

Quinones (Q) are a class of fully conjugated cyclic dione compounds that are widely distributed in nature.1 They can function as electron carriers in electron-transport chains (e.g. aerobic respiration)2 and undergo one- or two-electron reduction, coupled with protonation, leading to the corresponding semiquinones (QH.) or hydroquinones (QH2), respectively (Scheme 1). In biological systems, quinones are often reduced by coenzyme NAD(P)H (Scheme 2) in the presence of cellular reductases, such as NADH ubiquinone oxidoreductase, NADH cytochrome b5, and NADPH cytochrome P-450 reductase.3 Apart from enzymatic methods, the reduction of quinones by chemical, photochemical, and electrochemical methods has been investigated because of its biological and industrial importance.4 However, there are only a few reports of metal involvement in the reduction of quinones.5 For example, Sc3+ can form metal–quinone radical adducts using the cyclometalated iridium complex [Ir(ppy)3] (ppy=2-phenylpyridine) as an electron donor.5a Organometallic iridium complexes have potential for application in catalysis6 and biology.7 Coenzyme NADH can transfer hydride ions to cyclopentadienyl–IrIII complexes, generating iridium–hydride complexes.8 Here we investigate whether cyclopentadienyl–IrIII complexes can use NADH as a hydride source for the reduction of quinones, thus mimicking the action of reductases. We chose quinones vitamin K3 (menadione) and the related duroquinone as substrates (Scheme 2), because vitamin K is required for the synthesis of certain proteins that are involved in blood coagulation and metabolic pathways in bones and other tissues,9 and may play an important role in the treatment of cancer, Alzheimer’s and other diseases.10 This appears to be the first report of the catalytic reduction of quinones by an organometallic complex, and intriguingly, appears to involve the unusual IrII oxidation state in a novel mechanism.
Scheme 1.
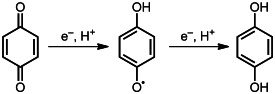
One- and two-electron reduction of a quinone (e.g. benzoquinone) leading to semiquinone and hydroquinone, respectively.
Scheme 2.
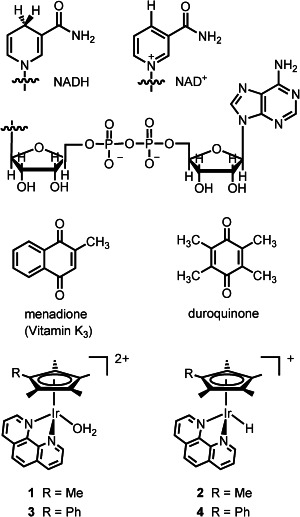
Structures of NADH, NAD+, quinones, and IrIII complexes studied herein.
Complexes [(η5-C5Me5)Ir(phen)(H2O)]2+ (1, phen=1,10-phenanthroline) and [(η5-C5Me4C6H5)Ir(phen)(H2O)]2+ (3; Scheme 2) were synthesized as PF6 salts and characterized by 1H NMR spectroscopy, ESI-MS, and CHN elemental analysis (see the Supporting Information for details).
First we studied the interactions of NADH, [(η5-C5Me5)Ir(phen)(H2O)]2+ (1) and menadione by 1H NMR spectroscopy. Upon the addition of two molar equivalents of NADH to 1 (1 mm) in [D4]MeOD/H2O (1:9), the colour of the solution changed from light to dark yellow immediately. The 1H NMR spectrum recorded at 298 K showed a sharp singlet at −11.3 ppm within the first 10 min, which is assignable to the iridium–hydride complex 2 (Scheme 2) together with a new set of signals that are attributable to NAD+ (Figure 1 b). When one molar equivalent of menadione was added to the reaction mixture, the resonances corresponding to 2 disappeared and were replaced by signals for the aqua complex 1 (Figure 1 c). No sharp signals for the added menadione were observed, only a set of broad resonances (Figure 1 c), thus suggesting that the menadione might be present as a paramagnetic species.
Figure 1.
