

Diabetes represents a ramping public health issue in the Western world owing to overnutrition and reduced physical activity coupled with genetic susceptibility (1,2). Nowadays, obesity is a major determinant of insulin resistance, which results in compensatory hyperinsulinemia with subsequent impairment of insulin secretion and rise of blood glucose levels. Over the last 30 years, the detrimental effects of hyperglycemia on the vessel wall have promoted major research efforts leading to the understanding that high glucose decreases availability of endothelium-derived nitric oxide (NO) and triggers vascular inflammation via an array of mechanisms involving the overproduction of reactive oxygen species (ROS) (3,4). Although the identification of different signaling pathways has opened several windows of opportunity to prevent diabetic vascular disease, we are still far from having found a thorough and, most of all, effective approach against cardiovascular burden of diabetes. Indeed, recent clinical trials have shown that normalization of blood glucose failed to reduce cardiovascular outcomes in the diabetic population (5–7). It is noteworthy that intensive glucose-lowering therapy in these trials was started after a median duration of diabetes ranging from 8 to 11 years (8). By contrast, glucose-lowering treatment of patients with new-onset diabetes was shown to be beneficial (9–12). These findings hint that early preservation of physiological metabolic environment is crucial for interfering with the natural history of diabetic vasculopathy. In this Perspective, the landmark Diabetes Control and Complications Trial (DCCT) and the follow-up study, Epidemiology of Diabetes Interventions and Complications (EDIC), demonstrated not only that intensive glycemic control in subjects with type 1 diabetes reduced the risk of microvascular complications but also that episodes of poor glycemic control can lead many years later to the long-term complications of diabetes (9,12). More recently, the 10-year posttrial monitoring follow-up of the UK Prospective Diabetes Study (UKPDS) study showed that early treatment of hyperglycemia significantly reduced the risk of myocardial infarction, diabetes-related deaths, and all-cause mortality (10). Collectively, these clinical observations imply that hyperglycemic environment may be remembered in vascular tissues.
Hyperglycemic memory
The persistence of hyperglycemic stress despite glucose normalization has been defined as “hyperglycemic memory” (13,14). This emerging concept strengthens the importance of early glycemic control and may explain why diabetic cardiovascular complications progress even in the presence of optimal glycemic control. The initial skepticism toward the concept of hyperglycemic memory, considered vague and not supported by solid evidence, has gradually given way to a growing interest in unmasking the underlying mechanisms. This phenomenon was initially described in mice with streptozotocin-induced diabetes, where normoglycemia restoration did not revert the expression of fibronectin mRNA in the kidney cortex, which was elevated for several weeks even after the maintenance of near-normal glucose levels by exogenous insulin administration (15). The putative mechanisms were investigated in human endothelial cells exposed to hyperglycemia followed by normal glucose restoration. This experiment revealed that cells previously exposed to high glucose continued to display elevated expression of fibronectin and collagen IV despite subsequent normalization of glucose concentration in the media (15). Other investigations demonstrated the irreversibility of microvascular damage also in the diabetic retina (16). More recently, it was postulated that ROS may be critically involved in the persistence of hyperglycemic stress in endothelial cells and experimental diabetes (17–19). This concept is in line with the notion that ROS generation plays a leading role in the development of diabetes complications (4). Mitochondrial accumulation of ROS as a result of hyperglycemia activates major pathways involved in the pathogenesis of cardiovascular complications including polyol pathway flux, increased production of advanced glycation end products (AGEs), protein kinase C (PKC) activation, and the hexosamine pathway (4). Although ROS are upstream molecules regulating a number of detrimental pathways in the vessels, we are still far from understanding the mechanisms responsible for persistent changes of gene expression despite restoration of normoglycemia in the setting of diabetes. Ihnat et al. (18) found that oxidative stress markers and upregulation of pro-oxidant enzymes, namely PKCβ and NAD(P)H oxidase, persist after restoring normoglycemia in human endothelial cells previously exposed to high glucose concentrations. Accordingly, only diabetic patients treated with a combination of insulin and vitamin C at high doses showed an improvement of the brachial artery flow-mediated vasodilation (20). Therefore, identification of ROS-generating machinery is the real challenge to reverse hyperglycemic memory and, eventually, prevent the development of cardiovascular complications.
Complex scenario involving epigenetic changes
A better understanding of the pathways leading to hyperglycemic memory may contribute to designing mechanism-driven strategies to blunt vascular damage and to improving cardiovascular outcome in subjects with diabetes. Although several studies have suggested that ROS plays a crucial role, only in most recent years have the molecular mechanisms of hyperglycemic memory been explored. These findings unravel a complex scenario of epigenetic changes that modulate transcription of ROS-generating and proinflammatory genes. Overproduction of ROS, in turn, maintains detrimental vicious cycles responsible for persistent vascular stress. Here, we provide a unifying context for the different pathways involved. While available knowledge supports single molecules as key mediators of this process, we proposed one intricate molecular pathway linking together the molecules implicated in chromatin remodeling, ROS generation, and vascular inflammation. Targeting individual components of this deleterious cascade may be the most promising option to dampen oxidative stress, reverse hyperglycemic memory, and, hence, prevent cardiovascular complications.
Set7/9 methyltransferase drives persistent vascular inflammation
In the last few years, several studies have identified critical molecular determinants of vascular hyperglycemic memory. Epigenetic mechanisms have been postulated and considered in metabolic memory only recently. Persistent epigenetic changes caused by hyperglycemia-induced mitochondrial ROS production have been advocated as the driving force underlying the progression of diabetic vasculopathy (21). Specifically, DNA methylation and histone modification represent a major breakthrough in understanding changes in gene expression occurring in disease states (21,22). Hypomethylation of CpG dinucleotides at the promoter region is generally related to enhanced gene activation. Promoter methylation interferes with transcription factors and facilitates the binding of methyl-CpG–binding domain proteins, which in turn recruit histone deacetylases, thereby repressing gene transcription (23). Methylation may also occur at lysine residue in the histone tail (24). This is a remarkably complex process, involving mono-, di-, or thrimethylation, which often clusters within specific regions resulting in the organization of chromosomes into distinct structural and functional domains (23). This process may lead to an open chromatin and gene activation rather than repression, as reported for CpG promoter methylation.
