

Abstract
The HLA-A*24 allele has shown negative associations with autoantibodies to islet antigen-2 (IA-2) and zinc transporter 8 (ZnT8) in patients with established type 1 diabetes. Understanding how this HLA class I allele affects humoral islet autoimmunity gives new insights into disease pathogenesis. We therefore investigated the epitope specificity of associations between HLA-A*24 and islet autoantibodies at disease onset. HLA-A*24 genotype and autoantibody responses to insulin (IAA), glutamate decarboxylase (GADA), IA-2, IA-2β, and ZnT8 were analyzed in samples collected from patients with recent-onset type 1 diabetes. After correction for age, sex, and HLA class II genotype, HLA-A*24 was shown to be a negative determinant of IA-2A and ZnT8A. These effects were epitope specific. Antibodies targeting the protein tyrosine phosphatase domains of IA-2 and IA-2β, but not the IA-2 juxtamembrane region, were less common in patients carrying HLA-A*24 alleles. The prevalence of ZnT8A specific or cross-reactive with the ZnT8 tryptophan-325 polymorphic residue, but not those specific to arginine-325, was reduced in HLA-A*24-positive patients. No associations were found between HLA-A*24 and IAA or GADA. Association of an HLA class I susceptibility allele with altered islet autoantibody phenotype at diagnosis suggests CD8 T-cell and/or natural killer cell–mediated killing modulates humoral autoimmune responses.
Autoantibodies to insulin (IAA), glutamate decarboxylase (GADA), islet antigen-2 (IA-2A), and zinc transporter 8 (ZnT8A) can appear many years before the diagnosis of type 1 diabetes and are powerful markers for predicting disease. IAA are generally the first antibodies to be detected in children at high genetic risk, followed by GADA; IA-2A and ZnT8A usually appear later (1). IA-2A responses often spread from the juxtamembrane (JM) region to the protein tyrosine phosphatase (PTP) region of IA-2 and IA-2β (2). ZnT8A epitopes are less well defined, but one major epitope includes the arginine-tryptophan polymorphism at position 325 (SLC30A8 single nucleotide polymorphism [SNP] rs1326663), which strongly influences ZnT8A responses (3).
HLA class II alleles confer the greatest genetic susceptibility for type 1 diabetes (4) but are also important determinants of humoral islet autoimmunity. Increased IAA and IA-2A prevalence at diagnosis is associated with HLA-DRB1*04 haplotypes (1,5), whereas GADA are more common in patients carrying HLA-DRB1*03 (6). Among IA-2A–positive patients, HLA-DQB1*02 haplotypes were negatively associated with JM autoantibodies (JMA) (5), and HLA-DR4-DQ8 haplotypes were positively associated with IA-2β autoantibodies (IA-2βA) (7). Associations between HLA class II alleles and ZnT8A, however, are less clear (8). HLA class I alleles also influence diabetes susceptibility and humoral autoimmunity. In patients with established diabetes, negative associations have been found between the diabetes susceptibility gene HLA-A*24 and IA-2A and between ZnT8A and the SNP rs9258750, which is in linkage with HLA-A*24, but not between HLA class I alleles and GADA (6,9,10). However, most of the patients in these studies had diabetes duration ≥5 years at the time of autoantibody testing, and these analyses may not have accounted fully for falls in antibody levels after diagnosis. Furthermore, neither study investigated the potential effect of HLA-A*24 on IA-2A epitope responses nor could they investigate potential associations of IAA with these alleles because IAA would be obscured by antibodies raised to exogenous insulin.
Our aim was therefore to investigate the influence of HLA-A*24 on islet autoantibody responses, including those to insulin and epitopes of IA-2, in a cohort of patients from whom samples were available close to diagnosis. Determining the effect of this HLA class I diabetes susceptibility allele on humoral islet autoimmunity at diabetes onset will give insights into the interaction between cytotoxic (CD8) and helper (CD4) components of the mature autoimmune response in type 1 diabetes.
RESEARCH DESIGN AND METHODS
Newly diagnosed patients.