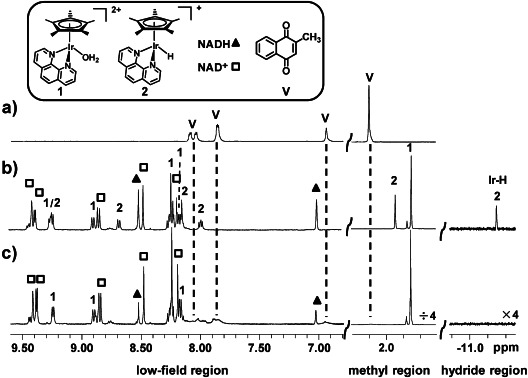
1H NMR spectra showing the formation of complex 2 [(η5-C5Me5)Ir(phen)(H)]+ and its reaction with menadione in [D4]MeOD/H2O (1:9) at 298 K. a) Menadione alone. b) Formation of hydrido complex 2 from reaction of [(η5-C5Me5)Ir(phen)(H2O)]2+ (1; 1 mm) with NADH (2 mm). c) Disappearance of signals of hydride 2 and broadening of signals of menadione 10 min after addition of menadione (1 mm) to mixture in (b).
To investigate the formation of paramagnetic products, EPR studies were carried out. Addition of menadione (1 mm) to a solution containing NADH (0.5 mm) and complex 1 (160 μm) in phosphate buffer (pH 7.2) resulted in an EPR spectrum that is consistent with the formation of the menadione semiquinone radical anion (M.−; Figure 2 a), whereas no EPR signal was detected in the absence of any of the three reactants. The simulated spectrum is characteristic of the menadione semiquinone radical anion (M.−) with hyperfine coupling constants aHH5=H6=H7=H8=3.03 G, aHH3=0.65 G (Figure 2 b), which agree with a previous report.11 The menadione radical anion formed here was stable for more than 20 h. Although some metal–quinone radical adducts have been reported,5a so far no superhyperfine splitting as a result of coupling with iridium nuclei was detected. Integration of the EPR signals (relative to a standard tempol (4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl) solution) showed that almost all the menadione (93 %) had been converted to menadione semiquinone.
Figure 2.
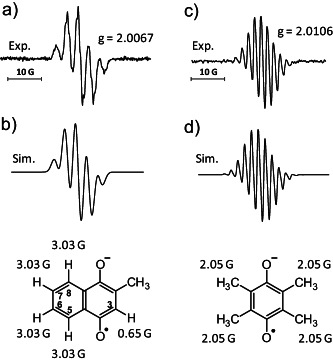
X-band EPR spectra at around 290 K. a) Menadione radical anion (M.−) generated by the reduction of menadione (1 mm) by NADH (0.5 mm) catalyzed by complex 1 or 3 (160 μm) in phosphate buffer (pH 7.2, 9 h). b) Simulated M.− EPR spectrum with hyperfine coupling constants. c) Durosemiquinone radical anion (D.−) generated by reduction of duroquinone (2 mm) by NADH (1 mm) catalyzed by complex 1 or 3 (330 μm) in phosphate buffer (pH 7.2, 10 h). d) Simulated D.− EPR spectrum with hyperfine coupling constants.
Strikingly, these data show the turnover of more than one molar equivalent of M.− per IrIII, thus suggesting that complex 1 can act as a catalyst. The catalytic reduction of menadione was further studied by EPR spectroscopy. Various molar excesses of menadione and NADH were added to a solution of complex 1 in phosphate buffer (pH 7.2; entries 1–4 in Figure 3) and EPR spectra were recorded. The turnover numbers (TON) and initial turnover frequencies (TOF, expressed as the number of moles of radical formed per mole of IrIII after the reaction was run for one hour) of these reactions increased with the concentrations of NADH and menadione (Figure 3). The maximum TON of 56.6 was observed for the reaction in entry 3 (molar ratio=1:30:80), and the maximum TOF of 12.4 h−1 for the reaction in entry 4 (1:60:120). Notably, a comparison of the TON with the amount of NADH present in entries 1–3 suggests that every molecule of NADH can reduce two quinone molecules (i.e. the semiquinone radical is formed). In entry 4, a similar TON was attained as in entry 3, although the concentrations of NADH and VK3 were increased, indicating that hydride transfer from NADH to 1 had reached saturation.
