

Summary
Pathogen‐associated molecular pattern molecules (PAMPs) are derived from microorganisms and recognized by pattern recognition receptor (PRR)‐bearing cells of the innate immune system as well as many epithelial cells. In contrast, damage‐associated molecular pattern molecules (DAMPs) are cell‐derived and initiate and perpetuate immunity in response to trauma, ischemia, and tissue damage, either in the absence or presence of pathogenic infection. Most PAMPs and DAMPs serve as so‐called ‘Signal 0s’ that bind specific receptors [Toll‐like receptors, NOD‐like receptors, RIG‐I‐like receptors, AIM2‐like receptors, and the receptor for advanced glycation end products (RAGE)] to promote autophagy. Autophagy, a conserved lysosomal degradation pathway, is a cell survival mechanism invoked in response to environmental and cellular stress. Autophagy is inferred to have been present in the last common eukaryotic ancestor and only to have been lost by some obligatory intracellular parasites. As such, autophagy represents a unifying biology, subserving survival and the earliest host defense strategies, predating apoptosis, within eukaryotes. Here, we review recent advances in our understanding of autophagic molecular mechanisms and functions in emergent immunity.
Keywords: PAMPs, DAMPs, autophagy, apoptosis, immunity, inflammation
This article is part of a series of reviews covering Metabolism and Autophagy in the Immune System appearing in Volume 249 of Immunological Reviews.
Introduction
Danger is everywhere. The host recognizes so‐called danger signals with induction of an innate and then adaptive immune response (Fig. 1 ). In the setting of microbial infection, pathogen‐associated molecular patterns (PAMPs), present in diverse organisms but absent in the host, provide exogenous signals that alert the immune system to the presence of pathogens, thereby promoting immunity 1, 2, 3. The notion that a ‘signal 0’ for innate immunity was necessary, as postulated by Janeway, distinguished it from later signals to drive adaptive immunity 2. This led to the first identification of so‐called pattern recognition receptors (PRRs). In contrast, cells release damage‐associated molecular pattern molecules (DAMPs) as endogenous danger signals that alert the innate immune system to unscheduled cell death, to microbial invasion, and in response to stress 4, 5, 6, 7.
Figure 1.
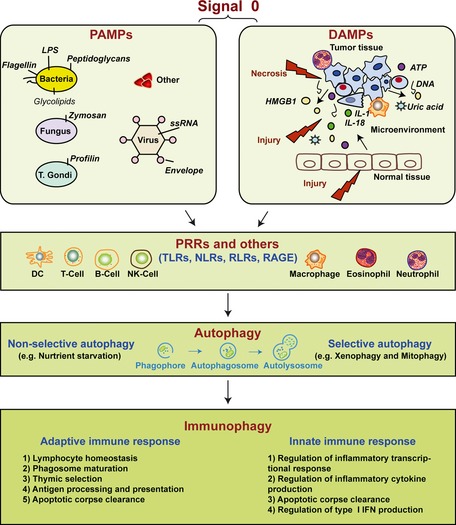
Signal 0s play critical roles in autophagy and immunity. Pathogen‐associated molecular patterns (PAMPs) and damage‐associated molecular patterns (DAMPs) serve as signal 0s, inducing autophagy and immunophagy in the emergent immune response before the later Signal 1 (antigenic peptide and major histocompatability molecules), Signal 2 (costimulatory molecules such as CD80 and CD86), both present on the surface of DCs recruited by the signal 0s. Signal 3 represents the DC provided IL‐6 family cytokine expression such as IL‐12 and IL‐23 which promote polarization of emergent T‐cell response. Signal 4 represents the integrin expression on DCs, defining the origin of the DCs and driving specialized molecules on T‐cells promoting T‐cell traffic to tissues. LPS, lipopolysaccharide; HMGB1, high mobility group box 1; ATP, adenosine‐5′‐triphosphate; PRRs, pattern recognition receptors; TLRs, Toll‐like receptors; NLRs, NOD‐like receptors; RLRs, RIG‐I‐like receptors; RAGE, receptor for advanced glycation end products.
Autophagy is a process by which cytoplasmic components, including soluble macromolecules (nucleic acids, proteins, carbohydrates, and lipids) and organelles (e.g. mitochondria, peroxisomes, and endoplasmic reticulum) are degraded by the lysosome 8, 9. It likely evolved as a cell stress response to starvation and subsequently to limit damage and maintain cellular homeostasis as a means to exert protein/organelle quality control 10. Autophagic dysfunction is linked to several human diseases. For example, it can exert tumor suppressing 11, 12 as well as tumor promoting functions 13, 14, in a context and cell type‐specific manner. Several studies reveal a crucial role for autophagy in adaptive and innate immunity 15, 16, 17, 18, 19, 20, 21, 22, 23, with the term ‘immunophagy’ 22 referring to all such processes collectively (Fig. 1 ). Autophagy degrades microbes (such as viruses, bacteria, and protozoa) that invade and gain access to the cytosol 16, 24, 25. This process is termed ‘xenophagy’ 26. The precise membrane dynamics and mediators of xenophagy, however, are not fully understood. In this review, we provide a brief overview of the process and function of autophagy and focus on the complex relationship between immunophagy and the initiating signal 0s with specific emphasis on PAMPs and DAMPs.
The processes and functions of autophagy: a brief overview
Lysosomes contain acid hydrolases, enzymes that function to break up waste materials and cellular debris and regulate cell death. Nutrient sensor complexes such as mTORC1 and mTORC2 sit within the lysosomal membrane, able to initiate anabolism and mitosis when the cell is nutritionally replete. Within the immune system, lysosomes link exocytosis, endocytosis, and phagocytosis 27. The ubiquitin‐proteasome system (UPS) and autophagy are two functionally linked major degradation pathways. Impairment of the UPS is compensated by upregulation of autophagy 28. In contrast, impairment of autophagy prevents ubiquitinated protein delivery to the proteasome for degradation 29. Some proteins, such as histone deacetylase‐6 (HDAC‐6) 30, p53 31, and p62/SQSTM1 29, play roles in the cross‐talk between the UPS and autophagy.
In mammals, three primary forms of autophagy have been described: chaperone‐mediated autophagy, microautophagy, and macroautophagy, which differ in their mechanism and function (Fig. 2 ). Among these, macroautophagy has been the most extensively studied 8. However, the precise mechanisms mediating microautophagy in mammalian cells are still unclear 32. Macroautophagy most commonly (hereafter referred to as ‘autophagy’) is initiated by the formation of the phagophore, followed by a series of steps, including the elongation and expansion of the phagophore and the closure and completion of a double‐membrane autophagosome, which sequesters cytosolic material. Autophagosome maturation includes several vesicular fusion events that originates from early and late endosomes (amphisomes) and lysosomes (autolysosomes), followed by breakdown and degradation of the autophagosome and amphisomes through acid hydrolases inside the autolysosome. Recycling of the resulting macromolecules is mediated through permeases. Several recent studies suggest that autophagosomes may also fuse with cell membranes, promoting exocytosis, secretion, or extrusion of autophagic contents 33, 34, 35.
Figure 2.
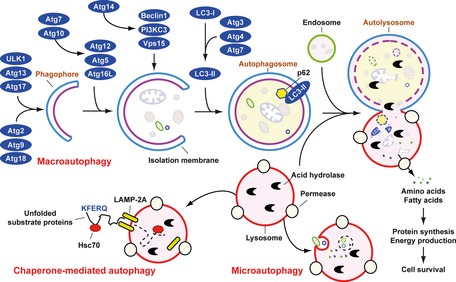
Means by which autophagy delivers antigen into the autolysosome. Microautophagy refers to the sequestration of cytosolic components directly by lysosomes through invaginations within their limiting membrane. Chaperone‐mediated autophagy involves direct translocation of unfolded substrate proteins (KFERQ‐like motif) across the lysosome membrane through the action of a cytosolic and lysosomal chaperone heat shock cognate protein of 70 kDa (Hsc70), and the integral membrane receptor lysosome‐associated membrane protein type 2A (LAMP‐2A). In the case of macroautophagy, the cargo is sequestered within a unique double membrane cytosolic vesicle, an autophagosome. The autophagosome itself is formed by expansion of the phagophore. The autophagosome undergoes fusion with a late endosome or lysosome to form an autolysosome, in which the sequestered material is degraded. Degradation of membrane lipids and proteins by the autolysosome generates free fatty acids, nucleotides, and amino acids that can be reused by the cell to maintain mitochondrial ATP energy production, protein synthesis, and thereby promote cell survival. The molecular machinery of macroautophagy was largely discovered in yeast and the centrally important proteins referred to as autophagy‐related (ATG) proteins although some similar proteins in mammals have disparate names (Beclin‐1 = ATG6, LC3 = ATG8 for example).
Autophagy involves a series of dynamic membrane‐rearrangement reactions mediated by a core set of proteins – the autophagy‐related (ATG) proteins 36 and other autophagy interaction network components 37. ATG proteins are composed of four functional groups (Fig. 2 ), including a protein serine/threonine kinase complex that responds to upstream signals (Atg1/ULK1, Atg13, and Atg17), a lipid kinase signaling complex that mediates vesicle nucleation (Atg6/Beclin1, Atg14, Vps34/PI3KC3, and Vps15), and two ubiquitin‐like conjugation pathways that mediate vesicle expansion (the Atg8/LC3 and Atg12 systems). Atg9 is a transmembrane protein and may provide lipids to the isolation membrane by cycling between distinct subcellular compartments 38. Beclin 1/Atg6 has an important role in autophagy and tumorigenesis 11. It interacts with several cofactors to regulate the class III phosphatidylinositol 3‐kinase (PI3KC3) and promote formation of Beclin 1‐PI3KC3 core complexes, thereby inducing autophagy 39, 40.
Autophagy can degrade substrates in a selective manner such as mitochondria, in a process termed mitophagy. Mitophagy is now a well‐established mechanism necessary for elimination of dysfunctional mitochondria and regulation of mitochondrial quality by specific mediators such as Nix/BNIP3L, Parkin, and Atg32 41. Cells may also remove damaged mitochondria to prevent the accumulation of reactive oxygen species (ROS). The induced ROS from mitochondria or NADPH oxidases have recently been shown to be important signals linking immunity with autophagy 42, 43, 44.
Autophagy and apoptosis are both tightly regulated biological processes (Fig. 3 ) that play a central role in cell survival and cell death 45. These two pathways are regulated by common factors such as Bcl‐2 family members and various transcription factors (Fig. 3 ). In addition, expressions of autophagy gene products are required for clearance of apoptotic cells and the prevention of tissue inflammation 46. Compared with apoptosis (‘programed cell death’), autophagy is primarily a cell survival process (‘programed cell survival’). In some cases, when apoptosis is compromised, such as in the setting of Bax/Bak deficiency, activation of autophagy leads to cell death 47, presumably via self‐cannibalization or bioenergetic failure.
Figure 3.
