

Abstract
Conformational changes of proteins are involved in all aspects of protein function in biology. Almost all classes of proteins respond to changes in their environment, ligand binding, and interaction with other proteins and regulatory agents through undergoing conformational changes of various degrees and magnitudes. Membrane channels and transporters are the major classes of proteins that are responsible for mediating efficient and selective transport of materials across the cellular membrane. Similar to other proteins, they take advantage of conformational changes to make transitions between various functional states. In channels, large-scale conformational changes are mostly involved in the process of “gating”, i.e., opening and closing of the pore of the channel protein in response to various signals. In transporters, conformational changes constitute various steps of the conduction process, and, thus, are more closely integrated in the transport process. Owing to significant progress in developing highly efficient parallel algorithms in molecular dynamics simulations and increased computational resources, and combined with the availability of high-resolution, atomic structures of membrane proteins, we are in an unprecedented position to use computer simulation and modeling methodologies to investigate the mechanism of function of membrane channels and transporters. While the entire transport cycle is still out of reach of current methodologies, many steps involved in the function of transport proteins have been characterized with molecular dynamics simulations. Here, we present several examples of such studies from our laboratory, in which functionally relevant conformational changes of membrane channels and transporters have been characterized using extended simulations.
Keywords: Membrane channels, membrane transporters, protein conformational changes, molecular dynamics simulation, biological membranes
Introduction
Transport across the cellular membrane constitutes one of the most fundamental and highly regulated processes in all living organisms. This basic function is mainly carried out by membrane proteins acting as molecular machines which are broadly known as membrane channels and transporters. The opening and closing of membrane channels involve large-scale protein conformational changes and are regulated by various signals in the cell, including ligand binding, membrane electrical potential, or mechanical stress. Once opened in response to such stimuli, membrane channels allow passive diffusion of their substrates through selective pores from one side of the membrane to the other, along the concentration gradient. Active membrane transporters, on the other hand, couple various sources of cellular energy to vectorial translocation of their substrates, often against the chemical gradient (pumping). This coupling requires a much more complex architecture and mechanism, and, thus, membrane transporters are significantly slower and mechanistically more complex than channels. While the energy is provided by ATP in primary transporters, secondary transporters couple substrate transport to co-transport (symport or antiport) of ionic species (most prominently H+ and Na+ ions). The transport cycle in membrane transporters is composed of many steps, most of which involve significant protein conformational changes whose nature and magnitude are largely unknown and often difficult to characterize experimentally.
Protein conformational changes are, therefore, central to the function of membrane channels and transporters. The dynamics of these proteins have diverse scales, ranging from localized side-chain conformational changes, to loop flipping motions, and up to extensive subdomain/domain structural transitions. In channels, such protein conformational changes constitute the mechanism of gating (response to signal), and the process of substrate conduction itself usually does not involve significant protein motions. In active transporters, on the other hand, protein conformational changes of various forms and magnitudes are an integral part of the transport process. Molecular dynamics (MD) simulation offers a method with sufficient temporal and spatial resolutions to characterize functionally relevant molecular events in proteins. Several such motions in membrane channels and transporters have been successfully captured through simulations.1–14
For transporters, a general mechanistic model named the “alternating-access mechanism” has been proposed.15 This model ensures that the substrate is only accessible from one side of the membrane at a given time, thus, preventing the formation of a leak (a channel-like structure that would allow the free diffusion of the substrate from one side to the other) during the transport cycle. The alternating-access model, requires at least two major conformational states of the protein, namely, the inward-facing (IF) and outward-facing (OF) states, whose inter-conversion switched the substrate accessibility from one side of the membrane to the other (Fig. 1). The complete transport cycle could involve many other intermediate states; for instance, it is known in many transporters that the IF state can exist in either open (IF-o) or occluded (IF-occ) states. The same applies to the OF state. Unfortunately, for most transporters, only one conformational state has been characterized structurally. The other states, and the conformational changes involved in their transitions, therefore, have to be studied using other methodologies that would yield a dynamical characterization of the process in these complex proteins.
Figure 1.

The alternating-access model proposed for the transporter function, including two major open states, outward-facing-open (OF-o) and inward-facing-open (IF-o), and two intermediate substrate-occluded states (OF-occ and IF-occ). “Out” and “In” represent outside and inside the cell, respectively. The access to the substrate (red dot) from the two sides of the membrane is controlled by protein conformational changes of different nature and magnitudes.
The size and complexity of the function of membrane transport proteins pose a great challenge for computational studies. Simulation of membrane channels and transporters also requires the inclusion of the embedding lipid bilayer, water and ions explicitly in the system. This often results in very large system sizes, in the range of 100,000 – 500,000 atoms, which can be computationally prohibitive. More importantly, characterizing the complete transport cycle in transporters and the entire gating motion of channels would require simulations on the order of at least μs-ms time scale, which are currently not possible. Despite these technical limitations, recent computational studies have demonstrated that extended, large-scale MD simulations of membrane transporters and channels can be very effective in describing some molecular events and processes involved in the function of these proteins. 3–14, 16–29 These studies show that simulations can, indeed significantly advance our understanding of the molecular mechanism of activation/deactivation in channels, and energy coupling and transport phenomena in transporters.
In this review, we use four systems, viz. acid sensing ion channel (ASIC), an ATP-binding cassette transporter (ABCT), glycerol-3-phosphate transporter (GlpT), and glutamate transporter (GlT), to showcase the results of application of MD simulations to membrane channels and transporters (Fig. 2). In all these systems, the system setup uses experimentally solved, atomic-resolution protein structures, while water, lipid (membrane), and ions are added by modeling. The program NAMD230 was adopted for the reported simulations. The system temperature is maintained at 310 K via Langevin dynamics with a damping coefficient, γ, of 0.5 ps−1. The pressure is maintained at 1 atm using the Langevin Nosé-Hoover method. 31, 32 These simulations employ Particle Mesh Ewald (PME)33 for calculation of long-range electrostatic forces without truncation. Missing parameters and topology files for ligands are included either by adopting similar parameters from the available force field or by quantum mechanical calculations in a manner consistent with the employed force field. The simulations involve a brief initial equilibration period, wherein the lipid tails are allowed to “melt”, while the lipid head groups and the protein are held constrained. This is followed by an unconstrained equilibration of the lipids and the protein, after which the area of the lipid bilayer is held constant. This is done to prevent excessive shrinking of the lipid bilayer, which may affect the intrinsic motion of the protein. The initial equilibration period typically ranges between 1–5 nanoseconds (ns), while the production simulations are carried out for 50–100 ns. As will be demonstrated, these time scales are able to capture fast motions such as water and ion diffusion, as well as side chain re-orientations and loop movements, and, in some cases, larger conformational changes such as domain motions.
Figure 2.

The four membrane proteins discussed in this review- ASIC, ABCT, GlT and GlpT. The proteins are shown embedded in lipid bilayer (grey), and surrounded by explicit water (blue) and ions (Na+, blue, Cl−, red), as a representation of the simulation setup for each system. “OUT” represents the extracellular region (outside the cell), and “IN” represents the intracellular region (inside the cell).
1 H+-Induced Gating of Acid Sensing Ion Channels
Acid sensing ion channels (ASICs) are pH sensors present in cell membranes in the nervous system of mammals. When the pH outside the cell shows a transient drop, these channels open in response, and allow cation (mainly Na+) influx into cells. Such pH drops are associated with perception of pain (nociception), cell signaling, and other important processes. ASICs are, thus, implicated as drug targets for treatment of pain and neurological disorders such as Alzheimer’s disease and multiple sclerosis, and also associated with key functions such as learning, memory, and mechanosensation.34–42
The presence of pH sensors in neurons was first detected by Krishtal and Pidoplichko37, 43 and these were subsequently cloned and characterized.34, 44–46 The sensors were identified as being pH-gated cation channels, belonging to the degenerin/epithelial (DEG/ENaC) sodium channel family34, 35, 45 which also include channels responding to mechanical stimuli or peptide binding. Detailed characterization of ASICs has revealed six ASIC isoforms. These isoforms can form homo- and heteromeric channels distributed in the mammalian nervous system and differ in their pH sensitivities and activation and desensitization patterns.47, 48 Structural determinants for these differences in behavior have not yet been assigned, but it is reasonable to assume that it is due to differences in the composition of pH sensing regions in these proteins. Though several mutagenesis experiments, functional studies as well as a computational study10, 49–54 have examined the involvement of a group of residues in channel activation, the complete set of residues that bind H+, and hence sense the pH, has not been determined. Also, the exact mechanism of ASIC channel function remains unknown, though several hypotheses based on structural and functional studies have been presented.54–57 The answers to these interlinked questions may be revealed upon characterization of the dynamic structural (conformational) changes upon H+ binding.
Crystal structures of only one member of the DEG/ENaC family are known, that of chicken ASIC1 at a resolution of 1.9 Å, for a truncated inactive form (ΔASIC1) 54 and 3 Å, for the functional channel.57 The crystal structures reveal ASIC1 to be a homotrimer. As seen in Fig. 3, each ASIC monomer has a large extracellular domain, connected to two transmembrane helices each. The N- and C- termini of each monomer are on the cytosolic side, but are not resolved in the crystal structure. The ΔASIC1 structure carries a high overall negative charge (−52) and has 176 acidic (aspartate and glutamate), 124 basic (lysine and arginine) and 15 histidine residues distributed on the surface as well as in the interior of the protein. The structure thus contains several possible H+ or cation binding sites. The extracellular domain in each monomer appears “stapled” by seven disulphide bonds formed by highly conserved cysteine residues in each monomer, of which five lie on a domain referred to as the “thumb domain” (Fig. 3). The monomers arrange to form a “tree-like” trimer, with the six transmembrane domains forming the “trunk”. The structure shows interesting cavities, notably, an “acidic pocket”10, 54 near the thumb domain and an “inner chamber” on the three-fold axis of symmetry, both with several negatively charged residues (Fig. 3). Mutagenesis experiments have reported the participation of “acidic pocket” residues in H+ sensing.50, 54 A computational study by our group detected persistent cation binding in the “inner chamber” and proposed it to be a temporary reservoir of ions that may diffuse to the pore of the channel.10 Another interesting structural feature of ASIC is the possible portals near the channel pore formed between monomers, which may serve as entry points for ion access to the pore (Fig. 3).
Figure 3.

Right: The trimeric ASIC structure with the three monomers colored in blue, orange, and gray respectively. The ‘thumb’ domain is highlighted in pink. Two highly acidic cavity surfaces are highlighted in red - the cavity near the ‘thumb’ domain is the ‘acidic’ pocket, and that in the protein interior is the ‘inner’ chamber. Possible portals for ion access to the pore are highlighted in green. Left, upper panel: magnified view of the thumb domain and the acidic pocket. Key residues in the latter are shown as sticks. Left, lower panel: Magnified view of the inner chamber, with key acidic residues shown as sticks.