Figure 3.
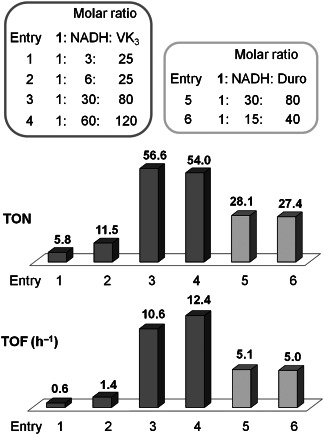
Reduction of menadione (VK3, entries 1–4) and duroquinone (Duro, entries 5 and 6) by NADH catalyzed by complex 1 (80 μm).
Similar NMR (Figure S1 in the Supporting Information) and EPR results (Figure 2 c and d) were obtained for the catalytic reduction of duroquinone by NADH/complex 1 (entries 5 and 6 in Figure 3), with a maximum TON of 28.1 and a maximum TOF of 5.1. These values are about half those for the reduction of menadione (entry 3). The presence of three additional electron-donating methyl groups makes duroquinone more difficult to reduce, which is in line with reported half-wave potentials.12 As with menadione, one mole of NADH reduces two moles of duroquinone (entry 6).
We also studied the reduction of quinones by NADH/[(η5-C5Me4C6H5)Ir(phen)(H2O)]2+ (3). The reaction pathways were similar to those of complex 1 (Figure 2, and Figure S2 in the Supporting Information), showing that the phenyl substituent on the Cp* has little effect on the reaction.
Since NADH is effectively a two-electron (hydride) donor, it is surprising that the reduction of quinone appears to involve two quantitative one-electron donor steps to give the semiquinone product. Although one-step hydride transfer from NADH and analogues to quinones (NADH+Q→NAD++QH−), and multi-step hydride transfer (electron transfer followed by proton/electron transfer, e−+H++e−) have been previously described,13 no mechanism for the reduction of quinone by metal-based hydrides has been reported.
DFT calculations (see the Supporting Information for details) were used to characterize various possible stationary points to provide insight into the likely mechanism of the iridium-catalyzed reduction of these quinones. A possible mechanism for the catalytic reduction of quinone to semiquinone by NADH/complex 1 is shown in Scheme 3 a. In the first step, a hydride ion is transferred from NADH to complex 1 to form the iridium–hydride complex 2 and NAD+. One-electron transfer to the quinone together with proton transfer to the phosphate buffer then generates the deprotonated semiquinone radical (Q.−) and a transient IrII center. Next, an electron is transferred from IrII to a second quinone molecule, resulting in further formation of semiquinone radical anion (Q.−) and regeneration of IrIII. The assumed formation of semiquinone radical anions is based on their relatively low pKa value (ca. 5).14 Subsequent coordination of a water molecule to the IrIII center completes the catalytic cycle. This oxidation state (II) is unusual for iridium and there are only a few reports of IrII.15
Scheme 3.