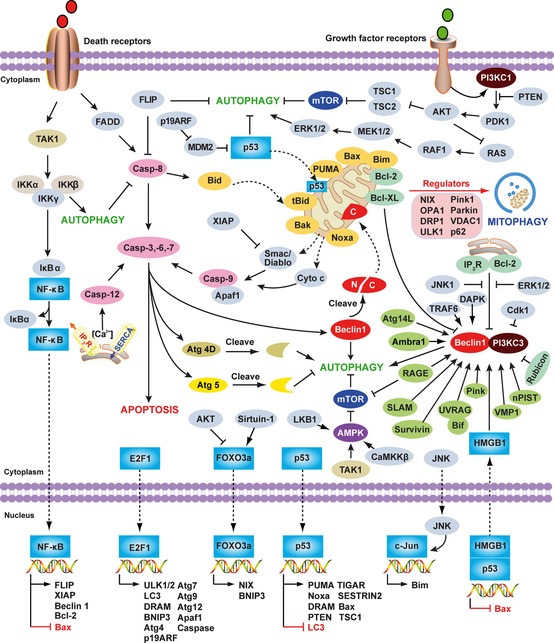
Overview of the major signal transduction pathways that regulate autophagy and apoptosis. Common molecular regulators include gene products that affect both autophagy and apoptosis and the pathways that they influence. The process of apoptotic cell death is mediated by two central pathways: an extrinsic pathway involving cell surface receptors (the death receptor pathway), and an intrinsic pathway using mitochondria and the endoplasmic reticulum (the mitochondrial pathway). A third pathway is mediated by cytolytic T and NK cells delivering perforin/granzymes to promote apoptosis. CD95 (also called Fas or APO‐1) induces apoptosis by forming a death‐inducing signaling complex (DISC) at the receptor that contains FADD, caspase‐8, and the caspase‐8 regulator. Autophagy can degrade active caspase‐8. The FLICE inhibitory protein (FLIP), a caspase‐like molecule without proteolytic activity, protects cells from CD95‐induced apoptosis. FLIP can suppress autophagy. The intrinsic mitochondrial pathway is activated by diverse apoptotic signals such as DNA damage, growth factor deprivation, and oxidative stress. Cytoplasmic translocation of mitochondrial proteins [such as cytochrome c (cyt c) and Smac/DIABLO] lead to activation of the caspase cascade and initiate apoptosis. A pivotal event in the mitochondrial pathway is mitochondrial outer membrane permeabilization (MOMP), which is mainly regulated by Bcl‐2 family members. Some of the Bcl‐2 family members (e.g. Bcl‐2, and Bcl‐XL) are anti‐apoptotic, whereas others (e.g. Bax, tBid, Bad, Bim, PUMA, and Noxa) are pro‐apoptotic. Mitophagy is a well established mechanism necessary for elimination of dysfunctional mitochondria and regulation of mitochondrial quality in yeast or mammalian cells associated with cytosolic mediators such as NIX, Atg32, optic atrophy 1 (OPA1), dynamin‐related protein 1 (DRP1), unc‐51‐like kinase 1 (ULK1), Parkin, Pink1, voltage‐dependent anion channel 1 (VDAC1), p62/Sequestosome 1 (SQSTM1), mitofusin 1, and mitofusin 2. Beclin 1, the mammalian ortholog of yeast Atg6, plays a central role in autophagy. It interacts with several cofactors (e.g. Atg14L, UVRAG, Bif‐1, Rubicon, Ambra1, HMGB1, nPIST, VMP1, SLAM, IP 3R, PINK, and Survivin) to regulate the formation of Beclin 1‐PI3KC3 complexes, thereby inducing autophagy. In contrast, the BH3 domain of Beclin 1 is bound to and inhibited by Bcl‐2 or Bcl‐XL. This interaction can be disrupted by phosphorylation of Bcl‐2 and Beclin 1, or ubiquitination of Beclin 1. Interestingly, caspase‐mediated cleavage of Beclin 1 promotes cross‐talk between apoptosis and autophagy. Although apoptosis‐associated cleavage of Beclin 1 and Atg5 inactivates autophagy, the cleavage of Atg4D by caspase‐3 generates a fragment with increased autophagic activity. In the presence of growth factors, growth factor receptor signaling activates PI3KC1 at the plasma membrane. PI3KC1 activates the downstream target AKT, leading to activation of mammalian target of rapamycin (mTOR) by inhibiting the tuberous sclerosis complex 1/2 (TSC1–TSC2), which results in inhibition of autophagy. Overexpression of the phosphatase and tensin homolog (PTEN) gene, by an inducible promoter, antagonizes PI3KC1 to induce autophagy. RAS has a dual effect on autophagy. When it activates PI3KC1, autophagy is inhibited, but when it selectively activates the RAF1–mitogen‐activated protein kinase kinase (MEK)–extracellular signal‐regulated kinase (ERK) cascade, autophagy is stimulated. AMPK monitors the energy status of the cell by sensing the AMP:ATP ratio. Several upstream kinases, including liver kinase B1 (LKB1, which is activated by energy depletion), calcium/calmodulin kinase kinase‐β (CaMKKβ, which is activated by cytosolic Ca2+ levels), and TGFβ activated kinase‐1 (TAK‐1, which is also involved in IKK activation), can activate AMPK by phosphorylation. Activated AMPK promotes inhibition of mTOR kinase, which induces autophagy. NF‐κB and p53 play a double role in regulating autophagy in a transcription dependent and/or independent fashion. In contrast, E2F1 and FOXO3 positively regulate autophagy in a transcription dependent fashion. Nuclear HMGB1 inhibits the p53‐dependent transactivation from the Bax promoter. Accumulation of cytosolic HMGB1 sustains autophagy by liberating the Beclin 1 and PI3KC3 complexes. RAGE‐induced autophagy is associated with decreased phosphorylation of mTOR and increased Beclin1‐PI3KC3 interactions.
PAMPs: the exogenous signal 0s
In 1989, Charles Janeway 1 proposed that the immune system evolved to protect the host, not against innocuous foreign antigens but rather against infectious pathogens, and postulated that receptors on antigen‐presenting cells of the innate immune system recognize so‐called signal 0s, now termed PAMPs. Shortly thereafter, when we (MTL) asked him about the role of signal 0s in the setting of sterile inflammation, including cancer, he indicated that their role was indeed a concern but was at that time unclear. PAMPs are essential functional components of microorganisms that direct the targeted host cell to distinguish ‘self’ from ‘non‐self’ (‘stranger hypothesis’) and promote signals associated with innate immunity 48. Major PAMPs are microbial nucleic acids, including DNA (e.g. unmethylated CpG motifs), double‐stranded RNA (dsRNA), single‐stranded RNA (ssRNA), and 5′‐triphosphate RNA, as well as lipoproteins, surface glycoproteins, and membrane components [peptidoglycans, lipoteichoic acid, lipopolysaccharide (LPS), and glycosylphosphatidylinositol]. They are recognized by Toll‐like receptors (TLRs) and other PRRs, such as retinoid acid‐inducible gene I (RIG‐I)‐like receptors (RLRs), AIM2 like receptors (ALRs), and nucleotide‐binding oligomerization domain (NOD)‐like receptors (NLRs) 48, 49, 50. Most of the TLRs are believed to be homodimers, although heterodimers exist in the cases of TLR1:TLR2 and TLR2:TLR6. The Toll/interleukin‐1 receptor homologous region (TIR) adapter proteins [myeloid differentiation factor 88 (MyD88), TIR adapter protein (TIRAP)/MyD88 adapter‐like (MAL), translocating chain‐associated membrane protein (TRAM), and TIR‐domain‐containing adapter‐inducing interferon‐β (TRIF)] also appear to associate with one another and are often illustrated as homodimers or heterodimers (Fig. 4 A).
Figure 4.
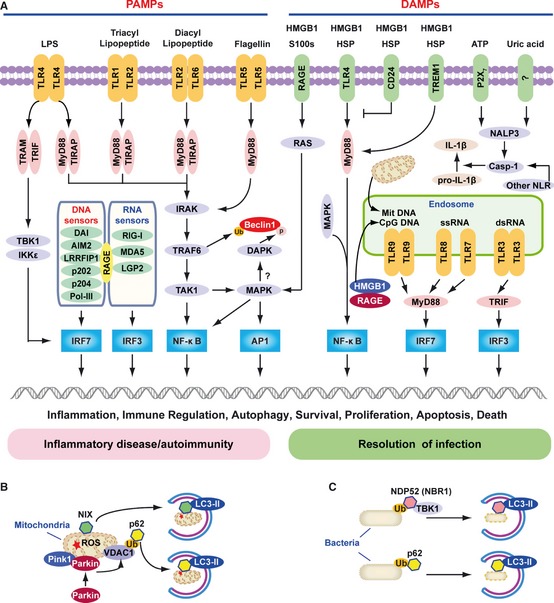
The role of TLR s, RLR s, and NLR s in PAMP and DAMP recognition. (A) Signaling pathways triggered by pathogen‐associated molecular pattern (PAMPs) and damage‐associated molecular pattern molecules (DAMPs). Lipopolysaccharide (LPS) activates both the myeloid differentiation factor 88 (MyD88)‐dependent and TIR‐domain‐containing adapter‐inducing interferon‐β (TRIF)‐dependent Toll‐like receptor 4 (TLR4) pathways. The MyD88‐dependent pathway is responsible for NF‐κB and mitogen‐activated protein kinase (MAPK) activation, which controls induction of proinflammatory cytokines. The TRIF‐dependent pathway activates IRF3 by TANK‐binding kinase 1 (TBK1)/IKKε, which is required for the induction of IFN‐inducible genes. TLR1‐TLR2 and TLR2‐TLR6 recognize bacterial triacylated lipopeptide or diacyl lipopeptide, respectively, and recruit TIR adapter protein (TIRAP) and MyD88 at the plasma membrane to activate the MyD88‐dependent pathway. TLR5 recognizes flagellin and activates the MyD88‐dependent pathway. TLR3, TLR7, TLR8, and TLR9 reside in the endosome and recognize dsRNA, ssRNA, CpG DNA, or mitochondrial DNA (Mit DNA), respectively. They recruit TRIF or MyD88 to activate the IRF3 or IRF7 pathway. All immunogenic nucleic acids bind indicated cytosolic DNA sensors or RNA sensors, including retinoid acid‐inducible gene I (RIG‐I)‐like receptors (RLRs), which are required for subsequent recognition by specific pattern recognition receptors to activate innate immune responses. DAMPs such as HMGB1, S100 proteins (S100s), and heat shock proteins (HSPs) recognize the receptor for advanced glycation end products (RAGE), TLR4, or triggering receptor expressed on myeloid cells‐1 (TREM‐1) and activate the MyD88‐MAPK‐NF‐κB pathway. HMGB1 and RAGE activate the TLR9‐MyD88 dependent pathway, which contributes to autoimmune pathogenesis. CD24 is a negative receptor to inhibit the DAMP‐induced TLR4 pathway. ATP binding of the P2X7 receptor and uric acid, as well as asbestos and alum, increase activation of caspase‐1 by the NALP3 inflammasome and other nucleotide‐binding oligomerization domain (NOD)‐like receptors (NLRs) to promote secretion of IL‐1β and IL‐18. PAMP and DAMP‐mediated signaling and induction of an innate immune response usually results in resolution of infection, but may also cause chronic inflammation or autoimmunity by altering various cell death and survival mechanisms. (B) Mitochondria in mammalian cells are removed by autophagy via the NIX adapter during developmental elimination of mitochondria, or via PTEN‐induced putative kinase 1 (Pink1) and Parkin‐mediated ubiquitination (Ub) of voltage‐dependent anion channel 1 (VDAC1), recognized by the adapter p62 for removal of stressed (e.g. depolarized or damaged) mitochondria. (C) Intracellular bacteria exposed to the cytosol are modified by ubiquitin (Ub) and recognized by the autophagic adapters p62 or nuclear dot protein 52 (NDP52)/TBK1 for sequestration into autophagosomes and subsequent elimination.
