

Abstract
Objective:
To report a series of 11 patients on the severe end of the spectrum of ryanodine receptor 1 (RYR1) gene–related myopathy, in order to expand the clinical, histologic, and genetic heterogeneity associated with this group of patients.
Methods:
Eleven patients evaluated in the neonatal period with severe neonatal-onset RYR1-associated myopathy confirmed by genetic testing were ascertained. Clinical features, molecular testing results, muscle imaging, and muscle histology are reviewed.
Results:
Clinical features associated with the severe neonatal presentation of RYR1-associated myopathy included decreased fetal movement, hypotonia, poor feeding, respiratory involvement, arthrogryposis, and ophthalmoplegia in 3 patients, and femur fractures or hip dislocation at birth. Four patients had dominant RYR1 mutations, and 7 had recessive RYR1 mutations. One patient had a cleft palate, and another a congenital rigid spine phenotype—findings not previously described in the literature in patients with early-onset RYR1 mutations. Six patients who underwent muscle ultrasound showed relative sparing of the rectus femoris muscle. Histologically, all patients with dominant mutations had classic central cores on muscle biopsy. Patients with recessive mutations showed great histologic heterogeneity, including fibrosis, variation in fiber size, skewed fiber typing, very small fibers, and nuclear internalization with or without ill-defined cores.
Conclusions:
This series confirms and expands the clinical and histologic variability associated with severe congenital RYR1-associated myopathy. Both dominant and recessive mutations of the RYR1 gene can result in a severe neonatal-onset phenotype, but more clinical and histologic heterogeneity has been seen in those with recessive RYR1 gene mutations. Central cores are not obligatory histologic features in recessive RYR1 mutations. Sparing of the rectus femoris muscle on imaging should prompt evaluation for RYR1-associated myopathy in the appropriate clinical context.
Central core disease (CCD) is a form of congenital myopathy due mostly to dominant, and occasionally to recessive, mutations in the skeletal muscle ryanodine receptor 1 (RYR1) gene, characterized clinically by a static to slowly progressive course beginning with congenital hypotonia.1–3 RYR1 is a 106 exon gene that encodes the skeletal muscle ryanodine receptor, an intracellular calcium-release channel that is crucial to excitation-contraction coupling in muscle.1,4–6
Patients with classic CCD typically present in infancy or early childhood with mild to moderate hypotonia, motor-development delay, proximal muscle weakness, and occasionally with congenital hip dislocation.1,4 A subset of patients with RYR1-associated myopathy have a more severe or even lethal neonatal form of the disorder, with features such as decreased fetal movement, polyhydramnios, arthrogryposis, kyphoscoliosis, or respiratory distress.1,4,7–9
Typical histologic findings on muscle biopsy include well-defined cores of varying size, centrally or peripherally located, or uneven areas of clearing with oxidative enzyme stains due to absence or low number of mitochondria in those regions.1 Other reported histologic findings include type 1 fiber predominance and fiber-type disproportion, multi-minicores, and centrally placed nuclei.1,10–12 The degree of pathologic changes may vary based on sampling site and age of the patient; very young cases in particular may not show clear-cut cores, suggesting an age-related development of the cores.1,2,13
Herein, we present a series of patients with a severe neonatal RYR1-associated myopathy caused by both dominant and recessive mutations of the RYR1 gene to broaden the known clinicopathologic spectrum in this disorder.
METHODS
Clinical.
Eleven patients with confirmed RYR1 mutations and neonatal or infantile onset of disease were included; 10 were seen at the Children's Hospital of Philadelphia from 2003 to present and 1 patient was identified on remote consultation and review of his biopsy.
Standard protocol approvals, registrations, and patient consents.
This study was reviewed by the local institutional review board and was determined not to involve human subjects research and therefore did not require further approval. Written consent to disclose for recognizable persons in photographs was obtained from patients' parents or guardians.
Genetic analysis.
Full gene sequencing of RYR1 was performed at Prevention Genetics laboratory in Marshfield, WI for patients 1–5 and 11; partial gene analysis of N- and C-terminal exons was done in patient 7. UCH, Grenoble, France performed testing in patients 8 and 9 by full sequencing of cDNA transcript from muscle, and in patients 6 and 10 by targeted analysis of N- and C-terminal exons.
Muscle imaging.
