

Abstract
G-quadruplex (GQ) is a noncanonical nucleic acid structure that is formed by guanine rich sequences. Unless it is destabilized by proteins such as replication protein A (RPA), GQ could interfere with DNA metabolic functions, such as replication or repair. We studied RPA-mediated GQ unfolding using single-molecule FRET on two groups of GQ structures that have different loop lengths and different numbers of G-tetrad layers. We observed a linear increase in the steady-state stability of the GQ against RPA-mediated unfolding with increasing number of layers or decreasing loop length. The stability demonstrated by different GQ structures varied by at least three orders of magnitude. Those with shorter loops (less than three nucleotides long) or a greater number of layers (more than three layers) maintained a significant folded population even at physiological RPA concentration (≈1 μM), raising the possibility of physiological viability of such GQ structures. Finally, we measured the transition time between the start and end of the RPA-mediated GQ unfolding process to be 0.35 ± 0.10 s for all GQ constructs we studied, despite significant differences in their steady-state stabilities. We propose a two-step RPA-mediated GQ unfolding mechanism that is consistent with our observations.
Introduction
Nucleic acid sequences rich in guanosine are capable of adopting four-stranded noncanonical structures called G-quadruplex (GQ) (1–4). GQ structures consist of an arrangement in which each guanosine occupies a corner of a G-tetrad and the G-tetrad layers stack together to form the GQ structure. GQ structures are stabilized by several mechanisms, including Hoogsteen hydrogen bonding between the four guanosines of each G-tetrad, stacking of the G-tetrad layers, shielding of repulsions between guanosines (due to negative charges on the O6 molecules) by monovalent cations that intercalate in or between the tetrad layers, and hydration (5–8). Genome-wide computational analysis has identified several hundred thousand potential GQ-forming sites (PQS) in the human genome, and in vitro assays have demonstrated GQ formation by these PQSs (9–11). In particular, PQSs are overrepresented in or near the promoter regions of numerous genes and at the ends of chromosomes (telomeres) (11–15). The telomeric GQ and capping proteins associated with telomeres are considered to protect the chromosome ends (17), whereas the roles of the nontelomeric GQ sequences have not been well characterized. Over-representation of these nontelomeric sequences in or near promoter sites suggests that they might be involved in transcription-level gene-expression regulation. RNA sequences can also form GQ structures. In particular, it has been demonstrated that RNA GQs located in the 5′-UTR play a role in translational level gene-expression regulation (18–21). The unique structure of GQ has motivated studies in which GQ has been used as a specific drug target (22,23).
Despite the abundance of PQSs in the genome, particularly nontelomeric GQ, and various in vitro demonstrations of GQ formation, it has been more challenging to unambiguously prove the existence and relevance of GQ structures in vivo. Most recently, GQs were visualized in human cells at both telomeric and nontelomeric locations, and their formation was shown to be modulated during the cell cycle (24). In addition, recent genome-wide studies have provided significant evidence on the formation and function of nontelomeric GQ structures in vivo. These studies demonstrate that eliminating certain helicases that are known to have GQ unfolding activity, such as Pif1 or BLM, results in increased DNA breaks in regions of the genome containing PQSs and severe retardation of DNA replication (9,25,26). Hence, in the absence of proteins that unfold them, GQ structures form and act as blocks to the replication system. These results are important indicators of the significance of protein-GQ interactions and their effects on genomic stability. Generic sequence constraints and in vitro thermal stability measurements have traditionally been used to characterize stability of GQ structures. However, within the context of cellular environment, the main factors that destabilize nontelomeric GQ are the proteins interacting with them and competition with the C-rich complementary strand. In particular, stability against GQ-unfolding proteins does not necessarily correlate with thermal stability (27). Thermal stability measurements provide a measure of the free energy difference between the folded GQ and the unfolded conformation, which has to be overcome by the protein while it is unfolding the GQ. However, these measurements are not necessarily sensitive to structural complexities that could be very important in the context of protein-GQ interactions. For example, the footprint of one of the DNA-binding domains of replication protein A (RPA) is about three nucleotides long. Therefore, having a loop size of two or four nucleotides would affect the efficiency of establishing an initial contact in a way that may not be adequately described by the thermal stability of the GQ. After an initial contact is established, the thermal stability of the GQ would then determine the stability of the GQ against protein-mediated unfolding. Therefore, it is essential to measure the stability of GQ structures against destabilizing proteins for better estimates of their physiological viability. The abundance of proteins with GQ unfolding activity presents a considerable challenge for in vivo viability of these structures. With an emerging interest in GQ structures as potential drug targets and their role in gene-expression regulation (22,23), it is essential to probe protein-GQ interactions in systematic studies of GQ constructs with different structural properties.
In this study, we probed protein-GQ interactions by studying the unfolding of systematically varied GQ structures by RPA. The sequences under study were selected with the aim of quantifying the influence of layer and loop structures on the stability of GQ against protein-mediated unfolding. Such variations in the structure are particularly important in the case of nontelomeric GQ, which could be formed by a broad class of different sequences. RPA was selected as the model protein because it is the most abundant single-strand DNA (ssDNA)-binding protein in eukaryotes (∼1 μM concentration in vivo (28)) and plays important roles in DNA metabolism, including DNA replication and repair (29–31). In particular, RPA protects the ssDNA created during replication or repair from enzymatic attack and prevents Watson-Crick pairing with the complementary strand before the completion of the process. In addition, RPA is involved in resolving certain secondary DNA structures that are formed during replication or repair, either by directly unfolding them or by initially binding to such structures and recruiting other proteins for unfolding (32,33). Unless resolved, such structures, including GQ, might act as roadblocks against the normal progression of replication or repair. RPA has very high affinity for ssDNA and GQ, with a dissociation constant, kD, on the order of 1 nM, and its affinity for dsDNA is approximately three orders of magnitude lower (32). Previous bulk studies have shown that RPA can unfold several different telomeric and nontelomeric GQs (32,34,35); however, no study has systematically compared this unfolding activity in various GQ structures. The number of G-tetrad layers, loop and tail length (overhang sequences at the end of GQs) and their sequence, and folding conformation are some of the potentially important parameters that could determine the stability of GQ against protein-mediated unfolding.