TLR4 recognizes LPS 51, a major cell wall component of Gram‐negative bacteria that activates the innate immune system. Recognition of LPS requires CD14 in addition to TLR4. The responsiveness of the TLR4 and CD14 complex to LPS is enhanced by MD2. While the recognition of extracellular DNA primarily involves TLR9 52, recognition of cytosolic DNA appears to involve several sensors (Fig. 4 A) such as DNA‐dependent activator of interferon (IFN)‐regulatory factors (DAI) 53, hematopoietic IFN‐ inducible nuclear protein with the 200‐amino‐acid repeat (HIN‐200) family members [e.g. absent in melanoma 2 (AIM2), p202, p204 (IFN‐inducible IFI16 protein)] 54, 55, 56, DNA‐dependent RNA polymerase III (Pol‐III) 57, and leucine rich repeat (in FLII) interacting protein 1 (LRRFIP1) 58. dsRNA is recognized by TLR3 59, RIG‐I 60, melanoma differentiation‐associated gene 5 (MDA5) 61, and laboratory of genetics and physiology 2 (LGP2) 62 (Fig. 4 A).
Following PAMP recognition, activated TLRs and other PRRs localized to the cell surface, the cytoplasm, and/or intracellular vesicles provide signals to the host indicating the presence of a microbial infection and trigger proinflammatory and anti‐microbial responses by activating a multitude of intracellular signaling pathways, including adapter molecules, kinases, and transcription factors such as nuclear factor‐κB (NF‐κB), activator protein‐1 (AP‐1), and IFN regulatory factors (IRFs) (Fig. 4 A). PAMP‐induced signal transduction pathways ultimately result in the activation of gene expression and the synthesis of a broad range of molecules, including cytokines, chemokines, cell adhesion molecules, and immunoreceptors that direct the adaptive immune response to invading pathogens by sensing microorganisms.
DAMPs: the endogenous signal 0s
In 1994, Polly Matzinger 4 proposed that the immune system is more concerned with ‘danger’ or ‘damage’ than with the distinction between self and non‐self. The model starts with the idea that the immune system defines danger as anything that causes tissue stress or destruction 63, 64. In this model, antigen‐presenting cells are activated by PAMPs and DAMPs from stressed or damaged tissues or microbes 65. Matzinger's ‘danger model’ suggests why potent immune responses are initially elicited in the setting of sterile inflammation.
DAMPs are cell‐derived molecules that can initiate and perpetuate immunity in response to trauma, ischemia, cancer, and other settings of tissue damage in the absence of overt pathogenic infection (Fig. 4 A). DAMPs are localized within the nucleus and cytoplasm (HMGB1), cytoplasm alone (S100 proteins), exosomes [heat shock proteins (HSPs)], the extracellular matrix (hyaluronic acid), and in plasma components such as complement (C3a, C4a, and C5a). Examples of non‐protein DAMPs include ATP, uric acid, heparin sulfate, RNA, and DNA. DAMPs can also be mimicked by release of intracellular mitochondria, consisting of formyl peptides and mitochondrial DNA (with CpG DNA repeats), to activate human polymorphonuclear neutrophils through activation of TLR9 66, which reveals an important link between trauma and inflammation. Following interaction between DAMPs and DAMP receptors [e.g. TLRs and the receptor for advanced glycation end products (RAGE)], activation of mitogen‐activated protein kinases (MAPKs), NF‐κB, and PI3K/AKT signaling pathways ensues thus mediating a potent response to cell survival and cell death (Fig. 4 A). Increased serum levels of these DAMPs are associated with inflammatory diseases, including sepsis, arthritis, atherosclerosis, systemic lupus erythematosus, Crohn's disease, and cancer.
The chromatin‐associated protein HMGB1 is considered to be one of the prototypical DAMPs. Release of HMGB1 extracellularly is a common denominator in the response to both cell or tissue injury including organ harvest and associated ischemia/reperfusion insults, and microbial invasion 67, 68, 69. The redox/thiol‐reducing protein HMGB1 mediates the response to infection, inflammation, migration, proliferation, and differentiation 70, 71, 72. It is specifically recognized by several cell surface receptors including RAGE, TLR4, TLR2, triggering receptor expressed on myeloid cells‐1 (TREM‐1), and CD24 (Fig. 5 ). CD24 serves as a negative signaling molecule to limit DAMP‐ but not PAMP‐mediated inflammation 73. The HMGB1 protein induces migration and activation of human dendritic cells (DCs), eosinophil, natural killer (NK)‐DC cross‐talk, and T‐cell activation 70, 71. HMGB1 causes TLR4‐dependent activation of NADPH oxidase as well as increased ROS production 74. Interestingly, a cysteine at position 106 (Cys106) within HMGB1 is required for binding to TLR4 and activation of cytokine release in macrophages 75. The oxidation of HMGB1 Cys106 alone is sufficient to block the immunogenic activity of HMGB1 for DCs 76. These findings suggest that redox regulates HMGB1 function in the setting of emergent immunity and inflammation (Fig. 5 ). In addition, HMGB1 is an essential component of DNA‐containing immune complexes that stimulate cytokine production through a TLR9‐MyD88 pathway involving RAGE 77. In contrast, HMGB1‐containing nucleosomes from apoptotic cells induce anti‐double‐stranded DNA (dsDNA) and anti‐histone IgG responses in a TLR2‐dependent manner 78. Indeed, HMGB family members function as universal sentinels for nucleic acids in innate immune signaling 79.
Figure 5.
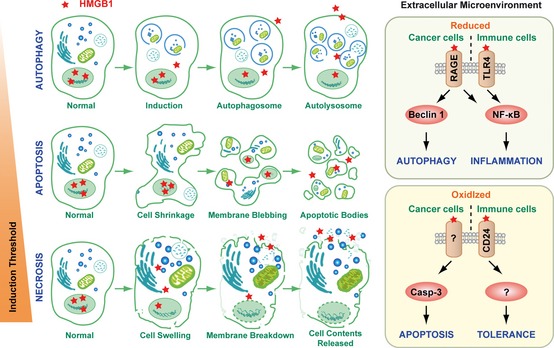
Cellular changes and HMGB 1 release observed with autophagy, apoptosis, and necrosis. The prototypical DAMP, high mobility group box 1 protein (HMGB1), is released with sustained autophagy, late apoptosis, and necrosis. Reducible HMGB1 binds to the receptor for advanced glycation end products (RAGE), inducing Beclin 1‐dependent autophagy, and binds both RAGE and Toll‐like receptor 2 (TLR2), TLR4, and TLR9, activating NF‐κB and promoting inflammation. In contrast, oxidized HMGB1 increases caspase 3 dependent apoptosis and tolerance by binding CD24 or other unknown receptors.
PAMPs and autophagy
A diverse array of pathogens interact with components of the autophagic pathway including Brucellus abortus 80, 81, Coxiella burnetii 82, Porphyromonas gingivalis 83, Salmonella enterica 84, Chlamydia trachomatis 85, Listeria monocytogenes 86, Group A Streptococcus 87, 88, Mycobacterium tuberculosis 89, Leishmania Mexicana 90, Shigella flexneri 91, poliovirus 92, herpes simplex virus 93, 94, sindbis virus 95, dengue virus 96, and coronavirus 97. As such, autophagy is likely the most ancient of the immune mechanisms, predating integration of mitochondria, generation of reactive oxygen species, and all innate and adaptive immune responses 15, 16, 17, 18, 19, 20, 24, 98, 99. Autophagy is not only a surveillance system to detect cytosolic microbes but also a mechanism for immune cells such as macrophages to capture, document, and digest microbes. Several of these components engage lipid rafts 96, 100. Recent studies have uncovered key ubiquitin‐binding adapters, such as p62 101, 102, 103, nuclear dot protein 52 kDa (NDP52) 104, neighbor of BRCA1 gene 1 (NBR1) 104, 105, and NIX 34, 106, in targeting bacteria (e.g. Salmonella, Shigella, Streptococci, Listeria, and Sindbis virus) or mitochondria to autophagosomes by binding to LC3 (Fig. 4 B,C). Other molecular species such as diacylglycerol serve as lipid signals that can also target bacteria to the autophagosome by activation of protein kinase C 107. In addition, the mammalian Atg8/LC3 family has many confirmed or likely interactions with other proteins 37, suggesting that these novel partners may be involved in xenophagy or other forms of selective autophagy.
LPS, a prototypical PAMP, directly induces autophagy in macrophages by activating the p38 MAPK and PI3KC3 pathways 108. The major signaling target of PAMPs during infection is the transcription factor NF‐κB. The relationship between NF‐κB and its regulation of autophagy is an area of great interest, emerging as a negative regulator of autophagy induced by tumor necrosis factor 109. NF‐κB p65 also directly regulates Beclin 1 expression 110. Other studies suggest that inhibitor of NF‐κB kinase (IKK), a kinase upstream of NF‐κB, is directly involved in the induction of autophagy (Fig. 3 ) and shows no strict NF‐κB correlation with control of autophagy 111, 112. Moreover, TGFβ‐activated kinase 1 (TAK1)‐binding proteins 2 and 3 (TAB 2 and TAB 3), two upstream activators of IKK, inhibit autophagy by binding Beclin 1 113. Autophagy is required for the activation of NF‐κB in mouse embryonic fibroblasts 114. Interestingly, LPS results in the activation of nuclear factor erythroid 2‐related factor 2 (Nrf2), which controls autophagic degradation by p62 115. p62 accumulation, in turn, results in hyperactivation of Nrf2 116. This raises the intriguing question of whether the p62‐Nrf2 pathway is an important means to activate autophagy during infection.
Selective viral autophagy plays a crucial role in antiviral host defense 117. Autophagy is essential for delivering cytoplasmic viral RNA to the endosomal pathway, extinguishing infection 118. The precise mechanisms underlying type I IFN production in autophagy are unknown but have been postulated to involve Atg5‐Atg12 conjugation 119. ROS accumulates in mitochondria in Atg5−/− cells without autophagy, amplifying RLR signaling pathways 44. Atg5 also participates in immunity and intracellular killing of pathogens via autophagosome‐independent processes, promoting immunity‐related GTPase (IRG) trafficking 120. Autophagy is an anti‐microbial effector of IRG. Murine IRG, Irgm1, promotes autophagy and sustains CD4+ T‐cell viability 121. The human IRG (IRGM) can eliminate mycobacteria through induction of autophagy 122, requiring IRGM expression in mitochondria 123. Moreover, IRGM is a common target of RNA viruses‐mediated autophagy, which regulates viral particle production 124. Recent study suggests that Rubicon, as part of a Beclin‐1‐Vps34‐containing autophagy complex, positively regulates NADPH oxidase (NOX2) assembly for superoxide generation in TLR2 signaling, and negatively regulates CARD9/Bcl10‐MALT‐1 complex and cytokine production in Dectin‐1 and RIG‐I signaling 125, 126, suggesting a direct impact of autophagy protein on pathogen‐specific host defense.