Patients were recruited between 1985 and 2002 as part of the Bart’s-Oxford (BOX) study of childhood diabetes (11). Sera collected within 3 months of diagnosis (median 1 day [range −61 to 90]) and genetic samples for HLA-A*24 testing were available from 589 of these patients (median age, 11.0 years [0.7–20.9]). GADA, IA-2A, and ZnT8A had already been tested in all 589 sera and IA-2βA in 588 sera. IAA results were available for 405 sera collected before or within 2 weeks after diagnosis (12). JMA and PTP autoantibodies (PTPA) were tested in 460 IA-2A–positive patients and considered negative in IA-2A–negative patients. The BOX study was approved by local research ethics committees.
Autoantibody assays.
Autoantibodies to insulin, full-length GAD65, the intracytoplasmic (606-979) or JM (609-631) regions of IA-2, IA-2β (723-1015), and ZnT8 (268-369) were measured by radioimmunoassay, as previously described (12). PTPA were measured, using the same protocol, against IA-2 (687-979). ZnT8A were tested in two separate assays using labels made with plasmids encoding arginine (ZnT8R) or tryptophan (ZnT8W) at position 325, provided by Dr. Vito Lampasona (San Raffaele Scientific Institute, Milan, Italy). Results were expressed in units derived from standard curves, except those for JMA, which were expressed as an index. Assay thresholds were set at the 97.5th percentile of schoolchild sera; 2860 for IAA, GADA, and IA-2A; 523 for ZnT8A; and 270 for IA-2βA, JMA, and PTPA.
Genotyping.
HLA class I-A typing was performed on blood or mouth swab DNA with sequence-specific PCR using published primer sets (13). Patients were screened for HLA-A*24, and if HLA-A*24 was present, were fully typed to establish HLA class I-A genotype; HLA-A*24/A*24 or HLA-A*24/A*Y (where Y is any other HLA-A genotype). HLA-A*24 four-digit typing was not performed because 98% of Caucasians carry HLA-A*2402 (14). This cohort had already been typed for HLA class II DRB1, DQA1, and DQB1 using sequence-specific primers (4). Taqman SNP Genotyping Assays were used for analysis of rs13266634 (encoding the SLC30A8 ZnT8-R325 W polymorphism) and rs9258750 (in linkage with HLA-A*24 [10]). A restriction fragment–length polymorphism method was previously used to genotype 194 samples for rs13266634. Identical results were obtained in 105 patients typed using both methods. Allele distributions were in Hardy-Weinberg equilibrium.
Statistical analysis.
The χ2 test was used to compare autoantibody prevalence between patients according to HLA-A*24 or HLA class II haplotype and the Mann-Whitney U test to compare autoantibody levels. HLA class II genetic risk was analyzed as DR3-DQ2/DR4-DQ8, DR4-DQ8/DR4-DQ8, DR3-DQ2/DR3-DQ2, DR4-DQ8/X, DQ3-DQ2/X, and X/X (where X was not DR3-DQ2 or DR4-DQ8). Logistic regression models were used to adjust for factors associated with islet autoantibodies (sex, age at diagnosis, DR-DQ genotype, and duration of disease at sampling). Age at diagnosis was modeled as four age bands (age younger than 5, 5–9, 10–14, and older than 15 years) or as age older and younger than 5 years. Analyses were performed using SPSS 16 software (SPSS Inc., Chicago, IL).