Muscle ultrasound was performed in 6 patients on a Sonoline Adara or Acuson S2000 machine (Siemens, Erlangen, Germany) using linear array transducers VFX13-5 at 12 MHz or 18L6 HD at 15 MHz, respectively, operated by an experienced clinician (C.G.B.).
Pathology.
Muscle biopsy was performed in all 11 patients at ages ranging from 3 weeks to 4 years. Patient 11 had a repeat biopsy at age 16 years. Muscle biopsies were snap-frozen in isopentane cooled in liquid N2. Routine stains included hematoxylin & eosin, modified Gömöri trichrome, picrosirius, Oil Red O, periodic acid-Schiff, alkaline phosphatase, acid phosphatase, adenosine triphosphatase at pH 9.4 and 4.3, nicotinamide adenine dinucleotide dehydrogenase (NADH)–tetrazolium reductase (TR), succinic dehydrogenase (SDH), cytochrome oxidase (COX), and combined COX/SDH.14 Immunohistochemistry for fast and slow myosin was performed in 7 cases and for developmental, neonatal myosin and neural cell adhesion molecule (N-CAM) in 7 cases (patients 1, 3, 4, 6, 7, 10, and 11).
RESULTS
Clinical presentation.
Nine unrelated patients and 1 pair of siblings from nonconsanguineous families were included, all presenting with symptoms in the neonatal period. Nine had symptoms noted in utero, including decreased perceived fetal movements, polyhydramnios, and intrauterine growth restriction (table 1). Features present at birth (figure 1) included hypotonia (n = 11), feeding difficulties (n = 8), arthrogryposis manifesting as adducted thumb (figure 1, E and F), finger flexor contractures, foot contractures (n = 6), and other orthopedic complications such as hip dislocations or long-bone fractures (n = 4). Congenital or early-onset kyphoscoliosis and early rapid progression of scoliosis was seen in 5 patients (figure 1, G–I). Respiratory function was moderately to severely impaired in 5 patients, ranging from mild hypoxia at birth to an inability to wean from the ventilator (figure 1B). One patient died at 24 days of life after withdrawal of support because of inability to wean, and 1 patient died at 4 years of age. Of the living patients, 1 remains on ventilatory support at the age of 5 years, 6 months. The remaining patients range in current age from 2 years, 4 months to 22 years. Only patient 1 has a mildly affected parent, which was detected in the setting of the evaluation of the much more affected child. Creatine kinase levels were all within normal range.
Table 1.
Review of genetic features
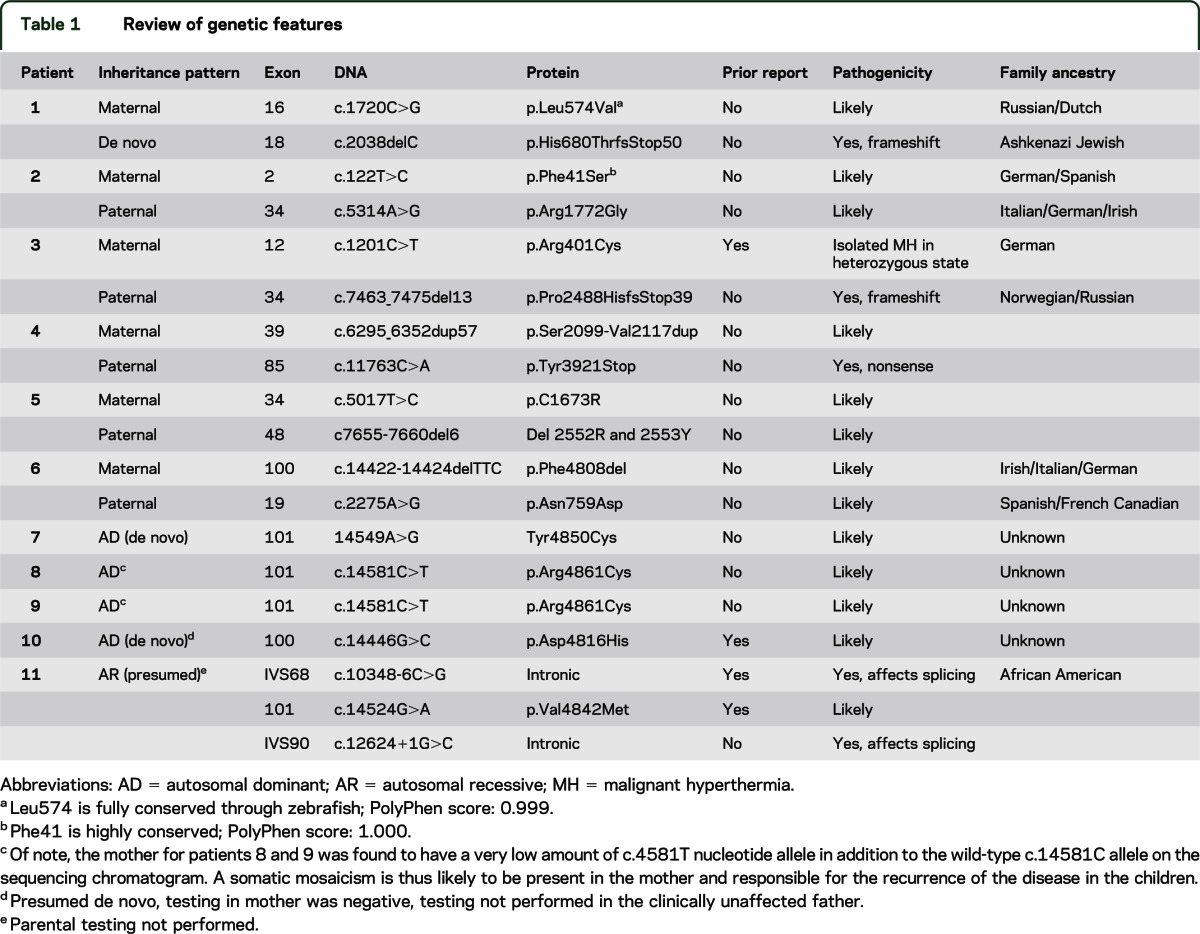
Figure 1. Clinical features.