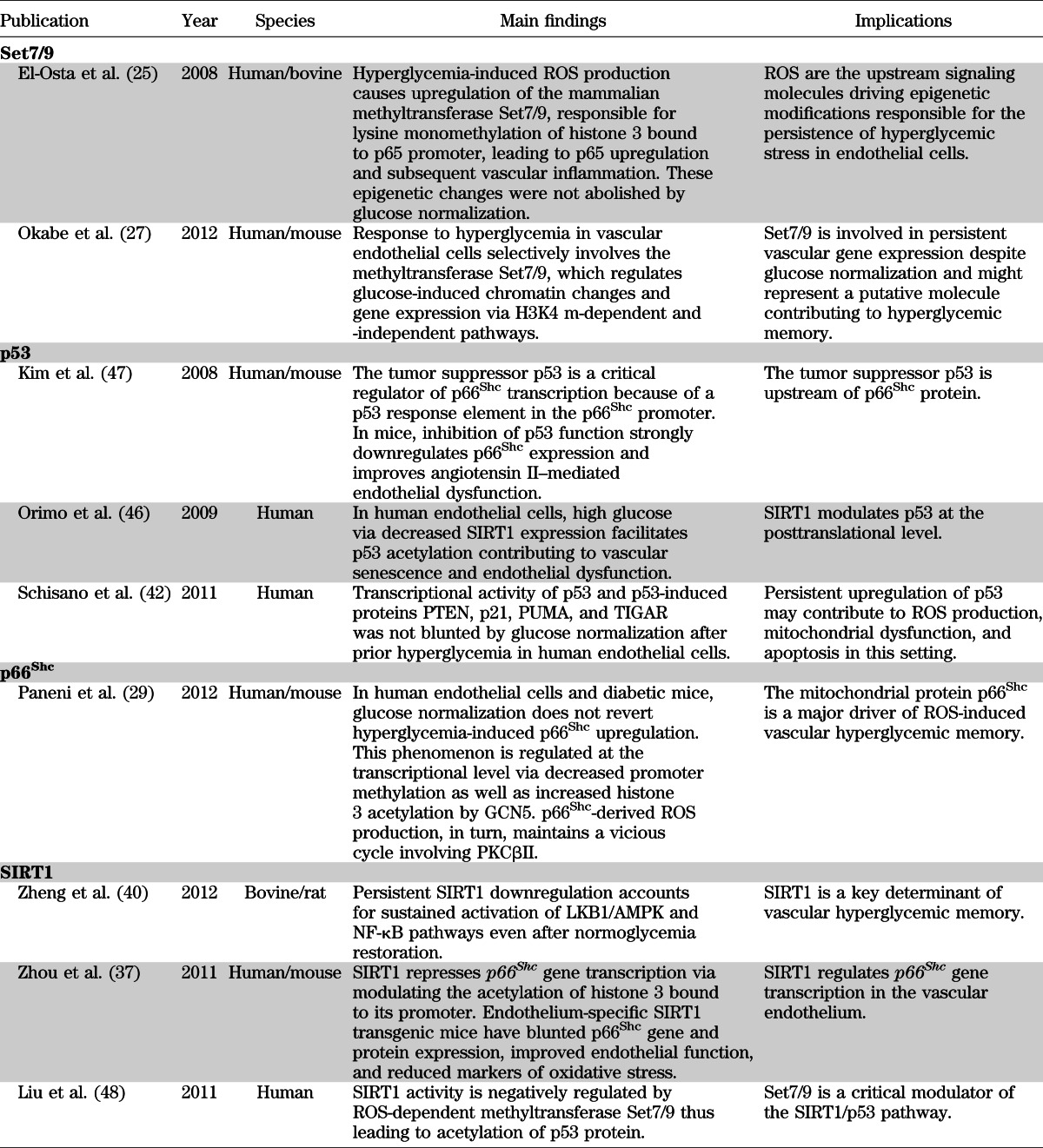
In 2008, El-Osta et al. (25) found that transient hyperglycemia in aortic endothelial cells was able to induce long-lasting activating epigenetic changes in the promoter of the nuclear factor κB (NF-κB) subunit p65. This epigenetic deregulation explains persistent p65 gene transcription and subsequent overexpression of the inflammatory genes monocyte chemoattractant protein-1 (MCP-1) and vascular cell adhesion molecule-1 (VCAM-1) after 6 days of glucose normalization (25). Interestingly, normalization of mitochondrial superoxide production dislodged the epigenetic markers at p65 promoter, clearly indicating that ROS still remain the upstream key molecules involved in the pathophysiology of diabetic vascular disease despite glucose control. Indeed, hyperglycemia-dependent ROS production was responsible for monomethylation of histone 3 at lysine 4 amino residue (H3K4 m) by the mammalian methyltransferase Set7/9. Methylation of H3K4 is a critical posttranslational modification favoring gene transcription in mammals (26) and is associated with persistent vascular inflammation when such methylation occurs on histone 3 binding proximal promoter region of NF-κB subunit p65 (25,27) (Fig. 1). Interestingly, knockdown of Set7/9 prevented H3K4 m and, hence, glucose-induced upregulation of p65 as well as of MCP-1 and VCAM-1 genes (25). For the first time, this study demonstrated that overproduction of ROS leads to activation of key enzymes involved in chromatin remodeling and persistent transcription of inflammatory genes.