RESULTS
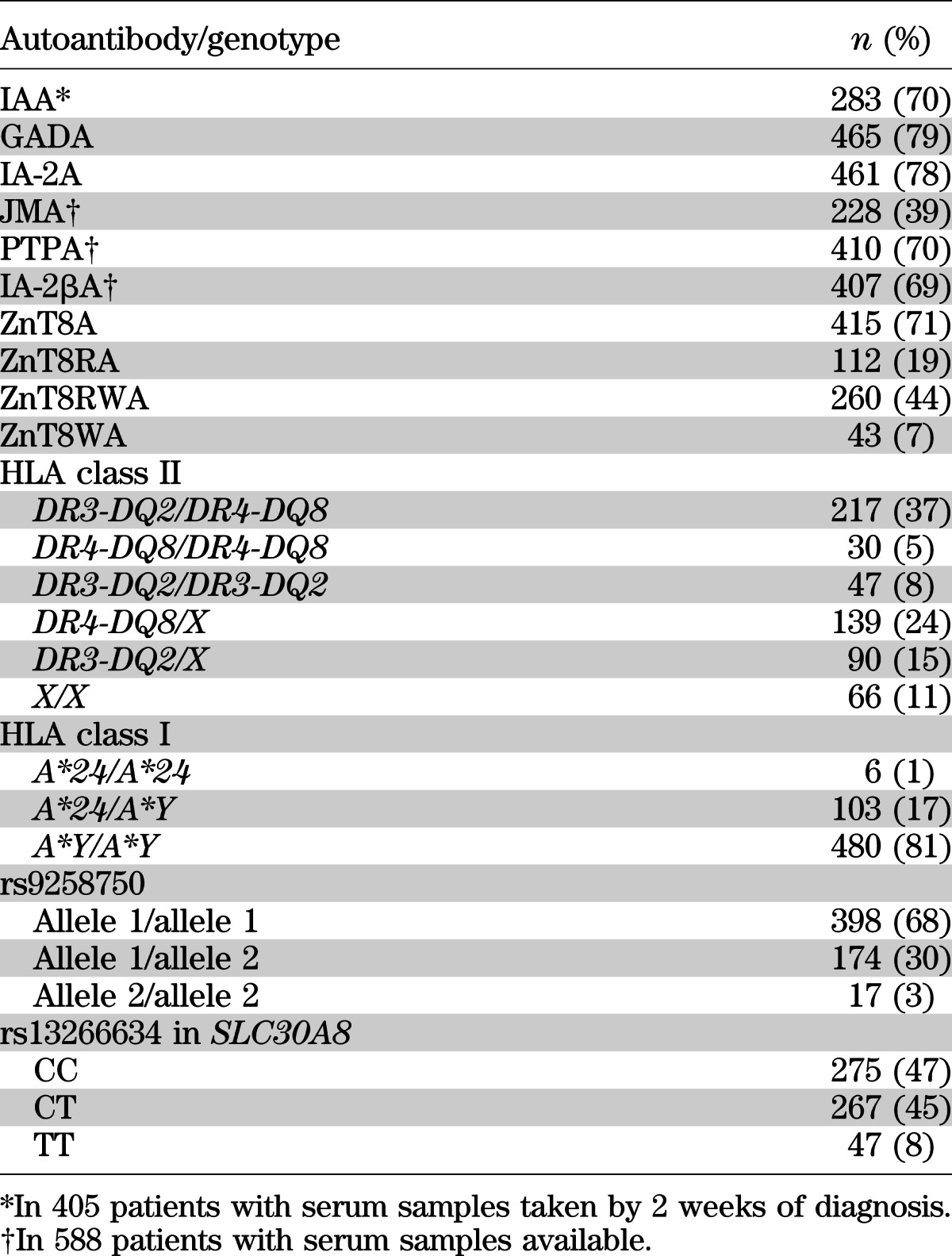
The distribution of islet autoantibodies, HLA class I A*24 alleles, and HLA class II DR3-DQ2 and DR4-DQ8 haplotypes in the patients is reported in Table 1. Of 589 patients, 109 carried at least one HLA-A*24 allele.
TABLE 1.
The distribution of autoautoantibodies, HLA class II, and HLA class I A*24 in the 589 patients
IA-2A was less common in patients carrying HLA-A*24 (71%) than in those without an HLA-A*24 allele (80%, P = 0.003 after adjustment; Table 2). IA-2A levels were also lower in IA-2A–positive patients carrying HLA-A*24 (P < 0.001, Fig. 1). rs9258750 was not associated with IA-2A (P = 0.456). PTPA and IA-2βA (P < 0.001 for both), but not JMA (P = 0.098), were negatively associated with HLA-A*24 (Table 2). IA-2A epitope responses were also strongly dependent on HLA class II genotype (Supplementary Fig. 1). IAA and GADA were not associated with HLA-A*24 (Table 2).