(A and B) Myopathic facies, ophthalmoplegia, and severe respiratory involvement in patient 5. (C) Mild myopathic facies and feeding difficulties requiring nasogastric tube in patient 3. (D) Cleft palate in patient 1. Arthrogryposis is seen with characteristic extended fingers and adducted thumb in patient 3 (E), patient 1 (F), and patient 6 (J). Early-onset scoliosis is seen in patient 1 (G) and in patient 2 (H and I). (K) Muscle ultrasound in patient 4 showing involvement of the vastus lateralis (VL), intermedius (VI), and medialis (VM) with a dense, mostly granular pattern, with relative sparing of the rectus femoris (RF).
Noteworthy neonatal findings included a cleft palate in patient 1 (figure 1D), congenital rigid spine in patient 10, and near complete ophthalmoparesis in patients 4, 5, and 11.
All patients had delayed motor milestones. Only 2 patients achieved independent ambulation. There was no evidence of intellectual impairment in any of the patients. Patient 11, the oldest patient in this cohort, never achieved the ability to ambulate, but retains the ability to sit independently.
Genetic analysis.
Dominantly acting mutations were determined in 4 patients. Six patients were compound heterozygous for RYR1 gene mutations (table 1), including compound heterozygosity for a null allele and a missense mutation (patients 1, 3, and 4), for an in-frame deletion and a missense mutation (patients 5 and 6), for 2 missense changes (patient 2), and for 3 mutations: a previously reported missense mutation and splice-site mutation, plus a novel splice-site mutation (patient 11). Patient 1 had 1 maternally inherited missense change and a de novo frameshift mutation on the other allele. The mother was affected by minimal proximal weakness, but had ultrasound findings compatible with RYR1-associated myopathy (i.e., myopathic changes in the quadriceps with relative sparing of the rectus femoris).
Muscle imaging.
In the 6 patients with muscle ultrasound, a consistent finding of relative sparing of involvement of the rectus femoris muscle in comparison to the other components of the quadriceps was seen (patients 1, 2, 3, 4, 7, and 10; figure 1K). Patients 1, 4, and 10 showed equal involvement of the tibialis anterior, gastrocnemius, and soleus, whereas patient 2 showed striking involvement of the gastrocnemius muscle with relative sparing of the soleus and tibialis anterior. Imaging in the upper extremities demonstrated more variability without uniform features. In patients 2 and 10, paraspinal muscles were also affected. No fasciculations were seen in any patients.
Pathology.