FIG. 1.

Signaling network of vascular hyperglycemic memory. Hyperglycemia via SIRT1 downregulation leads to acetylation of p53, NF-κB subunit p65, and histone 3 bound to p66Shc promoter. Activation of p53 leads to increased p66Shc transcription. In addition, glucose-induced GCN5 downregulation causes H3 acetylation and subsequent p66Shc transcription through chromatin remodeling. p53 protein as well as epigenetic-driven upregulation of p66Shc leads to persistent mitochondrial ROS production, which maintains hyperglycemia-induced PKCβII overexpression and PKCβII-dependent eNOS inhibitory phosphorylation at Thr495 residue even after glucose normalization. p66Shc also downregulates MnSOD, further increasing ROS accumulation. These changes underlie endothelial dysfunction and apoptosis via reduced NO availability and activation of caspase 3 and PARP cleavage, respectively. SIRT1-p53-p66Shc networking via ROS production increases activity of the methyltransferase Set7/9, responsible for promoter monomethylation (H3K4 m) of NF-κB subunit p65 leading to its persistent transcription and subsequent upregulation of MCP-1 and VCAM-1 inflammatory genes. Ac, acetylation; p, phoshorylation; RAGE, receptor for AGEs.
Mitochondrial adaptor p66Shc drives ROS-induced vascular memory
With such a background, we recently investigated whether mitochondrial adaptor p66Shc, a critical modulator of intracellular redox state, is the source of ROS-inducing hyperglycemic memory. The p66Shc adaptor protein functions as a redox enzyme implicated in mitochondrial ROS generation and translation of oxidative signals into apoptosis. Mice lacking p66Shc gene (p66Shc−/−) display prolonged lifespan and increased resistance to oxidative stress and are protected against hyperglycemia-induced endothelial dysfunction (28). High glucose levels lead to increased p66Shc gene transcription and protein phosphorylation, favoring mitochondrial translocation, cytochrome c oxidation, and subsequent ROS generation (29). The clinical relevance of p66Shc in the setting of dysglycemia is supported by the notion that p66Shc gene expression is increased in mononuclear cells obtained from patients with type 2 diabetes and correlates with plasma 8-isoprostane levels, an in vivo marker of oxidative stress (30). In human endothelial cells exposed to high glucose and aortas of diabetic mice, we recently found that activation of p66Shc by PKCβII was sustained even after returning to normoglycemia (29). Persistent p66Shc upregulation and its mitochondrial translocation were associated with ROS production, reduced NO bioavailability, and apoptosis. Interestingly, p66Shc gene overexpression was epigenetically regulated by promoter CpG demethylation and acetylation of histone 3 operated by acetyl-transferase general control nonderepressible 5 (GCN5). These experiments also showed that p66Shc-derived ROS production maintained PKCβII upregulation and PKCβII-dependent inhibitory phosphorylation of endothelial NO synthase at Thr495, contributing to a detrimental vicious cycle despite restoration of normoglycemia (Fig. 1). Importantly, in vivo gene silencing of p66Shc, performed at the time of glucose normalization, blunted ROS production, restored endothelium-dependent vasorelaxation, and attenuated apoptosis by limiting cytochrome c release, caspase 3 activity, and cleavage of poly(ADP-ribose) polymerase (PARP). These findings strongly suggest that the adaptor p66Shc is a critical source of mitochondrial ROS, and its downregulation clearly reverses the pathological features of hyperglycemic memory in vascular tissues. Indeed, p66Shc knockdown after normoglycemia restoration was associated with downregulation of PKCβII and eNOS dephosphorylation at Thr495, as well as reduced synthesis of the AGEs precursor methylglyoxal (29) (Fig. 1). This latter finding suggests that long-lasting p66Shc activation may account for increased AGEs turnover, which has been considered a key factor underlying the glycemic memory (14). Methylglyoxal readily reacts with arginine, lysine, and sulfhydryl groups of proteins and nucleic acids, inducing the formation of a variety of structurally identified AGEs, which exert an inhibitory effect on mitochondrial respiration (31). AGEs represent an important pathway in the alteration of vascular structure and function. Specifically, the AGEs pentosidine and carboxymethyl-lysine strongly correlate with indices of arterial stiffness in patients with type 1 diabetes (32,33). The clinical relevance of AGEs in the context of hyperglycemic memory is supported by the notion that an early glycemic control significantly reduced AGE levels in the DCCT trial, and this finding was paralleled by a consistent reduction of cardiovascular events (34). In this Perspective, recent evidence suggests that soluble forms of AGE receptors (sRAGEs) bind ligands including AGEs and can antagonize RAGE signaling in vitro and in vivo, thus prospecting an attractive therapeutic option to reverse hyperglycemic memory (35).
Collectively, these results are in line with seminal studies supporting the pivotal role of ROS in the pathogenesis of diabetes complications and suggest the perspective that, even when glucose levels have been normalized, elevated concentrations of mitochondrial superoxide maintain the activation of intracellular pathways involved in endothelial dysfunction, vascular inflammation, and apoptosis. The role of epigenetic modifications in this setting is confirmed by our observation that glucose-induced p66Shc overexpression is driven by decreased promoter methylation and increased histone 3 acetylation (Fig. 1). Indeed, at the time of glucose normalization siRNA-induced downregulation or pharmacological inhibition of acetyltransferase GCN5 reported p66Shc expression to control levels (29). Our study identified a specific molecule responsible for ROS generation and may assist in defining novel therapeutic targets to reduce the long-lasting deleterious effects of hyperglycemia on the vasculature. This aspect deserves attention, since available antioxidants only partially scavenge cellular ROS but do not target intracellular redox signaling. Such an assumption is confirmed by the negative results of major trials with oral supplementation of high-dose vitamins (36).