TABLE 2.
Association of HLA-A*24 with autoantibodies before and after correction for age, sex, HLA class II genotype, and duration of disease at sampling
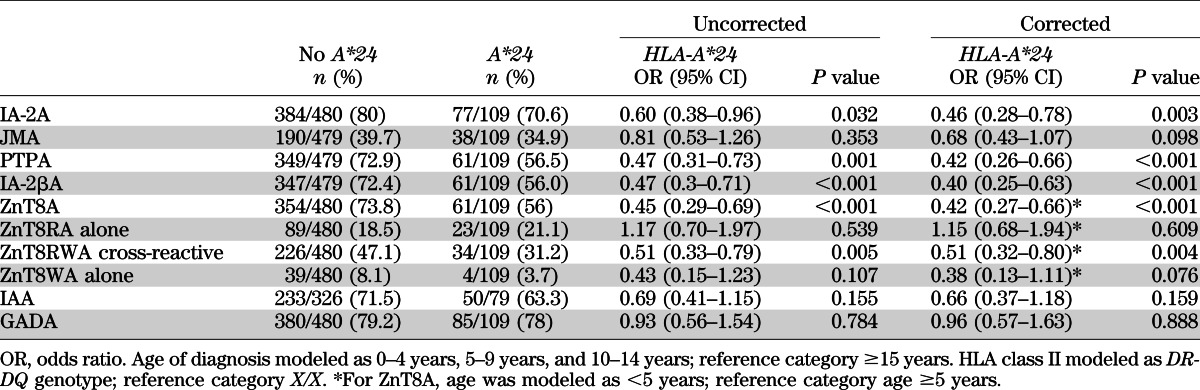
FIG. 1.

Box plots show the differences in autoantibody epitope levels among 460 IA-2A–positive patients according to HLA-A*24 genotype (n = 77 with HLA-A*24 and n = 383 without HLA-A*24). IA-2A (P < 0.001) (A), JMA (P = 0.661) (B), PTPA (P = 0.022) (C), and IA-2βA (P = 0.003) (D). The horizontal line in the middle of each box indicates the median; the top and bottom borders of the boxes represent the upper and lower quartiles, respectively, and the whiskers represent lower or upper quartile ± 1.5 times the interquartile range.
ZnT8A was less prevalent in patients carrying HLA-A*24 (56%) than in those without HLA-A*24 (74%, P < 0.001; Table 2). ZnT8As were also negatively associated with rs9258750 (P = 0.026), but this was explained by linkage to HLA-A*24 because rs9258750 did not add to models, including HLA-A*24 (P = 0.662). In contrast, the addition of HLA-A*24 significantly improved the model containing rs9258750 (P = 0.005). Autoantibodies that recognized ZnT8W or cross-reacted with both ZnT8 polymorphic variants (ZnT8RWA) were less common in patients carrying at least one HLA-A*24 allele (35 vs. 55%, P < 0.001 by χ2), but the proportion of sera with antibodies specific to ZnT8R alone was similar in HLA-A*24–positive and –negative patients (21 vs. 19%, P = 0.539; Table 2). Levels of ZnT8RA in ZnT8RA-positive patients and levels of ZnT8WA in ZnT8WA-positive patients showed no associations with HLA-A*24 (data not shown). As expected, the SLC30A8 SNP rs13266634 was associated with ZnT8A epitope specificity (Supplementary Table 1), but the distribution of rs13266634 genotypes was similar in patients with and without HLA-A*24 (Supplementary Table 2).
DISCUSSION
IA-2A and ZnT8A were less common at clinical onset of type 1 diabetes in patients carrying at least one HLA-A*24 allele, even after correction for age at onset and HLA class II genetic risk. Furthermore, the effects were epitope-specific; PTPA and IA-2βA prevalence and levels showed strong negative associations with HLA-A*24, whereas JMA were not affected significantly. Antibodies to ZnT8W and ZnT8RW, but not those to ZnT8R alone, were reduced in patients carrying HLA-A*24. IAA and GADA showed no significant association with the presence of HLA-A*24.