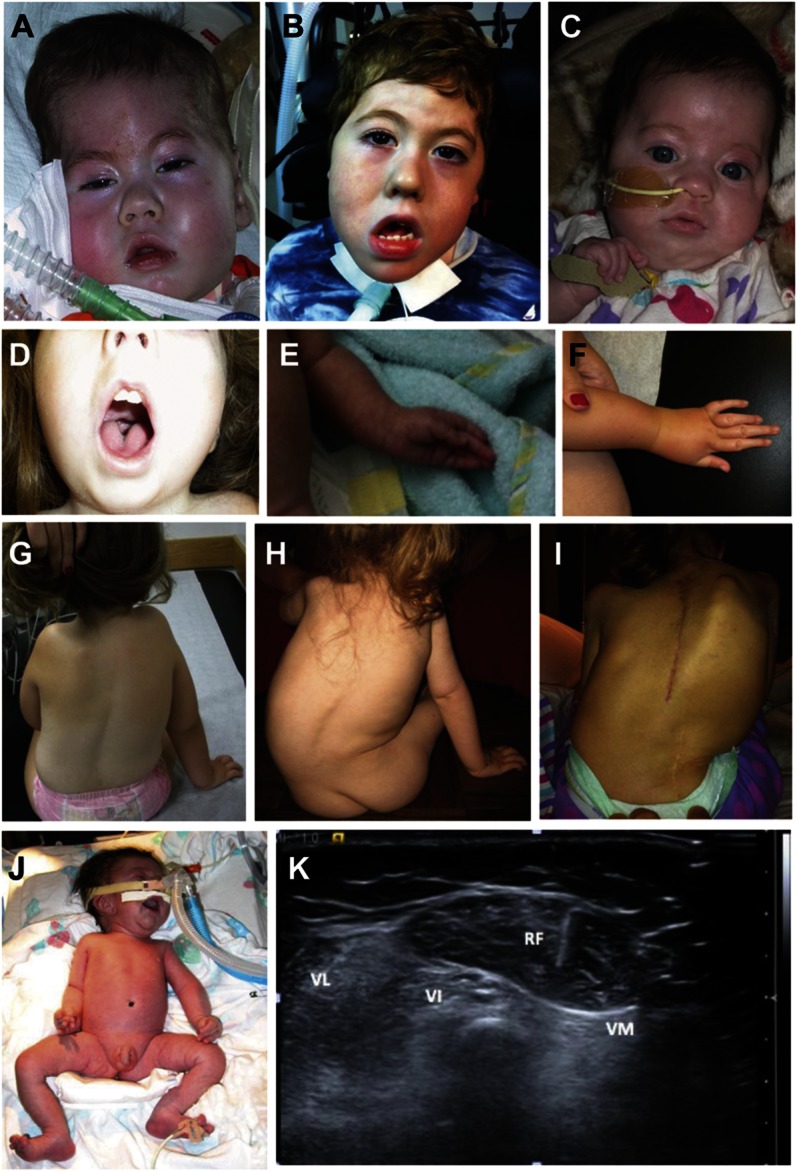
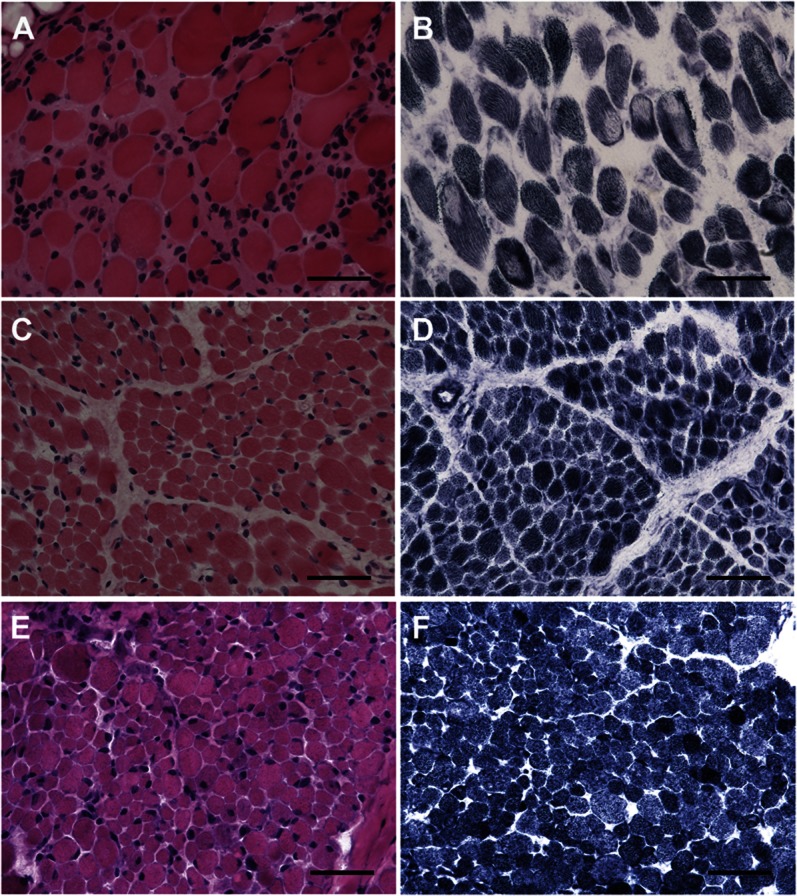
Classic cores on NADH-TR, SDH, and COX stains were seen in the majority of dominant cases (patients 7, 8, and 10), but in only 1 of the recessive cases (patient 6, figure 2B). Multi-minicores were seen in patient 9. The recessive cases had extremely diverse pathology (figures 2 and 3; table e-1 on the Neurology® Web site at www.neurology.org). Patient 3 was dominated by fibrosis, adipose tissue infiltration, strikingly small size of fibers, and occasional fibers with ill-defined cores on NADH-TR staining (figure 3, E and F). Very small fibers were also seen in patients 2 and 6. The very small fibers were predominantly fast fibers, and were largely positive for neonatal and developmental myosin, and less so for N-CAM (table e-1, figure e-1). Patients 2 and 11 appeared reminiscent of a dystrophic myopathy, except for the absence of prominent degenerating and clearly regenerating fibers (figure 3C). Fibers in patient 4 exhibited frequent internally placed nuclei with irregularities on NADH, including ill-defined cores and unevenness of stain (figure 3, A and B). Patients 1 and 5 demonstrated only mild fibrosis, and nonspecific myopathic changes without cores were seen with oxidative enzyme stains (figure 2, C–F). A striking predominance of type 1 staining fibers with the associated properties of slow fibers was seen in patients 1, 3, 4, 6, 7, 10, and 11 in both dominant and recessive cases (not shown). If cores were not seen with oxidative enzyme stains, there was also no evidence of core equivalents on electron microscopy.
Figure 2. Histologic features.