SIRT1 regulates p66Shc transcription
A recent study reported that vascular p66Shc gene transcription may be the result of decreased promoter deacetylation due to the downregulation of class III histone deacetylase SIRT1 (37). Expression of p66Shc gene transcript and protein was significantly increased by different kinds of class III histone deacetylase inhibitors in human endothelial cells. Consistently, SIRT1 overexpression inhibited high glucose–induced p66Shc upregulation, whereas SIRT1 knockdown exerted opposite effects. Moreover, endothelium-specific SIRT1 transgenic mice had blunted p66Shc gene and protein expression and improved endothelial function, as well as reduced accumulation of oxidative stress markers, compared with wild-type littermates (37). This study demonstrated that SIRT1 binds to the p66Shc promoter (−508 to −250 bp) where it deacetylates histone 3, thus suppressing gene transcription of the mitochondrial adaptor (37). Hence, increased acetylation by GCN5 and decreased SIRT1-dependent deacetylation are the main epigenetic changes responsible for persistent p66Shc overexpression in the vasculature despite glucose normalization (29,37) (Fig. 1). In addition, p66Shc upregulation in human endothelial cells is associated with a significant reduction of the antioxidant enzyme manganese superoxide dismutase (MnSOD) (38,39). This latter finding suggests that p66Shc is not only a critical source of mitochondrial ROS but is also involved in the downregulation of the scavenging enzyme, leading to unopposed ROS accumulation in the vascular endothelium (28) (Fig. 1).
In parallel with the notion that SIRT1 controls p66Shc expression, a recent work reported that glucose-induced SIRT1 downregulation has a strong memory effect after restoration of normoglycemia in bovine endothelial cells (40). In this study, in vitro and in vivo SIRT1 overexpression associated with optimal glucose control was able to interrupt the memory of hyperglycemic stress via ROS normalization, thus suppressing NF-κB activation and cleavage of PARP (40). Overexpression of SIRT1 also restored hyperglycemia-induced dephosphorylation of LKB1 and AMP kinase (AMPK), two critical regulators of energy balance in mammalian cells (41). Interestingly, SIRT1 activation during subsequent normoglycemia restored normal expression levels of MnSOD (40).
These studies provide the molecular background to conclude that even after optimal glycemic control, persistent SIRT1 downregulation may account for p66Shc overexpression leading to mitochondrial superoxide accumulation, reduced expression of scavenging enzymes, activation of NF-κB, and subsequent vascular dysfunction (Fig. 1 and Table 1). SIRT1 and GCN5 are the upstream regulators of p66Shc gene transcription by modulating the binding of histone 3 acetylation to p66Shc promoter (Fig. 1). The pharmacological modulation of enzymes involved in the epigenetic regulation of p66Shc is becoming an attractive therapeutic goal. In this regard, metformin through SIRT1 activation abolished oxidative stress and inflammation, reverting the pathological abnormalities in the retina of diabetic rats (40).
TABLE 1.
Experimental evidence linking detrimental pathways of vascular hyperglycemic memory
Tumor suppressor p53 is a critical intermediate between SIRT1 and p66Shc
Another study found that glycemic control does not affect the upregulation of the tumor suppressor transcription factor p53 (42). Indeed, 7 days of glucose normalization after 14 days of prior hyperglycemia were not able to interrupt the activation of p53 as well as p53-induced proteins PTEN, p21, PUMA, and TIGAR. Persistent p53 overexpression was associated with ongoing ROS production and DNA damage, suggesting the important role of this transcription factor in the maintenance of oxidative stress (43,44). Molecular studies indicate that p53 might perform its senescence and proapoptotic functions by directly signaling the mitochondria and inducing cytochrome c release (44). Additional evidence supports the hypothesis that p53 may be part of a signaling pathway linking SIRT1 and p66Shc. It was recently reported that SIRT1 inhibition increases p53 acetylation and transcriptional activity (45,46). Moreover, p53 was found to transcriptionally regulate p66Shc (47). Accordingly, downregulation of p66Shc expression as well as inhibition of p53 function in mice restored impairment of acetylcholine-induced vascular relaxation and increased NO bioavailability (47). Taken together, these observations strongly suggest that p53 is a critical intermediate between the upstream regulator SIRT1 and its downstream target p66Shc (Fig. 1 and Table 1).
In conclusion, SIRT1-p53- p66Shc pathway might be responsible for self-maintenance of vascular damage after restoration of normal glucose levels. Indeed, recent evidence clearly demonstrated that these mediators remain upregulated despite glucose control (29,40,42). The activation of such a pathway, in turn, leads to increased mitochondrial superoxide production triggering Set7/9-related epigenetic changes on the promoter of NF-κB component p65 and subsequent vascular inflammation (Fig. 1 and Table 1). Furthermore, Set7/9 has been recently found to negatively regulate SIRT1 activity thus maintaining acetylation of p53 protein and activation of its downstream targets (48).