Although autophagy serves as a potent cellular strategy to clear pathogens, several viruses have evolved to exploit autophagic signaling to promote their replication, including dengue virus 96, hepatitis C virus 127, and poliovirus 128, among others. The interaction between human immunodeficiency virus (HIV) and autophagy are indeed quite complicated. HIV‐1 infects CD4+ T cells as well as macrophages. Autophagy promotes HIV‐1 proliferation within macrophages 129, 130. HIV‐1 is also targeted for elimination by autophagy, countered by the virus assembly proteins Nef 129 and Env 130. Autophagy is inhibited in HIV‐1‐infected CD4+ cells 130. HIV‐1‐infected cells inhibit autophagy in non‐infected macrophages/monocytic cells through induction of Akt and STAT3 signaling 131. Further studies are required to better understand the contribution of autophagy to HIV pathogenesis.
Autophagy also plays a role in viral antigen processing and presentation, mediating major histocompatibility complex (MHC) class I or II presentation 132, 133, 134. This requires both TLR and Atg5 signaling in DCs 134, 135, 136, 137. In contrast, Atg5, Atg7, and Atg16L1 are all required for NOD2‐induced autophagy and antigen presentation 138. Stimulation of the B‐cell receptor (BCR) by DNA‐containing antigens results in the translocation of both the BCR and TLR9 to autophagosomes 139. It is not clear whether autophagy mediates TLR9 signaling following translocation of CpG DNA from the endoplasmic reticulum (ER)/Golgi apparatus into the lysosome or endolysosome 140, 141. However, Atg9a, but not Atg7, controls dsDNA‐driven dynamic translocation of stimulator of IFN genes (STING) from the ER to the Golgi 142.
Recent evidence suggests that autophagy is likely to play a prominent role in the pathogenesis of Crohn's inflammatory bowel disease (CD) 143. Genome‐wide association studies have identified CD‐associated susceptibility genes, such as Atg16L1, NOD2, and IRGM 144, which function to regulate autophagy. Mice engineered with deficiencies in the Atg16L1 gene displayed gut inflammatory phenotypes not previously associated with autophagy 145, 146. A causal relationship clearly exists linking environmental factors, ATG16L1 genetic susceptibility, and the development of CD 147, suggesting that the interaction between host defects in autophagy and environmental stressors such as infection may be crucial for the pathogenesis of certain inflammatory diseases. NOD2 variants have also been linked with CD 138. In mammalian cells, NOD1 and NOD2 signal to induce autophagy and functionally interact with Atg16L1 138, 148. ATG16L1 genetic variation also modulates NOD2‐induced pro‐inflammatory cytokine responses 149. Autophagy is involved in other inflammatory disorders including cystic fibrosis 150, obesity 151, and sepsis 152.
DAMPs and autophagy
HMGB1 is one of the best characterized DAMPs, expressed largely in the nucleus as a chromatin‐associated protein. HMGB1 release from the nucleus and from the cell is dependent on different types of stress (Fig. 5 ). In response to exogenous bacterial products (such as endotoxin or CpG‐DNA) 67, 153 or endogenous inflammatory stimuli (e.g. TNF, IFN‐γ, or hydrogen peroxide) 67, 154 or apoptotic cells 155, innate immune cells actively release HMGB1. In addition, HMGB1 can be passively released from necrotic cells 68 or cells infected by viruses 156, triggering an inflammatory response (Fig. 5 ). HMGB1 can also be released from chemotherapy drug‐induced apoptosis at later stages of tumor development 157.
There is a complex reciprocal relationship between autophagy and HMGB1. Autophagy, not apoptosis, is a major regulator of HMGB1 localization and release by ROS in the early events following cell stress 158, 159. Notably, both endogenous and exogenous HMGB1 are important regulators of autophagy 158, 159, 160, 161. Endogenous HMGB1 competes with Bcl‐2 for interaction with Beclin 1 and orients Beclin 1 toward autophagosomes 159. Interaction between HMGB1 and Beclin1 relies upon the autophagic complex ULK1‐mAtg13‐FIP200 162. In addition, HMGB1 may be involved in the regulation of Bcl‐2 phosphorylation by the extracellular signal‐regulated kinase (ERK)/MAPK pathway 159. The intramolecular disulfide bridge (C23/45) of HMGB1 is required for binding to Beclin 1 and sustaining autophagy (Fig. 6 ). Reducible HMGB1 binds to RAGE, induces Beclin 1‐dependent autophagy (Fig. 5 ), and promotes resistance to chemotherapeutic agents or ionizing radiation 158. In contrast, oxidized HMGB1 increases the cytotoxicity of these agents and induces apoptosis via the mitochondrial pathway 158. There is a direct molecular interaction between HMGB1 and p53 in colorectal cancer to regulate apoptosis and autophagy 163. Loss of p53 increases cytosolic HMGB1 leading to increased binding to Beclin 1, thereby promoting autophagy, and decreasing apoptosis. In contrast, loss of HMGB1 increases cytosolic p53 and apoptosis and decreases autophagy. HDACs regulate HMGB1 nuclear versus cytosolic localization within monocytic cells 164. HMGB1 within the nucleus enhances DNA repair and chromatin modification following DNA damage 165. It is unknown whether HMGB1 mediates the HDAC‐autophagy pathway in DNA‐damage repair 166, but this represents a distinct possibility. In addition, HMGB1 and the downstream mediator heat shock protein β‐1 (HSPB1/HSP27) modulates mitochondrial respiration and morphology by sustaining autophagy/mitophagy 167, as we have shown, suggesting that HMGB1 is essential for mitochondrial quality control. HMGB1 forms highly inflammatory complexes with ssDNA, LPS, IL‐1β, and nucleosomes 168, which interact with TLR9, TLR4, IL‐1R, and TLR2 receptors, respectively. Thus HMGB1 may mediate PAMP‐induced autophagy, as a ‘universal’ factor important in host defense and immune homeostasis.
Figure 6.
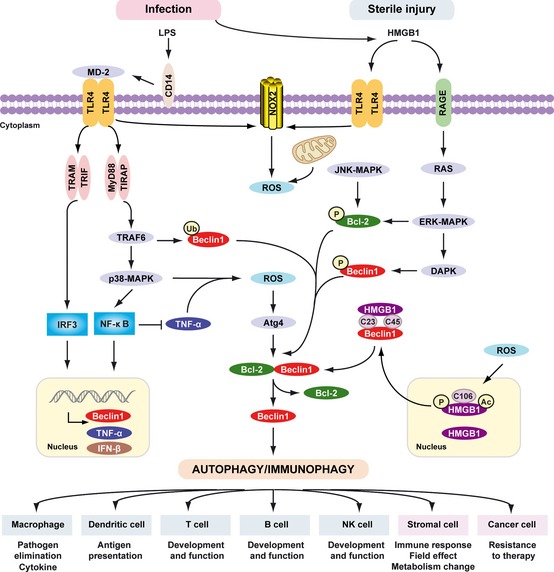
Signaling pathways triggered by LPS and HMGB 1 in autophagy/immunophagy. The inflammatory response, including the recruitment and migration of immune cells to the site of infection and release of cytokines, is mediated by lipopolysaccharide (LPS). LPS bound to LPS‐binding protein (LBP) is presented to CD14. CD14 maneuvers the LPS‐LBP complex to TLR4, and LPS, in combination with accessory molecule MD2, activates TLR4 signaling. LPS induces activation of the tumor necrosis factor receptor (TNFR)‐associated factor 6 (TRAF6)‐p38 MAPK pathway and induces the expression of TNF‐α and Beclin 1 by NF‐κB. NF‐κB inhibits TNF‐α‐induced autophagy. Multiple means to promote ROS production converge on the mitochondria or alternatively, NADPH oxidases such as NOX2 and NOX4. These in turn results in activation of autophagy through Atg4 activation, ultimately reducing the binding of Beclin 1 to Bcl‐2. TRIF‐dependent and/or MyD88‐dependent TLR4 pathway is required for LPS‐induced autophagy in macrophages. Moreover, TRAF6‐mediated ubiquitination of Beclin1 amplifies TLR4‐induced autophagy. HMGB1 links sterile injury and infection‐induced immunity. Stimuli that enhance reactive oxygen species promote cytosolic translocation of HMGB1 and thereby enhance autophagic flux. HMGB1 directly interacts with Beclin1, displacing Bcl‐2 requiring the cysteines at positions C23 and C45 within HMGB1. The HMGB1/RAGE interaction activates parallel signaling pathways, including ERK1/2 and NF‐ κB activation. Mitogen‐activated protein kinases (MAPKs), such as JNK1 and ERK1/2, also phosphorylate Bcl‐2 driving subsequent dissociation of the Beclin 1‐Bcl‐2 complex. Notably, DAPK phosphorylates Beclin 1, promoting the dissociation of Beclin 1 from Bcl‐2 like proteins, which in turn induces autophagy. Activation of phagocytic NADPH oxidase (NOX2) by HMGB1 requires TLR4 expression; the activation of other NADPH oxidases and interaction with TLRs is less clear. Thus PAMPs (LPS) and DAMPs (HMGB1) drive autophagy/immunophagy, regulating immune, stromal, and tumor cell functions.
In addition to HMGB1, other DAMPs such as ATP, S100, and host‐DNA induce autophagy in several cellular systems 139, 169, 170, 171. ATP‐induced autophagy is associated with the rapid killing of intracellular mycobacteria within human monocytes/macrophages 169, thus supporting the notion that autophagy plays a key role in the control of mycobacterial infections. Moreover, ATP‐induced autophagy is dependent on the mobilization of extracellular calcium and the P2X7 receptor 171 (Fig. 4 A). Recent study suggests chemotherapy‐induced autophagy causes the release of ATP from tumor cells, thereby stimulating anti‐tumor immune responses including recruitment of dendritic cells and CD4+ and CD8+ T cells 172. However, autophagy can also limit T cell‐mediated cytotoxicity 173. Thus, the process of autophagy plays dual roles in regulation of effective chemotherapy and the host‐derived anti‐cancer immune responses 174.
S100 proteins or calgranulins are a group of more than 20 related calcium‐binding proteins. S100A8, S100A9, and S100A12 are all expressed by phagocytes and secreted at sites of inflammation. S100A8/A9 induced autophagy plays a key role in the removal of damaged mitochondria in the setting of apoptosis 170. Although there is no direct evidence that uric acid, the NALP3 inflammasome inducer, is involved in autophagy, a close link exists between autophagy and inflammasome activation 42, 146, 175.