(A) Skeletal muscle of patient 6 demonstrates a large majority of rounded fibers with severe variation in size and occasional fibers with internally placed nuclei. (C) Patient 1 demonstrates mild increase in connective tissue and significant variation in fiber size, but no internally placed nuclei. (E) Patient 5, similarly to patient 1, demonstrates variation in fiber size and shape, but not significant fibrosis; in addition, scattered fibers have internally placed nuclei. Whereas oxidative enzyme stain (NADH-TR) reveals no cores in patients 1 (D) and 5 (F), there are numerous, well-defined cores centrally and peripherally located present in patient 6 (B). A, C, E: hematoxylin & eosin; B, D, F: NADH-TR; A–F calibration bar 50 µm. NADH-TR = nicotinamide adenine dinucleotide dehydrogenase–tetrazolium reductase.
Figure 3. Histologic features.
(A) Mild fibrosis and adipose tissue infiltration with moderate variation in size and shape but scattered fibers with internally placed nuclei are seen in patient 4. (C) Patient 2 demonstrates rounded fibers, most of which are surrounded by excess connective tissue, occasional dark, hypercontracted-appearing fibers, and severe variation in size with clusters of small fibers. (E) In patient 3, there is pronounced increase in adipose and connective tissue with numerous minute fibers and rounded hypertrophic fibers. Oxidative enzyme stain (NADH-TR) reveals scattered fibers with ill-defined cores in patients 2 (D) and 3 (F) and unevenness of stain with ill-defined cores in patient 4 (B). A, C, E: hematoxylin & eosin; B, D, F: NADH-TR; C, D, F calibration bar 100 μm; E, A, B calibration bar 200 μm. NADH-TR = nicotinamide adenine dinucleotide dehydrogenase–tetrazolium reductase.