Clinical perspectives
Since cardiovascular risk burden is not eradicated by intensive glycemic control, new mechanism-based therapeutic strategies are needed. The recent identification of epigenetically regulated genes in the setting of hyperglycemia may allow targeted approaches to reprogram these modifications (49). Plastic alterations of the chromatin are indeed responsible for the regulation of DNA-templated phenomena and may be amenable to pharmacological intervention. There are many examples suggesting the possibility of interference with gene expression by modulating acetylation and methylation of histone/DNA complexes (50,51). Folate treatment has shown to repress gene activation via increasing DNA methylation, and a correlation between homocysteine levels and such epigenetic modification was observed in healthy humans (51) (Fig. 2). Consistently, a recent work found that homocysteine stimulates p66Shc transcription in human endothelial cells via specific CpG dinucleotides demethylation in the p66Shc promoter (52). Interestingly, p66Shc promoter CpG methylation was significantly reduced in peripheral blood leukocytes of patients with coronary artery disease and high plasma homocysteine levels, thus strengthening the relevance of p66Shc-related epigenetic changes in the context of cardiovascular disease. On the other hand, SIRT1 activation by resveratrol improves vascular function via increasing NO availability and attenuates dyslipidemia and obesity-induced metabolic alterations in human subjects (53–55). SIRT1-dependent improvement of flow-mediated dilation can be partially explained by increased deacetylation of p66Shc promoter as well as posttranslational and transcriptional regulation of endothelial NO synthase (eNOS) (37,56) (Fig. 2). By contrast, a recent study of 24 healthy obese men showed that resveratrol supplementation had no significant effects on insulin sensitivity, blood pressure, visceral fat content, systemic inflammation, or lipid oxidation (57). However, resveratrol has been tested only in a limited number of small human clinical trials of efficacy outcomes, and we are still far from understanding whether the persuasive results of experimental research may translate to the human setting.
FIG. 2.

Mechanism-based pharmacological approaches to revert vascular hyperglycemic memory in subjects with diabetes. ET-1, endothelin-1; H2, histone 2; VEGF, vascular endothelial growth factor.
Therefore, the activation of SIRT1 may be only a part of a much more complex scenario in which deacetylation of histones may play a role. In this Perspective, metformin, a widely used antidiabetes drug, restores SIRT1 expression/activity in the retina of diabetic rats (40). Indeed, it was demonstrated that 4-week metformin treatment, after 2 weeks of diabetes, significantly increased SIRT1 expression, preserved activation of its downstream targets LKB1 and AMPK, and blunted persistent ROS formation. Interestingly, metformin also suppressed NF-κB activation (40). Altogether, these findings are in agreement with the notion that SIRT1 is an upstream mediator regulating p66Shc expression, ROS formation, and epigenetic-driven vascular inflammation (Fig. 1). Based on these experimental findings, one can postulate that metformin may contribute to reverse vascular hyperglycemic damage in patients with diabetes (Fig. 2). However, the enthusiasm of this pharmacological approach has to deal with the controversial results of the UKPDS (10) and Action to Control Cardiovascular Risk in Diabetes (ACCORD) (6) trials, where metformin was associated with either reduced or increased cardiovascular events, respectively. However, it is important to underline that despite the fact that 94.7% of patients in the intensive arm of the ACCORD trial were on metformin therapy, the drug was always given in association with other classes of glucose-lowering agents, namely, secretagogues (86.6%), thiazolidinediones (91.7%), and insulin (77.3%). Hence, it is not possible to derive any metformin-related increase in cardiovascular risk. Moreover, metformin by itself does not increase the risk of hypoglycemia, and the recent position statement of the American Diabetes Association and the European Association for the Study of Diabetes indicates metformin as the first-choice treatment for the management of hyperglycemia in type 2 diabetes (58).
Inhibitors of histone acetyltransferases have also shown to revert abnormalities linked to overexpression of pro-oxidant and inflammatory genes. The dietary compound curcumin, an inhibitor of the histone acetyltransferase CBP/p300, has recently been reported to prevent hyperglycemia-induced endothelial dysfunction and cardiac hypertrophy, indicating that the pharmacological removal of histone acetylation might be a promising therapeutic tool (59,60). Specifically, removal of CBP/p300-mediated histone 2XA acetylation blunts hyperglycemia-induced transcription of endothelin-1, vascular endothelial growth factor, and fibronectin, thus restoring endothelial homeostasis (59) (Fig. 2). Similarly, inhibition of the acetyltransferase GCN5 prevents angiotensin II–mediated downregulation of catalase (61), upregulation of NADPH subunit Nox2 (62), and hyperglycemia-induced p66Shc overexpression (29). Moreover, CBP/p300 and GCN5 are important cell cycle regulators playing a critical role in cancer development. Phase II and III studies are currently investigating the efficacy of curcumin in this setting (63). Another interesting approach linking epigenetic modifications to diabetic vascular dysfunction is represented by the agonists of peroxisome proliferator–activated receptors (PPARs). PPARs are crucial in metabolism and adipogenesis. PPARγ ligands such as thiazolidinediones exert insulin-sensitizing and anti-inflammatory effects, primarily through action on adipocytes. Recent studies have identified a number of PPARγ-interacting partners, many of which are known epigenetic regulators, including enzymes for histone acetylation/deacetylation and histone methylation/demethylation (Fig. 2). However, their functional roles in the PPARγ transcriptional pathway are not well defined. Thanks to the advances in ChIP-based and deep sequencing technology, epigenomic mechanisms and therapeutic potentials of this nuclear receptor pathway are emerging. Epigenetic reprogramming of oxidant genes may contribute to explaining the beneficial effect of PPARγ agonists on vascular function and cardiovascular outcomes in subjects with type 2 diabetes (64). Although a clear benefit of PPARγ agonists can be postulated, cardiovascular safety of the different compounds is controversial (58). Pioglitazone may modestly reduce cardiovascular events but may also increase the risk of bladder cancer. Rosiglitazone has been shown to increase the risk of myocardial infarction and has been withdrawn in Europe and restricted in the U.S. (64). Fibrates improve cardiovascular outcomes only in diabetic patients with metabolic syndrome and dyslipidemia (58,64). Finally, the cardiovascular safety of the new pan agonist aleglitazar, currently in phase II trials, remains to be determined. The critical question of why PPARγ agonists seem to improve cardiovascular risk factors without significantly improving cardiovascular outcomes requires further investigation.