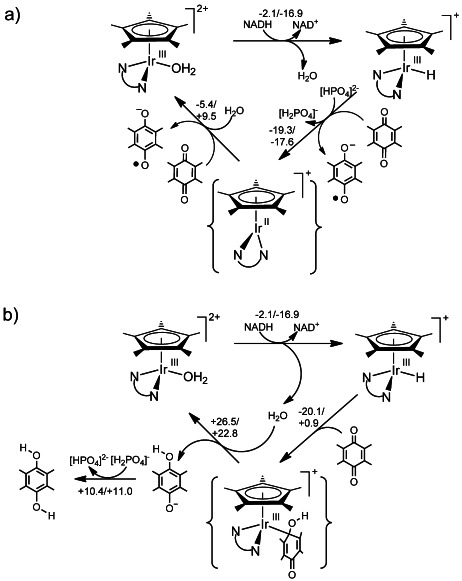
Two possible mechanisms for the catalytic reduction of duroquinone by complex 1. a) Reduction through two sequential one-electron transfers. Figure S3 in the Supporting Information shows a plot of the singly occupied molecular orbital of the IrII d7 complex. b) Reduction through a single two-electron transfer. The pairs of numbers associated with each step are potential-energy/free-energy couples (in kcal mol−1). The semiquinone has a pKa of around 5 and is therefore negatively charged at pH 7.2, whereas the quinol is protonated (pKa higher than 11).14
The results of DFT calculations on an alternative two-electron reduction pathway, which would give rise to the fully reduced dihydroquinone, are shown in Scheme 3 b. The mechanism involves a one-step hydride transfer from complex 2 to the quinone via an intermediate with an Ir–C(quinone) bond. In this case, the quinol is released and protons are taken up from the buffer because of the higher pKa value of the dihydroquinol.
In both cases, the regeneration of the aqua complex is the critical step. In Scheme 3 a, this step appears favored on the potential-energy surface, but disfavored on the free-energy surface, and in Scheme 3 b, this steps appears to be strongly disfavored on both surfaces. Thus, notwithstanding the difficulties of modeling H+/e− transfers and the need to invoke the phosphate buffer to balance the various reaction steps, the DFT calculations generally support two consecutive one-electron transfers (pathway in Scheme 3 a), generating the transient IrII complex and the experimentally-detected semiquinone. The alternative dihydroquinone pathway (Scheme 3 b), in which only IrIII species are involved, appears not to be viable theoretically.
In conclusion, organometallic cyclopentadienyl–iridium complexes offer the prospect of carrying out catalytic reductions of quinones without an enzyme through hydride transfer from NADH. These reactions produce semiquinones rather than the two-electron reduced products, the dihydroquinones. DFT calculations suggest that the reactions occur through a novel mechanism that involves an unusual transient IrII state, in which the iridium center plays a key role in promoting this one-electron pathway. This appears to be the first report of a catalytic reduction of a quinone by an organometallic complex, and may have promising potential for the design of catalytic metallodrugs and for activation of electron transfer in biological systems.16 Such organometallic complexes may therefore be valuable for the modulation of the redox status of cells (a potential drug target), as enzyme mimics, and for biocoupled hydrogenation reactions.
Supplementary material
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
References
- 1.Dey PM. In: Methods in Plant Biochemistry. Dey PM, Harborne JB, editors. London: Academic Press; 1989. [Google Scholar]
- 2a.Soballe B, Poole RK. Microbiology. 1999;145:1817–1830. doi: 10.1099/13500872-145-8-1817. [DOI] [PubMed] [Google Scholar]