ASIC function involves cycling between three main states (Fig. 4). In its resting state at physiological pH, the channel is closed and non-conducting. When the extracellular pH drops, the channel is activated by H+ binding, and moves to the open state. If low pH persists, the channel moves to the desensitized state, where the ligand remains bound but the channel is non-conducting. When physiological pH is restored, the channel returns to the closed state. The crystal structures54, 57 are reported in the desensitized state, hence structural information about the open and closed states and the transitions between these states is limited.
Figure 4.

The putative mechanism of ASIC. The channel is closed at physiological pH, opens upon drop in pH due to a coupled movement of the extracellular and transmembrane domains, and moves to the desensitized state, which returns to the closed state when the pH increases. Dotted lines indicate states whose structures are unknown, and solid lines represent the known (desensitized) state.
Previously proposed hypotheses represent differing views on the extent of conformational changes involved in ASIC function. An earlier model proposed minimal conformational changes in the protein, presenting removal of channel block as the activation mechanism.55 It is well-established that ASIC activity is modulated by the concentration of extracellular Ca2+.43, 46, 47, 58 and it was proposed that ASIC gating involves simple displacement of Ca2+ ions which are known to bind to and block the pore, by H+ at low pH conditions.55 However, subsequent studies based on kinetics of ASIC activation favored an allosteric mechanism.56 It was proposed that displacement of Ca2+ by H+ induced conformational changes linked to channel gating.54, 56 Large conformational changes have also been implicated in the open-to desensitized-state transition in a separate study on ASIC3.53 Based on these studies, a mechanism has been discussed for ASIC function. It was proposed that acid sensing involves residues in the “acidic pocket”, which are protonated when the pH drops. This induces a bending-away of the “thumb domain”, which forms part of this acidic pocket. This motion transfers its effect to the transmembrane helices, via a coupling loop, which lies at the hinge region between the extracellular and transmembrane domains. This finally results in transmembrane helix movement that opens the channel.54 Preliminary analysis of large-scale motions in ASIC indicate that such extracellular-vs-transmembrane motion is plausible (Shaikh and Tajkhorshid, unpublished results). A recent computational study also proposes a similar mechanism, though they argue that it is not the bending away of the thumb domain, but rather, its increased attraction with a neighbouring domain, that induces this cascade of motions.59 However, the exact mechanism has not yet been confirmed through any extensive structural or functional analyses.
A series of MD simulations were carried out to describe conformational changes associated with ASIC function.10 During system design for these simulations, the aim was to mimic possible open, closed and desensitized states of ASIC1. For this, a set of residues which had been proposed in earlier studies to form part of the H+ sensor on ASIC, were protonated or deprotonated. Lipids, water and ions were explicitly included in the setup (Fig. 3). Apart from conformational changes, other potential cation and ligand (H+) binding sites of the protein were also examined using Na+ or Ca2+ localization patterns. These simulations were designed to provide dynamic information, not only about which regions of the protein could be directly involved in the coordinated motion of the extracellular and transmembrane regions, but also on other aspects of channel function, such as acid sensing and gating.
The simulations revealed several pairs of acidic residues where Na+ or Ca2+ ions localized. Influenced by the presence of bound ligand (H+) or cation(s), these sites exhibited local conformational changes. For these residues, the extent of motion was measured as root mean squared deviations (RMSDs) from the original crystal structure (Fig. 5a). Of the five systems studied, three systems were designed (numbered 1, 3, 4) to represent the H+ bound i.e. desensitized state, and two were designed (numbered 2 and 5) as H+-free i.e. closed state. It was observed that the RMSDs were larger in the closed-like systems, compared to the desensitized-like systems, expectedly, since the crystal structure is reportedly in the desensitized state.
Figure 5.

(a) Root mean square deviations (Å) of residue pairs involved in persistent cation binding in ASIC. These residues are acidic pocket residues (black), inner chamber residues (red), surface residues (blue) and intracellular residues (green), and the residue numbers are indicated. The RMSDs are compared for five systems (numbered 1 to 5) including desensitized-like (3 and 4) and closed-like (2 and 5) states. Notable structural fluctuations caused by some of these residues are illustrated in (b), (c) and (d), comparing final snapshots from simulations of the desensitized-like state (yellow, corresponds to system 3) with all acidic residues H+-bound and the closed-like state (maroon, corresponding to system 5) with all acidic residues H+-free, as compared to the crystal structure (gray). In all cases, the desensitized state remains similar to the crystal structure. (b) In the acidic pocket, the loop and helix carrying key acidic pocket residues show a large movement in the closed state. (c) Top view of the highly acidic inner chamber, shows that the chamber expands in the closed-like state. (d) Slanting of the thumb domain occurs upon Ca2+ binding in the closed-like state.
Local conformational changes observed in the “acidic pocket” hinted at some mechanistic details of channel activation. Six acidic pocket residues have been proposed to participate in acid sensing from mutagenesis studies50, 54, hence some or all of these residues were modeled as H+-bound (uncharged) to mimic the desensitized state. In this region of the protein, which is distant from the transmembrane region, it is expected that the open state is similar to the desensitized state, since both are ligand-bound states. To mimic the closed state, these six acidic pocket residues were modeled as H+-free (charged). Upon simulation it was observed that the acidic pocket structure is closely maintained when the ligand (H+) is bound (desensitized/open-like state), but is perturbed when the acidic residues are ligand-free (closed-like state) (Fig. 5b). Also, multiple cation binding was observed in the pocket in the closed-like state. This indicates that substitution of cations by H+ in the acidic pocket results in the local structure to adopt the desensitized/open-like form.10 This is consistent with earlier experimental hypotheses where it was proposed that H+ displaces bound Ca2+ resulting in conformational changes involved in channel opening.56
Structural fluctuations in the “inner chamber” provided the first hints at a possible role of this chamber in channel function. In the closed-like systems, where all acidic residues are uncharged, the highly acidic inner chamber, expands possibly due to repulsion among the acidic residues (Fig. 5c). However, this effect was reversed when cations entered the chamber. Also, in the open/desensitized-like systems, some of these acidic residues were uncharged. Thus, this suggests that cations or H+ are required to maintain the structure of the chamber. Na+ ions access the acidic inner chamber in the protein which Ca2+ does not enter, and the chamber may be a temporary reservoir for Na+ with possible access to the transmembrane pore in the open state of the channel.10
Movement of the “thumb domain” captured in the simulations, provided hints about the response of the extracellular domain to cation/H+ binding. Ca2+ binding was observed to cause three pairs of surface acidic residues, which were initially distant, to move together and form a cation binding pair. This motion is associated with significant slanting of the thumb domain. This was an effect not observed with Na+. This observation strongly suggests that substitution of bound Ca2+ in this area, by H+, would result in movement of the thumb domain. This is consistent with experimental observations of Ca2+ displacement by H+ being important for channel function.54, 56 Also, movement of the thumb domain has been proposed in hypotheses on channel function.54, 59 Thus, this observation from the simulations is particularly notable since it connects cation/H+ binding to the acidic pocket with conformational change in the extracellular domain.
These simulations were, thus, able to capture the dynamic behavior of ASIC, and its initial response to change in protonation state within a short time span of 50 ns. While diffusion of Na+ and Ca2+ ions is fast enough to be described well at these time scales, the conformational changes in the protein can only be described partially. Despite this limitation, simulations have provided deep insight about the effect of protonation on the putative acid-sensing sites, the response and movement of the extracellular domain, and have even revealed yet-unknown cation localization sites.
The use of molecular dynamics simulations in studying ASIC mechanism is clearly promising. The description of larger motions such as extracellular-vs-transmembrane motion, pore opening/closure etc., require studies at longer time scales or with more advanced simulation techniques. Recent technological advances resulting in increased computational power, and developments in simulation methodologies, are now making it possible to monitor conformational changes at longer time scales and hence MD simulations may now be employed to test several hypotheses about channel behavior.
2 ATP-Induced Conformational Changes in ABC Transporters
ATP-binding cassette (ABC) transporters are ATP powered transporters ubiquitously expressed in all life forms. At least four basic building blocks are required for a functional ABC transporter, two transmembrane domains (TMDs) providing the physical pathway for the substrate permeation, and two nucleotide binding domains (NBDs) located at the cytoplasmic side of the transporter, and serving as the motor to drive the transport (Fig. 6). ABC transporters can function as importers or exporters, with members in the importer family equipped with an additional substrate binding protein (BP) bound to the extracellular side of the transporter (Fig. 6).
Figure 6.

The general mechanism of ABC transporters and representative crystal structures for different conformational states of different types of ABC transporters. Left: the E. coli maltose transporter in the IF70 and the OF66 states. Right: the lipid flippase MsbA of E. coli in the IF67 state and the same transporter of S. typhimurium in the OF67 state.
ABC transporters are powered by the two highly conserved NBDs, regardless of their function as importers or exporters, and despite the completely distinct structures of their TMDs (which is different even among importers). The NBDs bind and hydrolyze ATP to provide energy to drive the transport. In response to ATP binding and hydrolysis in the NBDs, the substrate accessibility of the TMDs is switched between either opening toward inside or outside the cell (alternating access model, refer to Fig. 1).
Several crystal structures of intact ABC transporters have been reported in recent years60–71 (for a list of crystal structures of ABC transporters, refer to Moussatova et al.72). Several aspects of the transport mechanism in ABC transporters have been revealed by these structures, as well as those on isolated NBDs of various ABC transporters. Based on these structural studies, a universal mechanism of transport has been proposed, despite different structures and directions of transport.73–78
The general scheme of alternating access model for all transporters (Fig. 1) also applies to ABC transporters. When ABC transporters are not bound by a nucleotide, the central opening of the TMDs, where the substrate translocates across the membrane, is accessible from the cytoplasmic side and completely sealed from the extracellular/periplasmic side. In this nucleotide-free configuration, the NBDs appear as separate monomers (open dimer). This conformational state is therefore termed “the resting state”. Upon ATP binding, the two NBDs associate with each other to form a nucleotide-sandwiched, closed dimer (Fig. 6), which renders the TMDs open toward outside the cell. After ATP hydrolysis in the NBDs, they separate from each other and the transporter returns to the resting state. While substrate transport occurs during the transition between different conformational states of the TMDs (outward-facing or inward-facing), the transport process is controlled by the dimerization states of the NBDs, which in turn is determined by the nucleotide species that is bound within the active sites at the NBD dimer interface. In other words, the transport mechanism is a conformational inversion of the TMDs controlled by the nucleotide binding and hydrolysis in the NBDs, through the conformational coupling between the TMDs and the NBDs (Fig. 6).
