated Cells Using a UV Picosecond Laser Diode. Journal of Fluorescence. 2004;14:649–654. doi: 10.1023/b:jofl.0000039351.09916.cc. [DOI] [PubMed] [Google Scholar]
- 17.Laursen BW, Krebs FC. Synthesis of a Triazatriangulenium Salt. Angewandte Chemie International Edition. 2000;39:3432–3434. doi: 10.1002/1521-3773(20001002)39:19<3432::aid-anie3432>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 18.Laursen BW, Krebs FC. Synthesis, structure, and properties of azatriangulenlenium salts. Chemistry. 2001;7:1773–1783. doi: 10.1002/1521-3765(20010417)7:8<1773::aid-chem17730>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 19.Rich RM, Stankowska DL, Maliwal BP, Sørensen TJ, Laursen BW, Krishnamoorthy RR, Gryczynski Z, Borejdo J, Gryczynski I, Fudala R. Elimination of autofluorescence background from fluorescence tissue images by use of time-gated detection and the AzaDiOxaTriAngulenium (ADOTA) fluorophore. Anal Bioanal Chem. 2013;405:2065–2075. doi: 10.1007/s00216-012-6623-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eliseeva SV, Bünzli J-CG. Lanthanide luminescence for functional materials and bio-sciences. Chem Soc Rev. 2009;39:189–227. doi: 10.1039/b905604c. [DOI] [PubMed] [Google Scholar]
- 21.Dileesh S, Gopidas KR. Photoinduced electron transfer in azatriangulenium salts. Journal of Photochemistry and Photobiology A: Chemistry. 2004;162:115–120. [Google Scholar]
- 22.Madan AK, Pang Y, Wilkiemeyer MB, Yu D, Beech DJ. Increased hyaluronidase expression in more aggressive prostate adenocarcinoma. Oncol Rep. 1999;6:1431–1433. doi: 10.3892/or.6.6.1431. [DOI] [PubMed] [Google Scholar]
- 23.Pham HT, Block NL, Lokeshwar VB. Tumor-derived hyaluronidase: A diagnostic urine marker for high-grade bladder cancer. Cancer research. 57:778–783. [PubMed] [Google Scholar]
- 24.Franzmann EJ, Schroeder GL, Goodwin WJ, Weed DT, Fisher P, Lokeshwar VB. Expression of tumor markers hyaluronic acid and hyaluronidase (HYAL1) in head and neck tumors. International Journal of Cancer. 2003;106:438–445. doi: 10.1002/ijc.11252. [DOI] [PubMed] [Google Scholar]
- 25.Liu D, Pearlman E, Diaconu E, Guo K, Mori H, Haqqi T, Markowitz S, Willson J, Sy MS. Expression of hyaluronidase by tumor cells induces angiogenesis in vivo. PNAS. 1996;93:7832–7837. doi: 10.1073/pnas.93.15.7832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lokeshwar VB, Cerwinka WH, Lokeshwar BL. HYAL1 Hyaluronidase: A Molecular Determinant of Bladder Tumor Growth and Invasion. Cancer Res. 2005;65:2243–2250. doi: 10.1158/0008-5472.CAN-04-2805. [DOI] [PubMed] [Google Scholar]
- 27.Xu H, Ito T, Tawada A, Maeda H, Yamanokuchi H, Isahara K, Yoshida K, Uchiyama Y, Asari A. Effect of Hyaluronan Oligosaccharides on the Expression of Heat Shock Protein 72. J Biol Chem. 2002;277:17308–17314. doi: 10.1074/jbc.M112371200. [DOI] [PubMed] [Google Scholar]
- 28.Rich RM, Mummert M, Foldes-Papp Z, Gryczynski Z, Borejdo J, Gryczynski I, Fudala R. Detection of hyaluronidase activity using fluorescein labeled hyaluronic acid and Fluorescence Correlation Spectroscopy. Journal of Photochemistry and Photobiology B: Biology. 2012;116:7–12. doi: 10.1016/j.jphotobiol.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin JC, Smith RG. Factors Influencing the Basicities of Triarylcarbinols. The Synthesis of Sesquixanthydrol. J Am Chem Soc. 1964;86:2252–2256. [Google Scholar]
- 30.Gendron P-O, Avaltroni F, Wilkinson KJ. Diffusion Coefficients of Several Rhodamine Derivatives as Determined by Pulsed Field Gradient–Nuclear Magnetic Resonance and Fluorescence Correlation Spectroscopy. J Fluoresc. 2008;18:1093–1101. doi: 10.1007/s10895-008-0357-7. [DOI] [PubMed] [Google Scholar]
- 31.Kapusta P, Wahl M, Benda A, Hof M, Enderlein J. Fluorescence Lifetime Correlation Spectroscopy. J Fluoresc. 2007;17:43–48. doi: 10.1007/s10895-006-0145-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.