A strength of this study is that the patients were well characterized, with detailed genetic data available. Our regression model showed that the influence of HLA-A*24 was not explained by linkage to HLA class II DRB1*03-DQ2 or DRB1*04-DQ8 haplotypes. Furthermore, we were able to test for all major islet autoantibodies, including the major IA-2A–related epitopes and both of the main polymorphic variants of ZnT8A. In contrast to previous studies, the sera from our cohort were collected within a few months of diagnosis, which removed the confounding effect of disease duration on antibody levels (15,16). Our power to test for associations between HLA-A*24 and IAA was limited, however, because fewer patients had been sampled within the 14-day window from diagnosis allowed for IAA testing. A negative association between HLA-A*24 and IAA may be revealed in larger cohorts.
In agreement with previous studies, we found a negative association of IA-2A prevalence with HLA-A*24 (6,9). We showed that this negative association was reflected in the level of the IA-2A response and was largely explained by reduced reactivity to epitopes in the PTP regions of IA-2 and IA-2β. In contrast to Howson et al. (10), however, we found that HLA-A*24 was the primary influence on ZnT8A prevalence rather than the linked locus identified by rs9258750. This suggests that a common mechanism may explain the reduced prevalence of ZnT8A and IA-2A. Because HLA-A is a major determinant of peptide presentation to cytotoxic CD8 T cells and can also modulate natural killer cell activity (17), the HLA-A*24 variant itself may mediate the observed effects, even though HLA class II CD4 T-helper cells are more commonly associated with controlling antibody responses (18).
One potential explanation for reduced autoantibody responses could be a loss of antigenic stimulus caused by more complete destruction of β-cells by diabetes onset. Consistent with this mechanism, long-term residual β-cell function was shown to be lower in patients with long-standing type 1 diabetes carrying HLA-A*24 (19). Stimulated C-peptide close to diagnosis was lower in patients homozygous for the T allele encoding ZnT8-325W, but no correlation was found between loss of ZnT8A and residual C-peptide during the early years of clinical disease (15,16). The relationship between residual antigenic stimulus and autoantibody responses therefore remains unclear.
Another possibility is that if β-cell killing is more rapid in HLA-A*24–positive individuals, inter- and intramolecular spreading of the autoimmune response could be attenuated. IA-2A often appears after IAA and GADA, and like ZnT8A, may identify a new phase in disease progression. If autoreactive CD8 T cells are less potent at killing target cells than their antiviral counterparts (20), IA-2A and ZnT8A may play a more important role in amplifying and maintaining T-cell responses in individuals with weaker HLA class I genetic susceptibility (21). HLA-A*24 is associated with younger type 1 diabetes onset (22), but early studies suggesting that diabetes progression may be quicker in individuals carrying this allele (18,19) have not yet been confirmed (23).
We are unable to explain why the influence of HLA-A*24 on ZnT8A is dependent on the polymorphic 325 residue. Differential expression of ZnT8R and ZnT8W could affect major histocompatibility complex class I presentation of these antigens to CD8 T cells in the thymus or in the periphery (8). The C-allele (encoding ZnT8-325R) confers a minor risk of type 2 diabetes and has been associated with reduced insulin secretion (3), but no direct evidence that this polymorphism alters ZnT8 expression is available. Alternatively, the polymorphism at position 325 could alter the T-cell epitope repertoire (8), thereby modifying the influence of HLA-A*24 on humoral autoimmunity (24), but so far, no association has been found between the breadth of the ZnT8 CD4 T-cell responses in patients and ZnT8A (25).