DISCUSSION
With the availability of complete genetic analysis of the RYR1 gene, the phenotypic spectrum of RYR1-related myopathy is evolving to include patients with a more severe neonatal onset.8,12 Our series expands the striking heterogeneity in clinical, genetic, and histologic features at the severe end of RYR1-associated myopathy. Histologically, compared with the dominant cases, the recessive cases showed the greatest histopathologic variability to include findings suggestive of RYR1-associated myopathy such as central cores, multi-minicores,15 or more subtle unevenness of stains, but also central nuclei, extensive fatty and connective tissue infiltration in the absence of prominent degeneration, and regeneration. Marked type 1 fiber predominance approaching type 1 uniformity was common. The occurrence of extremely small fibers was striking in 3 of our recessive cases. The small fibers are conspicuous for their expression of neonatal and developmental myosin. These small fibers are unlikely regenerating fibers following a necrotic or degenerating process, as there was little evidence for muscle fiber degeneration as a trigger for regeneration. Rather, this type of expression may reflect persistence or recapitulation of a developmental program, but it is not possible to determine with certainty whether the fibers represent a primary growth retardation or secondary atrophy. It is possible that the extremely small fibers lack individual neuromuscular junctions, leading to expression of N-CAM. Clinical severity may not correlate with the degree of histologic change. One very severe patient with complete ophthalmoplegia and respiratory insufficiency from birth had a largely nonspecific myopathic-appearing biopsy with very occasional fibers with centrally placed nuclei and no irregularities on the oxidative enzyme stains (patient 5). Thus, the absence of cores should not preclude consideration of an RYR1-associated myopathy, as age at biopsy and site of biopsy may influence the detection of classic cores.
We find that the degree of clinical variability in patients with a neonatal-onset RYR1-associated myopathy appears to be even greater than previously described. Findings such as decreased fetal movement, polyhydramnios, hypotonia, arthrogryposis, respiratory insufficiency, and feeding difficulties were seen in nearly all patients in this series, and are consistent with previously reported cases of the severe neonatal phenotype.9,16 Patient 10 with a de novo dominant mutation had clinically impressive spinal rigidity of neonatal onset. In addition, early-onset kyphoscoliosis as well as a rapid and early progression of scoliosis was seen in 4 patients, 1 of whom demonstrated paraspinal muscle involvement on muscle ultrasound. Cleft palate as seen in patient 1 has also not previously been described in patients with RYR1 mutations, but has been reported in the context of other severe congenital disorders of muscle.17 Ophthalmoplegia in association with RYR1 myopathy has been associated with histologic findings of centronuclear or multi-minicore myopathy12,15,18–20; it was present in 3 of the 7 patients with recessive RYR1 mutations including in patient 5 without these typical histologic findings. Notably, not all severe recessive patients in our series had ophthalmoplegia. Despite this great clinical variability, we find the consistent sparing of the rectus femoris on muscle ultrasound imaging a very useful clinical sign to suggest an RYR1-related myopathy as a possibility.
The molecular mechanisms by which dominant vs recessive mutations lead to ryanodine receptor dysfunction and myopathy are not completely clear. Dominant mutations associated with CCD are clustered in the C-terminal portion of RYR1 and likely result in functional abnormalities of the intracellular calcium-release channel.5 In contrast, a clear clustering of recessive RYR1 mutations is not yet clearly apparent. Recessive patients were typically compound heterozygous for a null allele in conjunction with a missense mutation or 2 frame-preserving mutations (missense mutations and/or in-frame deletions). Some but not all of these missense mutations are in the N-terminal portion of the RYR1 gene classically thought to be associated with the malignant hyperthermia phenotype, with obvious implications for counseling of clinically unaffected family members carrying the potential malignant hyperthermia predisposition allele. Interestingly, patient 1 was a compound heterozygote for a frameshifting c.2038delC deletion and a maternally inherited missense p.Leu574Val change. The carrier mother was mildly affected, suggesting that p.Leu574Val acts in a dominant fashion on its own, but occurring in conjunction with the second pathogenic allele would lead to a much more severe phenotype as observed in patient 1. Of interest among the dominant mutations were patients 8 and 9 (siblings) who inherited the mutation from their mother who was found to have a low degree of somatic mosaicism.16,21–24
With the rising awareness of the multifaceted clinical and histologic spectrum of RYR1-associated myopathy and the more common access to full molecular genetic analysis of the gene, the RYR1 spectrum will likely expand even further.