The molecular motions of ABC transporters have been demonstrated in MD simulations in several studies. Using the crystal structure of the isolated NBD dimer of maltose transporter, which was trapped in a conformation between the closed and open forms (termed “the semi-open form”), it has been established that the semi-open NBDs tend to open to a greater degree in their nucleotide-free form, while docking ATP into the active sites results in the closing movement of the NBD dimer.79 In the simulation of another ATP-bound NBD dimer, it was found that the dimer also separates when the two bound ATP molecules are replaced by ADP.80 In addition, MD simulations of full ABC transporters have been reported. One successful case was to combine perturbed anisotropic network model and essential dynamics sampling to predict the large-scale relative motions between the NBDs and the TMDs in a vitamin B12 transporter.81 Another attempt simulating the vitamin B12 transporter in full atomic models suggested that the transporter operates through an asymmetric manner.82 Moreover, the coupling mode between the NBDs and the TMDs in one type of ABC transporters has been investigated with normal mode analysis of the reduced models (anisotropic network model, ANM) of the vitamin B12 transporter and one of its homologs.83, 84
Biochemically, it is known that ATP hydrolysis triggers the conformational transition of the NBDs between the closed and open states. This is exemplified by the comparison of the structures of some NBDs in the ATP-bound form,85–88 and in the ADP-bound form.88–90 The nucleotide-dependent NBD arrangement is further supported in almost all crystal structures of intact ABC transporters, as the NBDs in the ATP-bound state are always found to exist along with outward-facing TMDs.63, 75 However, several processes may be involved in the transition between the ATP-bound and the ADP-bound states, including the hydrolysis reaction itself, and the dissociation of hydrolysis products, e.g., the inorganic phosphate (Pi), in addition to the rearrangement of the NBD dimerization state. The exact sequence of these events is not apparent from the crystal structures. Furthermore, the NBD dimer provides two active sites for ATP binding and hydrolysis, but whether ATP hydrolysis at both sites is required to induce dimer opening, or whether the two active sites take turns to convert ATP into ADP-Pi, remain unclear. To address these questions, it would be difficult to use traditional experimental approaches due to their limited temporal and spatial resolutions. Molecular dynamics simulations can serve as a tool to provide mechanistic details involved in the transition between different conformational states.
Using the crystal structure of the ATP-bound, dimeric NBD of the maltose transporter (MalK, PDB entry 1Q1286), four simulation systems were constructed with all possible nucleotide-bound states, namely ATP/ATP, ATP/ADP-Pi, ADP-Pi/ATP, and ADP-Pi/ADP-Pi..91 To simulate the effect of ATP hydrolysis, the bound ATP was converted as ADP-Pi in either or both of the active sites, with the Pi occupying the position of the γ-phosphate of the ATP molecule. Each of the four systems was simulated for at least 70 ns after initial equilibration in order to capture conformational changes under different nucleotide conditions.
The results of the simulations show that the closed dimer can only exist when both active sites are occupied by ATP (Fig. 7b), and that hydrolysis in one or both of the two active sites is able to induce the opening of the NBD dimer. Therefore, it is proposed that despite the existence of two active sites, one ATP hydrolysis is sufficient to trigger the transport mechanism. Moreover, since the ADP-Pi molecules in the post-hydrolysis systems all maintain their association with the binding sites over the entire simulation time span, the dimer opening is proposed to not require the dissociation of hydrolysis products. That is, the opening of the NBD dimer is a direct effect of the ATP hydrolysis, i.e., conversion of ATP to ADP and Pi. This implies that the product dissociation occurs later during the transport cycle after the NBDs have separated from each other.
Figure 7.

(a) The crystal structure of the E. coli maltose transporter in the ATP-bound, OF state, and a close-up view of the two nucleotide binding domains (NBDs) shown at the right panel. (b) Left: the final structures of the four simulations of the NBD dimer of the maltose transporter. Each of them has a different nucleotide configuration in the two active sites, either being ATP-Mg2+ or ADP-Pi with Mg2+. Right: the degree of dimer opening at each of the two active sites in the four simulation systems.
Comparing the three simulation trajectories containing post-hydrolysis active sites, it can be concluded that there is a delay between the ATP hydrolysis event and the opening of the active site at the dimer interface. However, there is no definite time between the two events (Fig. 7b). Also, hydrolysis reaction in one active site can result in the opening of any of the two active sites. The uncertainty in the time and location of the dimer opening after hydrolysis reaction indicates that the dimer opening is a stochastic process.
In the ATP-bound NBDs, the molecular interactions holding the two monomers together are solely mediated through ATP, especially the hydrogen bond network around the γphosphate that connects to both NBD monomers. Thus, ATP hydrolysis results in separation of the γ-phosphate of ATP from the β-phosphate, and, breaking the hydrogen bonds connecting to the NBD monomers. Therefore, the effect of ATP hydrolysis is simply to destabilize the dimer interface to allow the fluctuations of the protein and the nucleotides to break up the hydrogen bond network essential to the dimeric structure and result in the dimer opening. Because simultaneous rupture of several hydrogen bonds is required to open the NBD dimer, the NBD opening can only be captured in extended simulations.
Moreover, due to the stochastic nature of the hydrolysis-induced conformational changes, the systems require a certain amount of time (on the order of tens of nanoseconds in this case) to develop the opening event. Insufficient temporal coverage will result in incomplete conformational sampling, and failure in capturing the major events reported here.
With an increasing number of the crystal structures of intact ABC transporters being reported in recent years, as well as the rapid advances in computational algorithms and growing computational resources, we expect to be able to simulate intact ABC transporters in full atomic representation in explicit membrane/solution systems, up to a microsecond time scale. Such large scale, extended simulations continue to focus on mechanistic details of the transport cycle that are not readily resolved with experimental approaches, especially where dynamic processes and localized conformational changes within the systems are involved. It is therefore foreseeable that simulation approaches might be adopted to address some of the following questions for ABC transporters: what are the structural transitions between different conformational states and whether additional functionally relevant conformational states, e.g., outward-occluded or inward-occluded states, are involved in the mechanism; how does the substrate enter and leave the transporter during the transport cycle and how do they regulate the ATPase activity of the NBDs; and more importantly, how do the different domains couple to one another to yield various functional states involved in the transport cycle.
3 Substrate-Induced Rocker-Switch Motion in GlpT
One of the common strategies to account for the necessary free energy of transporting a molecule across the membrane against its concentration gradient is coupling the process to the transport of another molecule down its concentration gradient. This strategy is adopted by secondary active membrane transporters to accomplish their tasks. The largest and the most diverse group of secondary active membrane transporters, are the major facilitator superfamily, accounting for ~25% of all identified prokaryotic membrane transport proteins. Found ubiquitously in all three kingdoms of life, MFS contains a large number of medically relevant transporters.92–94 Despite the significance of this superfamily, atomic resolution structures have been reported only for four of its members95–101, due to technical difficulties associated with the crystallization of membrane transporters.102 One of the available MFS structures is that of glycerol-3-phosphate transporter (GlpT) from E.coli.95, 102, 103 GlpT is a member of organophosphate: phosphate antiporter family92–94, which facilitates the uptake of glycerol-3 phosphate (G3P) using inorganic phosphate gradient (Pi). 104–108 G3P is an essential molecule, which partakes in biosynthesis of phospholipids, the building blocks of the biological membranes, in addition to entering glycolysis metabolic pathway as an intermediate.109 The antibiotic fosfomycin, which bears a phosphate moiety and is structurally similar to G3P, was also shown to leak into the cell through GlpT.110–112 The structure of GlpT has served as a valuable model not only due to its role in nutrient uptake and antibiotic resistance, but also as a template to model its medically important eukaryotic MFS homologs.109, 113–116
GlpT shares a similar topology with other MFS transporters (Fig. 8a). The twelve transmembrane α-helices that compose GlpT are organized into two six-transmembrane helix bundles forming the N- and C-terminal halves (Fig. 8a). The N- and C-terminal halves connected by a loop unresolved in the crystal structure exhibit a pseudo-twofold symmetry with weak sequence homology.95, 109, 117 The inward-facing (IF) crystal structure features a lumen opening to the cytoplasm between the two halves. The apex of the lumen is conferred positive charges by two arginine residues (R45 and R269) which were suggested to constitute the putative substrate-binding site (Fig. 8b). Moreover, a highly conserved histidine residue (H165) located between the arginines has been proposed to be involved in substrate binding. Substrate-induced protonation of this histidine has been suggested as a possible trigger in the mechanism of the transporter.95, 109, 117 Several mutagenesis experiments, performed on GlpT118 and on its close homolog UhpT (hexose-6-phosphate transporter)95, 119, support the involvement of these residues in binding.
Figure 8.

(a) X-ray structure of GlpT. The N- and C-terminal halves are shown in green and pink, respectively. The location of the putative binding site in the apex of the lumen is indicated by a rectangular frame. (b) Zoomed in view of the putative binding site. (c) Alternating-access/Rocker-switch mechanism of GlpT. Pi binding from the cytoplasmic side results in closing of the cytoplasmic mouth of the lumen and subsequent opening of the periplasmic side. Once the binding site is exposed to the periplasm, Pi is replaced by G3P.
Based on the crystal structure of the IF state of GlpT, an “alternating access mechanism” (Fig. 1)120, in which the accessibility of the binding site from the two sides of the membrane is controlled through “rocker-switch ” type of conformational changes, was proposed (Fig. 8c).95, 109, 117, 121 It was proposed that Pi binding to the IF state of GlpT results in a series of conformational changes (predominantly internal helix motions122 and relative rigid rotation of the N- and C-terminal halves123), which close the cytoplasmic side and open the periplasmic side. Pi is, then, replaced in the outward-facing (OF) state by G3P taking the protein back to the starting configuration.95, 109, 117, 121 Since substrate binding is rapid, the rate limiting step in the transport is suggested to be large scale conformational changes involved in transition between IF and OF states.124 The role of substrate binding in this process appears to be lowering the activation energy of interconversion between the IF and OF states, through a mechanism in which the substrate pulls together the two arginines in the binding site (R45 and R269) (Fig. 8b), thus, bringing the N- and C-terminal halves together.95 A recent modeling study123 suggested that ~10 degrees of rigid rotation of each of the two halves, might be sufficient to obtain a functional state.94, 95, 109, 121
Despite the immense amount of information provided by the GlpT crystal structure95, the absence of bound substrates in the structure prevented complete insight into the molecular details of substrate binding and the transport mechanism. MD simulations have been recently applied successfully to characterize the interactions between transporters and their substrates and functionally relevant dynamics.125 In an effort to identify the binding site of GlpT and its conformational response to substrate binding, a set of simulations were performed on a membrane embedded GlpT in the presence of its natural substrates (monovalent or divalent Pi and G3P)9, 126. The simulations allowed identification of the substrate-binding site9, and revealed substrate-induced conformational changes in line with those expected from the rocker-switch model, i.e., closing at the cytoplasmic side of the lumen and reorganization of the periplasmic salt bridges.126
The model used for the simulations consisted of crystal structure of GlpT, embedded in a lipid bilayer, solvated and neutralized with Na+ and Cl− ions (Fig. 2). The substrate binding simulations were set up, in which the substrate was placed initially at the cytoplasmic mouth of the lumen in different simulations (Fig. 9a). GlpT was also simulated in the apo state to distinguish between the substrate-induced and random structural changes. This design not only minimizes the bias in determination of the binding site, since the substrates are allowed to diffuse into the binding site freely, but also allows the identification of the translocation mechanism, i.e., the sequence of contacts between the substrate and the protein.