IA-2A and ZnT8A responses were attenuated in patients with HLA-A*24 at the onset of type 1 diabetes in an epitope-specific manner. Most patients carrying HLA-A*24 were still positive for these autoantibodies, but the influence of this class I allele on antibody prevalence and levels was comparable to that observed previously with some class II alleles (5). Large prospective studies are required to show whether these reduced responses were caused by lack of spreading or loss of autoantibodies during the prodrome and whether this correlates with β-cell destruction. Determining how HLA-A*24 alters humoral islet autoimmunity will be important to our understanding of the interactions between CD4, CD8, and natural killer cells in disease pathogenesis and the potential contribution of islet autoantibodies to β-cell destruction. This may help to explain disease heterogeneity and identify pathways amenable to therapeutic modulations that halt disease progression.
Supplementary Material
ACKNOWLEDGMENTS
A.E.L. was funded by a PhD studentship provided by the Medical Research Council, U.K. The BOX study was supported by Diabetes UK and the Wellcome Trust.
No potential conflicts of interest relevant to this article were reported.
A.E.L. and A.J.K.W. researched data, contributed to discussion, and wrote the manuscript. K.M.G. researched data, contributed to discussion, and reviewed and edited the manuscript. R.J.A. and J.C.G. researched data and reviewed and edited the manuscript. P.J.B. researched data, contributed to discussion, reviewed and edited the manuscript, and coordinated the BOX study. A.J.K.W. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Parts of this study were presented in abstract form at the 12th International Congress of the Immunology of Diabetes Society, Victoria, British Columbia, Canada, 15–19 June 2012.
The authors thank James Pearson at the University of Bristol for his technical help and Dr. Vito Lampasona, San Raffaele Scientific Institute, Milan, Italy, for providing the ZnT8, IA-2, and IA-2β plasmids. The authors are also grateful to the diabetes team, pediatricians, physicians, and families in the Oxford region for participating in the BOX study.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db12-1468/-/DC1.
REFERENCES
- 1.Ziegler AG, Nepom GT. Prediction and pathogenesis in type 1 diabetes. Immunity 2010;32:468–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Naserke HE, Ziegler A-G, Lampasona V, Bonifacio E. Early development and spreading of autoantibodies to epitopes of IA-2 and their association with progression to type 1 diabetes. J Immunol 1998;161:6963–6969 [PubMed] [Google Scholar]
- 3.Wenzlau JM, Liu Y, Yu L, et al. A common nonsynonymous single nucleotide polymorphism in the SLC30A8 gene determines ZnT8 autoantibody specificity in type 1 diabetes. Diabetes 2008;57:2693–2697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lambert AP, Gillespie KM, Thomson G, et al. Absolute risk of childhood-onset type 1 diabetes defined by human leukocyte antigen class II genotype: a population-based study in the United Kingdom. J Clin Endocrinol Metab 2004;89:4037–4043 [DOI] [PubMed] [Google Scholar]
- 5.Williams AJ, Aitken RJ, Chandler MA, Gillespie KM, Lampasona V, Bingley PJ. Autoantibodies to islet antigen-2 are associated with HLA-DRB1*07 and DRB1*09 haplotypes as well as DRB1*04 at onset of type 1 diabetes: the possible role of HLA-DQA in autoimmunity to IA-2. Diabetologia 2008;51:1444–1448 [DOI] [PubMed] [Google Scholar]
- 6.Howson JM, Stevens H, Smyth DJ, et al. Evidence that HLA class I and II associations with type 1 diabetes, autoantibodies to GAD and autoantibodies to IA-2, are distinct. Diabetes 2011;60:2635–2644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vermeulen I, Weets I, Asanghanwa M, et al. Belgian Diabetes Registry Contribution of antibodies against IA-2β and zinc transporter 8 to classification of diabetes diagnosed under 40 years of age. Diabetes Care 2011;34:1760–1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delli AJ, Vaziri-Sani F, Lindblad B, et al. Better Diabetes Diagnosis Study Group Zinc transporter 8 autoantibodies and their association with SLC30A8 and HLA-DQ genes differ between immigrant and Swedish patients with newly diagnosed type 1 diabetes