- 2b.Hirst J. Biochem. Soc. Trans. 2005;33:525–529. doi: 10.1042/BST0330525. [DOI] [PubMed] [Google Scholar]
- 2c.Yagi T, Matsuno-Yagi A. Biochemistry. 2003;42:2266–2274. doi: 10.1021/bi027158b. [DOI] [PubMed] [Google Scholar]
- 3a.Matsushita K, Ohnishi T, Kaback HR. Biochemistry. 1987;26:7732–7737. doi: 10.1021/bi00398a029. [DOI] [PubMed] [Google Scholar]
- 3b.Schenkman JB, Jansson I. Pharmacol. Ther. 2003;97:139–152. doi: 10.1016/s0163-7258(02)00327-3. [DOI] [PubMed] [Google Scholar]
- 3c.Bachur NR, Gordon SL, Gee MV, Kon H. Proc. Natl. Acad. Sci. USA. 1979;76:954–957. doi: 10.1073/pnas.76.2.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3d.Li R, Bianchet MA, Talalay P, Amzel LM. Proc. Natl. Acad. Sci. USA. 1995;92:8846–8850. doi: 10.1073/pnas.92.19.8846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3e.Hirst J. Biochem. J. 2010;425:327–339. doi: 10.1042/BJ20091382. [DOI] [PubMed] [Google Scholar]
- 4a.Guin PS, Das S, Mandal PC. Int. J. Electrochem. 2011;2011:1–22. [Google Scholar]
- 4b.Müh F, Glöckner C, Hellmich J, Zouni A. Biochim. Biophys. Acta Bioenerg. 2012;1817:44–65. doi: 10.1016/j.bbabio.2011.05.021. [DOI] [PubMed] [Google Scholar]
- 4c.Haroon Y, Bacon DS, Sadowski JA. Biomed. Chromatogr. 1987;2:4–8. doi: 10.1002/bmc.1130020103. [DOI] [PubMed] [Google Scholar]
- 4d.Abraham I, Joshi R, Pardasani P, Pardasani RT. J. Braz. Chem. Soc. 2011;22:385–421. [Google Scholar]
- 5a.Yuasa J, Suenobu T, Fukuzumi S. ChemPhysChem. 2006;7:942–954. doi: 10.1002/cphc.200500640. [DOI] [PubMed] [Google Scholar]
- 5b.Fukuzumi S, Okamoto T. J. Am. Chem. Soc. 1993;115:11600–11601. [Google Scholar]
- 6a.Oro LA, Claver C. Iridium Complexes in Organic Synthesis. Weinheim: Wiley; 2008. [Google Scholar]
- 6b.Andersson PG. Iridium Catalysis. Berlin: Springer; 2011. [Google Scholar]
- 7a.Liu Z, Habtemariam A, Pizarro AM, Fletcher SA, Kisova A, Vrana O, Salassa L, Bruijnincx PCA, Clarkson GJ, Brabec V, Sadler PJ. J. Med. Chem. 2011;54:3011–3026. doi: 10.1021/jm2000932. [DOI] [PubMed] [Google Scholar]
- 7b.Liu Z, Habtemariam A, Pizarro AM, Clarkson GJ, Sadler PJ. Organometallics. 2011;30:4702–4710. [Google Scholar]
- 7c.Liu Z, Salassa L, Habtemariam A, Pizarro AM, Clarkson GJ, Sadler PJ. Inorg. Chem. 2011;50:5777–5783. doi: 10.1021/ic200607j. [DOI] [PubMed] [Google Scholar]
- 7d.Kastl A, Wilbuer A, Merkel AL, Feng L, Di Fazio P, Ocker M, Meggers E. Chem. Commun. 2012;48:1863–1865. doi: 10.1039/c1cc15378a. [DOI] [PubMed] [Google Scholar]
- 7e.Lo KK-W, Li SP-Y, Zhang KY. New J. Chem. 2011;35:265–287. [Google Scholar]
- 7f.Schäfer S, Sheldrick WS. J. Organomet. Chem. 2007;692:1300–1309. [Google Scholar]
- 8.Betanzos-Lara S, Liu Z, Habtemariam A, Pizarro AM, Qamar B, Sadler PJ. Angew. Chem. 2012;124:3963–3966. doi: 10.1002/anie.201108175. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. Int. Ed. 2012;51:3897–3900. doi: 10.1002/anie.201108175. [DOI] [PubMed] [Google Scholar]
- 9a.Weber P. Nutrition. 2001;17:880–887. doi: 10.1016/s0899-9007(01)00709-2. [DOI] [PubMed] [Google Scholar]