TLRs, NLRs, RLRs, and autophagy
Crosstalk between autophagy and TLRs results in the activation of innate immune responses 176. TLRs promoting autophagy include the TLR2/TLR1 heterodimer 177, TLR3 178, TLR4 108, 178, 179, 180, TLR5 180, TLR6 180, TLR7/8 178, and TLR9 139, 180 in various cell types including macrophages, DCs, and neutrophils. Direct stimulation of TLR7, however, does not lead to induction of autophagy in plasmacytoid DCs 118. Conventional DCs demonstrate high levels of basal autophagy, and afford very little or no induction of autophagy on stimulation with other types of immunological agonists or TLR signals 146, 181. LPS stimulation increases the number of autophagosomes in primary human monocytes, although it fails to induce autophagy in primary mouse macrophages 108, 146. In addition, agonists of mouse TLR7 induce autophagy in RAW264.7 myeloid cell lines and weakly in murine primary bone marrow macrophages 178. These studies suggest that induction of autophagy following TLR stimulation is a cell‐type‐specific response.
TLR‐induced autophagy appears to depend on both MyD88 and TRIF 178, 180 (Fig. 6 ). Moreover, TLR signaling enhances the interaction of MyD88 and TRIF with Beclin 1 and reduces the binding of Beclin 1 to Bcl‐2 180. Interestingly, tumor necrosis factor receptor (TNFR)‐associated factor 6 (TRAF6)‐mediated ubiquitination of Beclin1 amplifies TLR4‐induced autophagy 182 (Fig. 6 ). In contrast, the deubiquitinating enzyme A20 reduces ubiquitination of Beclin 1 and limits the induction of autophagy 182. A similar process may be linked to p62 recognition of microbial targets 91, as TRAF6 binds to p62. In addition, phosphorylation by death‐associated protein kinase (DAPK) of Beclin 1 on Thr 119 in the BH3 domain promotes autophagy 183. Other protein modifications have been identified in Beclin 1‐PI3KC3 complex formation 40. Beclin 1 may play a critical role in TLR‐mediated autophagy by post‐translational modification.
Autophagy may also participate in the regulation of TLR‐mediated inflammation. The most direct way in which autophagy influences inflammation is the breakdown of invading microorganisms such as the Bacille Calmette‐Guerin 89, 184 or the centrally important adapter protein myeloid differentiation factor 88 (MyD88) 185. Phagocytosis is one of the basic tools of innate immunity. TLR signaling in macrophages links the autophagic pathway to phagocytosis 179. LC3‐associated phagocytosis (LAP) is required for the clearance of dead cells 186. Phagocytosis of cells dying through autophagy evokes a pro‐inflammatory response in macrophages 186, 187. Vitamin K3 reduces pancreatic inflammation in acute pancreatitis through inhibition of the autophagic pathway 188. Possible links between these two forms of cellular ‘eating’ represent a new dimension in host defense and inflammation, potentially accessible with novel therapeutics.
In mammalian cells, NLR family members such as NOD1 and NOD2 induce autophagy to control bacterial infection and promote antigen presentation 138, 148. In Drosophila, the NLR‐type PRR PGRP‐LE is crucial for the induction of autophagy of L. monocytogenes 189. Four NLR family members have been described as components of inflammasomes: NALP1, NALP3, NLRC4, and NAIP5 175. Blockade of autophagy by genetic ablation of the autophagy regulators Atg16L1 or Atg7 enables LPS‐dependent inflammasome activation including the processing of pro‐IL‐1β into IL‐1β 146. Various inflammasome stimuli trigger autophagy in macrophages by activating nucleotide exchange (the replacement of GDP by GTP) on RalB 190. Moreover, mitophagy/autophagy blockade leads to the accumulation of damaged, ROS‐generating mitochondria, and this in turn activates the NLRP3 inflammasome 42. Conversely, autophagic proteins regulate NALP3‐dependent inflammation by preserving mitochondrial integrity 191. These findings suggest that autophagy contributes to homeostatic regulation of the inflammasome through the clearance of dysfunctional mitochondria and ROS production. In addition, NLR members may negatively regulate maturation of the autophagosome through interact with Beclin1 192. Interestingly, autophagy not only inhibits IL‐1β release by targeting pro‐IL‐1β for p62‐mediated lysosomal degradation 193, 194 but also promotes IL‐1β release by unconventional secretory pathway 195, suggesting a dual roles of autophagy in regulation of IL‐1β signaling including inflammasome activation.
The RLR family members recognize RNA viruses within the cytosol and induce the expression of potent antiviral factors, such as type I IFN and proinflammatory cytokines. RLRs are negatively regulated by Atg5‐Atg12 119 and can activate autophagy 196. Further analysis reveals that ROS play a key role in autophagy‐mediated RLR signaling 44. In addition, the pathogen receptor CD46 197 and the T‐cell receptor CD40 on myeloid and other cells 198 can activate autophagy with microorganism recognition.
RAGE, AIM2/AIM2‐like receptors, and DAI as DNA receptors and autophagy
RAGE induces cellular inflammation signaling events upon binding of a variety of ligands, such as glycated proteins, amyloid‐β, HMGB1, and S100 proteins 199. Moreover, RAGE can directly bind to dsDNA and dsRNA in vitro 200, suggesting that RAGE may act as both a DNA and RNA receptor (Fig. 4 A). In addition, interaction between RAGE and TLR9 contributes to autoimmune pathogenesis 77, whereas interaction between RAGE and TLR2 limits inflammation 201. RAGE is linked functionally to outcome in several infectious diseases including cancer, diabetes, and Alzheimer's disease 199, 202. Blockade of RAGE suppresses inflammation, tumor growth, and metastasis in various tumor models 203, 204.
We have shown that RAGE positively regulates autophagy (Fig. 6 ). RAGE sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival 205, 206, 207. RAGE‐sustained autophagy is associated with decreased phosphorylation of the mammalian target of rapamycin (mTOR) and increased Beclin 1‐PI3KC3 interaction and ATG12‐ATG5 conjugation 205, 206, 207. In addition, knockdown of RAGE but not TLR4 diminishes HMGB1‐induced autophagy in cancer cells 158. In contrast, TLR4 is required for PAMPs such as LPS to induce autophagy in macrophages 108, suggesting that the induction of autophagy by DAMPs or PAMPs may have alternative receptor‐dependent pathways. Another RAGE ligand, the heterodimer S100A8/A9, also induces autophagy 170, although it is not clear whether RAGE mediates this process directly. RAGE is an important inflammatory mediator that modulates crosstalk between pro‐survival pathways: IL‐6/pSTAT3 and autophagy in pancreatic ductal adenocarcinoma tumor cells and contributes to early pancreatic intraepithelial neoplasia formation 205. RAGE also functions as a phosphatidylserine receptor and assists in the clearance of apoptotic cells by phagocytosis in macrophages 208. Further studies are required to explore the structural basis and protein modification(s) necessary for RAGE‐mediated autophagy and phagocytosis in immunity.
Cytosolic DNA derived from vaccinia virus can be sensed by AIM2 in a complex with Asc and caspase‐1, leading to the processing of pro‐IL‐1β to IL‐1β 54. The AIM2‐like receptors (ALR) including the recently identified IFI16 form a newly defined family activating a unique inflammasome. Their roles in regulation of autophagy are currently undefined. In certain cultured cell lines, AT‐rich dsDNA can also be sensed by the protein DAI, which drives IFN‐β production through activation of the protein kinase TBK‐1 53. The DNA sensor LRRFIP1 mediates the production of type I IFN via a β‐catenin‐dependent pathway 58. Cytosolic DNA‐dependent RNA polymerase III (Pol‐III) is the DNA sensor linking DNA release by pathogenic bacteria and viruses in the host cell cytosol to IFN‐β production and innate immunity 57 (Fig. 4 A). It is unknown whether these cytosolic DNA sensors, including p202 and p204 (IFI16), evoke a robust innate immune response in an autophagy‐dependent pathway.
HMGB1/RAGE‐mediated autophagy and energy metabolism
Autophagy functions in protein and organelle quality control under basal conditions and can be activated in response to stress. The breakdown products derived from autophagy have a dual role, providing substrates for both biosynthesis and energy generation 209. ATP is produced by cellular respiration (either glycolysis or oxidative phosphorylation/OXPHOS) in mitochondria. Mitophagy is important in maintaining mitochondrial homeostasis 41. Suppression of HMGB1 expression in fibroblasts and cancer cells significantly inhibits both OXPHOS and glycolysis, and ATP production is decreased in HMGB1‐deficient cells 167. We demonstrated that HMGB1 modulates the expression of HSPB1. As a cytoskeleton regulator, HSPB1 is critical for dynamic intracellular trafficking during autophagy and mitophagy. Phosphorylation of HSPB1 (both Ser15 and Ser86) is required for HMGB1‐dependent mitochondrial homeostasis. Loss of either HMGB1 or HSPB1 results in a phenotypically similar deficiency in mitophagy typified by mitochondrial fragmentation with decreased aerobic respiration and ATP production. In addition, the Pink1‐Parkin pathway is required for HMGB1/HSPB1 mediated mitophagy. Recent findings indicate that mitochondrial STAT3 sustains the altered glycolytic and OXPHOS activities characteristic of cancer cells 210. We found that RAGE‐mediated autophagy is required for IL‐6‐induced mitochondrial localization and function of STAT3 205. Knockdown of RAGE by shRNA in murine and human pancreatic tumor cell lines significantly decreases IL‐6/STAT3‐mediated mitochondrial respiratory chain complex I activity and ATP production. These findings reveal a novel pathway coupling autophagy and cellular energy metabolism. Further studies involved with assessment of adaptive immune responses induced by chemotherapy 174 or immunotherapy with IL‐2 211 or cytolytic cells 212 suggest that there will be a complex interplay between innate factors such as DAMPs and PAMPs and autophagy.
Conclusion
Multiple positive feedback loops between DAMPs and PAMPs and their overlapping receptors temporally and spatially drive immune regulatory functions (Fig. 6 ). Interestingly, these exogenous and endogenous signal 0s all induce and increase autophagic flux in an ROS‐dependent fashion. Interactions between immune and dying tumor cells likely determine the balance between immunity and tolerance to tumor cells. As a defense mechanism, autophagy limits damage, sustains cell viability, removes intracellular pathogens, and participates in antigen presentation. The organization of the cellular networks linking autophagy to other biologic processes are quite complicated 37. For example, mTOR‐ 213, Beclin 1‐ 214, and Atg5/Atg7‐independent 215 alternative autophagy‐activating pathways have been discovered. Although the role of autophagy in host defense responses has been extensively investigated in vitro, it is now important to more broadly assess its role in vivo. Moreover, the relationship between autophagy and other membrane trafficking systems including phagocytosis, endocytosis, and exocytosis and their relation to host defense remains largely unknown and needs more intensive study. Whether autophagy is required for generation of immunological memory following inflammation initiated by signal 0s and the subsequent recruitment and maturation of inflammatory/immune cells is unknown. Whether autophagy serves as a necessary feedback loop to allow emergence of innate and adaptive immune function and the recall response are similarly unclear. This knowledge is important in an era developing and applying autophagy‐inhibiting drugs. In addition, DAMPs such as HMGB1 may have either a pro‐tumor or anti‐tumor immune effect, depending on the tumor type, established immune suppressor and effector cells, the state of oxidation extracellularly, and the overall nature of the tumor microenvironment. More studies are needed to confirm whether HMGB1, as a DNA binding protein, couples with cytosolic DNA sensors as well as RAGE to regulate autophagy and innate immunity in response to pathogen‐derived DNA, mitochondrial DNA, or nuclear DNA damage. Clearly this evolutionarily ancient system of autophagy is connected to many emergent innate and adaptive immune responses, largely through the response to stress, DAMPs, and ROS.