Figure 9.
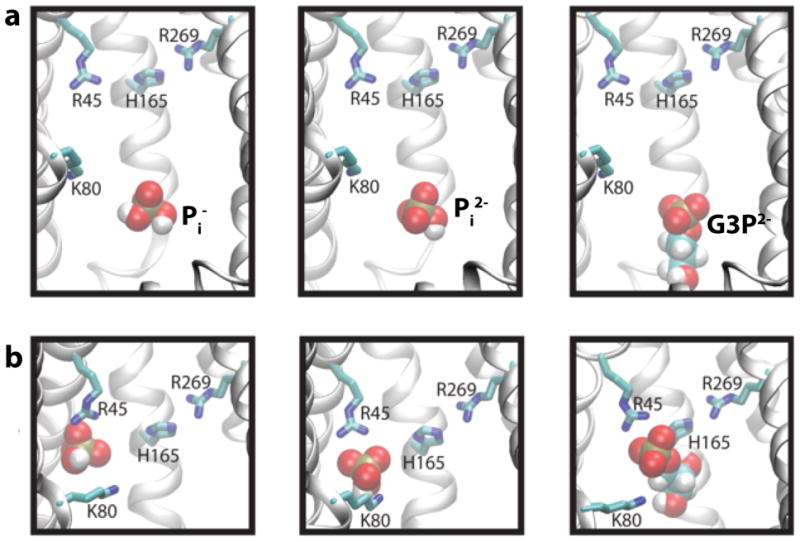
Translocation of the substrates from the mouth of the lumen to the binding site. (a) The substrate is initially placed so that the phosphorus atom of the substrate is ~15 Å away from the Cζ atom of R45. (b) The bound states at t = 50 ns for Pi −-, Pi2−-, and G3P2−-binding simulations.
All simulations revealed similar substrate translocation pathways inside the lumen and a common final binding site at the apex of the lumen. Spontaneous substrate binding is usually difficult to achieve in MD simulations given the time scale required for the process events are usually far beyond those accessible by MD. In GlpT, however, due to the presence of a strong luminal electrostatic potential and small size of the substrates, we were able to capture rapid spontaneous recruitment by GlpT. This observation is also in concordance with the kinetic data, which suggested substrate binding in GlpT is a rapid event.124 The observed spontaneous substrate binding involves two major steps: rapid recruitment of the substrate from the cytoplasmic inlet to the apex of the lumen facilitated by side chain motions of some residues inside a relatively rigid structure, followed by helical and side chain conformational changes in the protein while the substrate is coordinated stably by the binding site residues.
Small scale conformational changes were revealed to be important for substrate binding, from these simulations. Side chain motion of a highly conserved lysine (K80) that lines the lumen to be the key structural element in the recruitment of the substrate to the binding site. The main role of this lysine appears to be “fishing” the substrate from the entrance of the lumen and escorting it to the apex. K80, then, yields the substrate to one of the putative binding site arginines, R45 (Fig. 8b). Side chain motion of K80 while escorting the substrate to the apex of the lumen is the first important substrate induced conformational change.
These simulations also revealed the binding site of the substrate. Once K80 delivers the substrate, R45 tightly holds it in a “cage” formed by three tyrosine residues (Y38, Y42, Y76), which coordinate the phosphate moiety of the substrate with their hydroxyl groups (Fig. 9b). The function of tyrosine residues were originally thought to be limited to stabilization of the basicity of the lumen, but our simulations revealed that they are directly involved in substrate binding.94, 118 In the bound state, H165 also directly coordinates the substrate via hydrogen bonds. Interestingly, despite the position of R269 being symmetrical to R45, our simulations does not reveal any direct interaction between R269 and the substrate (Fig. 9b). Indeed, the mutagenesis experiments on binding site residues (R45, K80, H165, R269) reveal that only R45K mutation results in complete loss of binding in GlpT. R269K still retains the ability to bind with significantly reduced affinity.118 Our simulations further underline the functional difference between these two arginines. It is highly likely that R269 is involved in binding and transport in later stages.
While substrate recruitment (binding) is accompanied only with small scale conformational changes on the side-chain level, the bound substrate induces large scale helical conformational changes that are in line with the proposed rocker-switch mechanism over a longer time period. Thus, these simulations revealed that substrate binding induces closure of the cytoplasmic mouth of the lumen (Fig. 10b). Although the observed closure is incomplete, it is reproduced in all of the substrate binding simulations, while absent in the apo simulation. The closure is mainly due to cytoplasmic ends of helices 5 and 11 (Fig. 10a) approaching each other (Fig. 10c). This observation is in line with the proposed rocker-switch mechanism and describes the initial events in the formation of an occluded state via a rocker-switch mechanism.
Figure 10.

Substrate-induced partial closure of the cytoplasmic side of GlpT. (a) Helices 5 (pink) and 11 (green) colored on the ribbon representation of GlpT structure. The surface defined by dots indicates the lumen. (b) The radius of the lumen along the z-axis (parallel to the membrane normal).179 The closing region is indicated by dashed circles. (c) Superimposed frames from the trajectories showing the structural drift in the helices 5 and 11. Red and blue correspond to the beginning and the end of the simulation, respectively. Helices 5 and 11 come together at the cytoplasmic ends indicated by dashed rectangles upon binding of the substrates. This is not observed in the apo GlpT. (d) Substrate-induced salt bridge rearrangement in GlpT. The distribution of the distance between Cα–Nζ atoms of K46. The salt-bridge interactions that extended and compact K46 favors are shown in small panels.
The substrate-induced closure predominantly takes place much below the substrate binding site indicating that it is due to collective motion of the helices. Analysis of the simulation trajectories show that the helix motion is accompanied by the restriction of rotational freedom of H165 side chain, one of the residues in the identified binding site. In all the cases, the phosphate moiety confines the histidine side chain parallel to the plane of the membrane, whereas it can rotate freely in the absence of the substrate. It appears that H165, which is on helix 5, might act as a “pivot” for rotation of helix 5. Concordantly, when these helices were simulated individually in the membrane, it was seen that helix 5 and 11 exhibited the highest flexibility.122
Substrate binding also affects a periplasmic salt-bridge network which has been implicated as a switch in the rocker-switch mechanism118 of GlpT through side chain conformational changes of another conserved lysine residue (K46) and the binding-site histidine residue (H165). The salt-bridge network is composed of charged residues that link the N-and C-terminal halves (interdomain salt bridge) and those that are in the C-terminal half (intradomain salt bridge). The interdomain salt bridge is formed by a positively charged lysine (K46) on the N-terminal half interacting with either an aspartate (D274) located toward the periplasm or a glutamate (E299) near binding site on the C-terminal half (Fig. 10d). The same glutamate (E299) also forms the intradomain salt bridge with the putative binding site arginine (R299). These salt-bridges are thought to hold the N- and C-terminal halves of GlpT together, and their reorganization might act as a switch in the rocker-switch mechanism.94, 95, 118 All the residues except R269 are shown to be necessary for transport but are not directly involved in substrate binding.118 Simulations reveal side chain structural changes that reorganize the salt bridge network upon substrate binding.
The effect of the substrate on the interdomain salt bridge is mainly altering the side chain length of the lysine residue on the N-terminal domain. It indeed appears that the two negatively-charged residues on the C-terminal domain form two static charged spots, between which the lysine residue alternates. Substrate binding results in the extension of the lysine side chain, which seems to favor the K46–E299 salt bridge (Fig. 10d). On the other hand, the K46 side chain is compact in the apo state which favoring the K46–D274 salt bridge. This phenomenon is directly related to the electrostatic interaction between the substrate and K46, since the extension of the side chain of lysine depends on the titration state of the substrate, i.e., while the lysine side chain manifests similar extension in G3P2−- and Pi2−-binding simulations, it adopts a shorter side chain conformation in Pi−-binding simulation and even shorter in the apo state simulation (Fig. 10d).
The intradomain salt bridge (R269–E299) is stabilized by substrate binding indirectly through substrate-induced confinement of rotation of H165 side chain. It appears that as long as the hydrogen bonds can be maintained between R269 and H165, R269–E299 salt bridge can be stabilized. The side chain conformation that the substrate holds H165, i.e., parallel to the plane of the membrane, is ideal for hydrogen bond formation between the two residues.
The effect of substrate on the periplasmic interface might indeed be to destabilize and result in peeling of the tight junction between the two halves during the process of rocker-switch mechanism. Although our results do not provide direct evidence for salt-bridge reorganization resulting in destabilization of the interfacial interactions, they definitely indicate that the design of the transporter (the proximity of K46 to the binding site) allows the substrate manipulate distant interactions through K46 and H165.
The simulations captured the spontaneous binding of the substrate into the binding site along with associated structural changes. Substrate recruitment is facilitated by side chain motion of K80, which acts like a “fishing hook”. K80 delivers the substrate to R45, which keeps the substrate in a cage-like binding pocket formed by three tyrosine residues (Y38, Y42, and Y76). While H165 is also involved in the coordination of the phosphate moiety, the arginine on the C-terminal half (R269), which was suggested as a part of the putative substrate-binding site, does not form any direct interaction with the substrate. Moreover, capturing substrate binding allowed us to identify conformational response of GlpT to substrate binding and how it departs from the crystal structure. One of the significant conformational responses of GlpT to substrate binding is the partial closure of the cytoplasmic mouth of the lumen. The closure is determined mainly to originate from movement of helices 5 and 11 towards each other below the plane of the binding site. The other major affect of substrate binding appears to be on the periplasmic salt bridge network. Substrate binding results in extended conformation of the K46 side chain, and restriction of rotational freedom of H165 side chain resulting in reorganization of the interactions on the periplasmic side.
4 Extracellular Gate in Glutamate Transporter
Communication between neurons in the central nervous system is accomplished primarily by neurotransmitters. These chemicals are released into the synaptic cleft by presynaptic neurons in response to electrical activities, then detected and converted back into electrical signals by postsynaptic neurons. In order to maintain recurrent and selective signaling, the neurotransmitters must be rapidly removed after release.127, 128 Glutamate is the predominant excitatory neurotransmitter in the central nervous system that plays critical roles in fundamental processes such as learning and memory.129 Glutamate transporters (GlT) are membrane transporters in neurons and glial cells that catalyze the uptake of the neurotransmitter glutamate from the synapses.130, 131 The GlT family includes five human Excitatory Amino Acid Transporter (EAAT) subtypes, two neutral amino acid transporters, and a large number of bacterial amino acid and dicarboxylic acid transporters.132, 133 Malfunction of these transporters has been implicated in several neurodegenerative diseases, such as schizophrenia,129 Alzheimer’s disease,133 Huntington’s disease,134 and Parkinsonism-dementia complex.135
GlT belongs to the family of secondary membrane transporters, which couple “uphill” translocation of the substrate across the membrane to the energetically favorable flow of ions down their concentration gradient. By coupling to the co-transport of three Na+ and one H+, and the counter-transport of one K+, mammalian GlT transports one negatively charged glutamate across the membrane during each transport cycle.136–139 In contrast to the mammalian GlT, substrate transport in the bacterial homolog (Gltph)140 is not H+-coupled. Gltph, therefore, delivers substrate and Na+ ions during the transport cycle. According to this stoichiometry, glutamate transport via mammalian GlT or bacterial GlT (Gltph) is an electrogenic process meaning that it is associated with net charge transport across the membrane.