An important question to answer before the implementation of mechanism-based therapeutic strategies is whether similar pathways of hyperglycemic memory are activated in micro- and macrovasculature. Clinical observations suggest that an early intensive glycemic control may reduce first microvascular and then macrovascular complications with a timely order, suggesting microvascular disease as a prerequisite for macrovascular complications in patients with diabetes (65). Moreover, in the Atherosclerosis Risk in Communities (ARIC) study microcirculatory changes significantly correlated with macrovascular atherosclerosis and the occurrence of cardiovascular events, particularly in women (66). These findings are supported by the notion that histopathological changes of the retina and coronary arteries are similar in patients with arterial hypertension (65). Similarly, microangiopathic processes within the vessel wall of conduit arteries resemble that of retinal changes. These observations imply a close interaction between micro- and macrovasculature and suggest the possibility that different vascular beds may share similar molecular pathways (67). In this Perspective, metformin has shown similar benefits on micro- and macrovascular complications in the UKPDS trial (10). However, further research is needed to address these important aspects.
Conclusions
Hyperglycemic memory may explain why vascular diabetes complications progress despite intensive glycemic control, and the understanding of its mechanisms represents a real challenge to reverse persistent vascular damage and, eventually, improve cardiovascular outcome in diabetes. Additional investigations are needed to unmask the epigenetic regulation of specific genes altered by hyperglycemia. Moreover, the role of hyperglycemia in type 2 diabetes as a determinant of persistent vascular dysfunction remains to be clarified. Indeed, other important factors, namely, insulin resistance, participate in eNOS deregulation and endothelial dysfunction in this setting. Accordingly, the expression of SIRT1 and p66Shc is significantly affected in peripheral blood mononuclear cells of insulin-resistant individuals with type 2 diabetes (30,68). Hence, the removal of epigenetic tags resulting from SIRT1, GCN5, and Set7/9 deregulation may not be that unrealistic and may be a promising option to dampen oxidative stress and vascular inflammation and thus prevent cardiovascular complications in people with diabetes.
ACKNOWLEDGMENTS
This study was supported by grants from the Swiss Heart Foundation, Fondazione Roma, Italy (to F.C.), and the Swiss National Research Foundation (to T.F.L.) (3100-06811802/1). F.P. was the recipient of a PhD Program in Experimental Medicine from the University of Rome “La Sapienza.”
No potential conflicts of interest relevant to this article were reported.
F.P. developed the concept, wrote the manuscript, and conceived the figures. M.V. and T.F.L. provided critical feedback on earlier versions of the manuscript. F.C. developed the concept, wrote the manuscript, and conceived the figures. F.C. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
REFERENCES
- 1.American Diabetes Association Diagnosis and classification of diabetes mellitus. Diabetes Care 2012;35(Suppl. 1):S64–S71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hossain P, Kawar B, El Nahas M. Obesity and diabetes in the developing world—a growing challenge. N Engl J Med 2007;356:213–215 [DOI] [PubMed] [Google Scholar]
- 3.Creager MA, Lüscher TF, Cosentino F, Beckman JA. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: Part I. Circulation 2003;108:1527–1532 [DOI] [PubMed] [Google Scholar]
- 4.Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res 2010;107:1058–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patel A, MacMahon S, Chalmers J, et al. ADVANCE Collaborative Group Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 2008;358:2560–2572 [DOI] [PubMed] [Google Scholar]
- 6.Gerstein HC, Miller ME, Byington RP, et al. Action to Control Cardiovascular Risk in Diabetes Study Group Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008;358:2545–2559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duckworth W, Abraira C, Moritz T, et al. VADT Investigators Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med 2009;360:129–139 [DOI] [PubMed] [Google Scholar]
- 8.Boussageon R, Bejan-Angoulvant T, Saadatian-Elahi M, et al. Effect of intensive glucose lowering treatment on all cause mortality, cardiovascular death, and microvascular events in type 2 diabetes: meta-analysis of randomised controlled trials. BMJ 2011;343:d4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.The Diabetes Control and Complications Trial Research Group The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 1993;329:977–986 [DOI] [PubMed] [Google Scholar]
- 10.Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008;359:1577–1589 [DOI] [PubMed] [Google Scholar]
- 11.Writing Team for the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group Effect of intensive therapy on the microvascular complications of type 1 diabetes mellitus. JAMA 2002;287:2563–2569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Writing Team for the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group Sustained effect of intensive treatment of type 1 diabetes mellitus on development and progression of diabetic nephropathy: the Epidemiology of Diabetes Interventions and Complications (EDIC) study. JAMA 2003;290:2159–2167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.El-Osta A. Glycemic memory. Curr Opin Lipidol 2012;23:24–29 [DOI] [PubMed] [Google Scholar]
- 14.Ceriello A. Hypothesis: the “metabolic memory”, the new challenge of diabetes. Diabetes Res Clin Pract 2009;86(Suppl. 1):S2–S6 [DOI] [PubMed] [Google Scholar]