Acknowledgements
We thank the numerous colleagues in the field of autophagy, who through their animated discussions have helped shape this review. Thoughtful discussions and review of this work with Bennett Van Houten, Guido Kroemer, Douglas Green, Vojo Deretic, Charleen Chu, Russell Salter, and Beth Levine are much appreciated. We also thank Christine Heiner for careful editing of the manuscript. Work in our laboratory is generously supported by the University of Pittsburgh Department of Surgery (Dr. Timothy Billiar), the University of Pittsburgh Cancer Institute (Dr. Nancy Davidson), and the National Institutes of Health via a grant from the National Cancer Institute (1P01 CA 101944 to Michael T. Lotze, MD). The authors have no conflicts of interest to declare.
Immunological Reviews 2012. Vol.249: 158–175 22889221
References
- 1. Janeway CA Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol 1989;54:1–13. [DOI] [PubMed] [Google Scholar]
- 2. Janeway C. Immunogenicity signals 1,2,3… and 0. Immunol Today 1989;10:283–286. [DOI] [PubMed] [Google Scholar]
- 3. Akira S, Hemmi H. Recognition of pathogen‐associated molecular patterns by TLR family. Immunol Lett 2003;85:85–95. [DOI] [PubMed] [Google Scholar]
- 4. Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol 1994;12:991–1045. [DOI] [PubMed] [Google Scholar]
- 5. Rubartelli A, Lotze MT. Inside, outside, upside down: damage‐associated molecular‐pattern molecules (DAMPs) and redox. Trends Immunol 2007;28:429–436. [DOI] [PubMed] [Google Scholar]
- 6. Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol 2007;81:1–5. [DOI] [PubMed] [Google Scholar]
- 7. Lotze MT, et al. The grateful dead: damage‐associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol Rev 2007;220:60–81. [DOI] [PubMed] [Google Scholar]
- 8. Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol 2010;12:814–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010;140:313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell 2010;40:280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liang XH, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999;402:672–676. [DOI] [PubMed] [Google Scholar]
- 12. Liang C, et al. Autophagic and tumour suppressor activity of a novel Beclin1‐binding protein UVRAG. Nat Cell Biol 2006;8:688–699. [DOI] [PubMed] [Google Scholar]
- 13. Yang S, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev 2011;25:717–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Altman BJ, et al. Autophagy is essential to suppress cell stress and to allow BCR‐Abl‐mediated leukemogenesis. Oncogene 2011;30:1855–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 2011;469:323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Munz C. Enhancing immunity through autophagy. Annu Rev Immunol 2009;27:423–449. [DOI] [PubMed] [Google Scholar]
- 17. Delgado M, et al. Autophagy and pattern recognition receptors in innate immunity. Immunol Rev 2009;227:189–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Virgin HW, Levine B. Autophagy genes in immunity. Nat Immunol 2009;10:461–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol 2007;7:767–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Deretic V. Autophagy in innate and adaptive immunity. Trends Immunol 2005;26:523–528. [DOI] [PubMed] [Google Scholar]
- 21. Saitoh T, Akira S. Regulation of innate immune responses by autophagy‐related proteins. J Cell Biol 2010;189:925–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Deretic V. Autophagy as an immune defense mechanism. Curr Opin Immunol 2006;18:375–382. [DOI] [PubMed] [Google Scholar]
- 23. Kuballa P, Nolte WM, Castoreno AB, Xavier RJ. Autophagy and the immune system. Annu Rev Immunol 2012;30:611–646. [DOI] [PubMed] [Google Scholar]
- 24. Deretic V. Autophagy in immunity and cell‐autonomous defense against intracellular microbes. Immunol Rev 2011;240:92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Deretic V, Levine B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe 2009;5:527–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Levine B. Eating oneself and uninvited guests: autophagy‐related pathways in cellular defense. Cell 2005;120:159–162. [DOI] [PubMed] [Google Scholar]
- 27. Blott EJ, Griffiths GM. Secretory lysosomes. Nat Rev Mol Cell Biol 2002;3:122–131. [DOI] [PubMed] [Google Scholar]
- 28. Ding WX, et al. Linking of autophagy to ubiquitin‐proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol 2007;171:513–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin‐proteasome pathway substrates. Mol Cell 2009;33:517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pandey UB, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007;447:859–863. [DOI] [PubMed] [Google Scholar]
- 31. Du Y, et al. An insight into the mechanistic role of p53‐mediated autophagy induction in response to proteasomal inhibition‐induced neurotoxicity. Autophagy 2009;5:663–675. [DOI] [PubMed] [Google Scholar]
- 32. Chen Y, Klionsky DJ. The regulation of autophagy – unanswered questions. J Cell Sci 2011;124:161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Duran JM, Anjard C, Stefan C, Loomis WF, Malhotra V. Unconventional secretion of Acb1 is mediated by autophagosomes. J Cell Biol 2010;188:527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schweers RL, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USA 2007;104:19500–19505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre‐autophagosomal structures. Nat Cell Biol 2010;12:747–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Klionsky DJ, et al. A unified nomenclature for yeast autophagy‐related genes. Dev Cell 2003;5:539–545. [DOI] [PubMed] [Google Scholar]
- 37. Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature 2010;466:68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. He C, Baba M, Klionsky DJ. Double duty of Atg9 self‐association in autophagosome biogenesis. Autophagy 2009;5:385–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. He C, Levine B. The Beclin 1 interactome. Curr Opin Cell Biol 2010;22:140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 2011;18:571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol 2011;12:9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011;469:221–225. [DOI] [PubMed] [Google Scholar]
- 43. Huang J, et al. Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci USA 2009;106:6226–6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tal MC, Sasai M, Lee HK, Yordy B, Shadel GS, Iwasaki A. Absence of autophagy results in reactive oxygen species‐dependent amplification of RLR signaling. Proc Natl Acad Sci USA 2009;106:2770–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self‐eating and self‐killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007;8:741–752. [DOI] [PubMed] [Google Scholar]
- 46. Qu X, et al. Autophagy gene‐dependent clearance of apoptotic cells during embryonic development. Cell 2007;128:931–946. [DOI] [PubMed] [Google Scholar]
- 47. Shimizu S, et al. Role of Bcl‐2 family proteins in a non‐apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol 2004;6:1221–1228. [DOI] [PubMed] [Google Scholar]
- 48. Janeway CA Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol 2002;20:197–216. [DOI] [PubMed] [Google Scholar]
- 49. Creagh EM, O'Neill LA. TLRs, NLRs and RLRs: a trinity of pathogen sensors that co‐operate in innate immunity. Trends Immunol 2006;27:352–357. [DOI] [PubMed] [Google Scholar]
- 50. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell 2006;124:783–801. [DOI] [PubMed] [Google Scholar]
- 51. Poltorak A, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 1998;282:2085–2088. [DOI] [PubMed] [Google Scholar]
- 52. Hemmi H, et al. A Toll‐like receptor recognizes bacterial DNA. Nature 2000;408:740–745. [DOI] [PubMed] [Google Scholar]
- 53. Takaoka A, et al. DAI (DLM‐1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007;448:501–505. [DOI] [PubMed] [Google Scholar]
- 54. Hornung V, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase‐1‐activating inflammasome with ASC. Nature 2009;458:514–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Roberts TL, et al. HIN‐200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 2009;323:1057–1060. [DOI] [PubMed] [Google Scholar]
- 56. Unterholzner L, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol 2010;11:997–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chiu YH, Macmillan JB, Chen ZJ. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG‐I pathway. Cell 2009;138:576–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yang P, et al. The cytosolic nucleic acid sensor LRRFIP1 mediates the production of type I interferon via a beta‐catenin‐dependent pathway. Nat Immunol 2010;11:487–494. [DOI] [PubMed] [Google Scholar]
- 59. Salio M, Cerundolo V. Viral immunity: cross‐priming with the help of TLR3. Curr Biol 2005;15:R336–339. [DOI] [PubMed] [Google Scholar]
- 60. Pichlmair A, et al. RIG‐I‐mediated antiviral responses to single‐stranded RNA bearing 5′‐phosphates. Science 2006;314:997–1001. [DOI] [PubMed] [Google Scholar]
- 61. Kato H, et al. Length‐dependent recognition of double‐stranded ribonucleic acids by retinoic acid‐inducible gene‐I and melanoma differentiation‐associated gene 5. J Exp Med 2008;205:1601–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Komuro A, Horvath CM. RNA‐ and virus‐independent inhibition of antiviral signaling by RNA helicase LGP2. J Virol 2006;80:12332–12342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Matzinger P. The danger model: a renewed sense of self. Science 2002;296:301–305. [DOI] [PubMed] [Google Scholar]
- 64. Matzinger P. Friendly and dangerous signals: is the tissue in control? Nat Immunol 2007;8:11–13. [DOI] [PubMed] [Google Scholar]
- 65. Seong SY, Matzinger P. Hydrophobicity: an ancient damage‐associated molecular pattern that initiates innate immune responses. Nat Rev Immunol 2004;4:469–478. [DOI] [PubMed] [Google Scholar]
- 66. Zhang Q, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010;464:104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wang H, et al. HMG‐1 as a late mediator of endotoxin lethality in mice. Science 1999;285:248–251. [DOI] [PubMed] [Google Scholar]
- 68. Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002;418:191–195. [DOI] [PubMed] [Google Scholar]
- 69. Tsung A, et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia‐reperfusion. J Exp Med 2005;201:1135–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lotze MT, Tracey KJ. High‐mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol 2005;5:331–342. [DOI] [PubMed] [Google Scholar]
- 71. Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol 2011;29:139–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tang D, Kang R, Zeh HJ 3rd, Lotze MT. High‐mobility group box 1, oxidative stress, and disease. Antioxid Redox Signal 2011;14:1315–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chen GY, Tang J, Zheng P, Liu Y. CD24 and Siglec‐10 selectively repress tissue damage‐induced immune responses. Science 2009;323:1722–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Fan J, et al. Hemorrhagic shock induces NAD(P)H oxidase activation in neutrophils: role of HMGB1‐TLR4 signaling. J Immunol 2007;178:6573–6580. [DOI] [PubMed] [Google Scholar]
- 75. Yang H, et al. A critical cysteine is required for HMGB1 binding to Toll‐like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci USA 2010;107:11942–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kazama H, Ricci JE, Herndon JM, Hoppe G, Green DR, Ferguson TA. Induction of immunological tolerance by apoptotic cells requires caspase‐dependent oxidation of high‐mobility group box‐1 protein. Immunity 2008;29:21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tian J, et al. Toll‐like receptor 9‐dependent activation by DNA‐containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol 2007;8:487–496. [DOI] [PubMed] [Google Scholar]
- 78. Urbonaviciute V, et al. Induction of inflammatory and immune responses by HMGB1‐nucleosome complexes: implications for the pathogenesis of SLE. J Exp Med 2008;205:3007–3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Yanai H, et al. HMGB proteins function as universal sentinels for nucleic‐acid‐mediated innate immune responses. Nature 2009;462:99–103. [DOI] [PubMed] [Google Scholar]