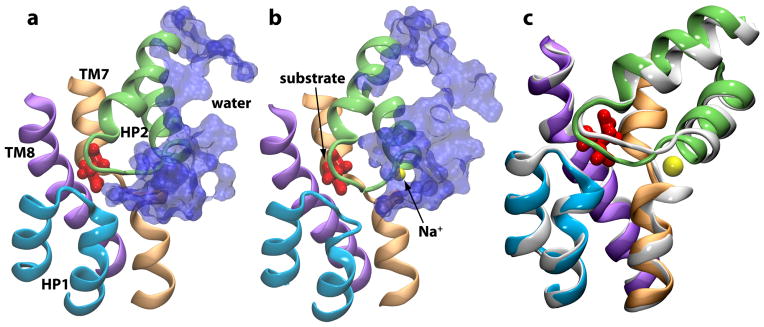
Similar to other transporters, substrate transport by GlT involves an alternating-access mechanism (Fig. 1) in which a conformational transition switches the access to the substrate between the intracellular and extracellular sides (Fig. 11 b).15 The transport cycle of GlT is proposed to involve four major states (Fig. 11 b): outward-facing open (OF-open), outward-facing occluded (OF-occluded), inward-facing occluded (IF-occluded), and inward-facing open (IF-open). The crystal structure of Gltph141 provided an opportunity to understanding the relationship between its structure and function, as well as the structural basis of its transport mechanism. Gltph shares about 36 % amino acid identity with mammalian GlTs, and many residues that have been implicated in substrate and ion binding or translocation are highly conserved throughout the GlT family, suggesting that it can serve as a structural model for understanding transport for the whole family.139, 142 The structure of Gltph141, 143 reveals a trimeric architecture for the transporter with a solvent-accessible extracellular basin extending halfway across the membrane, and captures it in the OF-occluded state. Each monomer is composed of eight transmembrane helices (TM1–TM8) and two highly conserved helical hairpins (HP1 and HP2), which are directly involved in the binding sites for the substrate and Na+ ions (Fig. 11 a). Each substrate binding site is cradled by these two helical hairpins reaching from the opposite sides of the membrane (Fig. 11 a). Crystallographic and thermodynamic studies of Gltph143 provided insightful structural information on the positions and the binding sites of the substrate and of two Na+ ions (termed Na1 and Na2 in the crystal structure, Fig. 11 a).
Figure 11.

Structure and hypothetical transport cycle of GlT. (a) The crystal structure of GlT with the bound substrate and two Na+ ions. Helical hairpins HP1 and HP2 that form the binding site are highlighted. (b) Schematic transport cycle: (i) the apo state (unbound) with HP2 in an open conformation; (ii) binding of three Na+ ions and the substrate induces the closure of HP2, yielding the outward facing-occluded state; (iii) the inward facing-occluded state is formed by conformational changes in the protein that switches the accessibility of the binding site to the cytoplasmic side of the membrane; and (iv) opening of cytoplasmic gates allows the release of the Na+ ions and the substrate into the cytoplasm.
A large number of experimental studies141–173 have investigated various structural and functional properties of GlTs. Based on the measurement of transport current, fluorescence signal and temperature dependence of the steady- and pre-steady-state kinetics during the transport cycle, it has been shown that substrate binding induces conformational changes in GlT. However, the limited spatial resolution of these studies made it difficult to draw specific conclusions about the nature and magnitude of such conformational changes. While earlier models suggested a rocker-switch mechanism174, 175 with large conformational changes for GlT, recent models154, 155 propose that localized, small-scale motions (serving as gates) alternate the accessibility of the substrate-binding site to the cytoplasmic and the extracellular solution. It is likely that both, gate mechanisms and large-scale motions, are involved in the mechanism. Fluorimetric measurements of conformational changes154, 155 and the study of the pre-steady-state kinetics170 in GlT suggest that binding of H+ precedes the binding of the substrate. Based on the X-ray structure of Gltph,141 Grewer et al. proposed a structural model for Na+ and glutamate binding to a homolog of mammalian GlT in which one Na+ ion binds to the empty transporter before glutamate binds.161 They also proposed that conformational changes take place in two glutamate-dependent half-cycles: glutamate-induced closing of an extracellular gate, and the subsequent opening of an unknown cytoplasmic gate that allows glutamate dissociation and diffusion into the cytoplasm.158 By determining the steady-and pre-steady-state kinetics of reverse glutamate transport, Grewer et al. recently proposed a kinetic model, which is based on a “first-in-first-out” mechanism, suggesting that glutamate association to its extracellular binding site precedes association of at least one of the co-transported Na+ ions, and that dissociation of glutamate from its intracellular binding site precedes dissociation of at least one Na+ ion.162
Although numerous experiments have provided insightful information about relevant features and aspects of the putative transport cycle of GlT (Fig. 11 b), the details of the mechanism that couples the opening and closing of the extracellular gate to substrate and ions, and the sequence of binding of the substrate and cotransported Na+ ions in the OF-open state are fundamental unanswered questions. In order to address these questions, a set of MD simulations of membrane-embedded trimeric models of GlT have been performed.176 Different combinations of the substrate and the two structurally resolved Na+ ions (Na1 and Na2)143 were used to investigate equilibrium dynamics of GlT at different bound states and the coupling of binding of Na+ ions and the substrate.176 The system was simulated under eight different conditions, each simulation lasting 20–30 ns,176 revealing two highly relevant mechanistic details regarding the transport cycle in GlT.
Comparison of the dynamics of the substrate-bound and the substrate-free (apo) states of GlT in our simulations suggests that the helical hairpin HP2 plays the role of the extracellular gate.176 Invariably in all the simulations performed in the presence of the substrate, HP2 has a very stable conformation (Fig. 12). After removing the substrate, however, HP2 undergoes a large opening motion resulting in the complete exposure of the substrate binding site to the extracellular solution (Fig. 12). Opening of the binding site is accompanied by its full hydration. These results suggest that HP2 plays the role of the extracellular gate, and that, more importantly, its opening and closure of the gate is controlled by substrate binding.176 A gating role for HP2 is supported by the structure of GlT in the presence of an inhibitor,143 and the results of rapid solution exchange and laser-pulse photolysis experiments.161 Furthermore, very recent inhibition studies in a mutant homolog of mammalian GlT using oxidative cross-linking of engineered cysteine pairs177 suggest that HP2 serves as the extracellular gate of the transporter and that substrate induces distinct conformations of HP2. A recent MD simulation study13 has also provided support for this idea.
Figure 12.

Dynamics of the extracellular gate in GlT. Left and right panels show the results of the simulations performed in the presence and in the absence of the substrate, respectively. (left, top panel) Substrate-bound state. Closing of HP2 (the extracellular gate) and formation of the Na2 binding site (marked with a circle; focusing of the helical dipole moments). (right, top panel) Substrate-free state. Opening of HP2 and exposure of the binding site. Note the significant misalignment of the dipole moments of helices TM7a and HP2a (blue arrows). The bottom panel shows time evolution of the RMSDs of the helical hairpins HP1 and HP2.
Interestingly, despite its apparent structural symmetry to HP2, helical hairpin HP1 was found to exhibit a high level of conformational stability regardless of the presence of the substrate (Fig. 12).176 This result, which might be attributed either to the shorter length of the loop of HP1 (when compared to HP2), or to its more closer contact with TM2, suggests that, at least during the extracellular half of the transport cycle, HP1 does not play a direct role, and its involvement might be limited to stabilization of the structure of HP2 upon substrate binding. The possibility of a gating role of HP1 in the cytoplasmic side will have to await the determination of the structure of a GlT in the IF conformation.
Substrate binding to GlT brings the HP2 and HP1 loops together, through establishing direct interactions between the charged groups of the substrate and the backbone groups of HP2. It should be noted that, upon substrate binding, only one half of HP2 (the Gly359 side) is sealed, a state that might be best characterized as a partially occluded state (Fig. 12). In this state, although the binding site is largely shielded from the extracellular region, water molecules can still move in and out of the binding pocket since the other half of HP2 (the Gly351 side) is not fully sealed (Fig. 13 a and 13 c). Therefore, a complete occlusion of the binding site requires additional steps, likely, binding of Na+ ion(s), that will bring the extracellular gate to a completely closed state (Fig. 13 b).
Figure 13.
Na2-induced formation of the occluded state. Binding of Na2 to GlT results in complete closure of the substrate binding site to the extracellular solution. In the absence of Na2 (a), the binding pocket is accessible to water, whereas upon Na2 binding (b), the binding site is completely sealed. An overlay of the two states (white, before Na2 binding; colored, after Na2 binding) in (c) highlights the small change in the conformation of HP2 upon Na2 binding.
Another major consequence of substrate binding revealed by the simulations is the formation of a new Na+ binding site.176 In the crystal structure, one of the Na+ ions (Na2) is bound to a binding site formed between two half-helical structures (HP2a and TM7a, see Fig. 12). In the apo state, the dipole moments of these half-helices were found to be totally misaligned (Fig. 12). Upon substrate binding, the two opposing half-helices align such that their dipole moments converge on a single point resulting in the formation of the Na2 binding site (Fig. 12). These results have direct implications with regard to the sequence and the coupling of binding of Na+ ions and the substrate; they strongly suggest that Na2 binding can only take place after binding of the substrate.
Na2 binding further stabilizes HP2, resulting in a completely occluded form of GlT, in which water molecules (and, therefore, H+ and Na+ ions) can no longer access the binding site from the extracellular side (Fig. 13 b). Thus, Na2 binding results in a complete closure of the extracellular gate. These simulation results are strongly supported by various experiments, including crystallographic and thermodynamic studies,143 determination of the steady- and pre-steady kinetics,162 and measurements of transporter currents associated with stoichiometric and anion charge movements in GlT,178 which have suggested that substrate binding enables the binding of one of the co-transported Na+ ions.