In this study, we concentrated on the effects of two variables on the stability of GQs against RPA-mediated unfolding: the number of tetrad layers and the length of the loops. Four different constructs that have the same loop length and sequence (TTT) but different numbers of G-tetrad layers—two, three, four, and five—were used to determine the effect of number of layers on GQ stability against RPA-mediated unfolding. These GQ structures are named L2–L5 and have sequences of the form TT(GnT3)3GnTT, where 2 ≤ n ≤ 5. In addition, five GQ constructs with a fixed number of G-tetrad layers (three) but with loop lengths varying between one and five nucleotides (T to TTTTT) were used to determine the effect of loop length on GQ stability. These GQ structures are named O1–O5 and have sequences of the form TT(G3Tn)3G3TT where 1 ≤ n ≤ 5. We employed single-molecule Förster resonance energy transfer (smFRET) in these studies (36). Our experiments were performed at room temperature, near physiological pH (pH 7.5) and ionic strength (150 mM KCl and 2 mM MgCl2), whereas RPA was titrated to its physiological concentration. Our results show that GQ steady-state stability against RPA-mediated unfolding systematically increases with increasing number of tetrad layers or decreasing loop length. We also monitored real-time unfolding of GQ by RPA and measured the unfolding time. Interestingly, all GQ constructs, L2–L5 and O1–O5, were unfolded within very similar times by RPA once an initial binding was established. Finally, we propose a two step model for RPA-mediated GQ unfolding that is consistent with these results.
Material and Methods
DNA constructs
The following DNA constructs were purchased from Integrated DNA Technologies (Coralville, IA) and used for FRET studies:
Stem: 5′-biotin-GCCTCGCTGCCGTCGCCA-Cy5-3′
L2: 5′-Cy3-TTGGTTTGGTTTGGTTTGGTTTGGCGACGGCAGCGAGGC-3′,
L3: 5′-Cy3-TTGGGTTTGGGTTTGGGTTTGGGTTTGGCGACGGCAGCGAGGC-3′,
L4: 5′-Cy3-TTGGGGTTTGGGGTTTGGGGTTTGGGGTTTGGCGACGGCAGCGAGGC-3′,
L5: 5′-Cy3-TTGGGGGTTTGGGGGTTTGGGGGTTTGGGGGTTTGGCGACGGCAGCGAGGC-3′,
O1: 5′-Cy3-TTGGGTGGGTGGGTGGGTTTGGCGACGGCAGCGAGGC-3′,
O2: 5′-Cy3-TTGGGTTGGGTTGGGTTGGGTTTGGCGACGGCAGCGAGGC-3′,
O3: 5′-Cy3-TTGGGTTTGGGTTTGGGTTTGGGTTTGGCGACGGCAGCGAGGC-3′,
O4: 5′-Cy3-TTGGGTTTTGGGTTTTGGGTTTTGGGTTTGGCGACGGCAGCGAGGC-3′,
O5: 5′-Cy3-TTGGGTTTTTGGGTTTTTGGGTTTTTGGGTTTGGCGACGGCAGCGAGGC-3′,
The DNA sequences shown in bold letters are the GQ-forming segment. Note that L3 and O3 are the same constructs. The underlined thymines are used as spacers to minimize the interaction between the GQ structures and the nearby fluorophore. The stem strand was annealed to L2–L5 and O1–O5 and a partial duplex DNA with an 18-basepair (bp) duplex stem was formed (see Fig. 2). The two DNA oligonucleotides were mixed at 2.0 μM concentration in 10 mM Tris buffer, pH 7.5, and 50 mM NaCl. The mixture was heated and maintained at 95°C for 5 min and cooled down gradually by placing a heated block at room temperature until the sample reached room temperature.
Figure 2.
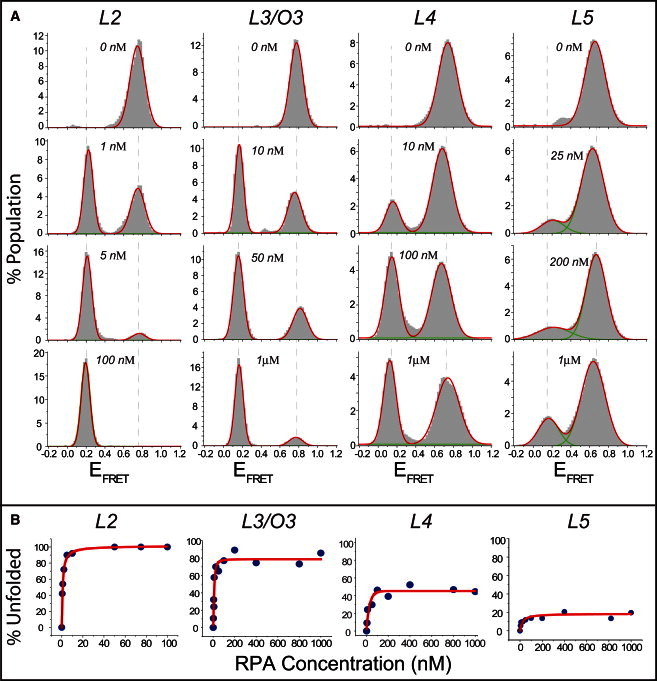
(A) smFRET data on unfolding of L2–L5 GQ constructs by varying concentrations of RPA. The concentration quoted on each histogram represents the concentration of RPA used for that measurement. High-FRET peaks represent the folded structure and low-FRET peaks the RPA-bound unfolded DNA. Each histogram represents RPA-mediated GQ unfolding at a particular RPA concentration. (B) Percentage of GQ molecules unfolded and bound by RPA as a function of RPA concentration for L2–L5. The red line shows a Langmuir binding isotherm fit to the data. The stability of GQ structures systematically increases as the number of G-tetrad layers is increased. L3–L5 molecules maintain a certain fraction of folded GQ molecules at RPA concentrations as high as 1 μM, whereas all L2 molecules are unfolded by 10 nM of RPA.
smFRET assay
Cleaned optical quartz slides and glass coverslips were used for preparing the imaging chambers. Surfaces were coated with a mixture of polyethylene-glycol and biotinylated polyethylene-glycol (m-PEG-5000 and biotin-PEG-5000, respectively; Laysan Bio, Arab, AL) to prevent nonspecific binding of DNA and RPA to the surface. Biotinylated DNA, at 15 pM concentration, was immobilized onto the biotin-PEG surface via neutravidin at 0.05 mg/ml concentration. For RPA titration studies, the GQ structures were first formed by incubating the surface-immobilized DNA constructs in 150 mM KCl for 15 min. RPA was then added to the chamber in an imaging solution (50 mM Tris, pH 7.5, 0.8 mg/ml glucose, 0.1 mg/ml bovine serum albumin, 140 mM β-mercaptoethanol, 0.1 mg/ml glucose oxidase, 0.02 mg/ml catalase, Trolox at saturating concentration, 2 mM MgCl2, and 150 mM KCl). Images were acquired after 15 min of incubation.
Imaging and data analysis
A prism-type total internal reflection fluorescence microscope built around an Olympus IX-71 microscope was used for these measurements. Movies 1000–2000 frames long were collected using an Andor Ixon electron-multiplying CCD camera (iXon DV 887-BI, Andor Technology, South Windsor, CT). An integration time of 100 ms was used for RPA titration experiments, and an integration time of 35 ms was used for flow experiments. Control flow experiments were also performed at 18-ms acquisition time. FRET time traces for individual molecules were analyzed to generate FRET histograms using a custom analysis program, and Origin Pro 8 was used for statistical analysis and curve fitting of FRET histograms.