- 80. Pizarro‐Cerda J, Moreno E, Sanguedolce V, Mege JL, Gorvel JP. Virulent Brucella abortus prevents lysosome fusion and is distributed within autophagosome‐like compartments. Infect Immun 1998;66:2387–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Starr T, et al. Selective subversion of autophagy complexes facilitates completion of the Brucella intracellular cycle. Cell Host Microbe 2012;11:33–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Beron W, Gutierrez MG, Rabinovitch M, Colombo MI. Coxiella burnetii localizes in a Rab7‐labeled compartment with autophagic characteristics. Infect Immun 2002;70:5816–5821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Dorn BR, Dunn WA, . Jr , Progulske‐Fox A. Porphyromonas gingivalis traffics to autophagosomes in human coronary artery endothelial cells. Infect Immun 2001;69:5698–5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Birmingham CL, Smith AC, Bakowski MA, Yoshimori T, Brumell JH. Autophagy controls Salmonella infection in response to damage to the Salmonella‐containing vacuole. J Biol Chem 2006;281:11374–11383. [DOI] [PubMed] [Google Scholar]
- 85. Al‐Younes HM, Brinkmann V, Meyer TF. Interaction of Chlamydia trachomatis serovar L2 with the host autophagic pathway. Infect Immun 2004;72:4751–4762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Birmingham CL, et al. Listeria monocytogenes evades killing by autophagy during colonization of host cells. Autophagy 2007;3:442–451. [DOI] [PubMed] [Google Scholar]
- 87. Nakagawa I, et al. Autophagy defends cells against invading group A Streptococcus. Science 2004;306:1037–1040. [DOI] [PubMed] [Google Scholar]
- 88. Nozawa T, Aikawa C, Goda A, Maruyama F, Hamada S, Nakagawa I. The small GTPases Rab9A and Rab23 function at distinct steps in autophagy during Group A Streptococcus infection. Cell Microbiol 2012; doi: 10.1111/j.1462-5822.2012.01792.x. [DOI] [PubMed] [Google Scholar]
- 89. Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004;119:753–766. [DOI] [PubMed] [Google Scholar]
- 90. Williams RA, Tetley L, Mottram JC, Coombs GH. Cysteine peptidases CPA and CPB are vital for autophagy and differentiation in Leishmania mexicana. Mol Microbiol 2006;61:655–674. [DOI] [PubMed] [Google Scholar]
- 91. Dupont N, et al. Shigella phagocytic vacuolar membrane remnants participate in the cellular response to pathogen invasion and are regulated by autophagy. Cell Host Microbe 2009;6:137–149. [DOI] [PubMed] [Google Scholar]
- 92. Suhy DA, Giddings TH Jr, Kirkegaard K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy‐like origin for virus‐induced vesicles. J Virol 2000;74:8953–8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Talloczy Z, et al. Regulation of starvation‐ and virus‐induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci USA 2002;99:190–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Orvedahl A, et al. HSV‐1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 2007;1:23–35. [DOI] [PubMed] [Google Scholar]
- 95. Orvedahl A, MacPherson S, Sumpter R Jr, Talloczy Z, Zou Z, Levine B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 2010;7:115–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Heaton NS, Randall G. Dengue virus‐induced autophagy regulates lipid metabolism. Cell Host Microbe 2010;8:422–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Prentice E, Jerome WG, Yoshimori T, Mizushima N, Denison MR. Coronavirus replication complex formation utilizes components of cellular autophagy. J Biol Chem 2004;279:10136–10141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Deretic V. Multiple regulatory and effector roles of autophagy in immunity. Curr Opin Immunol 2009;21:53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Deretic V. Autophagy in infection. Curr Opin Cell Biol 2010;22:252–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Singh R, et al. Autophagy regulates lipid metabolism. Nature 2009;458:1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Pankiv S, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007;282:24131–24145. [DOI] [PubMed] [Google Scholar]
- 102. Ponpuak M, et al. Delivery of cytosolic components by autophagic adaptor protein p62 endows autophagosomes with unique antimicrobial properties. Immunity 2010;32:329–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Zheng YT, Shahnazari S, Brech A, Lamark T, Johansen T, Brumell JH. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J Immunol 2009;183:5909–5916. [DOI] [PubMed] [Google Scholar]
- 104. Thurston TL, Ryzhakov G, Bloor S, von N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin‐coated bacteria. Nat Immunol 2009;10:1215–1221. [DOI] [PubMed] [Google Scholar]
- 105. Kirkin V, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell 2009;33:505–516. [DOI] [PubMed] [Google Scholar]
- 106. Sandoval H, et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008;454:232–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Shahnazari S, et al. A diacylglycerol‐dependent signaling pathway contributes to regulation of antibacterial autophagy. Cell Host Microbe 2010;8:137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll‐like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 2007;27:135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Djavaheri‐Mergny M, et al. NF‐kappaB activation represses tumor necrosis factor‐alpha‐induced autophagy. J Biol Chem 2006;281:30373–30382. [DOI] [PubMed] [Google Scholar]
- 110. Copetti T, Bertoli C, Dalla E, Demarchi F, Schneider C. p65/RelA modulates BECN1 transcription and autophagy. Mol Cell Biol 2009;29:2594–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Criollo A, et al. The IKK complex contributes to the induction of autophagy. EMBO J 2010;29:619–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Comb WC, Cogswell P, Sitcheran R, Baldwin AS. IKK‐dependent, NF‐kappaB‐independent control of autophagic gene expression. Oncogene 2011;30:1727–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Criollo A, et al. Inhibition of autophagy by TAB 2 and TAB 3. EMBO J 2011;30:4908–4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Criollo A, et al. Autophagy is required for the activation of NFkappaB. Cell Cycle 2012;11:194–199. [DOI] [PubMed] [Google Scholar]
- 115. Fujita K, Maeda D, Xiao Q, Srinivasula SM. Nrf2‐mediated induction of p62 controls Toll‐like receptor‐4‐driven aggresome‐like induced structure formation and autophagic degradation. Proc Natl Acad Sci USA 2011;108:1427–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Komatsu M, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 2010;12:213–223. [DOI] [PubMed] [Google Scholar]
- 117. Sumpter R Jr, Levine B. Selective autophagy and viruses. Autophagy 2011;7:260–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. Autophagy‐dependent viral recognition by plasmacytoid dendritic cells. Science 2007;315:1398–1401. [DOI] [PubMed] [Google Scholar]
- 119. Jounai N, et al. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc Natl Acad Sci USA 2007;104:14050–14055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Zhao Z, et al. Autophagosome‐independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe 2008;4:458–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Feng CG, et al. The immunity‐related GTPase Irgm1 promotes the expansion of activated CD4+ T cell populations by preventing interferon‐gamma‐induced cell death. Nat Immunol 2008;9:1279–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 2006;313:1438–1441. [DOI] [PubMed] [Google Scholar]
- 123. Singh SB, et al. Human IRGM regulates autophagy and cell‐autonomous immunity functions through mitochondria. Nat Cell Biol 2010;12:1154–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Gregoire IP, et al. IRGM is a common target of RNA viruses that subvert the autophagy network. PLoS Pathog 2011;7:e1002422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Yang CS, et al. Autophagy protein Rubicon mediates phagocytic NADPH oxidase activation in response to microbial infection or TLR stimulation. Cell Host Microbe 2012;11:264–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Yang CS, et al. The autophagy regulator rubicon is a feedback Inhibitor of CARD9‐Mediated Host Innate Immunity. Cell Host Microbe 2012;11:277–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Dreux M, Gastaminza P, Wieland SF, Chisari FV. The autophagy machinery is required to initiate hepatitis C virus replication. Proc Natl Acad Sci USA 2009;106:14046–14051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Jackson WT, et al. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol 2005;3:e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Kyei GB, et al. Autophagy pathway intersects with HIV‐1 biosynthesis and regulates viral yields in macrophages. J Cell Biol 2009;186:255–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Espert L, et al. Differential role of autophagy in CD4 T cells and macrophages during X4 and R5 HIV‐1 infection. PLoS ONE 2009;4:e5787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Van GrolJ, Subauste C, Andrade RM, Fujinaga K, Nelson J, Subauste CS. HIV‐1 inhibits autophagy in bystander macrophage/monocytic cells through Src‐Akt and STAT3. PLoS ONE 2010;5:e11733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Nedjic J, Aichinger M, Emmerich J, Mizushima N, Klein L. Autophagy in thymic epithelium shapes the T‐cell repertoire and is essential for tolerance. Nature 2008;455:396–400. [DOI] [PubMed] [Google Scholar]
- 133. Paludan C, et al. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 2005;307:593–596. [DOI] [PubMed] [Google Scholar]
- 134. English L, et al. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV‐1 infection. Nat Immunol 2009;10:480–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Blanchet FP, et al. Human immunodeficiency virus‐1 inhibition of immunoamphisomes in dendritic cells impairs early innate and adaptive immune responses. Immunity 2010;32:654–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Lee HK, et al. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity 2010;32:227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Ireland JM, Unanue ER. Autophagy in antigen‐presenting cells results in presentation of citrullinated peptides to CD4 T cells. J Exp Med 2011;208:2625–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Cooney R, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med 2010;16:90–97. [DOI] [PubMed] [Google Scholar]
- 139. Chaturvedi A, Dorward D, Pierce SK. The B cell receptor governs the subcellular location of Toll‐like receptor 9 leading to hyperresponses to DNA‐containing antigens. Immunity 2008;28:799–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Latz E, et al. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol 2004;5:190–198. [DOI] [PubMed] [Google Scholar]
- 141. Chockalingam A, Brooks JC, Cameron JL, Blum LK, Leifer CA. TLR9 traffics through the Golgi complex to localize to endolysosomes and respond to CpG DNA. Immunol Cell Biol 2009;87:209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Saitoh T, et al. Atg9a controls dsDNA‐driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci USA 2009;106:20842–20846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Cadwell K, Stappenbeck TS, Virgin HW. Role of autophagy and autophagy genes in inflammatory bowel disease. Curr Top Microbiol Immunol 2009;335:141–167. [DOI] [PubMed] [Google Scholar]
- 144. Barrett JC, et al. Genome‐wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet 2008;40:955–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Cadwell K, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008;456:259–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Saitoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin‐induced IL‐1beta production. Nature 2008;456:264–268. [DOI] [PubMed] [Google Scholar]