Supplementary Material
GLOSSARY
- COX
cytochrome oxidase
- NADH
nicotinamide adenine dinucleotide dehydrogenase
- N-CAM
neural cell adhesion molecule
- RYR1
ryanodine receptor 1
- SDH
succinic dehydrogenase
- TR
tetrazolium reductase
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
D.X. Bharucha-Goebel: study design, data acquisition. M. Santi: data analysis and interpretation, revision of the manuscript. L. Medne: data analysis and interpretation, revision of the manuscript. K. Zukosky: acquisition of data. J. Dastgir: acquisition of data (imaging). P.B. Shieh: acquisition of data. T. Winder: data analysis and interpretation. G. Tennekoon: acquisition of data. R.S. Finkel: acquisition of data. J.J. Dowling: critical revision of the manuscript. N. Monnier: data analysis and interpretation. C.G. Bönnemann: study design, study supervision, data acquisition, revision of the manuscript.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
D.X. Bharucha-Goebel, M. Santi, L. Medne, K. Zukosky, and J. Dastgir report no disclosures. P.B. Shieh is on the speaker's bureau for Grifols and has also served on the Neurology Advisory Board for Grifols. T. Winder is employed by the clinical laboratory–Prevention Genetics. G. Tennekoon reports no disclosures. R.S. Finkel serves on an advisory board for PTC Therapeutics, DuchenneConnect, Families of SMA, TREAT-NMD, and AVI (DSMB); consults with Isis Pharmaceuticals; receives commercial research support from PTC Therapeutics, Genzyme Corp., and Santhera Pharmaceuticals; receives government funding grants U54AR0526446-03, 1U54NS065712-01, 5R21NS058926-02, 5U54AR052646-03, R01NS043264, NHHSN265200423611C, and RO1AR056973; and receives research support from the Spinal Muscular Atrophy Foundation, Families of Spinal Muscular Atrophy, Foundation for the Eradication of Duchenne, Muscular Dystrophy Association, and Charcot-Marie-Tooth Association. His spouse (Dr. T.H. Finkel) receives commercial research support from Merck Pharmaceuticals; receives government funding grants (1) RC1AR058606, PI, 09/30/2009–11; (2) 1R21 AI078387, PI, 07/01/2008–10; (3) 1R21 AI078387-S1, PI, 06/05/09–10/31/09; (4) 1R41 A1071927, PI, 8/15/2006–10; (5) R01 A1063623, Co-I, 12/1/2004–09; (6) 1U19AIO82726, Co-I, 5/8/2009–14; (7) NIH, Program Project Core Director, 10% effort, 07/01/99–06/30/14 “Mechanism of Autoreactivity in SLE”; (8) R01AI063623 Co-I, 7.5% effort, 12/01/04–11/30/09 “High-throughput Selection for HIV-1 Resistant Viruses”; (9) NIH1U19AI082726 Program Project Co-I, 05/08/09–04/30/14 “Regulation of Type I IFNs in Tolerance and Autoimmunity”; (10) NIH R01 Co-I, 5% effort, 04/01/09–03/31/13 “Role of Small G Proteins in TCR-Cytoskeleton Interaction”; and (11) NIH T32 training grant Project Leader, 07/01/09–06/30/12; and receives licensing fee payments for technology or inventions from Southern Biotechnology Associates, Upstate Pharmaceuticals, and Santa Cruz Biotechnology. J.J. Dowling, N. Monnier, and C.G. Bönnemann report no disclosures. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Jungbluth H. Central core disease. Orphanet J Rare Dis 2007;2:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sewry CA, Müller C, Davis M, et al. The spectrum of pathology in central core disease. Neuromuscul Disord 2002;12:930–938 [DOI] [PubMed] [Google Scholar]
- 3.Jungbluth H, Müller CR, Halliger-Keller B, et al. Autosomal recessive inheritance of RYR1 mutations in a congenital myopathy with cores. Neurology 2002;59:284–287 [DOI] [PubMed] [Google Scholar]
- 4.Jungbluth H, Sewry CA, Muntoni F. What’s new in neuromuscular disorders? The congenital myopathies. Eur J Paediatr Neurol 2003;7:23–30 [DOI] [PubMed] [Google Scholar]
- 5.Treves S, Anderson AA, Ducreux S, et al. Ryanodine receptor 1 mutations, dysregulation of calcium homeostasis and neuromuscular disorders. Neuromuscul Disord 2005;15:577–587 [DOI] [PubMed] [Google Scholar]