A widely accepted view with regard to GlTs has been the alternating access mechanism in which the binding sites for the substrate and Na+ ions are alternatively exposed to the extracellular and intracellular sides via large-scale conformational changes of the transporter. The present structure-based simulations shed light on the mechanisms of the opening and closure of extracellular gate in GlT. The helical hairpin HP2 undergoes large conformational changes exposing the substrate binding site to the extracellular solution in the apo state, providing direct evidence for dynamical role of this loop in the gating of the substrate binding site in GlT. Although we have investigated the extracellular gating mechanism and the coupling between substrate and one of Na+ ions, the mechanisms of the transition between the OF-occluded and the IF-occluded states (Fig. 11 b) and of release of the substrate and Na+ ions from the IF-occluded state into the cytoplasm are completely unknown.143, 154, 158, 161
Conclusion
Protein conformational changes are one of the fundamental aspects of protein function in biology. Given the advances in structural biology resulting in a continually increasing number of protein structures at an atomic resolution and significant developments in algorithms and parallel computing, we have been able, over the past few years, to extend the scope of computer simulation into the realm of protein domain motions, permitting us to capture more functionally relevant conformational changes of proteins. In this review, we discussed the results of application of extended large-scale molecular dynamics simulations to membrane channel and transporter proteins. We demonstrated that such simulations are able to capture various forms and degrees of conformational changes involved in the function of these mechanistically complex proteins. Combining atomic representations of the proteins and their surrounding (lipid, water, ions, etc.) with extended simulations, we have been able to characterize motions ranging from side chain rotations and hydrogen-bond breaking events, all the way to flipping of subdomains and even domain separation in different proteins. Most importantly, we showed that these are all of functional significance, i.e., such motions are induced in the proteins in response to various events and elements that are involved in their function, and not random processes. In most cases, we have also demonstrated that such events are reproducible and are observed in independent simulations.
The results of these studies have produced novel hypotheses regarding the function of membrane channels and transporters. Many of these hypotheses can be used to design new experiments that can verify the proposed mechanisms. Along with the growing computational power, we will be able to extend further the time scale of the simulations and improve our sampling and statistics. Therefore, we should expect more examples of biomolecular simulations in which key functional dynamical events have been captured. In the near future, we also expect to have a larger number of high-resolution structures for intermediates and functional states, which, combined with extended simulations, will allow us to provide a complete description of the gating and conduction processes in membrane channels and to capture the dynamics of the entire transport cycle in membrane transporters.
Acknowledgments
The studies reported in this review were supported by grants from NIH (R01-GM086749, R01-GM067887, and P41-RR05969). The authors acknowledge computer time at TeraGrid resources (grant number MCA06N060), as well as computer time from the DoD High Performance Computing Modernization Program at the Arctic Region Supercomputing Center, University of Alaska at Fairbanks. The authors thank Dr. James C. Gumbart and Dr. Y. Zenmei Ohkubo for assistance in preparing Figures 2 and 11.
References
- 1.Law RJ, Henchman RH, McCammon JA. Proc Natl Acad Sci USA. 2005;102:6813. doi: 10.1073/pnas.0407739102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hung A, Tai K, Sansom M. Biophys J. 2005;88:3321. doi: 10.1529/biophysj.104.052878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arkin IT, Xu H, Jensen M, Arbely E, Bennett ER, Bowers KJ, Chow E, Dror RO, Eastwood MP, Flitman-Tene R, Gregersen BA, Klepeis JL, Kolossváry I, Shan Y, Shaw DE. Science. 2007;317:799. doi: 10.1126/science.1142824. [DOI] [PubMed] [Google Scholar]
- 4.Olkhova E, Padan E, Michel H. Biophys J. 2007;92:3784. doi: 10.1529/biophysj.106.098269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng X, Ivanov I, Wang H, Sine SM, McCammon JA. Biophys J. 2007;93:2622. doi: 10.1529/biophysj.107.109843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cordero-Morales JF, Jogini V, Lewis A, Vasquez V, Cortes DM, Roux B, Perozo E. Nat Struct Mol Biol. 2007;14:1062. doi: 10.1038/nsmb1309. [DOI] [PubMed] [Google Scholar]
- 7.Sonne J, Kandt C, Peters GH, Hansen FY, Jansen MO, Tieleman DP. Biophys J. 2007;92:2727. doi: 10.1529/biophysj.106.097972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Celik L, Schiott B, Tajkhorshid E. Biophys J. 2008;94:1600. doi: 10.1529/biophysj.107.117580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Law CJ, Enkavi G, Wang DN, Tajkhorshid E. Biophys J. 2009;97:1346. doi: 10.1016/j.bpj.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shaikh SA, Tajkhorshid E. Biophys J. 2008;95:5153. doi: 10.1529/biophysj.108.141606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang Z, Tajkhorshid E. Biophys J. 2008;95:2292. doi: 10.1529/biophysj.108.133421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wen PC, Tajkhorshid E. Biophys J. 2008;95:5100. doi: 10.1529/biophysj.108.139444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shrivastava IH, Jiang J, Amara SG, Bahar I. J Biol Chem. 2008;283:28680. doi: 10.1074/jbc.M800889200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gumbart JC, Weiner M, Tajkhorshid E. J Mol Biol. 2009 doi: 10.1016/j.jmb.2009.09.004. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jardetzky O. Nature. 1966;211:2406. doi: 10.1038/211969a0. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, Tajkhorshid E. J Nutr. 2007;137:1509S. doi: 10.1093/jn/137.6.1509S. [DOI] [PubMed] [Google Scholar]
- 17.Gumbart J, Wiener MC, Tajkhorshid E. Biophys J. 2007;93:496. doi: 10.1529/biophysj.107.104158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hashido M, Kidera A, Ikeguchi M. Biophys J. 2007;93:373. doi: 10.1529/biophysj.106.101170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang X, Zhan CG. Biophys J. 2007;93:3627. doi: 10.1529/biophysj.107.110924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holyoake J, Sansom MSP. Structure. 2007;15:873. doi: 10.1016/j.str.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 21.Klauda JB, Brooks BR. J Mol Biol. 2007;367:1523. doi: 10.1016/j.jmb.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anishkin A, Kamaraju K, Sukharev S. J Gen Physiol. 2008;132:67. doi: 10.1085/jgp.200810000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hub J, de Groot B. Proc Natl Acad Sci USA. 2008;105:1198. doi: 10.1073/pnas.0707662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Henin J, Tajkhorshid E, Schulten K, Chipot C. Biophys J. 2008;94:832. doi: 10.1529/biophysj.107.115105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi L, Quick M, Zhao Y, Weinstein H, Javitch JA. Mol Cell. 2008;30:667. doi: 10.1016/j.molcel.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noskov SY, Roux B. J Mol Biol. 2008;377:804. doi: 10.1016/j.jmb.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vasquez V, Sotomayor M, Cordero-Morales J, Schulten K, Perozo E. Science. 2008;321:1210. doi: 10.1126/science.1159674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y, Tajkhorshid E. Proc Natl Acad Sci USA. 2008;105:9598. doi: 10.1073/pnas.0801786105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li J, Tajkhorshid E. Biophys J. 2009 doi: 10.1016/j.bpj.2009.09.005. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. J Comp Chem. 2005;26:1781. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martyna GJ, Tobias DJ, Klein ML. J Chem Phys. 1994;101:4177. [Google Scholar]
- 32.Feller SE, Zhang YH, Pastor RW, Brooks BR. J Chem Phys. 1995;103:4613. [Google Scholar]
- 33.Darden T, York D, Pedersen L. J Chem Phys. 1993;98:10089. [Google Scholar]
- 34.Waldmann R, Lazdunski M. Curr Opin Neur. 1998;8:418. doi: 10.1016/s0959-4388(98)80070-6. [DOI] [PubMed] [Google Scholar]
- 35.Kellenberger S, Schild L. Physiol Rev. 2002;82:735. doi: 10.1152/physrev.00007.2002. [DOI] [PubMed] [Google Scholar]
- 36.Chen LY, Jiunn Pan C, Shieh JJ, Chou JY. Hum Mol Gen. 2002;11 doi: 10.1093/hmg/11.25.3199. [DOI] [PubMed] [Google Scholar]
- 37.Krishtal O, Pidoplichko V. Neurosci Lett. 1981;24:243. doi: 10.1016/0304-3940(81)90164-6. [DOI] [PubMed] [Google Scholar]
- 38.Wemmie JA, Chen J, Askwith CC, Hruska-Hageman AM, Price MP, Nolan BC, Yoder PG, Lamani E, Hoshi T, Freeman JH. Neuron. 2002;34:463. doi: 10.1016/s0896-6273(02)00661-x. [DOI] [PubMed] [Google Scholar]
- 39.Krishtal O. Trends in Neurosciences. 2003;26:477. doi: 10.1016/S0166-2236(03)00210-8. [DOI] [PubMed] [Google Scholar]
- 40.Wemmie JA, Price MP, Welsh MJ. Trends in Neurosciences. 2006;29:578. doi: 10.1016/j.tins.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 41.Friese M, Craner M, Etzensperger R, Vergo S, Wemmie J, Welsh M, Vincent A, Fugger L. Nat Med. 2007;13:1483. doi: 10.1038/nm1668. [DOI] [PubMed] [Google Scholar]