- 9b.Suttie JW. Annu. Rev. Nutr. 1995;15:399–417. doi: 10.1146/annurev.nu.15.070195.002151. [DOI] [PubMed] [Google Scholar]
- 10a.Saxena SP, Israels ED, Israels LG. Apoptosis. 2001;6:57–68. doi: 10.1023/a:1009624111275. [DOI] [PubMed] [Google Scholar]
- 10b.Nimptsch K, Rohrmann S, Linseisen J. Am. J. Clin. Nutr. 2008;87:985–992. doi: 10.1093/ajcn/87.4.985. [DOI] [PubMed] [Google Scholar]
- 10c.Allison AC. Med. Hypotheses. 2001;57:151–155. doi: 10.1054/mehy.2001.1307. [DOI] [PubMed] [Google Scholar]
- 11.Kubata BK, Kabututu Z, Nozaki T, Munday CJ, Fukuzumi S, Ohkubo K, Lazarus M, Maruyama T, Martin SK, Duszenko M, Urade Y. J. Exp. Med. 2002;196:1241–1252. doi: 10.1084/jem.20020885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frontana C, Vázquez-Mayagoitia Á, Garza J, Vargas R, González I. J. Phys. Chem. A. 2006;110:9411–9419. doi: 10.1021/jp060836+. [DOI] [PubMed] [Google Scholar]
- 13a.Gȩbicki J, Marcinek A, Zielonka J. Acc. Chem. Res. 2004;37:379–386. doi: 10.1021/ar030171j. [DOI] [PubMed] [Google Scholar]
- 13b.Zhu X-Q, Yang Y, Zhang M, Cheng J-P. J. Am. Chem. Soc. 2003;125:15298–15299. doi: 10.1021/ja0385179. [DOI] [PubMed] [Google Scholar]
- 13c.Yuasa J, Yamada S, Fukuzumi S. Angew. Chem. 2008;120:1084–1087. [Google Scholar]
- Angew. Chem. Int. Ed. 2008;47:1068–1071. doi: 10.1002/anie.200704136. [DOI] [PubMed] [Google Scholar]
- 13d.Yuasa J, Yamada S, Fukuzumi S. J. Am. Chem. Soc. 2006;128:14938–14948. doi: 10.1021/ja064708a. [DOI] [PubMed] [Google Scholar]
- 14.Rich PR, Bendall DS. Biochim. Biophys. Acta Bioenerg. 1980;592:506–518. doi: 10.1016/0005-2728(80)90095-x. [DOI] [PubMed] [Google Scholar]
- 15a.Hetterscheid DGH, de Bruin B. J. Mol. Catal. A. 2006;251:291–296. [Google Scholar]
- 15b.Cheung CW, Chan KS. Organometallics. 2011;30:4269–4283. [Google Scholar]
- 15c.Kimura T, Ishiwata K, Kuwata S, Ikariya T. Organometallics. 2012;31:1204–1207. [Google Scholar]
- 15d.Takahashi Y, Fujita K-i, Yamaguchi R. Eur. J. Inorg. Chem. 2008:4360–4368. [Google Scholar]
- 15e.Meiners J, Scheibel MG, Lemée-Cailleau M-H, Mason SA, Boeddinghaus MB, Fässler TF, Herdtweck E, Khusniyarov MM, Schneider S. Angew. Chem. 2011;123:8334–8337. doi: 10.1002/anie.201102795. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. Int. Ed. 2011;50:8184–8187. doi: 10.1002/anie.201102795. [DOI] [PubMed] [Google Scholar]
- 15f.Teets TS, Cook TR, McCarthy BD, Nocera DG. Inorg. Chem. 2011;50:5223–5233. doi: 10.1021/ic2005248. [DOI] [PubMed] [Google Scholar]
- 16a.Sasmal PK, Streu CN, Meggers E. Chem. Commun. 2013;49:1581–1587. doi: 10.1039/c2cc37832a. [DOI] [PubMed] [Google Scholar]
- 16b.Cowan JA. Pure Appl. Chem. 2008;80:1799–1810. [Google Scholar]
- 16c.Hocharoen L, Cowan JA. Chem. Eur. J. 2009;15:8670–8676. doi: 10.1002/chem.200900821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16d.Kuznetsov AM, Ulstrup J. Electron transfer in chemistry and biology: an introduction to the theory. Chichester: Wiley; 1999. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.