Circular dichroism assay
Circular dichroism (CD) measurements were performed at room temperature using a Jasco J-810 spectrophotometer and a cuvette with a 0.1-cm pathlength. The measurements were performed at 150 mM K+ and 2 mM Mg2+. In addition, control measurements were performed at 150 mM Li+ and 2 mM Mg2+ to demonstrate effective GQ folding in the presence of K+ but not Li+. The DNA concentration was kept at 4 μM in all measurements.
RPA preparation
The RPA purification procedure was adapted from previous works (37,38). Briefly, Escherichia coli cells were transformed with a p11d-tRPA construct containing the coding sequences of RPA 70, RPA 14, and RPA 32. Upon reaching an OD600 of 0.6, protein expression was induced by adding isopropylthio-β-galactoside to a final concentration of 0.4 mM. Cells were lysed by pelleting and sonication. Cellular debris was pelleted by centrifugation at 12,000 rpm for 30 min. The supernatant was loaded onto an Affi-Gel Blue column (Bio-Rad, Hercules, CA). Protein was eluted using 1.5 M NaSCN in Hepes-Inositol buffer, pH 7.8. Eluted fractions containing protein were loaded onto a hydroxyapatite column to further concentrate the protein and eluted with Hepes-Inositol-80 mM phosphate buffer, pH 7.5. RPA purity was assayed by sodium dodecyl sulfate polyacrylamide gel electrophoresis. RPA functionality was confirmed in the context of its role in a DNA checkpoint complex using the assay described by Choi et al. (39).
Results and Discussion
Circular dichroism measurements
We performed CD measurements to confirm GQ formation by L2–L5 and O1–O5. Fig. 1 shows the results of these measurements, which were performed under physiologically germane ionic-strength and pH conditions (150 mM K+, 2 mM Mg2+, pH 7.5). A peak at 260 nm and a trough at 240 nm in the ellipticity measured by CD is consistent with parallel GQ conformation, whereas a peak at 290 nm and a trough at 260 nm is consistent with antiparallel GQ conformation (40). On the other hand, an ellipticity that has a peak around 290 nm and a shoulder around 260 nm has been interpreted as the hybrid conformation or a mixture of GQ molecules possessing parallel or antiparallel conformations (41,42). Given these interpretations, L2, O4, and O5 have antiparallel GQ conformation, whereas O1 and O2 have parallel conformation. On the other hand L3–L5 data are consistent with the hybrid conformation (a mixture of parallel and antiparallel conformations). The data on O1–O5 show an interesting trend in which the short loop constructs (O1 and O2) are consistent with the parallel GQ conformation, whereas the longer loop constructs (O4 and O5) are consistent with the antiparallel conformation. The O3 construct, with intermediate loop length, is the intermediate state between these two conformations and has an ellipticity consistent with that of the hybrid conformation. Control CD measurements in the absence of salt or in the presence of 150 mM Li+ were performed for all constructs (see Fig. S1 in the Supporting Material). These measurements show no signature of GQ formation for some constructs or a significantly weaker signal for others, compared to incubation at 150 mM K+. Therefore, these results are consistent with K+ being a more efficient stabilizer of the GQ structure than Li+. The CD data are merely used here to confirm GQ formation. Studying all the folding conformations independently is beyond the resolution of the methods used in this work, both CD and smFRET. For example, NMR measurements have shown as many as five different folding conformations for a construct similar to L3 (43). In particular, smFRET is sensitive to distance between donor and acceptor fluorophores that are placed at the ends of the GQ-forming sequence so as to cause minimal disturbance to the structure. However, different folding conformations typically result from different arrangements of the loops without a significant change in the end-to-end distance of GQ. All the results presented for a given GQ construct should be considered as the average of all possible conformations of that GQ construct. For demonstration purposes, we illustrate one of the possible folding conformations for all the GQ constructs (Fig. S2, B and C).
Figure 1.
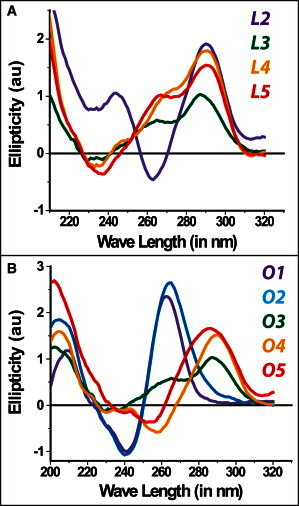
CD measurements on all the GQ constructs studied at 150 mM K+ and 2 mM Mg2+. (A) L2–L5 constructs. (B) O1–O5 constructs. Data on O1 and O2 are consistent with parallel GQs, whereas that on L2, O4, and O5 are consistent with antiparallel GQ conformations. Data on L3–L5 have signatures of both parallel and antiparallel GQ (the hybrid conformation).
Steady-sate smFRET measurements of RPA-mediated GQ unfolding
Prism-type total internal reflection fluorescence (TIRF) microscopy was used to perform smFRET measurements, as schematically shown in Fig. S2 A. The DNA constructs are in the form of a partial duplex with a double-stranded stem and a single-stranded overhang that folds into a GQ structure. The donor fluorophore (Cy3) is placed at the 5′ end of the single-stranded extension and the acceptor fluorophore (Cy5) is placed at the 3′ end of the double-stranded stem. When structured as GQ, the construct brings the donor-acceptor pair into close proximity, resulting in high FRET efficiency. On the other hand, unfolding of the GQ by RPA increases the distance between the donor-acceptor pair, resulting in low FRET efficiency. This low FRET efficiency peak is clearly distinguishable from the donor-only peak that is due to leakage through the dichroic (see Fig. S3 for an example). The duplex stem sequence is used to reduce the interaction of the GQ-forming sequence with the surface. As affinity of RPA to dsDNA is about three orders of magnitude smaller than its affinity to ssDNA or GQ structures, the interaction between RPA and the duplex stem is negligible (32,44,45). A control measurement was performed to demonstrate that RPA does not bind or modify this 18-bp duplex DNA (Fig. S4). A similar single-molecule assay has been used to study GQ formation of human telomeric repeats (27,46,47).
Proper folding of the oligonucleotides into G-quadruplex structure was established by monitoring the increase in FRET signal between the donor and acceptor fluorophores with increasing K+ ion concentration. As the GQ structure is stabilized with increasing K+ concentration, the donor and acceptor molecules move closer to each other, resulting in higher FRET. With a gradual increase in K+ concentration, various secondary structures form, which are manifested as different FRET peaks. All of these peaks eventually converge to a specific high-FRET state as K+ concentration is increased to 150 mM. We established formation of a stable structure by monitoring this progressive folding for all the constructs we studied. As shown in the example in Fig. S5, various structures are stabilized at different ionic strengths before the GQ becomes the dominant stable structure at 150 mM K+.