- 15.Roy S, Sala R, Cagliero E, Lorenzi M. Overexpression of fibronectin induced by diabetes or high glucose: phenomenon with a memory. Proc Natl Acad Sci USA 1990;87:404–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Engerman RL, Kern TS. Progression of incipient diabetic retinopathy during good glycemic control. Diabetes 1987;36:808–812 [DOI] [PubMed] [Google Scholar]
- 17.Inoguchi T, Battan R, Handler E, Sportsman JR, Heath W, King GL. Preferential elevation of protein kinase C isoform beta II and diacylglycerol levels in the aorta and heart of diabetic rats: differential reversibility to glycemic control by islet cell transplantation. Proc Natl Acad Sci USA 1992;89:11059–11063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ihnat MA, Thorpe JE, Kamat CD, et al. Reactive oxygen species mediate a cellular ‘memory’ of high glucose stress signalling. Diabetologia 2007;50:1523–1531 [DOI] [PubMed] [Google Scholar]
- 19.Zhong Q, Kowluru RA. Epigenetic changes in mitochondrial superoxide dismutase in the retina and the development of diabetic retinopathy. Diabetes 2011;60:1304–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ceriello A, Kumar S, Piconi L, Esposito K, Giugliano D. Simultaneous control of hyperglycemia and oxidative stress normalizes endothelial function in type 1 diabetes. Diabetes Care 2007;30:649–654 [DOI] [PubMed] [Google Scholar]
- 21.Cooper ME, El-Osta A. Epigenetics: mechanisms and implications for diabetic complications. Circ Res 2010;107:1403–1413 [DOI] [PubMed] [Google Scholar]
- 22.Ling C, Groop L. Epigenetics: a molecular link between environmental factors and type 2 diabetes. Diabetes 2009;58:2718–2725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jayaraman S. Epigenetic mechanisms of metabolic memory in diabetes. Circ Res 2012;110:1039–1041 [DOI] [PubMed] [Google Scholar]
- 24.Brasacchio D, Okabe J, Tikellis C, et al. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes 2009;58:1229–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.El-Osta A, Brasacchio D, Yao D, et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med 2008;205:2409–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell 2007;25:15–30 [DOI] [PubMed] [Google Scholar]
- 27.Okabe J, Orlowski C, Balcerczyk A, et al. Distinguishing hyperglycemic changes by Set7 in vascular endothelial cells. Circ Res 2012;110:1067–1076 [DOI] [PubMed] [Google Scholar]
- 28.Cosentino F, Francia P, Camici GG, Pelicci PG, Lüscher TF, Volpe M. Final common molecular pathways of aging and cardiovascular disease: role of the p66Shc protein. Arterioscler Thromb Vasc Biol 2008;28:622–628 [DOI] [PubMed] [Google Scholar]
- 29.Paneni F, Mocharla P, Akhmedov A, et al. Gene silencing of the mitochondrial adaptor p66(Shc) suppresses vascular hyperglycemic memory in diabetes. Circ Res 2012;111:278–289 [DOI] [PubMed] [Google Scholar]
- 30.Pagnin E, Fadini G, de Toni R, Tiengo A, Calò L, Avogaro A. Diabetes induces p66shc gene expression in human peripheral blood mononuclear cells: relationship to oxidative stress. J Clin Endocrinol Metab 2005;90:1130–1136 [DOI] [PubMed] [Google Scholar]
- 31.Prasad A, Bekker P, Tsimikas S. Advanced glycation end products and diabetic cardiovascular disease. Cardiol Rev 2012;20:177–183 [DOI] [PubMed] [Google Scholar]
- 32.Schram MT, Schalkwijk CG, Bootsma AH, Fuller JH, Chaturvedi N, Stehouwer CD, EURODIAB Prospective Complications Study Group Advanced glycation end products are associated with pulse pressure in type 1 diabetes: the EURODIAB Prospective Complications Study. Hypertension 2005;46:232–237 [DOI] [PubMed] [Google Scholar]
- 33.Yoshida N, Okumura K, Aso Y. High serum pentosidine concentrations are associated with increased arterial stiffness and thickness in patients with type 2 diabetes. Metabolism 2005;54:345–350 [DOI] [PubMed] [Google Scholar]
- 34.Monnier VM, Bautista O, Kenny D, et al. Skin collagen glycation, glycoxidation, and crosslinking are lower in subjects with long-term intensive versus conventional therapy of type 1 diabetes: relevance of glycated collagen products versus HbA1c as markers of diabetic complications. DCCT Skin Collagen Ancillary Study Group. Diabetes Control and Complications Trial. Diabetes 1999;48:870–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramasamy R, Yan SF, Schmidt AM. Receptor for AGE (RAGE): signaling mechanisms in the pathogenesis of diabetes and its complications. Ann N Y Acad Sci 2011;1243:88–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sesso HD, Buring JE, Christen WG, et al. Vitamins E and C in the prevention of cardiovascular disease in men: the Physicians’ Health Study II randomized controlled trial. JAMA 2008;300:2123–2133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou S, Chen HZ, Wan YZ, et al. Repression of P66Shc expression by SIRT1 contributes to the prevention of hyperglycemia-induced endothelial dysfunction. Circ Res 2011;109:639–648 [DOI] [PubMed] [Google Scholar]
- 38.Guo J, Gertsberg Z, Ozgen N, Steinberg SF. p66Shc links alpha1-adrenergic receptors to a reactive oxygen species-dependent AKT-FOXO3A phosphorylation pathway in cardiomyocytes. Circ Res 2009;104:660–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cai W, He JC, Zhu L, Chen X, Striker GE, Vlassara H. AGE-receptor-1 counteracts cellular oxidant stress induced by AGEs via negative regulation of p66shc-dependent FKHRL1 phosphorylation. Am J Physiol Cell Physiol 2008;294:C145–C152 [DOI] [PubMed] [Google Scholar]
- 40.Zheng Z, Chen H, Li J, et al. Sirtuin 1-mediated cellular metabolic memory of high glucose via the LKB1/AMPK/ROS pathway and therapeutic effects of metformin. Diabetes 2012;61:217–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Woods A, Dickerson K, Heath R, et al. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab 2005;2:21–33 [DOI] [PubMed] [Google Scholar]