- 6.Rossi D, DeSmet P, Lyfenko A, et al. A truncation in the RYR1 gene associated with central core lesions in skeletal muscle fibres. J Med Genet 2007;44:e67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goebel HH. Congenital myopathies in the new millennium. J Child Neurol 2005;20:94–101 [DOI] [PubMed] [Google Scholar]
- 8.Hernandez-Lain A, Husson I, Monnier N, et al. De novo RYR1 heterozygous mutation (I4898T) causing lethal core-rod myopathy in twins. Eur J Med Genet 2011;54:29–33 [DOI] [PubMed] [Google Scholar]
- 9.Romero NB, Monnier N, Viollet L, et al. Dominant and recessive central core disease associated with RYR1 mutations and fetal akinesia. Brain 2003;126:2341–2349 [DOI] [PubMed] [Google Scholar]
- 10.Wilmshurst JM, Lillis S, Zhou H, et al. RYR1 mutations are a common cause of congenital myopathies with central nuclei. Ann Neurol 2010;68:717–726 [DOI] [PubMed] [Google Scholar]
- 11.Bevilacqua JA, Monnier N, Bitoun M, et al. Recessive RYR1 mutations cause unusual congenital myopathy with prominent nuclear internalization and large areas of myofibrillar disorganization. Neuropathol Appl Neurobiol 2011;37:271–284 [DOI] [PubMed] [Google Scholar]
- 12.Klein A, Lillis S, Munteanu I, et al. Clinical and genetic findings in a large cohort of patients with ryanodine receptor 1 gene–associated myopathies. Hum Mutat 2012;33:981–988 [DOI] [PubMed] [Google Scholar]
- 13.Monnier N, Laquerrière A, Marret S, et al. First genomic rearrangement of the RYR1 gene associated with an atypical presentation of lethal neonatal hypotonia. Neuromuscul Disord 2009;19:680–684 [DOI] [PubMed] [Google Scholar]
- 14.Dubowitz V, Sewry CA. Muscle Biopsy: A Practical Approach, 3rd ed Philadelphia: Saunders Elsevier; 2007 [Google Scholar]
- 15.Jungbluth H, Zhou H, Hartley L, et al. Minicore myopathy with ophthalmoplegia caused by mutations in the ryanodine receptor type 1 gene. Neurology 2005;65:1930–1935 [DOI] [PubMed] [Google Scholar]
- 16.Monnier N, Marty I, Fauré J, et al. Null mutations causing depletion of the type 1 ryanodine receptor (RYR1) are commonly associated with recessive structural congenital myopathies with cores. Hum Mutat 2008;29:670–678 [DOI] [PubMed] [Google Scholar]
- 17.Vasjar J, Baskin B, Swoboda K, et al. Walker-Warburg Syndrome with POMT1 mutations can be associated with cleft lip and cleft palate. Neuromuscul Disord 2008;18:675–677 [DOI] [PubMed] [Google Scholar]
- 18.Jungbluth H, Zhou H, Sewry CA, et al. Centronuclear myopathy due to a de novo dominant mutation in the skeletal muscle ryanodine receptor (RYR1) gene. Neuromuscul Disord 2007;17:338–345 [DOI] [PubMed] [Google Scholar]
- 19.Romero NB. Centronuclear myopathies: a widening concept. Neuromuscul Disord 2010;20:223–228 [DOI] [PubMed] [Google Scholar]
- 20.Jungbluth H, Sewry CA, Muntoni F. Core myopathies. Semin Pediatr Neurol 2011;18:239–249 [DOI] [PubMed] [Google Scholar]
- 21.Robinson R, Carpenter D, Shaw MA, et al. Mutations in RYR1 in malignant hyperthermia and central core disease. Hum Mutat 2006;10:977–989 [DOI] [PubMed] [Google Scholar]
- 22.Tchernitchko D, Goossens M, Wajcman H. In silico prediction of the deleterious effect of a mutation: proceed with caution in clinical genetics. Clin Chem 2004;11:1974–1978 [DOI] [PubMed] [Google Scholar]
- 23.Davis M, Brown R, Dickson A, et al. Malignant hyperthermia associated with exercise-induced rhabdomyolysis or congenital abnormalities and a novel RYR1 mutation in New Zealand and Australian pedigrees. Br J Anaesth 2002;88:508–515 [DOI] [PubMed] [Google Scholar]
- 24.Wu S, Ibarra MC, Malicdan MC, et al. Central core disease is due to RYR1 mutations in more than 90% of patients. Brain 2006;129:1470–1480 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.