In the steady-state smFRET experiments, the constructs were incubated at 150 mM K+ and 2 mM Mg2+ for 15 min to attain a stable GQ conformation, except in the case of the O5 construct, which was incubated for 1 h. After proper folding of the GQ, different concentrations of RPA were introduced to the sample chamber while maintaining the same ionic strength and pH. Data were collected after 15 min of RPA incubation to attain a steady-state unfolding of GQ and binding of RPA to the unfolded DNA. Incubation times of RPA with DNA were varied between 5 and 30 min to determine the time necessary for attaining the steady state. Incubation times beyond 15 min did not make a difference in the fraction of GQ unfolded and bound by RPA.
Fig. 2 summarizes RPA-mediated GQ unfolding for L2–L5 constructs at different RPA concentrations. The high-FRET peaks (EFRET ≈ 0.7–0.8) in these data (Fig. 2 A) represent folded GQ structures, whereas the low-FRET peaks (EFRET ≈ 0.1–0.2) represent the RPA-bound unfolded conformations. The unfolded DNA that is bound by RPA demonstrates a significantly lower FRET efficiency compared to the unfolded DNA that is not bound by RPA (EFRET ≈ 0.3–0.6 depending on the length of the DNA construct) (48). The two states are distinguishable, as shown in the example in Fig. S6. Therefore, the low FRET peak we observe in Fig. 2 A represents the RPA-bound unfolded DNA. Larger fractions of GQ structures were unfolded and bound by RPA as the RPA concentration was increased, and eventually a saturating fraction was reached at a certain RPA concentration. We observed large variations among different GQ constructs in terms of their stability against RPA-mediated unfolding. As shown in Fig. 2 A, all of the L2 molecules were unfolded by <20 nM RPA, whereas a certain fraction of molecules in L3–L5 remain stably folded even at 1 μM RPA concentration. To quantify the unfolding phenomena, we plotted the percentage of GQ molecules unfolded and bound by RPA as a function of RPA concentration (Fig. 2 B). These curves were analyzed using a Langmuir binding isotherm of the form y = α[RPA]/([RPA] + Keq), where y describes the percentage of GQs unfolded and bound by RPA, [RPA] is the RPA concentration, Keq is the equilibrium constant, and α represents the percentage of unfolded and RPA-bound DNA at saturating RPA concentration. The α parameter accounts for the incomplete unfolding of L3, L4, and L5 at saturating RPA concentration. A summary of the fitting parameters for different constructs is given in Table 1. The steady-state stability of the GQ structures systematically increases as the number of G-tetrad layers is increased. In addition, the percentage of GQ molecules unfolded and bound by RPA at saturation (α parameter) systematically decreases as the number of G-tetrad layers is increased (see Fig. 4 A). The slope of the linear fit (see Fig. 4 A) suggests that each additional G-tetrad layer decreases the α parameter by 27%.
Table 1.
Summary of Langmuir binding isotherm analysis for all GQ constructs studied
| Fitting equation: y = α[RPA]/([RPA] + Keq) | ||
|---|---|---|
| Construct | α (%) | Keq (nM) |
| L2 | 100 | 0.8 ± 0.1 |
| L3 (O3) | 78.1 ± 3.5 | 11.5 ± 2.2 |
| L4 | 44.7 ± 2.6 | 23.1 ± 7.8 |
| L5 | 18.6 ± 1.0 | 22.1 ± 7.5 |
| O1 | 16.0 ± 0.9 | 81.2 ± 19.7 |
| O2 | 56.9 ± 2.9 | 7.1 ± 2.3 |
| O4 | 100 | 1.0 ± 0.1 |
| O5 | 100 | 0.6 ± 0.1 |
Figure 4.

(A) The α parameter, the percentage of GQ molecules unfolded and bound by RPA at saturating RPA concentration, is plotted for L2–L5, which have two to five G-tetrad layers, respectively. (B) The α parameter for O1–O5 constructs, which have two to five nucleotides, respectively, in each loop. The α parameter is obtained from the Langmuir binding isotherm analysis, as shown in Figs. 2B and 3B. The red lines are linear fits to the data, which are consistent with both data sets. The O5 data are not included in the fit, as 100% unfolding is already attained at the O4 construct.
The O1–O5 constructs were analyzed using methods similar to those presented for the L2–L5 constructs, as shown in Fig. 3. All of the O4 and O5 molecules were unfolded by 20 nM RPA, whereas some O1–O3 molecules remained folded even at 1 μM RPA (Fig. 3 A). Langmuir binding isotherm analysis was performed on these data and the results (α and Keq) are summarized in Table 1. The steady-state stability of the GQ structures systematically increases as the length of the loops is decreased and the α parameter systematically decreases as the length of the loops is decreased, as shown in Fig. 4 B. A linear fit to α parameters of O1–O4 constructs demonstrates a 26% increase with one nucleotide added to each loop (Fig. 4 B). O5 data were not included in the fitting, as 100% of O4 molecules were already unfolded and bound by RPA. Interestingly, for the GQ structures we studied, the effect on steady-state stability of reducing the length of each loop by one nucleotide is similar to that of adding a G-tetrad layer (26% vs. 27% change in α). Determining whether this is a coincidence due to the structures studied or a general feature of a broader class of GQ constructs would require systematic studies on a larger number of GQ constructs.
Figure 3.
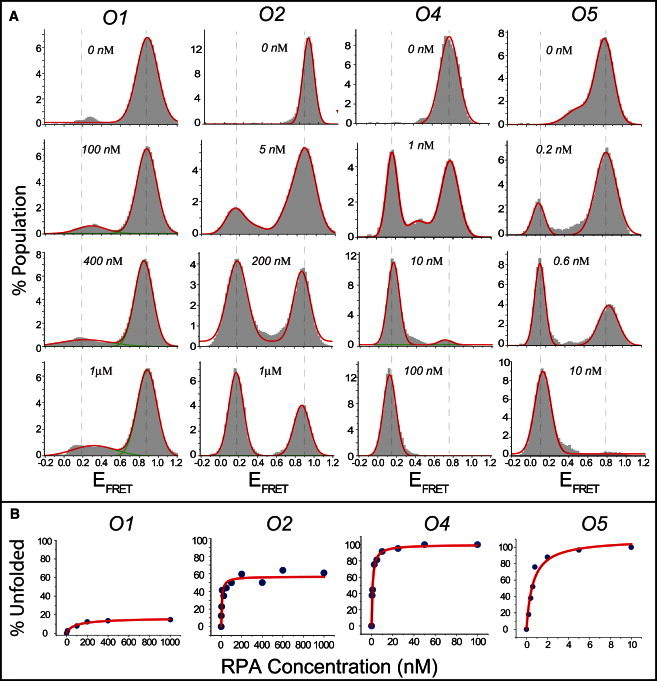
(A) smFRET data on unfolding of O1–O5 GQ constructs by varying concentrations of RPA. The concentration quoted on each histogram represents the concentration of RPA used for that measurement. (B) Percentage of GQ molecules unfolded and bound by RPA as a function of RPA concentration for O1–O5. The red line shows a Langmuir binding isotherm fit to the data. The stability of GQ structures systematically decreases as the loop length is increased. O1–O3 molecules maintain a certain fraction of folded GQ molecules at RPA concentrations as high as 1 μM, whereas all of the O4 and O5 molecules are unfolded by <20 nM of RPA.