- 42.Schisano B, Tripathi G, McGee K, McTernan PG, Ceriello A. Glucose oscillations, more than constant high glucose, induce p53 activation and a metabolic memory in human endothelial cells. Diabetologia 2011;54:1219–1226 [DOI] [PubMed] [Google Scholar]
- 43.Liu D, Xu Y. p53, oxidative stress, and aging. Antioxid Redox Signal 2011;15:1669–1678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao Y, Chaiswing L, Velez JM, et al. p53 translocation to mitochondria precedes its nuclear translocation and targets mitochondrial oxidative defense protein-manganese superoxide dismutase. Cancer Res 2005;65:3745–3750 [DOI] [PubMed] [Google Scholar]
- 45.Li L, Wang L, Li L, et al. Activation of p53 by SIRT1 inhibition enhances elimination of CML leukemia stem cells in combination with imatinib. Cancer Cell 2012;21:266–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Orimo M, Minamino T, Miyauchi H, et al. Protective role of SIRT1 in diabetic vascular dysfunction. Arterioscler Thromb Vasc Biol 2009;29:889–894 [DOI] [PubMed] [Google Scholar]
- 47.Kim CS, Jung SB, Naqvi A, et al. p53 impairs endothelium-dependent vasomotor function through transcriptional upregulation of p66shc. Circ Res 2008;103:1441–1450 [DOI] [PubMed] [Google Scholar]
- 48.Liu X, Wang D, Zhao Y, et al. Methyltransferase Set7/9 regulates p53 activity by interacting with Sirtuin 1 (SIRT1). Proc Natl Acad Sci USA 2011;108:1925–1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Handy DE, Castro R, Loscalzo J. Epigenetic modifications: basic mechanisms and role in cardiovascular disease. Circulation 2011;123:2145–2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Napoli C, Crudele V, Soricelli A, et al. Primary prevention of atherosclerosis: a clinical challenge for the reversal of epigenetic mechanisms? Circulation 2012;125:2363–2373 [DOI] [PubMed] [Google Scholar]
- 51.Yi P, Melnyk S, Pogribna M, Pogribny IP, Hine RJ, James SJ. Increase in plasma homocysteine associated with parallel increases in plasma S-adenosylhomocysteine and lymphocyte DNA hypomethylation. J Biol Chem 2000;275:29318–29323 [DOI] [PubMed] [Google Scholar]
- 52.Kim CS, Kim YR, Naqvi A, et al. Homocysteine promotes human endothelial cell dysfunction via site-specific epigenetic regulation of p66shc. Cardiovasc Res 2011;92:466–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Timmers S, Konings E, Bilet L, et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab 2011;14:612–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lekakis J, Rallidis LS, Andreadou I, et al. Polyphenolic compounds from red grapes acutely improve endothelial function in patients with coronary heart disease. Eur J Cardiovasc Prev Rehabil 2005;12:596–600 [DOI] [PubMed] [Google Scholar]
- 55.Wong RH, Howe PR, Buckley JD, Coates AM, Kunz I, Berry NM. Acute resveratrol supplementation improves flow-mediated dilatation in overweight/obese individuals with mildly elevated blood pressure. Nutr Metab Cardiovasc Dis 2011;21:851–856 [DOI] [PubMed] [Google Scholar]
- 56.Breen DM, Dolinsky VW, Zhang H, et al. Resveratrol inhibits neointimal formation after arterial injury through an endothelial nitric oxide synthase-dependent mechanism. Atherosclerosis 2012;222:375–381 [DOI] [PubMed] [Google Scholar]
- 57.Poulsen MM, Vestergaard PF, Clasen BF, et al. High-dose resveratrol supplementation in obese men: an investigator-initiated, randomized, placebo-controlled clinical trial of substrate metabolism, insulin sensitivity, and body composition. Diabetes 2013;62:1186–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Inzucchi SE, Bergenstal RM, Buse JB, et al. American Diabetes Association (ADA); European Association for the Study of Diabetes (EASD). Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2012;35:1364–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen S, Feng B, George B, Chakrabarti R, Chen M, Chakrabarti S. Transcriptional coactivator p300 regulates glucose-induced gene expression in endothelial cells. Am J Physiol Endocrinol Metab 2010;298:E127–E137 [DOI] [PubMed] [Google Scholar]
- 60.Morimoto T, Sunagawa Y, Kawamura T, et al. The dietary compound curcumin inhibits p300 histone acetyltransferase activity and prevents heart failure in rats. J Clin Invest 2008;118:868–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xiong S, Salazar G, San Martin A, et al. PGC-1 alpha serine 570 phosphorylation and GCN5-mediated acetylation by angiotensin II drive catalase down-regulation and vascular hypertrophy. J Biol Chem 2010;285:2474–2487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kikuchi H, Kuribayashi F, Kiwaki N, Takami Y, Nakayama T. GCN5 regulates the superoxide-generating system in leukocytes via controlling gp91-phox gene expression. J Immunol 2011;186:3015–3022 [DOI] [PubMed] [Google Scholar]
- 63.Mai A. The therapeutic uses of chromatin-modifying agents. Expert Opin Ther Targets 2007;11:835–851 [DOI] [PubMed] [Google Scholar]
- 64.Cariou B, Charbonnel B, Staels B. Thiazolidinediones and PPARγ agonists: time for a reassessment. Trends Endocrinol Metab 2012;23:205–215 [DOI] [PubMed] [Google Scholar]
- 65.Jax TW. Metabolic memory: a vascular perspective. Cardiovasc Diabetol 2010;9:51–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wong TY, Klein R, Sharrett AR, et al. Retinal arteriolar narrowing and risk of coronary heart disease in men and women. The Atherosclerosis Risk in Communities Study. JAMA 2002;287:1153–1159 [DOI] [PubMed] [Google Scholar]
- 67.Sax FL, Cannon RO, 3rd, Hanson C, Epstein SE. Impaired forearm vasodilator reserve in patients with microvascular angina. Evidence of a generalized disorder of vascular function? N Engl J Med 1987;317:1366–1370 [DOI] [PubMed] [Google Scholar]
- 68.Song R, Xu W, Chen Y, Li Z, Zeng Y, Fu Y. The expression of Sirtuins 1 and 4 in peripheral blood leukocytes from patients with type 2 diabetes. Eur J Histochem 2011;55:e10. [DOI] [PMC free article] [PubMed] [Google Scholar]