in the Better Diabetes Diagnosis study. Diabetes 2012;61:2556–2564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qu HQ, Polychronakos C. The effect of the MHC locus on autoantibodies in type 1 diabetes. J Med Genet 2009;46:469–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Howson JM, Krause S, Stevens H, et al. Genetic association of zinc transporter 8 (ZnT8) autoantibodies in type 1 diabetes cases. Diabetologia 2012;55:1978–1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bingley PJ, Gale EA. Incidence of insulin dependent diabetes in England: a study in the Oxford region, 1985-6. BMJ 1989;298:558–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Long AE, Gillespie KM, Rokni S, Bingley PJ, Williams AJ. Rising incidence of type 1 diabetes is associated with altered immunophenotype at diagnosis. Diabetes 2012;61:683–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bunce M, O’Neill CM, Barnardo MC, et al. Phototyping: comprehensive DNA typing for HLA-A, B, C, DRB1, DRB3, DRB4, DRB5 & DQB1 by PCR with 144 primer mixes utilizing sequence-specific primers (PCR-SSP). Tissue Antigens 1995;46:355–367 [DOI] [PubMed] [Google Scholar]
- 14.Noble JA, Valdes AM, Varney MD, et al. Type 1 Diabetes Genetics Consortium HLA class I and genetic susceptibility to type 1 diabetes: results from the Type 1 Diabetes Genetics Consortium. Diabetes 2010;59:2972–2979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wenzlau JM, Walter M, Gardner TJ, et al. Kinetics of the post-onset decline in zinc transporter 8 autoantibodies in type 1 diabetic human subjects. J Clin Endocrinol Metab 2010;95:4712–4719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nielsen LB, Vaziri-Sani F, Pörksen S, et al. Hvidoere Study Group on Childhood Diabetes Relationship between ZnT8Ab, the SLC30A8 gene and disease progression in children with newly diagnosed type 1 diabetes. Autoimmunity 2011;44:616–623 [DOI] [PubMed] [Google Scholar]
- 17.Stern M, Ruggeri L, Capanni M, Mancusi A, Velardi A. Human leukocyte antigens A23, A24, and A32 but not A25 are ligands for KIR3DL1. Blood 2008;112:708–710 [DOI] [PubMed] [Google Scholar]
- 18.Tait BD, Colman PG, Morahan G, et al. HLA genes associated with autoimmunity and progression to disease in type 1 diabetes. Tissue Antigens 2003;61:146–153 [DOI] [PubMed] [Google Scholar]
- 19.Nakanishi K, Kobayashi T, Murase T, Naruse T, Nose Y, Inoko H. Human leukocyte antigen-A24 and -DQA1*0301 in Japanese insulin-dependent diabetes mellitus: independent contributions to susceptibility to the disease and additive contributions to acceleration of beta-cell destruction. J Clin Endocrinol Metab 1999;84:3721–3725 [DOI] [PubMed] [Google Scholar]
- 20.Knight RR, Kronenberg D, Zhao M, et al. Human β-cell killing by autoreactive preproinsulin-specific CD8 T cells is predominantly granule-mediated with the potency dependent upon T-cell receptor avidity. Diabetes 2013;62:205–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allen JS, Pang K, Skowera A, et al. Plasmacytoid dendritic cells are proportionally expanded at diagnosis of type 1 diabetes and enhance islet autoantigen presentation to T-cells through immune complex capture. Diabetes 2009;58:138–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Valdes AM, Erlich HA, Carlson J, Varney M, Moonsamy PV, Noble JA. Use of class I and class II HLA loci for predicting age at onset of type 1 diabetes in multiple populations. Diabetologia 2012;55:2394–2401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lipponen K, Gombos Z, Kiviniemi M, et al. Effect of HLA class I and class II alleles on progression from autoantibody positivity to overt type 1 diabetes in children with risk-associated class II genotypes. Diabetes 2010;59:3253–3256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kronenberg D, Knight RR, Estorninho M, et al. Circulating preproinsulin signal peptide-specific CD8 T cells restricted by the susceptibility molecule HLA-A24 are expanded at onset of type 1 diabetes and kill β-cells. Diabetes 2012;61:1752–1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dang M, Rockell J, Wagner R, et al. Human type 1 diabetes is associated with T cell autoimmunity to zinc transporter 8. J Immunol 2011;186:6056–6063 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.