Several control measurements were performed to assure the validity of the analysis, and conclusions presented in Figs. 2 and 3. In one of these measurements, we confirmed that all secondary DNA structures were removed in the RPA-bound unfolded state (see Fig. S7). A GQ-forming DNA (the L4 construct) and a polythymine DNA of very similar length (30 vs. 29 nucleotides long, respectively) were used for this study. The polythymine DNA was selected because it does not form any secondary structure. We demonstrated that the FRET peak representing the unfolded and RPA-bound state of L4 is identical to that of the RPA-bound polythymine DNA, suggesting that all secondary structures have been removed from the L4 construct. In another measurement, of the γ parameter for the Cy3-Cy5 pair before and after introducing RPA, we confirmed that the lower FRET peak observed upon introduction of RPA is not due to interactions of the fluorophores with RPA or the GQ structure (see Fig. S8).
An interesting aspect of the steady-state smFRET measurements is that a certain fraction of L3–L5 and O1–O3 molecules remain stably folded even at the highest RPA concentration studied. For all these constructs, a steady state is reached at ∼100 nM RPA, and adding more RPA to the environment does not give rise to any further significant unfolding of GQ. This suggests a dynamic binding/unbinding of RPA, although such was not observed in our 2- to 3-min-long single-molecule time traces. In particular, RPA binding to the unfolded DNA is very stable and essentially irreversible within our 2- to 3-min observation time. It is possible that RPA would dissociate and GQ would refold over longer time periods. To test this idea, movies 10–20 min long with 0.5-s integration time and very low laser power were acquired. Back-and-forth transitions between the folded, unfolded, and RPA-bound states were rarely observed even in these long movies. Examples of these rare transitions and stability of RPA binding are shown in Fig. S9.
A related issue is the stoichiometry of RPA binding. The length of our DNA constructs varies between 19 and 33 nucleotides, all of which lengths support stable binding of a single RPA, which can bind to 8- to 30-nucleotide-long ssDNA (as discussed in more detail below). The only possible exception to this is the L5 construct, which is 33 nucleotides long and therefore might possibly accommodate stable binding of one RPA and binding of one of the DNA-binding domains of a second RPA. In the smFRET histograms in Fig. 2 A, a single peak is observed for the RPA-bound and unfolded state of L5, suggesting that binding of a second RPA either does not take place or is a very rare event.
Dynamics of the RPA-mediated GQ unfolding process
Finally, single-molecule buffer-exchange measurements (called RPA flow) were performed to measure transition time from the folded GQ state to the RPA-bound unfolded state. In RPA flow experiments, the DNA constructs were incubated in a buffer that contains 150 mM K+ (in the absence of RPA) for 15 min to ensure proper folding of GQ, with the exception of O5, which was incubated for 1 h. A buffer containing 100 nM RPA and 150 mM K+ was flowed into the chamber by a microfluidic syringe pump while the folded GQ construct was being imaged, enabling us to monitor RPA-mediated GQ unfolding in real time. A representative single-molecule time trace of this unfolding process is shown in Fig. 5 A. The initial high-FRET state represents the folded GQ conformation, and the low-FRET state that follows corresponds to the RPA-bound unfolded state. The duration of time between these two states is called the unfolding time for brevity. The low-FRET state matches with the RPA-bound state obtained in the steady-state experiments, and it is different from the donor-only level (the FRET level after acceptor photobleaching takes place (Fig. 5 A)).
Figure 5.
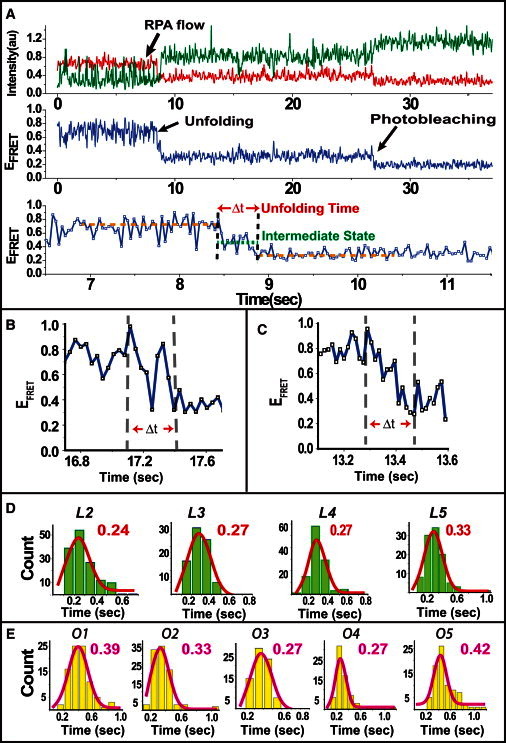
(A) An RPA flow experiment showing unfolding of a GQ by RPA in real time. As GQ is unfolded by RPA, FRET efficiency transitions from a high level, corresponding to the folded GQ structure, to a low level, corresponding to RPA-bound unfolded DNA. The bottom panel shows the section of the time trajectory that is in the vicinity of RPA binding and unfolding of the GQ. The unfolding time is marked by red arrows. We occasionally observed intermediate states during unfolding (green dotted line). (B) Example of an unfolding event in which the GQ molecule transitions back and forth between the folded and unfolded states. (C) Example of an unfolding event in which the transition from the folded to the unfolded state occurs gradually, without a distinct stable intermediate state. (D) Histograms of unfolding times (Δt) for L2–L5. (E) Histograms of unfolding times (Δt) for O1–O5. The red curves in D and E are Gaussian fits to the data. Interestingly all the constructs have similar unfolding times despite the large difference in their steady-state stability.