- 147. Cadwell K, et al. Virus‐plus‐susceptibility gene interaction determines Crohn's disease gene Atg16L1 phenotypes in intestine. Cell 2010;141:1135–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Travassos LH, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol 2010;11:55–62. [DOI] [PubMed] [Google Scholar]
- 149. Plantinga TS, et al. Crohn's disease‐associated ATG16L1 polymorphism modulates pro‐inflammatory cytokine responses selectively upon activation of NOD2. Gut 2011;60:1229–1235. [DOI] [PubMed] [Google Scholar]
- 150. Luciani A, et al. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS‐mediated autophagy inhibition. Nat Cell Biol 2010;12:863–875. [DOI] [PubMed] [Google Scholar]
- 151. Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab 2010;11:467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Watanabe E, et al. Sepsis induces extensive autophagic vacuolization in hepatocytes: a clinical and laboratory‐based study. Lab Invest 2009;89:549–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Ivanov S, et al. A novel role for HMGB1 in TLR9‐mediated inflammatory responses to CpG‐DNA. Blood 2007;110:1970–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Tang D, et al. Hydrogen peroxide stimulates macrophages and monocytes to actively release HMGB1. J Leukoc Biol 2007;81:741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Qin S, et al. Role of HMGB1 in apoptosis‐mediated sepsis lethality. J Exp Med 2006;203:1637–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Chen LC, Yeh TM, Wu HN, Lin YY, Shyu HW. Dengue virus infection induces passive release of high mobility group box 1 protein by epithelial cells. J Infect 2008;56:143–150. [DOI] [PubMed] [Google Scholar]
- 157. Bell CW, Jiang W, Reich CF, 3rd, Pisetsky DS. The extracellular release of HMGB1 during apoptotic cell death. Am J Physiol Cell Physiol 2006;291:C1318–1325. [DOI] [PubMed] [Google Scholar]
- 158. Tang D, et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 2010;29:5299–5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Tang D, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol 2010;190:881–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Tang D, Kang R, Livesey KM, Zeh HJ, Lotze MT. High mobility group box 1 (HMGB1) activates an autophagic response to oxidative stress. Antioxid Redox Signal 2011;15:2185–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Liu L, et al. HMGB1‐induced autophagy promotes chemotherapy resistance in leukemia cells. Leukemia 2011;25:23–31. [DOI] [PubMed] [Google Scholar]
- 162. Huang J, et al. HMGB1 promotes drug resistance in osteosarcoma. Cancer Res 2012;72:230–238. [DOI] [PubMed] [Google Scholar]
- 163. Livesey K, et al. p53/HMGB1 complexes regulate autophagy and apoptosis. Cancer Res 2012;72:1996–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Bonaldi T, et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J 2003;22:5551–5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Lange SS, Mitchell DL, Vasquez KM. High mobility group protein B1 enhances DNA repair and chromatin modification after DNA damage. Proc Natl Acad Sci USA 2008;105:10320–10325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Robert T, et al. HDACs link the DNA damage response, processing of double‐strand breaks and autophagy. Nature 2011;471:74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Tang D, et al. High‐mobility group box 1 is essential for mitochondrial quality control. Cell Metab 2011;13:701–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Bianchi ME. HMGB1 loves company. J Leukoc Biol 2009;86:573–576. [DOI] [PubMed] [Google Scholar]
- 169. Biswas D, Qureshi OS, Lee WY, Croudace JE, Mura M, Lammas DA. ATP‐induced autophagy is associated with rapid killing of intracellular mycobacteria within human monocytes/macrophages. BMC Immunol 2008;9:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Ghavami S, et al. S100A8/A9 induces autophagy and apoptosis via ROS‐mediated cross‐talk between mitochondria and lysosomes that involves BNIP3. Cell Res 2010;20:314–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Takenouchi T, et al. The activation of P2X7 receptor impairs lysosomal functions and stimulates the release of autophagolysosomes in microglial cells. J Immunol 2009;182:2051–2062. [DOI] [PubMed] [Google Scholar]
- 172. Michaud M, et al. Autophagy‐dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011;334:1573–1577. [DOI] [PubMed] [Google Scholar]
- 173. Noman MZ, et al. Blocking hypoxia‐induced autophagy in tumors restores cytotoxic T‐cell activity and promotes regression. Cancer Res 2011;71:5976–5986. [DOI] [PubMed] [Google Scholar]
- 174. Weiner LM, Lotze MT. Tumor‐cell death, autophagy, and immunity. N Engl J Med 2012;366:1156–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Schroder K, Tschopp J. The inflammasomes. Cell 2010;140:821–832. [DOI] [PubMed] [Google Scholar]
- 176. Delgado MA, Deretic V. Toll‐like receptors in control of immunological autophagy. Cell Death Differ 2009;16:976–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Shin DM, et al. Mycobacterial lipoprotein activates autophagy via TLR2/1/CD14 and a functional vitamin D receptor signalling. Cell Microbiol 2010;12:1648–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll‐like receptors control autophagy. EMBO J 2008;27:1110–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Sanjuan MA, et al. Toll‐like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007;450:1253–1257. [DOI] [PubMed] [Google Scholar]
- 180. Shi CS, Kehrl JH. MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J Biol Chem 2008;283:33175–33182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Schmid D, Pypaert M, Munz C. Antigen‐loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity 2007;26:79–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Shi CS, Kehrl JH. TRAF6 and A20 regulate lysine 63‐linked ubiquitination of Beclin‐1 to control TLR4‐induced autophagy. Sci Signal 2010;3:ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Zalckvar E, et al. DAP‐kinase‐mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl‐XL and induction of autophagy. EMBO Rep 2009;10:285–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Jagannath C, Lindsey DR, Dhandayuthapani S, Xu Y, Hunter RL Jr, Eissa NT. Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nat Med 2009;15:267–276. [DOI] [PubMed] [Google Scholar]
- 185. Puneet P, et al. The helminth product ES‐62 protects against septic shock via Toll‐like receptor 4‐dependent autophagosomal degradation of the adaptor MyD88. Nat Immunol 2011;12:344–351. [DOI] [PubMed] [Google Scholar]
- 186. Martinez J, et al. Microtubule‐associated protein 1 light chain 3 alpha (LC3)‐associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci USA 2011;108:17396–17401. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 187. Petrovski G, et al. Clearance of dying autophagic cells of different origin by professional and non‐professional phagocytes. Cell Death Differ 2007;14:1117–1128. [DOI] [PubMed] [Google Scholar]
- 188. Chinzei R, et al. Vitamin K3 attenuates cerulein‐induced acute pancreatitis through inhibition of the autophagic pathway. Pancreas 2011;40:84–94. [DOI] [PubMed] [Google Scholar]
- 189. Yano T, et al. Autophagic control of listeria through intracellular innate immune recognition in drosophila. Nat Immunol 2008;9:908–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Shi CS, et al. Activation of autophagy by inflammatory signals limits IL‐1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol 2012;13:255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Nakahira K, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 2011;12:222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Jounai N, Kobiyama K, Shiina M, Ogata K, Ishii KJ, Takeshita F. NLRP4 negatively regulates autophagic processes through an association with beclin1. J Immunol 2011;186:1646–1655. [DOI] [PubMed] [Google Scholar]
- 193. Lee J, et al. Autophagy suppresses interleukin‐1beta (IL‐1beta) signaling by activation of p62 degradation via lysosomal and proteasomal pathways. J Biol Chem 2012;287:4033–4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Harris J, et al. Autophagy controls IL‐1beta secretion by targeting pro‐IL‐1beta for degradation. J Biol Chem 2011;286:9587–9597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy‐based unconventional secretory pathway for extracellular delivery of IL‐1beta. EMBO J 2011;30:4701–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Tormo D, et al. Targeted activation of innate immunity for therapeutic induction of autophagy and apoptosis in melanoma cells. Cancer Cell 2009;16:103–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Joubert PE, et al. Autophagy induction by the pathogen receptor CD46. Cell Host Microbe 2009;6:354–366. [DOI] [PubMed] [Google Scholar]
- 198. Andrade RM, Wessendarp M, Gubbels MJ, Striepen B, Subauste CS. CD40 induces macrophage anti‐Toxoplasma gondii activity by triggering autophagy‐dependent fusion of pathogen‐containing vacuoles and lysosomes. J Clin Invest 2006;116:2366–2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Sparvero LJ, et al. RAGE (receptor for advanced glycation endproducts), RAGE ligands, and their role in cancer and inflammation. J Transl Med 2009;7:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Park H, Boyington JC. The 1.5 A crystal structure of human receptor for advanced glycation endproducts (rage) ectodomains reveals unique features determining ligand binding. J Biol Chem 2011;285:40762–40770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Sorci G, et al. The danger signal S100B integrates pathogen‐ and danger‐sensing pathways to restrain inflammation. PLoS Pathog 2011;7:e1001315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol 2010;28:367–388. [DOI] [PubMed] [Google Scholar]
- 203. Taguchi A, et al. Blockade of RAGE‐amphoterin signalling suppresses tumour growth and metastases. Nature 2000;405:354–360. [DOI] [PubMed] [Google Scholar]
- 204. Gebhardt C, et al. RAGE signaling sustains inflammation and promotes tumor development. J Exp Med 2008;205:275–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Kang R, et al. The expression of the receptor for advanced glycation endproducts (RAGE) is permissive for early pancreatic neoplasia. Proc Natl Acad Sci USA 2012;109:7031–7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Kang R, Tang D, Livesey KM, Schapiro N, Lotze MT, Zeh HJ. The receptor for advanced glycation end‐products (RAGE) protects pancreatic tumor cells against oxidative injury. Antioxid Redox Signal 2011;15:2175–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Kang R, et al. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ 2010;17:666–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. He M, et al. Receptor for advanced glycation end products binds to phosphatidylserine and assists in the clearance of apoptotic cells. EMBO Rep 2011;12:358–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Rabinowitz JD, White E. Autophagy and metabolism. Science 2010;330:1344–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Wegrzyn J, et al. Function of mitochondrial Stat3 in cellular respiration. Science 2009;323:793–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Liang X, et al. Inhibiting autophagy during interleukin 2 immunotherapy promotes long term tumor regression. Cancer Res 2012;72:2791–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Buchser WJ, Laskow TC, Pavlik PJ, Lin HM, Lotze MT. Cell‐mediated autophagy promotes cancer cell survival. Cancer Res 2012;72:2970–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Yamamoto A, Cremona ML, Rothman JE. Autophagy‐mediated clearance of huntingtin aggregates triggered by the insulin‐signaling pathway. J Cell Biol 2006;172:719–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Scarlatti F, Maffei R, Beau I, Codogno P, Ghidoni R. Role of non‐canonical Beclin 1‐independent autophagy in cell death induced by resveratrol in human breast cancer cells. Cell Death Differ 2008;15:1318–1329. [DOI] [PubMed] [Google Scholar]
- 215. Nishida Y, et al. Discovery of Atg5/Atg7‐independent alternative macroautophagy. Nature 2009;461:654–658. [DOI] [PubMed] [Google Scholar]