- 42.Drummond HA, Jernigan NL, Grifoni SC. Hypertension. 2008;51:1265. doi: 10.1161/HYPERTENSIONAHA.107.093401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krishtal O, Pidoplichko V. Neuroscience. 1980;5:2325. doi: 10.1016/0306-4522(80)90149-9. [DOI] [PubMed] [Google Scholar]
- 44.Price MP, Snyder PM, Welsh NJ. J Biol Chem. 1996;271:7879. doi: 10.1074/jbc.271.14.7879. [DOI] [PubMed] [Google Scholar]
- 45.García-Anoveros J, Derfler B, Neville-Golden J, Hyman BT, Corey DP. Proc Natl Acad Sci USA. 1997;94:1459. doi: 10.1073/pnas.94.4.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Waldmann R, Champigny G, Bassilana F, Heurteaux C, Lazdunski M. Nature. 1997;386:173. doi: 10.1038/386173a0. [DOI] [PubMed] [Google Scholar]
- 47.Zhang P, Canessa CM. J Gen Physiol. 2002;120:553. doi: 10.1085/jgp.20028574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hesselager M, Timmermann DB, Ahring PK. J Biol Chem. 2004;279:11006. doi: 10.1074/jbc.M313507200. [DOI] [PubMed] [Google Scholar]
- 49.Smith ESJ, Zhang X, Cadiou H, McNaughton PA. Neurosci Lett. 2007;426:12. doi: 10.1016/j.neulet.2007.07.047. [DOI] [PubMed] [Google Scholar]
- 50.Paukert M, Chen X, Polleichtner G, Schindelin H, Gründer S. J Biol Chem. 2008;283:572. doi: 10.1074/jbc.M706811200. [DOI] [PubMed] [Google Scholar]
- 51.Coric T, Zhang P, Todorovic N, Canessa CM. J Biol Chem. 2003;278:45240. doi: 10.1074/jbc.M304441200. [DOI] [PubMed] [Google Scholar]
- 52.Pfister Y, Gautschi I, Takeda AN, van Bemmelen M, Kellenberger S, Schild L. J Biol Chem. 2006;281:11787. doi: 10.1074/jbc.M513692200. [DOI] [PubMed] [Google Scholar]
- 53.Cushman KA, Marsh-Haffner J, Adelman JP, McCleskey EW. J Gen Physiol. 2007;129:345. doi: 10.1085/jgp.200709757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jasti J, Furukawa H, Gonzales EB, Gouaux E. Nature. 2007;449:316. doi: 10.1038/nature06163. [DOI] [PubMed] [Google Scholar]
- 55.Immke DC, McCleskey EW. Neuron. 2003;37:75. doi: 10.1016/s0896-6273(02)01130-3. [DOI] [PubMed] [Google Scholar]
- 56.Zhang P, Sigworth FJ, Canessa CM. J Gen Physiol. 2006;127:109. doi: 10.1085/jgp.200509396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gonzalez EB, Kawate T, Gouaux E. Nature. 2009;460:599. doi: 10.1038/nature08218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Immke DC, McCleskey EW. Nature Neurosci. 2001;4:869. doi: 10.1038/nn0901-869. [DOI] [PubMed] [Google Scholar]
- 59.Yang H, Yu Y, Li WG, Yu F, Cao H, Xu TL, Jiang H. PLoS Biol. 2009;7:e1000151. doi: 10.1371/journal.pbio.1000151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Locher KP, Lee AT, Rees DC. Science. 2002;296:1091. doi: 10.1126/science.1071142. [DOI] [PubMed] [Google Scholar]
- 61.Dawson RJ, Locher KP. Nature. 2006;443:180. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]
- 62.Pinkett HW, Lee AT, Lum P, Locher KP, Rees DC. Science. 2007;315:373. doi: 10.1126/science.1133488. [DOI] [PubMed] [Google Scholar]
- 63.Dawson RJ, Locher KP. FEBS Lett. 2007;581:935. doi: 10.1016/j.febslet.2007.01.073. [DOI] [PubMed] [Google Scholar]
- 64.Hollenstein K, Frei DC, Locher KP. Nature. 2007;446:213. doi: 10.1038/nature05626. [DOI] [PubMed] [Google Scholar]
- 65.Hvorup RN, Goetz BA, Niederer M, Hollenstein K, Perozo E, Locher KP. Science. 2007;317:1387. doi: 10.1126/science.1145950. [DOI] [PubMed] [Google Scholar]
- 66.Oldham ML, Khare D, Quiocho FA, Davidson AL, Chen J. Nature. 2007;450:515. doi: 10.1038/nature06264. [DOI] [PubMed] [Google Scholar]
- 67.Ward A, Reyes CL, Yu J, Roth CB, Chang G. Proc Natl Acad Sci USA. 2007;104:19005. doi: 10.1073/pnas.0709388104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gerber S, Comellas-Bigler M, Goetz BA, Locher KP. Science. 2008;321:246. doi: 10.1126/science.1156213. [DOI] [PubMed] [Google Scholar]
- 69.Kadaba NS, Kaiser JT, Johnson E, Lee A, Rees DC. Science. 2008;321:250. doi: 10.1126/science.1157987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Khare D, Oldham ML, Orelle C, Davidson AL, Chen J. Mol Cell. 2009;33:528. doi: 10.1016/j.molcel.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, Harrell PM, Trinh YT, Zhang Q, Urbatsch IL, Chang G. Science. 2009;323:1718. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Moussatova A, Kandt C, O’Mara ML, Tieleman DP. Biochim Biophys Acta. 2008;1778:1757. doi: 10.1016/j.bbamem.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 73.Dawson RJ, Hollenstein K, Locher KP. Mol Microbiol. 2007;65:250. doi: 10.1111/j.1365-2958.2007.05792.x. [DOI] [PubMed] [Google Scholar]
- 74.Hollenstein K, Dawson RJ, Locher KP. Curr Opin Struct Biol. 2007;17:412. doi: 10.1016/j.sbi.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 75.Oldham ML, Davidson AL, Chen J. Curr Opin Struct Biol. 2008;18:726. doi: 10.1016/j.sbi.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Locher KP. Phil Trans R Soc Lond B. 2009;364:239. doi: 10.1098/rstb.2008.0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rees DC, Johnson E, Lewinson O. Nat Rev Mol Cell Biol. 2009;10:218. doi: 10.1038/nrm2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kos V, Ford RC. Cell Mol Life Sci. 2009 doi: 10.1007/s00018-009-0064-9. in Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Oloo EO, Fung EY, Tieleman DP. J Biol Chem. 2006;281:28397. doi: 10.1074/jbc.M513614200. [DOI] [PubMed] [Google Scholar]
- 80.Jones MK, Catte A, Patterson JC, Gu F, Chen J, Li L, Segrest JP. Biophys J. 2009;96:954. doi: 10.1016/j.bpj.2008.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sonne J, Kandt C, Peters GH, Hansen FY, Jansen MO, Tieleman DP. Biophys J. 2007;92:2727. doi: 10.1529/biophysj.106.097972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ivetac A, Campbell JD, Sansom MS. Biochemistry. 2007;46:2767. doi: 10.1021/bi0622571. [DOI] [PubMed] [Google Scholar]
- 83.Weng J, Ma J, Fan K, Wang W. Biophys J. 2008;94:612. doi: 10.1529/biophysj.107.110734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Weng J, Ma J, Fan K, Wang W. Biophys J. 2009;96:1918. doi: 10.1016/j.bpj.2008.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Smith PC, Karpowich N, Millen L, Moody JE, Rosen J, Thomas PJ, Hunt JF. Mol Cell. 2002;10:139. doi: 10.1016/s1097-2765(02)00576-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chen J, Lu G, Lin J, Davidson AL, Quiocho FA. Mol Cell. 2003;12:651. doi: 10.1016/j.molcel.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 87.Zaitseva J, Jenewein S, Jumpertz T, Holland IB, Schmitt L. EMBO J. 2005;24:1901. doi: 10.1038/sj.emboj.7600657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zaitseva J, Oswald C, Jumpertz T, Jenewein S, Wiedenmann A, Holland IB, Schmitt L. EMBO J. 2006;25:3432. doi: 10.1038/sj.emboj.7601208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yuan YR, Blecker S, Martsinkevich O, Millen L, Thomas PJ, Hunt JF. J Biol Chem. 2001;276:32313. doi: 10.1074/jbc.M100758200. [DOI] [PubMed] [Google Scholar]
- 90.Lu G, Westbrooks JM, Davidson AL, Chen J. Proc Natl Acad Sci USA. 2005;102:17969. doi: 10.1073/pnas.0506039102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wen PC, Tajkhorshid E. Biophys J. 2008;95:5100. doi: 10.1529/biophysj.108.139444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Marger M, Saierjr M. Trends Biochem Sci. 1993;18:13. doi: 10.1016/0968-0004(93)90081-w. [DOI] [PubMed] [Google Scholar]
- 93.Pao SS, Paulsen IT, Saier MH. Microbiol Mol Biol Rev. 1998;62:1. doi: 10.1128/mmbr.62.1.1-34.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Law CJ, Maloney PC, Wang DN. Annu Rev Microbiol. 2008;62:289. doi: 10.1146/annurev.micro.61.080706.093329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Huang Y, Lemieux MJ, Song J, Auer M, Wang DN. Science. 2003;301:616. doi: 10.1126/science.1087619. [DOI] [PubMed] [Google Scholar]
- 96.Abramson J, Smirnova I, Kasho V, Verner G, Kaback HR, Iwata S. Science. 2003;301:610. doi: 10.1126/science.1088196. [DOI] [PubMed] [Google Scholar]
- 97.Mirza O, Guan L, Verner G, Iwata S, Kaback HR. EMBO J. 2006;25:1177. doi: 10.1038/sj.emboj.7601028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Guan L, Mirza O, Verner G, Iwata S, Kaback HR. Proceedings of the National Academy of Sciences. 2007;104:15294. doi: 10.1073/pnas.0707688104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Heymann JAW, Hirai T, Shi D, Subramaniam S. J Struct Biol. 2003;144:320. doi: 10.1016/j.jsb.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 100.Hirai T, Subramaniam S. Biophys J. 2004;87:3600. doi: 10.1529/biophysj.104.049320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yin Y, He X, Szewczyk P, Nguyen T, Chang G. Science. 2006;312:741. doi: 10.1126/science.1125629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lemieux MJ, Song J, Kim MJ, Huang Y, Villa A, Auer M, Li XD, Wang DN. Prot Sci. 2003;12:2748. doi: 10.1110/ps.03276603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Luckey M. Membrane structural biology: with biochemical and biophysical foundations. Cambridge University Press; 2008. [Google Scholar]
- 104.Hayashi S-i, Koch JP, Lin ECC. J Biol Chem. 1964;239:3098. [PubMed] [Google Scholar]
- 105.Silhavy TJ, Hartig-Beecken I, Boos W. J Bacteriol. 1976;126:951. doi: 10.1128/jb.126.2.951-958.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Larson TJ, Schumacher G, Boos W. J Bacteriol. 1982;152:1008. doi: 10.1128/jb.152.3.1008-1021.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Elvin CM, Hardy CM, Rosenberg H. J Bacteriol. 1985;161:1054. doi: 10.1128/jb.161.3.1054-1058.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ambudkar SV, Larson TJ, Maloney PC. J Biol Chem. 1986;261:9083. [PubMed] [Google Scholar]