The unfolding time, shown as Δt in Fig. 5, A–C, is measured from the start of FRET decrease. The unfolding is considered to be complete when the FRET signal stabilizes at a low-FRET state representing the RPA-bound DNA structure. As shown in Fig. 5, A–C, several different types of unfolding patterns are observed in these measurements. In some cases, a clear intermediate FRET state is observed (Fig. 5 A, lower), whereas in other cases, the FRET signal transitions back and forth between the folded and unfolded states before stabilizing at the unfolded state (Fig. 5 B). Finally, in some cases, the FRET signal gradually reduces from the folded state to the unfolded state without an obvious intermediate state, as shown in Fig. 5 C. These intermediate states might be giving rise to the FRET populations between the folded and RPA-bound unfolded states in the steady-state histograms in Figs. 2 and 3. Regardless of the details of the unfolding process, the unfolding time is determined as the time between the stable high-FRET state and the stable low-FRET state, as shown in Fig. 5, A–C.
The movies for these measurements were acquired at a rate of 0.035 s/frame. However, the uncertainty in determining the beginning and end of the transition reduces our time resolution to 0.10 s for these measurements. Fig. 5 D shows the unfolding-time histograms for the constructs L2–L5, and Fig. 5 E shows similar histograms for O1–O5. The unfolding time represents a multistep process including GQ unfolding and RPA binding. The histograms in Fig. 5 D were fit by Gaussian curves, and the peak values were as follows: 0.24 ± 0.10 s, 0.27 ± 0.10 s, 0.27 ± 0.10 s, and 0.33 ± 0.10 s for L2–L5, respectively. Gaussian fits to histograms in Fig. 5 E resulted in peaks of 0.39 ± 0.10 s, 0.33 ± 0.10 s, 0.27 ± 0.10 s, 0.27 ± 0.10 s, and 0.42 ± 0.10 s for O1–O5, respectively. Given the uncertainty in determining the transition time, all constructs are essentially unfolded by RPA within very similar times of ∼0.35 ± 0.10 s. To ascertain whether the observed unfolding time was due to a limitation of the image acquisition time used for these measurements, we used half the CCD screen for image acquisition and reduced the image acquisition time to 0.018 s. The RPA flow experiment for L2 was repeated using this improved time resolution, and an unfolding time of 0.27 ± 0.05 s was found, which is consistent with the measurements at longer image-acquisition time (see Fig. S10 for a histogram and a sample trace). An example of a trace at this higher time resolution is shown in Fig. 5 C.
A model of RPA-mediated GQ unfolding
Unfolding of all the GQ constructs within very similar times is an interesting result, especially considering the orders-of-magnitude difference in their steady-state stabilities. It is important to consider the specifics of the RPA structure to better interpret these results. RPA is a heterotrimeric protein with subunits RPA1 (70 kDa), RPA2 (32 kDa), and RPA3 (14 kDa), and it has six ssDNA-binding domains (DBDs). Four of these DBDs (DBD-A, DBD-B, DBD-C, and DBD-D) are active and can bind to 8- to 30-nucleotide-long ssDNAs depending on the number of DBDs involved (49–52). For a recent review of the structure, DNA binding properties, and role in replication of RPA, see Prakash and Borgstahl (53). It is widely accepted that initial binding of RPA to ssDNA is achieved via binding of DBD-A and DBD-B, both in RPA1, and this initial interaction is further stabilized by successive binding of DBD-C and DBD-D. DBD-A and DBD-B each have a footprint of three nucleotides, and these three-nucleotide stretches are separated by a two-nucleotide stretch (52). This binding mode effectively enables RPA to stably bind to ssDNAs as short as eight nucleotides. However, three nucleotides may be enough for achieving an initial binding of DBD-A or DBD-B to ssDNA. In the rest of the discussion, we assume that DBD-A binds first and DBD-B binds second to make the description of our model easier to follow.
Based on the variation of stabilities we observed for different GQ constructs, and taking into account the structure of RPA, we propose a two-step model for RPA-mediated GQ unfolding (Fig. 6). The first step starts with RPA establishing contact with the GQ structure via binding of DBD-A to the available single-stranded regions, e.g., either the loops or the overhangs. The longer the loops or the overhangs, the easier it is to establish this initial contact. After this initial contact is achieved, RPA interacts with the folded GQ structure, which eventually leads to destabilization of the GQ and binding of DBD-B. The thermal stability of the GQ is an important parameter to consider here, as it would be more difficult for RPA to destabilize a GQ with higher thermal stability, e.g., more layers or shorter loops. The other important parameter is the affinity of DBD-B for ssDNA, which determines the level of the interactions between RPA and GQ. The second step of this model starts with destabilization of the GQ and is followed by binding of DBD-C and DBD-D to ssDNA as the GQ unfolds. Destabilization of the GQ is the onset of the FRET change we observe in RPA flow measurements. This model is consistent with the findings of SELEX studies suggesting that binding of either DBD-A or DBD-B results in a weakly bound state that would then enable RPA to invade longer stretches of DNA and destabilize the GQ (35). GQ structures that have four or five nucleotide loops (O4 and O5) are efficiently and completely unfolded by RPA, whereas those that have three or fewer nucleotide loops (O1–O3) are unfolded less efficiently and cannot attain complete unfolding of all molecules. For the cases of 1- to 2-nucleotide-long loops or overhangs, transient melting of the hydrogen bonds between the guanines that form the G-tetrad would be necessary before DBD-A or DBD-B can bind to the GQ. Such a melting event could take place due to thermal fluctuations or possibly due to RPA-GQ interactions. For the case of L2–L5 constructs, which all have three nucleotide loops, the stability of the GQ structure against thermal fluctuations or RPA-induced destabilization would increase with each additional layer. This would result in an increased stability against RPA-mediated GQ unfolding, as demonstrated in Fig. 2.
Figure 6.
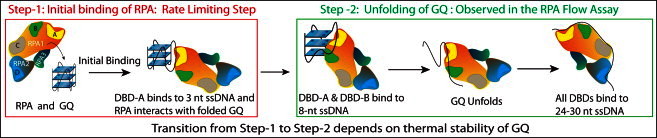
Cartoon of a model describing RPA-mediated GQ unfolding as a two-step process.
The observed dynamics of RPA-mediated GQ unfolding and unfolding of all the GQ constructs within very similar times can also be understood in the context of the two-step model proposed in Fig. 6. The unfolding time we measure in these measurements is essentially a measure of the second step of this interaction (Fig. 6, right). This step starts with measurable destabilization of GQ followed by binding of DBD-C and DBD-D as more ssDNA becomes available. The first step is not detected, as the GQ is still folded and FRET efficiency does not change significantly. As we do not observe any variation in the duration of the second step of the unfolding mechanism, we conclude that the first step determines the stability of GQ against RPA-mediated unfolding. The stability of GQ that is described in the first step of this model depends on both the thermal stability of the GQ and the length of available ssDNA in the loops or overhangs, which enable RPA to position in the vicinity of GQ and interact with it.
Conclusion
In conclusion, this study provides a systematic way to characterize the stability of GQs in terms of protein-mediated unfolding. RPA, the most abundant ssDNA-binding protein in eukaryotes, is expected to frequently interact with nontelomeric PQSs. Our data identify the effects of number of tetrad layers and loop lengths on the stability of GQ against RPA-mediated unfolding. Clearly, live-cell environment is significantly more complicated than the environment in in vitro assays, and there are various other factors that are known to destabilize GQ structures, e.g., helicases or competition with the complementary DNA strand. It is also possible that there might be direct or indirect mechanisms or factors that could stabilize the GQ in the context of chromatin. Nevertheless, this study provides what we believe to be a new perspective on the interactions of an abundant and very important protein with systematically varied GQ structures. Our results impose strict constraints on physiologically viable GQ structures. For example, GQs with four- or five-nucleotide-long loops are completely unfolded by RPA, suggesting that GQ structures with such long loops are not viable in a physiological setting. Finally, we propose a two-step model of RPA-GQ interactions that is consistent with our data. The quantitative data we present would be particularly important in providing guidelines for computational work on modeling the interaction of an arbitrary GQ structure with RPA. Extension of these studies to a broader class of GQs with systematically varied structures or with inclusion of multiprotein complexes would have the potential to make better estimates for the physiological viability of GQ structures.
Acknowledgments
We thank Prof. Aziz Sancar and members of his group for providing us with RPA. M.H.Q. thanks ICAM for funding during his training in the laboratory of Prof. Aziz Sancar. H.B. designed the experiments, S.R., M.H.Q., D.W.M., J.B.B., and U.Ç. performed the experiments and the analysis and edited the manuscript, and H.B. and S.R. wrote the article.
This work was supported by the start-up funds of H.B. from Kent State University.
Supporting Material
References
- 1.Blackburn E.H. Structure and function of telomeres. Nature. 1991;350:569–573. doi: 10.1038/350569a0. [DOI] [PubMed] [Google Scholar]
- 2.Gellert M., Lipsett M.N., Davies D.R. Helix formation by guanylic acid. Proc. Natl. Acad. Sci. USA. 1962;48:2013–2018. doi: 10.1073/pnas.48.12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gilbert D.E., Feigon J. Multistranded DNA structures. Curr. Opin. Struct. Biol. 1999;9:305–314. doi: 10.1016/S0959-440X(99)80041-4. [DOI] [PubMed] [Google Scholar]
- 4.Williamson J.R. G-quartet structures in telomeric DNA. Annu. Rev. Biophys. Biomol. Struct. 1994;23:703–730. doi: 10.1146/annurev.bb.23.060194.003415. [DOI] [PubMed] [Google Scholar]
- 5.Chaires J.B. Human telomeric G-quadruplex: thermodynamic and kinetic studies of telomeric quadruplex stability. FEBS J. 2010;277:1098–1106. doi: 10.1111/j.1742-4658.2009.07462.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gray R.D., Chaires J.B. Kinetics and mechanism of K+- and Na+-induced folding of models of human telomeric DNA into G-quadruplex structures. Nucleic Acids Res. 2008;36:4191–4203. doi: 10.1093/nar/gkn379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lane A.N., Chaires J.B., Trent J.O. Stability and kinetics of G-quadruplex structures. Nucleic Acids Res. 2008;36:5482–5515. doi: 10.1093/nar/gkn517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stegle O., Payet L., Leon J.H. Predicting and understanding the stability of G-quadruplexes. Bioinformatics. 2009;25:i374–i382. doi: 10.1093/bioinformatics/btp210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lipps H.J., Rhodes D. G-quadruplex structures: in vivo evidence and function. Trends Cell Biol. 2009;19:414–422. doi: 10.1016/j.tcb.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 10.Verma A., Yadav V.K., Chowdhury S. Evidence of genome-wide G4 DNA-mediated gene expression in human cancer cells. Nucleic Acids Res. 2009;37:4194–4204. doi: 10.1093/nar/gkn1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burge S., Parkinson G.N., Neidle S. Quadruplex DNA: sequence, topology and structure. Nucleic Acids Res. 2006;34:5402–5415. doi: 10.1093/nar/gkl655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reference deleted in proof.
- 13.Eddy J., Maizels N. Gene function correlates with potential for G4 DNA formation in the human genome. Nucleic Acids Res. 2006;34:3887–3896. doi: 10.1093/nar/gkl529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eddy J., Maizels N. Conserved elements with potential to form polymorphic G-quadruplex structures in the first intron of human genes. Nucleic Acids Res. 2008;36:1321–1333. doi: 10.1093/nar/gkm1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huppert J.L., Balasubramanian S. Prevalence of quadruplexes in the human genome. Nucleic Acids Res. 2005;33:2908–2916. doi: 10.1093/nar/gki609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reference deleted in proof.
- 17.Blackburn E.H., Greider C.W., Szostak J.W. Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging. Nat. Med. 2006;12:1133–1138. doi: 10.1038/nm1006-1133. [DOI] [PubMed] [Google Scholar]
- 18.Kumari S., Bugaut A., Balasubramanian S. An RNA G-quadruplex in the 5′ UTR of the NRAS proto-oncogene modulates translation. Nat. Chem. Biol. 2007;3:218–221. doi: 10.1038/nchembio864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morris M.J., Basu S. An unusually stable G-quadruplex within the 5′-UTR of the MT3 matrix metalloproteinase mRNA represses translation in eukaryotic cells. Biochemistry. 2009;48:5313–5319. doi: 10.1021/bi900498z. [DOI] [PubMed] [Google Scholar]
- 20.Morris M.J., Negishi Y., Basu S. An RNA G-quadruplex is essential for cap-independent translation initiation in human VEGF IRES. J. Am. Chem. Soc. 2010;132:17831–17839. doi: 10.1021/ja106287x. [DOI] [PubMed] [Google Scholar]