- 109.Lemieux M. Res Microbiol. 2004;155:623. doi: 10.1016/j.resmic.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 110.Argast M, Ludtke D, Silhavy TJ, Boos W. J Bacteriol. 1978;136:1070. doi: 10.1128/jb.136.3.1070-1083.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nilsson AI, Berg OG, Aspevall O, Kahlmeter G, Andersson DI. Antimicrob Agents Chemother. 2003;47:2850. doi: 10.1128/AAC.47.9.2850-2858.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sakamoto Y, Furukawa S, Ogihara H, Yamasaki M. Biosci Biotechnol Biochem. 2003;67:2030. doi: 10.1271/bbb.67.2030. [DOI] [PubMed] [Google Scholar]
- 113.Salasburgos A, Iserovich P, Zuniga F, Vera J, Fischbarg J. Biophys J. 2004;87:2990. doi: 10.1529/biophysj.104.047886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Holyoake J, Caulfeild V, Baldwin S, Sansom M. Biophys J. 2006;91:L84. doi: 10.1529/biophysj.106.093971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lemieux MJ. Mol Membr Biol. 2007;24:333. doi: 10.1080/09687680701496507. [DOI] [PubMed] [Google Scholar]
- 116.Blodgett DM, Graybill C, Carruthers A. J Biol Chem. 2008;283:36416. doi: 10.1074/jbc.M804802200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lemieux M, Huang Y, Wang D. Curr Opin Struct Biol. 2004;14:405. doi: 10.1016/j.sbi.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 118.Law CJ, Almqvist J, Bernstein A, Goetz RM, Huang Y, Soudant C, Laaksonen A, Hovmlle S. J Mol Biol. 2008;378:828. doi: 10.1016/j.jmb.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fann MC, Davies AH, Varadhachary A, Kuroda T, Sevier C, Tsuchiya T, Maloney PC. J Membr Biol. 1998;164:187. doi: 10.1007/s002329900404. [DOI] [PubMed] [Google Scholar]
- 120.Stroud RM. Proc Natl Acad Sci USA. 2007;104:1445. doi: 10.1073/pnas.0610349104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lemieux MJ, Huang Y, Wang DN. Journal of Electron Microscopy. 2005:54. doi: 10.1093/jmicro/54.suppl_1.i43. [DOI] [PubMed] [Google Scholar]
- 122.D’rozario RSG, Sansom MSP. Mol Membr Biol. 2008;25:571. doi: 10.1080/09687680802549113. [DOI] [PubMed] [Google Scholar]
- 123.Tsigelny IF, Greenberg J, Kouznetsova V, Nigam SK. J Bioinformatics Comput Biol. 2008;6:885. doi: 10.1142/s0219720008003801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Law CJ, Yang Q, Soudant C, Maloney PC, Wang DN. Biochemistry. 2007:46. doi: 10.1021/bi701383g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yan Q. Membrane transporters: methods and protocols. Humana Press; 2003. [Google Scholar]
- 126.Enkavi G, Tajkhorshid E. 2009 submitted. [Google Scholar]
- 127.Clements JD. Trends Neurosci. 1996;19:163. doi: 10.1016/s0166-2236(96)10024-2. [DOI] [PubMed] [Google Scholar]
- 128.Bergles DE, Diamond JS, Jahr CE. Curr Opin Neurobiol. 1999;9:293. doi: 10.1016/s0959-4388(99)80043-9. [DOI] [PubMed] [Google Scholar]
- 129.Dingledine R, Borges K, Bowie D, Traynelis SF. Pharmacol Rev. 1999;51:7. [PubMed] [Google Scholar]
- 130.Chen NH, Reith ME, Quick MW. Pflug Arch Eur J Physiol. 2004;447:519. doi: 10.1007/s00424-003-1064-5. [DOI] [PubMed] [Google Scholar]
- 131.Grewer C, Rauen T. J Membr Biol. 2005;203:1. doi: 10.1007/s00232-004-0731-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Slotboom DJ, Konings WN, Lolkema JS. Microbiol Mol Biol Rev. 1999;63:293. doi: 10.1128/mmbr.63.2.293-307.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Danbolt NC. Prog Neurobiol. 2001;65:1. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- 134.Behrens PF, Franz P, Woodman B, Lindenberg KS, Landwehrmeyer GB. Brain. 2002;125:1908. doi: 10.1093/brain/awf180. [DOI] [PubMed] [Google Scholar]
- 135.Yi JH, Hazell AS. Neurochem Int. 2006;48:394. doi: 10.1016/j.neuint.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 136.Zerangue N, Kavanaugh MP. Nature. 1996;383:634. doi: 10.1038/383634a0. [DOI] [PubMed] [Google Scholar]
- 137.Levy LM, Warr O, Attwell D. J Neurosci. 1998;18:9620. doi: 10.1523/JNEUROSCI.18-23-09620.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pines G, Kanner BI. Biochemistry. 1990;29:11209. doi: 10.1021/bi00503a008. [DOI] [PubMed] [Google Scholar]
- 139.Kavanaugh MP, Bendahan A, Zerangue N, Zhang Y, Kanner BI. J Biol Chem. 1997;272:1703. doi: 10.1074/jbc.272.3.1703. [DOI] [PubMed] [Google Scholar]
- 140.Ryan RM, Compton EL, Mindell JA. J Biol Chem. 2009;284:17540. doi: 10.1074/jbc.M109.005926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yernool D, Boudker O, Jin Y, Gouaux E. Nature. 2004;431:811. doi: 10.1038/nature03018. [DOI] [PubMed] [Google Scholar]
- 142.Bendahan A, Armon A, Madani N, Kavanaugh MP, Kanner BI. J Biol Chem. 2000;275:37436. doi: 10.1074/jbc.M006536200. [DOI] [PubMed] [Google Scholar]
- 143.Boudker O, Ryan RM, Yernool D, Shimamoto K, Gouaux E. Nature. 2007;445:387. doi: 10.1038/nature05455. [DOI] [PubMed] [Google Scholar]
- 144.Grewer C, Balani P, Weidenfeller C, Bartusel T, Tao Z, Tauen T. Biochemistry. 2005;44:11913. doi: 10.1021/bi050987n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Grunewald M, Kanner BI. J Biol Chem. 1995;270:17017. doi: 10.1074/jbc.270.28.17017. [DOI] [PubMed] [Google Scholar]
- 146.Grunewald M, Bendahan A, Kanner BI. Neuron. 1998;21:623. doi: 10.1016/s0896-6273(00)80572-3. [DOI] [PubMed] [Google Scholar]
- 147.Grunewald M, Kanner BI. J Biol Chem. 2000;275:9684. doi: 10.1074/jbc.275.13.9684. [DOI] [PubMed] [Google Scholar]
- 148.Slotboom DJ, Lolkema JS, Konings WN. J Biol Chem. 1996;271:31317. doi: 10.1074/jbc.271.49.31317. [DOI] [PubMed] [Google Scholar]
- 149.Slotboom DJ, Konings WN, Lolkema JS. J Biol Chem. 2001;276:10775. doi: 10.1074/jbc.M011064200. [DOI] [PubMed] [Google Scholar]
- 150.Seal RP, Amara SG. Neuron. 1998;21:1487. doi: 10.1016/s0896-6273(00)80666-2. [DOI] [PubMed] [Google Scholar]
- 151.Seal RP, Leighton BH, Amara SG. Neuron. 2000;25:695. doi: 10.1016/s0896-6273(00)81071-5. [DOI] [PubMed] [Google Scholar]
- 152.Zarbiv R, Grunewald M, Kavanaugh MP, Kanner BI. J Biol Chem. 1998;273:14231. doi: 10.1074/jbc.273.23.14231. [DOI] [PubMed] [Google Scholar]
- 153.Borre L, Kavanaugh MP, Kanner BI. J Biol Chem. 2002;277:13501. doi: 10.1074/jbc.M110861200. [DOI] [PubMed] [Google Scholar]
- 154.Koch HP, Larsson PH. J Neurosci. 2005;25:1730. doi: 10.1523/JNEUROSCI.4138-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Larsson PH, Tzingounis AV, Koch HP, Kavanaugh MP. Proc Natl Acad Sci USA. 2004;101:3951. doi: 10.1073/pnas.0306737101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Leighton BH, Seal RP, Watts SD, Skyba MO, Amara SG. J Biol Chem. 2006;281:29788. doi: 10.1074/jbc.M604991200. [DOI] [PubMed] [Google Scholar]
- 157.Boudker O, Ryan RM, Yernool D, Shimamoto K, Gouaux E. Nature. 2007;445:387. doi: 10.1038/nature05455. [DOI] [PubMed] [Google Scholar]
- 158.Mim C, Tao Z, Grewer C. Biochemistry. 2007;46:9007. doi: 10.1021/bi7005465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Slotboom DJ, Sobczak I, Konings WN, Lolkema JS. Proc Natl Acad Sci USA. 1999;96:14282. doi: 10.1073/pnas.96.25.14282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Leighton BH, Seal RP, Shimamoto K, Amara SG. J Biol Chem. 2002;277:29847. doi: 10.1074/jbc.M202508200. [DOI] [PubMed] [Google Scholar]
- 161.Tao Z, Zhang Z, Grewer C. J Biol Chem. 2006;281:10263. doi: 10.1074/jbc.M510739200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Zhang Z, Tao Z, Gameiro A, Barcelona S, Braams S, Rauen T, Grewer C. Proc Natl Acad Sci USA. 2007;104:18025. doi: 10.1073/pnas.0704570104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Haugeto O, Ullensvang K, Levy LM, Chaudhry FA, Honore T, Mielsen M, Lehre KP, Danbolt NC. J Biol Chem. 1996;271:27715. doi: 10.1074/jbc.271.44.27715. [DOI] [PubMed] [Google Scholar]
- 164.Kanai Y, Stelzner M, Nussberger S, Khawaja S, Hebert SC, Smith CP, Hediger MA. J Biol Chem. 1994;269:20599. [PubMed] [Google Scholar]
- 165.Kanner BI, Borre L. Biochim Biophys Acta. 2002;1555:92. doi: 10.1016/s0005-2728(02)00260-8. [DOI] [PubMed] [Google Scholar]
- 166.Zhang Y, Kanner B. Proc Natl Acad Sci USA. 1999;96:1710. doi: 10.1073/pnas.96.4.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Leighton BH, Seal RP, Shimamoto K, Amara SG. J Biol Chem. 2002;277:29847. doi: 10.1074/jbc.M202508200. [DOI] [PubMed] [Google Scholar]
- 168.Ryan RM, Vandenberg RJ. J Biol Chem. 2002;277:13494. doi: 10.1074/jbc.M109970200. [DOI] [PubMed] [Google Scholar]
- 169.Amara SG, Fontana ACK. Neurochem Int. 2002;41:313. doi: 10.1016/s0197-0186(02)00018-9. [DOI] [PubMed] [Google Scholar]
- 170.Watzke N, Rauen T, Bamberg E, Grewer C. J Gen Physiol. 2000;11:609. doi: 10.1085/jgp.116.5.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Grewer C, Watzke N, Wiessner M, Rauen T. Proc Natl Acad Sci USA. 2000;97:9706. doi: 10.1073/pnas.160170397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Grewer C, Watzke N, Rauen T, Bicho A. J Biol Chem. 2003;278:2585. doi: 10.1074/jbc.M207956200. [DOI] [PubMed] [Google Scholar]
- 173.Rosental N, Bendahan A, Kanner BI. J Biol Chem. 2006;281:27905. doi: 10.1074/jbc.M600331200. [DOI] [PubMed] [Google Scholar]
- 174.Abramson J, Smirnova I, Kasho V, Verner G, Kaback HR, Iwata S. Science. 2003;301:610. doi: 10.1126/science.1088196. [DOI] [PubMed] [Google Scholar]
- 175.Huang Y, Lemieux MJ, Song J, Auer M, Wang DN. Science. 2003;301:616. doi: 10.1126/science.1087619. [DOI] [PubMed] [Google Scholar]
- 176.Huang ZJ, Tajkhorshid E. Biophys J. 2008;95:2292. doi: 10.1529/biophysj.108.133421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Qu S, Kanner BI. Proc Natl Acad Sci USA. 2008;283:26391. [Google Scholar]
- 178.Bergles DE, Tzingounis AV, Jahr CE. J Neurosci. 2002;22:10153. doi: 10.1523/JNEUROSCI.22-23-10153.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Smart O, Goodfellow J, Wallace B. Biophys J. 1993;65:2455. doi: 10.1016/S0006-3495(93)81293-1. [DOI] [PMC free article] [PubMed] [Google Scholar]