- 21.Huppert J.L., Bugaut A., Balasubramanian S. G-quadruplexes: the beginning and end of UTRs. Nucleic Acids Res. 2008;36:6260–6268. doi: 10.1093/nar/gkn511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Balasubramanian S., Hurley L.H., Neidle S. Targeting G-quadruplexes in gene promoters: a novel anticancer strategy? Nat. Rev. Drug Discov. 2011;10:261–275. doi: 10.1038/nrd3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Balasubramanian S., Neidle S. G-quadruplex nucleic acids as therapeutic targets. Curr. Opin. Chem. Biol. 2009;13:345–353. doi: 10.1016/j.cbpa.2009.04.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Biffi G., Tannahill D., Balasubramanian S. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 2013;5:182–186. doi: 10.1038/nchem.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paeschke K., Capra J.A., Zakian V.A. DNA replication through G-quadruplex motifs is promoted by the Saccharomyces cerevisiae Pif1 DNA helicase. Cell. 2011;145:678–691. doi: 10.1016/j.cell.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paeschke K., McDonald K.R., Zakian V.A. Telomeres: structures in need of unwinding. FEBS Lett. 2010;584:3760–3772. doi: 10.1016/j.febslet.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qureshi M.H., Ray S., Balci H. Replication protein A unfolds G-quadruplex structures with varying degrees of efficiency. J. Phys. Chem. B. 2012;116:5588–5594. doi: 10.1021/jp300546u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim C., Wold M.S. Recombinant human replication protein A binds to polynucleotides with low cooperativity. Biochemistry. 1995;34:2058–2064. doi: 10.1021/bi00006a028. [DOI] [PubMed] [Google Scholar]
- 29.Wold M.S. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 1997;66:61–92. doi: 10.1146/annurev.biochem.66.1.61. [DOI] [PubMed] [Google Scholar]
- 30.Iftode C., Daniely Y., Borowiec J.A. Replication protein A (RPA): the eukaryotic SSB. Crit. Rev. Biochem. Mol. Biol. 1999;34:141–180. doi: 10.1080/10409239991209255. [DOI] [PubMed] [Google Scholar]
- 31.Oakley G.G., Patrick S.M. Replication protein A: directing traffic at the intersection of replication and repair. Front. Biosci. 2010;15:883–900. doi: 10.2741/3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fan J.H., Bochkareva E., Gray D.M. Circular dichroism spectra and electrophoretic mobility shift assays show that human replication protein A binds and melts intramolecular G-quadruplex structures. Biochemistry. 2009;48:1099–1111. doi: 10.1021/bi801538h. [DOI] [PubMed] [Google Scholar]
- 33.Wu Y., Rawtani N., Brosh R.M., Jr. Human replication protein A melts a DNA triple helix structure in a potent and specific manner. Biochemistry. 2008;47:5068–5077. doi: 10.1021/bi702102d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salas T.R., Petruseva I., Saintomé C. Human replication protein A unfolds telomeric G-quadruplexes. Nucleic Acids Res. 2006;34:4857–4865. doi: 10.1093/nar/gkl564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prakash A., Natarajan A., Borgstahl G.E. Identification of the DNA-binding domains of human replication protein A that recognize G-quadruplex DNA. J. Nucleic Acids. 2011;2011:896947. doi: 10.4061/2011/896947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ha T., Enderle T., Weiss S. Probing the interaction between two single molecules: fluorescence resonance energy transfer between a single donor and a single acceptor. Proc. Natl. Acad. Sci. USA. 1996;93:6264–6268. doi: 10.1073/pnas.93.13.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Binz S.K., Dickson A.M., Wold M.S. Functional assays for replication protein A (RPA) Methods Enzymol. 2006;409:11–38. doi: 10.1016/S0076-6879(05)09002-6. [DOI] [PubMed] [Google Scholar]
- 38.Henricksen L.A., Umbricht C.B., Wold M.S. Recombinant replication protein A: expression, complex formation, and functional characterization. J. Biol. Chem. 1994;269:11121–11132. [PubMed] [Google Scholar]
- 39.Choi J.H., Lindsey-Boltz L.A., Sancar A. Reconstitution of RPA-covered single-stranded DNA-activated ATR-Chk1 signaling. Proc. Natl. Acad. Sci. USA. 2010;107:13660–13665. doi: 10.1073/pnas.1007856107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hazel P., Huppert J., Neidle S. Loop-length-dependent folding of G-quadruplexes. J. Am. Chem. Soc. 2004;126:16405–16415. doi: 10.1021/ja045154j. [DOI] [PubMed] [Google Scholar]
- 41.Phan A.T., Patel D.J. Two-repeat human telomeric d(TAGGGTTAGGGT) sequence forms interconverting parallel and antiparallel G-quadruplexes in solution: distinct topologies, thermodynamic properties, and folding/unfolding kinetics. J. Am. Chem. Soc. 2003;125:15021–15027. doi: 10.1021/ja037616j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang N., Phan A.T., Patel D.J. (3 + 1) Assembly of three human telomeric repeats into an asymmetric dimeric G-quadruplex. J. Am. Chem. Soc. 2005;127:17277–17285. doi: 10.1021/ja0543090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heddi B., Phan A.T. Structure of human telomeric DNA in crowded solution. J. Am. Chem. Soc. 2011;133:9824–9833. doi: 10.1021/ja200786q. [DOI] [PubMed] [Google Scholar]
- 44.Lao Y., Lee C.G., Wold M.S. Replication protein A interactions with DNA. 2. Characterization of double-stranded DNA-binding/helix-destabilization activities and the role of the zinc-finger domain in DNA interactions. Biochemistry. 1999;38:3974–3984. doi: 10.1021/bi982371m. [DOI] [PubMed] [Google Scholar]
- 45.Walther A.P., Gomes X.V., Wold M.S. Replication protein A interactions with DNA. 1. Functions of the DNA-binding and zinc-finger domains of the 70-kDa subunit. Biochemistry. 1999;38:3963–3973. doi: 10.1021/bi982370u. [DOI] [PubMed] [Google Scholar]
- 46.Lee J.Y., Okumus B., Ha T. Extreme conformational diversity in human telomeric DNA. Proc. Natl. Acad. Sci. USA. 2005;102:18938–18943. doi: 10.1073/pnas.0506144102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Okumus B., Ha T. Real-time observation of G-quadruplex dynamics using single-molecule FRET microscopy. Methods Mol. Biol. 2010;608:81–96. doi: 10.1007/978-1-59745-363-9_6. [DOI] [PubMed] [Google Scholar]
- 48.Murphy M.C., Rasnik I., Ha T. Probing single-stranded DNA conformational flexibility using fluorescence spectroscopy. Biophys. J. 2004;86:2530–2537. doi: 10.1016/S0006-3495(04)74308-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fanning E., Klimovich V., Nager A.R. A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res. 2006;34:4126–4137. doi: 10.1093/nar/gkl550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bastin-Shanower S.A., Brill S.J. Functional analysis of the four DNA binding domains of replication protein A. The role of RPA2 in ssDNA binding. J. Biol. Chem. 2001;276:36446–36453. doi: 10.1074/jbc.M104386200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bochkarev A., Bochkareva E. From RPA to BRCA2: lessons from single-stranded DNA binding by the OB-fold. Curr. Opin. Struct. Biol. 2004;14:36–42. doi: 10.1016/j.sbi.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 52.Cai L., Roginskaya M., Zou Y. Structural characterization of human RPA sequential binding to single-stranded DNA using ssDNA as a molecular ruler. Biochemistry. 2007;46:8226–8233. doi: 10.1021/bi7004976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Prakash A., Borgstahl E.O. The structure and function of replication protein A in DNA replication. In: MacNeill S., editor. The Eukaryotic Replisome: A Guide to Protein Structure and Function. Springer; New York: 2012. pp. 171–196. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.